Introduction
A total of ~1% of the global population suffers from
atrial fibrillation (AF), which is the most common clinically
significant arrhythmia in adults (1). Hypertension, congestive heart
failure, rheumatic and ischemic heart disease, and older age are
the risk factors for AF. The 3 most important modes of treatment
are rhythm control, rate control and anticoagulation therapy.
Patients with AF generally suffer from increased morbidity and
mortality rates due to events including ischemic stroke, heart
failure and sudden cardiac death (2). Apart from increased risk of
all-cause mortality (3),
increased morbidity and cardiovascular mortality, AF also has a
significant adverse effects on patient quality of life (4). In 2010, it was estimated that 5.2
million people in the United States of America suffered from AF,
and a projected 12.1 million people are expected to suffer from AF
by the year 2030 (5). Although
atrial fibrosis has been associated with the AF process, little is
known about the differing expression of the external cellular
matrix (ECM) components collagen I, collagen III and fibronectin in
the various forms of AF.
In endothelial cells (ECs), laminar shear stress
(LSS) is a physiological regulator of a number of important
homeostatic functions. It provokes a rapid elevation in the
intracellular Ca2+ concentration in ECs that originates
from intracellular stores and from Ca2+ influx (6). By controlling the production of
major EC-derived relaxing factors including nitric oxide (NO) and
prostacyclin, endothelial Ca2+-activated K+
channels (KCa) serve an important role in regulating vasomotor tone
and blood pressure (7). There are
5 KCa subtypes: KCa1.1, KCa2.1, KCa2.2, and KCa2.3, and KCa3.1
(8). In general, ECs in different
regions of the vascular systems of several species constitutively
and predominantly express KCa2.3 and KCa3.1 (9). Previous studies have indicated that
NO-mediated vasodilation is associated with KCa2.3 activation
(10-12). Therefore, additional clarification
of the effect of shear patterns on the expression of KCa.23 and the
potential regulatory mechanisms in myocardial cells is required. In
cardiac tissues, KCa2.3 channel blockers function to prevent atrial
fibrillation (13,14).
Previous studies have demonstrated that
phosphoinositide 3-kinase (PI3K) is a cardioprotective protein that
inhibits pathological signaling cascades downstream of G
protein-coupled receptors (15);
therefore, loss of PI3K may increase the likelihood of
cardiotoxicity (16). In
addition, previous data have also suggested that ibrutinib
increases the risk of AF, potentially by inhibiting cardiac
PI3K-protein kinase B (Akt) signaling (17). The PI3K/Akt signaling pathway has
certain potential advantages in this regard, due to intracellular
dialysis of cardiomyocytes with phosphatidylinositol (3,4,5)-trisphosphate (PIP3) normalizes ion
channels and eliminates arrhythmias. However, as the pathway
regulates cell proliferation and survival by activating PIP3
downstream signals, transfusion of PIP3 at supraphysiological
levels may result in abnormal cell growth that is detrimental to
the cardiomyocytes. As the PI3K inhibitors used in the treatment of
cancer have been identified to cause proarrhythmia, they represent
a potential avenue for the study and evaluation of the potential
effectiveness of a range of antiarrhythmic and anticancer drugs
that are either presently in use or under development. PI3K may
regulate cardiac ion channels when arrhythmias occur; however,
limited information is available for PI3K/Akt signaling and
arrhythmias. This demonstrates that there is a requirement to
identify novel ways to improve the testing of antiarrhythmic drugs
and increase understanding of PI3K/Akt signaling and arrhythmia
(18).
Based on the aforementioned data, we hypothesized
that the expression of major ECM proteins in left atrial tissue
differs in patients with alterations in sinus rhythm (SR), AF alone
and AF with underlying mitral valve disease (MVD). In addition, we
hypothesized that KCa2.3 and the PI3K/Akt pathway may serves
crucial roles in the AF process, and we hypothesized that the
aberrant expression of KCa2.3 induced by LS is regulated by the
PI3K/Akt signaling pathway.
Materials and methods
Patients
Patients with AF alone (n= 8; 4 females and 4 males,
age range, 48-72; left atrial diameter range, 43-57 mm) and
patients with AF combined with MVD (n=6; 3 MVD- paroxysmal AF and 3
MVD-chronic AF, age range, 33-74; left atrial diameter range, 47-59
mm) were included. The control group consisted of patients with a
normal SR (n=6; 3 females and 3 males; age range, 41-69; left
atrial diameter range, 30-41 mm) and were matched to the AF groups
according to age, left atrial size and left ventricular function.
All patients provided written informed consent to participate in
the present study. The Institutional Ethical Committee of the First
Affiliated Hospital of Kunming Medical University approved the
study, and the study was performed in concordance with the
principles outlined in the Declaration of Helsinki. Atrial tissue
from all patients was obtained from the left atrial free wall near
the interatrial septum during cardiac surgery and was quickly
placed in formaldehyde solution at room temperature, or frozen in
liquid nitrogen and stored at -80°C until use.
Hematoxylin and eosin (H&E) staining,
Masson staining and immunofluorescence
Left atrial tissue was fixed in 4% paraformaldehyde
for 30 min at 4°C, embedded in paraffin, and transected into
4-µm thick sections along the center of the tissue. Under a
light microscope (magnification, ×20) formalin-fixed
paraffin-embedded tissues were respectively stained with H&E
and Masson's trichrome stain according to the manufacturer's
protocols (cat. no. G134; Beijing Solarbio Science &
Technology, Beijing, China). Following this, all collagen fibers
were stained blue, and cardiomyocytes appeared red. Fibrous tissue
areas were quantified using Image-Pro Plus 6.0 software (Media
Cybernetics, Inc., Rockville, MD, USA) (19). The tissue expression of KCa2.3 was
assessed by immunofluorescence. Briefly, glass covers were fixed on
the tissue cross-section with 4% paraformaldehyde for 15 min at
room temperature. Samples were then permeated by 0.3% Triton™ X-100
for 20 min at room temperature and blocked using PBS containing 5%
donkey serum (ab7475; Abcam, Cambridge, MA, USA) and 1% BSA for 1 h
at room temperature to reduce nonspecific reactions. Then, an
antibody against KCa2.3 (1:500, ab28631; Abcam) overnight followed
by incubation at 4°C with a fluorescein isothiocyanate-conjugated
rabbit anti-human antibody (1:100; Anttene, Wuhan, China) for 2 h.
DAPI at a final concentration of 0.5 µg/ml (Beyotime
Institute of Biotechnology, Haimen, China) was used to stain the
cell nuclei for 5 min at room temperature. Immunofluorescence was
visualized using a confocal microscope (magnification, ×20; Leica
Microsystems, Inc., Buffalo Grove, Il, USA) to detect the
expression of KCa2.3 in the left atrial tissue using ImageJ
software [v.1.8; National Institutes of Health (NIH), Bethesda, MD,
USA].
H9c2 cell culture
H9c2 cells were purchased from the American Type
Culture Collection (Manassas, VA, USA) and maintained in a standard
humidified incubator at 37°C and 5% CO2 in Dulbecco's
modified Eagle's medium (SH3008101, HyClone; GE Healthcare Life
Sciences, Logan, UT, USA) for a 48 h starvation period prior to use
in experiments to eliminate KCa induction by growth factors and to
minimize background signaling. H9c2 cells at passages 2-4 were used
in the experimental protocols.
Shear stress studies
H9c2 cells grown to 60% confluence as monolayers
were exposed to static culture conditions (ST) or to a laminar
shear stress condition (LS) using a computer-controlled shearing
device at 15 dynes/cm2 for 12 h. In certain experiments,
the cells were pretreated for 30 min with 20 nM dactolisib, a
specific PI3K inhibitor, 10 nM GSK690693, a specific Akt inhibitor
or 400 nM C646, a histone acetyltransferase p300 (p300) inhibitor
binding to transcription factors, prior to exposure to LS.
RNA isolation and reverse transcription
quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cells using the
RNAsimple Total RNA kit (Tiangen Biotech Co., Ltd., Beijing, China)
according to the manufacturer's protocol. β-actin was used as a
control. Genomic DNA was amplified by RT-qPCR using specific gene
amplification primers. The forward and reverse PCR primers for
measuring gene expression are summarized in Table I. The samples were amplified in an
RT-qPCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) with a SYBR®-Green detection system
(cat. no. 204141; Qiagen, Inc., Valencia, CA, USA). The PCR
parameters were denaturation at 95°C for 5 min; followed by 40
cycles of (95°C for 15 sec, 55°C for 30 s and 72°C for 15 sec) and
another 60°C for 5 min. To assess the amplification of specific
transcripts, melting curve profiles were generated at the end of
each PCR assay. All reactions were in triplicate, including
controls without added template. The relative expression of gene of
interest was calculated using the 2−ΔΔCq method
(20), where ΔΔCt is the
difference between the ΔCq of the two cDNA samples being
compared.
 | Table IPrimers sequence used in the present
study. |
Table I
Primers sequence used in the present
study.
| Gene name | Primers | Product length,
bp |
|---|
| PI3K | Forward:
5′-GATTGGTTCTTTCCTGTCTCTG-3′ | 324 |
| Reverse:
5′-CCACCCTATCAATTTACAACCA-3′ | |
| Akt | Forward:
5′-GCACAACGAGGGGAGTACAT-3′ | 113 |
| Reverse:
5′-CCTCACGTTGGTCCACATC-3′ | |
| p300 | Forward:
5′-CAGTCCAGTAAATCAGCCTGCC-3′ | 255 |
| Reverse:
5′-AATCCTGTTTGTCCTCCCATCTG-3′ | |
| KCa2.3 | Forward:
5′-CTTAATCACAGAACTCAATG-3′ | 178 |
| Reverse:
5′-TTAGCAACTGCTTGAACTTG-3′ | |
| β-actin | Forward:
5′-TTCCAGCCTTCCTTCTTG-3′ | 153 |
| Reverse:
5′-GGAGCCAGAGCAGTAATC-3′ | |
Western blot analysis
Western blot analysis was used to evaluate the
expression of KCa2.3 protein. In brief, the cells were washed three
times with ice-cold TBS and lysed in 200 µl lysis buffer
(cat. no. N8031; Beijing Solarbio Science & Technology).
Through a single round of freeze-thawing, the lysates were
additionally homogenized. The protein concentration of each sample
was quantified using an ND-1000 spectrophotometer (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). A total of 10 µl
proteins/lane from each sample was resolved on 10% SDS-PAGE gels
and subsequently transferred to a polyvinylidene difluoride
membrane (Bio-Rad Laboratories, Inc.). The membrane were blocked in
TBS-T containing 5% fat-free dry milk for 30 min at room
temperature and then incubated with primary antibodies (1:2,000,
sc-28621; Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
overnight at 4°C followed by incubation with horseradish
peroxidase-conjugated secondary antibody (1:1,000, SE134; Beijing
Solarbio Science & Technology) at room temperature for 1 h.
Protein expression was detected by chemiluminescence (cat. no.
PE0010; Beijing Solarbio Science & Technology). The primary
antibodies used were directed against phosphorylated (p)-Akt
(1:1,500, cat. no. 9271; Cell Signaling Technologies, Inc.,
Danvers, MA, USA), total Akt (1:2,000, cat. no. 4691; Cell
Signaling Technologies, Inc.), p-p300 (1:500, cat. no. PA5-12652;
Thermo Fisher Scientific, Inc.) and total p300 (1:2,000, cat. no.
86377; Cell Signaling Technologies, Inc.) and β-actin (1:1,500,
sc-47778; Santa Cruz Biotechnology, Inc.). Representative
immunoblots from ≥3 experiments are presented with the results of
the densitometric analyses (ImageJ v.1.8; NIH).
Chromatin immunoprecipitation (ChIP)
assay
To determine the binding of nuclear proteins to the
KCa2.3 promoter, ChIP assays were performed as described previously
(21). Briefly, 80% confluent
H9c2 cells were treated with 4% paraformaldehyde for 20 min at room
temperature and dissolved in ChIP buffer (50 mM Tris-HCl pH 7.9,
150 mM NaCl, 5 mM EDTA, 0.1% sodium deoxycholate, 1% TritonX-100,
0.5% Nonidet P-40, 1 mM PMSF and protease inhibitor). These fixed
cells were washed and lysed in SDS-lysis buffer (cat. no. 20-163;
EMD Merck Millipore, Billerica, MA, USA) and sonicated with a
frequency of 22.5KHz on ice until the DNA size was decreased to
200-800 bp. Following centrifugation at 100 × g for 2 min for 30
min at 4°C with rotation, a portion of the pre-cleared solution was
used as a DNA input control; the remaining portion was divided into
aliquots and incubated with specific primary antibodies against
p300 (1:2,000, cat. no. 86377; Cell Signaling Technologies, Inc.)
or rabbit pre-immune IgG (1 µg/1 mg lysate) at 4°C for 8 h.
The immunoprecipitated complexes of Ab-protein-DNA were collected
and washed with low-salt buffer, high-salt buffer, LiCl buffer and
Tris-EDTA, and then buffered with an elution buffer. The
cross-linking of protein-DNA complexes was reversed by incubation
with 5 M NaCl at 65°C for 4 h, and the DNA was digested with
proteinase K for 1 h at 45°C. The DNA was extracted with a 1:1
phenol-chloroform, and the purified DNA was precipitated with
isopropanol at a concentration of 15%. Following washing, the DNA
pellet was resuspended in H2O and subjected to PCR
amplification as aforementioned using the forward
(5′-AGGACTGGGAACTGTAGTTTTTG-3′) and reverse primers
(5′-GAAAATGTCTACTTTTAA-3′) (-688 to -203 bp). These primers were
specifically designed based on the sequence of the KCa2.3 promoter
region (supplied by Generay Biotech Co., Ltd., Shanghai, China).
The PCR products were analyzed on agarose gels stained with
ethidium bromide and analyzed with ImageJ (v.1.8; NIH).
Statistical analysis
All data are presented as the mean ± standard
deviation. Statistical analysis was assessed by Student's t-test or
one-way analysis of variance with a Dunnett's post hoc test and a
nonparametric Kruskal-Wallis for data that are not distributed
normally, where appropriate. All statistical procedures were
performed using GraphPad Prism 5.0. (GraphPad Software, Inc., La
Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
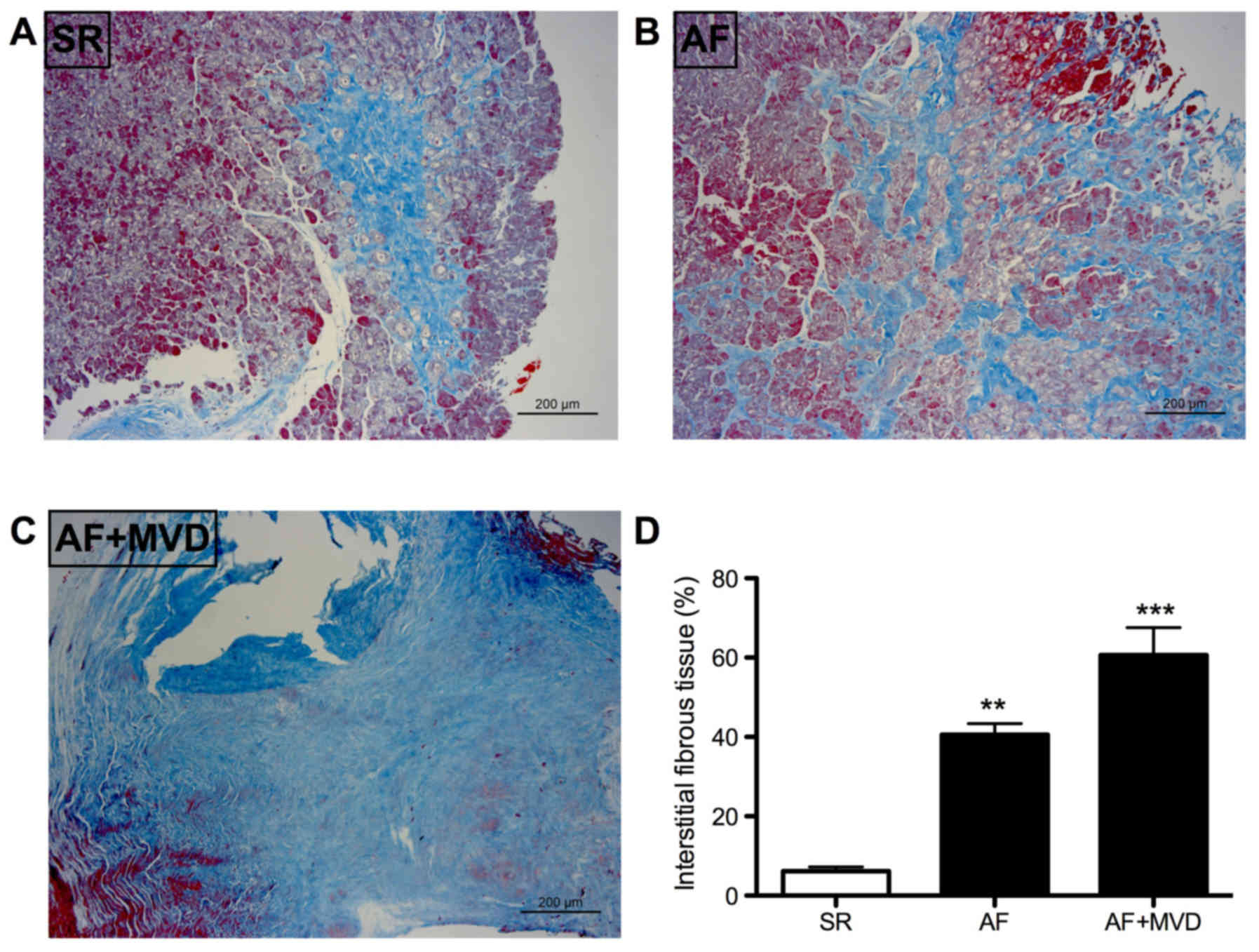
Atrial structural remodeling
As presented in Fig.
1, according to the Masson trichrome staining, patients with AF
exhibited significant interstitial fibrosis. The degree of atrial
fibrosis in the AF and AF combined with MVD groups was
significantly increased compared with that in the SR group.
Specifically, the average degree of atrial fibrosis in patients
with SR, AF and AF combined with MVD was 6.17±1.07, 40.6±2.8 and
60.63±6.93, respectively (P<0.05; Fig. 1).
KCa2.3 is highly expressed in patients
with AF and AF combined with MVD
To investigate KCa2.3 gene expression, RNA was
extracted from clinical tissues. KCa2.3 expression was initially
investigated in samples from the three groups; significantly
increased expression levels in the AF and AF combined with MVD
groups compared with the SR group [a 2.66-fold increase (P=0.006)
and a 3.1-fold increase (P=0.005), respectively] were observed
(Fig. 2A). As the KCa2.3 protein
has a specific function in regulating biological processes, KCa2.3
protein expression in the three groups was additionally
investigated by western blot analysis. As indicated in Fig. 2B, the expression of KCa2.3 protein
was significantly increased in the AF and AF combined with MVD
groups compared with the control. Then, KCa2.3 protein expression
was detected by immunofluorescence. As demonstrated in Fig. 2C, the expression of KCa2.3 was
upregulated in the AF and AF combined with MVD groups compared with
the control. In addition, to elucidate the molecular mechanisms
underlying LS-induced atrial remodeling in patients with AF, the
levels of the pivotal signal pathway initiator PI3K were
investigated. Compared with the SR control, AF significantly
altered the levels of PI3K protein expression. In the patients in
the AF and AF combined with MVD groups, PI3K expression was
significantly increased by 2.38- and 2.93-fold, respectively,
compared with the control group (both P<0.05; Fig. 2D).
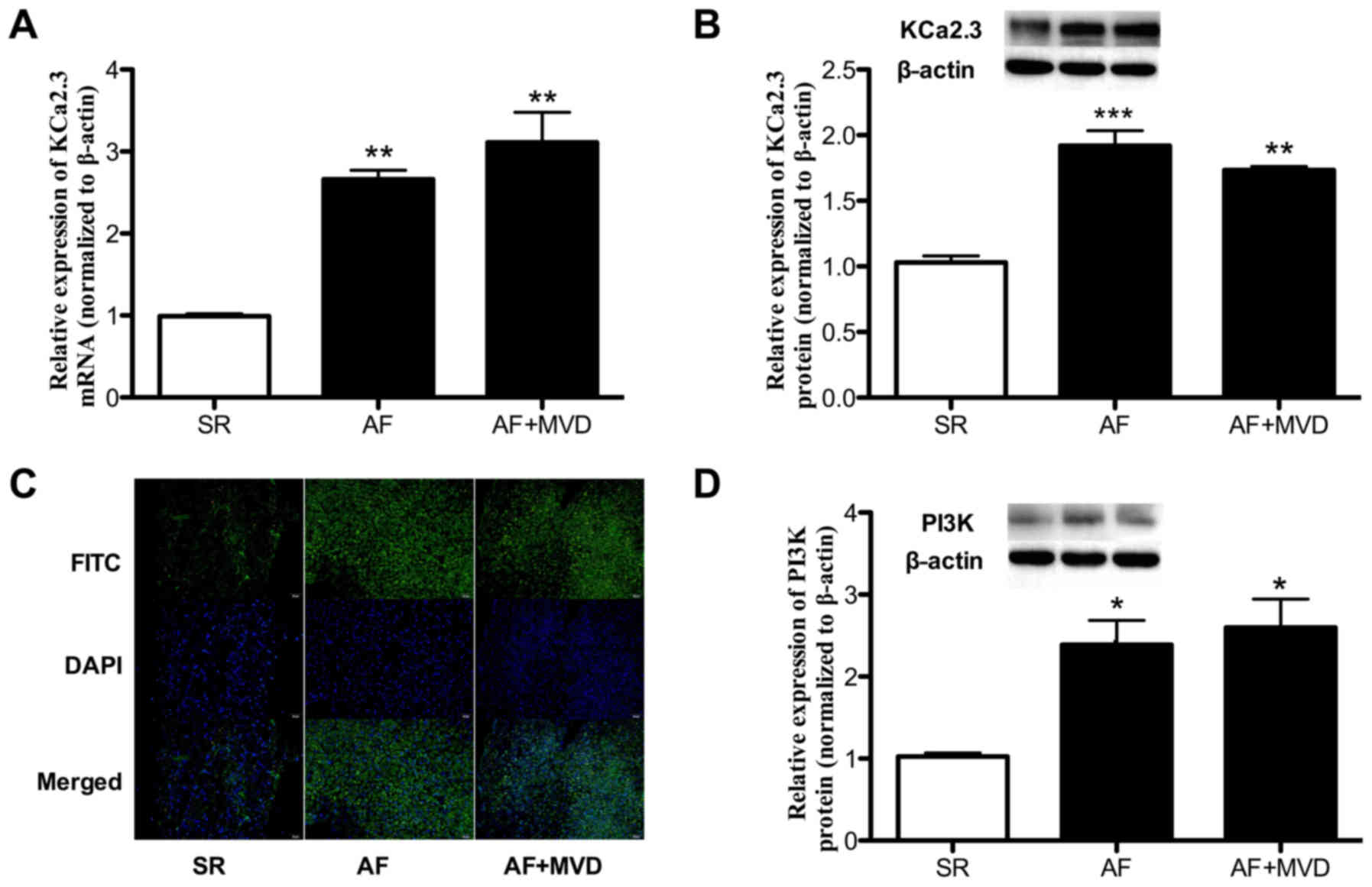 | Figure 2KCa2.3 gene expression analysis in
clinical samples. (A) 2.66- and 3.1-fold increases in KCa2.3
expression levels were observed in the AF (n = 8) and AF combined
with MVD (n=6) groups compared to the SR groups (n=6; P=0.006 and
0.005, respectively). (B) Increases in 85% and 63% in KCa2.3
expression was observed in the AF (n=8) and AF combined with MVD
(n=6) groups compared to the SR group (n=6; P<0.001 and P=0.004,
respectively). (C) Expression levels of KCa2.3 in SR, AF and AF
combined with MVD groups were examined by immunofluorescence.
Fluorescence was observed by laser-scanning microscopy. Scale bars,
50 µm. (D) Western blot analysis was performed to detect the
protein expression of PI3K in patients with SR, AF and AF combined
with MVD; β-actin was used as an internal reference protein.
Densitometric quantification of the western blot analysis results
are presented; the bars represents the mean ± standard deviation of
three independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. SR group.
KCa, Ca2+-activated K+ channels; SR, sinus
rhythm; AF, atrial fibrillation; MVD, mitral valve disease. |
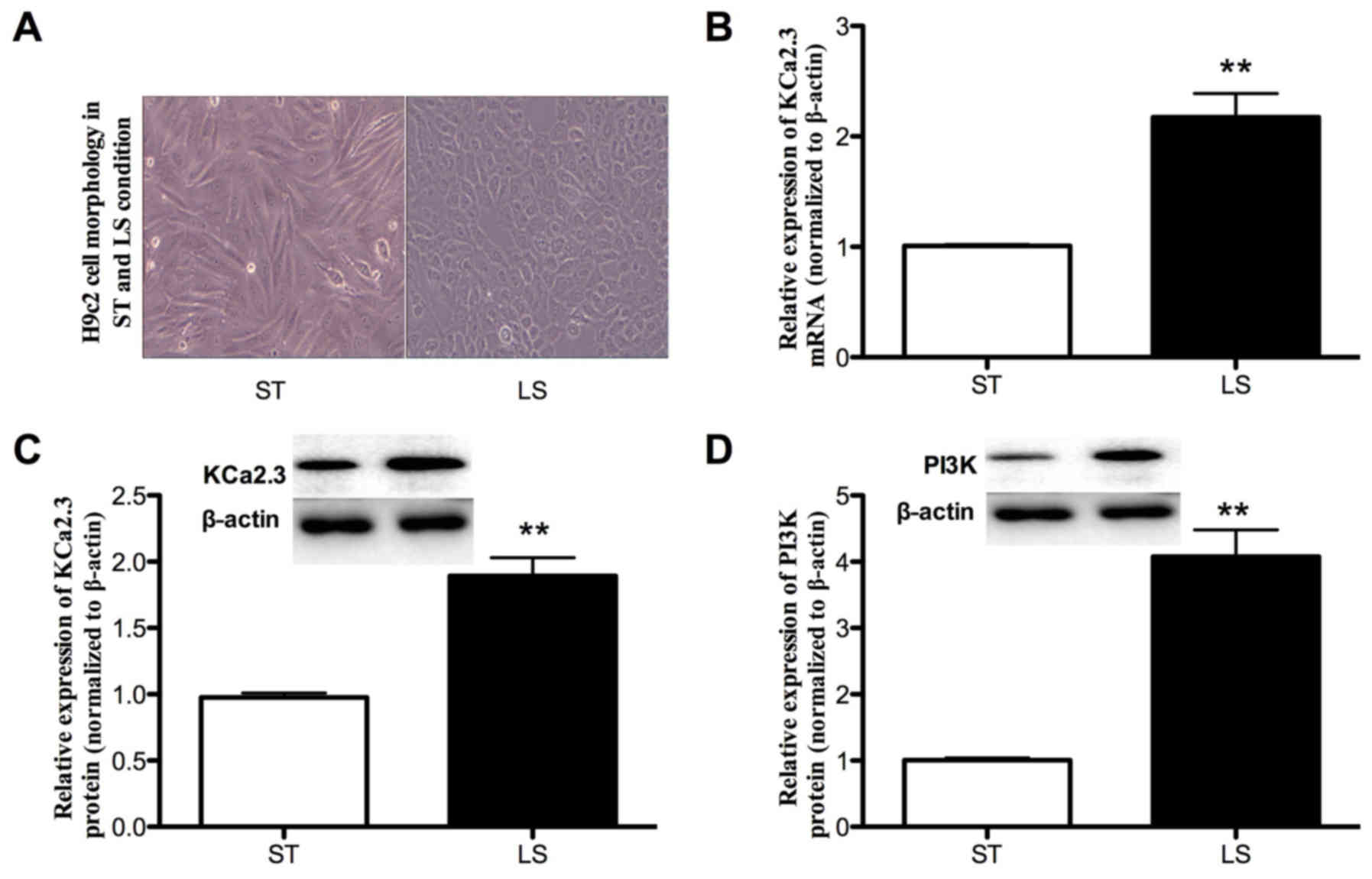
LS increases PI3K and KCa2.3
expression
The effect of shear stress on the expression of
KCa2.3 in H9c2 cells was determined by RT-qPCR and western blot
analysis. Exposure to LS (15 dynes/cm2) for 12 h
markedly changed the morphology of the cells (Fig. 3A) and increased KCa2.3 mRNA and
protein expression (1.16- and 0.91-fold, respectively; both
P<0.01; Fig. 3B and C). The
effect of LS on KCa2.3 expression was dose- and time-dependent;
exposure to lower LS (5 dynes/cm2) for 12 h resulted in
a moderate induction of KCa2.3, whereas exposure to LS at 15
dynes/cm2 for 6 or 24 h decreased KCa2.3 expression
(data not shown). To elucidate the potential mechanisms by which
the LS induced the KCa2.3 expression, based on the data obtained
from the clinical samples, the PI3K expression between ST and LS
exposure was also detected. As indicated in Fig. 3D, exposure to LS (15
dynes/cm2) for 12 h upregulated PI3K protein expression
by 3.07-fold (P=0.0016) compared to the ST condition. Taken
together, these data indicate that LS increases PI3K and KCa2.3
expression levels. Next, the subsequent studies were performed at
the arterial level of LS (15 dynes/cm2) to determine the
signaling pathway responsible for the physiological induction of
these channels.
PI3K/Akt/p300 axis activation mediates
the LS-induced increase in KCa2.3
The role of the PI3K/Akt/p300 signaling pathway in
the LS-induced upregulation of KCa2.3 was examined. H9c2 cells were
exposed to LS for 12 h in the absence or presence of 20 nM
dactolisib, a specific inhibitor of PI3K, 10 nM GSK690693, a
specific Akt inhibitor or 400 nM C646, an inhibitor of p300
transcription factor binding. As demonstrated in Fig. 4A, dactolisib markedly inhibited
PI3K protein expression; it decreased the p-Akt/Akt and p-p300/p300
ratios and KCa2.3 expression levels by 27, 22 and 33%, respectively
(all P<0.05; Fig. 4B).
GSK690693 decreased Akt expression and had no effect on PI3K
expression under LS conditions (P=0.063; Fig. 4C and D). However, GSK690693
attenuated the p-p300/p300 ratio and KCa2.3 protein expression by
48 and 37%, respectively (both P<0.01; Fig. 4D). By contrast, following the
addition of C646, an inhibitor of p300, western blot analysis data
suggested that the KCa2.3 protein level decreased by 38%
(P<0.001) with no change in the expression of PI3K or in the
ratio of p-Akt/Akt ratio (Fig. 4E and
F). These results indicate that the PI3K/Akt/p300 signal axis
serves an important role in mediating LS-induced upregulation of
the KCa2.3 channel in H9c2 cells.
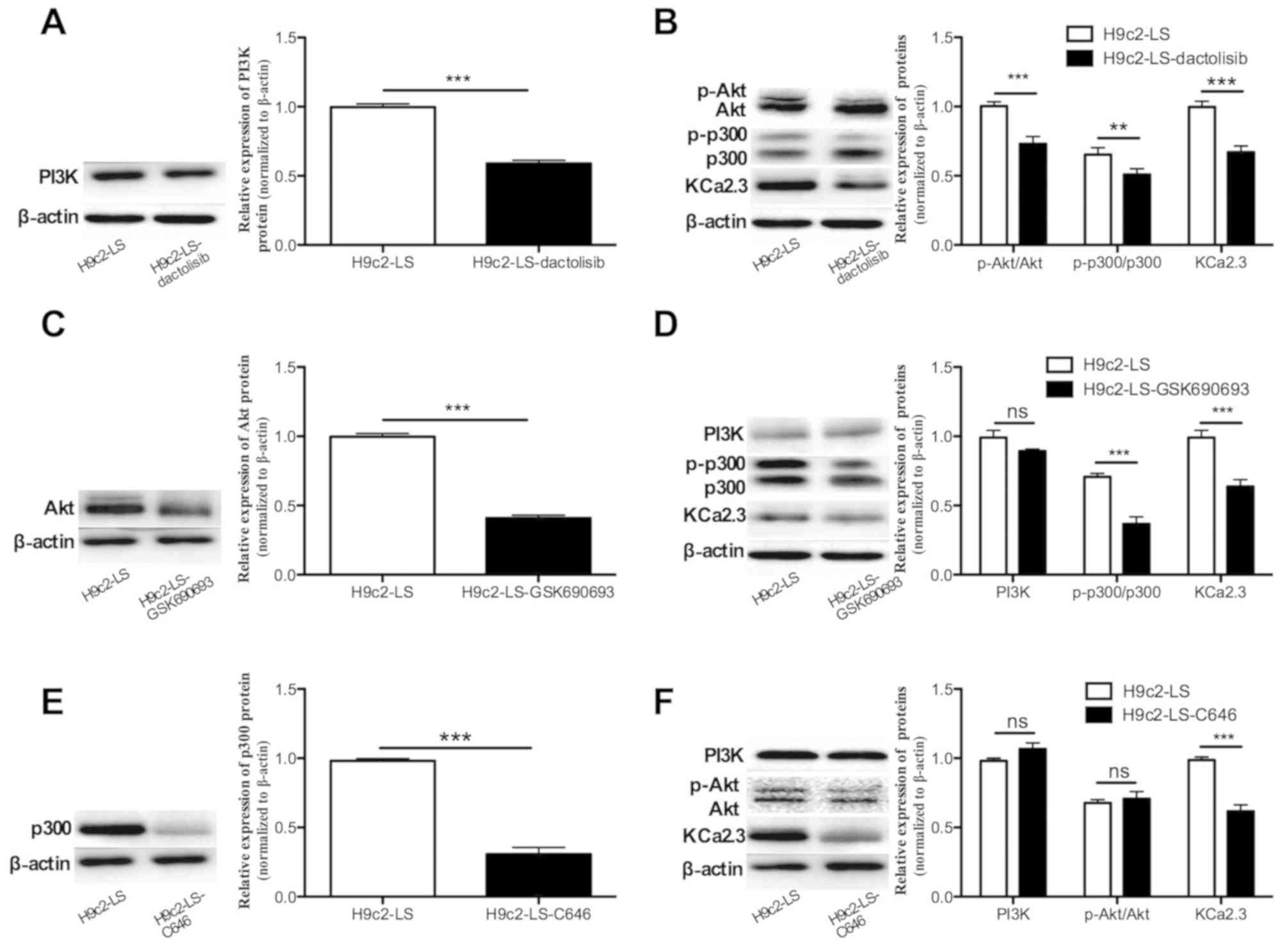 | Figure 4Activation of the PI3K/Akt/p300 axis
mediates the LS-induced increase in KCa2.3. H9c2 cells were exposed
to LS for 12 h in the absence or presence of 20 nM dactolisib, a
PI3K inhibitor, 10 nM GSK690693, a specific Akt inhibitor or 400 nM
C646, a p300 inhibitor. (A) Dactolisib decreased the ratio of PI3K.
(B) Dactolisib decreased the ratios of p-Akt/Akt and p-p300/p300
and KCa2.3 expression levels by 27, 22 and 33%, respectively (all
P<0.05). (C) GSK690693 decreased Akt expression. (D) GSK690693
had no effect on PI3K expression under LS conditions (P=0.063).
However, GSK690693 decreased the ratio of p-p300/p300 ratio and
KCa2.3 protein expression by 48 and 37% respectively (both
P<0.001). (E) Following the addition of C646, an inhibitor of
p300, western blot analysis indicated that p300 protein levels
decreased by 38% (P<0.001). (F) Following treatment with C464,
no change in the PI3K expression or in the ratio of p-Akt/Akt was
observed. **P<0.01 and ***P<0.001 vs.
H9c2-LS group. PI3K, phosphoinositide 3-kinase; Akt, protein kinase
B; p300, histone acetyltransferase p300; p, phosphorylated; LS,
laminar shear stress condition; KCa, Ca2+-activated
K+ channels. |
Recruitment of p300 to the KCa2.3
promoter is essential for LS-induced KCa2.3 upregulation
A previous study has suggested that phosphorylation
of p300 is required for KCa2.3 transcription activation (8). In addition, activation of p300 may
be regulated by Akt (22).
Another study has demonstrated that p300 is required for the
expression of certain genes (21). As demonstrated in Fig. 4F, pretreatment of H9c2 cells with
C646 significantly inhibited LS-induced KCa2.3 expression in a
time-dependent manner, suggesting that p300 may be involved in the
regulation of the H9c2 promoter. Therefore, the aim of the present
study was to investigate the hypothesis that recruitment of p300 is
regulated by the PI3K/Akt cascade in LS-stimulated H9c2 cells. To
investigate whether LS stimulated p300 recruitment to the KCa2.3
promoter, recruitment of p300 was examined using a ChIP-PCR assay
(Fig. 5A). A total of 31 cycles
of PCR amplifications were conducted using equal amounts of
immunoprecipitated DNA in the presence of specific primer pairs
encompassing the region between positions -688 and -203 region of
the rat KCa2.3 promoter (Fig.
5B-a). Following LS treatment, recruitment of p300 to the
KCa2.3 promoter region was induced in a time-dependent manner;
recruitment increased within 4 h and was sustained until at least
12 h (Fig. 5B-b). These results
indicated that LS-induced KCa2.3 expression required the
recruitment of p300 in H9c2 cells.
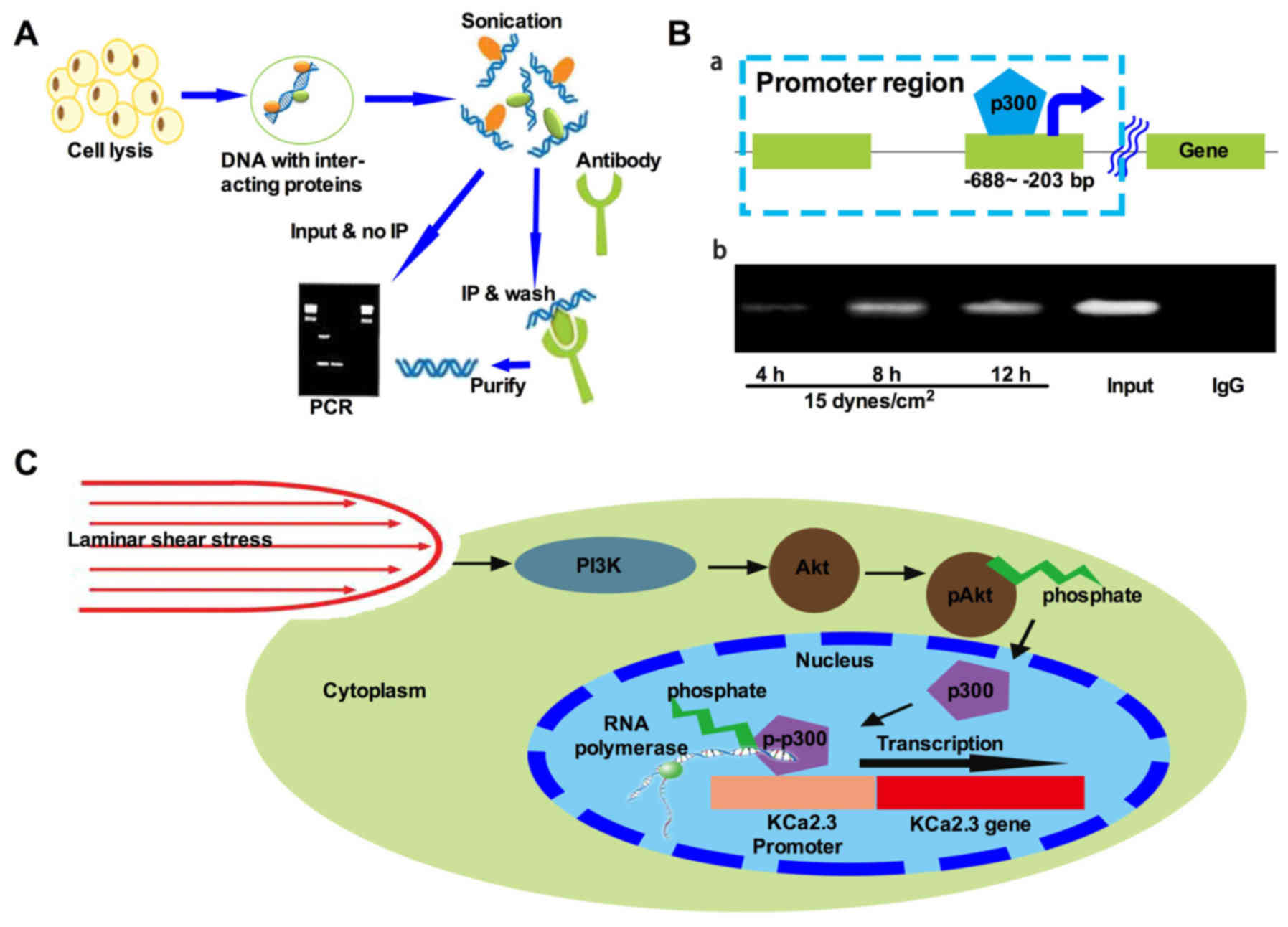 | Figure 5Transcriptional cofactor p300 is
required for LS-induced expression of KCa2.3. (A) An overview of
the ChIP procedure. Generally, cells are initially treated with a
cross-linking agent that covalently links DNA-interacting proteins
to the DNA. The genomic DNA is then isolated and sheared by
sonication to yield a suitable fragment size distribution. An
antibody that specifically recognizes the protein of interest is
then added, and immunoprecipitation is used to isolate specific
protein-DNA complexes. The cross-links are then reversed, and the
DNA fragments are purified and subjected to PCR analysis. (B-a)
Schematic of the KCa2.3 genomic DNA region. The square boxes
indicate the DNA region amplified by PCR in the ChIP assay. (B-b)
Time dependence of LS-stimulated recruitment of p300 to the KCa2.3
promoter as measured in ChIP assays. The cells were treated with LS
for the indicated times. The precipitated KCa2.3 promoter region
(-688 to -203 bp) was amplified by PCR. The figure represents one
of at least three individual experiments. The proximal KCa2.3
promoter region (-688 to -203 bp) was amplified by PCR following
ChIP. Mouse IgG represents negative control. The ChIP experiments
were repeated three times. (C) Proposed model of LS-induced KCa2.3
activation in H9c2 cells. Under normal conditions, KCa2.3 is
expressed at a basal level. When cells are subjected to LS, p300 is
recruited to the KCa2.3 promoter, where it activates KCa2.3
transcription. LS results in activation of the PI3K/Akt cascade and
recruitment of p300 to the promoter of the KCa2.3 promoter. KCa2.3
transcription is regulated in a p300-dependent manner by a PI3K/Akt
cascade. This signaling pathway may contribute to the sustained
expression of KCa2.3 in H9c2 cells. p300, histone acetyltransferase
p300; ChIP, chromatin immunoprecipitation; PCR, polymerase chain
reaction; LS, laminar shear stress condition; PI3K,
phosphoinositide 3-kinase; Akt, protein kinase B; KCa,
Ca2+-activated K+ channels. |
Discussion
There were 5 major novel insights gained from the
present study. Firstly, KCa2.3 was upregulated in patients with AF
and in patients with AF combined with MVD. Secondly, LS induced a
marked upregulation of KCa2.3 mRNA and protein expression in H9c2
cells. Thirdly, PI3K activation was associated with LS-induced
upregulation of the KCa2.3 channel. Fourthly, this upregulation was
mediated by PI3K/Akt-dependent Akt activation. Finally, LS
induction of KCa2.3 involved the binding of p300 to transcription
factors in the promoter region of the KCa2.3 gene.
AF is the most common arrhythmia in humans. It
affects 5% of the population >65 years of age, and its incidence
is projected to increase as the mean population age increases
(23). Experimental data from
animal models of AF indicate that AF is associated with progressive
structural and electrical remodeling of the atria. Atrial
structural remodeling is characterized by atrial enlargement and
interstitial fibrosis (24) and
has been considered a major contributor to AF (25). Increased fibrosis has been
observed in the atria of patients with AF (26). It is characterized by enhanced
deposition of matrix collagen proteins; this leads to inhomogeneous
atrial electrical conduction, and results in electrical reentry
circuits that result in AF (27).
Atrial fibrosis alters atrial electrical conduction and
excitability and provides a substrate for AF maintenance. As a
hallmark of atrial structural remodeling, atrial fibrosis serves a
critical role in the maintenance of chronic AF. However, whether
fibrosis is causally associated with AF or an epiphenomenon, and
the precise mechanisms underlying atrial fibrosis, remain
uncertain. The results of the present study suggest that the
percentage of fibroblasts in patients with AF and AF combined with
MVD is increased compared that in patients with SR (~10-fold),
suggesting a complicated association between atrial fibrosis and
AF, consistent with the results of previous studies (28).
In the present study, it was demonstrated that PI3K
was upregulated in patients with AF and in patients with AF
combined with MVD, indicating that PI3K may be involved in the
increased KCa2.3 expression observed in these patients. An
overexpression of the KCa2.3 channel may affect the vascular
structure of the heart during development (29). In cardiac muscle, blockers of
KCa2.3 channels have been demonstrated to prevent atrial
fibrillation (13,14), but at present it is unknown
whether specific openers of KCa2.3 channels will incur
pro-arrhythmic effects. It was noted that previous studies
conducted by Pretorius et al (30), revealed that PI3K activity was
decreased in atrial samples from patients with acute or chronic AF
compared with patients without AF, which is markedly different from
the results from the present study. The disparity between the data
from Pretorius et al (30)
and the present study were examined, and the potential explanations
include, but are not limited to: i) A small sample size in the
present study and that of Pretorius et al (30). Only 4 patients with AF and 6
patients with SR were included in the study by Pretorius et
al (30), and 8 patients with
AF and 6 patients with SR were included in the present study.
Statistically, larger sample sizes would yield more accurate
results, and studies with small sample sizes may provide more
deviation; ii) the disparate sex distribution of the study samples
may have contributed to the differences in the results. In the
present study, the ratios of females to males in the AF and SR
groups were 1:1, whereas in the study of Pretorius et al
(30), the sex distribution was
markedly disparate (3 females and 1 male in the AF group, and 1
female and 5 males in the SR group). This may lead to markedly
different results, as sex is an independent contributory factor in
AF (30-32); iii) race and ethnicity
differences. The present study was performed in a Chinese
population, whilst the study by Pretorius et al (30), was undertaken in an Australian
population; this may have resulted in different findings as race
may also contribute to the AF process (33). Other studies have demonstrated
that mice with decreased cardiac PI3K-Akt activity were highly
susceptible to AF and concluded that ibrutinib increased the risk
of AF partially via inhibition of the cardiac PI3K-Akt pathway
(17,30), was also considered. We
hypothesized that additional data should be collected to achieve an
improved understanding of the potential association between AF and
PI3K as the specific electrophysiological properties of cardiac
cells may lead to major functional differences between human and
nonhuman hearts (34,35).
In fact, although cyclic stretch and hydrostatic
pressure affect the function of ECs, Ohashi et al (36) concluded that physiological shear
stress is dominant to physiological hydrostatic pressure up to 100
mmHg, suggesting the relative contribution of physiological
hydrostatic pressure and fluid shear stress to endothelial
monolayer integrity. Concomitantly, the aim of the present study
was to reveal the potential role of shear stress in inducing AF;
therefore, all experiments were focused on shear stress only.
Previous studies have indicated that shear stress may induce
mechanotransduction in vascular endothelial cells (37) and three-way activation of
mechanically sensitive ion channels, including direct physical
effects mediated by shear stress, changes in the mechanical tension
of the cytoskeleton and the changes in cell membrane fluidity
induced by shear stress (38).
Akt is a major regulator of vital cellular processes including cell
cycle progression, growth and survival, and activation of
extracellular signal-regulated PI3K/Akt signaling pathways has been
suggested to be critical for increased KCa2.3 channels expression.
As calcium-activated, voltage-gated SK3 potassium channels, KCa2.3
channels function as major regulators of Ca2+ transport
through the cell membrane (39,40). As demonstrated in the present
study, treating H9c2 cells with an Akt inhibitor decreased Akt
protein expression and subsequently decreased LS-induced KCa2.3
expression in H9c2 cells. These results are consistent with those
of other previous studies, in which it was demonstrated that Akt is
a key regulator for expression of KCa2.3 (39,40).
Although novel therapeutics for the treatment of AF
are undergoing experimental and clinical evaluation, small molecule
signal transduction inhibitors are the most promising class of
agents. However, the involvement of PI3K/Akt/p300 in LS-induced
KCa2.3 expression in H9c2 cells remains unknown. To the best of our
knowledge, the results of the present study demonstrated for the
first time that in H9c2 cells, LS-induced KCa2.3 expression was
mediated through the activation of PI3K/Akt and downstream
transcriptional co-factor p300, as revealed by the pharmacological
inhibitors dactolisib, GSK690693 and C646. These data not only
confirm the results of previous studies indicating that LS may
induce KCa2.3 expression (8,13),
but also reveal the potential underlying mechanisms by which LS
induces its expression. Dai et al (41) suggested that the PI3K-dependent
pathway participates in the regulation of nuclear factor erythroid
2-related factor 2 nuclear translocation and binding to cofactors
in the LS-induced condition, which provides insight into the
potential roles of LS in inducing transcription factor
translocation or recruitment. Therefore, the present study utilized
ChIP assays to determine whether LS-induced KCa2.3 expression is
regulated via p300, one of the cofactors in KCa2.3 transcription.
The results suggested that, under the condition of 15
dynes/cm2, the amount of PCR product increased gradually
in a time-dependent manner, suggesting that p300 is the key
regulator of KCa2.3 gene expression. Based on the data from the
present study, useful clinical trials aiming to assess the efficacy
of PI3K/Akt/p300 inhibitors in patients with AF should be
performed, and we propose that these data will provide substantial
information regarding the importance of this novel pathway in
patients with AF and AF combined with MVD.
In conclusion, the present study revealed that
atrial fibrosis in patients with AF and AF combined with MVD is
more serious than that in patients with SR, and that KCa2.3 was
upregulated in patients with AF and in those with AF combined with
MVD. In addition, based on the data obtained from clinical samples,
the significant upregulation of KCa2.3 mRNA and protein expression
in H9c2 cells was identified. Subsequent experiments revealed that
this upregulation was mediated by PI3K/Akt-dependent Akt
activation, and that LS induction of KCa2.3 involves the binding of
p300 to transcription factors in the promoter region of KCa2.3
gene. Taken together, these data suggest that a PI3K/Akt/p300
cascade mediates LS induction of KCa2.3. Through understanding this
potentially crucial pathway involved in AF and AF combined with
MVD, rational drug designs may be proposed for these molecules,
which are critical to cell signaling. Based on the specific
expression of molecular targets, clinicians will then be able to
appropriately individualize treatment for patients with AF.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant no. 31360227) and the Yunnan
Province-Kunming Medical University Joint Foundation for Applied
Basic Research (grant no. 2017NS015).
Availability of data and materials
The datasets used in the present study are available
from the corresponding author on reasonable request.
Authors' contributions
GL, QY and YY contributed to the design of the study
and drafted the manuscript. GY and LD performed the experiments.
JW, ZM, YS and GZ made substantial contributions to the analysis of
data, revised and amended the manuscript. All authors have read and
approved this manuscript.
Ethics approval and consent to
participate
All patients provided written informed consent to
participate in the present study. The Institutional Ethical
Committee of the First Affiliated Hospital of Kunming Medical
University approved the study, and the study is in concordance with
the principles outlined in the Declaration of Helsinki.
Patient consent for publication
All patients provided written informed consent to
participate in the present study.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Go AS, Hylek EM, Phillips KA, Chang Y,
Henault LE, Selby JV and Singer DE: Prevalence of diagnosed atrial
fibrillation in adults: national implications for rhythm management
and stroke prevention: the AnTicoagulation and Risk Factors in
Atrial Fibrillation (ATRIA) Study. JAMA. 285:2370–2375. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chugh SS, Havmoeller R, Narayanan K, Singh
D, Rienstra M, Benjamin EJ, Gillum RF, Kim YH, McAnulty JH Jr,
Zheng ZJ, et al: Worldwide epidemiology of atrial fibrillation: A
Global Burden of Disease 2010 study. Circulation. 129:837–847.
2014. View Article : Google Scholar :
|
|
3
|
Benjamin EJ, Wolf PA, D'Agostino RB,
Silbershatz H, Kannel WB and Levy D: Impact of atrial fibrillation
on the risk of death: The Framingham Heart Study. Circulation.
98:946–952. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ynsaurriaga FA, Peinado RP and Ormaetxe
Merodio JM: Atrial fibrillation and quality of life related to
disease and treatment: Focus on anticoagulation. Future Cardiol.
10:381–393. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Colilla S, Crow A, Petkun W, Singer DE,
Simon T and Liu X: Estimates of current and future incidence and
prevalence of atrial fibrillation in the U.S. adult population. Am
J Cardiol. 112:1142–1147. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kwan HY, Leung PC, Huang Y and Yao X:
Depletion of intracellular Ca2+ stores sensitizes the
flow-induced Ca2+ influx in rat endothelial cells. Circ
Res. 92:286–292. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grgic I, Kaistha BP, Hoyer J and Köhler R:
Endothelial Ca+-activated K+ channels in
normal and impaired EDHF-dilator responses–relevance to
cardiovascular pathologies and drug discovery. Br J Pharmacol.
157:509–526. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Takai J, Santu A, Zheng H, Koh SD, Ohta M,
Filimban LM, Lemaître V, Teraoka R, Jo H and Miura H: Laminar shear
stress upregulates endothelial Ca2+-activated
K+ channels KCa2.3 and KCa3.1 via a
Ca2+/calmodulin-dependent protein kinase kinase/Akt/p300
cascade. Am J Physiol Heart Circ Physiol. 305:H484–H493. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ledoux J, Werner ME, Brayden JE and Nelson
MT: Calcium-activated potassium channels and the regulation of
vascular tone. Physiology (Bethesda). 21:69–78. 2006.
|
|
10
|
Brähler S, Kaistha A, Schmidt VJ, Wölfle
SE, Busch C, Kaistha BP, Kacik M, Hasenau AL, Grgic I, Si H, et al:
Genetic deficit of SK3 and IK1 channels disrupts the
endothelium-derived hyperpolarizing factor vasodilator pathway and
causes hypertension. Circulation. 119:2323–2332. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Milkau M, Köhler R and de Wit C: Crucial
importance of the endothelial K+ channel SK3 and
connexin40 in arteriolar dilations during skeletal muscle
contraction. FASEB J. 24:3572–3579. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Si H, Heyken WT, Wölfle SE, Tysiac M,
Schubert R, Grgic I, Vilianovich L, Giebing G, Maier T, Gross V, et
al: Impaired endothelium-derived hyperpolarizing factor-mediated
dilations and increased blood pressure in mice deficient of the
intermediate-conductance Ca2+-activated K+
channel. Circ Res. 99:537–544. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Diness JG, Skibsbye L, Jespersen T,
Bartels ED, Sørensen US, Hansen RS and Grunnet M: Effects on atrial
fibrillation in aged hypertensive rats by
Ca(2+)-activated K(+) channel inhibition.
Hypertension. 57:1129–1135. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qi XY, Diness JG, Brundel BJ, Zhou XB,
Naud P, Wu CT, Huang H, Harada M, Aflaki M, Dobrev D, et al: Role
of small-conductance calcium-activated potassium channels in atrial
electrophysiology and fibrillation in the dog. Circulation.
129:430–440. 2014. View Article : Google Scholar
|
|
15
|
McMullen JR, Amirahmadi F, Woodcock EA,
Schinke-Braun M, Bouwman RD, Hewitt KA, Mollica JP, Zhang L, Zhang
Y, Shioi T, et al: Protective effects of exercise and
phosphoinositide 3-kinase(p110alpha) signaling in dilated and
hypertrophic cardiomyopathy. Proc Natl Acad Sci USA. 104:612–617.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rossello X, Riquelme JA, He Z, Taferner S,
Vanhaesebroeck B, Davidson SM and Yellon DM: The role of PI3Kα
isoform in cardioprotection. Basic Res Cardiol. 112:662017.
View Article : Google Scholar
|
|
17
|
McMullen JR, Boey EJ, Ooi JY, Seymour JF,
Keating MJ and Tam CS: Ibrutinib increases the risk of atrial
fibrillation, potentially through inhibition of cardiac PI3K-Akt
signaling. Blood. 124:3829–3830. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ezeani M and Elom S: Necessity to evaluate
PI3K/Akt signalling pathway in proarrhythmia. Open Heart.
4:e0005962017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seyed Jafari SM and Hunger RE: IHC Optical
Density Score: A new practical method for quantitative
immunohistochemistry image analysis. Appl Immunohistochem Mol
Morphol. 25:e12–e13. 2017. View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Wu CY, Hsieh HL, Sun CC, Tseng CP and Yang
CM: IL-1 beta induces proMMP-9 expression via c-Src-dependent
PDGFR/ PI3K/Akt/p300 cascade in rat brain astrocytes. J Neurochem.
105:1499–1512. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang WC and Chen CC: Akt phosphorylation
of p300 at Ser-1834 is essential for its histone acetyltransferase
and transcriptional activity. Mol Cell Biol. 25:6592–6602. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zoni-Berisso M, Lercari F, Carazza T and
Domenicucci S: Epidemiology of atrial fibrillation: European
perspective. Clin Epidemiol. 6:213–220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nattel S and Harada M: Atrial remodeling
and atrial fibrillation: Recent advances and translational
perspectives. J Am Coll Cardiol. 63:2335–2345. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Iwasaki YK, Nishida K, Kato T and Nattel
S: Atrial fibrillation pathophysiology: Implications for
management. Circulation. 124:2264–2274. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Frustaci A, Chimenti C, Bellocci F,
Morgante E, Russo MA and Maseri A: Histological substrate of atrial
biopsies in patients with lone atrial fibrillation. Circulation.
96:1180–1184. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Friedrichs K, Baldus S and Klinke A:
Fibrosis in atrial fibrillation - role of reactive species and MPO.
Front Physiol. 3:2142012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tan AY and Zimetbaum P: Atrial
fibrillation and atrial fibrosis. J Cardiovasc Pharmacol.
57:625–629. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wandall-Frostholm C, Skaarup LM, Sadda V,
Nielsen G, Hedegaard ER, Mogensen S, Köhler R and Simonsen U:
Pulmonary hypertension in wild type mice and animals with genetic
deficit in KCa2.3 and KCa3.1 channels. PLoS One. 9:e976872014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pretorius L, Du XJ, Woodcock EA, Kiriazis
H, Lin RC, Marasco S, Medcalf RL, Ming Z, Head GA, Tan JW, et al:
Reduced phosphoinositide 3-kinase (p110alpha) activation increases
the susceptibility to atrial fibrillation. Am J Pathol.
175:998–1009. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fang MC, Singer DE, Chang Y, Hylek EM,
Henault LE, Jensvold NG and Go AS: Gender differences in the risk
of ischemic stroke and peripheral embolism in atrial fibrillation:
The AnTicoagulation and Risk factors In Atrial fibrillation (ATRIA)
study. Circulation. 112:1687–1691. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dagres N, Nieuwlaat R, Vardas PE, Andresen
D, Lévy S, Cobbe S, Kremastinos DT, Breithardt G, Cokkinos DV and
Crijns HJ: Gender-related differences in presentation, treatment,
and outcome of patients with atrial fibrillation in Europe: A
report from the Euro Heart Survey on Atrial Fibrillation. J Am Coll
Cardiol. 49:572–577. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shen AY, Yao JF, Brar SS, Jorgensen MB and
Chen W: Racial/ethnic differences in the risk of intracranial
hemorrhage among patients with atrial fibrillation. J Am Coll
Cardiol. 50:309–315. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nass RD, Aiba T, Tomaselli GF and Akar FG:
Mechanisms of disease: Ion channel remodeling in the failing
ventricle. Nat Clin Pract Cardiovasc Med. 5:196–207. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Byrd JC, Hillmen P and James DF: Response:
Additional data needed for a better understanding of the potential
relationship between atrial fibrillation and ibrutinib. Blood.
125:16732015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ohashi T, Sugaya Y, Sakamoto N and Sato M:
Relative contribution of physiological hydrostatic pressure and
fluid shear stress to endothelial monolayer integrity. Biomed Eng
Lett. 6:31–38. 2016. View Article : Google Scholar
|
|
37
|
Li X, Yang Q, Wang Z and Wei D: Shear
stress in atherosclerotic plaque determination. DNA Cell Biol.
33:830–838. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Butler PJ, Norwich G, Weinbaum S and Chien
S: Shear stress induces a time- and position-dependent increase in
endothelial cell membrane fluidity. Am J Physiol Cell Physiol.
280:C962–C969. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zamanian M, Veerakumarasivam A, Abdullah S
and Rosli R: Calreticulin and cancer. Pathol Oncol Res. 19:149–154.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fresno Vara JA, Casado E, de Castro J,
Cejas P, Belda-Iniesta C and González-Barón M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dai G, Vaughn S, Zhang Y, Wang ET,
Garcia-Cardena G and Gimbrone MA Jr: Biomechanical forces in
atherosclerosis-resistant vascular regions regulate endothelial
redox balance via phosphoinositol 3-kinase/Akt-dependent activation
of Nrf2. Circ Res. 101:723–733. 2007. View Article : Google Scholar : PubMed/NCBI
|