Introduction
Periodontal disease is a chronic inflammatory
disease and the major cause of teeth loss in adults (1). The pathogenesis of periodontal
disease is due to the microorganisms that adhere to and grow on the
surfaces of teeth or periodontal tissues, triggering chronic
inflammatory reactions and subsequently damaging the connection of
periodontal soft tissues to facilitate the absorption of alveolar
bone. This leads to destruction of the structures supporting the
teeth and eventual tooth loss (2,3).
Currently, there are no effective treatment methods for periodontal
disease. The periodontal ligament contains a group of pluripotent
periodontal stem cells, which can express surface markers of
mesenchymal stem cells with the characteristics of self-renewal and
pluripotency (4)..
Human periodontal ligament stem cells (hPDLSCs) can differentiate
to form bones, cartilage, neurons, adipocytes and blood vessel
tissues via in vitro induction, and can form a
cementum-periodontal ligament-alveolar bone-like structure
following in vivo implantation (5-8).
This indicates that hPDLSCs have significant involvement in the
reconstruction, regeneration and fixation of periodontal tissue.
hPDLSCs were isolated for the first time from the third molar
periodontal ligament by Seo et al (9). hPDLSCs are also found on the inner
surface of the tooth socket following tooth extraction (10). Highly purified hPDLSCs clones have
also been extracted from periodontal tissues (11); studies have purified hPDLSCs from
inflammatory periodontal ligament tissue and confirmed their
potential to regenerate cementum and periodontal ligament (12). Studies have revealed novel ways of
obtaining purified hPDLSCs from suppressed periodontal ligaments
(13-16). Furthermore, studies have shown
that the periodontal inflammatory microenvironment can destroy
periodontal tissue by suppressing the regeneration ability of
hPDLSCs; however, the underlying mechanism remains to be elucidated
(17).
Due to their proliferation and pluripotent
differentiation ability, and their capacity to form Sharpey's
fibres and cementum-like structures, hPDLSCs are considered the
optimal seeding cells in periodontal engineering (18). The co-implantation of hPDLSCs with
hydroxyapatite or β-tricalcium phosphate scaffold in nude mice
allowed the formation of periodontal ligament- and cementum-like
structures (9). In addition,
animal experiments have revealed that multiple PDLSCs from
different sources, including human, canine and swine sources, can
initiate the homing effects and facilitate the regeneration of
periodontal tissue following implantation (19-21). Dentin non-collagen proteins can
increase the proliferation and adhesion abilities, facilitate
morphological changes, increase alkaline phosphatase (ALP) activity
and induce the differentiation of hPDLSCs into cementoblasts
(22). As hPDLSCs cannot express
human leukocyte antigen II or co-stimulatory molecules, it is
hypothesized that the underlying mechanism of hPDLSC
immunosuppression may be achieved by prostaglandin 2-mediated T
cell non-responsiveness (23).
Therefore, studies investigating periodontal disease should focus
on investigating the molecular mechanisms involved in the
osteogenic differentiation of hPDLSCs in the chronic inflammatory
microenvironment, in order to provide a theoretical basis for
optimizing the stem cell-mediated treatment of periodontal
inflammatory diseases.
Toll-like receptors (TLRs) are a class of
transmembrane receptors, which can recognize and bind the
corresponding pathogen-associated molecular patterns, activate the
signal transduction pathway and induce the expression of certain
immune effector molecules, including inflammatory cytokines
(24). TLRs are type I
transmembrane proteins and can be divided into three structural
parts: Extracellular, cytoplasmic and transmembrane. The
extracellular domain of TLRs are responsible for receptor
recognition and the binding to other auxiliary receptors to form a
receptor complex (25,26). TLR4, the first identified
mammalian TLR, is expressed in all cell lines. Findings indicate
that TLR4 is important in pro-inflammatory reactions, promoting the
maturation and differentiation of immune cells, and regulating
immune responses (27). TLR4 is
involved in the inflammatory response to ischemia-reperfusion
injury in vivo. TLR4 is also involved in the apoptosis of
various cell types, including immune cells, hepatocytes,
cardiomyocytes, gastric cancer cells, breast cells, renal tubular
epithelial cells and airway smooth muscle cells (28). Studies have demonstrated that mice
with deficient TLR4 function exhibit significantly lower ischemia
damage compared with wild-type mice. The inhibition of TLR4 can
reduce ischemia-reperfusion injury by regulating apoptosis
(29). As a member of the TLR
family, TLR4 is a receptor of lipopolysaccharide (LPS) and lipid A
in Gram-negative bacteria. When LPS is bound together with TLR4, a
cascade of signaling pathways is triggered which can activate the
expression of type I interferons (IFN-α and IFN-β),
pro-inflammatory cytokines, including interleukin (IL)-1, IL-6 and
tumor necrosis factor (TNF)-α, and chemotactic cytokine IL-8
(30). Previous studies have
indicated that the LPS-induced activation of hPDLSCs is followed by
activation of the TLR4/MyD88 complex, which causes the release of
cytokines, including IL-1α, IL-8 and TNF-α (31). However, the exact role of TLR4 in
the differentiation of hPDLSCs, particularly under inflammatory
conditions, and the underlying molecular mechanisms have not been
investigated in detail.
The present study aimed to investigate the
regulatory roles of TLR4 in the osteogenic and adipogenic
differentiation of hPDLSCs under inflammatory conditions, in order
to reveal the regulatory role of the TLR4 signaling pathway in stem
cell osteogenic differentiation. This may lay a foundation for the
diagnosis and treatment of the chronic inflammatory processes and
tissue engineering.
Materials and methods
Isolation and primary culture of
hPDLSCs
Specimens were collected from patients who underwent
orthodontic teeth extraction in the Department of Oral and
Maxillofacial Surgery of Hospital of Stomatology, Tongji
University. The specimens were divided into two groups of 10
samples: The control group comprised patients without decayed
teeth, apical periodontitis or periodontitis, and the experimental
group comprised patients with periodontal inflammation. Briefly,
following orthodontic extraction, the teeth were transferred into
the culture medium and the tooth roots were repeatedly washed with
antibiotic-containing PBS to remove the clotted blood.
Subsequently, two-thirds of the periodontal membranes in the roots
were scraped, and transferred into centrifuge tubes following
thorough cutting with scissors. To each tube, 3 ml of 0.1%
collagenase was added, and the tube was shaken and digested at 37°C
for 50-60 min. Subsequently, 1% trypsin was added for further
digestion for 25 min. Thereafter, a small volume of 10% FBS was
added to terminate the digestion. Following centrifugation at 700 ×
g for 10 min at room temperature, the supernatant was discarded and
then inoculated into a culture flask. An appropriate quantity of
α-MEM culture medium supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) was added and placed in
an incubator containing 5% CO2 at 37°C in saturated
humidity. The medium was replaced every 2-3 days and the cells were
sub-cultured when they grew to confluence. Finally, the original
medium was discarded and the cells were washed with PBS; the cells
were then examined under an inverted microscope. In order to
examine changes between the normal and malignant/differentiation
conditions, only hPDLSCs from healthy subjects were used in the
subsequent experiments.
Purification of periodontal membrane stem
cells by flow cytometry
Third passage hPDLSCs were collected, washed with
PBS and digested with trypsin. A cell suspension was prepared at a
density of 1×106 cells/ml and dispensed into a
sterilized EP tube (1.5 ml). The fluorescently marked anti-human
Stro-1-APC (cat. no. MA5-28635), CD146-FITC (cat. no. 11-1469-42),
CD90-APC (cat. no. 17-0909-42), CD105-PE (cat. no. 12-1051-82),
CD29-FITC (cat. no. 11-0291-82), CD31-APC (cat. no. 17-0311-82) and
CD45-PE (cat. no. 12-0459-41) antibodies (1:10; all from Thermo
Fisher Scientific, Inc.) were added to each tube, separately. The
cells were incubated on ice for 1.5 h and subsequently centrifuged
at 700 × g for 3 min at room temperature. Finally, the supernatant
was discarded and the cells were washed with PBS three times,
re-suspended in 3% FBS and subjected to flow cytometric
analysis.
Immunohistochemistry and
immunofluorescence of hPDLSCs
The well-grown cells were collected to prepare
slides and fixed in 4% paraformaldehyde for 30 min, and TLR4 and
Stro-1 were stained for using the immunochemical SABC method.
Briefly, following washing three times with PBS (2 min each time),
the cells were incubated with 3% Triton-100 solution for 15 min to
increase cell permeability. Subsequently, 3% hydrogen
peroxide-methanol solution was added for 15 min to eliminate the
effect of endogenous peroxidase. The cells were then washed three
times with PBS, and normal goat serum (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) blocking solution was added for 15 min to
eliminate non-specific staining. Following this, primary antibodies
against TLR4 (polyclonal antibody; cat. no. sc-293072; 1:300; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA), keratin (polyclonal
antibody; cat. no. PA5-68042; 1:300; Thermo Fisher Scientific,
Inc.); Stro-1 (rabbit anti-human antibody; cat. no. sc-47733;
1:100; Santa Cruz Biotechnology, Inc.) were added. For the negative
control, PBS was used instead of primary antibody and transferred
to a refrigerator at 4°C overnight. Following rewarming in a 37°C
incubator for 1 h and washing with PBS (5 min, three times),
biotinylated secondary antibody (cat. no. ab6788; 1:1,000; Abcam,
Cambridge, UK) was added at 37°C for 1 h. Following this, a 1:50
dilution of FITC-labeled goat anti-rabbit IgG (cat. no. sc-2040;
1:50; Santa Cruz Biotechnology, Inc.) was added in the Stro-1
immunofluorescence group. Following washing with PBS and rinsing
with distilled water, 500 ml of non-fluorescent buffer glycerol was
used for sealing in the Stro-1 immunofluorescence group, whereas
SABC reagent was added to the other two groups, and incubated in a
37°C incubator for 20 min. Following DAB staining and hematoxylin
staining, examination was performed under an optical
microscope.
MTT assay
The fourth passage isolated hPDLSCs were
sub-cultured in 96-well plates at a density of 2×103
cells/ml per well (six replicates) in an incubator containing 5%
CO2 at 37°C. Therefore, 20 µl of 0.5% MTT
solution was added every day at a fixed time point, followed by
incubation for 4 h. This step was repeated for 14 days. In each
case, when culture was stopped, the medium was carefully removed
and 150 µl of DMSO was added into each well. The cultures
were then shaken at a low speed for 10 min to completely dissolve
the crystals. Finally, the absorbance was measured at 490 nm using
a spectrophotometric approach.
Cell cycle analysis
Cells were collected in the logarithmic growth
phase, counted with a hemocytometer and inoculated into 6-well
plates at a density of 5×105 cells/ml. From this,
~1×106 cells were collected and transferred into a
centrifuge tube. Following centrifugation at 700 × g for 5 min at
room temperature, the supernatant was discarded and the cells were
washed with PBS and resuspended with 0.5 ml PBS in the tube.
Subsequently, 75% cold ethanol was added and mixed, followed by
storage at −20°C for 24 h. During the assay, the ethanol was
discarded by centrifugation, washed with PBS and resuspended with 1
ml PBS. RNase (5 µl, 10 mg/ml) was added and followed by
incubation in a water bath at 37°C for 1 h. Propidium iodide (PI; 5
µl, 10 mg/ml) was then added, followed by incubation at room
temperature for 30 min in the dark. Finally, the cell cycle was
detected by flow cytometry.
Pluripotency induction
Cells at logarithmic growth phase were cultured at a
density of 2×104 cells/ml in a six-well plate with
coverslips in an incubator at 37°C containing 5% CO2.
The medium was replaced every 3 days, and when the cells reached
60% confluence, the culture medium was replaced with osteogenic,
chondrogenic and adipogenic culture medium. The conventional
standard medium was used as a control. The cells were collected on
the 14th day and the medium was discarded. Fixation was performed
using 4% formaldehyde (for Alizarin Red S staining) or formalin
(Oil Red O staining) for 15 min at room temperature followed by
washing (twice) with PBS. The cells were finally stained with
Alizarin Red S and Oil Red O, respectively, and were visualized and
analyzed under a microscope.
ALP assays
ALP activity was assessed using an ALP assay kit.
The cells were washed and, following measurement of the cell
concentration, 50 µl of the assay buffer was added and
followed by centrifugation at 11,900 × g for 3 min at 4°C.
Following this, 50 µl of 5 mM PNPP reaction solution was
added to each well and reacted at 60°C for 60 min in the dark.
Finally, 20 µl of the stop solution was added and mixed, and
the values of absorbance were measured at 405 nm. Each assay
condition was performed in triplicate and the results were repeated
in at least three independent experiments. ALP activity was
normalized by total cellular protein concentrations among the
samples.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the hPDLSCs of the
normal and inflammation groups using TRIzol reagent and reverse
transcribed into cDNA. PCR primers (Table I) were designed using the Primer3
program (http://bioinfo.ut.ee/primer3-0.4.0/). SYBR-Green-based
qPCR analysis was performed to amplify the genes of interest using
the RG-3000 Real-Time DNA Detection system (Corbett Research;
Qiagen, Inc., Valencia, CA, USA). Triplicate reactions were
performed for each sample. The amplification system was as follows:
Power SYBR®-Green Master Mix 10 µl, Forward
Primer (10 µM) 0.5 µl, Reverse Primer (10 µM)
0.5 µl, cDNA 1 µl, ddH2O up to 20
µl. The amplification conditions were as follows: 94°C for 2
min for one cycle and 30 cycles at 92°C for 20 sec, 57°C for 30
sec, and 72°C for 20 sec, followed by extension at 78°C for each
cycle. The relative expression levels of OCN, RUNX2, LPL, COLI,
TLR4, NF-κBP65, ALP, PPARγ were calculated using the
2−ΔΔCq method (32).
All samples were run in triplicate and normalized by the endogenous
expression level of GAPDH.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Primer | Sequence
(5′-3′) | Temperature
(°C) | Gene size (bp) |
|---|
| GAPDH-F CC |
TGCACCACCAACTGCTTA | 60.54 | 144 |
| GAPDH-R C |
ATCACGCCACAGCTTTCCA | 61.24 | |
| OCN-F |
ATTGTGACGAGCTAGCGGAC | 60.18 | 131 |
| OCN-R |
TCGAGTCCTGGAGAGTAGCC | 60.11 | |
| RUNX2-F |
TGGCCGGGAATGATGAGAAC | 60.11 | 79 |
| RUNX2-R |
TTGAACCTGGCCACTTGGTT | 60.03 | |
| LPL-F |
CATGCAAGAACGGTGCCAAG | 59.96 | 169 |
| LPL-R C |
TCACAGTGATGGCCTGTGT | 59.96 | |
| COLI-F |
TGCGACAGTCTGGATGTCTT | 59.03 | 184 |
| COLI-R |
TGAAGGACCCGGATTTCACG | 60.04 | |
| TLR4-F |
TGTATCGGTGGTCAGTGTGC | 60.04 | 172 |
| TLR4-R |
CAGCTCGTTTCTCACCCAGT | 59.97 | |
| NF-κBP65-F |
GATCCTTTCGGAACTGGGCA | 60.04 | 113 |
| NF-κBP65-R |
AGGTATGGGCCATCTGTTGAC | 59.79 | |
| ALP-F |
GCAGCACTCCTTCCGGTATT | 60.11 | 156 |
| ALP F-R |
GTACAATCTTCACGCCCGGA | 60.11 | |
| PPARr-F |
ATTCGGCTAAAGCTGGCGTA | 59.82 | 106 |
| PPARγ-R |
TGCATTGTGTGACATCCCGA | 59.96 | |
Stimulation and culture of hPDLCs
The cells were divided into a control group, TLR4
activation group (LPS-PG, 10 ng/ml, 24 h), TLR4 antagonist group
(TLR4 antagonist, 1 µg/ml, 24 h), osteogenic induction
control group, osteo-genic induction and agonist group, and
osteogenic induction and antagonist group. Each group of cells were
cultured in α-MEM medium (D-glucose 1,000 mg/l, L-glutamine 292
mg/l, sodium pyruvate 110 mg/l, NaHCO3 2200 mg/l,
Penicillin 100 U/ml, Streptomycin 100 µg/ml, pH 7.0-7.4)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.),
and placed in an incubator containing 5% CO2 at 37°C in
a saturated humidity.
Scratch assay
The back of a 6-well plate was marked with uniformly
horizontal lines, separated by 0.5-1 cm. Each well included at
least five lines. The cells (~5×105) were added to each
well and incubated overnight. The following day, a pipette tip and
ruler were used to scratch perpendicularly to the back horizontal
line. The cells were washed three times with PBS, added to
serum-free cell culture medium and incubated for 24 h in a 37°C
incubator containing 5% CO2. Images were captured by
using an inverted phase microscope (Nikon Corporation, Tokyo,
Japan) at 0, 24 and 48 h.
Enzyme-linked immunosorbent assay
(ELISA)
Briefly, 96-well polysorp plates (Nalge Nunc
International, Penfield, NY, USA) were coated with recombinant
tubulin-α-1c protein at a concentration of 10 µg/ml in 0.05
M carbonate buffer at 4°C overnight. The wells were then washed
with PBS containing 0.05% Tween-20 (PBS-T) four times and blocked
with 3% BSA-PBS for 3.5 h at 37°C. The samples were diluted at
1:100 with PBS-T containing 1% BSA and were added to the 96-well
plate. Wells filled with PBS-T containing 1% BSA without samples
were set up to examine non-specific background. Following
incubation for 1.5 h at 37°C, the wells were washed six times with
PBS-T. Subsequently, 100 µl of goat anti-human IgG
conjugated to peroxidase, (cat. no. 109-001-003; 1:5,000; Jackson
Immunoresearch Laboratories, West Grove, PA, USA), was added to
each well and incubated for 30 min at 37°C. Following washing with
PBS-T four times, the bound antibodies were detected with
o-phenylenediamine as substrate. The reaction was terminated by
adding 100 µl of 2 M sulfuric acid to each well. The values
of absorbance were measured at 450 nm using a Bio-Rad plate
reader.
Apoptosis detection
The cells were collected and re-suspended in PBS.
The cells (1×104) were centrifuged at 700 × g for 5 min
at room temperature and the supernatant discarded. The cells were
gently re-suspended by adding 500 µl of Annexin V-FITC
conjugate. The cells were then mixed and incubated at room
temperature for 10 min in the dark. Following centrifugation at 700
× g for 5 min at room temperature, the supernatant was discarded
and the cells were gently re-suspended by adding 500 µl of
Annexin V-FITC Binding Solution. Following this, 10 µl of PI
staining solution was added, followed by gentle mixing in an ice
bath in the dark. Flow cytometry was then used for the detection of
apoptosis.
Western blot analysis
Briefly, the cells were collected and lysed in RIPA
buffer. The BCA method was used to detect protein concentration,
and 30 µg of protein was loaded onto a 10% gradient SDS-PAGE
gel. Following electrophoretic separation, the proteins were
transferred onto an Immobilon-P membrane. The membranes were
blocked with Super-Block Blocking Buffer, and probed with primary
antibodies targeting NF-κBP65 (cat. no. sc-156137), TLR4 (cat. no.
sc-293072), and GAPDH (cat. no. sc-47724; all 1:1,000; all from
Santa Cruz Biotechnology, Inc.) followed by incubation with a
secondary antibody conjugated to horseradish peroxidase (cat. no.
sc-2354; 1:5,000; Santa Cruz Biotechnology, Inc.). The proteins
were detected using the SuperSignal West Pico Chemiluminescent
Substrate kit (Thermo Fisher Scientific, Inc.) and quantified using
ImageJ Software (version 1.48; National Institutes of Health,
Bethesda, MD, USA).
Statistical analysis
GraphPad Prism software version 6 (GraphPad
Software, Inc., La Jolla, CA, USA) for Windows was used to complete
the statistical analysis. The data are expressed as the mean ±
standard deviation. One-way analysis of variance was performed and
followed by Tukey's multiple comparison post hoc test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Isolation of hPDLSCs
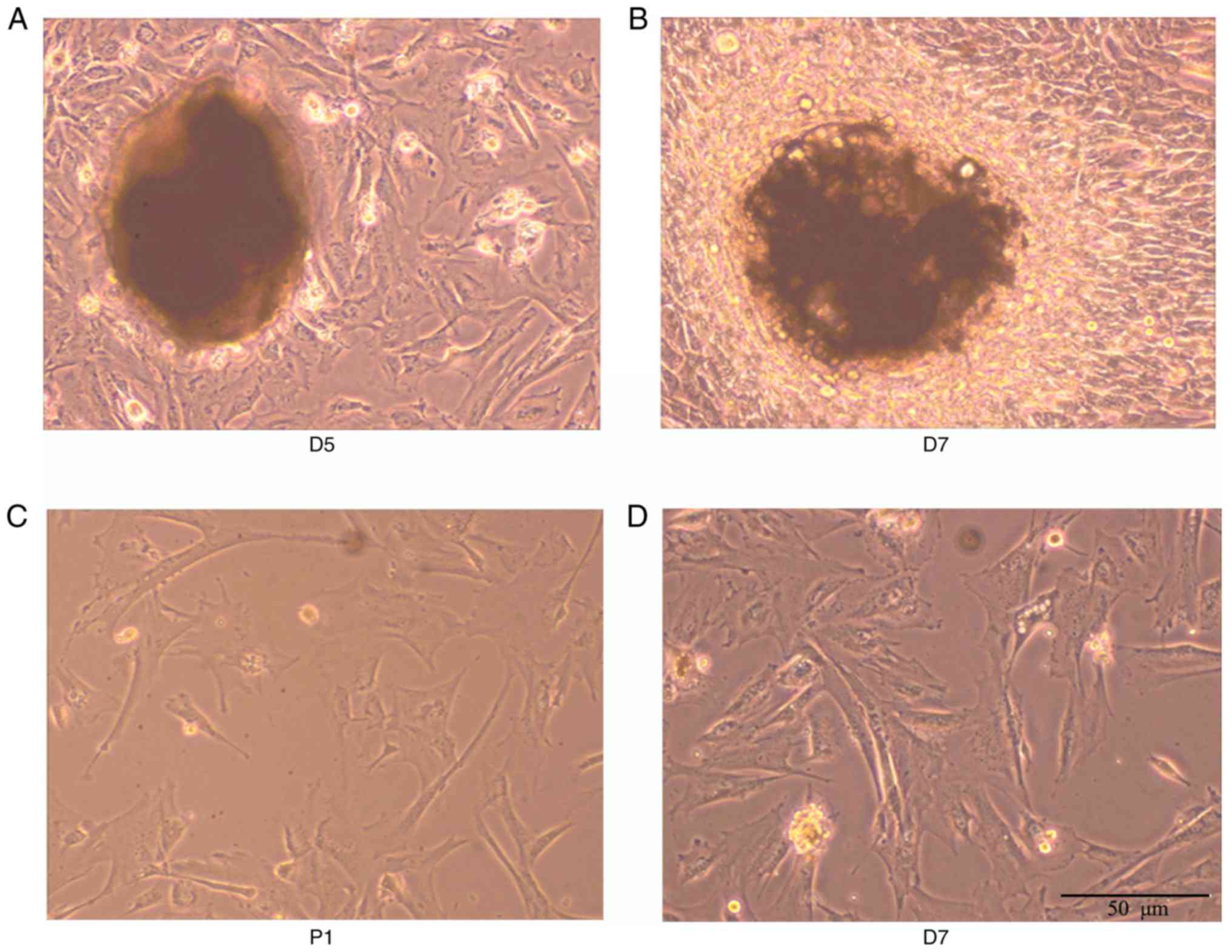
As shown in Fig.
1A-D, from the 5th day of culture adhesion, cells emerged from
the edge of the tissue block, most of which showed structures
including long spindle-shaped or irregular polygons. Cells in the
center of the tissue block were condensed, whereas peripheral cells
were radial in growth. The cell body was full and the nucleus
clear. With subculture, cell density was increased. The
proliferation rates of the passage 1 and passage 2 cells were
accelerated. Following inoculation at the ratio of 1:3, the
appearance of the cells was monolayer cell fusion, and the cells
were long spindle-shaped and arranged in bundles. The flow
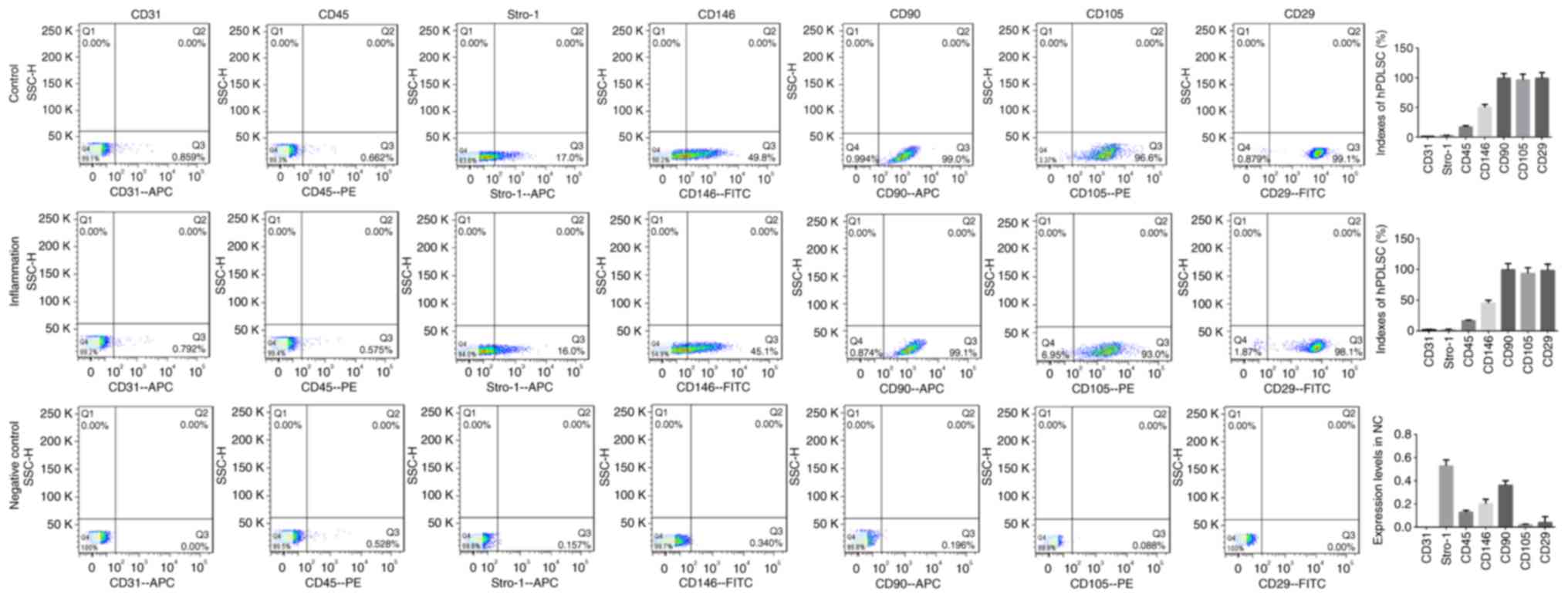
cytometry results (Fig. 2) showed
that the expression levels of CD31, CD45, CD146, CD90, CD105, CD29
and Stro-1 in the negative control group were all <1%, and the
indices of the hPDLSCs in the inflammatory groups (CD146, CD90,
CD105, CD29 and Stro-1) were all positive, indicating that
isolation of the hPDLSCs had been successful.
LPS induces the inflammation of hPDLSCs
and increases the expression of TLR4
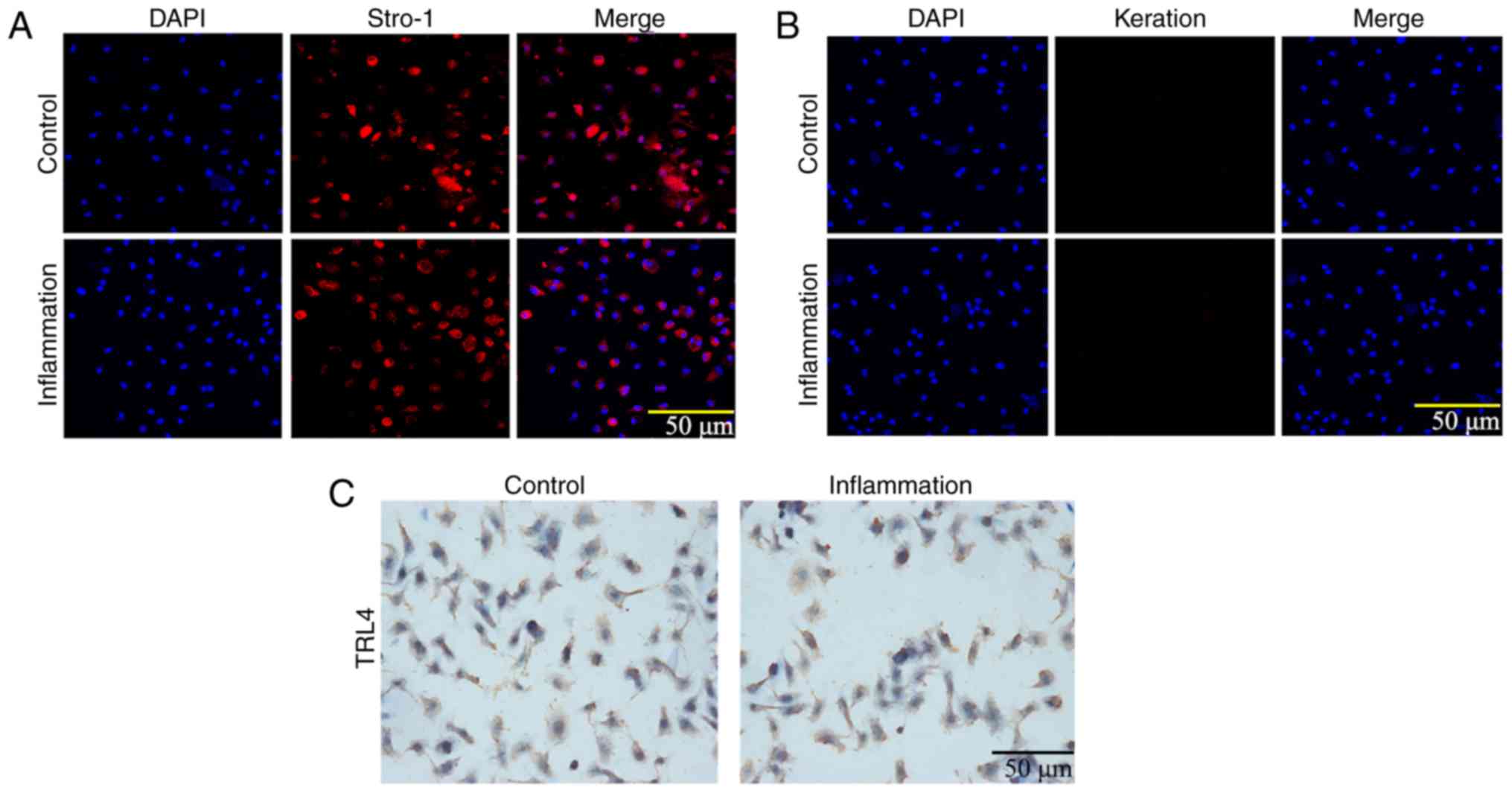
The expression levels of Stro-1 and keratin were
detected by immunofluorescence. As shown in Fig. 3A and B, Stro-1 was positively
expressed in the control and inflammation groups, whereas keratin
was not expressed. The expression of Stro-1 in the control group
was increased compared with that in the inflammation group,
indicating that inflammation affected cell growth. In addition, the
immunohistochemistry analysis showed that the expression of TLR4
was increased in the inflammation group (Fig. 3C). The MTT assay showed that the
logarithmic cell growth phase was obtained between 3 and 8 days,
whereas the stable growth phase was reached in 8-10 days (Fig. 4A). The absorbance in the
inflammation group was lower than that in the control group,
indicating that inflammation affected the cell proliferation rate.
Flow cytometric analysis of the cell cycle (Fig. 4B and C) showed that the frequency
of hPDLSCs in the G1 phase in the control group was higher than
that in the inflammation group. By contrast, the frequency of
hPDLSCs in the G2 phase was lower compared with that in the
inflammation group, indicating that inflammation affected the cell
cycle. The Alizarin Red staining showed that hPDLSCs in the control
and inflammation groups were positively stained and the number of
calcium nodules in the control group was higher compared with that
in the inflammation group (Fig.
4D), suggesting that inflammation inhibited osteogenic
differentiation. The number of oil droplets in the control group
was lower than that in the inflammation group (Fig. 4D), suggesting that there was
induction of adipo-differentiation by LPS. The activity of ALP in
the hPDLSCs in the control group was higher than that in the
inflammation group, indicating decreased osteogenic differentiation
in the inflammation group (P<0.05; Fig. 4E). The RT-qPCR results indicated
that the expression levels of OCN, Runx2 and COL1 in the control
group were higher than those in the inflammatory group, and the
expression levels of the adipogenesis-related genes, including poly
(ADP-ribose) polymerase (PPAR)γ and lipoprotein lipase (LPL), were
lower than those in the inflammatory group, indicating that the
cells in the control group had higher osteogenic capacity than
those in the inflammatory group, whereas lipogenic capacity was
lower in the control group than in the inflammation group
(P<0.05; Fig. 4F).
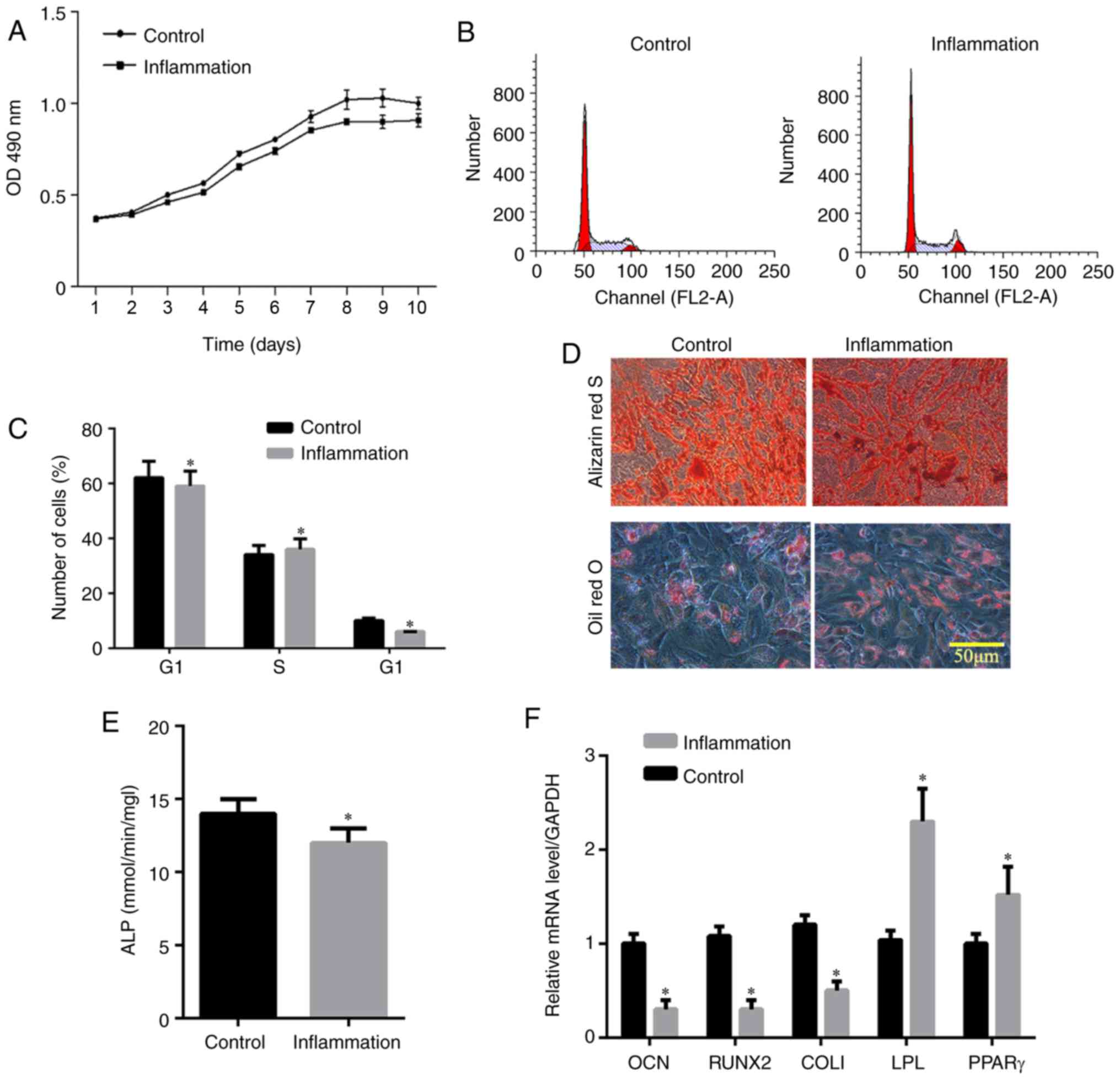 | Figure 4Effect of inflammation on human
periodontal ligament stem cells. (A) Cell proliferation detected
using an MTT assay; (B) cell cycle analysis; (C) distribution of
cell cycle detected by flow cytometry; (D) pluripotency induction
(osteogenic differentiation and adipogenic differentiation); (E)
relative content of ALP; (F) relative content of osteogenic
induction genes (OCN, Runx2 and COL1) and adipogenesis-related
genes (PPARγ and LPL). *P<0.05, vs. Control group.
LPS, lipopolysaccharide; ALP, alkaline phosphatase; OCN,
osteocalcin; Runx2, Runt-related transcription factor 2; Col1,
collagen I; PPAR, poly (ADP-ribose) polymerase; LPL, lipoprotein
lipase; D, day. |
Long-term TLR4 activation inhibits cell
proliferation and migration but increases inflammation and
apoptosis of hPDLSCs
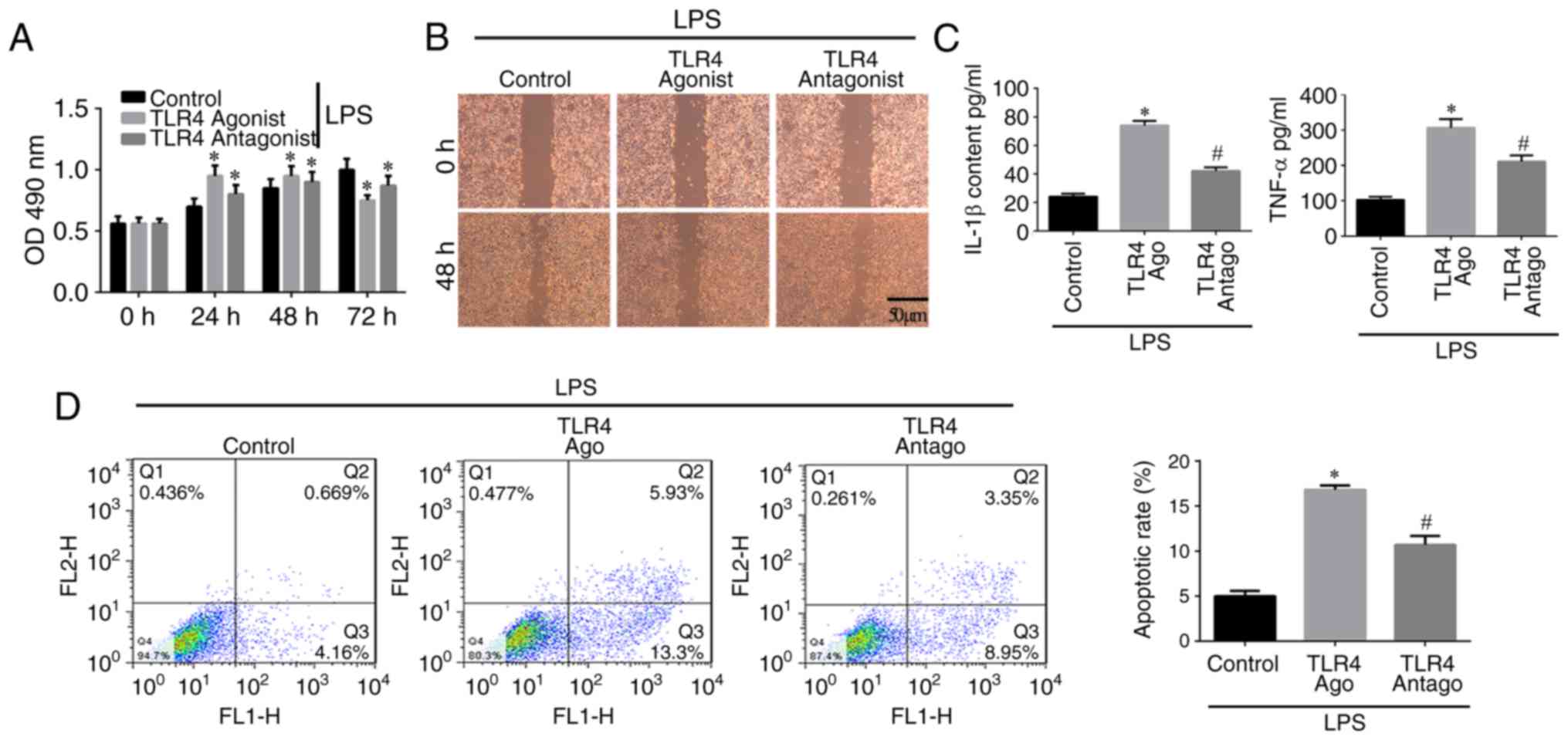
The results of the MTT assay (Fig. 5A) showed that cell proliferation
in the control group increased with time. Cell proliferation in the
TLR4 activation group was increased between 0 and 48 h but
decreased at 72 h, indicating that TLR4 activation stimulated cell
proliferation within a certain period of time, following which the
proliferation ability was reduced. The scratch assay (Fig. 5B) showed that a large number of
cells migrated into the scratch area in the control group, whereas
fewer cells had migrated in the TLR4 activation group. In addition,
in the TLR4 antagonist group, an increased number of cells was
observed in the scratch area, but this was relatively lower than
the control group. The ELISA assay indicated that the
concentrations of IL-1β and TNF-α in the TLR4 activation group were
higher compared with those in the other groups (P<0.05; Fig. 5C). The levels of IL-1β and TNF-α
in the control group were lower compared with those in the other
groups. In the TLR4 antagonist group, the levels of IL-1β and TNF-α
were higher than in the control group but lower that in the TLR4
activation group (Fig. 5C). The
flow cytometric assay of apoptosis indicated that the apoptotic
rate of cells in the TLR4 activation group was higher compared with
the rates in the other groups (P<0.05), but the rate in the TLR4
antagonist group was higher compared with that in the control group
(Fig. 5D). Therefore, TLR4
activation inhibits the proliferation and migration, but induces
the inflammation and apoptosis of hPDLSCs.
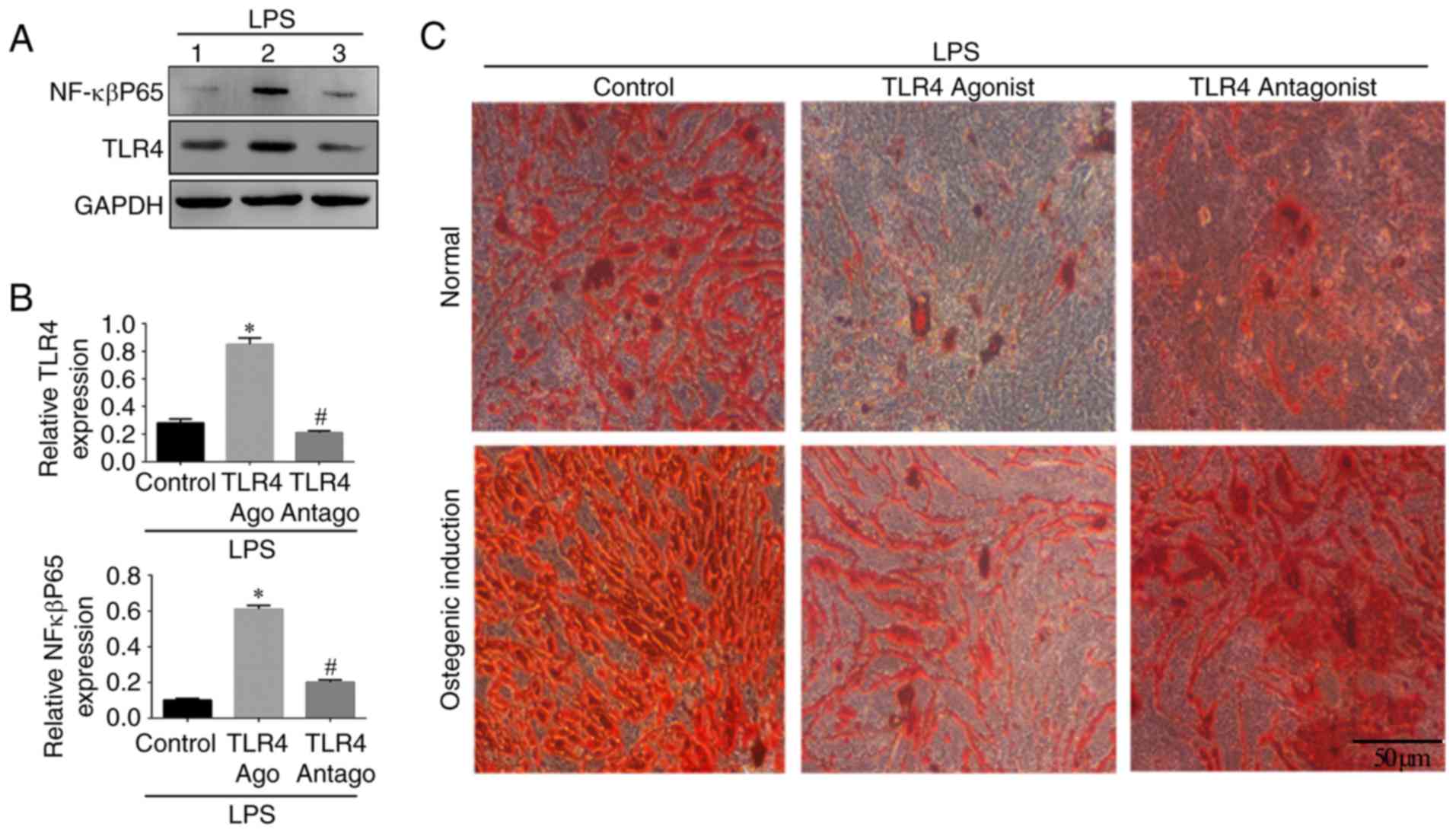
NF-κBP65/TLR4 axis is involved in the
osteogenic differentiation of hPDLSCs
In order to investigate the involvement of the
NF-κBP65/TLR4 axis, western blotting was used to detect the
expression of NF-κBp65 and TLR4 in the different groups. The
expression levels of NF-κBp65 and TLR4 were increased in the TLR4
activation group compared with those in the other groups
(P<0.05; Fig. 6A and B). The
expression of NF-κBp65 in the control group was significantly lower
compared with that in the other groups. No significant difference
in the expression of TLR4 was found between the TLR4 antagonist
group and the control group. The number of Alizarin Red S-stained
mineralized nodules in the LPS-induced cells cultured in normal
medium in presence of TLR4 antagonist and in the control group was
higher compared with that in the TLR4 agonist group (Fig. 6C). The same trend was observed in
the osteogenic medium, in which the number of mineralized nodules
was higher compared with that in the normal medium (Fig. 6C). The results of the RT-qPCR
analysis confirmed that the expression of NF-κBp65 in the TLR4
activation group was higher compared with that in the other groups
(P<0.05) and the lowest expression levels were found in the
control group. Compared with the control group, the expression
levels of osteogenic genes (OCN, Runx2 and COL1) and ALP were
decreased by treatment of the LPS-induced cells with TLR4 agonist,
whereas the TLR4 antagonist reversed this effect. The inverse
results were observed for the adipogenic genes (PPARγ and LPL).
Similar trends in the expression levels of these genes were
recorded in the osteogenic induction medium (P<0.05; Fig. 7). These results indicated that the
NF-κBp65/TLR4 axis is involved and may inhibit the osteogenic
differentiation of hPDLSCs under inflammatory conditions.
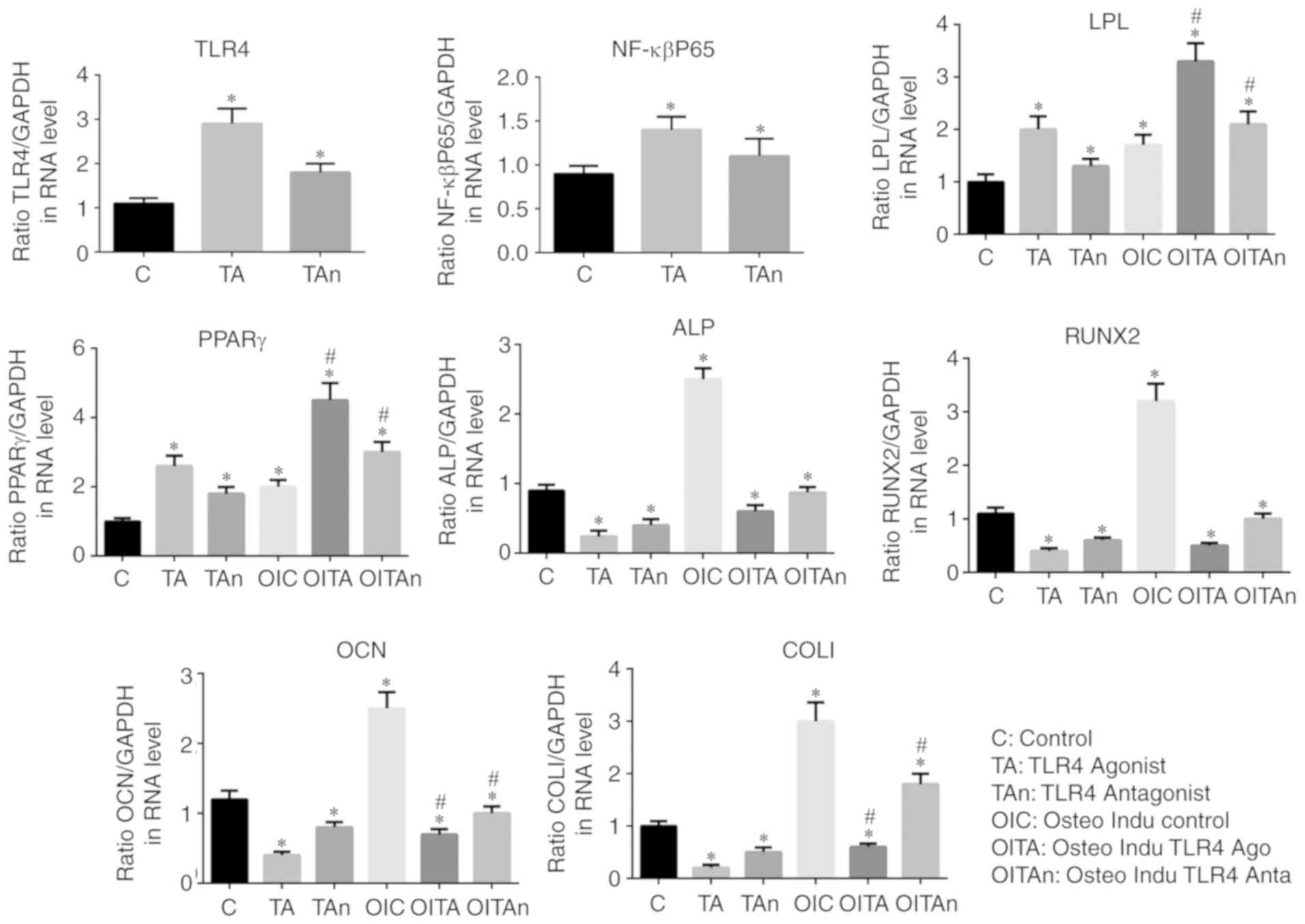 | Figure 7Expression levels of osteogenic
induction genes (OCN, Runx2 and COL1), adipogenesis-related genes
(PPARγ and LPL), ALP, NF-κBp65 and TLR4. *P<0.05 vs.
C group; #P<0.05 vs. TA group. TLR4, Toll-like
receptor 4; C, Control; TA, TLR4 agonist; Tan, TLR4 antagonist;
OIC, osteogenic induction control; OITA, osteogenic induction TLR4
agonist; OITAn, osteogenic induction TLR4 antagonist; ALP, alkaline
phosphatase; OCN, osteocalcin; Runx2, Runt-related transcription
factor 2; Col1, collagen I; PPAR, poly (ADP-ribose) polymerase;
LPL, lipoprotein lipase. |
Discussion
TLR4 is a known natural receptor for LPS, which is
significantly increased in tissues with severe periodontitis,
suggesting that TLR4 may be involved in periodontal tissue immune
processes (33,34). However, the regulatory role of the
TLR4 signaling pathway in hPDLSCs under inflammatory conditions has
not been elucidated. The present study aimed to investigate the
functional role of TLR4 signaling in hPDLSCs induced with LPS. It
was found that LPS induced inflammation of the hPDLSCs and
increased the expression of TLR4 in the cell. Additionally, the
TLR4 agonist decreased the levels of inflammatory markers in
hPDLSCs and inhibited the osteogenic differentiation of these
cells. Of note, the study revealed the NF-κBP65/TLR4 axis is a key
regulatory pathway driving the osteogenic differentiation of
hPDLSCs.
TNF-α and IL-1β are inflammatory factors produced
downstream of TLR4. When TLR4 is specifically activated by LPS on
hPDLSCs, its downstream signaling pathway is activated and
expresses various inflammatory factors, including TNF-α and IL-1β.
Wang et al found a positive correlation between the
expression of TLR4 and TNF-α on the surface of gingival fibroblasts
(35). Additionally, they found
that LPS of Porphyromonas gingivalis can be combined with
TLR4 on the gingival fibroblast membrane to transmit
intracellularly, stimulating gingival fibroblasts to produce
inflammatory cytokines IL-1 and IL-6, which can activate
osteoclasts and cause alveolar bone resorption. Sun et al
found that the expression levels of TLR4 and IL-6 were
significantly increased in hPDLSCs cultured in vitro
following LPS stimulation (36).
Similarly, Scheres et al found high mRNA expression levels
of TLR4, TLR7 and CD14 in periodontitis on culturing periodontal
cells in healthy and periodontitis groups (37). Chaudhari et al detected
that the IL-1β content in the gingival tissues of patients with
periodontitis was more than twice that of normal individuals,
confirming that IL-1β is involved in the immune response,
inflammation and alveolar bone resorption of periodontal tissues
(38). The present study found
that inflammation induced by LPS inhibited the osteogenic
differentiation, proliferation and migration of hPDLSCs. In
addition, it was found that only low levels of TNF-α and IL-1β were
expressed in hPDLSCs under normal conditions. Following LPS
stimulation, the expression levels of TNF-α, IL-1β and NF-κBp65
increased with the increased expression of TLR4, indicating that
LPS stimulated the hPDLSCs, and increased the expression of TLR4
and the release of inflammatory factors. These LPS-induced changes
caused inflammatory damage, subsequently leading to
periodontitis.
A limitation of the present study was that it did
not examine whether treatment with the TLR4 antagonist prevented or
reversed the harmful response induced by LPS or the agonist.
In conclusion, LPS-induced upregulation of the TLR4
signaling pathway induced the expression of proinflammatory
cytokines IL-1β, TNF-α, NF-κBP65, and inhibited osteogenic
differentiation, but induced adipogenesis of the hPDLSCs. The
NF-κBP65/TLR4 axis is a key regulatory pathway driving the
osteogenic differentiation of hPDLSCs under inflammatory
conditions, which can provide novel targets for controlling
periodontal inflammation and immune response, and potentially novel
methods for the clinical prevention and treatment of periodontal
disease.
Funding
The present study was supported by the Shanghai
Health Bureau Youth Research Project (grant no. 20144Y0256), the
Shanghai Medical Exploration Project (grant nos. 1641196201 and
17411972600), the National Natural Science Foundation of China
(grant no. 81700974) and the Natural Science Foundation of Shanghai
(grant no. 17ZR1432800).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
BY, QL and MZ made substantial contributions to the
conception and design of the research and acquisition of data. MZ
made substantial contributions to the analysis and interpretation
of data. BY and QL were involved in the drafting and revision of
the manuscript and acquisition of funding. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Institutional Review
Boards of Affiliated Stomatology Hospital of Tongji University
(Shanghai, China). All procedures involving human participants were
performed in accordance with the ethical standards of the
Institutional Review Boards of Affiliated Stomatology Hospital of
Tongji University, and with the 1964 Helsinki declaration and its
later amendments or comparable ethical standards. Written informed
consent was obtained from all individual participants included in
the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Kulkarni V, Bhatavadekar NB and Uttamani
JR: The effect of nutrition on periodontal disease: A systematic
review. J Calif Dent Assoc. 42:302–311. 2014.PubMed/NCBI
|
|
2
|
Al-Ghutaimel H, Riba H, Al-Kahtani S and
Al-Duhaimi S: Common periodontal diseases of children and
adolescents. Int J Dent. 2014:8506742014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
AlJehani YA: Risk factors of periodontal
disease: Review of the literature. Int J Dent. 2014:1825132014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu N, Gu B, Liu N, Nie X, Zhang B, Zhou X
and Deng M: Wnt/β-catenin pathway regulates cementogenic
differentiation of adipose tissue-deprived stem cells in dental
follicle cell-conditioned medium. PLoS One. 9:e933642014.
View Article : Google Scholar
|
|
5
|
Tran Hle B, Doan VN, Le HT and Ngo LT:
Various methods for isolation of multipotent human periodontal
ligament cells for regenerative medicine. In Vitro Cell Dev Biol
Anim. 50:597–602. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang H, Gao LN, An Y, Hu CH, Jin F, Zhou
J, Jin Y and Chen FM: Comparison of mesenchymal stem cells derived
from gingival tissue and periodontal ligament in different
incubation conditions. Biomaterials. 34:7033–7047. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Silvério KG, Rodrigues TL, Coletta RD,
Benevides L, Da Silva JS, Casati MZ, Sallum EA and Nociti FH Jr:
Mesenchymal stem cell properties of periodontal ligament cells from
deciduous and permanent teeth. J Periodontol. 81:1207–1215. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kadar K, Kiraly M, Porcsalmy B, Molnar B,
Racz GZ, Blazsek J, Kallo K, Szabo EL, Gera I, Gerber G and Varga
G: Differentiation potential of stem cells from human dental
origin-promise for tissue engineering. J Physiol Pharmacol.
60(Suppl 7): 167–175. 2009.
|
|
9
|
Seo BM, Miura M, Gronthos S, Bartold PM,
Batouli S, Brahim J, Young M, Robey PG, Wang CY and Shi S:
Investigation of multi-potent postnatal stem cells from human
periodontal ligament. Lancet. 364:149–155. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang L, Shen H, Zheng W, Tang L, Yang Z,
Gao Y, Yang Q, Wang C, Duan Y and Jin Y: Characterization of stem
cells from alveolar periodontal ligament. Tissue Eng Part A.
17:1015–1026. 2011. View Article : Google Scholar
|
|
11
|
Park JC, Kim JM, Jung IH, Kim JC, Choi SH,
Cho KS and Kim CS: Isolation and characterization of human
periodontal ligament (PDL) stem cells (PDLSCs) from the inflamed
PDL tissue: In vitro and in vivo evaluations. J Clin Periodontol.
38:721–731. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Diomede F, Caputi S, Merciaro I, Frisone
S, D'Arcangelo C, Piattelli A and Trubiani O: Pro-inflammatory
cytokine release and cell growth inhibition in primary human oral
cells after exposure to endodontic sealer. Int Endod J. 47:864–872.
2014. View Article : Google Scholar
|
|
13
|
Zhang J, An Y, Gao LN, Zhang YJ, Jin Y and
Chen FM: The effect of aging on the pluripotential capacity and
regenerative potential of human periodontal ligament stem cells.
Biomaterials. 33:6974–6986. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Monsarrat P, Vergnes JN, Nabet C, Sixou M,
Snead ML, Planat-Bénard V, Casteilla L and Kémoun P: Concise
review: Mesenchymal stromal cells used for periodontal
regeneration: A systematic review. Stem Cells Transl Med.
3:768–774. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Koori K, Maeda H, Fujii S, Tomokiyo A,
Kawachi G, Hasegawa D, Hamano S, Sugii H, Wada N and Akamine A: The
roles of calcium-sensing receptor and calcium channel in osteogenic
differentiation of undifferentiated periodontal ligament cells.
Cell Tissue Res. 357:707–718. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Navabazam AR, Sadeghian Nodoshan F,
Sheikhha MH, Miresmaeili SM, Soleimani M and Fesahat F:
Characterization of mesenchymal stem cells from human dental pulp,
preapical follicle and periodontal ligament. Iran J Reprod Med.
11:235–242. 2013.
|
|
17
|
Liu W, Konermann A, Guo T, Jäger A, Zhang
L and Jin Y: Canonical Wnt signaling differently modulates
osteogenic differentiation of mesenchymal stem cells derived from
bone marrow and from periodontal ligament under inflammatory
conditions. Biochim Biophys Acta. 1840:1125–1134. 2014. View Article : Google Scholar
|
|
18
|
Ozer A, Yuan G, Yang G, Wang F, Li W, Yang
Y, Guo F, Gao Q, Shoff L, Chen Z, et al: Domain of dentine
sialoprotein mediates proliferation and differentiation of human
periodontal ligament stem cells. PLoS One. 8:e816552013. View Article : Google Scholar
|
|
19
|
Park CH, Rios HF, Jin Q, Sugai JV,
Padial-Molina M, Taut AD, Flanagan CL, Hollister SJ and Giannobile
WV: Tissue engineering bone-ligament complexes using fiber-guiding
scaffolds. Biomaterials. 33:137–145. 2012. View Article : Google Scholar
|
|
20
|
Ding G, Liu Y, Wang W, Wei F, Liu D, Fan
Z, An Y, Zhang C and Wang S: Allogeneic periodontal ligament stem
cell therapy for periodontitis in swine. Stem Cells. 28:1829–1838.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Park CH, Rios HF, Jin Q, Bland ME,
Flanagan CL, Hollister SJ and Giannobile WV: Biomimetic hybrid
scaffolds for engineering human tooth-ligament interfaces.
Biomaterials. 31:5945–5952. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xie H and Liu H: A novel mixed-type stem
cell pellet for cementum/periodontal ligament-like complex. J
Periodontol. 83:805–815. 2012. View Article : Google Scholar
|
|
23
|
Li Z, Jiang CM, An S, Cheng Q, Huang YF,
Wang YT, Gou YC, Xiao L, Yu WJ and Wang J: Immunomodulatory
properties of dental tissue-derived mesenchymal stem cells. Oral
Dis. 20:25–34. 2014. View Article : Google Scholar
|
|
24
|
Kawasaki T and Kawai T: Toll-like receptor
signaling pathways. Front Immunol. 5:4612014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hemmati F, Ghasemi R, Mohamed Ibrahim N,
Dargahi L, Mohamed Z, Raymond AA and Ahmadiani A: Crosstalk between
insulin and Toll-like receptor signaling pathways in the central
nervous system. Mol Neurobiol. 50:797–810. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
He Q, Wang L, Wang F and Li Q: Role of gut
microbiota in a zebrafish model with chemically induced
enterocolitis involving toll-like receptor signaling pathways.
Zebrafish. 11:255–264. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Potnis PA, Dutta DK and Wood SC: Toll-like
receptor 4 signaling pathway mediates proinflammatory immune
response to cobalt-alloy particles. Cell Immunol. 282:53–65. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guijarro-Muñoz I, Compte M,
Álvarez-Cienfuegos A, Álvarez-Vallina L and Sanz L:
Lipopolysaccharide activates Toll-like receptor 4 (TLR4)-mediated
NF-κB signaling pathway and proinflammatory response in human
pericytes. J Biol Chem. 289:2457–2468. 2014. View Article : Google Scholar
|
|
29
|
Hunt JJ, Astley R, Wheatley N, Wang JT and
Callegan MC: TLR4 contributes to the host response to Klebsiella
intraocular infection. Curr Eye Res. 39:790–802. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang P, Liu W, Peng Y, Han B and Yang Y:
Toll like receptor 4 (TLR4) mediates the stimulating activities of
chitosan oligosaccharide on macrophages. Int Immunopharmacol.
23:254–261. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Diomede F, Zingariello M, Cavalcanti MFXB,
Merciaro I, Pizzicannella J, De Isla N, Caputi S, Ballerini P and
Trubiani O: MyD88/ERK/NFkB pathways and pro-inflammatory cytokines
release in periodontal ligament stem cells stimulated b.
Porphyromonas gingivalis Eur J Histochem. 61:27912017.
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
33
|
Qureshi ST, Larivière L, Leveque G,
Clermont S, Moore KJ, Gros P and Malo D: Endotoxin-tolerant mice
have mutations in Toll-like receptor 4 (Tlr4). J Exp Med.
189:615–625. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sakai A, Ohshima M, Sugano N, Otsuka K and
Ito K: Profiling the cytokines in gingival crevicular fluid using a
cytokine antibody array. J Periodontol. 77:856–864. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang PL, Oido-Mori M, Fujii T, Kowashi Y,
Kikuchi M, Suetsugu Y, Tanaka J, Azuma Y, Shinohara M and Ohura K:
Heterogeneous expression of Toll-like receptor 4 and downregulation
of Toll-like receptor 4 expression on human gingival fibroblasts by
Porphyromonas gingivalis lipopolysaccharide. Biochem Biophys Res
Commun. 288:863–867. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun Y, Shu R, Zhang MZ and Wu AP:
Toll-like receptor 4 signaling plays a role in triggering
periodontal infection. FEMS Immunol Med Microbiol. 52:362–369.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Scheres N, Laine ML, Sipos PM,
Bosch-Tijhof CJ, Crielaard W, de Vries TJ and Everts V: Periodontal
ligament and gingival fibroblasts from periodontitis patients are
more active in interaction with Porphyromonas gingivalis. J
Periodontal Res. 46:407–416. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chaudhari AU, Byakod GN, Waghmare PF and
Karhadkar VM: Correlation of levels of interleukin-1β in gingival
crevicular fluid to the clinical parameters of chronic
periodontitis. J Contemp Dent Pract. 12:52–59. 2011. View Article : Google Scholar : PubMed/NCBI
|