Introduction
Mitochondria play a key role in neuronal cell
survival and death through their function in ATP production
(1,2) and involvement in the intrinsic
pathway of apoptosis (3). In the
majority of cells, including neuronal cells, mitochondria form a
dynamic tubular network that is continuously remodeled; they may
either divide via binary fission and form separate entities, or
fuse and form a more continuous network (4). Both the fusion, due to collision,
and the fission, due to separation, of mitochondria are dependent
on the intracellular movement of these organelles. In specific
intracellular compartments, mitochondria are in close contact with
the endoplasmic reticulum (ER), albeit without fusion, particularly
in the ER mitochondria-associated membranes (MAM). ER-mitochondrial
contacts strongly affect mitochondrial dynamics, as they favor
mitochondrial constriction and consequent fission (5). Recent research results suggest that
mitochondrial function depends on mitochondrial morphology, which
can rapidly change in response to cellular conditions (6,7).
Fused mitochondria, prevailing in healthy metabolically active
cells, exhibit a higher ATP production that is attributed to
optimized exchange of metabolites and mitochondrial DNA within the
mitochondrial matrix. On the contrary, fragmented mitochondria,
which are encountered in quiescent and metabolically inactive
cells, exhibit reduced respiration and are associated with
different pathological conditions (8). However, mitochondrial fission is
crucial for cell division and for the generation of single
mitochondria that can be transported along axons by the motor
protein apparatus (9). The latter
function is particularly important for neuronal cells. In addition,
mitochondrial fission is crucial for the release of cytochrome
c and other intermembrane space proteins during apoptosis,
and for the elimination of damaged organelles from the
mitochondrial network by autophagy (10). The fission/fusion equilibrium is
controlled by specific mitochondrial proteins. Mitochondrial
fission is mediated by the cytosolic GTPase dynamin-related protein
1 (DRP1), which is activated by various post-translational
modifications (11) and
consequently translocates to the outer mitochondrial membrane
(12). Mitochondrial fusion
involves two membranes that must be rearranged in a coordinated
manner in order to maintain mitochondrial integrity. Inner
mitochondrial membrane fusion is mediated by the mitochondrial
dynamin-like GTPase (OPA1), whereas two mitofusins (Mfn), namely
Mfn1 and 2, are involved in the process of mitochondrial outer
membrane fusion. Despite Mfn1 and Mfn2 displaying high homology
(81%) and identity (~60%), they have non-redundant functions
(13). In addition to its role in
fusion, Mfn2 plays a role in the control of the ER-mitochondria
interaction (14), although the
exact function of Mfn2 in this inter-organelle interplay remains a
subject of intense discussion (15,16).
With respect to MAM, the first complex that tethers
the ER and mitochondria was identified in mammalian cells and is
the tripartite complex between the cytosolic chaperone
glucose-regulated protein 75 (GRP75), the mitochondrial
voltage-dependent anion-selective channel 1 (VDAC1), and the
inositol 1,4,5-triphosphate receptor located at the ER membrane
(17). MAM play a key role in the
maintenance of lipid and Ca2+ homeostasis, in the
initiation of autophagy and mitochondrial fission, and in sensing
metabolic shifts (18).
Disturbances of mitochondrial dynamics and MAM are
associated with mitochondrial dysfunction and are considered to be
an important mechanism underlying several serious human diseases,
including neurodegenerative diseases (19,20), such as Parkinson's (21) and Alzheimer's (22) disease and ischemic
neurodegeneration (23).
Alterations at the protein level of Mfn2 (24,25), DRP1 (26), OPA1 (27,28) and VDCA1 (29) have been documented in various
models of brain ischemia. The aim of the present study was to
investigate the impact of transient global brain ischemia on the
expression of selected proteins involved in mitochondrial dynamics
and MAM. Previous studies have investigated the impact of different
types of brain ischemia on the total levels of the abovementioned
proteins, but without focusing on their intracellular localization.
Therefore, we focused on Mfn2, DRP1, VDAC1 and GRP75 with respect
to their intracellular localization, and performed western blot
(WB) analysis of both total cell extracts and mitochondria isolated
from either the cerebral cortex or the hippocampus. In addition,
Mfn2 intracellular localization was analyzed by laser scanning
confocal microscopy.
Materials and methods
Ischemia-reperfusion
Animal studies were performed according to the
guideline for Animal Care and Health of the State Veterinary and
Food Department of the Slovak Republic (approval no.
2414/06-221/3). The experiments were conducted in accordance with
Directive 2010/63/EU of the European Parliament and of the Council
for the protection of animals used for scientific purposes.
A total of 50 adult male Wistar rats (Velaz, Ltd.)
were used. All animals were maintained on a 12/12-h light/dark
cycle. Food and water were available ad libitum until the
beginning of the experiments. Animal health and behaviour were
monitored regularly by a doctor of veterinary medicine. Transient
global cerebral ischemia was produced using the four-vessel
occlusion model. Briefly, on day 1, both vertebral arteries were
irreversibly occluded by coagulation through the alar foramina
following anesthesia with a mixture of 2% halo-thane, 30%
O2 and 68% N2O. On day 2, both common carotid
arteries were occluded for 15 min by small clips under anesthesia
with a mixture of 2% halothane, 30% O2 and 68%
N2O. Two minutes prior to carotid occlusion, the
halothane was removed from the mixture. Body temperature was
maintained by means of a homeothermic blanket. Global ischemia was
followed by 1, 3, 24 or 72 h of reperfusion. During a short time of
reperfusion (1 or 3 h), the animals were monitored by the
experimenter. Animals surviving over a longer time of reperfusion
(24 or 72 h) were monitored by a doctor of veterinary medicine.
Control animals underwent the same procedure, apart from the
carotid occlusion. The duration of the experiment was 1-4 days,
depending on the time of reperfusion. With respect to WB analysis,
control animals and animals undergoing ischemia and reperfusion
were first anesthetized with a mixture of 2% halothane, 30%
O2 and 68% N2O and then sacrificed by
decapitation. The cerebral cortex and both hippocampi were
dissected and processed immediately. With respect to fluorescence
immunohistochemistry (FIHC), control and experimental animals were
anesthetized, perfused transcardially with ice-cold 0.1 mol/l
phosphate-buffered saline (PBS, pH 7.4) and fixed by perfusion with
ice-cold 4% paraformaldehyde in PBS. The brains were removed,
post-fixed with the same solution as mentioned above for 24 h at
4°C, and cryoprotected by infiltration using 30% sucrose for the
next 24 h at 4°C.
Experimental groups of animals
The rats were randomized into the following groups:
i) Control sham-operated rats (CNT; n=5 for WB and n=3 for FIHC);
ii) rats that underwent a 15-min global brain ischemia (ISCH; n=5
for WB and n=3 for FIHC); iii) rats that underwent a 15-min global
brain ischemia followed by 1 h of reperfusion (I1R; n=5); iv) rats
that underwent a 15-min global brain ischemia followed by 3 h of
reperfusion (I3R; n=5); v) rats that underwent a 15-min global
brain ischemia followed by 24 h of reperfusion (I24R; n=5); and vi)
rats that underwent a 15-min global brain ischemia followed by 72 h
of reperfusion (I72R; n=5 for WB and n=3 for FIHC).
Preparation of protein extracts and
isolation of mitochondria
Protein extracts were prepared by homogenization of
either the cerebral cortex or both hippocampi in homogenization
buffer (10 mM Tris-HCl, pH=7.4, 1 mM EDTA and 0.24 M sucrose) using
a Potter Teflon glass homogenizer. Total cell extracts were
prepared by the addition of an appropriate volume of 6X RIPA buffer
[6X PBS, 6% (v/v) Nonidet P-40, 3% (w/v) sodium deoxycholate, 0.6%
(w/v) sodium dodecyl sulphate (SDS)] to the homogenate.
Mitochondria from both whole hippocampi were
isolated by differential centrifugation as described previously
(30). Non-synaptic mitochondria
from the cerebral cortex were isolated by differential
centrifugation using one-step Percoll gradient (16% Percoll in 0.25
M sucrose), as described previously (31). The protein concentration was
determined by the protein Dc assay kit (Bio-Rad Laboratories, Inc.)
with bovine serum albumin (BSA) as a standard.
WB analysis
Isolated proteins were separated by 10% SDS-PAGE.
Following electrophoresis, the separated proteins were transferred
onto nitrocellulose membranes using a semi-dry transfer protocol.
The membranes were controlled for even load and possible transfer
artefacts by staining with Ponceau Red solution. After blocking
with BSA blocking buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.05%
Tween-20 and 2% BSA), the membranes were first incubated for 90 min
with primary mouse monoclonal antibodies against GRP75 (1:1,000,
sc-133137), VDAC1 (1:1,000, sc390996), β-actin (1:2,000, sc-47778)
(all from Santa Cruz Biotechnology, Inc.) and cytochrome c
oxidase subunit 1 (COXI; 1 µg/ml, 459600, Invitrogen; Thermo
Fisher Scientific, Inc.), rabbit polyclonal antibodies raised
against Mfn2 (1:500; sc-50331), or goat polyclonal antibodies
raised against DRP1 (1:500, sc-21804) (all from Santa Cruz
Biotechnology, Inc.) dissolved in BSA blocking solution. Membranes
incubated with primary antibodies were washed in TBS-T solution (50
mM Tris-Cl, pH 7.5, 150 mM NaCl and 0.05% Tween 20) and then
incubated with secondary antibodies conjugated with horseradish
peroxidase (1:5,000, Santa Cruz Biotechnology, Inc). After
extensive washes with TBS-T solution (4 times, 15 min), the
membranes were incubated in SuperSignal West Pico Chemiluminescent
Substrate solution (Thermo Fisher Scientific, Inc.) for 3 min.
Following exposure of the membranes to Chemidoc XRS (Bio-Rad
Laboratories, Inc.), the intensities of the corresponding bands
were quantified using Quantity One software (BioRad Laboratories,
Inc.). The intensities of the bands of interest were normalized to
the corresponding band intensities of either β-actin or COXI.
Detection of β3 tubulin and Mfn2 by
FIHC
The brains from control and experimental rats were
frozen and cut with a cryostat into 30-µm sections; the
sections were mounted on Superfrost Plus glass slides (Thermo
Fisher Scientific, Inc.). Mounted brain sections were permeabilized
with a permeabilization solution (0.1% Triton X-100 with 10% BSA)
for 1 h. Mouse monoclonal antibody against β3 tubulin (1:50;
sc-80005; Santa Cruz Biotechnology, Inc.), as a specific marker of
neuronal cell body cytoplasm and axon guidance, was used as a
primary antibody. Mfn2 rabbit polyclonal antibodies (1:50;
sc-50331; Santa Cruz Biotechnology, Inc.) were used to detect Mfn2.
The tissue sections were incubated at 4°C overnight in primary
antibodies diluted in permeabilization solution. Alexa Fluor 488
goat-anti-rabbit IgG (1:50; cat. no. 4412, Cell Signaling
Technology, Inc.) was applied as a secondary antibody for Mfn2, and
Alexa Fluor 594 goat-anti-mouse IgG (1:100, cat. no. 8890, Cell
Signaling Technology, Inc.) was applied as a secondary antibody for
β3 tubulin. Finally, the brain sections were cover-slipped with
Fluoromount-G® medium containing
4′,6-diamidino-2-phenylindole (DAPI, CA 0100-20, SouthernBiotech).
In the absence of primary antibody, no immunoreactivity was
observed. The slides were examined under an Olympus FluoView FV10i
confocal laser scanning microscope (Olympus Corporation) equipped
with an objective of ×10 with a zoom up to a magnification of ×40
and filters for fluorescein isothiocyanate for Alexa Fluor 488
(excitation: 499 nm; emission: 520 nm) and Texas Red (excitation:
590 nm; emission: 618 nm). Images were captured using Olympus
Fluoview FV10-ASW software, version 02.01 (Olympus Corporation) and
Quick Photo Micro software, version 2.3 (Promicra, s.r.o.) and
further processed in Adobe Photoshop CS3 Extended, version 10.0 for
Windows (Adobe Systems, Inc.).
The brightness and contrast of each image file were
uniformly calibrated using Adobe Photoshop CS3 Extended, version
10.0 for Windows (Adobe Systems, Inc.). Values of background
staining were obtained and subtracted from the immunoreactive
intensities.
Statistical analysis
All statistical analyses were performed using
GraphPad InStat V2.04a (GraphPad Software, Inc.). For the
comparison of the ischemia-induced changes among all groups,
one-way analysis of variance was first performed to determine any
differences among all experimental groups. Additionally, an
unpaired Tukey's test was used to determine differences between
individual groups. The significance level was set at P<0.05.
Results
WB analysis of the levels of the selected
proteins in total cell extracts from the cerebral cortex and
hippocampus of control and experimental animals
In order to study the impact of global brain
ischemia and ischemia followed by reperfusion on the levels of
proteins involved in mitochondrial dynamics and MAM, WB analysis of
total cell extracts from the cerebral cortex and hippocampus of
control and experimental animals was performed. In total cell
extracts prepared form the cerebral cortex, significantly decreased
total levels of Mfn2 were observed after ischemia followed by 3 h
(69.5% of control, P<0.05), 24 h (58.3% of control, P<0.01),
and 72 h (65.9% of control, P<0.05) of reperfusion. In addition
to Mfn2, the total level of VDAC1 was significantly increased after
72 h of reperfusion (417.7% of control, P<0.05). The levels of
DRP1 were increased, whereas those of GRP75 were decreased after
ischemia and after ischemia followed by reperfusion, but the
observed changes were not statistically significant (Fig. 1). In hippocampal total cell
extracts, ischemia followed by reperfusion led to an increase in
the levels of the Mfn2 protein, although these changes were not
statistically significant (Fig.
2). A statistically significantly increased level of VDAC1 was
documented after 3 h of reperfusion (189.8% of control, P<0.01).
The levels of other investigated proteins in hippocampal total cell
extracts were unaltered after ischemia and after ischemia followed
by reperfusion (Fig. 2).
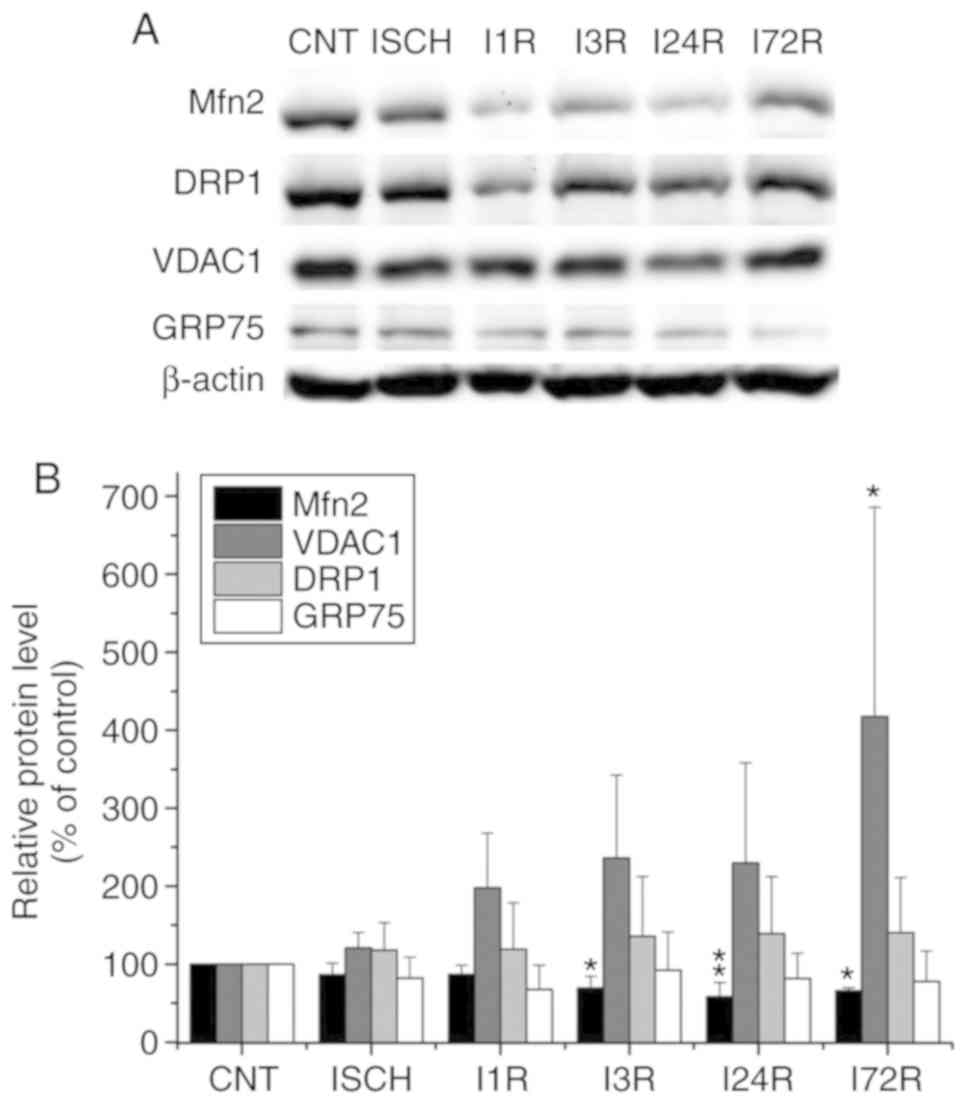 | Figure 1Effect of transient global brain
ischemia on the total levels of the Mfn2, DRP1, VDAC1 and GRP75
proteins in rat cerebral cortex. (A) Experimental rats were
subjected to 15 min of transient global brain ischemia (ISCH) or 15
min of transient global brain ischemia followed by 1 (I1R), 3
(I3R), 24 (I24R) or 72 h (I72R) of reperfusion. The pattern of
protein expression was evaluated by western blot analysis of total
cell extracts prepared from the cerebral cortex of control (CNT)
and experimental rats, as described in Materials and methods.
β-actin served as the loading control. (B) Quantification of the
post-ischemic changes in Mfn2, DRP1, VDAC1 and GRP75 protein levels
in total cell extracts isolated from rat cerebral cortex. The data
were normalized to the β-actin level and are expressed relative to
controls. Data are presented as means ± standard deviation (n=5 per
group). *P<0.05 and **P<0.01,
significantly different from control. Mfn2, mitofusin 2; DRP1,
dynamin-related protein 1; VDAC1, voltage-dependent anion-selective
channel 1; GRP75, glucose-regulated protein 75. |
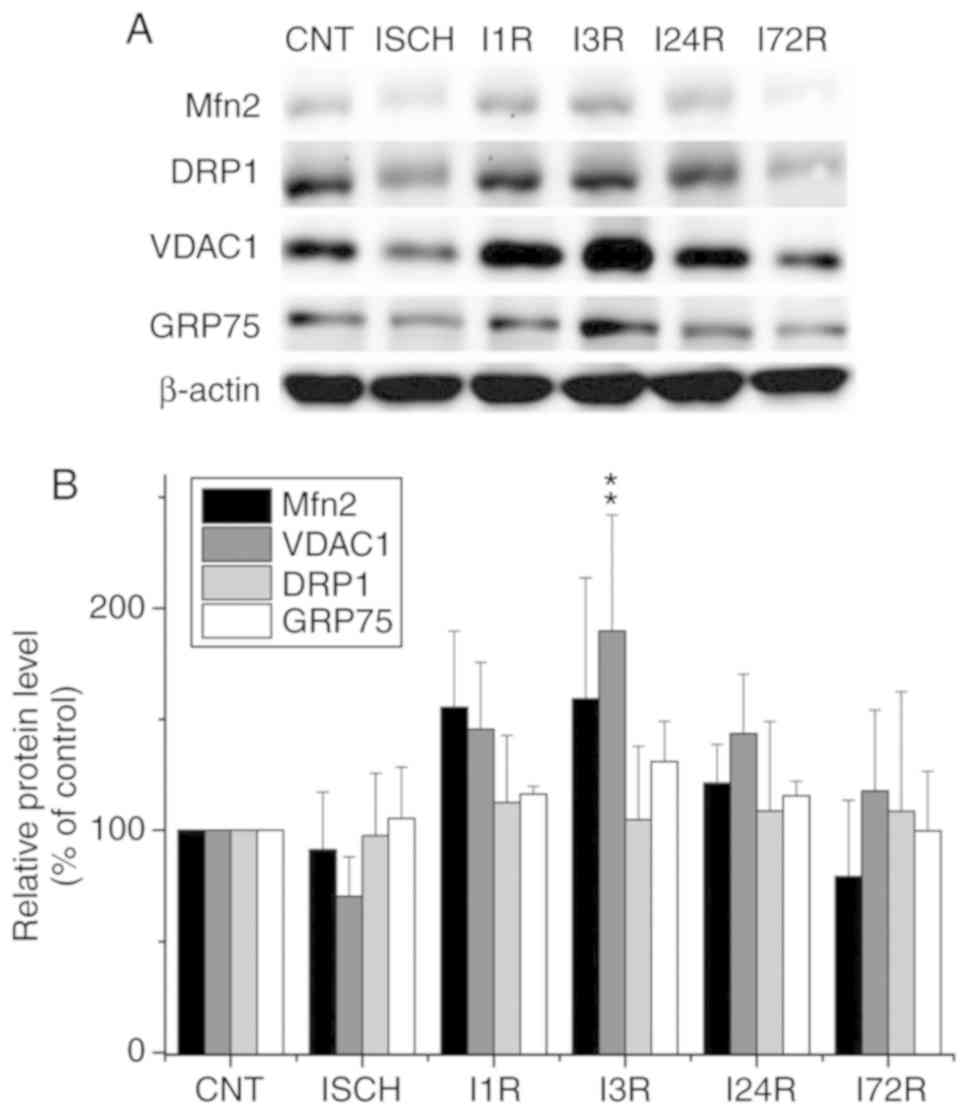 | Figure 2Effect of transient global brain
ischemia on the total levels of the Mfn2, DRP1, VDAC1 and GRP75
proteins in the rat hippocampus. (A) Experimental rats were
subjected to 15 min of transient global brain ischemia (ISCH) or 15
min of transient global brain ischemia followed by 1 (I1R), 3
(I3R), 24 (I24R), or 72 h (I72R) of reperfusion. The pattern of
protein expression was evaluated by western blot analysis of total
cell extracts prepared from both hippocampi of control (CNT) and
experimental rats, as described in Materials and methods. β-actin
served as the loading control. (B) Quantification of the
post-ischemic changes in Mfn2, DRP1, VDAC1 and GRP75 protein levels
in total cell extracts isolated from rat hippocampus. The data were
normalized to the β-actin level and are expressed relative to
controls. Data are presented as means ± standard deviation (n=5 per
group). **P<0.01, significantly different from
control. Mfn2, mitofusin 2; DRP1, dynamin-related protein 1; VDAC1,
voltage-dependent anion-selective channel 1; GRP75,
glucose-regulated protein 75. |
WB analysis of the levels of the selected
proteins in mitochondria isolated from the cortex and hippocampus
of control and experimental animals
In addition to the analysis of total cell extracts,
the levels of selected proteins were determined in mitochondria
isolated from the cortex and hippocampus of control and
experimental animals (Figs. 3 and
4). We observed that ischemia and
ischemia followed by reperfusion led to a decrease in the levels of
Mfn2 in mitochondria isolated from the cerebral cortex; this
decrease was significant after ischemia (60.8% of control,
P<0.001) and after 1 h (54.8% of control, P<0.001), 3 h
(65.2% of control, P<0.01), 24 h (60.6% of control, P<0.001),
and 72 h (66.7% of control, P<0.01) of reperfusion (Fig. 3). We also observed that ischemia
followed by reperfusion led to a decrease in the levels of the
VDAC1 protein in cortical mitochondria, although the changes were
not statistically significant (Fig.
3). The levels of the other investigated proteins in the
cortical mitochondria were not significantly altered after ischemia
and after ischemia followed by reperfusion (Fig. 3). In hippocampal mitochondria, the
levels of Mfn2, VDAC1 and GRP75 were increased after ischemia
followed by reperfusion, but the changes were not statistically
significant. The levels of DRP1 in hippocampal mitochondria were
unaltered after ischemia and after ischemia followed by reperfusion
(Fig. 4).
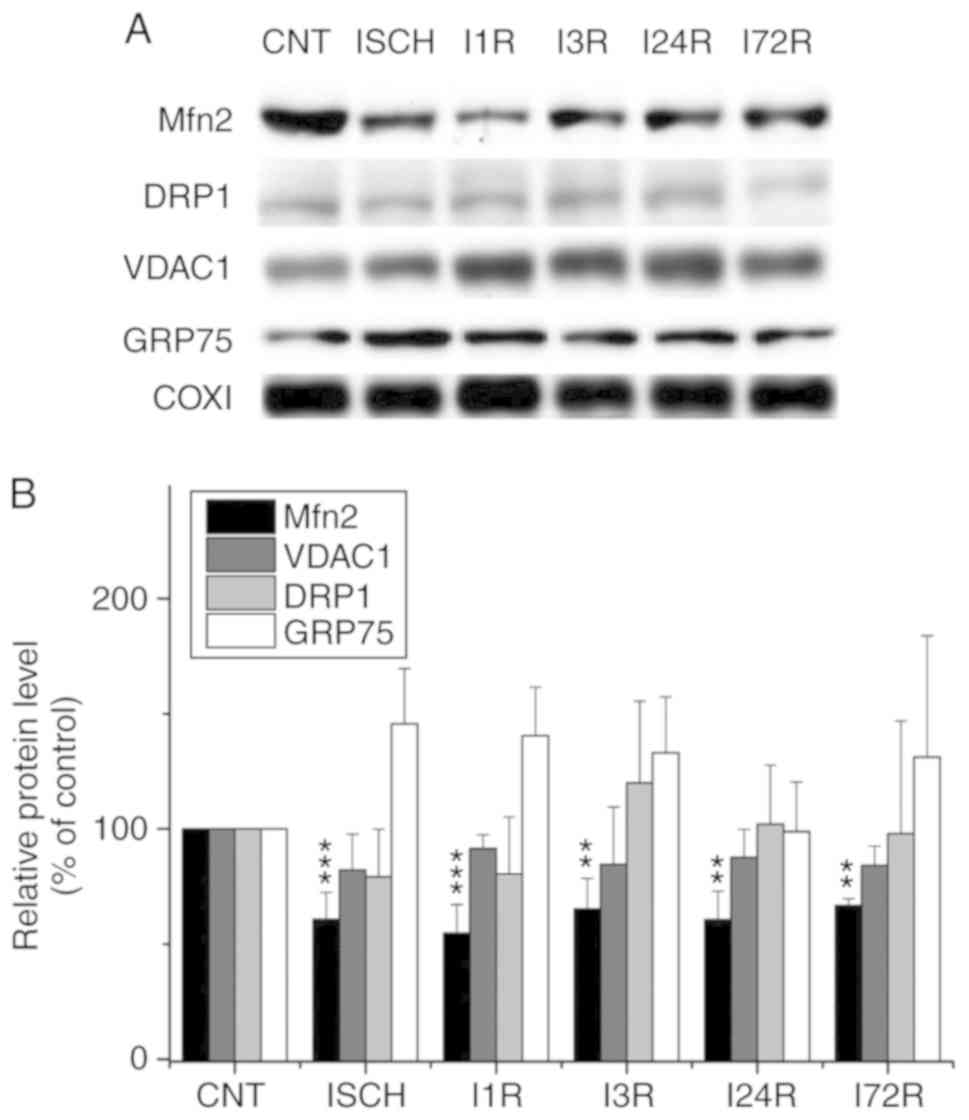 | Figure 3Effect of transient global brain
ischemia on the levels of Mfn2, DRP1, VDAC1 and GRP75 proteins in
mitochondria isolated from rat cerebral cortex. (A) Experimental
rats were subjected to 15 min of transient global brain ischemia
(ISCH) or 15 min of transient global brain ischemia followed by 1
(I1R), 3 (I3R), 24 (I24R), or 72 h (I72R) of reperfusion. The
mitochondrial levels of Mfn2, Drp1, VDAC1 and GRP75 were determined
by western blot analysis of mitochondria isolated from cerebral
cortex of control (CNT) and experimental rats as described in
Materials and methods. COXI served as the loading control. (B)
Quantification of the post-ischemic changes of Mfn2, DRP1, VDAC1
and GRP75 protein levels in mitochondria isolated from rat cerebral
cortex. The data were normalized to the COXI level and are
expressed relative to controls. Data are presented as means ±
standard deviation (n=5 per group). **P<0.01 and
***P<0.001: significantly different from control.
Mfn2, mitofusin 2; DRP1, dynamin-related protein 1; VDAC1,
voltage-dependent anion-selective channel 1; GRP75,
glucose-regulated protein 75; COXI, cytochrome c oxidase
subunit 1. |
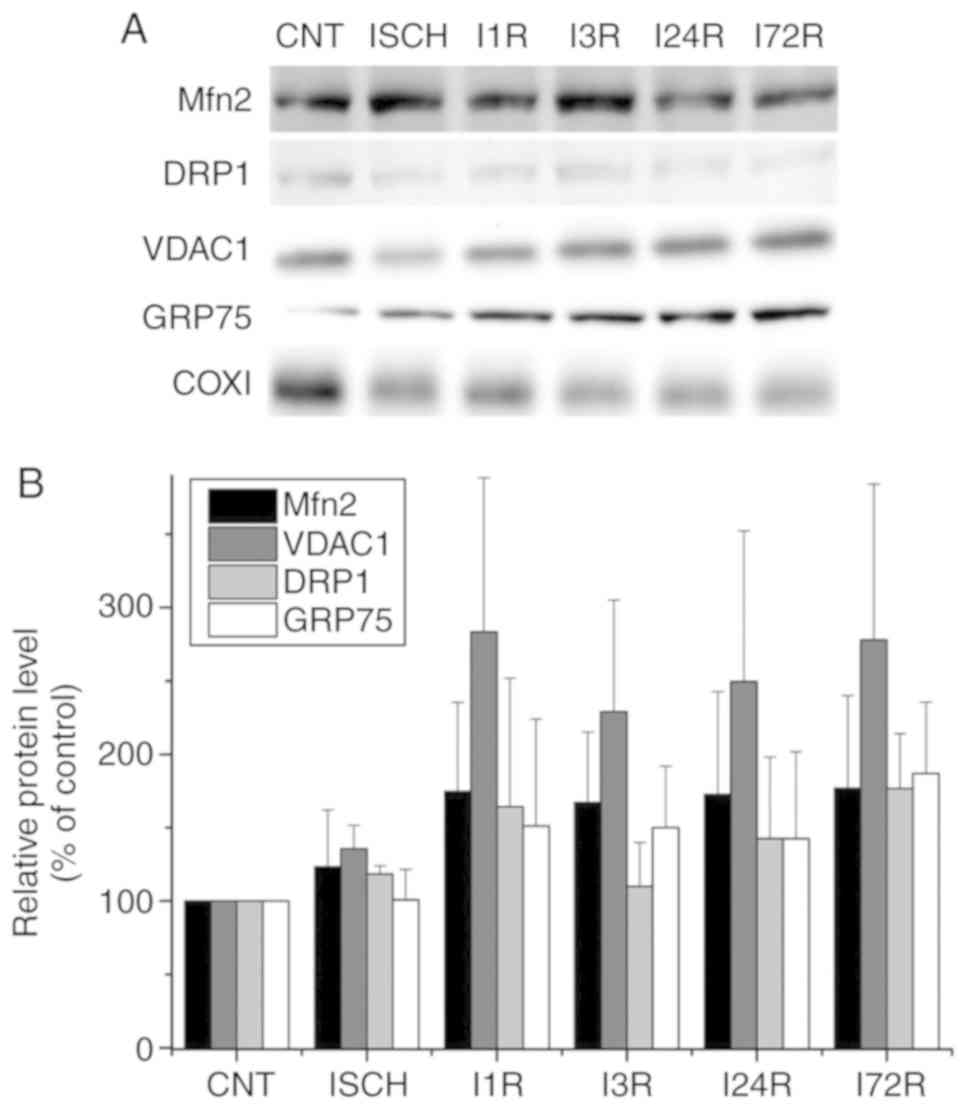 | Figure 4Effect of transient global brain
ischemia on the levels of the Mfn2, Drp1, VDAC1 and GRP75 proteins
in mitochondria isolated from rat hippocampus. (A) Experimental
rats were subjected to 15 min of transient global brain ischemia
(ISCH) or 15 min of transient global brain ischemia followed by 1
(I1R), 3 (I3R), 24 (I24R), or 72 h (I72R) of reperfusion. The
mitochondrial levels of Mfn2, DRP1, VDAC1 and GRP75 were determined
by western blot analysis of mitochondria isolated from hippocampus
of control and experimental rats as described in Materials and
methods. COXI served as the loading control. (B) Quantification of
the post-ischemic changes of Mfn2, DRP1, VDAC1 and GRP75 protein
levels in hippocampal mitochondria. The data were normalized to the
COXI level and are expressed relative to controls. Data are
presented as means ± standard deviation (n=5 per group). Mfn2,
mitofusin 2; DRP1, dynamin-related protein 1; VDAC1,
voltage-dependent anion-selective channel 1; GRP75,
glucose-regulated protein 75; COXI, cytochrome c oxidase
subunit 1. |
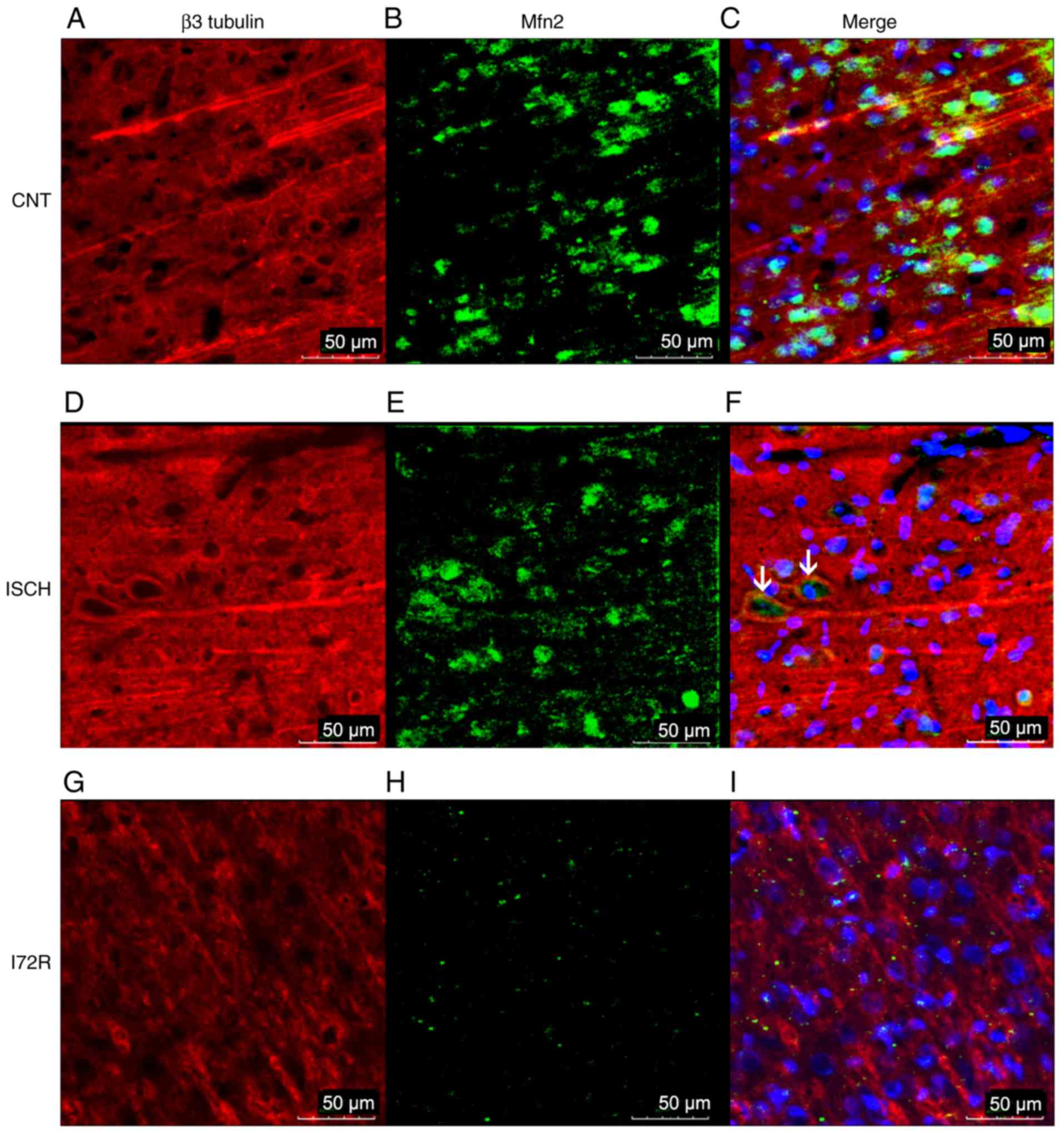
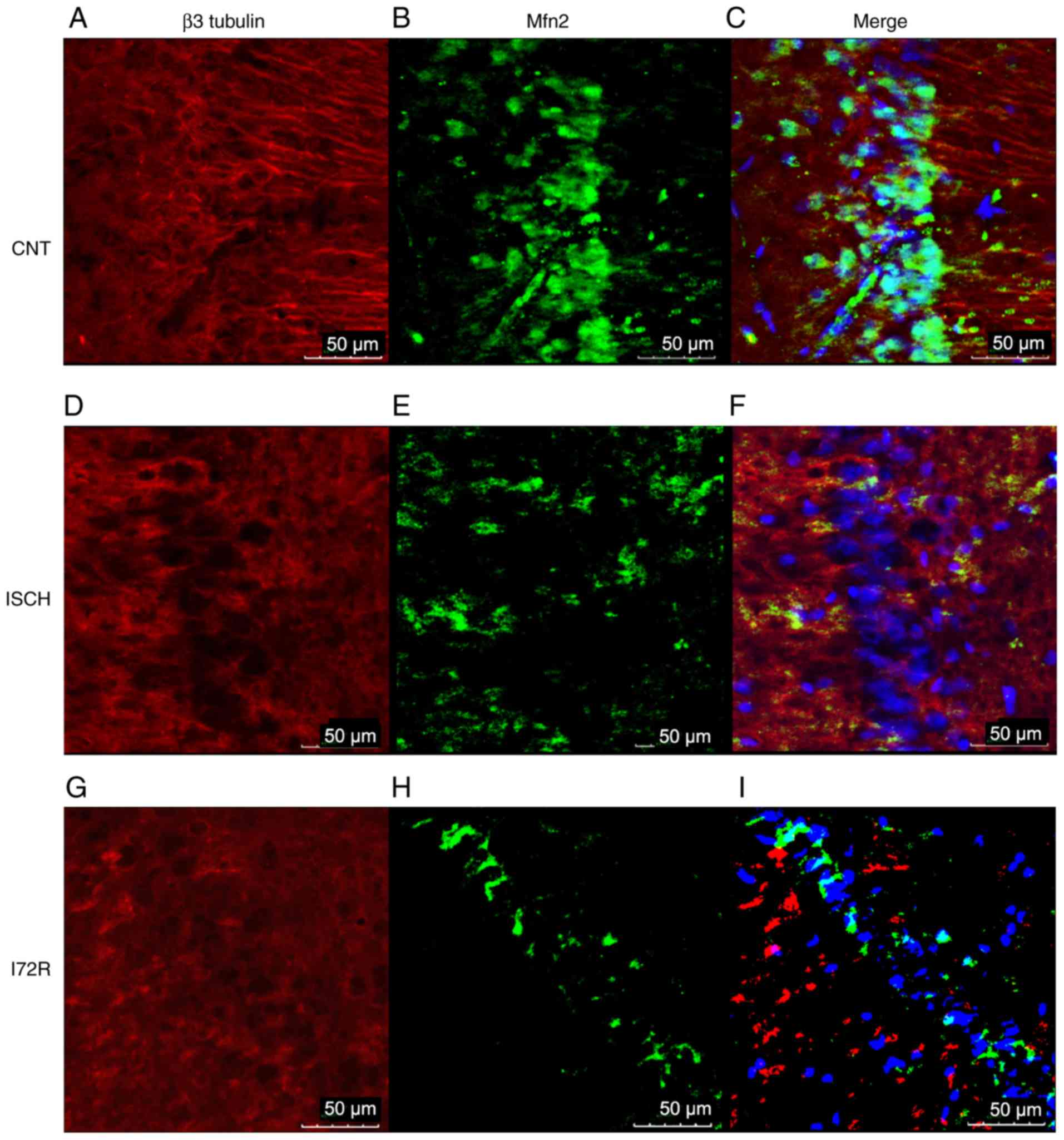
Immunoreactivity of Mfn2 and β3 tubulin
in the hippocampus and in the M1 region of the rat brain
cortex
To further confirm the WB results,
immunofluorescence was used to detect any immunoreactivity of Mfn2
and β3 tubulin, as a neuronal cytoskeletal marker, in the
hippocampal area and in the M1 region of the rat brain cortex.
Representative images from control rats (CNT), rats that were
subjected to global brain ischemia for 15 min (ISCH), and from rats
that underwent reperfusion with a duration of 72 h after 15 min of
global brain ischemia (I72R) are shown in Figs. 5 and 6. Signals corresponding to Mfn2 were
predominantly located within the perikaryal cytoplasm, in the
processes of histologically normal tissue in the M1 region of the
cortex (Fig. 5B), and in the CA1
layer of the hippo-campus (Fig.
6B). In the M1 region of ischemic rats (Fig. 5E), the intensity of the Mfn2
signal was slightly decreased. The localization of Mfn2 was
shifted, and Mfn2 and β3 tubulin signals were co-localized in some
neurons (Fig. 5F, arrows). In the
CA1 layer of the hippocampus, the signal for Mfn2 was reduced
compared with that of the control, and the localization of Mfn2 was
transferred to the neuronal processes and the neurophil (Fig. 6E). In the M1 region of the cortex,
a 15-min ischemia followed by 72 h of reperfusion resulted in a
diminution of the Mfn2 signal, a finding that was consistent with
the data from the WB analysis. The localization of Mfn2 was limited
to the neuronal processes and the neurophil (Fig. 5H). In the CA1 layer of the
hippocampus, Mfn2 immunoreactivity was decreased after 15 min of
ischemia followed by 72 h of reperfusion (Fig. 6H), and was relocated to the
periphery of the perikaryon, with minimal intersection with the
neuronal processes or neurophil. The cytoskeleton of neurons in CA1
was highly disintegrated with no specific morphology in this group
of rats (Fig. 6G).
Discussion
The focus of the present study was the effect of
transient global brain ischemia on the expression and intracellular
distribution of selected proteins involved in mitochondrial
dynamics and MAM. It was demonstrated that ischemia for 15 min, as
well as a 15-min ischemia followed by 1, 3, 24 and 72 h of
reperfusion, were associated with a significant decrease of the
Mfn2 protein in mitochondria isolated from the cerebral cortex, but
not in hippocampal mitochondria. Moreover, the translocation of the
Mfn2 protein to the cytoplasm immediately after global brain
ischemia was documented in the neurons of the cerebral cortex using
laser scanning confocal microscopy. The translocation of Mfn2 was
followed by decreased expression of Mfn2 during reperfusion. In
addition, significantly elevated levels of the VDAC1 protein were
detected in total cell extracts isolated from the hippocampus of
rats that had undergone 15 min of global brain ischemia followed by
3 h of reperfusion, and from the cerebral cortex of rats that had
undergone 15 min of global brain ischemia followed by 72 h of
reperfusion. The mitochondrial and total levels of DRP1 and GRP75
exhibited no significant changes in either the hippocampus or the
cortex in all experimental groups.
Mfn2 is an important protein involved in the process
of mitochondrial outer membrane fusion, which plays an important
role in several intracellular pathways and in the pathogenesis of
neurodegenerative diseases, metabolic disorders, cardiomyopathies
and cancer (32). In addition,
Mfn2 is involved in the process of mitophagy (33), which represents an important
mechanism of mitochondria quality control that is often
dysregulated in neurodegenerative diseases and ischemia-reperfusion
injury (23). Various models of
brain ischemia have been used to document the alterations in the
total levels of the Mfn2 protein (24,25). As shown recently, Mfn2
downregulation causes mitochondrial dysfunction, altered
Ca2+ homeostasis and enhanced Bax translocation to the
mitochondria, resulting in delayed neuronal death (24). Since a significant decrease in
Mfn2 was observed 6 h after middle cerebral artery occlusion for 90
min, Mfn2 reduction was suggested to be a late event during
reperfusion, and its targeting may help reduce ischemic damage and
expand the currently narrow therapeutic window in stroke (24). In the present study, the data of
WB and laser scanning confocal microscopy analysis are in favor of
the view that Mfn2 is released from mitochondria to the cytoplasm
even after transient global brain ischemia for 15 min. The release
of Mfn2 from mitochondria resulting in decreased mitochondrial
level of Mfn2 was persistent during all investigated periods of
reperfusion, and was followed by a decrease in the total Mfn2 level
that was significant after 3 h of reperfusion. Interestingly, these
changes in the level and mitochondrial localization of the Mfn2
protein were observed in the cerebral cortex but not in the
hippocampus, although pyramidal neurons of the CA1 layer belong to
the most vulnerable cells with respect to global brain ischemia
(34,35). The mechanism controlling
mitochondrial localization and the release of Mfn2 from the
mitochondria following global brain ischemia remains elusive. With
respect to the total level of Mfn2, the suppression of the
transcription of the Mfn2 gene has been described in a model of
focal brain ischemia and N-methyl-D-aspartate-induced
excitotoxicity of primary cortical neurons (24). On the contrary,
Ca2+-dependent activation of the cysteine protease
calpain in response to glutamate resulting in the degradation of
Mfn2 and Mfn2-mediated mitochondrial fragmentation that precedes
glutamate-induced neuronal death has been shown in primary spinal
cord motor neurons (36). In a
model of global ischemia, Mfn2 was found to be upregulated in the
mouse hippocampus after 2 and 72 h of reperfusion (26). In the present study, we also
observed increased levels of Mfn2 in the rat hippocampus during
early reperfusion (1 and 3 h), although the changes were not
statistically significant. The upregulation of Mfn2 observed in the
mouse hippocampus was paralleled with the upregulation of the inner
mitochondrial membrane fusion protein OPA1 during the same
reperfusion time (26). Despite
the upregulation of the fusion proteins Mfn2 and OPA1, the
mitochondria in the CA1 neurons of the hippocampus were fragmented,
which has been attributed to the increased phosphorylation of DRP1
observed during late reperfusion (24 and 72 h) (26). In the present study, we did not
observe significant changes in either the mitochondrial or total
levels of DRP1 in either the hippocampus or the cortex in all
experimental groups. However, the results of recent studies
(37,38) have clearly demonstrated that the
process of mitochondrial fragmentation is regulated by
post-translational modifications of DRP1. In the CA1 neurons of the
rat hippocampus, fragmentation of mitochondria during late
reperfusion after transient global brain ischemia has also been
documented, but has been attributed to the release of OPA1 from the
mitochondria (28). The
fragmentation of mitochondria, caused by either the disruption of
mitochondrial fusion resulting from the downregulation of Mfn2 and
OPA1, or the stimulation of mitochondrial fission resulting from
post-translation modifications of DRP1, appears to be a common
result of ischemic insult.
In addition to the decreased Mfn2 level in cerebral
cortex mitochondria, we observed increased total levels of VDAC1 in
the hippocampus and cortex at 3 and 72 h after global brain
ischemia lasting 15 min. In the hippocampus, increased levels of
VDAC1 have also been documented by immunofluorescence at 72 h after
transient global brain ischemia lasting 10 min (29). An increase of VDAC1 in the
hippocampus has been observed following the same reperfusion time,
resulting in the increased mitochondrial levels of p53 (30,31) and BAD (39) observed after global brain
ischemia. The involvement of VDAC1 in ischemic neuronal cell injury
is unclear. In general, the involvement of VDAC1 in the mechanisms
underlying cell death remains unclear (40,41). With respect to cell death, the
results of recent research indicate that post-translational
modifications of VDAC1 are even more important than the levels of
this protein (42). In accord
with this suggestion, the oligomerization of VDCA1 has been shown
to play an important role in the process of mitochondrial
fragmentation and dysfunction associated with the glutamate
excitotoxicity that has often been implicated in the mechanisms
underlying brain ischemia-reperfusion injury (43).
In conclusion, the results of the present study
indicate that global brain ischemia is associated with the fast
release of Mfn2 from mitochondria to the cytoplasm, followed by a
decrease of the total Mfn2 level in the cerebral cortex. The
release of Mfn2 from mitochondria, as observed during early
reperfusion, may represent an important mechanism of mitochondrial
dysfunction associated with the neuronal dysfunction or death
induced by global brain ischemia.
Funding
The present study was supported by the Slovak
Research and Development Agency under contract no. APVV-16-0033 (to
PR) and by grants VEGA 1/0171/18 (to MK) from the Ministry of
Education of the Slovak Republic.
Availability of materials and data
All the datasets generated and analyzed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
KK, MK, MC and IP performed the experiments; KK and
MK analyzed the data; ZT and PK analyzed the data and revised the
manuscript for important intellectual content; PR initiated and
supervised the study, designed the experiments and wrote the paper.
All the authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Animal studies were performed according to the
guideline for Animal Care and Health of the State Veterinary and
Food Department of the Slovak Republic (approval no.
2414/06-221/3). The experiments were conducted in accordance with
Directive 2010/63/EU of the European Parliament and of the Council
for the protection of animals used for scientific purposes.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests to disclose.
Abbreviations:
|
WB
|
western blot
|
|
FIHC
|
fluorescence immunohistochemistry
|
Acknowledgments
The authors would like to thank Dr Theresa Jones for
linguistic corrections. The authors are grateful to Mrs. Greta
Kondekova and Mrs. Agata Resetarova for their valuable help with
immunohistochemical procedures.
References
|
1
|
Erecinska M, Cherian S and Silver IA:
Energy metabolism in mammalian brain during development. Prog
Neurobiol. 73:397–445. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hyder F, Patel AB, Gjedde A, Rothman DL,
Behar KL and Shulman RG: Neuronal-glial glucose oxidation and
glutamatergic-GABAergic function. J Cereb Blood Flow Metab.
26:865–877. 2004. View Article : Google Scholar
|
|
3
|
Fricker M, Tolkovsky AM, Borutaite V,
Coleman M and Brown GC: Neuronal cell death. Physiol Rev.
98:813–880. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chan DC: Fusion and fission: Interlinked
processes critical for mitochondrial health. Annu Rev Genet.
46:265–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Friedman JR, Lackner LL, West M,
DiBenedetto JR, Nunnari J and Voeltz GK: ER tubules mark sites of
mitochondrial division. Science. 334:358–362. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Campello S and Scorrano L: Mitochondrial
shape changes: Orchestrating cell pathophysiology. EMBO Rep.
11:678–684. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sebastián D and Zorzano A: Mitochondrial
dynamics and metabolic homeostasis. Curr Opin Physiol. 3:34–40.
2018. View Article : Google Scholar
|
|
8
|
Hu C, Huang Y and Li L: Drp1-dependent
mitochondrial fission plays critical roles in physiological and
pathological progresses in mammals. Int J Mol Sci. 18:E1442017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Detmer SA and Chan DC: Functions and
dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol.
8:870–879. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Twig G, Elorza A, Molina AJ, Mohamed H,
Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al:
Fission and selective fusion govern mitochondrial segregation and
elimination by autophagy. EMBO J. 27:433–446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liesa M, Palacín M and Zorzano A:
Mitochondrial dynamics in mammalian health and disease. Physiol
Rev. 89:799–845. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Smirnova E, Griparic L, Shurland DL and
van der Bliek AM: Dynamin-related protein Drp1 is required for
mitochondrial division in mammalian cells. Mol Biol Cell.
12:2245–2256. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ishihara N, Eura Y and Mihara K: Mitofusin
1 and 2 play distinct roles in mitochondrial fusion reactions via
GTPase activity. J Cell Sci. 117:6535–6546. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
de Brito OM and Scorrano L: Mitofusin 2
tethers endoplasmic reticulum to mitochondria. Nature. 456:605–610.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Filadi R, Greotti E, Turacchio G, Luini A,
Pozzan T and Pizzo P: Mitofusin 2 ablation increases endoplasmic
reticulum-mitochondria coupling. Proc Natl Acad Sci USA.
112:E2174–E2181. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Naon D, Zaninello M, Giacomello M,
Varanita T, Grespi F, Lakshminaranayan S, Serafini A, Semenzato M,
Herkenne S, Hernández-Alvarez MI, et al: Critical reappraisal
confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria
tether. Proc Natl Acad Sci USA. 113:11249–11254. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Szabadkai G, Bianchi K, Várnai P, De
Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T and Rizzuto
R: Chaperone-mediated coupling of endoplasmic reticulum and
mitochondrial Ca2+ channels. J Cell Biol. 175:901–911. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van Vliet AR, Verfaillie T and Agostinis
P: New functions of mitochondria associated membranes in cellular
signaling. Biochim Biophys Acta. 1843:2253–2262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Burté F, Carelli V, Chinnery PF and
Yu-Wai-Man P: Disturbed mitochondrial dynamics and
neurodegenerative disorders. Nat Rev Neurol. 11:11–24. 2015.
View Article : Google Scholar
|
|
20
|
Erpapazoglou Z, Mouton-Liger F and Corti
O: From dysfunctional endoplasmic reticulum-mitochondria coupling
to neurodegeneration. Neurochem Int. 109:171–183. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gómez-Suaga P, Bravo-San Pedro JM,
González-Polo RA, Fuentes JM and Niso-Santano M: ER-mitochondria
signaling in Parkinson's disease. Cell Death Dis. 9:3372018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Area-Gomez E, de Groof A, Bonilla E,
Montesinos J, Tanji K, Boldogh I, Pon L and Schon EA: A key role
for MAM in mediating mitochondrial dysfunction in Alzheimer
disease. Cell Death Dis. 9:3352018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Anzell AR, Maizy R, Przyklenk K and
Sanderson TH: Mitochondrial quality control and disease: Insights
into ischemia-reperfusion injury. Mol Neurobiol. 55:2547–2564.
2018. View Article : Google Scholar
|
|
24
|
Martorell-Riera A, Segarra-Mondejar M,
Muñoz JP, Ginet V, Olloquequi J, Pérez-Clausell J, Palacín M, Reina
M, Puyal J, Zorzano A and Soriano FX: Mfn2 downregulation in
excito-toxicity causes mitochondrial dysfunction and delayed
neuronal death. EMBO J. 33:2388–2407. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Peng C, Rao W, Zhang L, Wang K, Hui H,
Wang L, Su N, Luo P, Hao YL, Tu Y, et al: Mitofusin 2 ameliorates
hypoxia-induced apoptosis via mitochondrial function and signaling
pathways. Int J Biochem Cell Biol. 69:29–40. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Owens K, Park JH, Gourley S, Jones H and
Kristian T: Mitochondrial dynamics: Cell-type and hippocampal
region specific changes following global cerebral ischemia. J
Bioenerg Biomembr. 47:13–31. 2015. View Article : Google Scholar
|
|
27
|
Sanderson TH, Raghunayakula S and Kumar R:
Neuronal hypoxia disrupts mitochondrial fusion. Neuroscience.
301:71–78. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kumar R, Bukowski MJ, Wider JM, Reynolds
CA, Calo L, Lepore B, Tousignant R, Jones M, Przyklenk K and
Sanderson TH: Mitochondrial dynamics following global cerebral
ischemia. Mol Cell Neurosci. 76:68–75. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park E, Lee GJ, Choi S, Choi SK, Chae SJ,
Kang SW, Pak YK and Park HK: The role of glutamate release on
voltage-dependent anion channels (VDAC)-mediated apoptosis in an
eleven vessel occlusion model in rats. PLoS One. 5:e151922010.
View Article : Google Scholar
|
|
30
|
Racay P, Tatarkova Z, Drgova A, Kaplan P
and Dobrota D: Effect of ischemic preconditioning on mitochondrial
dysfunction and mitochondrial p53 translocation after transient
global cerebral ischemia in rats. Neurochem Res. 32:1823–1832.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Racay P, Chomova M, Tatarkova Z, Kaplan P,
Hatok J and Dobrota D: Ischemia-induced mitochondrial apoptosis is
significantly attenuated by ischemic preconditioning. Cell Mol
Neurobiol. 29:901–908. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Filadi R, Pendin D and Pizzo P: Mitofusin
2: From functions to disease. Cell Death Dis. 9:3302018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Palikaras K, Lionaki E and Tavernarakis N:
Mechanisms of mitophagy in cellular homeostasis, physiology and
pathology. Nat Cell Biol. 20:1013–1022. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pulsinelli WA: Selective neuronal
vulnerability: Morphological and molecular characteristics. Prog
Brain Res. 63:29–37. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Smith ML, Auer RN and Siesjö BK: The
density and distribution of ischemic brain injury in the rat
following 2-10 min of forebrain ischemia. Acta Neuropathol.
64:319–332. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang W, Zhang F, Li L, Tang F, Siedlak SL,
Fujioka H, Liu Y, Su B, Pi Y and Wang X: MFN2 couples glutamate
excitotoxicity and mitochondrial dysfunction in motor neurons. J
Biol Chem. 290:168–182. 2015. View Article : Google Scholar :
|
|
37
|
Martorell-Riera A, Segarra-Mondejar M,
Reina M, Martínez- Estrada OM and Soriano FX: Mitochondrial
fragmentation in excitotoxicity requires ROCK activation. Cell
Cycle. 14:1365–1369. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Flippo KH, Gnanasekaran A, Perkins GA,
Ajmal A, Merrill RA, Dickey AS, Taylor SS, McKnight GS, Chauhan AK,
Usachev YM and Strack S: AKAP1 protects from cerebral ischemic
stroke by inhibiting Drp1-dependent mitochondrial fission. J
Neurosci. 38:8233–8242. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pilchova I, Klacanova K, Chomova M,
Tatarkova Z, Dobrota D and Racay P: Possible contribution of
proteins of Bcl-2 family in neuronal death following transient
global brain ischemia. Cell Mol Neurobiol. 35:23–31. 2015.
View Article : Google Scholar
|
|
40
|
Baines CP, Kaiser RA, Sheiko T, Craigen WJ
and Molkentin JD: Voltage-dependent anion channels are dispensable
for mitochondrial-dependent cell death. Nat Cell Biol. 9:550–555.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Geisler S, Holmström KM, Skujat D, Fiesel
FC, Rothfuss OC, Kahle PJ and Springer W: PINK1/Parkin-mediated
mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol.
12:119–131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kerner J, Lee K, Tandler B and Hoppel CL:
VDAC proteomics: Post-translation modifications. Biochim Biophys
Acta. 1818:1520–1525. 2012. View Article : Google Scholar
|
|
43
|
Nagakannan P, Islam MI, Karimi-Abdolrezaee
S and Eftekharpour E: Inhibition of VDAC1 protects against
glutamate-induced oxytosis and mitochondrial fragmentation in
hippocampal HT22 cells. Cell Mol Neurobiol. 39:73–85. 2019.
View Article : Google Scholar
|