Introduction
Osteoarthritis (OA) is a type of degenerative joint
disease, which is primarily characterized by the progressive
destruction of articular cartilage and joint inflammation, leading
to physical disability and decreased quality of life (1). This disease affects ~80% of the
population aged ≥60 years and accounts for high annual
hospitalizations in the developed world (2). Chondrocytes, the only cells in
articular cartilage, are critical in the pathological progression
of OA via apoptosis and cytokine production (3,4).
Therefore, the alleviation of chondrocyte apoptosis and
inflammation can be an effective therapeutic intervention in
patients of OA.
MicroRNAs (miRNAs) are small (~22 nucleotides)
non-coding RNA molecules, which typically inhibit the translation
and target the degradation of mRNAs through partial complementarity
(5,6). A large body of evidence indicates
that miRNAs are frequently dysregulated in human inflammatory
diseases, including OA, and that these miRNAs are important in
cartilage degradation (7,8). Specifically, a list of miRNAs
differentially expressed in plasma in primary OA has been
identified in inflammatory disorders (9). For example, miRNA (miR)-9 was found
to be downregulated in the cartilage tissues of OA rats and the
upregulation of miR-9 inhibited the expression level of matrix
metalloproteinase-13 (MMP-13), decreased its suppressive effects on
collagen type II α1 (Col2A1), and contributed to antagonizing OA
(10). Another study showed that
the upregulation of miR-218-5p protected OA mice from cartilage
degradation by inhibiting the phosphoinositide
3-kinase/Akt/mammalian target of rapamycin signaling pathway
(11). miR-210 negatively
regulates the lipopolysaccharide (LPS)-induced production of
pro-inflammatory cytokines by targeting nuclear factor-κB (NF-κB)
in murine macrophages (12).
However, limited studies have focused on the role of miRNAs in the
inflammatory pathogenesis of OA.
In the present study, an miRNA microarray was
performed to investigate miRNA expression profiles in an OA mice
model. Subsequently, the regulatory mechanisms of miR-93 on
inflammatory response and apoptosis were examined in an LPS-induced
chondrocyte injury model. Subsequently, the improvement of the
inflammatory response and apoptosis induced by miR-93 in OA mice
was verified. Finally, it was found that miR-93 targeted TLR4 and
inhibited activation of the NF-κB pathway in vitro and in
vivo. These findings provide information for the future
direction of treatments for patients with OA.
Materials and methods
Animals
A total of 36 male (n=6/group), eight-week-old
C57BL/6 mice weighing 20-25 g (Joint Ventures Sipper BK
Experimental Animal Company, Shanghai, China) were kept under
standard animal room conditions (temperature, 23±1°C; humidity,
55±5%) in a 12-h light/dark cycle and fed a standard laboratory
diet and water ad libitum. The animal use and experimental
protocols were approved by the Animal Care Committee of the Huaihe
Hospital of Henan University (Kaifeng, China) in accordance with
the institutional guidelines.
In vivo experiment
The miR-93 agomir and agomir-negative control
(agomir-NC) were obtained from GenePharma. The mice were randomly
divided into four groups, a sham group, an OA group, an OA +
agomir-miR-93 group and an OA + agomir-NC group. In the OA group
(n=6/group), the mice were subjected to MMT. Mice subjected to sham
surgery were used in the sham group. In the OA + agomir-miR-93
group (n=6/group), the mice were treated with agomir-miR-93 (5
nmol) via intra-articular injection administered through a medial
parapatellar approach at 1 week post-surgery (13). The mice were subjected to MMT and
treated with agomir-NC as the negative control. After 2 weeks, all
mice were sacrificed following induction of deep anesthesia, and
the articular cartilages of the medial tibial plateau and the
synovial fluid were collected for further experiments.
Isolation of primary mouse articular
chondrocytes and treatment
The primary chondrocytes were harvested from
articular cartilage as previously described (7). Briefly, the articular cartilage were
cut finely into 1-3 mm3 slices with a scalpel blade,
digested with 0.25% trypsin including 0.01% EDTA for 30 min, and
rinsed twice in sterile PBS. Subsequently, 3 mg/ml collagenase D
(Roche Applied Science, Indianapolis, IN, USA) in culture medium
was added for 3.5-4.5 h until the majority of the chondrocytes were
visible. The digested cartilage was collected and centrifuged at
600 × g for 5 min at 4°C. The pellet was resuspended in Dulbecco’s
modified Eagle’s medium (DMEM) and filtered to remove the
undigested cartilage through 70-mm nylon mesh. Finally, the
chondrocyte cells were plated in a new Petri dish with DMEM
containing 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and 1% antibiotics at 37°C in a
5% CO2 incubator.
For the induction of inflammation and apoptosis in
the cultured chondrocytes, LPS (200 µg/100 µl,
Escherichia coli, 055:B5, Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) was used to treat the cells
(2×105/well) at a final concentration of 5 µg/ml
for 6 h at 37°C in a 5% CO2 incubator.
Establishment of the OA model
The OA model was induced by medial meniscectomy tear
(MMT) surgery as previously reported (14). The mice were anesthetized with an
intraperitoneal injection of sodium pentobarbital (100 mg/kg),
following which the mice were subjected to the MMT procedure. At 2
weeks post-surgery, the articular cartilages of the medial tibial
plateau were collected and stored at −80°C for subsequent
analysis.
miRNA microarray
Total RNAs were isolated from the articular
cartilage tissues using the miRNeasy mini kit (Qiagen UK, Crawley,
UK) according to the manufacturer’s protocol. Following
quantification and quality assurance with a NanoDrop ND-1000
spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and an Agilent 2100 Bioanalyzer, duplicate samples were
dephosphorylated and labeled using the miRCURY™Hy3™/Hy5™ Power
labeling kit (Exiqon; Qiagen AB, Sollentuna, Sweden) and were then
hybridized on the miRCURY™ LNA Array (v.16.0; Exiqon; Qiagen AB)
according to the manufacturer’s protocol. The procedure and image
process method were as described previously (15).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) validation
Total RNAs were isolated from the articular
cartilage tissues and articular chondrocytes using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) and reverse
transcribed into cDNA with a TaqMan MicroRNA Reverse Transcription
kit (Applied Biosystems; Thermo Fisher Scientific, Inc.) according
to the manufacturer’s protocol. To quantify TLR4 mRNA, ~1 mg of
total RNA was reverse-transcribed into cDNA using the Oligo dT
primer (Takara Biotechnology Co., Ltd., Dalian, China). qPCR
analysis was performed using the TaqMan miRNAs Quantitation kit
(Applied Biosystems, Foster City, CA, USA) on an Applied Biosystems
7300 real-time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). PCR reaction system was composed of 10 µl
RT-qPCR-Mix, 0.5 µl upstream primer, 0.5 µl
downstream primer, 2 µl cDNA template and 7 µl
ddH2O. qPCR amplification conditions were as follows:
Initial denaturation at 95°C for 10 min, 40 cycles of 95°C for 1
min, and annealing at 60°C for 30 sec. The following primers were
used: miR-93 forward, 5′-AGG CCC AAA GTG CTG TTC GT-3′ and reverse,
5′-GTG CAG GGT CCG AGG T-3′; TLR4 forward, 5′-AGT TGA TCT ACC AAG
CCT TGA GT-3′ and reverse, 5′-GCT GGT TGT CCC AAA ATC ACT TT-3′;
Collagen type XI α2 (Col11a2) forward, 5′-CCT GGA CCC CTT GGA
AAG-3′ and reverse, 5′-TCC CCC TTA GCT CCC TTC T-3′; Col2a1
forward, 5′-ACC CCC AGG TGC TAA TGG-3′ and reverse, 5′-GAA CAC CTT
TGG GAC CAT CTT-3′; Aggrecan (Acan) forward, 5′-GCC CTT CAC GTG TAA
AAA GG-3′ and reverse, 5′-CAG GGA GCT GAT CTC GTA GC-3′; U6
forward, 5′-GCT TCG GCA GCA CAT ATA CTA AAA T-3′ and reverse,
5′-CGC TTC ACG AAT TTG CGT GTC AT-3′; Glyceraldehyde phosphate
dehydrogenase (GAPDH) forward, 5′-CTG GGC TAC ACT GAG CAC C-3′ and
reverse, 5′-AAG TGG TCG TTG AGG GCA ATG-3′. The relative expression
of miR-93 was normalized with U6 and the relative expression of
TLR4, Col11a2, Col2a1 and Acan were normalized with GAPDH. All
reactions were performed in triplicate. The relative expression was
calculated using the 2−ΔΔcq method (16).
Transfection
The miR-93 mimics, mimics negative control (mimics
NC), miR-93 inhibitor and inhibitor NC were synthesized from
GenePharma (Shanghai, China). To overexpress TLR4 in chondrocytes,
the open reading frame region of human TLR4 gene was amplified and
inserted into the pcDNA3.1 eukaryotic expression vector
(Invitrogen; Thermo Fisher Scientific, Inc.). Lipofectamine 2000
was used for transfection according to the protocol of the
manufacturer until the chondrocytes reached 30-50% confluence.
Cell viability assay
Cell viability was determined using an MTT assay to
detect the effect of miR-93 on chondrocyte viability following LPS
treatment. At 48 h post-transfection, 25 µl of MTT (Sigma;
Merck KGaA) was added into each well (2×105/well) and
the cells were incubated for another 4 h at 37°C. The OD absorbance
at 450 nm was measured using a microplate reader (Infinite M200;
Tecan Group, Ltd., Mannedorf, Switzerland). All experiments were
performed in triplicate.
Detection of apoptosis by flow
cytometry
Following transfection for 48 h, cell apoptosis was
measured using an Annexin V-FITC Apoptosis Detection kit (BD
Biosciences, San Diego, CA, USA) according to the manufacturer’s
protocol. The cells and their supernatants were harvested and
washed twice with PBS. Annexin V was added to the suspended cells
and incubated at 4°C for 15 min in the dark. Propidium iodide was
then added for incubation for 5 min in the dark. The stained cells
were analyzed using a flow cytometer (FACSCalibur; BD
Biosciences).
Enzyme-linked immunosorbent assay
(ELISA)
To determine the release of inflammatory cytokines,
the levels of TNF-α, IL-1β, and IL-6 in the supernatants of
chondrocytes transfected with miRNA oligonucleotides or pcDNA-TLR4
were detected using the Valukine ELISA kit (R&D Systems, Inc.,
Minneapolis, MN, USA) according to the manufacturer’s protocol.
Terminal deoxynucleotidyl
transferase-mediated dUTP-biotin nick-end labeling (TUNEL)
staining
The articular cartilage tissues (10-mm thickness)
obtained in the above experimental procedure were subjected to
TUNEL staining. Following deparaffinization with xylene, the
sections of articular cartilage tissues were rehydrated with
ethanol at graded concentrations of 100-70% (v/v), followed by
washing with water. Subsequently, the tissues sections were
subjected to 100 µl proteinase K (20 µg/ml; Roche
Diagnostics, Basel, Switzerland) for 15 min at room temperature,
and then washed three times with PBS. TUNEL solution preparation
and staining were performed using a TUNEL Apoptosis Detection kit
(Alexa Fluor 488; Roche Diagnostics). Cell quantification was
performed using an inverted fluorescence microscope (DP73; Olympus
Corporation, Tokyo, Japan) at ×400 magnification. The
TUNEL-positive cells were counted in three fields of view per
section.
NF-κB activity assay
The chondrocytes were plated in 6-well tissue
culture plates at a concentration of 5×104 cells/ well
for 24 h. The cells were then transfected with 2.5 µg of a
NF-κB reporter luciferase construct. After 6 h, the cells were
washed and then co-transfected with miR-93 mimics and pcDNA-TLR4
for 24 h. The cells were then washed in PBS and harvested in 500
µl 1X passive lysis buffer. Luciferase activity was
quantified using a Promega luciferase assay kit on a luminometer.
The experimental values were recorded relative to those of
untreated control samples.
Dual-luciferase reporter assays
The predicted and mutated sequences targeting the
3′UTR of TLR4 were amplified and cloned into the pGL3 vector
(Promega Corporation, Madison, WI, USA). pGL3-TLR4-3′UTR wild-type
(Wt) and pGL3-TLR4-3′UTR mutated (Mut) were synthesized by
GenePharma. 293 cells (American Type Culture Collection, Manassas,
VA, USA) were seeded into 24-well plates at a density of
1-2×105 cells per well, and co-transfected with 20 nM
miR-93 mimics, 20 nM miR-93 inhibitor, or 20 nM miR-NC and 0.2
µg pGL3-TLR4-3′UTR Wt or 0.2 µg pGL3-TLR4-3′UTR Mut
using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific,
Inc.). The 293 cells were collected 48 h following transfection and
analyzed using the Dual-Luciferase Reporter Assay system (Promega
Corporation) The pRL-TK vector was used as an internal control. All
experiments were performed in triplicate and repeated three
times.
Western blot analysis
Protein samples from the tissues and cells were
prepared with lysis buffer and protease inhibitor (Roche
Diagnostics). The protein concentration was determined using a
bicinchoninic acid assay kit (Beyotime Institute of Biotechnology,
Haimen, China). The proteins (30 µg each sample) were
resolved on 10% SDS-PAGE gels, transferred onto PVDF membranes (EMD
Millipore, Billerica, MA, USA). Following blocking with
Tris-buffered saline and Tween (TBST) containing 5% skim milk, the
membranes were incubated overnight at 4°C with primary antibodies
against TLR4 (cat. no. 14358; 1:2,000), nuclear phosphorylated
(p)-p65 (cat. no. 3033; 1:2,000), p-inhibitor of NF-κB (IκB)α (cat.
no. 14358; 1:2,000) β-actin (cat. no. 4970; 1:2,000) and Histone H3
(cat. no. 9728; 1:2,000; all from Cell Signaling Technology, Inc.,
Danvers, MA, USA). Detection was performed by incubation with
peroxidase-conjugated secondary antibodies (cat. no. ab6734;
1:2,000; Abcam, Cambridge, UK) and chemiluminescence (EMD
Millipore) for 1 h at room temperature. Densitometric analysis was
performed using Quantity One software (v4.62; Bio-Rad Laboratories,
Inc., Hercules, CA, USA). β-actin protein was used as the inner
control of the cytoplasmic proteins; Histone H3 protein was used as
the inner control of the nuclear proteins. Each experiment was run
in triplicate.
Statistical analysis
Statistical analysis was performed using SPSS 15.0
(SPSS, Inc., Chicago, IL, USA). Quantitative data are presented as
the mean ± standard deviation. The comparison between data was
calculated using Student’s t-test and one-way analysis of variance
followed by Tukey’s post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Aberrant expression of miRNAs in the
articular cartilages of OA mice
To investigate the potential involvement of miRNAs
following OA, microarray analysis was performed to determine miRNA
levels in the articular cartilages (n=2/group). In the miRNA
microarray analysis, 16 miRNAs were upregulated and 34 miRNAs were
downregulated in the OA group compared with the sham group
(Fig. 1A). Among aberrant miRNAs,
miR-93 was one of the most significantly downregulated in the
articular cartilages of OA mice based on the microarray expression
data, and multiple studies have shown that miR-93 has
anti-apoptotic and anti-inflammatory effects in several types of
cells (17-19). Therefore, miR-93 was selected for
further investigation. The expression of miR-93 was validated using
RT-qPCR analysis and was shown to be significantly lower in the OA
group than in the sham group (Fig.
1B).
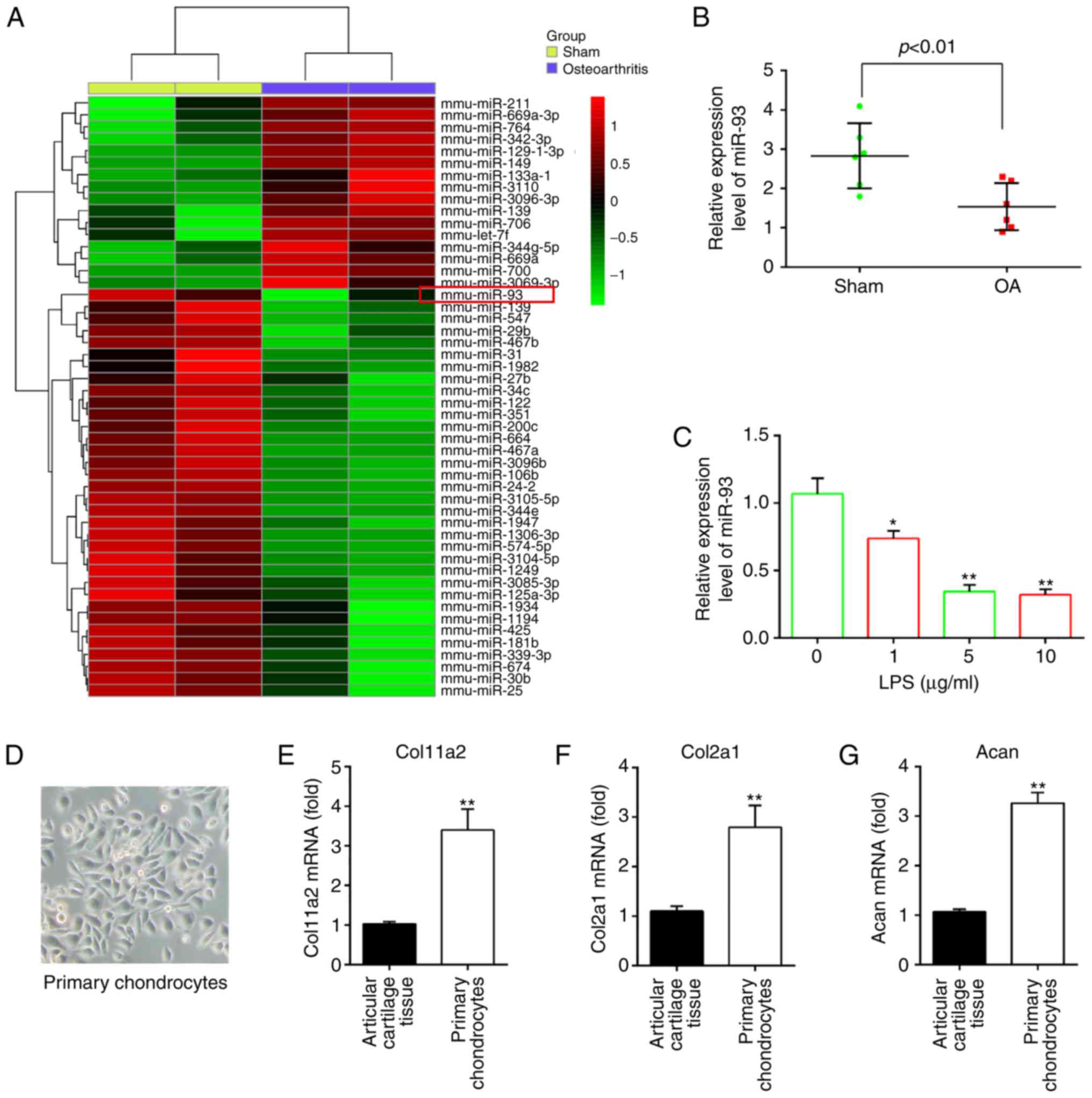 | Figure 1miR-93 is downregulated in OA mice
and LPS-treated chondrocytes. (A) Mice were randomly divided into
two groups, the sham group and OA group (n=2/group). The heat map
of miRNA profiles in the articular cartilages shows the
significantly dysregulated miRNAs. The color code in the heat maps
is linear, with green as the lowest and red as the highest.
Upregulated miRNAs are shown in red, whereas downregulated miRNAs
are shown in green. (B) Expression of miR-93 was validated by
RT-qPCR analysis in OA mice (n=6). P<0.01, vs. Sham group. (C)
Chondrocytes were stimulated with an increasing concentration
gradient of LPS (0, 1, 5, and 10 µg/ml) for 6 h, and the
expression of miR-93 was measured by RT-qPCR analysis. Data are
presented as the mean ± standard deviation of three independent
experiments. *P<0.05, **P<0.01, vs.
untreated group. (D) Image of primary chondrocytes cultured for 7
days (magnification, ×100). Expression of chondrocyte markers (E)
Col11a2, (F) Col2a1 and (G) Acan in articular cartilage tissues and
mouse primary chondrocytes were measured by RT-qPCR analysis. Data
are presented as the mean ± standard deviation of three independent
experiments. **P<0.01, vs. articular cartilage
tissues. OA, osteoarthritis; miRNA/miR, microRNA, LPS,
lipopolysaccharide; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction; Col11a2, collagen type XI α2; Col2a1,
collagen, type II, α1; chain; Acan, aggrecan. |
As the in vitro model of LPS-treated
chondrocytes is often used in OA investigations (20,21), the present study investigated the
functions of miR-93 in the development of OA using this cell model.
The expression of miR-93 was first examined in the LPS-induced OA
cell model. Consistent with the findings from analyzing the
expression of miR-93 in the articular cartilages, the expression of
miR-93 was found to be markedly decreased by LPS treatment, and the
levels of miR-93 levels were downregulated in a dose-dependent
manner (Fig. 1C). No significant
difference in the level of miR-93 was found between 5 and 10
µg/ml, therefore LPS at a dose of 5 µg/ml was used
for subsequent experiments, which has been used in a previous study
(22). The data suggested that
miR-93 may be important in the development of OA.
After 4 days, the primary mouse articular
chondrocytes began to appear spread out in the tissue and the cells
reached confluence ~7 days later. The cultured primary mouse
articular chondrocytes were spindle-shaped with a fibroblast-like
appearance (Fig. 1D). They were
characterized by the absence of chondrocyte markers, including
Col11a2, Col2a1 and Acan. The results of the RT-qPCR analysis
showed that the levels of these chondrocyte markers were higher in
the mouse primary chondrocytes than that in the articular cartilage
tissues, indicating that chondrocytes do not lose their
characteristics (Fig. 1E-G).
miR-93 inhibits LPS-induced apoptosis and
inflammatory cytokine production
To further investigate the role of miR-93 in OA,
chondrocytes were transfected with miR-93 mimics for 24 h, and then
treated with 5 µg/ml LPS for another 24 h. The miRNA
transfection efficiency was first evaluated via the RT-qPCR assay.
The frequent downregulation of miR-93 in the articular cartilages
of OA mice suggests that miR-93 may be important in the progression
of OA. The biological consequences of the overexpression of miR-93
in regulating apoptosis and the inflammatory response were then
examined using cell biology assays. The expression of miR-93 was
significantly increased in chondrocytes transfected with miR-93
mimics, followed by treatment with LPS (Fig. 2A). Subsequently, cell viability,
apoptosis and inflammatory cytokine production were assessed using
MTT, flow cytometry and ELISA, respectively. The results showed
that LPS treatment significantly decreased the cell viability and
induced cell apoptosis compared with the control group, whereas
these effects were inhibited following miR-93 mimics transfection.
(Fig. 2B-D). The effect of the
overexpression of miR-93 on the release of pro-inflammatory
cytokines that contribute to the clinical symptoms of OA was
further assayed (23). The
protein levels of cytokines, including TNF-α, IL-1β and IL-6, in
chondrocytes overexpressing miR-93 following LPS stimulation was
measured by ELISA. The upregulation of miR-93 suppressed the
LPS-induced expression of pro-inflammatory cytokines, compared with
chondrocytes transfected with the mimics NC (Fig. 2E-G). These results indicated that
miR-93 protected the chondrocytes from LPS-induced apoptosis and
inflammation.
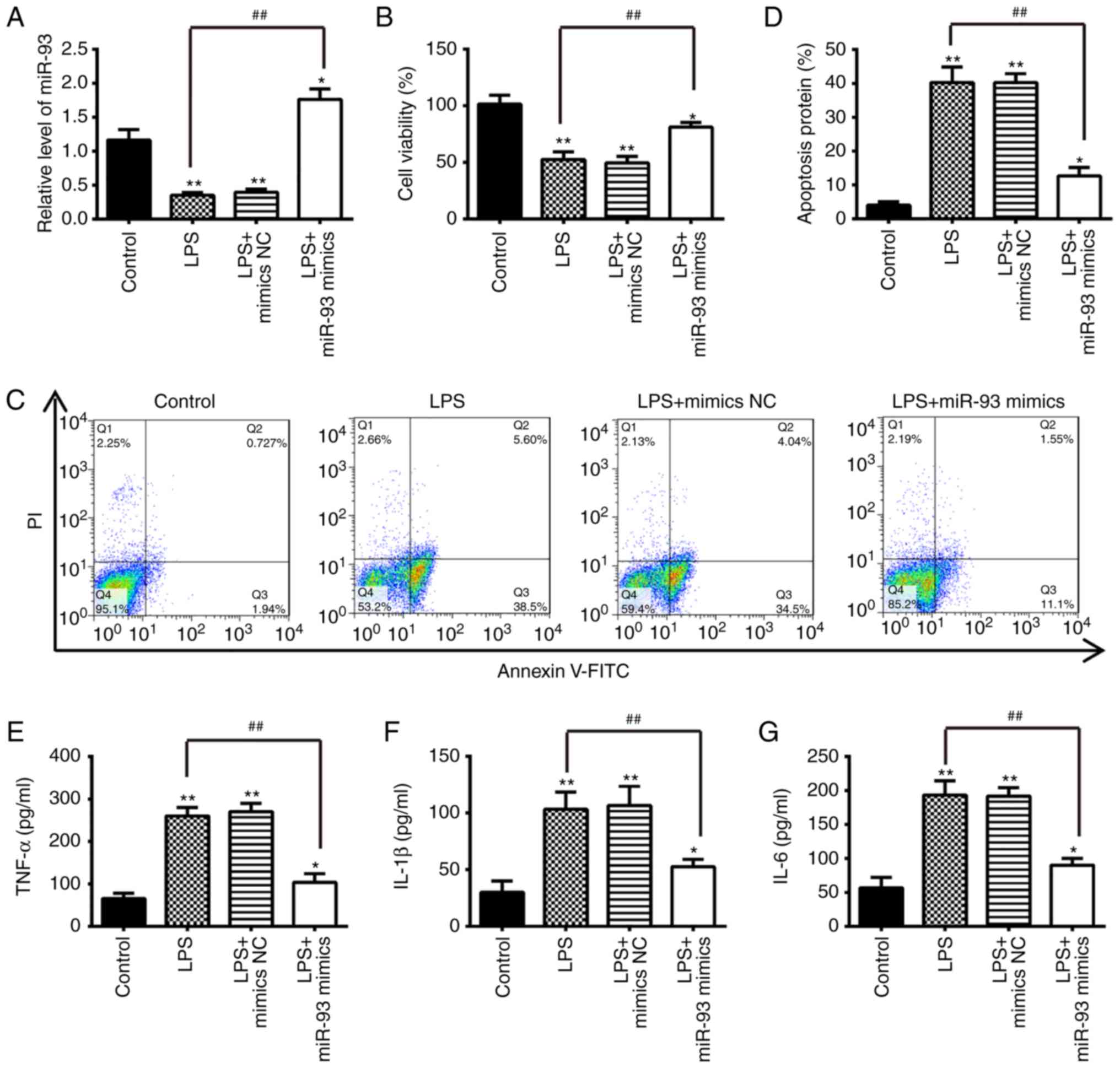 | Figure 2Overexpression of miR-93 suppresses
apoptosis and inflammation in LPS-treated chondrocytes.
Chondrocytes were transfected with miR-93 mimics or miR-NC for 24 h
and then treated with LPS (5 µg/ml) for 24 h. The cells and
cell supernatants were used for further analysis. (A) Expression
levels of miR-93 were assessed by reverse
transcription-quantitative polymerase chain reaction analysis. (B)
Cell viability was measured using an MTT assay. (C) Apoptosis was
detected by flow cytometry and (D) quantification. The levels of
(E) TNF-α, (F) IL-1β, and (G) IL-6 were measured using
enzyme-linked immunosorbent assay kits. Data are presented as the
mean ± standard deviation of three independent experiments.
*P<0.05, **P<0.01, vs. control group.
##P<0.01, vs. miR-NC; LPS alone group. miR, microRNA;
LPS, lipopolysaccharide; NC, negative control; TNF-α, tumor
necrosis factor; IL, interleukin; PI, propidium iodide. |
TLR4 is a direct target of miR-93
To investigate the potential molecular mechanism of
miR-93, the potential targets of miR-93 were predicted by
bioinformatics analysis. It was found that the 3′-UTR of TLR4, an
important regulator of the NF-κB pathway, possesses sequences
complementary to the miR-93 seed sequence (Fig. 3A and B). To verify whether miR-93
is directly bound to TLR4, a dual luciferase reporter assay was
performed. It was observed that the overexpression of miR-93
decreased relative luciferase activity in the presence of the Wt
3′-UTR, whereas the knockdown of miR-93 increased the relative
luciferase activity. Similarly, no significant change in luciferase
activity was observed when the targeted sequence of TLR4 was
mutated in the miR-93-binding site (Fig. 3C). To further confirm that TLR4
was negatively regulated by miR-93, the of mRNA and protein
expression levels of TLR4 were analyzed by RT-qPCR and western blot
analyses. The expression of TLR4 at the mRNA and protein levels was
significantly downregulated following the overexpression of miR-93
in chondrocytes, but upregulated following miR-93 knockdown
(Fig. 3D and E). In addition,
using an OA cell model, it was found that LPS treatment increased
the mRNA levels of TLR4 and this promoting effect was reversed by
transfection with the miR-93 mimics. By contrast, the miR-93
inhibitor enhanced the promoting effect of LPS on the mRNA levels
of TLR4, suggesting that miR-93 also negatively regulated the
expression of TLR4 in the LPS-induced chondrocytes (Fig. 3F). As TLR4/NF-κB signaling is
associated with inflammation in OA, miR-93 may exert its protective
effects on LPS-induced injury by targeting TLR4.
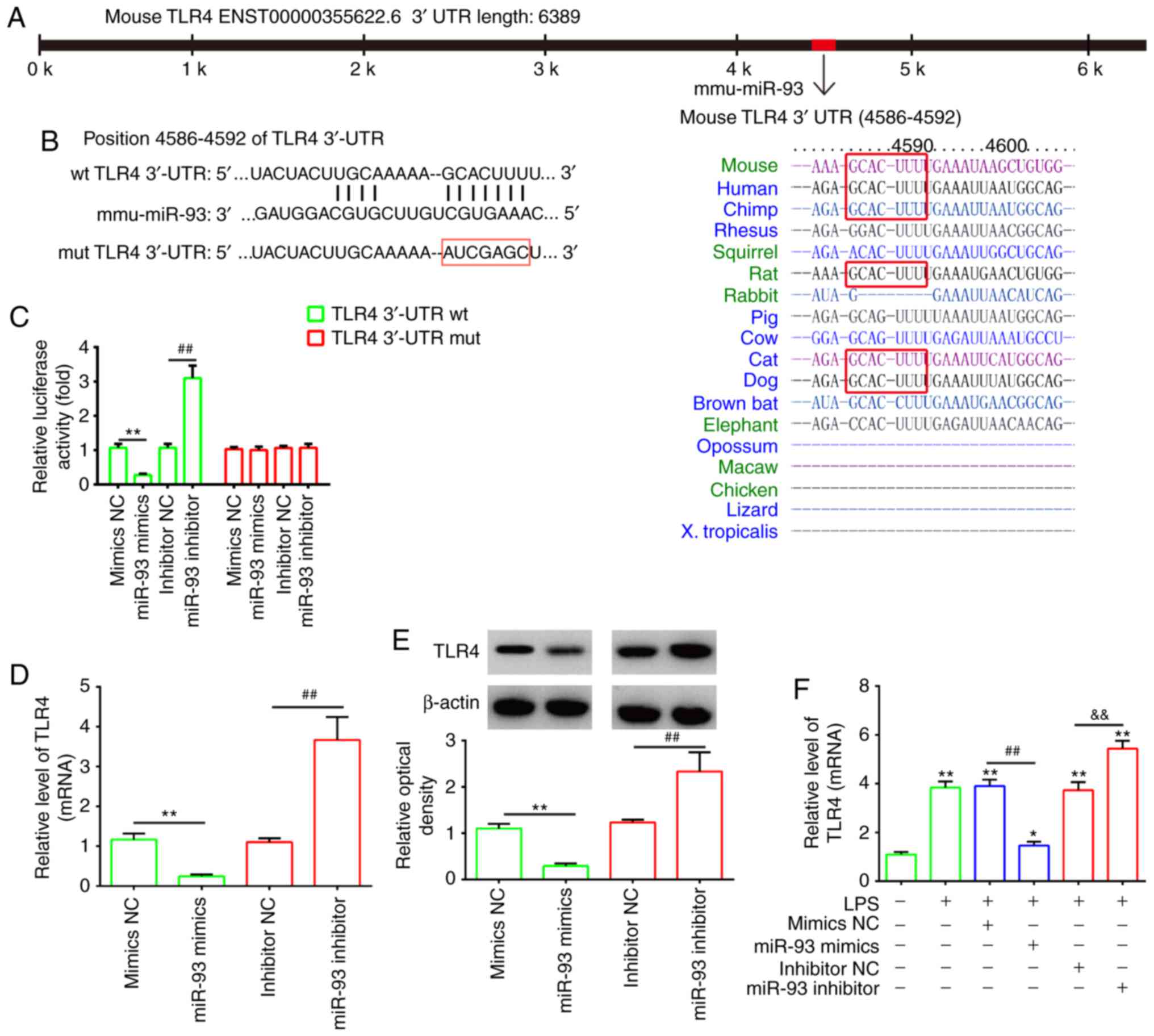 | Figure 3TLR4 is a direct target of miR-93.
(A) Putative binding site of miR-93 and TLR4. (B) Mutation was
generated on the TLR4 3′UTR sequence in the complementary site for
the seed region of miR-93. TLR4 3′UTR fragments containing the
wild-type or mutant miR-93-binding sequence were cloned downstream
of the luciferase reporter gene in pGL3-luc vector. (C) Luciferase
assay of 293 cells co-transfected with firefly luciferase
constructs containing TLR4 wt or mut 3′-UTRs and miR-93 mimics,
mimics NC, miR-93 inhibitor or inhibitor NC, as indicated (n=3).
Data are presented as the mean ± standard deviation of three
independent experiments. **P<0.01, vs mimics NC,
##P<0.01, vs inhibitor NC. (D) mRNA and (E) protein
expression of TLR4 following transfection with miR-93 mimics or
miR-93 inhibitor was measured by RT-qPCR and western blot analyses.
Data are presented as the mean ± standard deviation of three
independent experiments. **P<0.01, vs, inhibitor NC,
##P<0.01, vs mimics NC. (F) Chondrocytes were
transfected with miR-93 mimics or miR-93 inhibitor for 24 h and
then treated with LPS (5 µg/ml) for 24 h. Expression of TLR4
was assessed by RT-qPCR analysis. Data are presented as the mean ±
standard deviation of three independent experiments.
*P<0.05, **P<0.01, vs. control group;
##P<0.01, vs. LPS + mimics NC group;
&&P<0.01, vs. LPS + inhibitor NC group. TLR4,
Toll-like receptor 4; miR, microRNA; UTR, untranslated region; wt,
wild-type; mut, mutant; NC, negative control; LPS,
lipopolysaccharide; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction. |
miR-93 inhibits LPS-induced apoptosis and
inflammation through TLR4 in chondrocytes
To ascertain whether TLR4 is involved in miR-93
inhibiting the LPS-induced inflammatory response and apoptosis of
chondrocytes, the chondrocytes were transfected with miR-93 mimics
and pcDNA-TLR4 in addition to LPS stimulation. A western blot assay
was used to assess the transfection efficiency. The protein
expression level of TLR4 was significantly increased in the
LPS-treated chondrocytes following pcDNA-TLR4 transfection
(Fig. 4A). Subsequently, the
viability, apoptosis and the levels of pro-inflammatory cytokines
were assessed by MTT, flow cytometry and ELISA, respectively. As
shown in Fig. 4B, overexpression
of TLR4 significantly reduced the viability of the chondrocytes
transfected with miR-93 mimics. In addition, the cell apoptosis and
the production of pro-inflammatory cytokines (TNF-α, IL-1β and
IL-6) reduced by the miR-93 mimics were reversed by the
overexpression of TLR4 (Fig.
4B-G). Taken together, these data suggested that miR-93
suppressed LPS-stimulated apoptosis and pro-inflammatory cytokine
production through targeting TLR4.
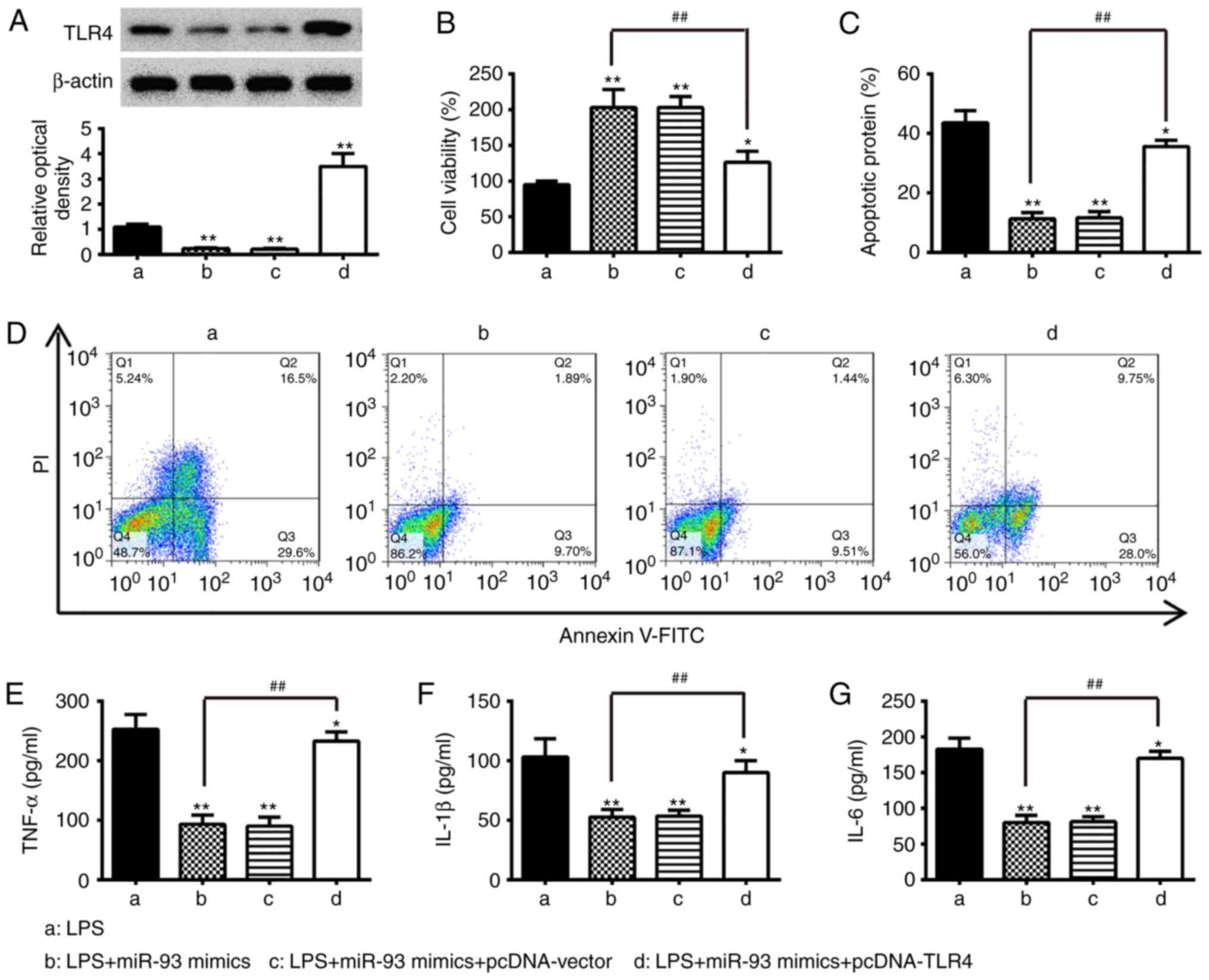 | Figure 4Overexpression of TLR4 abrogates the
inhibitory effects of miR-93 mimics on inflammation and apoptosis.
Chondrocytes were co-transfected with miR-93 mimics and pcDNA-TLR4
for 24 h and then exposed to 5 µg/ml LPS for 24 h. The cells
and cell supernatants were used for further analysis. (A) Protein
levels of TLR4 were detected by western blot analysis. (B) Cell
viability was measured using an MTT assay. Apoptosis was (C)
quantified following (D) detection by flow cytometry. Levels of (E)
TNF-α, (F) IL-1β, and (G) IL-6 were measured using ELISA kits. Data
are presented as the mean ± standard deviation of three independent
experiments. *P<0.05, **P<0.01, vs. LPS
alone group, ##P<0.01, vs. LPS + miR-93 mimics group.
TLR4, Toll-like receptor 4; miR, microRNA; LPS, lipopolysaccharide;
TNF, tumor necrosis factor; IL, interleukin; PI, propidium
iodide. |
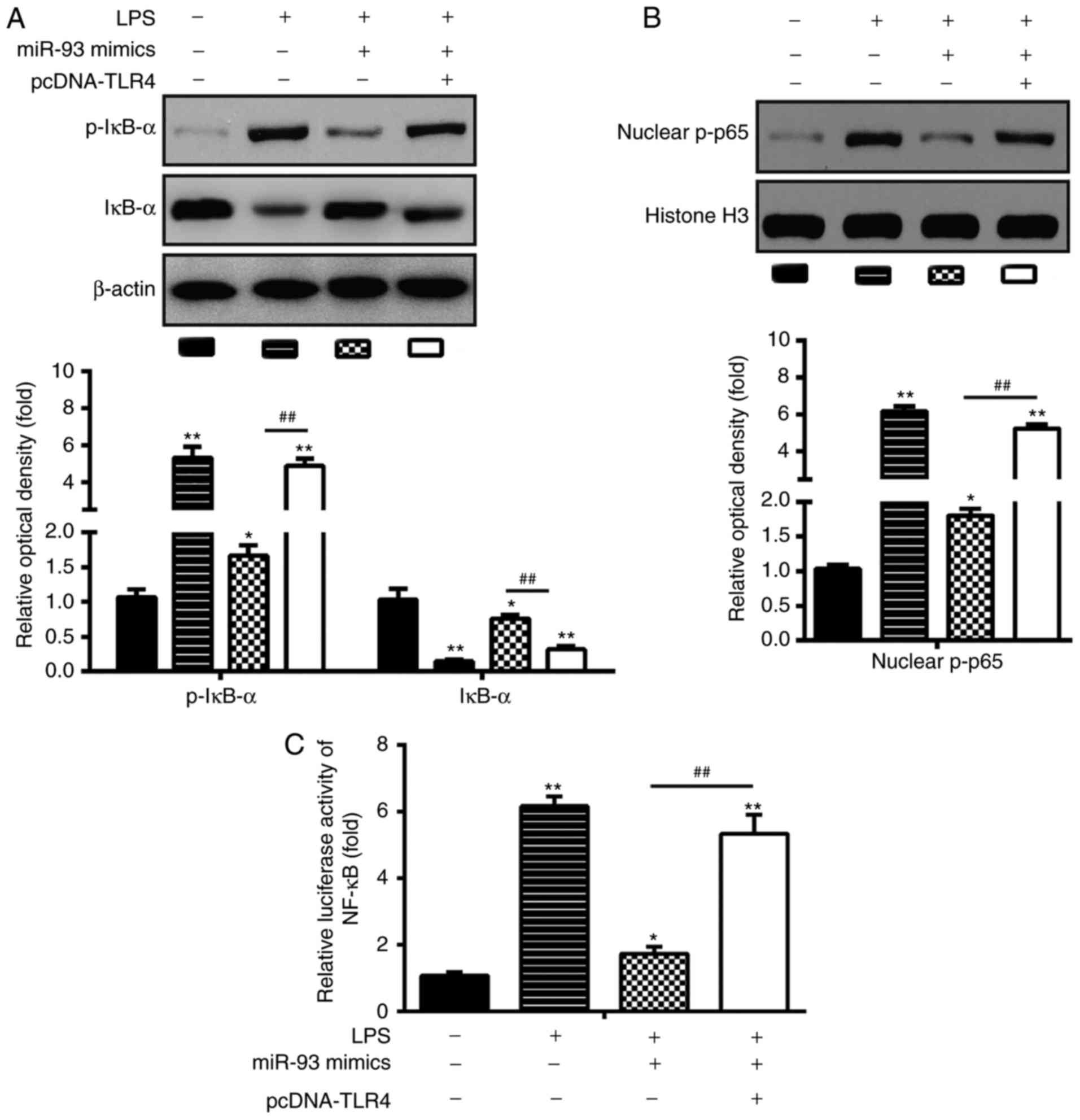
miR-93 inhibits activation of the
TLR4/NF-κB pathway in LPS-treated chondrocytes
Previous studies have shown that TLR4 can activate
the NF-κB pathway, and thus result in the secretion of
pro-inflammatory cytokines (24,25). In the present study, it was found
that the overexpression of miR-93 negatively influenced the
expression of TLR4 in LPS-treated chondrocytes. Therefore, the
effects of miR-93 on the LPS-induced expression of NF-κB
pathway-related core factors, nuclear p-p65 and p-IκB-α were TLR4.
As shown in Fig. 5A and B, in
LPS-induced chondrocytes, LPS promoted the activation of the NF-κB
pathway through increasing the expression of nuclear p-p65 and
reducing the expression of p-IκB-α, which was inhibited by
transfection with miR-93 mimics. However, the upregulation of TLR4
reactivated the NF-κB pathway inhibited the overexpression of
miR-93. To confirm the inhibitory effect of miR-93 in the
TLR4/NF-kB signaling pathway, the NF-κB activity assay was
performed. As shown in Fig. 5C,
the activity of NF-κB was reduced in the miR-93 mimics-transfected
chondrocytes, however, this inhibitory effect was reversed by the
overexpression of TLR4 (Fig. 5C).
These data suggest that miR-93 inhibited activation of the
TLR4/NF-κB pathway in the LPS-treated chondrocytes.
Agomir-93 improves apoptosis and
inflammation in OA mice
In order to confirm the functions of miR-93 in the
development of OA in vivo, a mouse model of OA was injected
with agomir-miR-93 (5 nmol) via intra-articular injection. The
overexpression efficiency of agomir-miR-93 was evaluated using
RT-qPCR analysis in the articular cartilage tissues (n=6/group). As
shown in Fig. 6A, the relative
expression of miR-93 was significantly upregulated compared with
that in the OA group. Subsequently, ELISA was performed to evaluate
pro-inflammatory cytokine production in the OA mouse model
following agomir-miR-93 injection. As shown in Fig. 6B-D, the upregulation of miR-93 in
OA mice caused a significant reduction of TNF-α, IL-1β and IL-6.
Furthermore, the effect of miR-93 on apoptotic cells in the OA mice
was analyzed by TUNEL staining. It was found that, compared with
the sham group, treatment with agomir-miR-93 significantly reduced
the positive cells and certain TUNEL-positive cells were larger in
size with dark-brown dots in the OA group (Fig. 6E). In addition, the expression of
NF-κB pathway-related core factors were measured in vivo. As
shown in Fig. 6F, the expression
levels of TLR4 and nuclear p-p65 were increased, and p-IκB-α was
decreased in OA mice compared with sham group, whereas the effects
were reversed by the overexpression of miR-93 (Fig. 6F). These findings suggested that
miR-93 inhibits apoptosis and inflammation by modulating the
TLR4/NF-κB signaling pathway in OA mice.
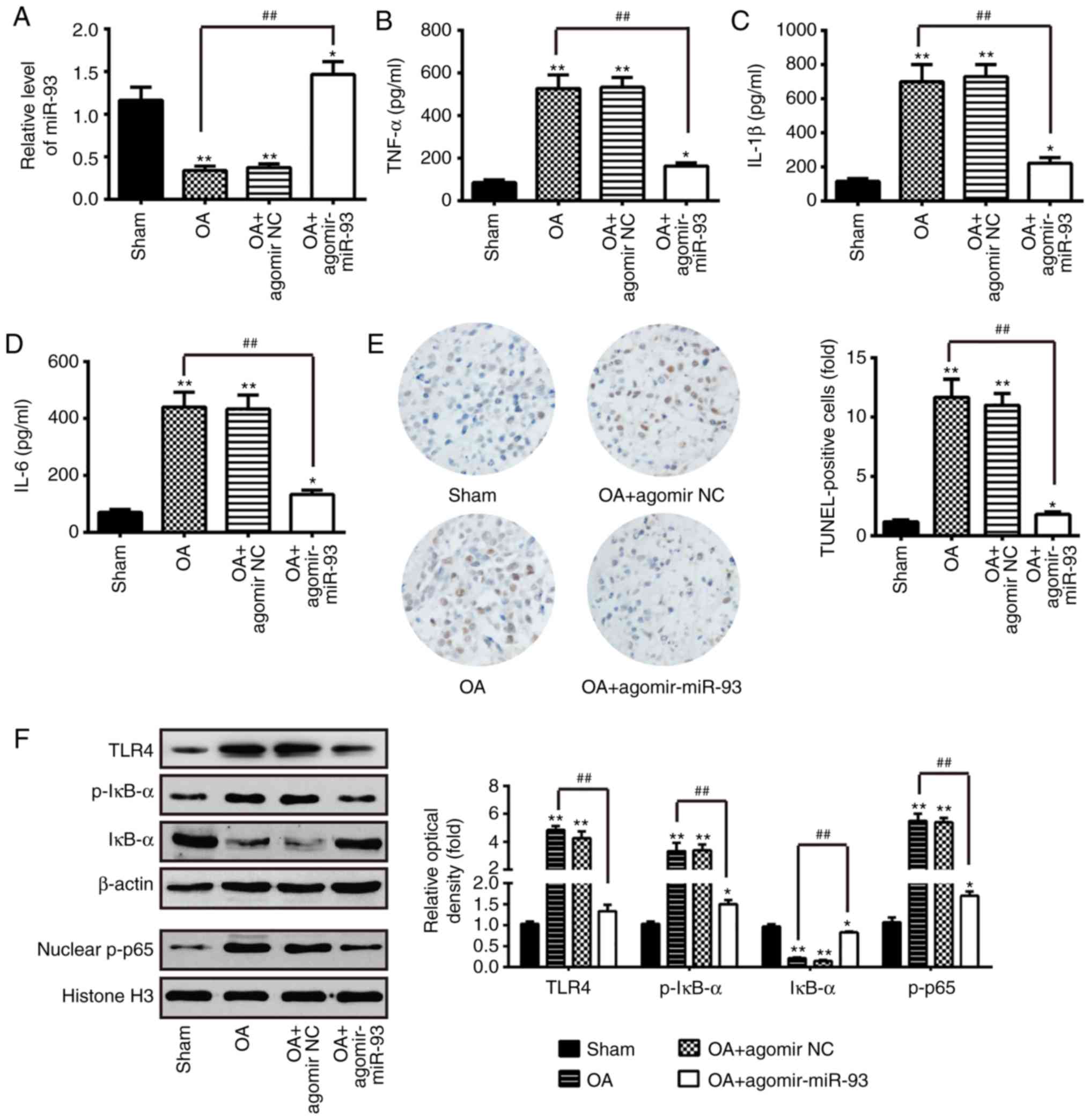 | Figure 6miR-93 attenuates inflammation and
apoptosis in OA mice. The mouse model of OA was injected with
antagomir-miR-93 (5 nmol) via intra-articular injection. The mice
were then subjected to medial meniscectomy tear surgery and treated
with agomir-NC as the negative control. After 2 weeks, all mice
were sacrificed. Subsequently, the articular cartilages of the
medial tibial plateau and the synovial fluid were collected for
further analysis. (A) Relative expression of miR-93 was determined
by reverse transcription-quantitative polymerase chain reaction
analysis. Release of (B) TNF-α, (C) IL-1β, and (D) IL-6
inflammatory cytokines were measured using ELISA kits. (E)
Apoptotic cells were determined using TUNEL staining
(magnification, ×100). (F) Levels of TLR4, nuclear p-p65 and
p-IκB-α were measured by western blot analysis. β-actin protein was
used as the inner control of the cytoplasmic proteins; Histone H3
protein was used as the inner control of nuclear proteins. Data are
presented as the mean ± standard deviation of three independent
experiments. *P<0.05, **P<0.01, vs.
Sham group; ##P<0.01, vs. OA group. Toll-like
receptor 4; miR, microRNA; OA, osteoarthritis; NC, negative
control; TNF, tumor necrosis factor; IL, interleukin; NF-κB,
nuclear factor-κB; IκB-α, inhibitor of NF-κB-α; p-, phosphorylated;
TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP-biotin
nick-end labeling. |
Antagomir-93 exacerbates apoptosis and
inflammation in OA mice
Subsequently, the present study evaluated the
effects of miR-93 antagomir in OA mice. The mouse model of OA was
injected with antagomir-miR-93 (5 nmol) via intra-articular
injection. As shown in Fig. 7A,
the expression of miR-93 was significantly lower in the articular
cartilage tissues from OA + antagomir-miR-93 group than that in OA
group. The results of the ELISA analysis showed that the expression
levels of pro-inflammatory cytokines (TNF-α, IL-1β and IL-6) were
markedly higher in the OA + antagomir-miR-93 group than levels in
the OA group (Fig. 7B-D).
Furthermore, compared with the OA group, more TUNEL-positive cells
were observed in the OA + antagomir-miR-93 group (Fig. 7E). These data indicated that
antagomir-93 exacerbates inflammation and apoptosis in OA mice.
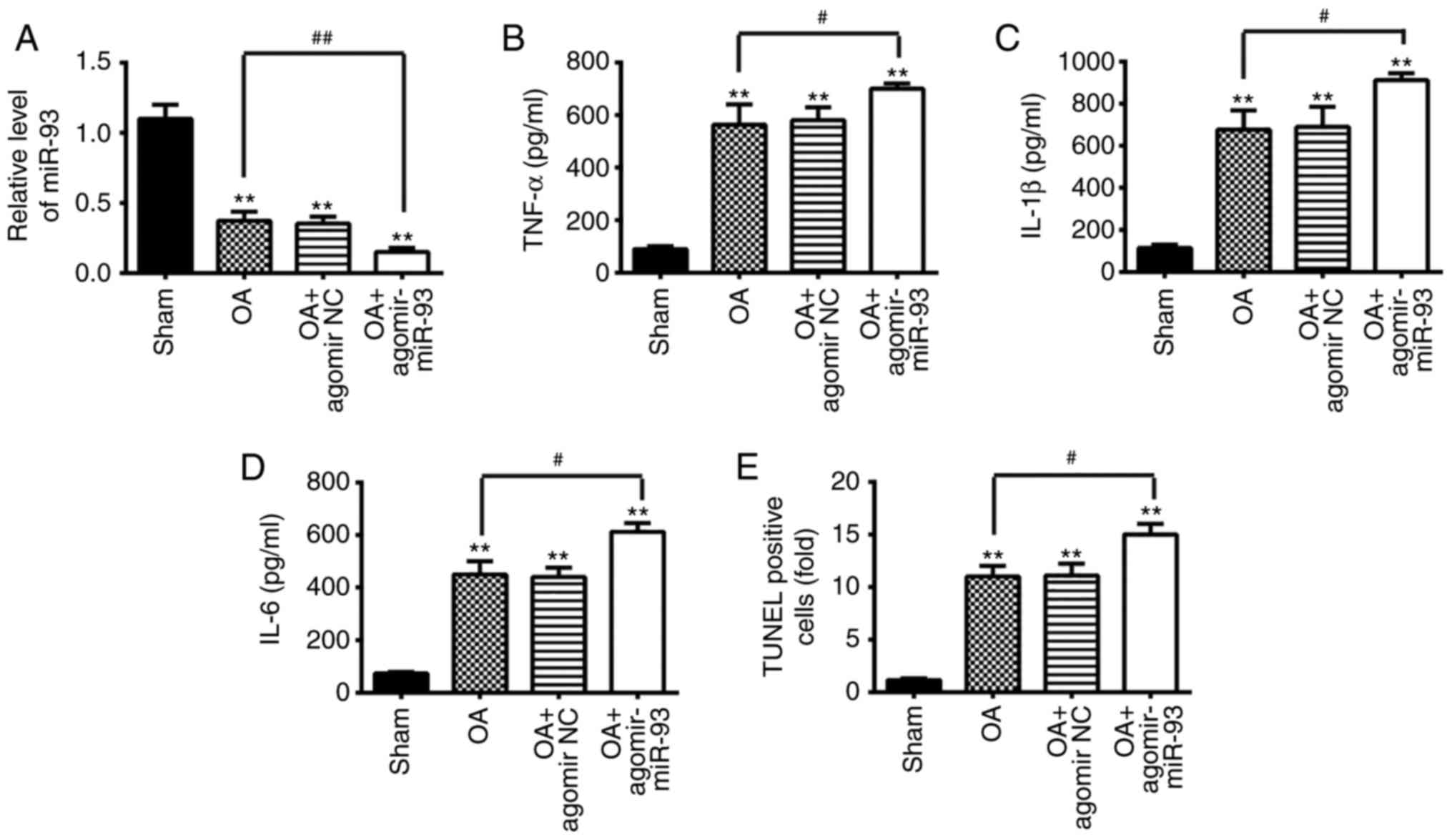 | Figure 7Antagomir-93 exacerbates apoptosis
and inflammation in OA mice. The mouse model of OA was injected
with antagomir-miR-93 (5 nmol) via intra-articular injection. The
mice were subjected to medial meniscectomy tear surgery and treated
with antagomir-negative control as the negative control. After 2
weeks, all mice were sacrificed. Subsequently, the articular
cartilages of the medial tibial plateau and the synovial fluid were
collected for further analysis. (A) Relative expression of miR-93
was determined by reverse transcription-quantitative polymerase
chain reaction analysis. Release of (B) TNF-α, (C) IL-1β, and (D)
IL-6 inflammatory cytokines was measured using ELISA kits. (E)
Apoptotic cells were determined using TUNEL staining. Data are
presented as the mean ± standard deviation of three independent
experiments. **P<0.01, vs. Sham group;
#P<0.05, ##P<0.01, vs. OA group.
Toll-like receptor 4; miR, microRNA; OA, osteoarthritis; NC,
negative control; TNF, tumor necrosis factor; IL, interleukin;
TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP-biotin
nick-end labeling. |
Discussion
The present study demonstrated that miR-93 was
significantly decreased in articular cartilage tissues from the OA
mice model and in the LPS-induced chondrocytes. The overex-pression
of miR-93 significantly inhibited LPS-induced cell apoptosis and
pro-inflammatory cytokine production in vitro and in
vivo. TLR4 was identified as a direct target of miR-93 and the
overexpression of TLR4 reversed the biological effects of miR-93.
In view of this, we speculate that miR-93 may potentially provide a
new strategy for the treatment of OA.
Increasing evidence has indicated that miRNAs are
involved in the pathogenesis of OA (4). For example, Wu et al showed
that the upregulation of miR-24 prevented the occurrence and
progression of OA through the mitogen-activated protein kinase
signaling pathway (26). miR-21
was found to be significantly increased in human OA tissues and the
overexpression of miR-21 improved chondrogenesis by targeting
growth differentiation factor 5 (27). Si et al found that the
expression of miR-140 was significantly reduced in human OA
chondrocytes, and intra-articular injection of miR-140 alleviated
the progression of OA by modulating cartilage extracellular matrix
homeostasis in rats (13). In the
present study, using a miRNA microarray, it was found that
miR-93was significantly downregulated in articular cartilage
tissues from the OA mice model and LPS-induced OA cell model. These
data suggest that miR-93 may be involved in the pathogenesis of
OA.
Previous studies have shown that miR-93 is important
in inflammatory diseases. For example, Ma et al found that
the upregulation of miR-93 reduced the inflammatory response by
negatively targeting SPP1 in mouse cardiac microvascular
endothelial cell injury (28).
Tian et al demonstrated that miR-93 was reduced in cerebral
ischemia reperfusion (CIR) mouse brains and that ago-miR-93
injection inhibited inflammatory responses and the rate of cell
apoptosis following CIR injury (19). Xu et al found that the
overexpression of miR-93 suppressed inflammatory cytokine
production in LPS-stimulated murine macrophages by targeting
interleukin-1 receptor-associated kinase 4 (29). To the best of our knowledge, no
other data are available on the roles of miR-93 in the regulation
of inflammatory responses associated with OA. In the present study,
the LPS-induced OA cell model was used to examine the regulatory
mechanism of miR-93 on inflammation and apoptosis. The results
showed that the overexpression of miR-93 suppressed the
inflammation and cell apoptosis induced by LPS in chondrocytes, and
in vivo. The findings also confirmed that miR-93 exerted its
protective effects against OA through suppressing inflammation and
apoptosis.
TLR4, one of the pathogen recognition receptors, has
received attention in OA for its ability to recognize microbial or
host-derived ligands found in OA (30,31). TLR4 has been shown to be expressed
in OA cartilage and on activated synoviocytes (32), and its expression in joint tissues
is increased with aging and with increasing severity of OA
(33). Li et al showed
that inhibiting the expression of TLR4 in cartilage lessened the
severity of OA in the rat model (34). miRNAs have been found to affect
the activation of TLR4 (35-38). For example, Chen et al
found that miR-20a negatively regulated TLR4 signaling under
atherosclerotic risk (39). A
previous study performed by Li et al showed that the
overexpression of miR-93 has a protective effect on an Angiotensin
II-induced cardiac hypertrophy model by directly targeting TLR4
(40). In the present study, TLR4
was identified as a target of miR-93 in the chondrocytes and
negatively regulated by miR-93. Therefore, it was hypothesized that
miR-93 protects chondrocytes from LPS-induced inflammation through
targeting TLR4 signaling. As expected, the overexpression of TLR4
significantly abrogated the inhibitory effects of miR-93 on
inflammation and apoptosis in LPS-induced chondrocytes. Taken
together, these results indicate that the miR-93/TLR4 axis may
represent a novel and promising target for the treatment of OA.
NF-κB is an important transcription factor and is
key in the induction of inflammatory injury (41). Upon stimulation by LPS, NF-κB
detaches from IκB and translocates into the nucleus to regulate
inflammatory cytokine expression, which induces destruction of the
articular joint, leading to the onset and progression of OA
(42). TLR4 has been reported as
an inducer of the NF-κB inflammatory signaling pathway (25,43). A previous study showed that the
TLR4/NF-kB signaling pathway is a vital mechanism for the
regulation of inflammatory responses in human OA chondrocytes
(44). Given the association
between TLR4 and miR-93, it was hypothesized that miR-93 is
important in inflammatory responses by mediating the TLR4/NF-κB
signaling pathway. In the present study, it was observed that the
overexpression of miR-93 significantly inhibited the LPS-induced
activation of NF-κB in vitro and in vivo. These
results suggested that miR-93 inhibited the LPS-induced
inflammatory response by inhibiting the TLR4/NF-κB signaling
pathway.
In conclusion, the present study demonstrated that
miR-93 inhibits LPS-induced inflammatory responses and cell
apoptosis via inhibition of the TLR4/NF-κB pathway. The
miR-93/TLR4/NF-κB axis may serve as a promising target for the
treatment of OA.
Funding
The present study was supported by the Fund of
Science and Technology Development Project of Kaifeng City (grant
no. 2017123) and the Scientific Research Project of Administration
of Traditional Chinese Medicine of Henan Province (grant no.
2017ZY3044).
Availability of data and materials
All data generated and analyzed during the present
study are included in this article.
Authors’ contributions
YD, LW and QZ performed the experiments, contributed
to data analysis and wrote the manuscript. YD, LW, QZ, ZW and LK
analyzed the data. QZ conceptualized the study design, and
contributed to data analysis and experimental materials. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All individuals provided informed consent for the
use of human specimens for clinical experiments. The present study
was approved by the Ethics Committees of Huaihe Hospital of Henan
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Taruc-Uy RL and Lynch SA: Diagnosis and
treatment of osteoarthritis. Prim Care. 40:821–836. vii2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fransen M, McConnell S, Harmer AR, Van der
Esch M, Simic M and Bennell KL: Exercise for osteoarthritis of the
knee: A Cochrane systematic review. Br J Sports Med. 49:1554–1557.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Aigner T, Söder S, Gebhard PM, McAlinden A
and Haag J: Mechanisms of disease: Role of chondrocytes in the
pathogenesis of osteoarthritis-structure, chaos and senescence. Nat
Clin Pract Rheumatol. 3:391–399. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Le LT and Clark IM: Review: The role of
microRNAs in osteoarthritis and chondrogenesis. Arthritis Rheum.
65:1963–1974. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang G, Sun Y, Wang Y, Liu R, Bao Y and
Li Q: MiR-502 5p inhibits IL-1β-induced chondrocyte injury by
targeting TRAF2. Cell Immunol. 302:50–57. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu C, Tian B, Qu X, Liu F, Tang T, Qin A,
Zhu Z and Dai K: MicroRNAs play a role in chondrogenesis and
osteoarthritis (review). Int J Mol Med. 34:13–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Junker A, Krumbholz M, Eisele S, Mohan H,
Augstein F, Bittner R, Lassmann H, Wekerle H, Hohlfeld R and Meinl
E: MicroRNA profiling of multiple sclerosis lesions identifies
modulators of the regulatory protein CD47. Brain. 132:3342–3352.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang H, Song B and Pan Z: Downregulation
of microRNA-9 increases matrix metalloproteinase-13 expression
levels and facilitates osteoarthritis onset. Mol Med Rep.
17:3708–3714. 2018.
|
|
11
|
Lu J, Ji ML, Zhang XJ, Shi PL, Wu H, Wang
C and Im HJ: MicroRNA-218-5p as a potential target for the
treatment of human osteoarthritis. Mol Ther. 25:2676–2688. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qi J, Qiao Y, Wang P, Li S, Zhao W and Gao
C: microRNA-210 negatively regulates LPS-induced production of
proinflammatory cytokines by targeting NF-κB1 in murine
macrophages. FEBS Lett. 586:1201–1207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Si HB, Zeng Y, Liu SY, Zhou ZK, Chen YN,
Cheng JQ, Lu YR and Shen B: Intra-articular injection of
microRNA-140 (miRNA-140) alleviates osteoarthritis (OA) progression
by modulating extracellular matrix (ECM) homeostasis in rats.
Osteoarthritis Cartilage. 25:1698–1707. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Glasson SS, Blanchet TJ and Morris EA: The
surgical destabilization of the medial meniscus (DMM) model of
osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage.
15:1061–1069. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang Q, Zhang D and Li Y and Li Y and Li
Y: Paclitaxel alleviated liver injury of septic mice by alleviating
inflammatory response via microRNA-27a/TAB3/NF-κB signaling
pathway. Biomed Pharmacother. 97:1424–1433. 2018. View Article : Google Scholar
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
17
|
Liu LJ, Yu JJ and Xu XL: MicroRNA-93
inhibits apoptosis and promotes proliferation, invasion and
migration of renal cell carcinoma ACHN cells via the TGF-beta/Smad
signaling pathway by targeting RUNX3. Am J Transl Res. 9:3499–3513.
2017.
|
|
18
|
Ke ZP, Xu P, Shi Y and Gao AM: MicroRNA-93
inhibits ischemia-reperfusion induced cardiomyocyte apoptosis by
targeting PTEN. Oncotarget. 7:28796–28805. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tian F, Yuan C, Hu L and Shan S:
MicroRNA-93 inhibits inflammatory responses and cell apoptosis
after cerebral ischemia reperfusion by targeting interleukin-1
receptor-associated kinase 4. Exp Ther Med. 14:2903–2910. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Canagarajah BJ, Khokhlatchev A, Cobb MH
and Goldsmith EJ: Activation mechanism of the MAP kinase ERK2 by
dual phosphorylation. Cell. 90:859–869. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang D, Zhao Q, Liu H, Guo Y and Xu H:
PPAR-α agonist WY-14643 inhibits LPS-induced inflammation in
synovial fibroblasts via NF-κB pathway. J Mol Neurosci. 59:544–553.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao C, Wang Y, Jin H and Yu T: Knockdown
of microRNA-203 alleviates LPS-induced injury by targeting MCL-1 in
C28/I2 chondrocytes. Exp Cell Res. 359:171–178. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Q, Rozelle AL, Lepus CM, Scanzello
CR, Song JJ, Larsen DM, Crish JF, Bebek G, Ritter SY, Lindstrom TM,
et al: Identification of a central role for complement in
osteoarthritis. Nat Med. 17:1674–1679. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen C, Chen W, Li Y, Dong Y, Teng X, Nong
Z, Pan X, Lv L, Gao Y and Wu G: Hyperbaric oxygen protects against
myocardial reperfusion injury via the inhibition of inflammation
and the modulation of autophagy. Oncotarget. 8:111522–111534. 2017.
View Article : Google Scholar
|
|
25
|
Cao C, Yin C, Shou S, Wang J, Yu L, Li X
and Chai Y: Ulinastatin protects against LPS-induced acute lung
injury by attenuating TLR4/NF-κB pathway activation and reducing
inflammatory mediators. Shock. 50:595–605. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu YH, Liu W, Zhang L, Liu XY, Wang Y, Xue
B, Liu B, Duan R, Zhang B and Ji Y: Effects of microRNA-24
targeting C-myc on apoptosis, proliferation and cytokine
expressions in chondrocytes of rats with osteoarthritis via MAPK
signaling pathway. J Cell Biochem. 119:7944–7958. 2018. View Article : Google Scholar
|
|
27
|
Zhang Y, Jia J, Yang S, Liu X, Ye S and
Tian H: MicroRNA-21 controls the development of osteoarthritis by
targeting GDF-5 in chondrocytes. Exp Mol Med. 46:e792014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma SX, Bai ZF, Wang W and Wu HY: Effects
of Microrna-93 on mouse cardiac microvascular endothelial cells
injury and inflammatory response by mediating SPP1 through the
NF-κB pathway. J Cell Biochem. Dec 12–2017. View Article : Google Scholar
|
|
29
|
Xu Y, Jin H, Yang X, Wang L, Su L, Liu K,
Gu Q and Xu X: MicroRNA-93 inhibits inflammatory cytokine
production in LPS-stimulated murine macrophages by targeting IRAK4.
FEBS Lett. 588:1692–1698. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Abella V, Scotece M, Conde J, López V,
Pirozzi C, Pino J, Gómez R, Lago F, González-Gay MÁ and Gualillo O:
The novel adipokine progranulin counteracts IL-1 and TLR4-driven
inflammatory response in human and murine chondrocytes via TNFR1.
Sci Rep. 6:203562016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang P, Zhu F, Tong Z and Konstantopoulos
K: Response of chondrocytes to shear stress: Antagonistic effects
of the binding partners Toll-like receptor 4 and caveolin-1. FASEB
J. 25:3401–3415. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim HA, Cho ML, Choi HY, Yoon CS, Jhun JY,
Oh HJ and Kim HY: The catabolic pathway mediated by Toll-like
receptors in human osteoarthritic chondrocytes. Arthritis Rheum.
54:2152–2163. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Goldring MB: Chondrogenesis, chondrocyte
differentiation, and articular cartilage metabolism in health and
osteoarthritis. Ther Adv Musculoskelet Dis. 4:269–285. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J, Xie ZG, Xie Y and Dong QR:
Calcitonin treatment is associated with less severe osteoarthritis
and reduced toll-like receptor levels in a rat model. J Orthop Sci.
19:1019–1027. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen Y, Wu Z, Yuan B, Dong Y, Zhang L and
Zeng Z: MicroRNA-146a-5p attenuates irradiation-induced and
LPS-induced hepatic stellate cell activation and hepatocyte
apoptosis through inhibition of TLR4 pathway. Cell Death Dis.
9:222018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bao CX, Zhang DX, Wang NN, Zhu XK, Zhao Q
and Sun XL: MicroRNA-335-5p suppresses lower extremity deep venous
thrombosis by targeted inhibition of PAI-1 via the TLR4 signaling
pathway. J Cell Biochem. 119:4692–4710. 2018. View Article : Google Scholar
|
|
37
|
Wang Y, Zheng F, Gao G, Yan S, Zhang L,
Wang L, Cai X, Wang X, Xu D and Wang J: MiR-548a-3p regulates
inflammatory response via TLR4/NF-kappaB signaling pathway in
rheumatoid arthritis. J Cell Biochem Jan. 6:2018. View Article : Google Scholar
|
|
38
|
Jiang W, Liu G and Tang W: MicroRNA-182-5p
ameliorates liver ischemia-reperfusion injury by suppressing
toll-like receptor 4. Transplant Proc. 48:2809–2814. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen M, Li W, Zhang Y and Yang J:
MicroRNA-20a protects human aortic endothelial cells from
Ox-LDL-induced inflammation through targeting TLR4 and TXNIP
signaling. Biomed Pharmacother. 103:191–197. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li Y, Wang J, Sun L and Zhu S: LncRNA
myocardial infarction-associated transcript (MIAT) contributed to
cardiac hypertrophy by regulating TLR4 via miR-93. Eur J Pharmacol.
818:508–517. 2018. View Article : Google Scholar
|
|
41
|
Oeckinghaus A and Ghosh S: The NF-kappaB
family of transcription factors and its regulation. Cold Spring
Harb Perspect Biol. 1:a0000342009. View Article : Google Scholar
|
|
42
|
Rigoglou S and Papavassiliou AG: The NF-κB
signalling pathway in osteoarthritis. Int J Biochem Cell Biol.
45:2580–2584. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cao SG, Chen R, Wang H, Lin LM and Xia XP:
Cryptotanshinone inhibits prostaglandin E2 production and COX-2
expression via suppression of TLR4/NF-kappaB signaling pathway in
LPS-stimulated Caco-2 cells. Microb Pathog. 116:313–317. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fu Y, Lei J, Zhuang Y, Zhang K and Lu D:
Overexpression of HMGB1 A-box reduced IL-1beta-induced MMP
expression and the production of inflammatory mediators in human
chon-drocytes. Exp Cell Res. 349:184–190. 2016. View Article : Google Scholar : PubMed/NCBI
|