Introduction
Non-alcoholic fatty liver disease (NAFLD) is the
most widespread metabolic syndrome characterized by aberrant lipid
accumulation in the hepatocyte cytoplasm, affecting ~25% of adults
worldwide (1). NAFLD refers to a
variety of liver diseases ranging from non-alcoholic simple fatty
liver, to liver fibrosis/cirrhosis, non-alcoholic steatohepatitis
(NASH) and NASH-associated hepatocellular carcinoma (NASH-HCC)
(2,3). NASH is a form of NAFLD characterized
by steatosis, hepatocellular necrosis and fibrosis, and is
associated with the development of obesity, metabolic syndrome and
diabetes (4,5). There is increasing evidence that
NASH can eventually lead to irreversible liver injury, or even the
development and progression of HCC (6). Therefore, basic and clinical studies
in the coming decades are focusing on the pathogenesis and latent
therapeutic targets of NAFLD.
Traditionally, the development of NAFLD has been
associated with genetics, systolic blood pressure, serum
cholesterol, fasting glucose, sex, age and waist circumference
(7). It has been gradually
realized that sirtuins and associated metabolic processes are also
involved in the development of NAFLD (8). Sirtuins (SIRT1-7) have a spectrum of
functions through regulating post-translational protein
modification. SIRT1 is closely associated with cellular metabolism
through deacetylating cellular proteins at the post-translational
level, and serves an important role in a variety of metabolic
diseases, including NAFLD (9).
Quaking (QKI) is a member of the STAR family of
RNA-binding proteins with diverse functions in mRNA stability
(10,11) and translation (12,13), microRNA processing (14,15) and alternative splicing (13,16–20). It was demonstrated that SIRT1
increased the acetylation level of QKI 5 through inhibiting SIRT1
activity. By contrast, increased SIRT1 activity resulted in QKI 5
deacetylation. The acetylation level of QKI 5 was increased in
liver cells in which the SIRT1 gene was silenced and in the liver
tissues of SIRT1-knockdown mice (13–15,18). These results implicate alternative
splicing as a key regulatory factor in the lipid metabolism of
hepatocytes.
In the present study, the effect of QKI 5 mediated
by SIRT1 on triglyceride synthesis of the liver in an NAFLD model
was investigated. The NAFLD model mice, induced by a high-fat diet
(HFD), was successfully established, and mouse hepatocytes were
isolated to characterize the effects of QKI 5 mediated by SIRT1 on
the synthesis of triglycerides in the liver and the activation of
the peroxisome proliferator-activated receptor (PPAR)α, Forkhead
box protein O1 (FoxO1) signaling pathway.
Materials and methods
Animal experiment
C57BL/6 mice (male, 8–10-weeks old) obtained from
the Animal Experimental Center at Tsinghua University (Beijing,
China) was housed with standard cages at constant room temperature
(22±2°C) and relative humidity of 45±15%, with free access to food
and water on a 12/12 h light/dark cycle. Then, these mouse were fed
with standard food as controls or the HFD (fat 60%, carbohydrate
20.6%, protein 19.4%; Research Diets, Inc., New Brunswick, NJ, USA)
for 8 weeks. All mice were then sacrificed. The liver wet weight
and body weight were measured, and alanine aminotransferase (ALT),
and aspartate aminotransferase (AST) were detected using an
automatic biochemical analyzer. The histopathological changes of
the liver were evaluated by hematoxylin and eosin (H&E)
staining. Furthermore, the level of SIRT1 in the serum was
determined using the ELISA method.
To investigate the functional roles of QKI 5 in
NAFLD, 24 C57BL/6 mice were randomized into four groups as follows
(n=6 per group): Control group, mice fed with a HFD (model group),
mice fed with a HFD and treated with 100 mg/kg/day per os of
SRT1720 (SRT1720 group) or vehicle (1% DMSO in 20% cyclodextrin;
vehicle group) (21). Following 8
weeks of continuous administration, the mice were sacrificed. Blood
samples were collected for the determination of serum SIRT1, ALT,
AST, total cholesterol (TC) and triglyceride (TG) levels.
Additionally, liver tissues were subjected to pathological H&E
staining, determination of NAFLD activity score (NAS), and an assay
of the synthesis of TG. All animal experiments were approved by the
Animal Ethics Committee of Tsinghua University.
Metabolic phenotyping
The mouse body composition, including fat mass, was
assessed with non-invasive quantitative magnetic resonance using an
EchoMRI700 instrument. Values are expressed as a percentage of body
weight (BW). All experiments were performed at Tsinghua University.
Homogenization and protein extraction from the liver
tissue-homogenization and extraction of individual liver sections
were performed in NP-40 lysis buffer.
Liver histology and analysis
To observe the liver histo-pathological changes,
standard H&E staining was performed. Briefly, following
dehydration with an ethanol gradient, the liver tissues fixed using
formalin (4%) were embedded in paraffin, then cut into 5-µm
sections. The sections were then stained with H&E or Oil Red O
staining, and immunostaining with anti-SIRT1 or anti-QKI 5 was
performed. The slides were mounted with aqueous mountant and viewed
with a fluorescent microscope (IX71; Olympus Corporation, Tokyo,
Japan). Hepatic TG and TC or TG in cells were measured using the
infinity TG and TC reagent kits (cat. nos. TR13421 and TR-22421;
Thermo Fisher Scientific, Inc., Waltham, MA, USA),
respectively.
Circulating TG, TC, ALT and AST
content
The serum levels of TG, TC, ALT and AST were
assessed with an automatic biochemical analyzer (Olympus
Corporation).
Serum SIRT1 measurement
Based on the manufacturer's protocol, the serum
SIRT1 concentration was measured by ELISA using an SIRT1 mouse
ELISA kit (Abbexa, Cambridge, UK).
Pathological examination and assessment
of disease severity
For liver biopsies of NAFLD, the novel NAS system is
suggested for the grading of steatosis, hepatocellular ballooning
and inflammatory activity, referring to a guide on the diagnosis
and treatment for NAFLD (22).
Mouse hepatocyte isolation and
treatment
Mouse hepatocytes were dissociated from the tissues
of the C57BL/6 mice using an in situ recirculating
collagenase perfusion method (23). The liver tissues were transferred
into Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher
Scientific, Inc.) on ice and filtered through a multi-layer gauze
to obtain a hepatocyte suspension. Following washing with PBS, the
supernatant was discarded. The primary hepatocytes were maintained
in DMEM with 10% fetal bovine serum (FBS; Thermo Fisher Scientific,
Inc.) for cell adherence with 5% CO2 at 37°C for 4–6
h.
The obtained cells were rinsed with fresh medium to
remove the dead cells and cell fragments, and then washed with PBS
and re-suspended in serum-free medium containing 50 ng/ml SRT1720
or 10 µM niacinamide for 48 h at 37°C, respectively. The
cells were collected for synthesis of TG at each time point using a
TG assay kit for quantification (cat. no. ab65336; Abcam,
Cambridge, MA, USA).
To initially investigate the effect of SRT1720
treatment on the FOXO1 and PPARα signaling pathway, the mouse
hepatocytes (5×105 cells/well) were cultured in a
12-well plate at 37°C, and treated with either DMSO (control),
SRT1720 (50 ng/ml), FOXO1 inhibitor AS1842856 (100 nM; cat. no.
344355; Merck KGaA, Darmstadt, Germany) or SRT1720 (50 ng/ml) +
FOXO1 inhibitor AS1842856 (10 nM). Following 48 h of treatment, the
protein expression levels of FOXO1 and PPARα were determined by
western blotting. The synthesis of TGs in the mouse hepatocytes was
detected.
Adenovirus construction and
transfection
Recombinant adenoviruses (Ads) containing SIRT1 were
produced with an AdEasy (Ad) Vector system (Stratagene; Agilent
Technologies, Inc., Santa Clara, CA, USA). The concentration of 10
plaque-forming units/hepatocyte was used to infect cells. AdGFP was
used as the infection control.
Cell transfection
Whether SIRT1 inhibited the activation of FOXO1 was
then determined, in addition to the PPARα signaling pathway, in
mouse hepatocytes. The mouse hepatocytes were transfected using
Invitrogen™ Lipofectamine® 2000 Transfection Reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) with either scrambled
small interfering (si)RNA or siRNA of SIRT1, followed by treatment
with or without SRT1720 (50 ng/ml). Western blotting was used to
quantify the protein levels of SIRT1, and the synthesis of TGs was
measured. The sequences of scrambled siRNA and siRNA of SIRT1 were
as follows: Sense, 5′-ACUUUGCUGUAACCCUGUAdTdT-3′ and antisense,
5′-UACAGGGUUACAGCAAAGUdTdT-3′ (scrambled siRNA); sense,
5′-CCUACGCCACCAAUUUCGU-3′ and antisense, 5′-ACGAAAUUGGUGGCGUAGG-3′
(siRNA of SIRT1).
Western blotting
A lysis buffer (150 mM NaCl, 0.1% SDS, 0.02%
NaN3, 1% NP-40, and 50 mM pH 8.0 Tris) containing a
cocktail of inhibitors for proteases and phenylmethylsulfonyl
fluoride (1 mM) was used to lyse cells. Western blotting was
performed using the Bio-Rad Protean II minigel system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The concentrations of total
proteins were quantified by bicinchoninic acid assay. A 50
µg protein sample was injected into every gel (12%) well,
and then transferred onto a polyvinylidene difluoride membrane (EMD
Millipore, Bedford, MA, USA) following electrophoresis.
Subsequently, 1× Tween-20 TBS (5% dry skimmed milk) was used to
block the non-specific binding. The membranes were then
sequentially incubated with primary at 4°C overnight, and secondary
antibodies for 1 h at room temperature. The membranes were
visualized with ECL chemiluminescence reagent using the Hyperfilm
ECL kit, and exposed using x-ray film. The primary antibodies
included anti-SIRT1 (D739; cat. no. CST 2493; Cell Signaling
Technology, Inc., Danvers, MA, USA), QKI 5 (cat. no. BL 1041;
Bethyl Laboratories, Inc., Montgomery, TX, USA), FOXO1 (L27; cat.
no. CST 9454; Cell Signaling Technology, Inc.), PPARα (467D1a; cat.
no. sc-130640; Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
with dilution 1:1,000, and β-actin (13E5; cat. no. CST 4970S;
1:4,000; Cell Signaling Technology, Inc.). The secondary antibody
was horseradish peroxidase-conjugated secondary antibody (cat. no.
CST 4410S; Cell Signaling Technology, Inc.).
Determination of total TG in cells
Mouse primary hepatocytes were seeded into a 12-well
cell culture plate, following treatment with SRT1720 for 48 h as
described above. The supernatant was discarded, then
3×105 cells were collected and lysed. The cell lysate
was transferred into a 1.5-ml centrifuge tube and a sample was
obtained to be analyzed. The standard protein concentration
gradient was performed according to the kit protocol. A total of 50
µl of gradient standard and 10 µl of the sample were
added into the 96-well microtiter plate. Following the addition of
lipase into the main reaction system, the absorbance at 570 nm was
measured with a microplate reader, and the absorbance of the sample
was calculated according to the standard curve. The concentration
of TG was normalized by the quantity of protein per unit
(µg).
Analysis of acetylation modification of
QKI 5 induced by SIRT1
The local tool of KA-predictor, which can be freely
downloaded (http://sourceforge.net/p/ka-predictor), was used to
predict the acetylation modification site of QKI 5.
Immunoprecipitation (IP) with acetylated
antibody
The cells/tissues were fully lysed with IP lysate
and then centrifuged at 6,720 × g for 10 min at 4°C to remove the
precipitate. The protein (1 mg) solution was added to 20 µl
agarose cross-linked acetylated lysine antibody (acetyl lysine
antibody, agarose) (cat. no. CST 9441S; Cell Signaling Technology,
Inc.; dilution, 1:800). Following mixing and incubation overnight
at 4°C, the supernatant was discarded. The sediment was washed five
times with ice pre-cooled PBS, following which 50 µl of
elution buffer was used to elute the proteins which were mixed
gently. Following centrifugation at 336 × g for 1 min at 4°C, the
IP product was assessed using the aforementioned western blotting
protocol. The primary antibody of QKI 5 (cat. no. BL 1041; Bethyl
Laboratories, Inc.; dilution, 1:500) was used to detect QKI 5
content in the IP products.
Statistical analysis
The differences between two groups were statistical
analyzed using Student's t-test. Normally distributed continuous
variables are presented as the mean ± standard deviation.
Abnormally distributed data among groups was analyzed with the
Kruskal-Wallis one-way analysis of variance method. SPSS (version
18.0) software (SPSS, Inc., Chicago, IL, USA) was used for all
statistical analyses. P<0.05 was considered to indicate a
statistically significant difference.
Results
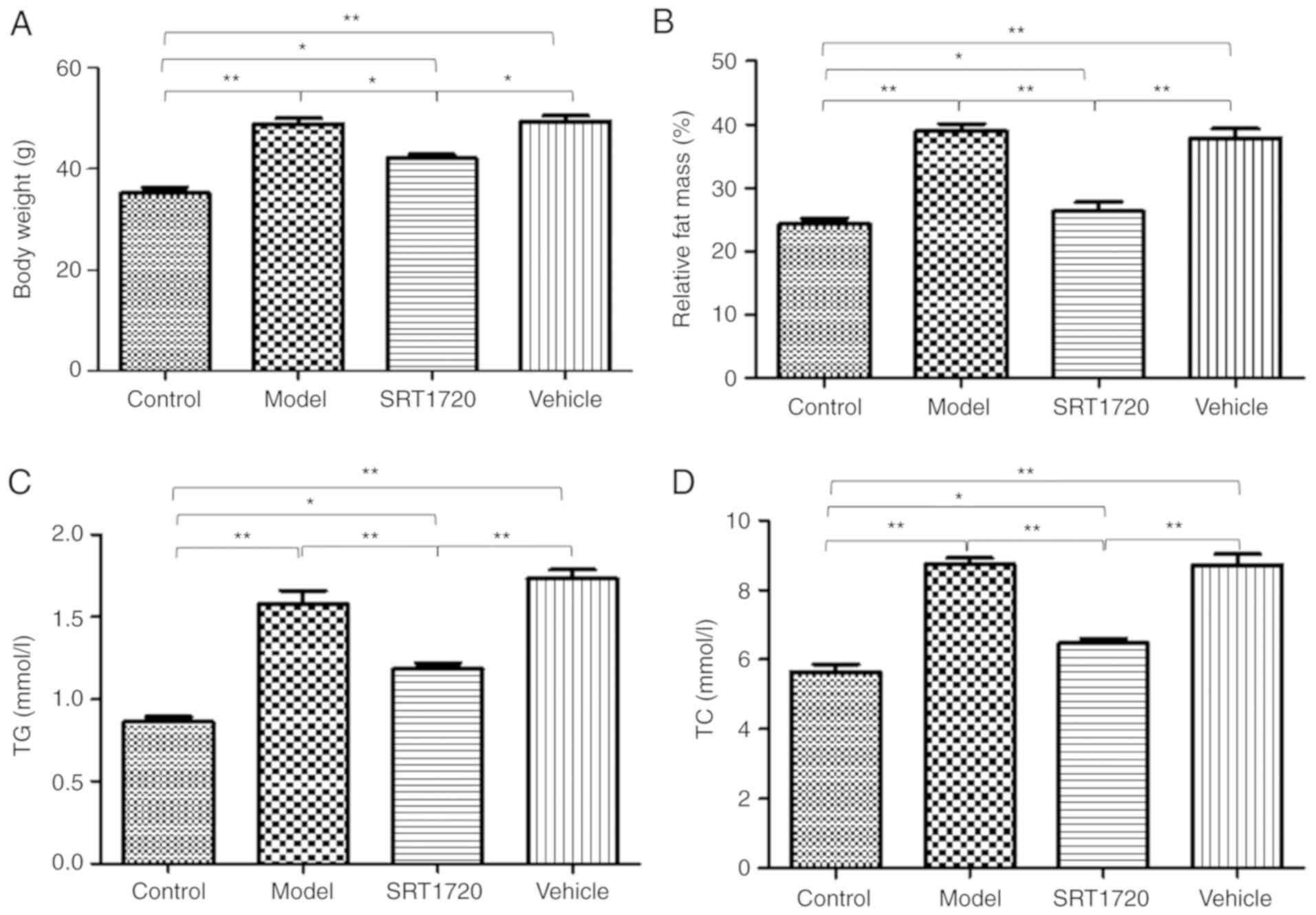
Metabolic disorders of HFD mice are
reversed by SRT1720
The mice fed the HFD became obese at 8 months of
age. The BW (Fig. 1A) and the
relative fat mass ratio (Fg. 1B)
of the mice in the model group and vehicle group increased
significantly, but were only enhanced ~20% in the mice treated with
SRT1720. Furthermore, the model and vehicle mice developed
hyperlipidemia, as measured by total plasma TG (Fig. 1C) and TC levels (Fg. 1D). Furthermore, these changes were
reversed by SRT1720. Consistent with an increase in plasma lipids,
the HFD model mice exhibited significantly enlarged fatty livers at
8 months of age (Fig. 1E and
1F), which was reduced by
SRT1720.
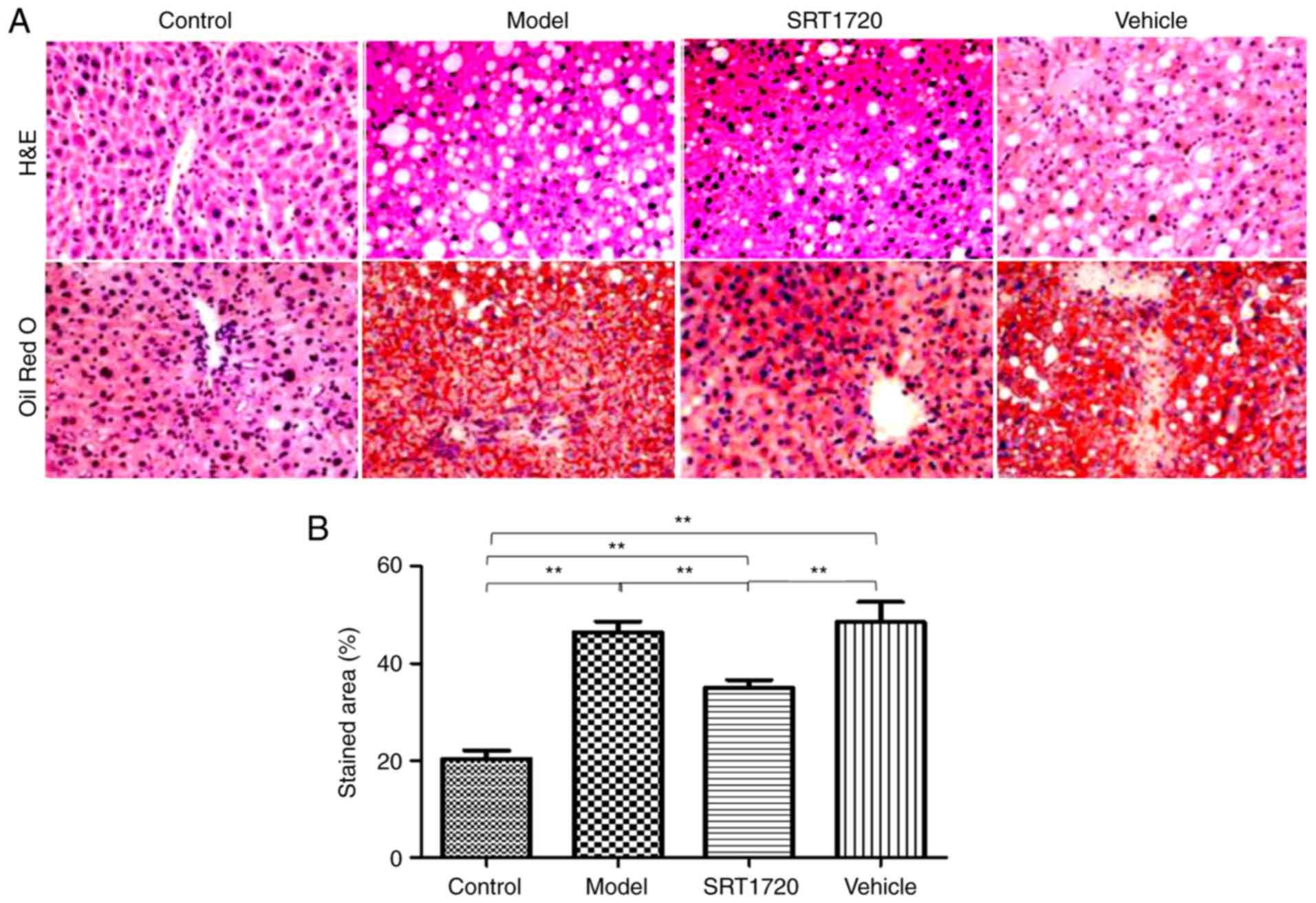
HFD mice develop NAFLD, which is reversed
by SRT1720
The hepatic lipid content of the HFD model mice at 8
months of age was significantly increased compared with their
litter-mate controls, as separately measured using H&E and Oil
Red O staining of liver sections (Fig. 2A), and quantification of Oil Red O
staining (Fig. 2B). These results
indicated the typical pathology of liver steatosis. Furthermore,
the TG (Fig. 2C) and TC (Fig. 2D) contents of the extracted liver
tissues from the HFD model were significantly increased compared
with those of the controls, but were reduced by SRT1720. This
demonstrated that the developmental process of liver steatosis was
reversed by SRT1720 (Fig. 2).
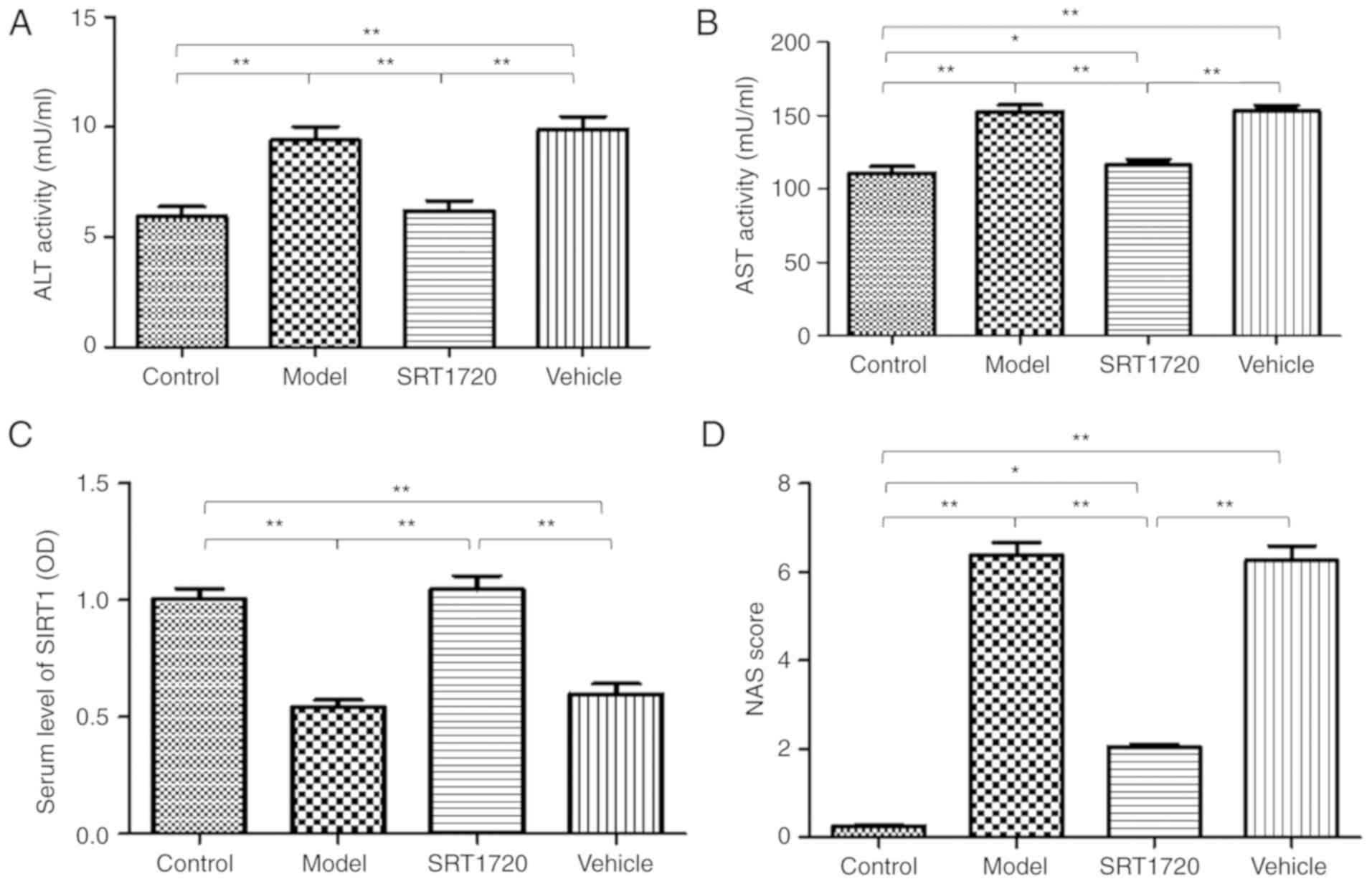
In addition, mildly increased plasma activities of
ALT (Fig. 3A) and AST (Fig. 3B) were identified in the model
mice. However, the mice treated with SRT1720 demonstrated no signs
of liver damage or inflammatory changes in plasma ALT or AST
activity. The serum level of SIRT1 in the model mice was
significantly decreased compared with that in the control group,
however, this was reversed by SRT1720 (Fig. 3C). Furthermore, the NAS was
elevated in the model mice; the results revealed significant
differences in the model and vehicle groups compared with the
control group mice. The NAS of the group treated with SRT1720 was
reduced compared with that in the model and vehicle groups
(Fig. 3D), however, the NAS score
of the SRT1720 group was increased compared with that in the
control mice. These results suggested that the administration of
SRT1720 improved the pathophysiological process of NAFLD in
mice.
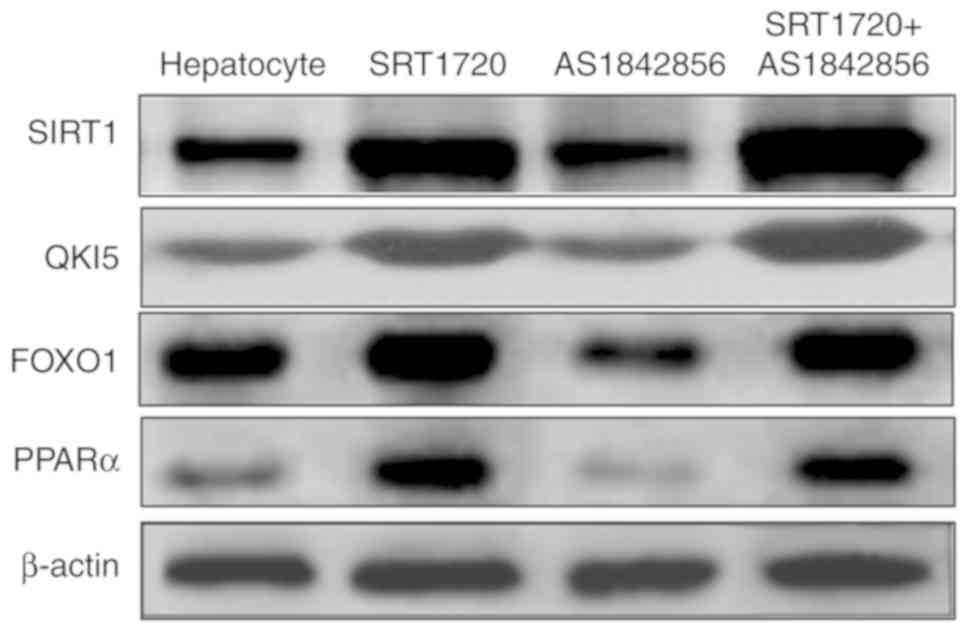
Hepatic SIRT1 regulates the synthesis of
TGs in NAFLD mice via the PPARα/FoxO1 signaling pathway in
vitro
An inhibitor of FOXO1 was also used in the present
study. The results, as shown in Fig.
4, demonstrated that the inhibitor of FOXO1 (AS1842856)
suppressed FOXO1 and PPARα, however, this was reversed by SRT1720.
AS1842856 did not inhibit the expression levels of SIRT1 or QKI 5.
This demonstrated that hepatic SIRT1 regulated the expression of
QKI 5 via the PPARα/FoxO1 signaling pathway.
SIRT1 regulates the acetylation level of
QKI 5
In the liver tissues of the model mice, the
acetylation level of QKI 5 increased, and this was reversed by
SRT1720 (Fig. 5A). An increase in
the acetylation level of QKI 5 was induced by the inhibitor of
SIRT1 (niacinamide), whereas the acetylation of QKI 5 decreased in
hepatocytes treated with SRT1720 (Fig. 5B). Furthermore, the increased
acetylation level of QKI 5 was induced by siRNA of SIRT1, whereas
the acetylation of QKI 5 decreased in hepatocytes treated with
Ad-SIRT1 (Fig. 5C). The results
of the protein interaction analysis confirmed the interaction
between SIRT1 and QKI 5, and its level in the SRT1720 group was
higher than in the model mice (Fig.
5D and E).
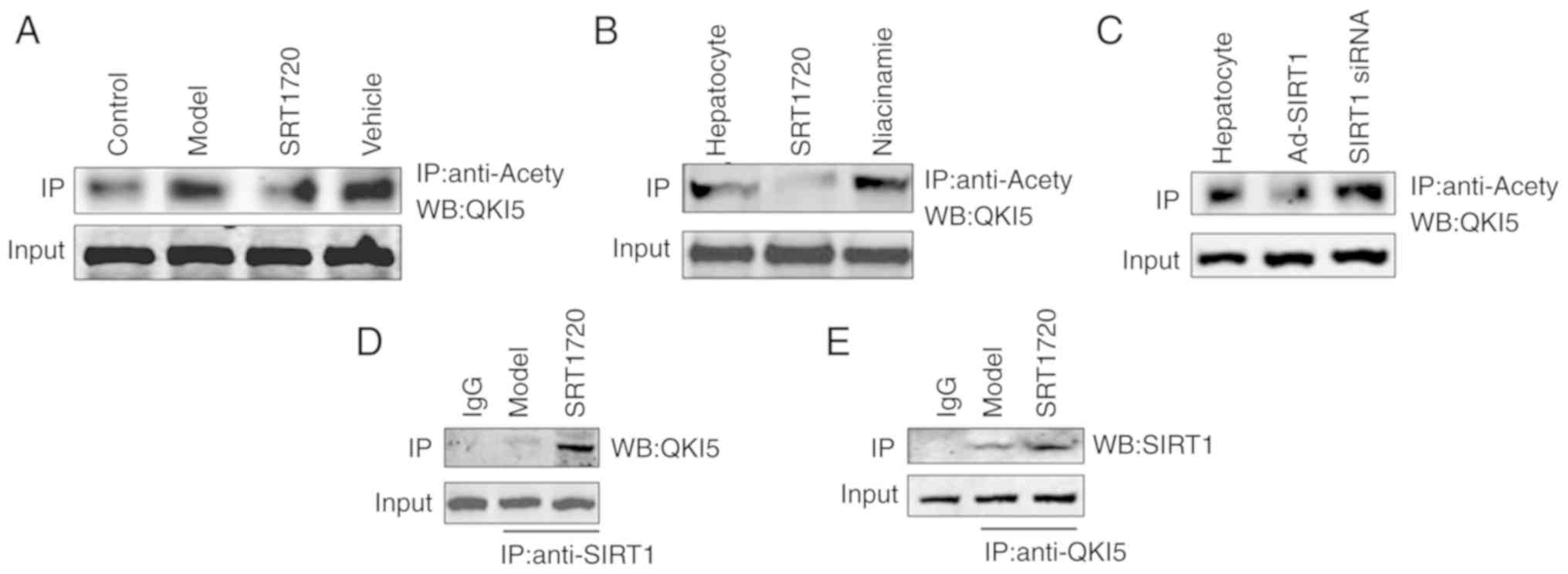 | Figure 5SIRT1 regulates the acetylation level
of QKI 5. (A) In the liver tissues of model mice, the acetylation
level of QKI 5 increased, but this was reversed by SRT1720. (B)
Increased acetylation of QKI 5 was induced by the inhibition of
SIRT1 (niacinamide), but was decreased in hepatocytes treated with
SRT1720. (C) Increasing acetylation of QKI 5 was induced by siRNA
of SIRT1, but was decreased in hepatocytes treated with Ad-SIRT1.
The results of protein interaction confirmed the interaction
between (D) SIRT1 and (E) QKI 5, and its level in the SRT1720 group
was increased compared with model mice. QKI 5, Quaking 5; SIRT1,
Sirtuin 1; siRNA, small interfering RNA; Ad, adenovirus; IP,
immunoprecipitation; WB, western blotting. |
Hepatic SIRT1 regulates the synthesis of
TGs in NAFLD mice via the PPARα/FoxO1 signaling pathway in
vivo
To evaluate the ability of hepatic SIRT1 to maintain
lipid homeostasis, the agonist (SRT1720) and inhibitor
(niacinamide) of SIRT1 was used in the present study. Furthermore,
Ad-mediated gene repletion of SIRT1 was used in mice primary
hepatocytes, and siRNA of SIRT1 was used to downregulate the
expression of SIRT1.
In the primary hepatocytes, SRT1720 enhanced the
expression of SIRT1, and the expression levels of QKI 5, FOXO1 and
PPARα were increased. However, the expression level of SIRT1 was
reduced by niacinamide, which also induced the downregulation of
QKI 5, FOXO1 and PPARα (Fig. 6A).
In addition, a decrease in intracellular TG content was induced by
SRT1720, whereas niacinamide increased the content of TG in the
primary hepatocytes (Fig. 6B).
Additionally, in the primary hepatocytes, Ad-SIRT1 enhanced the
expression of SIRT1 QKI 5, FOXO1 and PPARα, and this was inhibited
by SIRT1 siRNA (Fig. 6C). SRT1720
reduced the TG content of the primary hepatocytes, which was
promoted by SIRT1 siRNA (Fig.
6D). Therefore, SIRT1 mediated the synthesis of TGs in NAFLD
mice, and this was associated with the QKI 5 and the PPARα/FoxO1
signaling pathway.
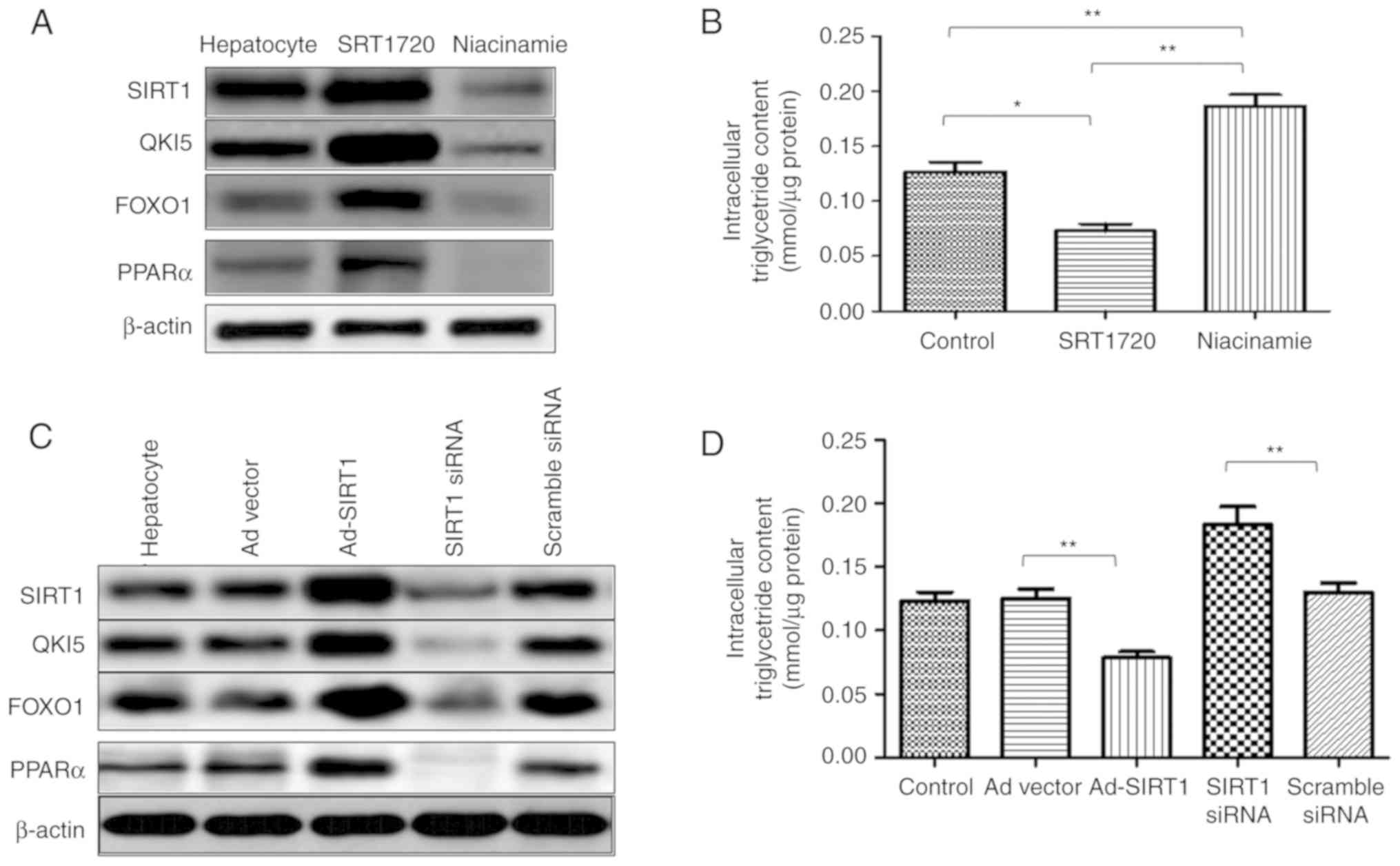 | Figure 6Hepatic SIRT1 regulates the synthesis
of triglycerides in non-alcoholic fatty liver disease mice via the
PPARα/FoxO1 signaling pathway in vivo. In primary
hepatocytes, SRT1720 enhanced the expression of SIRT1, QKI 5, FOXO1
and PPARα. (A) Expression of SIRT1 was reduced by niacinamide,
which also induced the downregulation of QKI 5, FOXO1 and PPARα.
(B) Decreased intracellular triglyceride content was caused by
SRT1720, whereas niacinamide enhanced the triglyceride content in
primary hepatocytes. (C) In primary hepatocytes, Ad-SIRT1 enhanced
the expression of SIRT1, QKI 5, FOXO1 and PPARα, which was
inhibited by siRNA of SIRT1. (D) SRT1720 reduced the triglyceride
content in primary hepatocytes, which was promoted by SIRT1 siRNA.
The data are presented as the mean ± standard deviation from three
independent experiments. *P<0.05 and
**P<0.01. PPARα, peroxisome proliferator-activated
receptor α; FOXO1, Forkhead box protein O1; QKI 5, Quaking 5;
SIRT1, Sirtuin 1; siRNA, small interfering RNA; Ad, adenovirus. |
The results of the immunohistochemistry assay
indicated that, the expression levels of SIRT1 and QKI 5 were
downregulated in the NAFLD model mice, but were promoted by SRT1720
(Fig. 7A). In addition, the
western blot assay demonstrated that the expression levels of
SIRT1, QKI 5, FOXO1 and PPARα were decreased in the model mice, and
this was also reversed by SRT1720 (Fig. 7B).
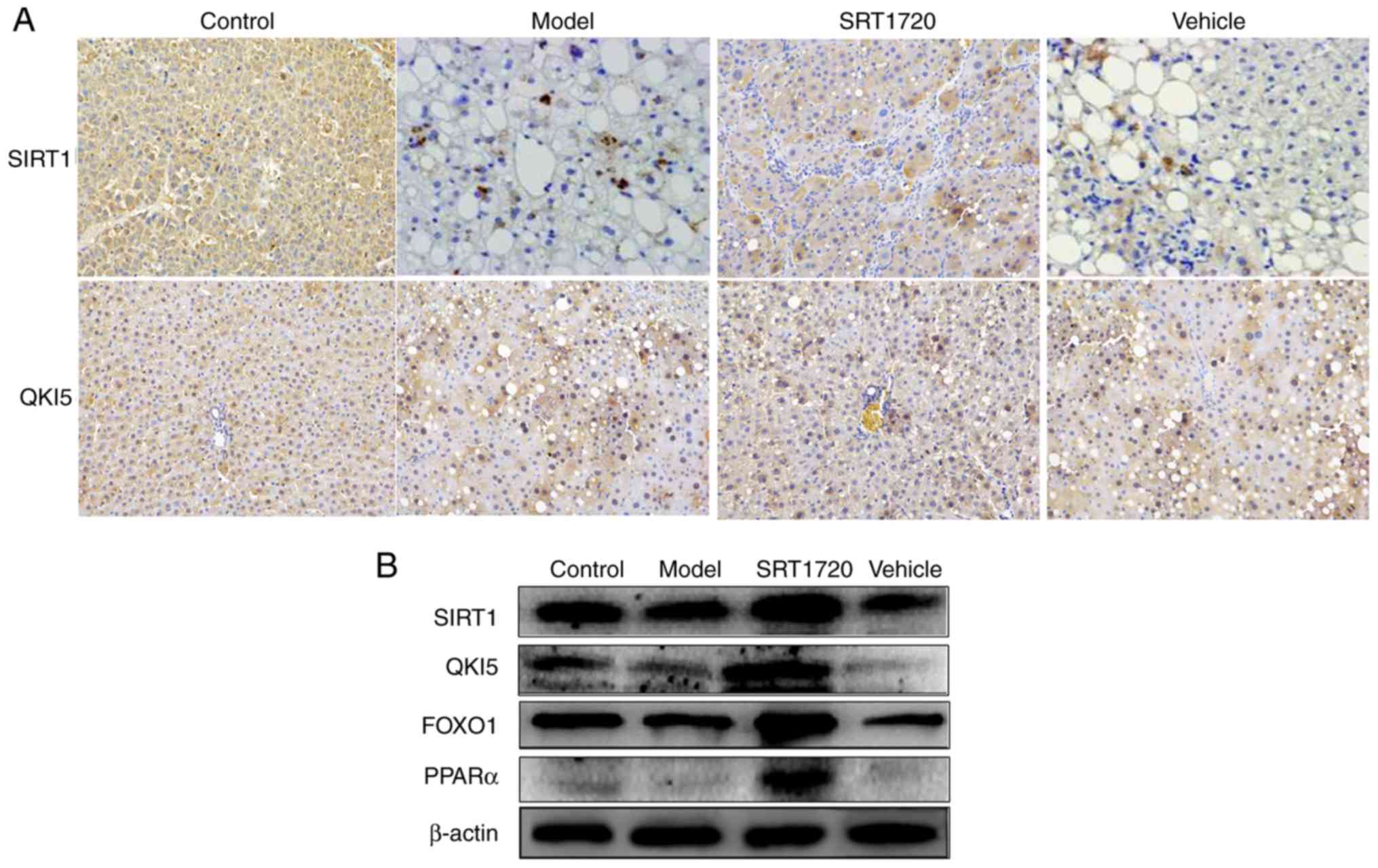 | Figure 7Expression levels of SIRT1, QKI 5,
FOXO1 and PPARα in mice using immunohistochemistry and western blot
assays. (A) Results of immunohisto-chemistry (magnification, ×400)
confirmed that, in non-alcoholic fatty liver disease model mice,
the expression levels of SIRT1 and QKI 5 were downregulated, but
were promoted by SRT1720. (B) Western blotting demonstrated that
the expression levels of SIRT1 and QKI 5 were decreased in the
model mice, and this was reversed by SRT1720. PPARα, peroxisome
proliferator-activated receptor α; FOXO1, Forkhead box protein O1;
QKI 5, Quaking 5; SIRT1, Sirtuin 1. |
Discussion
Hepatic lipid accumulation is a typical
characteristic of NAFLD. There is an ongoing research effort to
investigate the mechanism of hepatic steatosis in NAFLD to identify
potential novel therapeutic targets (24). Furthermore, de novo
lipogenesis has an important effect on the pathogenesis of NAFLD
(25). Therefore, in the present
study, the process and mechanism of TG synthesis in NAFLD were
investigated. NAFLD model mice were produced, with the mice fed an
HFD becoming obese at 8 months of age. The results indicated that
the BW and the relative fat mass ratio of mice in the model group
and vehicle group increased. Furthermore, the model and vehicle
mice developed hyperlipidemia, as measured by total plasma TG and
TC levels. Consistent with an increase in plasma lipids, the HFD
model mice exhibited enlarged fatty livers at 8 months of age. The
hepatic lipid content of the HFD model mice at 8 months of age was
increased compared with that in their littermate controls, as
separately measured using H&E and Oil Red O staining of liver
sections, and quantification of the Oil Red O staining. These data
indicate a typical pathology of liver steatosis. Furthermore, the
TG and TC content in the extracted livers from the NAFLD model mice
were increased compared with the non-NAFLD mice. In addition,
mildly increased plasma activities of ALT and AST were identified
in the model mice. The serum level of SIRT1 in the model mice was
reduced compared with that in the control group. Furthermore, the
NAS was elevated in the mice; the results exhibited a difference in
the model and vehicle group mice compared with the control group
mice. These results indicated that the NAFLD model was successfully
established.
Carbohydrate and lipid metabolism is regulated by
SIRT1, which is associated with the development of NAFLD (26–31). In vivo and in vitro
studies of NAFLD have confirmed that SIRT1 is a metabolic sensor,
and may improve NAFLD. Through the upregulation of
gluconeogenesis-associated genes and de novo lipogenesis,
the levels of intracellular lipid and glucose increase, induced by
suppressing the expression of SIRT1 (31). Furthermore, the downregulation of
SIRT1 in mice treated with small hairpin RNA caused hepatic
steatosis (32,33). Therefore, the synthesis of TG in
NAFLD associated with SIRT1 was investigated in the present
study.
The results of the immunohistochemistry in the
present study indicated the decreased expression level of SIRT1in
the NAFLD model mice. Furthermore, the western blot assay
demonstrated that the expression level of SIRT1 was decreased in
model mice. Therefore, to evaluate the ability of hepatic SIRT1 to
maintain lipid homeostasis, the agonist (SRT1720) and inhibitor
(niacinamide) of SIRT1 were used. In addition, adenovirus-mediated
gene repletion of SIRT1 was used in primary hepatocytes of mice,
and siRNA of SIRT1 was used to downregulate the expression of
SIRT1. The increased BW, relative fat mass ratio, hyperlipidemia
levels (total plasma TG and TC), enlarged fatty liver, the hepatic
lipid content of the NAFLD model mice were reversed by SRT1720.
Additionally, SRT1720 reduced the typical pathology of liver
steatosis, increased damage or inflammatory changes of plasma ALT
or AST activity, and NAS score. These results demonstrated that the
developmental processes of the NAFLD model were reversed by
SRT1720.
RNA-binding protein-mediated post-transcriptional
regulation is rarely investigated in liver metabolism. STAR family
member QKI is an RNA-binding protein that produces multiple
isoforms in the human body, and demonstrates a variety of
expression patterns in the cells of various types of tissues
(10). Among them, QKI 5 is
mainly located in the nucleus but can also shuttle to the cytoplasm
(13,14), whereas QKI 6 and QKI 7 are mainly
distributed in the cytoplasm. By binding to the specific
recognition element of the 3′ untranslated region of mRNA (11,12), QKI is involved in regulating the
cytoplasmic/nuclear localization, stability and translation
efficiency of mRNA (15,16). It has been demonstrated that QKI
is expressed in the liver, and the most commonly expressed isomer
is QKI 5, although its function in the liver has not been
reported.
SIRT1 is mediated by post-translational regulation,
and a number of transcription factors, including FOXO1, sterol
regulatory element-binding protein 1, carbohydrate-responsive
element-binding protein, PPARα and PPARγ coactivator 1α are
regulated at the transcriptional level (21). QKI is an RNA-binding protein that
is regulated at the post-transcriptional level of RNA. Specific to
RNA sequence binding, QKI is widely involved in variable splicing,
subcellular localization, stability maintenance and the
translational regulation of RNA.
In the present study, primary hepatocytes were
extracted to examine the mechanism associated with SIRT1. It was
demonstrated that SRT1720 and Ad-SIRT1 enhanced the expression of
SIRT1, QKI 5, FOXO1 and PPARα. However, the reduced expression
level of SIRT1 induced by niacinamide and siRNA of SIRT1, resulted
in the downregulation of QKI 5, FOXO1 and PPARα. In addition, a
decrease in intercellular TG content was caused by SRT1720 and
Ad-SIRT1, whereas niacinamide and siRNA of SIRT1 enhanced the TG
content of primary hepatocytes.
The results of the immunohistochemistry in the
present study indicated that the expression levels of SIRT1 and QKI
5 were downregulated in the NAFLD model mice, but were promoted by
SRT1720. Furthermore, the results of the western blot assay
demonstrated that the expression levels of SIRT1, QKI 5, FOXO1 and
PPARα were decreased in the model mice, and this was reversed by
SRT1720; therefore, the inhibitor of FOXO1 (AS1842856) was used.
The data indicated that AS1842856 suppressed FOXO1 and PPARα, and
that this was reversed by SRT1720. AS1842856 did not inhibit the
expression levels of SIRT1 or QKI 5. The results demonstrated that
hepatic SIRT1 regulated the expression of QKI 5 via the PPARα/FoxO1
signaling pathway. In addition, SIRT1 mediated the synthesis of TGs
in NAFLD mice, which was associated with QKI 5 and the PPARα/FoxO1
signaling pathway.
The role of SIRT1 in regulating the acetylation
level of QKI 5 was also investigated. In the liver tissues of the
model mice, the acetylation level of QKI 5 increased, but this was
reversed by SRT1720. In addition, the increasing acetylation level
of QKI 5 was induced by the inhibitor of SIRT1 (niacinamide),
whereas the acetylation of QKI 5 was decreased in the hepatocytes
treated with SRT1720. An increase in the acetylation level of QKI 5
was also induced by SIRT1 siRNA. In addition, the acetylation of
QKI 5 was decreased in hepatocytes treated with Ad-SIRT1. The
results of this protein interaction confirmed that the interaction
between SIRT1 and QKI 5, and its level in the SRT1720 group were
increased compared with the model mice.
In conclusion, it was shown that SIRT1 deacetylates
QKI 5, which was the RNA-binding protein affecting the synthesis of
TGs in the liver of NAFLD mouse model. Furthermore, it activated
the transcription factor FOXO1 through post-transcriptional
regulation of the expression of PPARα, and further inhibited the
synthesis of TG, thereby restricting the progression of NAFLD.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 30600524
and 81341067), the National Natural Science Foundation of Guangdong
Province, China (grant no. 2017A030313510), the Introduction of
Talent Fund of Guangdong Second Provincial General Hospital (grant
no. YY2016-006), the Capital Clinical Featured Applied Research and
Results Promotion projects (grant no. Z161100000516141) and
Zhejiang Provincial Medical Health Science Technology Project
(grant no. 2015KYB382). The study sponsors had no involvement in
the study.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
WZ and YS conducted the experiments. WL and JD were
involved in the experiments. JC designed the experiments and wrote
the manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Animal
Ethics Committee of Tsinghua University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Chang Y, Cho YK, Kim Y, Sung E, Ahn J,
Jung HS, Yun KE, Shin H and Ryu S: Non-heavy drinking and worsening
of non-invasive fibrosis markers in nonalcoholic fatty liver
disease: A cohort study. Hepatology. Jul 17–2018.Epub ahead of
print.
|
|
2
|
Wang J, Yang W, Chen Z, Chen J, Meng Y,
Feng B, Sun L, Dou L, Li J, Cui Q and Yang J: Long noncoding RNA
lncSHGL recruits hnRNPA1 to suppress hepatic gluconeogenesis and
lipogenesis. Diabetes. 67:581–593. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jeong HS, Kim KH, Lee IS, Park JY, Kim Y,
Kim KS and Jang HJ: Ginkgolide A ameliorates non-alcoholic fatty
liver diseases on high fat diet mice. Biomed Pharmacother.
88:625–634. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tarantino G, Saldalamacchia G, Conca P and
Arena A: Non-alcoholic fatty liver disease: Further expression of
the metabolic syndrome. J Gastroenterol Hepatol. 22:293–303. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Affo S, Yu LX and Schwabe RF: The role of
cancer-associated fibroblasts and fibrosis in liver cancer. Annu
Rev Pathol. 12:153–186. 2017. View Article : Google Scholar :
|
|
6
|
Estes C, Anstee QM, Arias-Loste MT, Bantel
H, Bellentani S, Caballeria J, Colombo M, Craxi A, Crespo J, Day
CP, et al: Modeling NAFLD disease burden in China, France, Germany,
Italy, Japan, Spain, United Kingdom, and United States for the
period 2016-2030. J Hepatol. 69:896–904. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Buzzetti E, Pinzani M and Tsochatzis EA:
The multiple-hit pathogenesis of non-alcoholic fatty liver disease
(NAFLD). Metabolism. 65:1038–1048. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nassir F and Ibdah JA: Sirtuins and
nonalcoholic fatty liver disease. World J Gastroenterol.
22:10084–10092. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pillman KA, Phillips CA, Roslan S, Toubia
J, Dredge BK, Bert AG, Lumb R, Neumann DP, Li X, Conn SJ, et al:
miR-200/375 control epithelial plasticity-associated alternative
splicing by repressing the RNA-binding protein Quaking. EMBO J.
37:pii. e990162018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Larocque D, Galarneau A, Liu HN, Scott M,
Almazan G and Richard S: Protection of p27(Kip1) mRNA by quaking
RNA binding proteins promotes oligodendrocyte differentiation. Nat
Neurosci. 8:27–33. 2005. View
Article : Google Scholar
|
|
11
|
Zhao L, Ku L, Chen Y, Xia M, LoPresti P
and Feng Y: QKI binds MAP1B mRNA and enhances MAP1B expression
during oligodendrocyte development. Mol Biol Cell. 17:4179–4186.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Saccomanno L, Loushin C, Jan E, Punkay E,
Artzt K and Goodwin EB: The STAR protein QKI-6 is a translational
repressor. Proc Natl Acad Sci USA. 96:12605–12610. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
de Bruin RG, Shiue L, Prins J, de Boer HC,
Singh A, Fagg WS, van Gils JM, Duijs JM, Katzman S, Kraaijeveld AO,
et al: Quaking promotes monocyte differentiation into
pro-atherogenic macrophages by controlling pre-mRNA splicing and
gene expression. Nat Commun. 7:108462016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Y, Vogel G, Yu Z and Richard S: The
QKI-5 and QKI-6 RNA binding proteins regulate the expression of
microRNA 7 in glial cells. Mol Cell Biol. 33:1233–1243. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang F, Song W, Zhao H, Ma Y, Li Y, Zhai
D, Pi J, Si Y, Xu J, Dong L, et al: The RNA-binding protein QKI5
regulates primary miR-124-1 processing via a distal RNA motif
during erythropoiesis. Cell Res. 27:416–439. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hall MP, Nagel RJ, Fagg WS, Shiue L, Cline
MS, Perriman RJ, Donohue JP and Ares M Jr: Quaking and PTB control
overlapping splicing regulatory networks during muscle cell
differentiation. RNA. 19:627–638. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van der Veer EP, de Bruin RG, Kraaijeveld
AO, de Vries MR, Bot I, Pera T, Segers FM, Trompet S, van Gils JM,
Roeten MK, et al: Quaking, an RNA-binding protein, is a critical
regulator of vascular smooth muscle cell phenotype. Circ Res.
113:1065–1075. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zong FY, Fu X, Wei WJ, Luo YG, Heiner M,
Cao LJ, Fang Z, Fang R, Lu D, Ji H and Hui J: The RNA-binding
protein QKI suppresses cancer-associated aberrant splicing. PLoS
Genet. 10:e10042892014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fagg WS, Liu N, Fair JH, Shiue L, Katzman
S, Donohue JP and Ares M Jr: Autogenous cross-regulation of Quaking
mRNA processing and translation balances Quaking functions in
splicing and translation. Genes Dev. 31:1894–1909. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hayakawa-Yano Y, Suyama S, Nogami M,
Yugami M, Koya I, Furukawa T, Zhou L, Abe M, Sakimura K,
Takebayashi H, et al: An RNA-binding protein, Qki5, regulates
embryonic neural stem cells through pre-mRNA processing in cell
adhesion signaling. Genes Dev. 31:1910–1925. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sonnemann J, Kahl M, Siranjeevi PM,
Blumrich A, Blümel L, Becker S, Wittig S, Winkler R, Krämer OH and
Beck JF: Reverse chemomodulatory effects of the SIRT1 activators
resveratrol and SRT1720 in Ewing's sarcoma cells: Resveratrol
suppresses and SRT1720 enhances etoposide- and vincristine-induced
anticancer activity. J Cancer Res Clin Oncol. 142:17–26. 2016.
View Article : Google Scholar
|
|
22
|
Bechmann LP, Kocabayoglu P, Sowa JP, Sydor
S, Best J, Schlattjan M, Beilfuss A, Schmitt J, Hannivoort RA,
Kilicarslan A, et al: Free fatty acids repress small heterodimer
partner (SHP) activation and adiponectin counteracts bile
acid-induced liver injury in superobese patients with nonalcoholic
steatohepatitis. Hepatology. 57:1394–1406. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Biel TG, Lee S, Flores-Toro JA, Dean JW,
Go KL, Lee MH, Law BK, Law ME, Dunn WA Jr, Zendejas I, et al:
Sirtuin 1 suppresses mitochondrial dysfunction of ischemic mouse
livers in a mitofusin 2-dependent manner. Cell Death Differ.
23:279–290. 2016. View Article : Google Scholar :
|
|
24
|
Kawano Y and Cohen DE: Mechanisms of
hepatic triglyceride accumulation in non-alcoholic fatty liver
disease. J Gastroenterol. 48:434–441. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Donnelly KL, Smith CI, Schwarzenberg SJ,
Jessurun J, Boldt MD and Parks EJ: Sources of fatty acids stored in
liver and secreted via lipoproteins in patients with nonalcoholic
fatty liver disease. J Clin Invest. 115:1343–1351. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu T, Liu YH, Fu YC, Liu XM and Zhou XH:
Direct evidence of sirtuin downregulation in the liver of
non-alcoholic fatty liver disease patients. Ann Clin Lab Sci.
44:410–418. 2014.PubMed/NCBI
|
|
27
|
Bruce KD, Szczepankiewicz D, Sihota KK,
Ravindraanandan M, Thomas H, Lillycrop KA, Burdge GC, Hanson MA,
Byrne CD and Cagampang FR: Altered cellular redox status, sirtuin
abundance and clock gene expression in a mouse model of
developmentally primed NASH. Biochim Biophys Acta. 1861:584–593.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Geng C, Zhang Y, Gao Y, Tao W, Zhang H,
Liu X, Fang F and Chang Y: Mst1 regulates hepatic lipid metabolism
by inhibiting Sirt1 ubiquitination in mice. Biochem Biophys Res
Commun. 471:444–449. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Colak Y, Yesil A, Mutlu HH, Caklili OT,
Ulasoglu C, Senates E, Takir M, Kostek O, Yilmaz Y, Yilmaz Enc F,
et al: A potential treatment of non-alcoholic fatty liver disease
with SIRT1 activators. J Gastrointestin Liver Dis. 23:311–319.
2014.PubMed/NCBI
|
|
30
|
Colak Y, Ozturk O, Senates E, Tuncer I,
Yorulmaz E, Adali G, Doganay L and Enc FY: SIRT1 as a potential
therapeutic target for treatment of nonalcoholic fatty liver
disease. Med Sci Monit. 17:HY5–HY9. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tobita T, Guzman-Lepe J, Takeishi K, Nakao
T, Wang Y, Meng F, Deng CX, Collin de l'Hortet A and Soto-Gutierrez
A: SIRT1 disruption in human fetal hepatocytes leads to increased
accumulation of glucose and lipids. PLoS One. 11:e01493442016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Purushotham A, Schug TT, Xu Q, Surapureddi
S, Guo X and Li X: Hepatocyte-specific deletion of SIRT1 alters
fatty acid metabolism and results in hepatic steatosis and
inflammation. Cell Metab. 9:327–338. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim KE, Kim H, Heo RW, Shin HJ, Yi CO, Lee
DH, Kim HJ, Kang SS, Cho GJ and Choi WS: Myeloid-specific SIRT1
deletion aggravates hepatic inflammation and steatosis in high-fat
diet-fed mice. Korean J Physiol Pharmacol. 19:451–460. 2015.
View Article : Google Scholar : PubMed/NCBI
|