Introduction
Acute cerebral infarction (ACI) is a common disease
with high morbidity, disability rate and mortality worldwide, as
well as in China (1). It affects
the quality of life of patients and adds to the economic burden on
the society and the patient's family (1). ACI is a disease with etiological
heterogeneity; therefore, extensively investigating blood
biomarkers reflecting the ACI pathophysiological changes has great
clinical significance and application value (2). These biomarkers can be used for
early diagnosis, etiology recognition and selection of early
clinical individualized treatment (2). In addition, they can predict disease
prognosis, assisting in the search for novel targets for disease
intervention (2,3).
MicroRNAs (miRNAs or miRs) are a class of small
endogenous non-coding single-stranded RNA molecules and one of the
primary regulatory factors of gene expression (4). miRNA interacts with the specific
sequence of its target gene to suppress the target gene activity or
promote its degradation following transcription (5). Furthermore, it regulates target gene
expression and participates in various biological processes,
including cell proliferation, differentiation, apoptosis and
metabolism. A brain-specific miRNA is abundant in the central
nervous system (6); however, it
is not expressed or is rarely expressed in other organs, and serves
a vital role in neurogenesis and function (6). It is estimated that miRNAs may be
passively leaked from broken tissue and cells or invasive cells,
similar to other substances. Alternatively, they may be actively
secreted into the blood circulation from the damaged tissue and
cells (6). Previous animal and
clinical research reported that miRNA expression profiles in the
brain tissue and blood are aberrantly altered following an ischemic
stroke (3). Such findings
indicate that stroke-associated miRNAs are involved in the
pathophysiological process occurring subsequent to cerebral
ischemia (7).
The transforming growth factor-β (TGF-β)/Smad
signaling transduction pathway is involved in the genesis and
development of Alzheimer's disease, hydrocephalus and neuroglioma
(8). It also participates in the
pathological process of rat cerebral ischemia reperfusion injury
(8). The TGF-β level has been
reported to be markedly higher in patients with ACI (8). This suggests that TGF-β participates
in the pathogenesis of ACI and is an important reference molecule
for diagnosing ACI recurrence (8). Smad proteins also participate in
multiple pathological processes, such as cerebral injury. Research
has been conducted to analyze cortical neuron morphological changes
caused by experimental communicating hydrocephalus, as well as the
effect of the TGF-β/Smad signaling pathway on these changes
(8). The results indicate that
abnormal Smad protein expression is involved in the neuron damage
process occurring in hydrocephalus (8). Therefore, the present study aimed to
examine the function of miR-323 in ACI and its underlying
mechanism.
Materials and methods
Animals and ACI model establishment
Male adult Sprague-Dawley rats (weight, 220-250 g;
n=12) were purchased from Beijing Vital River Laboratory Animal
Technology Co., Ltd. (Beijing, China). All rats were housed at
22-23°C at 55% humidity and a 12/12 h light/dark cycle. Food and
water were available ad libitum. The rats were randomly
divided into the sham surgery group (serving as the control) and
the ACI model group (n=6 rats per group). The animals were
anesthetized by pentobarbital sodium (30 ml/kg; intraperitoneal
injection), and then a surgical midline incision was made to expose
the internal carotid artery, external carotid artery and right
common carotid artery. A nylon suture was inserted into the right
common carotid artery and injected at 18 mm into the internal
carotid artery. After 2 h of the injection, the nylon sutures were
gently removed from the internal carotid artery and reperfusion was
performed for 1 h. Rats in the control group received the same
surgical procedures without the insertion of the monofilament nylon
suture. After 24 h, the animals were anesthetized by pentobarbital
sodium (30 ml/kg; intraperitoneal injection), sacrificed and
hippocampal tissues were extracted. The animal experiments were
approved by the Ethics Committee of the Beijing Luhe Hospital
Capital Medical University (Beijing, China).
H&E staining
Hippocampal tissues were fixed with 4%
para-formaldehyde for 24 h at room temperature. Following this,
tissues were dehydrated using ethanol, embedded in paraffin and
then cut into sections (10 µm). Sections were then stained
with H&E for 10 min at room temperature and observed using a
light microscope (magnification, ×100; Leica Microsystems GmbH,
Wetzlar, Germany) and analyzed using Image Lab 3.0 (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Gene microarray
Total protein was extracted using a
radio-immunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Nanjing, China) and then protein concentration was
determined using a bicinchoninic acid assay (Beyotime Institute of
Biotechnology). Total RNA (500 ng) obtained from hippocampal
tissues or transfected cells was hybridized to the SurePrint G3
Mouse Whole Genome GE 8 X60 K Microarray G4852A platform
(G4471A-021828; Agilent Technologies, Inc., Santa Clara, CA, USA),
to analyze signaling proteins, including IL-1β, IL-6 and PI3K.
Images were quantified using Agilent Feature Extraction software
(version A.10.7.3.1; Agilent Technologies, Inc.).
Cell culture and miRNA transfection
PC12 cells were purchased from the Institute of
Biochemistry and Cell Biology (Shanghai, China). The cells were
incubated in Dulbecco's modified Eagle's medium (DMEM; Hyclone; GE
Healthcare Life Sciences, Beijing, China) with 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 2
mmol/l glutamine (Invitrogen; Thermo Fisher Scientific, Inc.), 200
U/ml penicillin (Hyclone; GE Healthcare Life Sciences) and 100
µg/ml streptomycin (Hyclone; GE Healthcare Life Sciences) at
37°C with 5% CO2.
miR-323, anti-miR-323 and control mimics were
purchased from RiboBio Co., Ltd. (Guangzhou, China), and their
sequences were as follows: miR-323, 5'-AUG UAA UGA GAC GGU UCU UUU
UU-3'; anti-miR-323, 5'-AAA AAA GAA CCG UCU CAU UAC AU-3'; and
control, 5'-AAU AGC AUC GAA AAG UCC GG-3'. A total of 100 nM mimics
were transfected into the cells using Invitrogen
Lipofectamine® 3000 (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. The medium was changed
after 4 h of transfection in 5% CO2 incubator at 37°C,
and the cells were then incubated for 24, 48 and 72 h time
intervals. Following this, fresh DMEM was added into the cell
culture without glucose under hypoxic conditions (5% CO2
and 95% N2) and incubated for 6 h. Following
transfection at 37°C, SMAD3 inhibitor (1 µM SIS3;
MedChemExpress, Monmouth Junction, NJ, USA) or TGF-β1 inhibitor (20
nm, LY2109761) was added to the cells and subsequently incubated
for 48 h without glucose under hypoxic conditions (5%
CO2 and 95% N2).
Reverse transcription-quantitative polymerase
chain reaction (RT-qPCR). Total RNA samples were extracted from
the transfected PC12 cells using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). RNA concentrations were determined using
a microplate reader (Benchmark Plus; Bio-Rad Laboratories, Inc.) at
a wavelength of 260/280 nm. For the miRNA expression assay, RT was
performed using a TaqMan miRNA assay kit (Applied Biosystems;
Thermo Fisher Scientific, Inc.). Next, the PCR reaction was
performed using a Bio-Rad iCycler iQ RealTime PCR detection system
(Bio-Rad Laboratories, Inc.) and a SYBR® Premix Ex Taq™
II kit (Takara Biotechnology Co., Ltd., Dalian, China). The thermal
cycling conditions included denaturation at 95°C for 15 min,
followed by 40 cycles of 95°C for 30 sec and 60°C for 15 sec. The
following primers were used in qPCR: miR-323 sense, 5'-GCG CCC CAG
GAG GCT GAT GC-3', and antisense, 5'-CGT GGT GGT CCC GCC GCC 3-3';
and U6 sense, 5'-GTT TTG TAG TTT TTG GAG TTA GTG TTG TGT-3' and
antisense, 5'-CTC AAC CTA CAA TCA AAA ACA ACA CAA ACA-3'. miR-323
expression was normalized using the 2−ΔΔCq method
(8).
Dual-luciferase reporter assay
The 3'-untranslated region (3'-UTR) of SMAD3 mRNA
containing the predicted miR-323 binding sequences was amplified
and subcloned into a pGL3 luciferase promoter vector (Invitrogen;
Thermo Fisher Scientific, Inc.). pGL3 was then co-transfected with
AAV-pre-miR-323 or controls into PC12 cells. Following incubation
for 48 h, the luciferase report activity was detected using the
Dual-Luciferase Reporter Assay kit (Promega Corporation, Madison,
WI, USA) according to the manufacturer's protocol.
Cell viability assay
The viability of each cell group at 24, 48 and 72 h
post-transfection was measured using an MTT assay at 96 well plates
(1×103 cells/well). Briefly, 20 µl MTT solution
was added into the cells and incubated at 37°C for 4 h. Dimethyl
sulfoxide was then added for 20 min at 37°C, and the absorbance was
measured using a microplate reader (Benchmark Plus; Bio-Rad
Laboratories, Inc.) at a wavelength of 490 nm.
Lactate dehydrogenase (LDH) activity
A total of 48 h post-transfection, the LDH activity
of each cell group was measured using an LDH activity kit (Beyotime
Institute of Biotechnology), according to the manufacturer's
protocol. The absorbance was measured using a microplate reader
(Benchmark Plus) at a wavelength of 450 nm.
Flow cytometry
A total of 48 h post-transfection, the apoptosis
rate of cells was measured using an FITC Annexin V kit (cat. no.
556420; BD Biosciences, San Jose, CA, USA). Briefly, cells were
harvested, washed three times with PBS and resuspended with 150
µl binding buffer. Next, cells were stained with 5 µl
FITC-conjugated Annexin V and 5 µl PI for 15 min in the
dark. The apoptosis rate was assessed by flow cytometry with a
FACSCalibur and the CellQuest software (BD Biosciences).
Caspase-3/9 activity assay
Caspase-3 and caspase-9 activities were measured
using the Caspase 3 Activity Assay kit (cat. no. C1115; Beyotime
Institute of Biotechnology) or Caspase 9 Activity Assay kit (cat.
no. C1158; Beyotime Institute of Biotechnology), according to the
manufacturer's protocol. The absorbance was then measured using a
microplate reader (Benchmark Plus) at a wavelength of 405 nm.
Western blot analysis
Total protein in the cells was extracted using a
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology) and protein concentration was determined using a
bicinchoninic acid assay (Beyotime Institute of Biotechnology).
Protein (50 µg) was then separated by 10% SDS-PAGE and
blotted onto a pre-wet nitrocellulose membrane (GE Healthcare Life
Sciences). Subsequent to blocking with 5% skimmed milk in TBST for
1 h at 37°C, the membranes were immunoblotted with the following
primary antibodies at 4°C overnight: SMAD2 (1:1,000; sc-8332),
TGF-β1 (1:1,000; sc-9043), SMAD3 (1:1,000; sc-8332), B-cell
lymphoma 2-associated X protein (Bax; 1:1,000; sc-6236) and GAPDH
(1:5,000; sc-25778; all purchased from Santa Cruz Biotechnology,
Inc., Dallas, TX, USA). The membranes were then washed with TBST
for 15 min at and immunoblotted with a secondary antibody
conjugated to horseradish peroxidase (1:5,000; sc-2004 and sc-2005;
Santa Cruz Biotechnology, Inc.) for 1 h at 37°C. Immunolabeling was
visualized using an enhanced chemiluminescence system (GE
Healthcare Life Sciences) and analyzed using Image Lab 3.0 software
(Bio-Rad Laboratories, Inc.).
Immunocytochemistry
Cell was washed with PBS and then fixed with 4%
paraformaldehyde for 20 min at room temperature. Cells were then
blocked with 5% bovine serum albumin (Nanjing Sunshine
Biotechnology Co., Ltd., Nanjing, China) and 0.1% Tris-X100 in
Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h at
room temperature. Following this, cells were incubated with SMAD2/3
antibodies (1:100; sc-8332; Santa Cruz Biotechnology, Inc.) at 4°C
overnight and then washed with TBST for 15 min at room temperature.
Cells were subsequently incubated with goat anti-rabbit
IgG-CruzFluor™ 555 (1:100; sc-362272; Santa Cruz Biotechnology,
Inc.) for 1 h at 37°C and then washed with TBST for 15 min at room
temperature. Cell was then stained with 10 µg/ml of
4',6-diamidino-2-phenylindole for 15 min in darkness at room
temperature. Cells were subsequently analyzed at room temperature
using a Zeiss Axioplan 2 confocal microscope (magnification, ×200;
Carl Zeiss AG, Oberkochen, Germany) and analyzed using Image Lab
3.0 (Bio-Rad Laboratories, Inc.).
Statistical analysis
Data are presented as the mean ± standard deviation
(n=3) of at least three independent experiments using SPSS 17.0
(SPSS, Inc., Chicago, IL, USA). The difference between two
independent groups was assessed using Student's t-test. The
difference between three independent groups was assessed using
one-way analysis of variance and Tukey's post hoc test. P<0.05
was considered to indicate a statistically significant
difference.
Results
miR-323 expression in rats with cerebral
infarction
In the present study, cerebral infarction was
established in rat hippocampal neurons, followed by analysis of
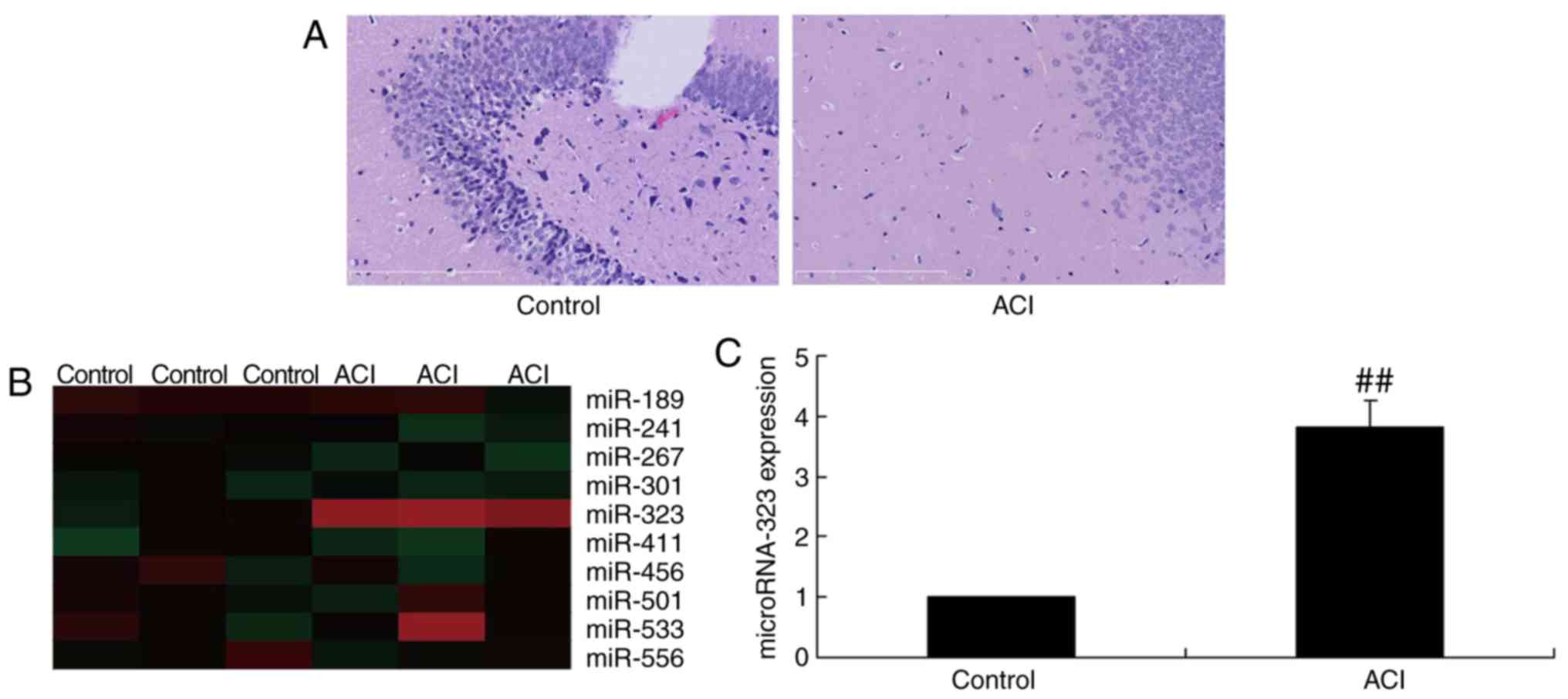
miR-323 expression. As shown in Fig.
1A, a higher apoptotic rate was observed in the nerve cells of
the ACI model group, as compared with that of the control group. As
displayed in Fig. 1B and C,
miR-323 expression was significantly upregulated in rats with
cerebral infarction, compared with that in the sham-control group.
These data suggested that miR-323 may participate in the nerve cell
apoptosis in ACI.
miR-323 regulates nerve cell toxicity in
the in vitro cell model
The function of miR-323 on nerve cell toxicity was
investigated in the in vivo model of cerebral infarction in
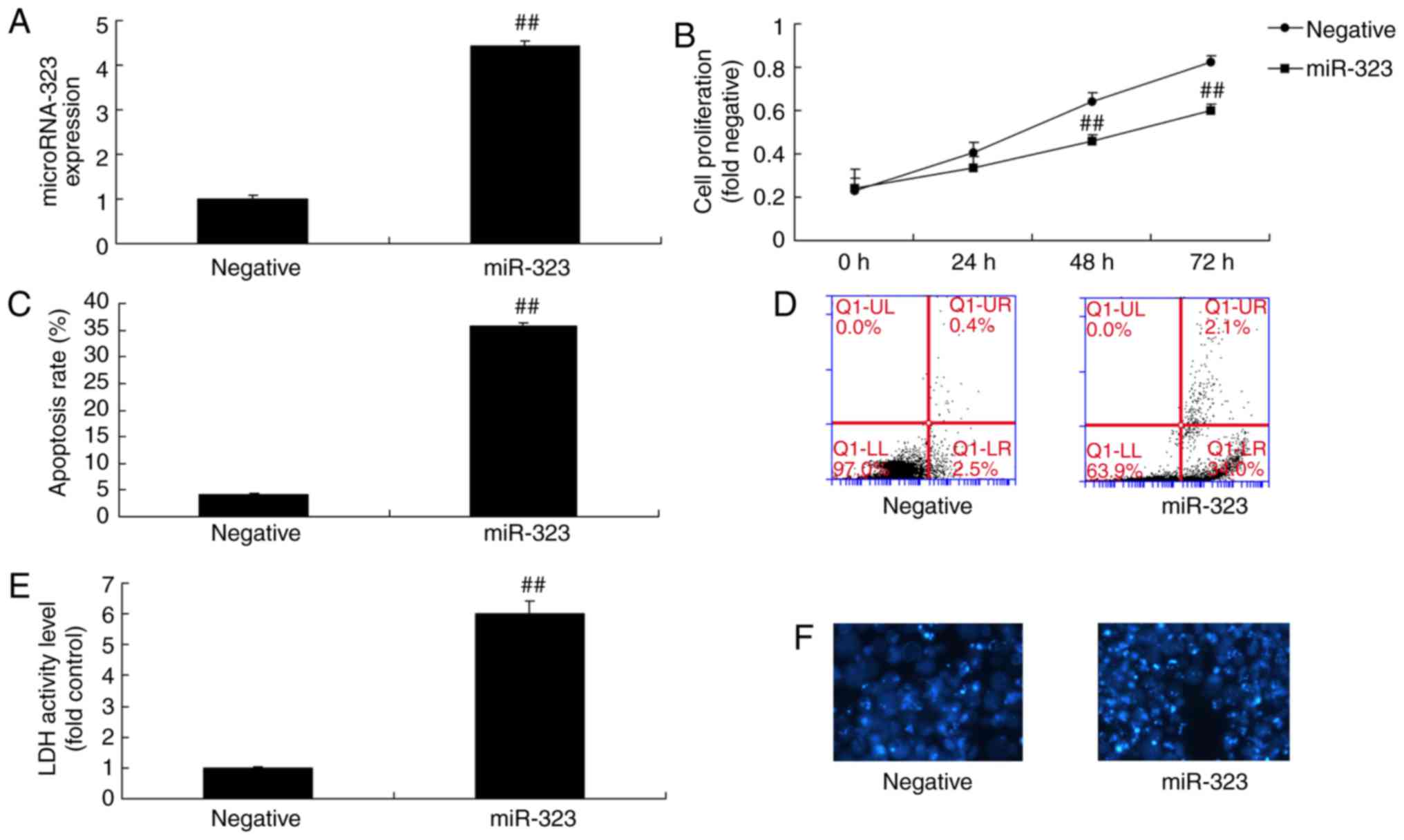
the present study. As shown in Fig.
2A, the expression level of miR-323 was significantly increased
in the in vivo model following transfection with miR-323
mimic for 24 h, compared with that in the control mimics group.
Next, it was observed that overexpression of miR-323 significantly
inhibited cell viability in the in vitro model and increased
the LDH activity level, as compared with the negative-control
mimics group (Fig. 2B-F). The
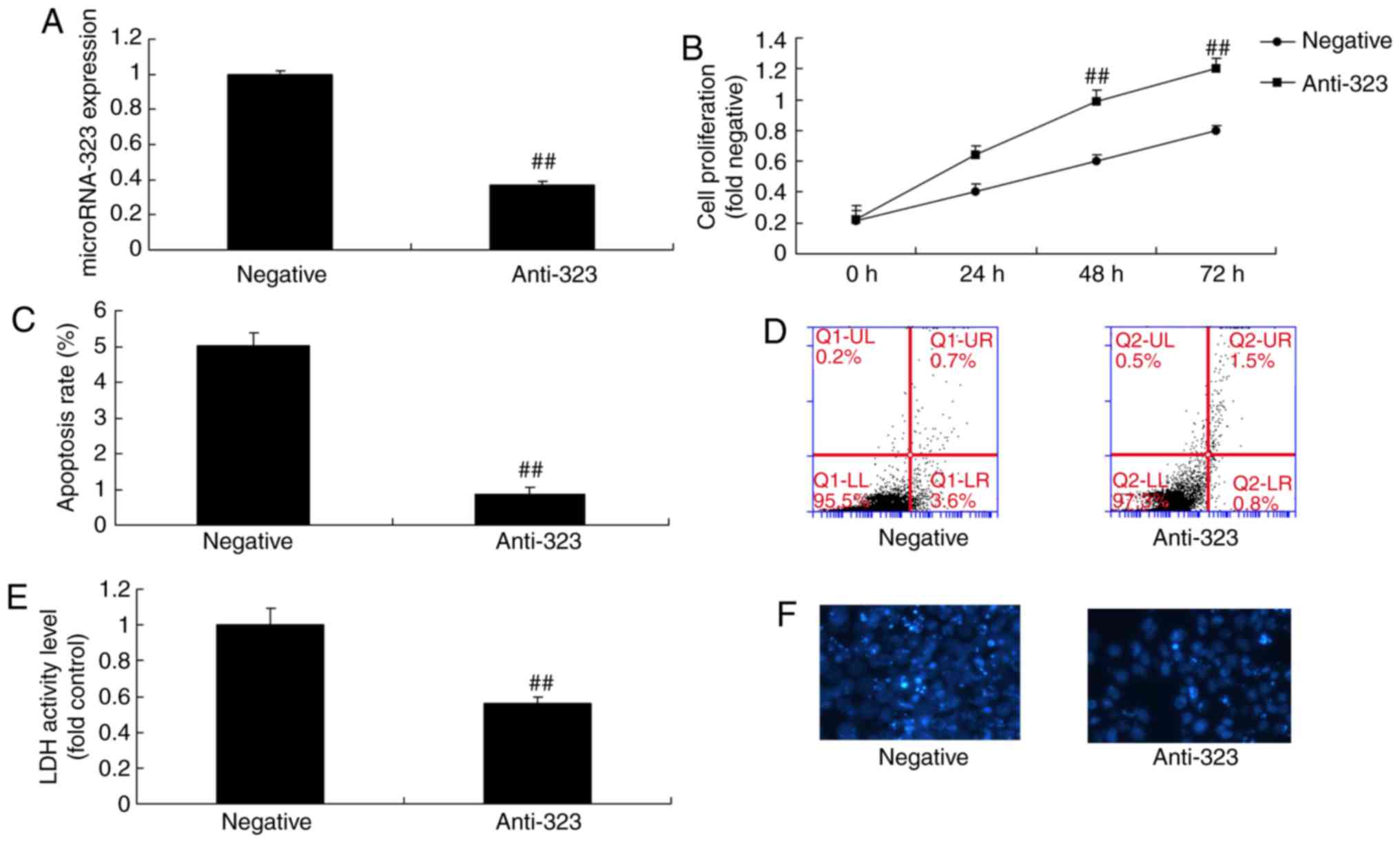
expression level of miR-323 was then significantly decreased in the
in vitro model by transfection with anti-miR-323 for 24 h,
compared with that in the group transfected with negative-control
mimics (Fig. 3A). Downregulation
of miR-323 significantly promoted cell viability and inhibited the
LDH activity level in the in vitro model, in comparison with
those in the negative-control mimics group (Fig. 3B-F). These results indicated that
miR-323 promoted nerve cell toxicity in cells.
miR-323 regulates Bax/caspase signaling
pathway in the in vivo model
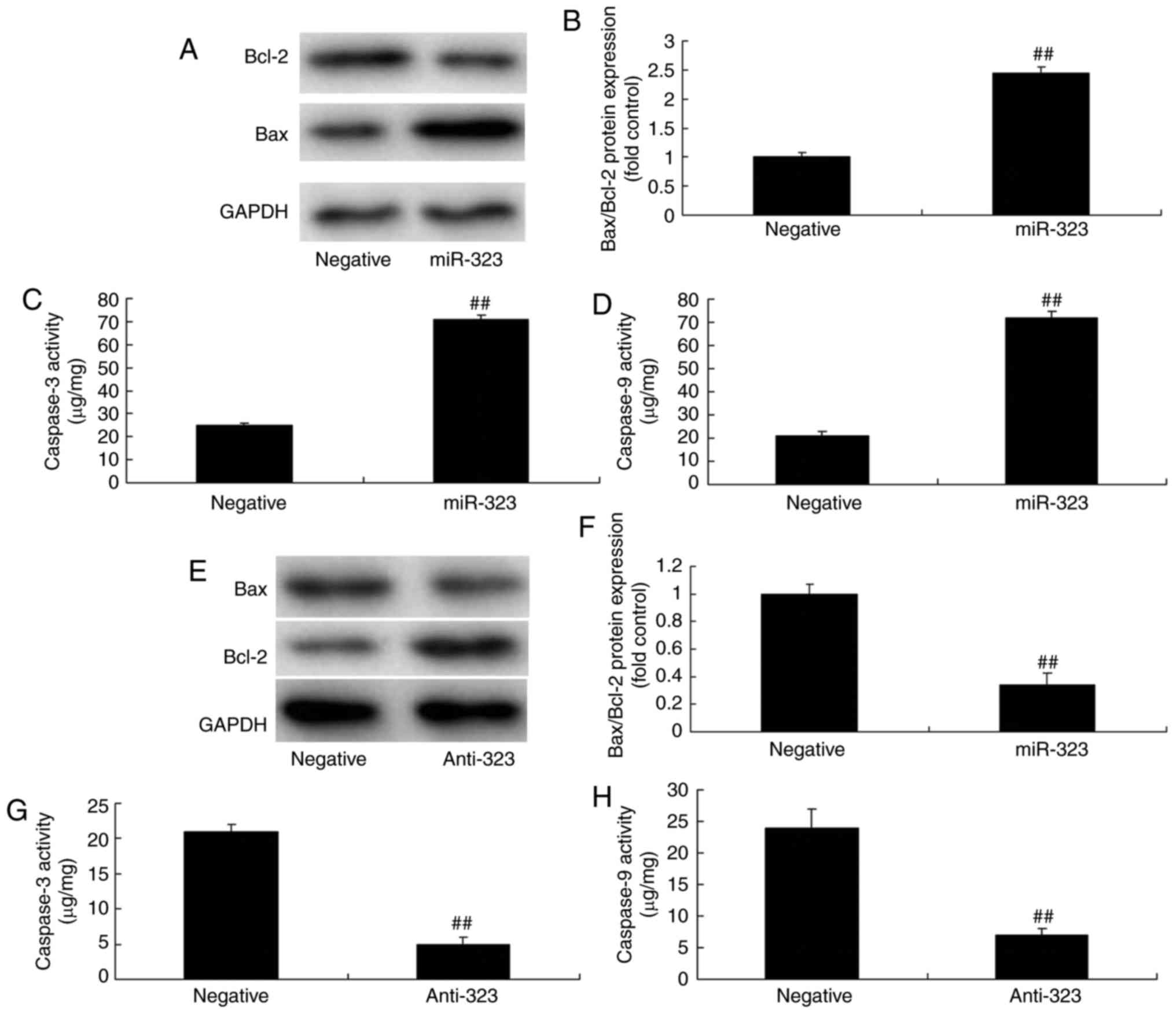
To demonstrate the contribution of miR-323 in
cerebral infarction-induced nerve cell toxicity, changes in the
Bax/caspase signaling pathway were analyzed. As revealed in
Fig. 4A-E, overexpression of
miR-323 significantly increases the ratio of Bax/Bcl-2, and
increased caspase-3 and caspase-9 activity levels in the in
vivo model, compared with negative-control mimics group. By
contrast, downregulation of miR-323 significantly suppressed the
protein expression of the ratio of Bax/Bcl-2, and decreased
caspease-3 and caspase-9 activity levels in the in vivo
model, compared with the negative-control mimics group (Fig. 4E-H). These results indicated that
miR-323 regulated the Bax/caspase signaling pathway in the in
vivo model.
miR-323 regulates TGF-β1/SMAD3 signaling pathway
in the in vivo model
Gene chip analysis was employed to analyze the
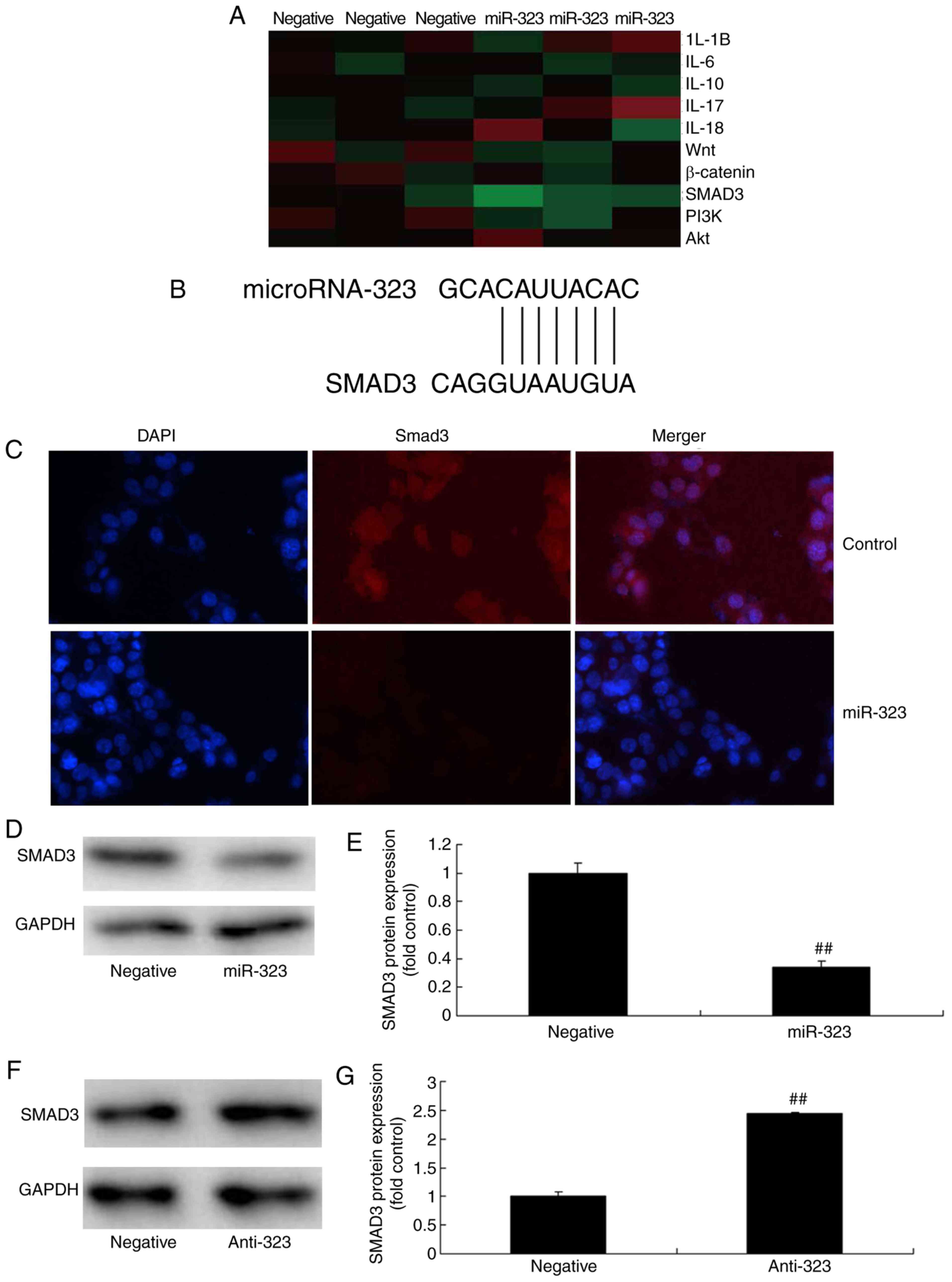
signaling pathway changes in the in vivo model of ACI. The
results revealed that SMAD3 was reduced upon over-expression of
miR-323, compared with the negative control group (Fig. 5A). A dual-luciferase reporter
assay was then performed to analyze the interaction of miR-323 and
SMAD3. As shown in Fig. 5B,
target sites were located in the 3'-UTR of SMAD3 mRNA. In addition,
immunofluorescence analysis revealed that overexpression of miR-323
suppressed SMAD3 protein expression in the in vitro model of
cerebral infarction, compared with that in the negative control
group (Fig. 5C). Similarly,
western blot analysis revealed that overexpression of miR-323
significantly suppressed SMAD3 protein expression in this cell
model (Fig. 5D and E). By
contrast, downregulation of miR-323 significantly induced SMAD3
protein expression in the in vitro model of cerebral
infarction, compared with negative-control mimics group, as
demonstrated by the immunoassay results (Fig. 5F and G). These results revealed
that miR-323 regulates the TGF-β1/SMAD3 signaling pathway, which
suppressed nerve cell toxicity in cerebral infarction.
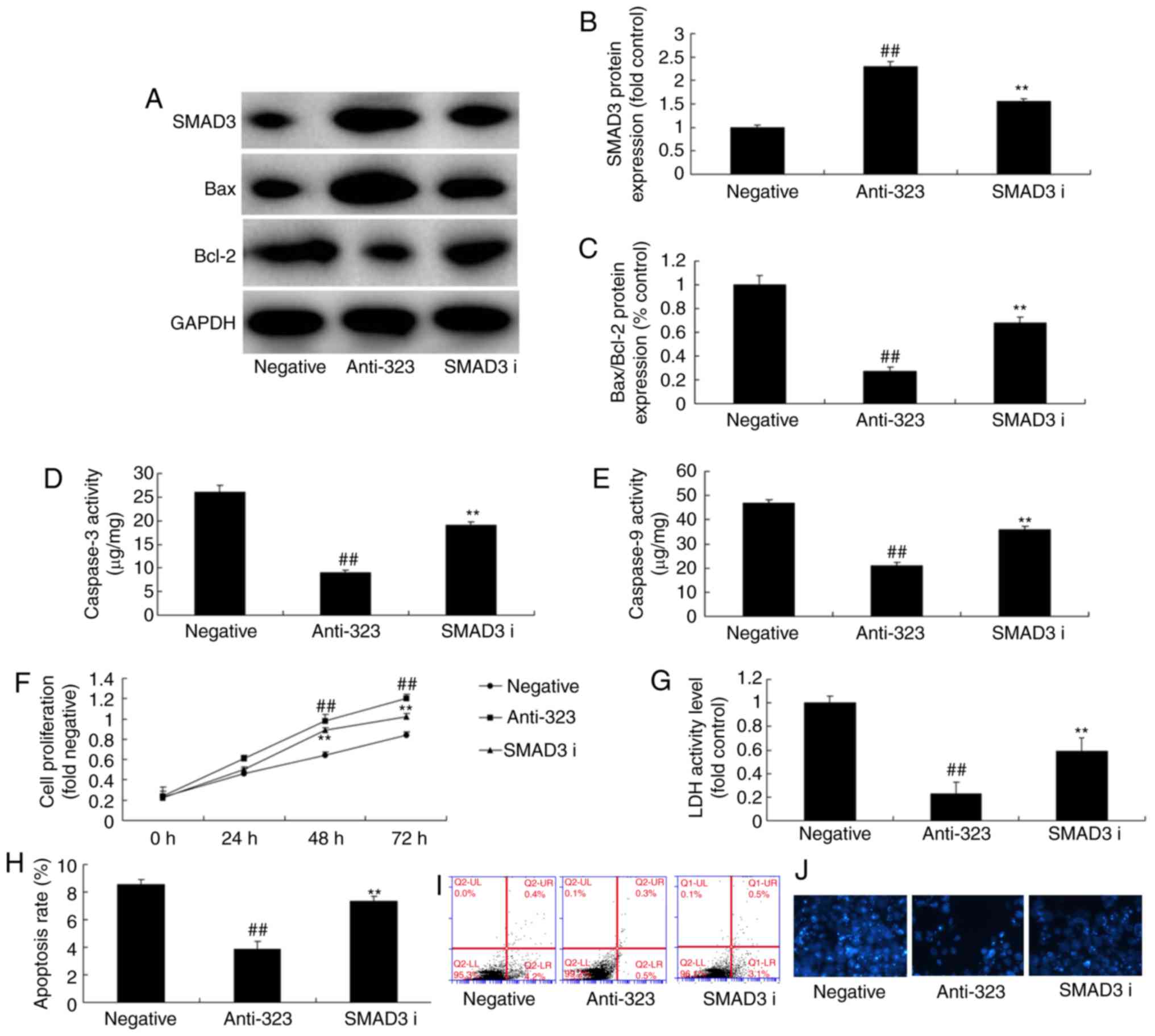
SMAD3 inhibitor participates in the
effect of miR-323 in cerebral infarction through the TGF-β1/SMAD3
signaling pathway
To further explore the function of SMAD3 in the
effect of miR-323 in cerebral infarction, a SMAD3 inhibitor (SIS3)
was used in the in vitro model. It was observed that SMAD3
inhibitor significantly suppressed SMAD3 protein expression,
induced Bax/Bcl-2 protein expression and increased
caspase-3/caspase-9 activity levels in in vitro model of
cerebral infarction following downregulation miR-323, compared with
the miR-323 downregulation group (Fig. 6A-E). In addition, the SMAD3
inhibitor in combination with miR-323 down-regulation reduced cell
viability and LDH activity level in the in vitro model,
compared with the downregulation miR-323 group (Fig. 6E-J). These results demonstrated
that SMAD3 is associated with the effects of miR-323 in cerebral
infarction.
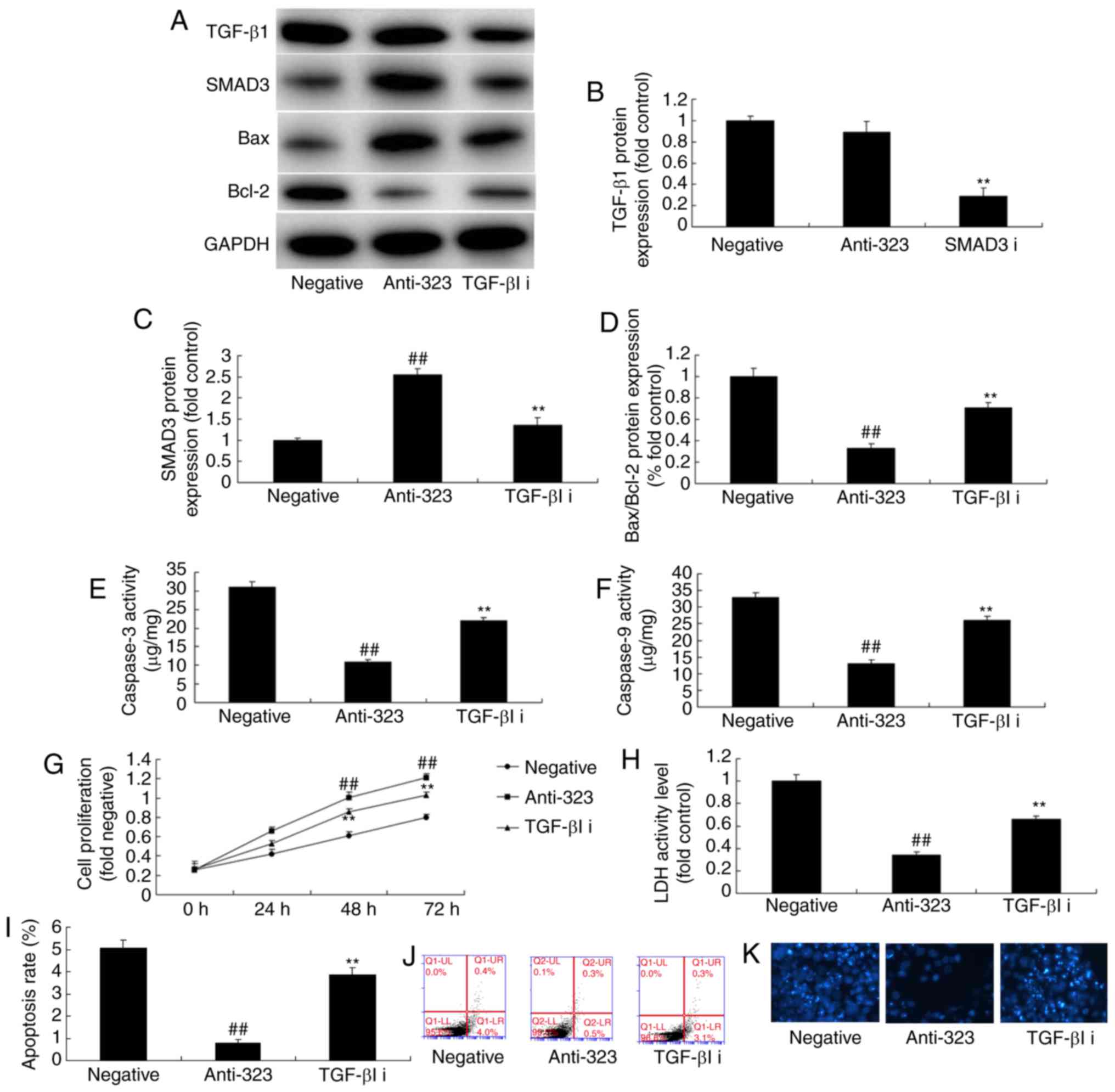
TGF-β1 inhibitor participates in the
effect of miR-323 in cerebral infarction through the TGF-β1/SMAD3
signaling pathway
To examine whether the TGF-β1/SMAD3 signaling
pathway is involved in the effect of miR-323 in cerebral
infarction, TGF-β1 inhibitor was utilized to suppress the
TGF-β1/SMAD3 signaling pathway. As a result, TGF-β1 inhibitor, 20
nm of LY2109761, significantly suppressed SMAD3 protein expression,
induced Bax/Bcl-2 protein expression, and increased
caspase-3/caspase-9 activity levels in the in vitro model of
cerebral infarction following the downregulation of miR-323, as
compared with the anti-miR-323 group (Fig. 7A-F). Meanwhile, TGF-β1 inhibitor
in combination with anti-miR-323 significantly reduced the effect
of miR-323 downregulation on cell viability and LDH activity level
in the in vitro model when compared with anti-miR-323 group
(Fig. 7G-K). These results
suggested that the miR-323/TGF-β1/SMAD3 signaling pathway regulated
cell death during cerebral infarction.
Discussion
ACI, also referred to as ischemic stroke, is a
pathological process of neural dysfunction or injury within brain
tissue that results from local cerebral ischemia and dysfunction
(9). The morbidity and mortality
of ACI patients is increasing annually in clinical practice
(2). This can be attributed to
improvement in diagnostic technologies (9). The data of the present study
demonstrated that miR-323 expression was upregulated in rats with
cerebral infarction as compared that in the sham-control group.
Yang et al (10) indicated
that miR-323 regulated ischemia/reperfusion-induced rat neuronal
cell death. The current study only used immunohistochemical
analysis to assess the effects of miR-323 on SMAD3 in the in
vivo model; however, this is an insufficient, and future
studies should use in situ hybridization histochemical and
immunohistochemical analyses to analyze the function of miR-323 in
ACI.
Certain miRNAs serve important roles in neurogenesis
and neuronal function, even at low concentrations. The majority of
miRNAs are located in the tissues and cells (11), while they can also stably exist in
body fluids, including blood, urine and amniotic fluid (12). Human tissue miRNA detection can
hardly be achieved or repeatedly used in clinical practice. A
previous study reported that there are stable miRNA molecules in
the peripheral blood (4).
Furthermore, different miRNA expression profiles are identified in
different diseases (12). These
discoveries have initiated the novel strategy of using peripheral
blood miRNAs for non-invasive disease diagnosis (4). Research reported that peripheral
blood miRNAs with specific expression changes can be found in
multiple diseases, including tumor, diabetes, myocardial
infarction, Parkinson' disease and Alzheimer's disease (11). In the present study,
overexpression of miR-323 was found to significantly increase LDH
activity level and inhibit cell viability in an in vitro
model of ACI. These results suggested that miR-323 induced nerve
cell apoptosis in ACI.
TGF-β exerts multiple effects, such as
anti-oxidation, blocking apoptosis, regulating the inflammatory
response, and regulating microglial cell and astrocyte responses
(13). It can also inhibit
central nervous system inflammatory response at the early stage of
ischemia (13), therefore
alleviating encephaledema, reducing the infarct size and promoting
microangiogenesis (14). Besides,
it serves an important role in the repair of brain tissue injury. A
study on a rat ischemia and hypoxia model verified that exogenous
TGF-β alleviated the microglial cell response and reduced neuron
death, as well as the infarct size (14). In the current study,
overexpression of miR-323 was found to significantly suppress SMAD3
protein expression in an in vitro model of cerebral
infarction. Wang et al (15) suggested that miR-323-3p inhibited
cell invasion and metastasis in pancreatic ductal adenocarcinoma
via SMAD2 and SMAD3.
TGF-β cytokine is a multi-functional protein that is
closely associated with cell proliferation, differentiation,
apoptosis and angiogenesis (16).
In the case of injury, the nonparenchymal cells release TGF-β,
which then binds with the cell surface receptor and promotes the
phosphorylation of other TGF-β receptors through a positive
feedback (16). TGF-β can also
activate the Smad pathway. The phosphorylated TGF-β receptor
renders spatial conformational changes in the intracellular
nucleus-cytoplasm signaling molecules SMAD2 and SMAD3 through
phosphorylation (17,18). Thereby, these molecules cross-link
with Smad4 to form an isotrimer complex, which subsequently enters
the nucleus to regulate target gene transcription (18). Finally, it will promote oxidative
stress, inflammatory response, fibrosis and apoptosis (18,19). In the current study, it was
demonstrated that the TGF-β1 inhibitor participated in the effect
of miR-323 in cerebral infarction through the TGF-β1/SMAD3
signaling pathway. Kärner et al (20) further suggested that increased
miR-323-3p in asthma served a role in the regulation of the TGF-β
pathway and interleukin-22 production. These results were in
agreement with the findings of present study, indicating that
miR-323 suppresses nerve cell toxicity in cerebral infarction
through the TGF-β1/SMAD3 signaling pathway.
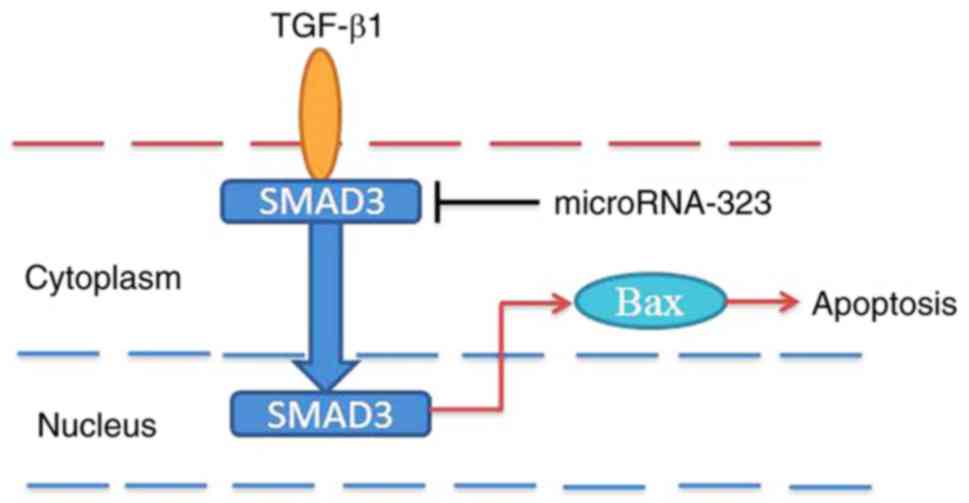
In conclusion, the current data demonstrated for the
first time, to the best of our knowledge, that miR-323 suppresses
nerve cell apoptosis in cerebral infarction via the TGF-β1/SMAD3
signaling pathway (Fig. 8). The
study expanded on the understanding of the pathogenic role of
miR-323 in cerebral infarction via TGF-β1/SMAD3 signaling pathway,
and this miRNA may represent a crucial target for potential
therapeutic intervention.
Funding
No funding was received.
Availability of data and materials
The analyzed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
FC designed the present study. HD, JW, WZ, ZC and YT
performed the experiments. FC and HD analyzed the data. FC wrote
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Animal experiments were approved by the Ethics
Committee of the Beijing Luhe Hospital Capital Medical University
(Beijing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Al-Ali F, Berkhemer OA, Yousman WP, Elias
JJ, Bender EN, Lingsma HF, van der Lugt A, Dippel DW, Roos YB, van
Oostenbrugge RJ, et al: The capillary index score as a marker of
viable cerebral tissue: Proof of concept-the capillary index score
in the MR CLEAN (multicenter randomized clinical trial of
endovascular treatment for acute ischemic stroke in the
Netherlands) trial. Stroke. 47:2286–2291. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maeshima S, Okamoto S, Okazaki H, Mizuno
S, Asano N, Tsunoda T, Maeda H, Masaki M and Sonoda S: Hemorrhagic
transformation in patients with cerebral infarction referred to a
rehabilitation hospital. Interv Neurol. 4:69–74. 2016. View Article : Google Scholar
|
|
3
|
Kim DH, Kim SU, Sung JH, Lee DH, Yi HJ and
Lee SW: Significances and outcomes of mechanical thrombectomy for
acute infarction in very elderly patients: A single center
experience. J Korean Neurosurg Soc. 60:654–660. 2017. View Article : Google Scholar
|
|
4
|
Fasanaro P, Greco S, Ivan M, Capogrossi MC
and Martelli F: microRNA: Emerging therapeutic targets in acute
ischemic diseases. Pharmacol Ther. 125:92–104. 2010. View Article : Google Scholar
|
|
5
|
Khanna S, Rink C, Ghoorkhanian R, Gnyawali
S, Heigel M, Wijesinghe DS, Chalfant CE, Chan YC, Banerjee J, Huang
Y, et al: Loss of miR-29b following acute ischemic stroke
contributes to neural cell death and infarct size. J Cereb Blood
Flow Metab. 33:1197–1206. 2013. View Article : Google Scholar
|
|
6
|
Kim JM, Jung KH, Chu K, Lee ST, Ban J,
Moon J, Kim M, Lee SK and Roh JK: Atherosclerosis-related
circulating MicroRNAs as a predictor of stroke recurrence. Transl
Stroke Res. 6:191–197. 2015. View Article : Google Scholar
|
|
7
|
Selvamani A, Williams MH, Miranda RC and
Sohrabji F: Circulating miRNA profiles provide a biomarker for
severity of stroke outcomes associated with age and sex in a rat
model. Clin Sci (Lond). 127:77–89. 2014. View Article : Google Scholar
|
|
8
|
Slevin M, Krupinski J, Slowik A, Kumar P,
Szczudlik A and Gaffney J: Serial measurement of vascular
endothelial growth factor and transforming growth factor-beta1 in
serum of patients with acute ischemic stroke. Stroke. 31:1863–1870.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dowling MM, Quinn CT, Plumb P, Rogers ZR,
Rollins NK, Koral K and Buchanan GR: Acute silent cerebral ischemia
and infarction during acute anemia in children with and without
sickle cell disease. Blood. 120:3891–3897. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang L, Xiong Y, Hu XF and Du YH:
MicroRNA-323 regulates ischemia/reperfusion injury-induced neuronal
cell death by targeting BRI3. Int J Clin Exp Pathol. 8:10725–10733.
2015.PubMed/NCBI
|
|
11
|
Dewdney B, Trollope A, Moxon J, Thomas
Manapurathe D, Biros E and Golledge J: Circulating MicroRNAs as
biomarkers for acute ischemic stroke: A Systematic review. J Stroke
Cerebrovasc Dis. 27:522–530. 2018. View Article : Google Scholar
|
|
12
|
Zhou J and Zhang J: Identification of
miRNA-21 and miRNA-24 in plasma as potential early stage markers of
acute cerebral infarction. Mol Med Rep. 10:971–976. 2014.
View Article : Google Scholar
|
|
13
|
Kim JS, Yoon SS, Kim YH and Ryu JS: Serial
measurement of interleukin-6, transforming growth factor-beta, and
S-100 protein in patients with acute stroke. Stroke. 27:1553–1557.
1996. View Article : Google Scholar
|
|
14
|
Ata KA, Lennmyr F, Funa K, Olsson Y and
Terent A: Expression of transforming growth factor-beta1, 2, 3
isoforms and type I and II receptors in acute focal cerebral
ischemia: An immunohistochemical study in rat after transient and
permanent occlusion of middle cerebral artery. Acta Neuropathol.
97:447–455. 1999. View Article : Google Scholar
|
|
15
|
Wang C, Liu P, Wu H, Cui P, Li Y, Liu Y,
Liu Z and Gou S: MicroRNA-323-3p inhibits cell invasion and
metastasis in pancreatic ductal adenocarcinoma via direct
suppression of SMAD2 and SMAD3. Oncotarget. 7:14912–14924.
2016.
|
|
16
|
Ronaldson PT, Demarco KM,
Sanchez-Covarrubias L, Solinsky CM and Davis TP: Transforming
growth factor-beta signaling alters substrate permeability and
tight junction protein expression at the blood-brain barrier during
inflammatory pain. J Cereb Blood Flow Metab. 29:1084–1098. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lan W, Yang F, Li Z, Liu L, Sang H, Jiang
Y, Xiong Y and Zhang R: Human urine kininogenase attenuates
balloon-induced intimal hyperplasia in rabbit carotid artery
through transforming growth factor β1/SMAD2/3 signaling pathway. J
Vasc Surg. 64:1074–1083. 2016. View Article : Google Scholar
|
|
18
|
Tinoco-Veras CM, Santos AAQA, Stipursky J,
Meloni M, Araujo APB, Foschetti DA, López-Ureña D, Quesada-Gómez C,
Leitão RFC, Gomes FCA and Brito GAC: Transforming growth factor
β1/SMAD signaling pathway activation protects the intestinal
epithelium from clostridium difficile toxin a-induced damage.
Infect Immun. 85:e00430–17. 2017. View Article : Google Scholar
|
|
19
|
Grand Moursel L, Munting LP, van der Graaf
LM, van Duinen SG, Goumans MTH, Ueberham U, Natté R, van Buchem MA,
van Roon-Mom WMC and van der Weerd L: TGFβ pathway deregulation and
abnormal phospho-SMAD2/3 staining in hereditary cerebral hemorrhage
with amyloidosis-Dutch type. Brain Pathol. 28:495–506. 2018.
View Article : Google Scholar
|
|
20
|
Kärner J, Wawrzyniak M, Tankov S, Runnel
T, Aints A, Kisand K, Altraja A, Kingo K, Akdis CA, Akdis M and
Rebane A: Increased microRNA-323-3p in IL-22/IL-17-producing T
cells and asthma: A role in the regulation of the TGF-β pathway and
IL-22 production. Allergy. 72:55–65. 2017. View Article : Google Scholar
|