Introduction
Lung cancer is the leading cause of cancer-related
deaths worldwide, imposing a heavy social and economic burden in
China (1). Non-small cell lung
cancer (NSCLC) accounts for ~85% of lung cancer histological
subtypes (2). The majority of
NSCLC cases are diagnosed at an advance stage (~70%), with patients
missing the chance for radical surgery and thus facing chemotherapy
as the main treatment option (3).
Despite the excitement after the shift from empirical therapy to
more effective and better tolerated regimens for NSCLC over the
past two decades, the 5-year overall survival rate remains as low
as 4-17% depending on the stage and regional differences (4). What makes the situation worse is
that among patients with NSCLC, after a transient response to
initial treatment, drug resistance of malignant cells occurs
frequently. Therefore, finding an effective and safe anti-tumour
agent is one of the highest-priority goals in lung cancer
research.
Signal transducer and activator of transcription 3
(STAT3) is an important member of the STAT family and is recognised
as a key oncogenic factor highly relevant to NSCLC (5). STAT3 plays crucial roles in cancer
initiation and progression by binding to the promoter regions of a
number of genes that encode regulators of cell proliferation (such
as cyclin D1 and MYC), survival (such as BCL-2
and myeloid cell leukemia-1 (MCL-1)], inflammation [such as
interleukin-6 (IL-6) and cyclooxygenase-2], and metastasis
[such as matrix metallopeptidase 9 (MMP-9) and vimentin]
(6-8). STAT3 activation is transient and
dynamic under physiological conditions. However, activated STAT3 is
persistently present in 50% of NSCLC tissue (9) due to stimulation of an upstream
signaling pathway (10),
suggesting that STAT3 is a lung cancer-specific target for
anticancer therapy. Activation of cell cycle and survival genes
enables STAT3-expressing cells to escape damage from conventional
chemotherapeutic drugs (11);
thus, the STAT3 oncogenic pathway is involved in drug resistance
(12). Similarly, a meta-analysis
has revealed that (13)
expression of STAT3 or phosphorylated STAT3 (p-STAT3) is a strong
predictor of poor prognosis among patients with NSCLC (14). Hence, interference with the STAT3
pathway might counteract the resistance to chemotherapy and exert
an adjuvant effect during the treatment of lung cancer.
Because current standard treatments are far from
ideal for patients with NSCLC, a natural compound with multiple
advantages has come to our attention (15). Triptolide (TP), a classical
natural compound, has been demonstrated to exert anti-tumour
effects on ovarian cancer (16),
breast cancer (17),
hepatocellular carcinoma (18)
and lung cancer (19). Studies
have revealed that low-dose TP helps to circumvent resistance,
enhances effectiveness and relieves adverse effects of anticancer
therapies (20,21). It has been reported that TP exerts
its anti-tumour activities in terms of cell proliferation, cell
death, angiogenesis, and metastasis (22). Nevertheless, at present, the
potential influence of TP on IL-6/STAT3 signalling in NSCLC is
still unknown.
In this study, we determined the effect of TP on the
proliferation, apoptosis, and invasiveness of NSCLC cells and
investigated the potential underlying mechanisms by evaluating low
dose TP's influence on the IL-6/STAT axis.
Materials and methods
Materials
TP (>98% purity) was purchased from Dalian Meilun
Co., Ltd. Human recombinant IL-6 (cat. no. 200-06; Peprotech, Inc.)
was resolved and stored according to the manufacturer's
instructions. 3-(4,5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium
bromide (MTT) and protease inhibitor cocktail (cat. no.
04693132001) were purchased from Sigma-Aldrich (Merck KGaA).
Antibodies against anti-cleaved caspase-3 (cat. no. GB11009) were
purchased from Servicebio. Antibodies against BCL-2 (cat. no.
182858) and MMP-9 (cat. no. 76003) were purchased from Abcam, MCL-1
(cat. no. 94296), cyclin D1 (cat. no. 2978S), C-myc (cat. no.
5605S), STAT3 (cat. no. 4904) and p-STAT3 (cat. no. 9145) were
acquired from Cell Signaling Technology Inc., and antibodies
against β-actin (cat. no. BB0710) were from Biossci Biotechnology
Co., Ltd. DAPI, Triton X-100 and an anti-fade mounting medium were
purchased from Servicebio. The Annexin V/PI Apoptosis Detection kit
(cat. no. KGA105-KGA108) was from Nanjing KeyGen Biotech Co., Ltd.
Propidium iodide (PI) and the JC-1 Staining kit were acquired from
Beyotime Institute of Biotechnology. DyLight 488-conjugated goat
anti-rabbit IgG antibody (cat. no. A24221) was from Abbkine
Scientific Co., Ltd.
Cell culture
Human lung adenocarcinoma PC9 cells sensitive to
epidermal growth factor receptor (EGFR) with low expression of IL-6
and human alveolar basal epithelial adenocarcinoma A549 cells with
higher expression of IL-6 were provided by the Caner Laboratory,
Tongji Medical College (Wuhan, China). The cells were cultured in
RPMI-1640 medium (HyClone; GE Healthcare Life Sciences)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.),
100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a
humidified incubator with 5% CO2 and 95% air.
MTT assay
Cells were seeded in 96-well plates at a density of
5×103 cells per well and cultured overnight. A series of
concentrations ranging to 500 nM TP were added into the medium.
After either 24 or 48 h, MTT was added to assay cell viability.
Absorbance at 570 nm was recorded on a microplate reader (Synergy
2; BioTek Instruments, Inc.). The inhibition ratio was calculated
as 1-optical density (OD)treated/ODcontrol. The IC50
values were calculated by plotting the drug concentration versus
the percent cell viability of the cells after TP intervention for
24 and 48 h.
Flow cytometry
At 3×105 cells per well were exposed to
TP in 12 wells for 24 h after overnight serum starvation.
Thereafter, the cells were washed with PBS and fixed with 70%
ethanol at 4°C overnight. Then, cell cycle analyses were performed
through flow cytometry according to the manufacturer's instructions
(Beyotime Institute of Biotechnology). Apoptosis was analyzed via
pre-treatment with a selected concentration of TP for 24 h (PC9
cells) or 48 h (A549 cells) and then was detected with the Annexin
V/PI Staining kit (Nanjing KeyGen Biotech Co., Ltd.) and assayed by
flow cytometry (BD FACS Calibur; BD Biosciences). Cells undergoing
early-stage apoptosis were categorized as Annexin
V+/PI−, whereas Annexin
V+/PI+ cells were assumed to be at a late
stage of apoptosis. Subsequently, mitochondrial trans-membrane
potential ΔΨ) was determined according to the manufacturer's
instructions using the JC-1 Apoptosis Detection kit (Beyotime
Institute of Biotechnology). Data analysis was conducted using the
FlowJo software (oxs 10.6; FlowJo LLC).
Transwell assay
Transwell migration assays were performed using
24-well Transwell inserts (8 μm pore size; Costar; Corning,
Inc.), as described previously (23). A total of 1×105 cells
were pre-treated with TP for 24 h, then harvested and resuspended
in RPMI-1640 (HyClone; GE Healthcare Life Sciences) at a
concentration of 105 cells/ml. Next, 200 μl of
the cell suspension mixed with 750 μl RPMI-1640 medium
supplemented with 10% FBS was added to the upper chamber, and
incubated for another 24 h at 37°C. Non-migratory cells were gently
wiped from the top surface of the membrane with a cotton swab.
Migratory cells on the bottom side of the membrane were rinsed with
PBS, fixed in 4% paraformaldehyde and stained with 0.1% (m/v)
crystal violet for 10 min at room temperature. Images were captured
under a ZEISS digital microscope (magnification ×100; Carl Zeiss
AG). The migration rate was quantified by counting the stained
cells in selected 3-5 fields of view.
Western blotting
Cells were treated with or without TP for 24 h, then
cells were washed twice with ice-cold PBS and lysed in RIPA buffer
containing a protease inhibitor cocktail (Thermo Fisher Scientific,
Inc.). Soluble proteins were collected after centrifugation at
12,000 x g for 15 min at 4°C. Protein concentration was determined
by the bicinchoninic acid (BCA) method. Subsequently, 50 μg
of each protein sample was separated by SDS-PAGE on a 10-12% gel
(120 V, 90 min) and transferred to a PVDF membrane (Bio-Rad
Laboratories, Inc.) at 280 mA for 90 min. After blocking with 5%
non-fat milk for 1 h at room temperature, the membranes were
incubated with specific primary antibodies (1:1,000) at 4°C
overnight. After three washes with TBS containing 0.1% Tween 20,
the membranes were incubated with fluorescently-labeled secondary
antibodies (1:20,000) for 1 h at room temperature. The bands were
visualized using a near-infrared fluorescence imaging system
(Odyssey; LI-COR Biosciences). Band densities were quantified in
the ImageJ software (version 1.52 National Institutes of Health).
In addition, cells were pretreated with TP or homoharringtonine
(HHT) for 24 h, then 50 nM IL-6 (cat. no. 200-06; PeproTech, Inc.)
was added for another 30 min, finally cells were harvested and
lysed for western blotting.
Immunofluorescence
Cells (105 cells/well) were seeded on
glass cover slides. After treatment with 60 nM TP or 4 μM
STAT3 inhibitor for 24 h, the cells were fixed with 4% buffered
formalin for 30 min at room temperature, permeabilized with 0.5%
Triton X-100, and blocked with 10% goat serum in PBS (Servicebio)
for 30 min at room temperature. After successive incubation with
the primary antibody (1:100) at 4°C for 12 h, a secondary antibody
(DyLight 488-conjugated goat anti-rabbit IgG; 1:8,000) was
incubated for another 1 h at room temperature, at last cells were
stained with DAPI (1:500) in the dark for 5 min. The fluorescence
signals were captured using a fluorescence microscope (Nikon
Eclipse CI; Nikon Corporation).
Statistical analysis
All experiments were conducted in triplicate and
repeated three times. Representative results are shown as the mean
± SD and were plotted using Prism 6.0 (GraphPad Software, Inc.).
Statistical calculations were performed via one-way ANOVA followed
by the Student-Newman-Keuls test. P<0.05 was considered to
indicate a statistically significant difference.
Results
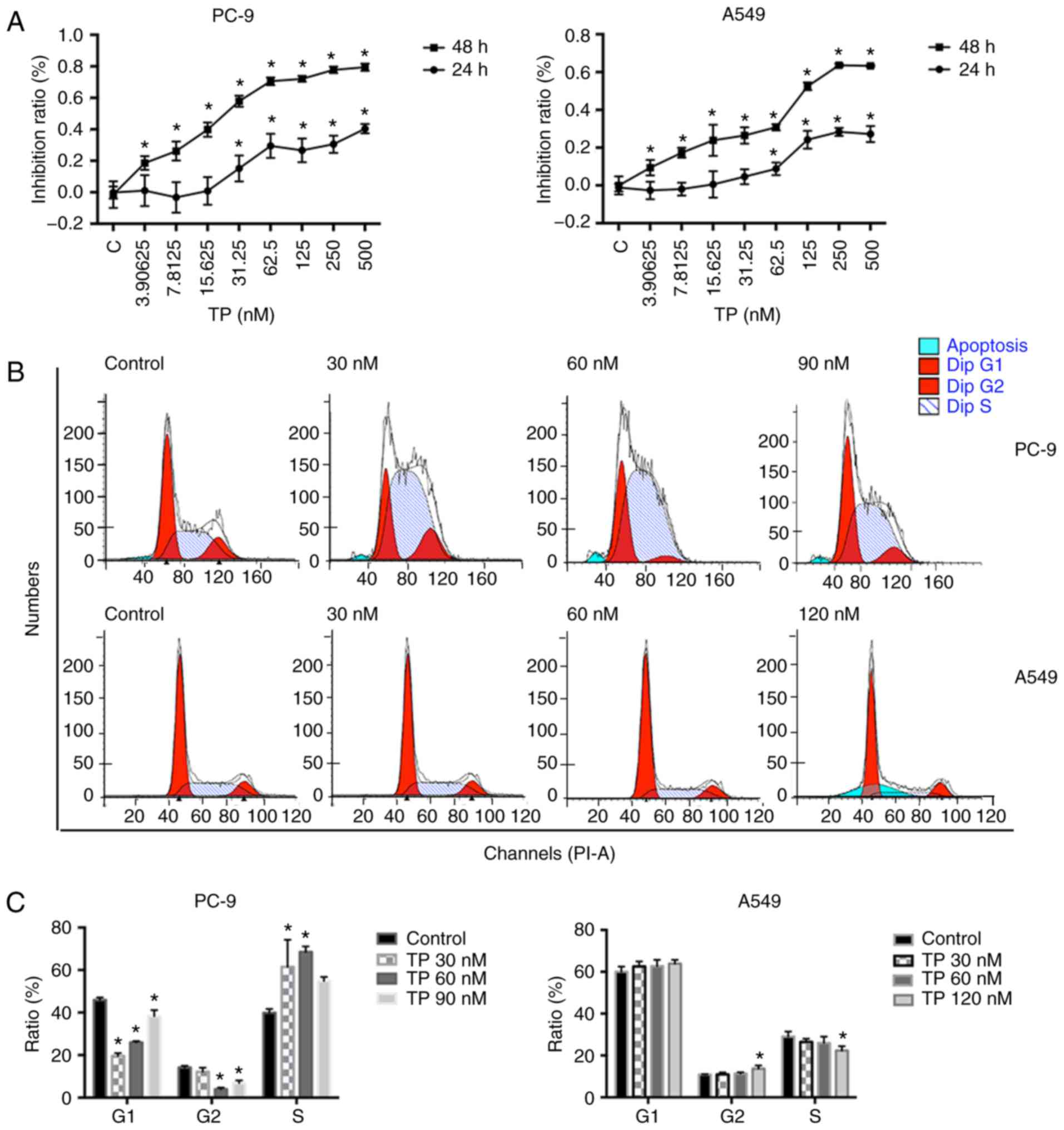
TP impairs viability of NSCLC cells by
inducing cell cycle arrest
To evaluate the cytotoxic action of TP, PC9 and A549
cells were treated with a series of TP concentrations ranging from
0 to 500 nM for either 24 or 48 h. The IC50 was
calculated using log formula. The IC50 of PC9 and was
1,238±243.8 nM and the IC50 of A549 was 1,339±150.2 nM.
After 48 h of treatment with TP, the corresponding values were
29.33±2.102 and 135.9±0.9328 nM. As shown in Fig. 1A, TP was cytotoxic after 48 h
treatment even at a low concentrations (P<0.05). TP inhibited
proliferation (according to an inhibition ratio) of PC9 and A549
cells at 48 h in a dose-dependent manner. During the 24 h
treatment, TP at concentrations <15.625 nM was not cytotoxic at
all toward PC9 and A549 cells. The inhibition of proliferation
started to be noticeable via a sharp slope in the inhibition ratio
(15.625-62.500 nM in PC9 cells and 15.625-125.000 nM in A549 cells)
(24) followed by a relatively
flat plateau at high TP concentrations. Therefore, these
comparatively low doses were selected to conduct subsequent
experiments.
Whether TP influences cell viability by affecting
the cell cycle was examined (Fig.
1B). On the basis of the inhibition ratio, two concentrations
within the sharp-slope region (30 and 60 nM) and one concentration
in the plateau region (90 nM, PC-9; 120 nM, A549) were selected. As
shown in Fig. 1C, TP at the
concentration in the sharp-slope region (30 and 60 nM) strongly
promoted the G1/S arrest in PC9 cells. There was a slight increase
in the number of cells in the S phase at 90 nM TP as well, but the
change was not statistically significant. Similar results were not
seen in corresponding experiments on A549 cells after treatment
with TP for 24 h. A slight decrease in the number of cells in the S
phase at 120 nM TP was observed, though it appeared that TP in
general did not affect the cell cycle in A549 cells (Fig. 1C).
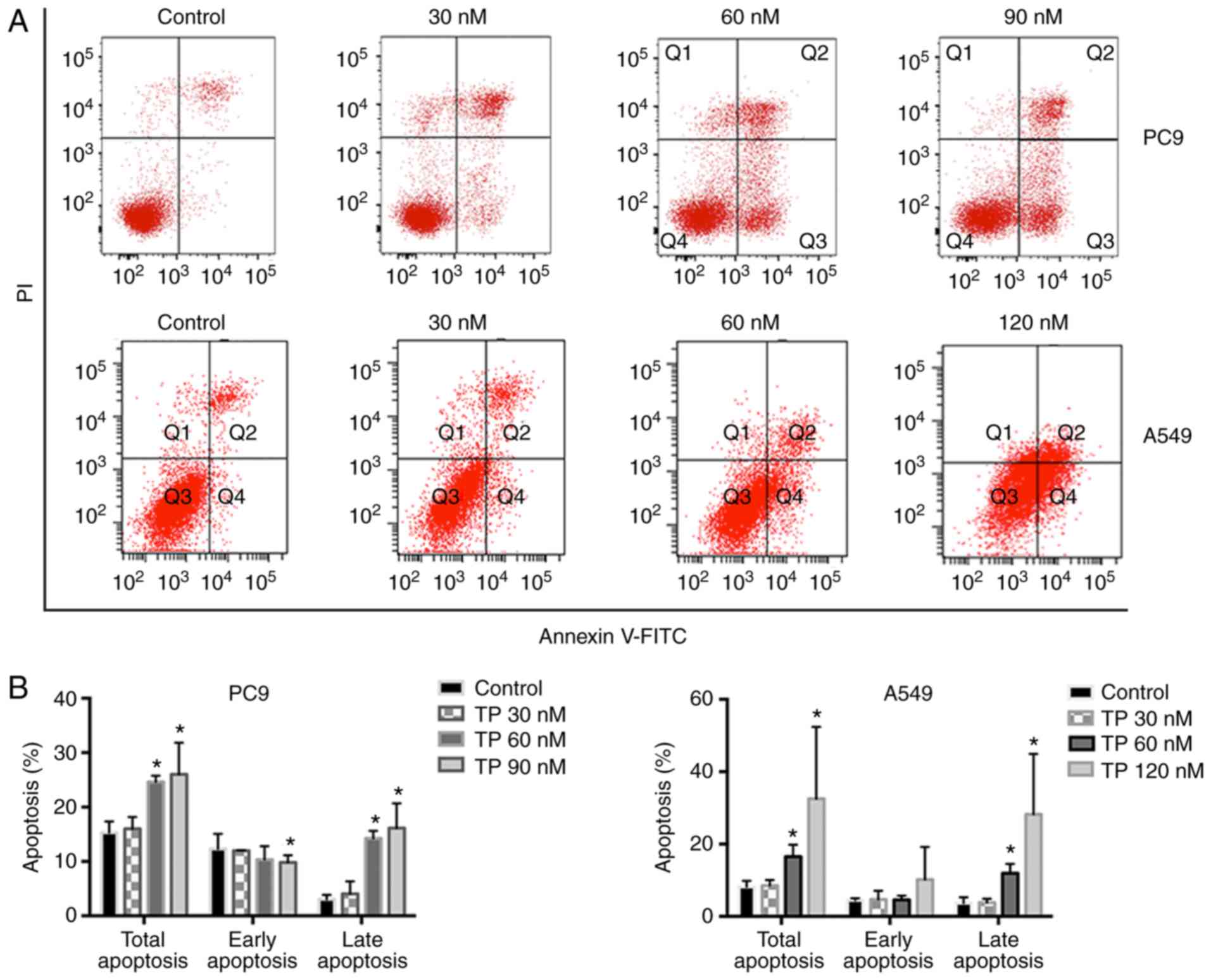
TP disrupts active mitochondria and
induces apoptosis in NSCLC cells
It was subsequently analyzed whether TP treatment
has any impact on apoptosis. Starting from the concentration of 60
nM, treatment of PC9 cells with TP for 24 h caused a significant
increase in total apoptosis (Fig. 2A
and B). In contrast, after 48 h treatment of A549 cells with
TP, total apoptosis increased strongly, and this change was
attributed to a late-apoptosis population (Fig. 2A and B). Then, ΔΨ was measured as
disruption of mitochondria is regarded as a distinctive feature of
the intrinsic pathway of apoptosis. As demonstrated by the JC-1
blue ratio, treatment with TP for 24 h slightly decreased ΔΨ
(Fig. 2C and D). These results
suggested that TP induced apoptosis at least partially at an early
stage by disrupting active mitochondria.
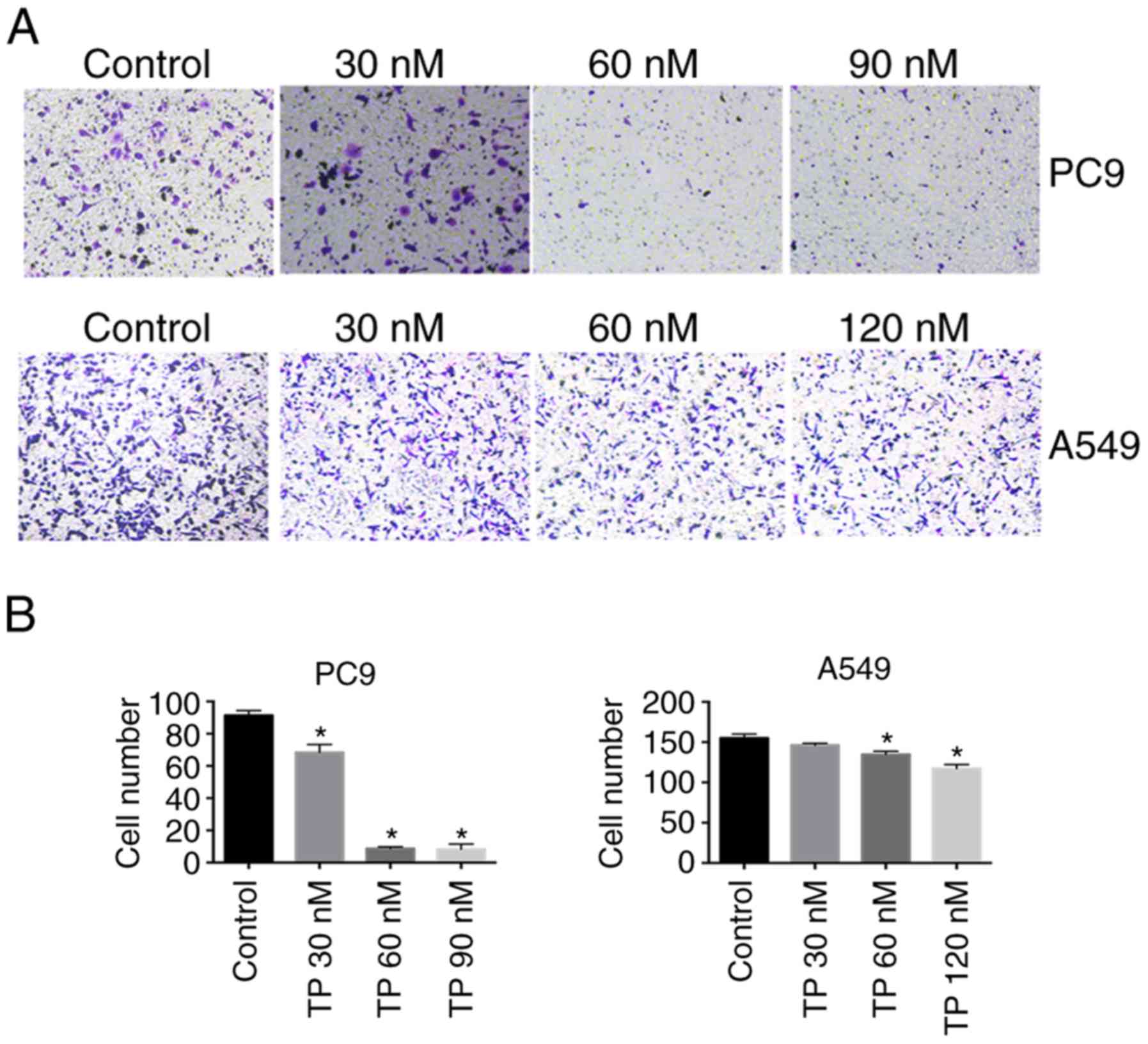
TP inhibits migration of NSCLC cells
Considering the anti-metastasis effect of TP, we an
in vitro chamber migration assay was performed to test
whether TP affects NSCLC cell migration. As presented in Fig. 3, the number of migratory PC9 cells
was decreased by TP treatment and dropped dramatically at 60 and 90
nM; whereas the number of migratory A549 cells stayed largely
unchanged upon TP treatment; however, a statistically significant
decrease in the number of migratory cells was observed after
treatment with 60 and 120 nM TP, but this was less dramatic than
the effect in PC9 cells.
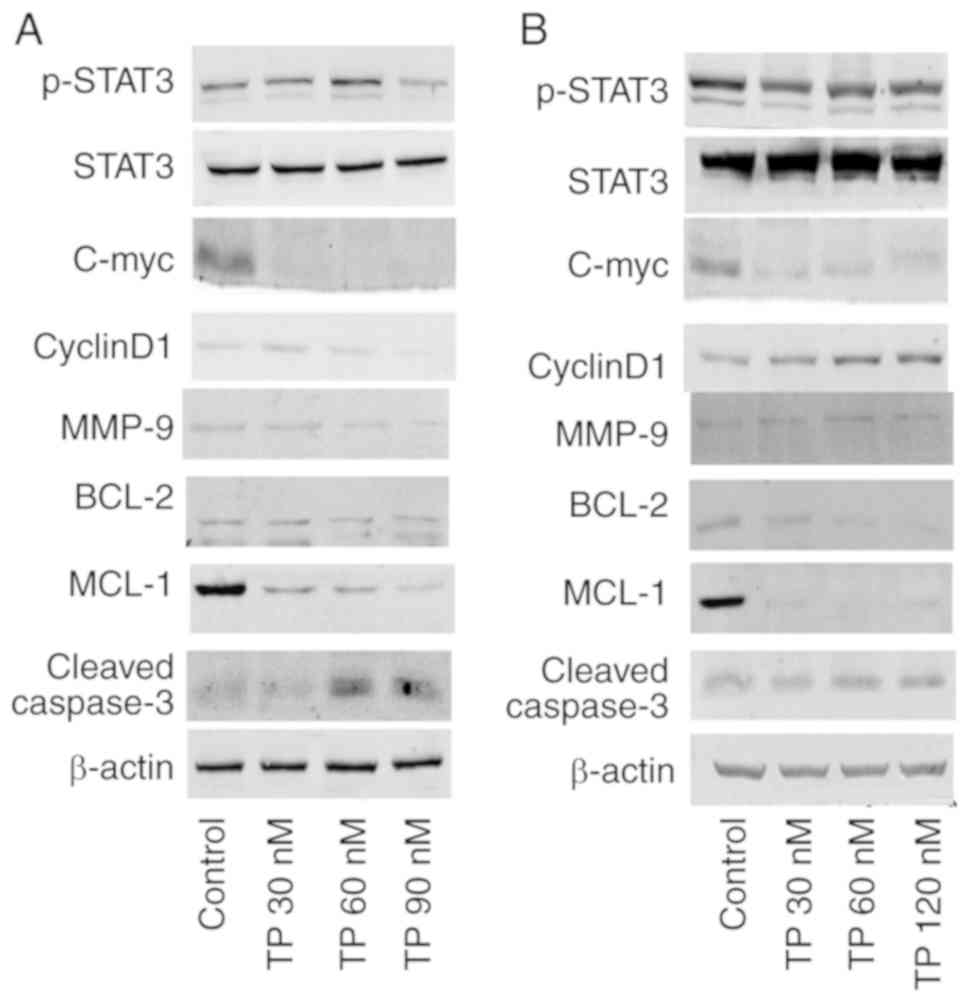
TP disturbs an activated STAT3 pathway in
NSCLC cells
Aberrant and constitutive activation of STAT3 in
NSCLC has been reported (9) and
is associated with tumor progression and a poor prognosis (14). After treatment with TP for 24 h,
p-STAT3 was decreased in PC9 (Fig.
4A) and A549 cells (Fig. 4B).
The mRNA expression of PC9 (Fig.
S1) and A549 (Fig. S2) was
consistent with protein expression. As shown in Fig. S3, the ratio of p-STAT3/total
STAT3 was marginally decreased by TP in A549 cells, with
statistical significance at 120 nM (P<0.05).
Expression levels of target genes of STAT3 were also
examined. C-myc expression was found to greatly decreased upon TP
treatment in PC9 (Fig. 4A) and
A549 cells (Fig. 4B). Although
cyclin D1 was downregulated by TP in PC9 cells (Fig. 4B), cyclin D1 was upregulated by TP
treatment in A549 cells (Fig.
4B). The overexpression of cyclin D1 may contribute to
resistance to TP (25), as well
as cell DNA damage (24).
Caspase-3 is a critical executioner of apoptosis, as it is either
partially or totally responsible for the proteolytic cleavage of
many key proteins, and cleaved caspase-3 is the active form of the
protein. As shown in PC9 cells (Fig.
4A), TP increased the level of cleaved caspase-3. Expression of
anti-apoptotic proteins MCL-1 and BCL-2 was decreased after TP
treatment in a dose-dependent manner in PC9 (Fig. 4A) and A549 cells (Fig. 4B). The two bands of BCL-2 seen in
Fig. 4A may be homedimers or poor
antibody specificity (26). TP
also downregulated MMP-9 expression in a dose-dependent manner in
PC9 (Fig. 4A) and A549 cells
(Fig. 4B). The above results
suggested that TP treatment inhibited cell proliferation, induced
apoptosis and inhibited cell migration by suppressing the
activation of STAT3.
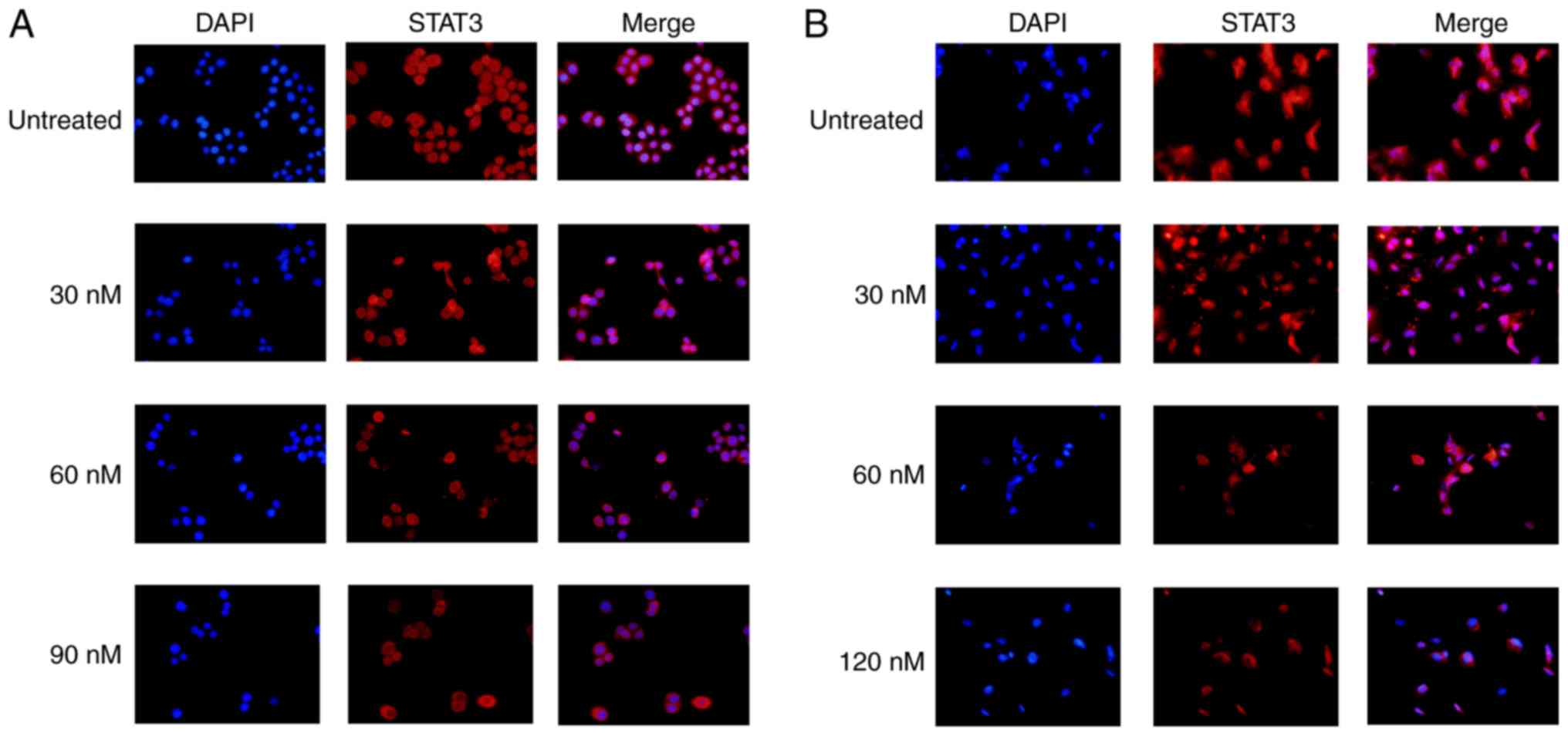
Signal transduction in the STAT3 pathway, as
visualized by STAT3 translocation into the nucleus, was inhibited
by TP treatment. Less prevalent and weaker STAT3 staining was
observed in cell nuclei in the TP treatment groups (Fig. 5).
TP attenuated STAT3 activation induced by
IL-6
Elevated IL-6 expression has been observed in many
patients with lung cancer (27).
A549 cells appear to be much less sensitive to TP compared with PC9
cells, thus it was speculated that the expression of IL-6 in these
cells is associated with the results. The effect of TP in the
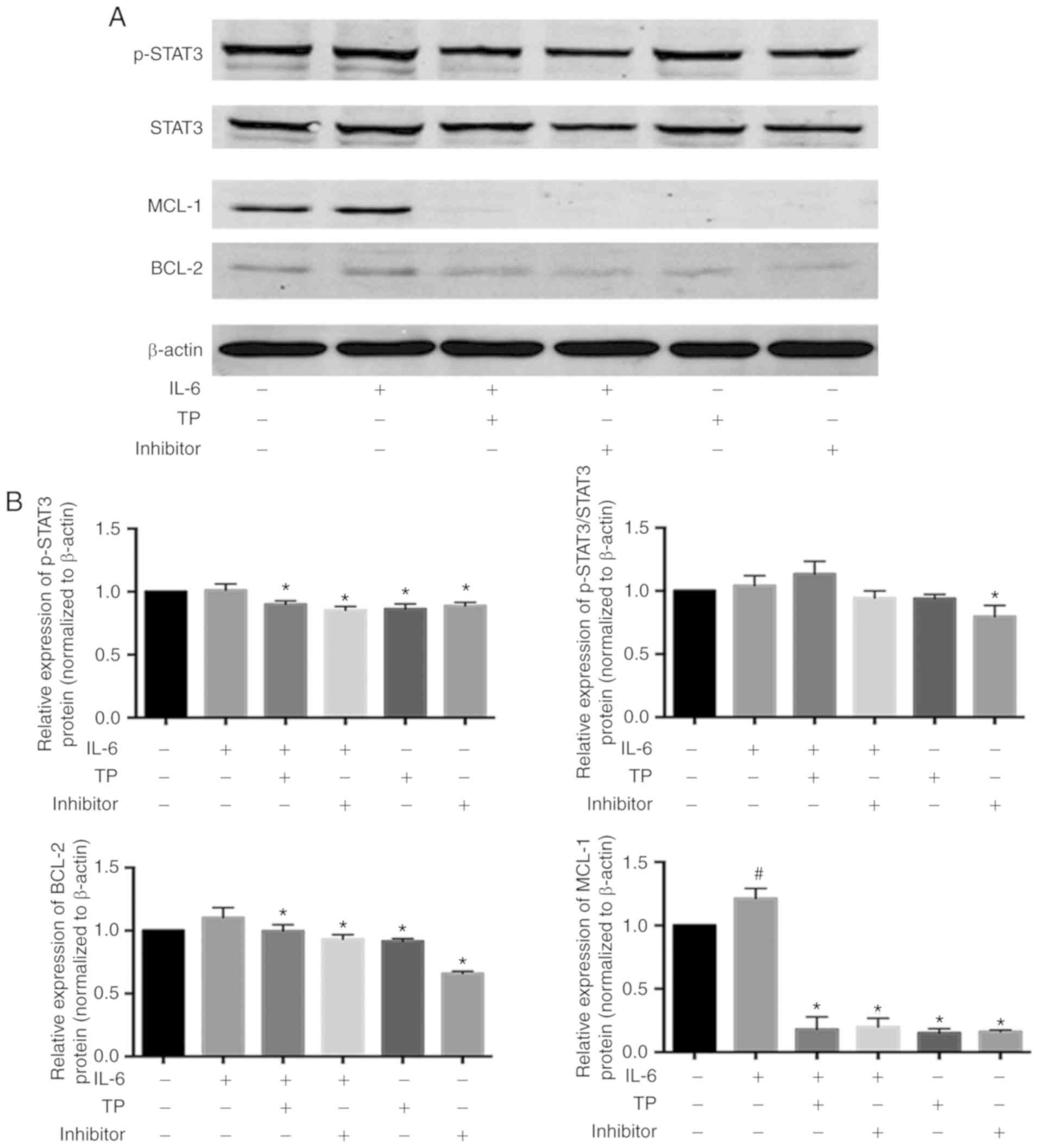
presence of exogenous IL-6 in PC9 cells was analyzed, with a
non-specific IL-6/STAT3 inhibitor HHT serving as a positive
control, which prevents the initial elongation step of protein
synthesis (Fig. 6). Simlar to Ham
et al (28), IL-6 seems
not significantly increase the phosphorylation of STAT3, but did
impair the ability of HHT to inhibit STAT3 phosphorylation. IL-6
appeared to induce expression of the STAT3 downstream target gene,
BCL-2, but there was no statistical difference (P>0.05). At the
same time, TP and HHT could reduce the expression of BCL-2 compared
with IL-6-stimulated cells. Compared with the control group, the
expression of MCL-1 was significantly increased by exogenous IL-6
(P<0.05). Similar to HHT, pretreated TP can almost completely
block MCL-1 expression. Briefly, TP might enhance the effects of
chemotherapy by inhibiting IL-6-induced MCL-1 activation.
Discussion
Even though treatment of NSCLC has changed
dramatically over the past two decades, only modest improvements
have been achieved in terms of the patient survival rate (4). Drug resistance and side effects are
still obstacles to successful cancer therapy. With the advantages
of low toxicity and cost, natural compounds have been regarded as a
promising adjuvant therapies for NSCLC (29). Initially prescribed as an
anti-inflammatory drug, TP is considered an alternative therapeutic
agent for the treatment of a wide range of cancers, such as
pancreatic cancer and colon cancer (30), ovarian cancer (31), breast cancer (32), hepatocellular carcinoma (33) and lung cancer (34). A distinct antitumour activity of
TP lies in its antagonism of drug resistance (20,31). This beneficial effect might be
mediated by changes in ATP-binding cassette transporters (35), induction of apoptosis pathways,
increase in tumor suppressors and decrease in oncogenic factors,
and interactions with the RNA polymerase II complex (36) by TP. Aside from mitogen-activated
protein kinase (32) and
Wnt/β-catenin pathways (33), the
IL-6/JAK/STAT3 axis (34,37) is also also involved in the
antitumor activity of TP in cancer types other than lung cancer.
However, at present, the influence of TP on IL-6/STAT3 signaling
during the treatment of NSCLC is still unclear.
Although the antitumor effect of TP has been
described previously, the current study further confirms how low
doses of TP exert anticancer effects or adjuvant to chemotherapy in
lung cancer through IL-6/STAT3. In this study, the antitumor
effects of TP on PC9 and A549 were analyzed. After balancing
effectiveness and possible toxicity, appropriate doses were
selected to conduct subsequent experiments. TP inhibited cell
proliferation, promoted cell apoptosis and cell migration at a
comparative dose in PC9 cells, while TP decreased the expression of
genes associated apoptosis resistance, such as MCL-1. Studies
indicated that MCL-1 is a key target of adjuvant chemotherapy to
reverse the cisplatin-resistance in NSCLC (38). The direct antitumor effect in PC9
and the adjuvant effect of circumventing resistance in A549 make it
superior to other traditional Chinese therapeutic agents. TP
exerted antiproliferative action on both NSCLC cell lines. In
agreement with other findings (39), the data showed significant S phase
cell cycle arrest in PC9 cells. Such S phase arrest was not
significant in A549 cells after treatment with TP for 24 h.
Unexpectedly, there was a decrease of DNA synthesis in the cells
treated with 120 nM TP; DNA synthesis change might be a consequence
of increased apoptosis observed in the same experimental setting.
In any case, the weaker effect of TP on cell cycle arrest in A549
cells compared with PC9 cells is worthy of further investigation.
C-myc and cyclin D1 are crucial factors that are associated with
STAT3 activation and cell proliferation. A decrease in both C-myc
expression and cyclin D1 expression was seen in PC9 cells treated
with TP; this result echoes the aforementioned findings and further
elucidates the TP-induced cell cycle arrest in S phase. In
parallel, C-myc was also found to be downregulated by TP in A549
cells, whereas TP elevated the cyclin D1 level in A549 cells.
Overexpression cyclin D1 may cause resistance to cytotoxic drugs
(40), thus limiting the effects
of TP. However, high levels of cyclin D1 prime cells for an
enhanced DNA damage response (24), causing sustained cyclin
D1-cyclin-dependent kinase activity, leading to inappropriate
replication of DNA and chromosomal damage (41,42). This result may be the reason why S
phase arrest was barely detectable among TP-treated A549 cells. On
the basis of the literature (24,43), it is speculated that TP enhances
DNA damage and arrests the cell cycle in the S phase to inhibit
cell proliferation.
Bypassing apoptosis is a critical step in
carcinogenesis. The apoptosis pathway, as a result, has been a hot
target for many anticancer therapies. As many as 22-65% of NSCLC
cells aberrantly express apoptotic proteins; this feature
malignantly enhances cell proliferation and promotes resistance to
chemotherapy. In this study, TP markedly increased the percentage
of cells undergoing early apoptosis in PC9 and A549 cells.
Activated caspase-3 is a typical hallmark of apoptosis (44). The findings of the present study
showed that TP increases the expression of cleaved caspase-3 in
NSCLC cells. Other authors (13,45) have also stated that inhibition of
STATs directly decreases the levels of anti-apoptotic proteins,
such as BCL-2 and MCL-1, which are known to facilitate tumor cell
survival. The results of the current study revealed that TP
treatment decreased the expression of BCL-2 and MCL-1. Given that
TP reduced the phosphorylation of STAT3 in PC9 and A549 cells, it
was concluded that TP induces apoptosis of NSCLC cells through the
STAT3/MCL-1 axis.
Aberrant activation of STAT3 contributes to cancer
progression in human tumors (46). It induces MMP-9, which has an
important role in cell invasion and metastasis (47). Activated MMP-9 degrades collagens
in the extracellular matrix, and this activity is closely
associated with the invasive and metastatic potential of numerous
types of solid tumor (48-50).
The findings of the current study clearly indicate that TP
downregulates MMP-9 expression and inhibits NSCLC cell migration,
thus indicating that TP inhibits the STAT3/MMP-9 axis.
The present study demonstrated that TP inhibited
cell proliferation and migration, and induced apoptosis by
suppressing STAT3 activation and downregulating targets of STAT3.
The finding that TP inhibited STAT3 phosphorylation and its nuclear
translocation is consistent with data from studies on
colitis-related colon cancer (37), multiple myeloma cells (51), and pancreatic cancer (52). The results of the current study
show that A549 cells are less responsive to TP, which may be
associated with the expression of IL-6 in these cells. NSCLC cells
carry mutations in oncogenes Egfr (45%) and Kras
(25%) (13); these mutations
drive the overexpression of IL-6. Indeed, IL-6 is overexpressed in
many patients with lung cancer (27). Hyperactivation of IL-6 could
activate STAT3, which in turn produces more IL-6 (5,13,53). In the present study, the influence
of TP on STAT3 activation by exogenous IL-6 was analyzed. IL-6
significantly upregulated MCL-1 expression in PC9 cells, and MCL-1
has been demonstrated to be associated with chemoresistance in oral
squamous cell carcinoma (54), so
IL-6 might be a factor involved in drug resistance. Additionally,
IL-6 significantly reduced the ability of HHT to inhibit STAT3
phosphorylation, that is possible reason why A549 showed smaller
response to TP than PC9. These data are indicative of a promising
application of TP in lung cancer management.
Although appropriate methods have been used to
illustrate the hypothesis, it is difficult to demonstrate the exact
mechanism of TP against NSCLC. The effect of TP on downstream
target genes of STAT3 indicated the possible existence of another
regulator, and more research is needed to confirm this conclusion.
This preliminary study illustrated that TP has promising effects on
NSCLC, a clinical investigation using combined TP with conventional
therapy is necessary determine the effects in recurrent advanced
NSCLC. In conclusion, the IL-6/STAT3 pathway appears to be another
therapeutic target in NSCLC, and TP may have antitumor effects or
at least an adjuvant effect.
Supplementary Data
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81573802).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YiH, ZC and ST conceived and designed the study.
YiH, YW, XB and YaH performed the experiments. YiH and ST wrote the
paper. YiH, ZC and ST reviewed and edited the manuscript. PS and HW
provided assistance for data acquirement, statistical analysis and
manuscript editing. All authors read and approved the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors acknowledge the gift of cells from
Tongji Cancer Laboratory.
References
|
1
|
Hong QY, Wu GM, Qian GS, Hu CP, Zhou JY,
Chen LA, Li WM, Li SY, Wang K, Wang Q, et al: Prevention and
management of lung cancer in China. Cancer. 121(Suppl 17):
S3080–S3088. 2015. View Article : Google Scholar
|
|
2
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
553:446–454. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shi Y, Sun Y, Ding C, Wang Z, Wang C, Wang
Z, Bai C, Bai C, Feng J, Liu X, et al: China experts consensus on
icotinib for non-small cell lung cancer treatment (2015 version). J
Thorac Dis. 7:E468–E472. 2015.PubMed/NCBI
|
|
4
|
Hirsch FR, Scagliotti GV, Mulshine JL,
Kwon R, Curran WJ Jr, Wu YL and Paz-Ares L: Lung cancer: Current
therapies and new targeted treatments. Lancet. 389:299–311. 2017.
View Article : Google Scholar
|
|
5
|
Gao SP, Mark KG, Leslie K, Pao W, Motoi N,
Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B and Bromberg
JF: Mutations in the EGFR kinase domain mediate STAT3 activation
via IL-6 production in human lung adenocarcinomas. J Clin Invest.
117:3846–3856. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: A leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bournazou E and Bromberg J: Targeting the
tumor microenvironment: JAK-STAT3 signaling. JAKSTAT.
2:e238282013.PubMed/NCBI
|
|
8
|
Bromberg JF, Wrzeszczynska MH, Devgan G,
Zhao Y, Pestell RG, Albanese C and Darnell JE Jr: Stat3 as an
oncogene. Cell. 98:295–303. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li CJ, Li YC, Zhang DR and Pan JH: Signal
transducers and activators of transcription 3 function in lung
cancer. J Cancer Res Ther. 9(Suppl 2): S67–S73. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grivennikov SI and Karin M: Dangerous
liaisons: STAT3 and NF-kappa B collaboration and crosstalk in
cancer. Cytokine Growth Factor Rev. 21:11–19. 2010. View Article : Google Scholar
|
|
11
|
Brown JM and Wilson G: Apoptosis genes and
resistance to cancer therapy: What does the experimental and
clinical data tell us? Cancer Biol Ther. 2:477–790. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Barré B, Vigneron A, Perkins N, Roninson
IB, Gamelin E and Coqueret O: The STAT3 oncogene as a predictive
marker of drug resistance. Trends Mol Med. 13:4–11. 2007.
View Article : Google Scholar
|
|
13
|
Yan X, Li P, Zhan Y, Qi M, Liu J, An Z,
Yang W, Xiao H, Wu H, Qi Y and Shao H: Dihydroartemisinin
suppresses STAT3 signaling and Mcl-1 and Survivin expression to
potentiate ABT-263-induced apoptosis in non-small cell lung cancer
cells harboring EGFR or RAS mutation. Biochem Pharmacol. 150:72–85.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu YH and Lu S: A meta-analysis of STAT3
and phospho-STAT3 expression and survival of patients with
non-small-cell lung cancer. Eur J Surg Oncol. 40:311–317. 2014.
View Article : Google Scholar
|
|
15
|
Mishra BB and Tiwari VK: Natural products:
An evolving role in future drug discovery. Eur J Med Chem.
46:4769–4807. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu L, Xiong X, Shen M, Ru D, Gao P, Zhang
X, Huang C, Sun Y, Li H and Duan Y: Co-delivery of triptolide and
curcumin for ovarian cancer targeting therapy via mPEG-DPPE/CaP
nanoparticle. J Biomed Nanotechnol. 14:1761–1772. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Klauber-DeMore N, Schulte BA and Wang GY:
Targeting MYC for triple-negative breast cancer treatment.
Oncoscience. 5:120–121. 2018.PubMed/NCBI
|
|
18
|
Li SG, Shi QW, Yuan LY, Qin LP, Wang Y,
Miao YQ, Chen Z, Ling CQ and Qin WX: C-Myc-dependent repression of
two oncogenic miRNA clusters contributes to triptolide-induced cell
death in hepatocellular carcinoma cells. J Exp Clin Cancer Res.
37:512018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mao X, Tong J and Wang Y, Zhu Z, Yin Y and
Wang Y: Triptolide exhibits antitumor effects by reversing
hypermethylation of WIF1 in lung cancer cells. Mol Med Rep.
18:3041–3049. 2018.PubMed/NCBI
|
|
20
|
Hou ZY, Tong XP, Peng YB, Zhang BK and Yan
M: Broad targeting of triptolide to resistance and sensitization
for cancer therapy. Biomed Pharmacother. 104:771–780. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Beglyarova N, Banina E, Zhou Y,
Mukhamadeeva R, Andrianov G, Bobrov E, Lysenko E, Skobeleva N,
Gabitova L, Restifo D, et al: Screening of conditionally
reprogrammed patient-derived carcinoma cells identifies ERCC3-MYC
interactions as a target in pancreatic cancer. Clin Cancer Res.
22:6153–6163. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Meng C, Zhu H, Song H, Wang Z, Huang G, Li
D, Ma Z and Ma J, Qin Q, Sun X and Ma J: Targets and molecular
mechanisms of triptolide in cancer therapy. Chin J Cancer Res.
26:622–626. 2014.PubMed/NCBI
|
|
23
|
Chang L, Lei X, Qin YU, Zhang X, Jin H,
Wang C, Wang X, Li G, Tan C and Su J: MicroRNA-133b inhibits cell
migration and invasion by targeting matrix metalloproteinase 14 in
glioblastoma. Oncol Lett. 10:2781–2786. 2015. View Article : Google Scholar
|
|
24
|
Li Z, Jiao X, Wang C, Shirley LA, Elsaleh
H, Dahl O, Wang M, Soutoglou E, Knudsen ES and Pestell RG:
Alternative cyclin D1 splice forms differentially regulate the DNA
damage response. Cancer Res. 70:8802–8811. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lamb J, Ramaswamy S, Ford HL, Contreras B,
Martinez RV, Kittrell FS, Zahnow CA, Patterson N, Golub TR and Ewen
ME: A mechanism of cyclin D1 action encoded in the patterns of gene
expression in human cancer. Cell. 114:323–334. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dremina ES, Sharov VS and Schöneich C:
Heat-shock proteins attenuate SERCA inactivation by the
anti-apoptotic protein Bcl-2: Possible implications for the ER
Ca2+-mediated apoptosis. Biochem J. 444:127–139. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Duan S, Tsai Y, Keng P and Chen Y, Lee SO
and Chen Y: IL-6 signaling contributes to cisplatin resistance in
non-small cell lung cancer via the up-regulation of anti-apoptotic
and DNA repair associated molecules. Oncotarget. 6:27651–27660.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ham IH, Oh HJ, Jin H, Bae CA, Jeon SM,
Choi KS, Son SY, Han SU, Brekken RA, Lee D and Hur H: Targeting
interleukin-6 as a strategy to overcome stroma-induced resistance
to chemotherapy in gastric cancer. Mol Cancer. 18:682019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sharma SB and Gupta R: Drug development
from natural resource: A systematic approach. Mini Rev Med Chem.
15:52–57. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yan P and Sun X: Triptolide: A new star
for treating human malignancies. J Cancer Res Ther. 14(Suppl):
S271–S275. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jiang N, Dong XP, Zhang SL, You QY, Jiang
XT and Zhao XG: Triptolide reverses the Taxol resistance of lung
adenocarcinoma by inhibiting the NF-κB signaling pathway and the
expression of NF-κB-regulated drug-resistant genes. Mol Med Rep.
13:153–159. 2016. View Article : Google Scholar
|
|
32
|
Teng F, Xu Z, Chen J, Zheng G, Zheng G, Lv
H, Wang Y, Wang L and Cheng X: DUSP1 induces apatinib resistance by
activating the MAPK pathway in gastric cancer. Oncol Rep.
40:1203–1222. 2018.PubMed/NCBI
|
|
33
|
Li X, Lu Q, Xie W, Wang Y and Wang G:
Anti-tumor effects of triptolide on angiogenesis and cell apoptosis
in osteosarcoma cells by inducing autophagy via repressing
Wnt/β-catenin signaling. Biochem Biophys Res Commun. 496:443–449.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fan D, He X, Bian Y, Guo Q, Zheng K, Zhao
Y, Lu C, Liu B, Xu X, Zhang G and Lu A: Triptolide modulates TREM-1
signal pathway to inhibit the inflammatory response in rheumatoid
arthritis. Int J Mol Sci. 17:4982016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen J, Gao J, Yang J, Zhang Y and Wang L:
Effect of triptolide on the regulation of ATP-binding cassette
transporter A1 expression in lipopolysaccharide-induced acute lung
injury of rats. Mol Med Rep. 10:3015–3020. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vispé S, DeVries L, Créancier L, Besse J,
Bréand S, Hobson DJ, Svejstrup JQ, Annereau JP, Cussac D, Dumontet
C, et al: Triptolide is an inhibitor of RNA polymerase I and
II-dependent transcription leading predominantly to down-regulation
of short-lived mRNA. Mol Cancer Ther. 8:2780–2790. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Z, Jin H, Xu R, Mei Q and Fan D:
Triptolide downregulates Rac1 and the JAK/STAT3 pathway and
inhibits colitis-related colon cancer progression. Exp Mol Med.
41:717–727. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ma J, Zhao Z, Wu K, Xu Z and Liu K: MCL-1
is the key target of adjuvant chemotherapy to reverse the
cisplatin-resistance in NSCLC. Gene. 587:147–154. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Oliveira A, Beyer G, Chugh R, Skube SJ,
Majumder K, Banerjee S, Sangwan V, Li L, Dawra R, Subramanian S, et
al: Triptolide abrogates growth of colon cancer and induces cell
cycle arrest by inhibiting transcriptional activation of E2F. Lab
Invest. 95:648–659. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Biliran H Jr, Wang Y, Banerjee S, Xu H,
Heng H, Thakur A, Bollig A, Sarkar FH and Liao JD: Overexpression
of cyclin D1 promotes tumor cell growth and confers resistance to
cispl-atin-mediated apoptosis in an elastase-myc
transgene-expressing pancreatic tumor cell line. Clin Cancer Res.
11:6075–6086. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Aggarwal P, Vaites LP, Kim JK, Mellert H,
Gurung B, Nakagawa H, Herlyn M, Hua X, Rustgi AK, McMahon SB and
Diehl JA: Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression
and triggers neoplastic growth via activation of the PRMT5
methyltransferase. Cancer Cell. 18:329–340. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shields BJ, Hauser C, Bukczynska PE, Court
NW and Tiganis T: DNA replication stalling attenuates tyrosine
kinase signaling to suppress S phase progression. Cancer Cell.
14:166–179. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jirawatnotai S, Hu Y, Michowski W, Elias
JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB,
et al: A function for cyclin D1 in DNA repair uncovered by protein
interactome analyses in human cancers. Nature. 474:230–234. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yao Z, Fenoglio S, Gao DC, Camiolo M,
Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V,
et al: TGF-beta IL-6 axis mediates selective and adaptive
mechanisms of resistance to molecular targeted therapy in lung
cancer. Proc Natl Acad Sci USA. 107:15535–15540. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang S, Long S, Xiao S, Wu W and Hann SS:
Decoction of chinese herbal medicine fuzheng kang-ai induces lung
cancer cell apoptosis via STAT3/Bcl-2/caspase-3 pathway. Evid Based
Complement Alternat Med. 2018:85679052018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yu H and Jove R: The stats of cancer-new
molecular targets come of age. Nat Rev Cancer. 4:97–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dechow TN, Pedranzini L, Leitch A, Leslie
K, Gerald WL, Linkov I and Bromberg JF: Requirement of matrix
metallopro-teinase-9 for the transformation of human mammary
epithelial cells by Stat3-C. Proc Natl Acad Sci USA.
101:10602–10607. 2004. View Article : Google Scholar
|
|
48
|
El-Badrawy MK, Yousef AM, Shaalan D and
Elsamanoudy AZ: Matrix metalloproteinase-9 expression in lung
cancer patients and its relation to serum mmp-9 activity,
pathologic type, and prognosis. J Bronchology Interv Pulmonol.
21:327–334. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Backstrom JR and Tökés ZA: The 84-kDa form
of human matrix metalloproteinase-9 degrades substance P and
gelatin. J Neurochem. 64:1312–1318. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ao N and Liu Y, Bian X, Feng H and Liu Y:
Ubiquitin-specific peptidase 22 inhibits colon cancer cell invasion
by suppressing the signal transducer and activator of transcription
3/matrix metalloproteinase 9 pathway. Mol Med Rep. 12:2107–2113.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kim JH and Park B: Triptolide blocks the
STAT3 signaling pathway through induction of protein tyrosine
phosphatase SHP-1 in multiple myeloma cells. Int J Mol Med.
40:1566–1572. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Niu F, Li Y, Lai FF, Ni L, Ji M, Jin J,
Yang HZ, Wang C, Zhang DM and Chen XG: LB-1 exerts antitumor
activity in pancreatic cancer by inhibiting HIF-1α and Stat3
signaling. J Cell Physiol. 230:2212–2223. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kim SM, Kwon OJ, Hong YK, Kim JH, Solca F,
Ha SJ, Soo RA, Christensen JG, Lee JH and Cho BC: Activation of
IL-6R/JAK1/STAT3 signaling induces de novo resistance to
irreversible EGFR inhibitors in non-small cell lung cancer with
T790M resistance mutation. Mol Cancer Ther. 11:2254–2264. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Maji S, Shriwas O, Samal SK, Priyadarshini
M, Rath R, Panda S, Das Majumdar SK, Muduly DK and Dash R: STAT3-
and GSK3β-mediated Mcl-1 regulation modulates TPF resistance in
oral squamous cell carcinoma. Carcinogenesis. 40:173–183. 2019.
View Article : Google Scholar
|