Introduction
Non-small cell lung cancer (NSCLC) accounts for
approximately 85% of lung cancer cases and 10-30% of patients with
NSCLC bear activating mutations in the epidermal growth factor
receptor gene (EGFR) (1).
A prospective, multinational PIONEER study confirmed that there is
an even higher EGFR mutation frequency (51.4% overall) in
tumors from Asian patients with lung adenocarcinoma compared with
their Caucasian counterparts (2).
Almost 75% of patients with activated EGFR mutations have a
longer median overall survival and better response rates when they
are treated with an EGFR-tyrosine kinase inhibitor (EGFR-TKI)
compared with only traditional platinum-based chemotherapy
(3-6). Regretfully, most invariably develop
or 'acquire' resistance to these agents during the treatment course
(7).
Icotinib (also known as BPI-2009H and Conmana) is
the first oral quinazoline compound that has an established
survival benefit and fewer side effects in Chinese patients with
NSCLC (8,9). A network meta-analysis demonstrated
that icotinib shares equivalent efficacies with erlotinib,
gefitinib and afatinib, but has a lower toxicity (10). The double-blind, head-to-head
phase III ICOGEN study indicated that icotinib demonstrated an
improved median progression-free survival compared with gefitinib
and was also associated with fewer adverse events compared
gefitinib when considering all grades of reactions together
(11).
By acting on signaling pathways, including
PI3K-AKT-mTOR, Ras-Raf-MEK-ERK and STAT, an EGFR-TKI regulates cell
proliferation, apoptosis, invasion, migration and angiogenesis
(12). A growing body of evidence
has elucidated the mechanism of EGFR-TKI resistance (13). Although almost half of all TKI
resistance is caused by a secondary T790M mutation (14), the abnormal activation,
independent of EGFR, of EGFR's downstream signaling pathways, such
as PI3K-AKT-mTOR (15), also
contributes to the acquisition of resistance.
The protein kinase casein kinase II (CK2) is an
evolutionary, highly conserved serine/threonine kinase that
phosphorylates and interacts with more than 300 proteins (16). It is noteworthy that several
members of the EGFR downstream singling pathways (Fig. 1), including PTEN, ribosomal
protein S6 kinase β-1 (S6) and AKT within the PI3K-AKT-mTOR
signaling pathway, have been previously reported to be
phosphorylated or modulated by CK2 (17,18). Quinalizarin is known as a potent,
selective and cell-permeable inhibitor of CK2 (19). A previous study revealed that
quinalizarin reduced cell viability, suppressed migration and
accelerated apoptosis in different human lung cancer cell lines
with wild-type EGFR and EGFR-resistant mutations, as well as
for those with an EGFR-sensitive mutation (20). Therefore, the authors of the
current study hypothesized that a top-down inhibition of EGFR,
combined with the lateral suppression of its multiple downstream
pathways by targeting CK2 would create a pharmacologic synthetic
lethal event and result in the resistance to EGFR-TKIs being
overcome. The purpose of the current study was to investigate the
effects of icotinib and quinalizarin on proliferation and apoptosis
in four human lung adenocarcinoma cell lines (A549, HCC827, H1650
and H1975) with different EGFR genotypes, as well as to
reveal quinalizarin's underlying mechanisms.
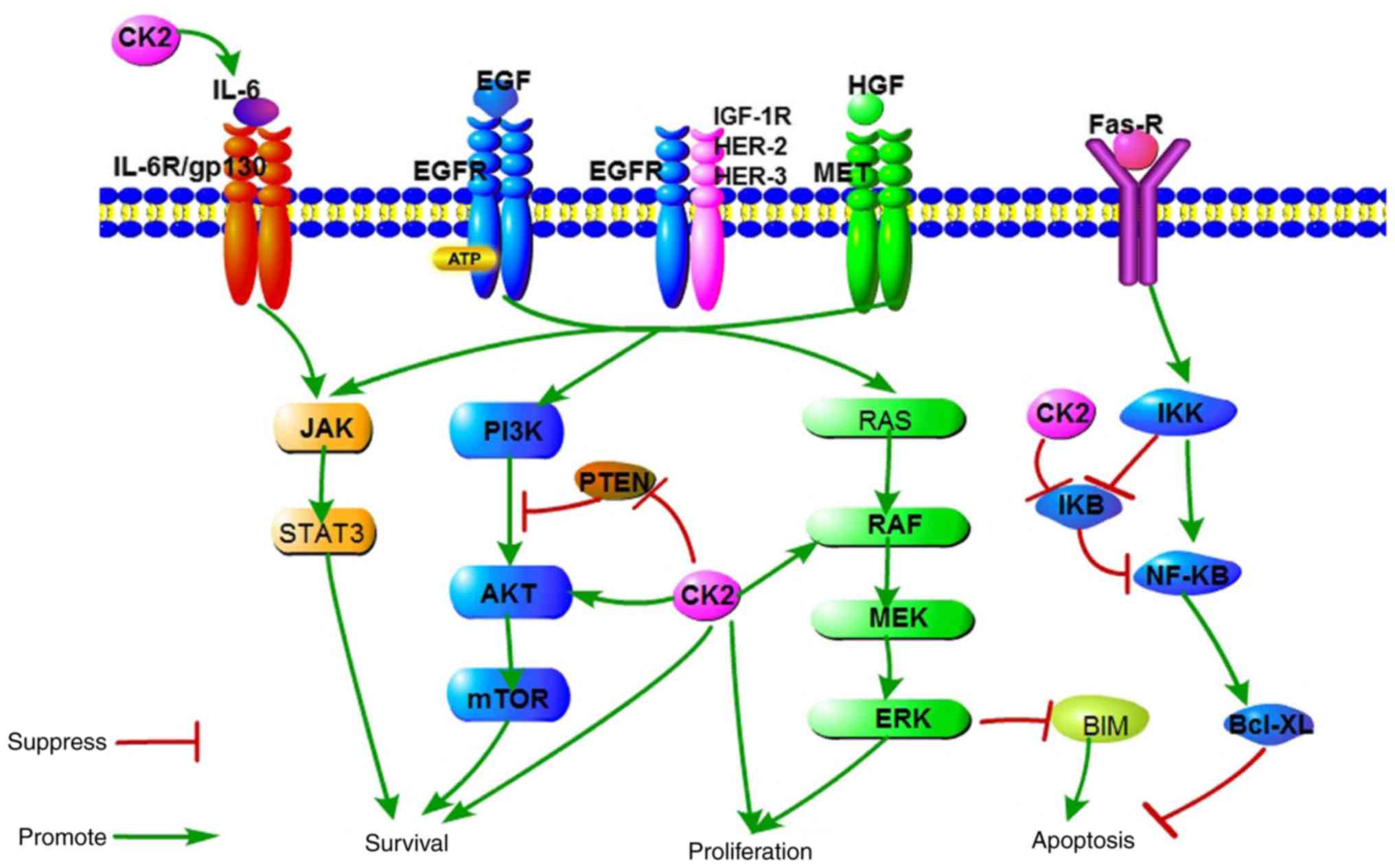 | Figure 1A schematic representation of
signaling pathways responsible for cell survival, proliferation and
apoptosis, which are regulated by EGFR and CK2. CK2, casein kinase
II; EGF, epidermal growth factor; EGFR, epidermal growth factor
receptor; MEK, dual specificity mitogen-activated protein kinase
kinase; IκB, NF-κ-B inhibitor; IKK, IκB kinase; BIM, Bcl-2-like
protein 11; IL-6R, interleukin-6 receptor; IGF-1R, insulin-like
growth factor 1 receptor; HER, receptor tyrosine-protein kinase
erbB-4; HGF, hepatocyte growth factor; MET, hepatocyte growth
factor receptor. |
Materials and methods
Cell lines
Human lung adenocarcinoma A549 (wild-type
EGFR), HCC827 (EGFR E716-A750del), NCI-H1975
(EGFR L858R+T790M), NCI-H1650 (EGFR E716-A750del and
PTEN lost) cells were purchased from the American Type Culture
Collection (Manassas, VA, USA) and were used within 3 months of
resuscitation. The cells were cultured in RPMI 1640 supplemented
with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin (all
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), which
will be termed culture medium henceforth, in a humidified
atmosphere of 5% CO2 and 95% air at 37°C.
Reagents
Icotinib was from Betta Pharmaceuticals Co., Ltd.
(Hangzhou, China). The specific CK2 inhibitor quinalizarin was
purchased from Merck KGaA (Darmstadt, Germany).
Cell viability assays
For each cell line, the cells were harvested at the
logarithmic phase and were then seeded into a 96-well plate at a
density of 6,000 cells per well with 100 µl culture medium.
The cells were cultured at 37°C with 5% CO2 overnight,
and then various concentrations of icotinib and/or quinalizarin
were added to each well (6.25, 12.5, 25, 50 and 100 µM),
with a total of 200 µl culture medium and were further
incubated for 48 h. Thereafter, 20 µl of the MTT solution
(Wuhan Boster Biological Technology, Ltd., Wuhan, China) was added
into each well. A total of 4 h later, 150 µl dimethyl
sulfoxide (DMSO) was used to dissolve the crystals. The plates were
placed on a low-speed shaker for 10 min to fully dissolve the
crystals. The optical density (OD) of each well was read at a
wavelength of 490 nm using a microplate reader. A cell viability
curve was plotted using the concentration as the x-axis and the
cell viability rate as the y-axis. The blank control group was
incubated in culture medium without treatment. The values were
calculated as follows: Cell survival rate=(OD value of experimental
well-OD value of blank control well)/(OD value of no drug control
well-OD value of blank control well) ×100%.
Each experiment was independently repeated at least
three times for each cell line. The dose-effect relationship was
fitted using curve regression models to obtain the 50% inhibitory
concentration of 6.25, 12.5, 25, 50 and 100 µM icotinib,
quinalizarin, and icotinib and quinalizarin combined by GraphPad
Prism software (version 5.0; GraphPad Software, Inc., San Diego,
CA, USA).
Apoptosis assays
Annexin V-enhanced green fluorescent protein
(EGFP)-propidium iodide (PI) staining and flow cytometry were
performed to detect cell apoptosis. Cells
(1×104/cm2) in the logarithmic growth phase
were plated into 12-well plates with fresh culture medium. After 24
h, for attachment, the cells were washed with PBS two to three
times, and then 6.25, 12.5, 25, 50 and 100 µM icotinib
and/or quinalizarin were added to EGFR-resistant cell lines. The
cells were cultured with a trypsin enzyme digesting technique 48 h
later and cell apoptosis was assessed using an Annexin V-EGFP
Apoptosis Detection kit (cat. no. C1067M; Beyotime Institute of
Biotechnology, Shanghai, China) following the product
specifications.
Cell morphology
H1650, H1975 or A549 cells
(1×104/cm2) were seeded into a 6-well plate
for 24 h, washed with PBS two to three times, and then 6.25, 12.5,
25, 50 and 100 µM icotinib and/or quinalizarin were added.
After 48 h, the cells were fixed with 4% paraformaldehyde at room
temperature for 20 min, morphology was observed by a light
microscope (IX71; Olympus Optical, Tokyo, Japan) at a magnification
of ×100 and images were taken.
Western blot analysis
The cells (1×104/cm2) were
seeded into a 6-well plate for 24 h. Then, different concentrations
of drugs (50 µM icotinib or/and quinalizarin) were applied
to the cells and they were further incubated 24 h. Thereafter, the
cells were lysed in radioimmunoprecipitation assay buffer,
phenylmethylsulfonyl fluoride and a phosphatase inhibitor cocktail,
which were purchased from Wuhan Google Biological Technology Co.,
Ltd. (Wuhan, China). The protein concentration was measured on a
microplate reader according to the bicinchoninic acid method.
Protein (50 µg/lane) from the cell lysates were
electrophoresed by SDS-PAGE on a 12% gel and were transferred to a
polyvinylidene difluoride membrane. The membrane was blocked with
5% dry milk at room temperature for 1 h and was then incubated with
anti-p-ERK (cat. no. 4370S), anti-EGFR (cat. no. ab289; Abcam,
Cambridge, MA, USA), anti-CK2α (#212), anti-CK2β (#269), anti-AKT
(cat. no. 1081-1; Epitomics; Abcam), anti-p-AKT (pS473; cat. no.
2118-1; Epitomics; Abcam) anti-ERK (cat. no. BS1112; Bioworld
Technology, Inc., St. Louis Park, MN, USA), anti-p-EGFR (cat. no.
AF3048, Affinity), anti-p-forkhead box protein O1 (FoxO1; cat. no.
9461), anti-p-S6 (cat. no. 2211; both Cell Signaling Technology,
Inc.) or anti-β-actin (cat. no. sc-1616r; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) antibodies diluted at 1:1,000 in 5% bovine
serum albumin (BSA; cat. no. AR1006; Wuhan Boster Biological
Technology, Ltd.) at 4°C overnight. After washing with 1X PBS, the
membrane was incubated with horseradish peroxidase-conjugated
anti-mouse (cat. no. 5450-0011) or anti-rabbit (cat. no. 5220-0336)
secondary antibodies (Kirkegaard & Perry Laboratories; SeraCare
Life Sciences, Inc., Milford, MA, USA) diluted at 1:500 in 5% BSA
at room temperature for 1 h. The proteins were observed with
Western Blotting Luminol Reagent (sc-2048; Santa Cruz
Biotechnology, Inc.). The intensity of the bands underwent
densitometric analysis and they were calculated using AlphaEase FC
software (version 5.0; Alpha Innotech Corporation, San Leandro, CA,
USA). Anti-CK2α (#212) and anti-CK2β (#269) antibodies were
generated as described previously (21) and were kind gifts from Professor
Mathias Montenarh from Medical Biochemistry and Molecular Biology,
Saarland University (Homburg, Germany).
Statistical analysis
Data are presented as mean ± standard deviation. All
the statistical analyses were performed with two-way analysis of
variance followed by Bonferroni test using GraphPad Prism software
(version 5.0). Statistical diagrams were generated using GraphPad
Prism software (version 5.0). P<0.05 indicated that the
difference between groups was statistically significant.
Results
Cell lines with different EGFR genotypes
have different basal expressions of CK2 and EGFR
To explore whether targeting CK2 eliminates
icotinib-resistance, the basal protein expression of the catalytic
CK2 subunit and EGFR were assessed in the four aforementioned lung
adenocarcinoma cell lines with different EGFR genotypes,
namely, A549 (wild-type EGFR), HCC827 (EGFR
E716-A750del), NCI-H1975 (EGFR L858R+T790M) and NCI-H1650
(EGFR E716-A750del and PTEN lost). HCC827 has been
demonstrated to be sensitive to EGFR-TKIs, while A549, NCI-H1975
and NCI-H1650 are known as EGFR-TKI resistant cell lines (22,23).
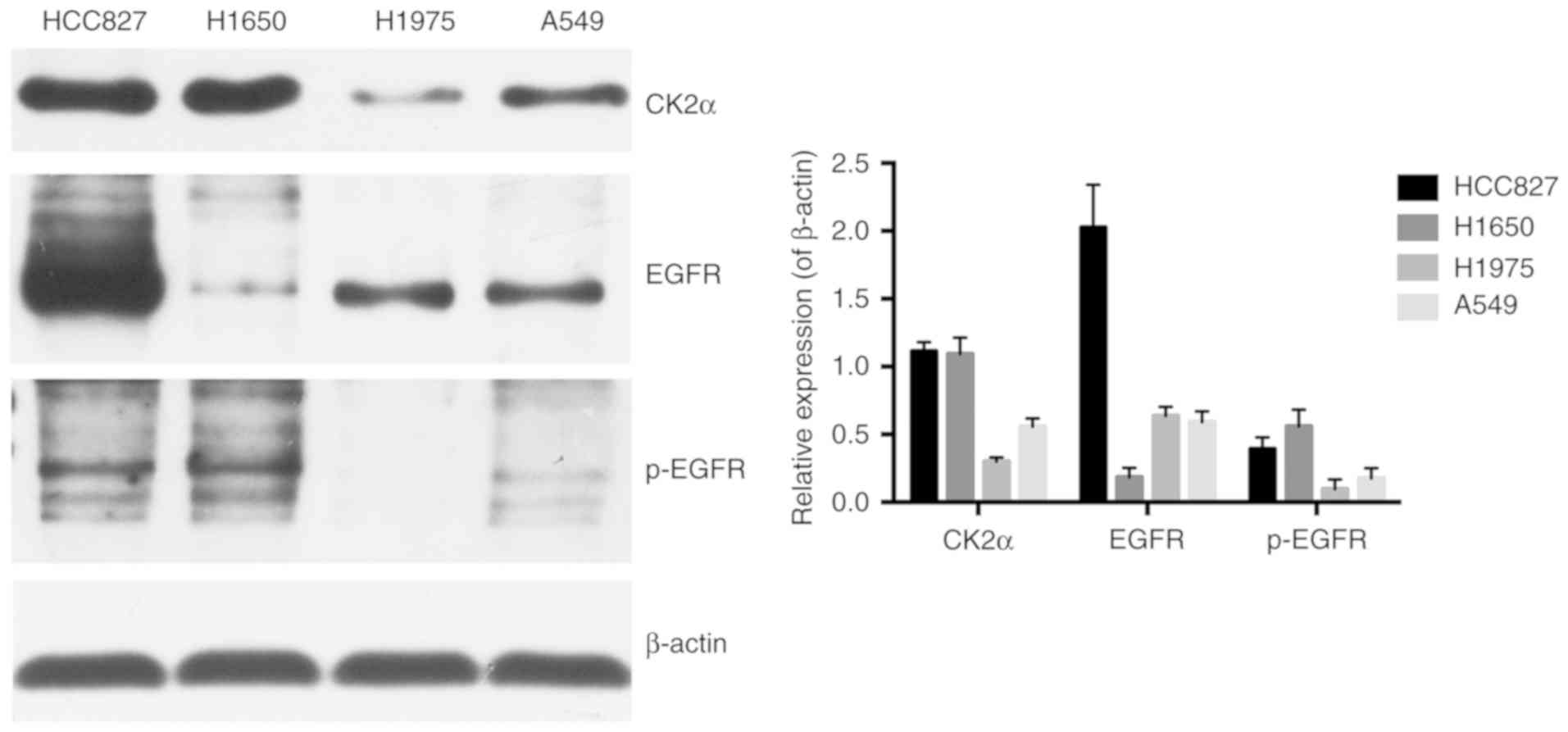
As presented in Fig.
2, the protein expression of the CK2 catalytic subunit α was
strong and was clearly overexpressed in the HCC827 and NCI-H1650
cells compared with the A549 cells. It was notable that in the
NCI-H1975 cells, there was markedly less CK2α protein expression
compared with the other cells. Total EGFR and p-EGFR expression
levels were also detected. The results revealed that EGFR
expression within those four cell lines differed greatly, with the
HCC827 cells containing the highest amount of total EGFR and the
second highest amount of the phosphorylated form of EGFR. On the
other hand, the lowest amount of total EGFR expression and the
highest amount of the phosphorylated form were observed in
NCI-H1650 cells. In addition, total EGFR expression in A549 and
NCI-H1975 cells was relatively the same, which were markedly lower
compared with in the HCC827 cells and markedly higher compared with
the NCI-H1650 cells. The amount of phosphorylated form of EGFR in
A549 and NCI-H1975 cells was clearly lower when compared with the
HCC827 and NCI-H1650 cells, with the least amount in the NCI-H1975
cells.
Sensitivity to icotinib is different in
various lung adenocarcinoma cell lines and is closely associated
with the EGFR genotype
To test the sensitivity of icotinib in different
human lung adenocarcinoma cell lines with various EGFR
genotypes, the cells were exposed to various concentrations of
icotinib for 48 h. Then, the MTT assay was used to determine the
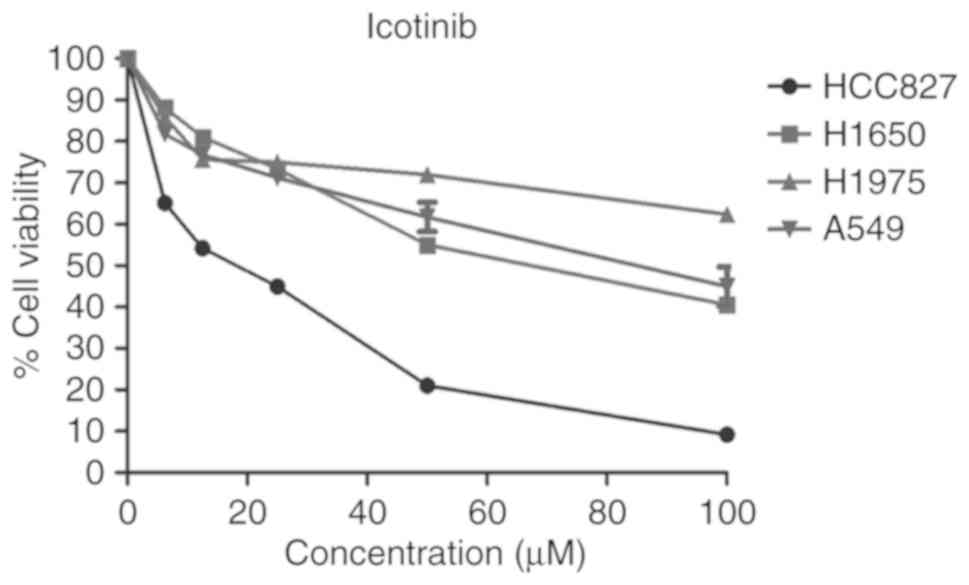
cell viability. Fig. 3
demonstrates that the HCC827 cells were sensitive to icotinib,
while the other three cell lines were resistant to icotinib. The
sensitivity of the cells to icotinib depended on their EGFR
genotypes, as described previously. Moreover, the levels of EGFR
and p-EGFR were also closely related with the different EGFR
genotypes. The H1975 cells, which are EGFR-mutant on both L858R and
T790M, showed the lowest level of p-EGFR (Fig. 2) and the least sensitivity to the
icotinib (Fig. 3).
Quinalizarin inhibits the cell viability
of all four cell lines and promotes apoptosis in cell lines with
wild-type EGFR and EGFR-resistant mutations
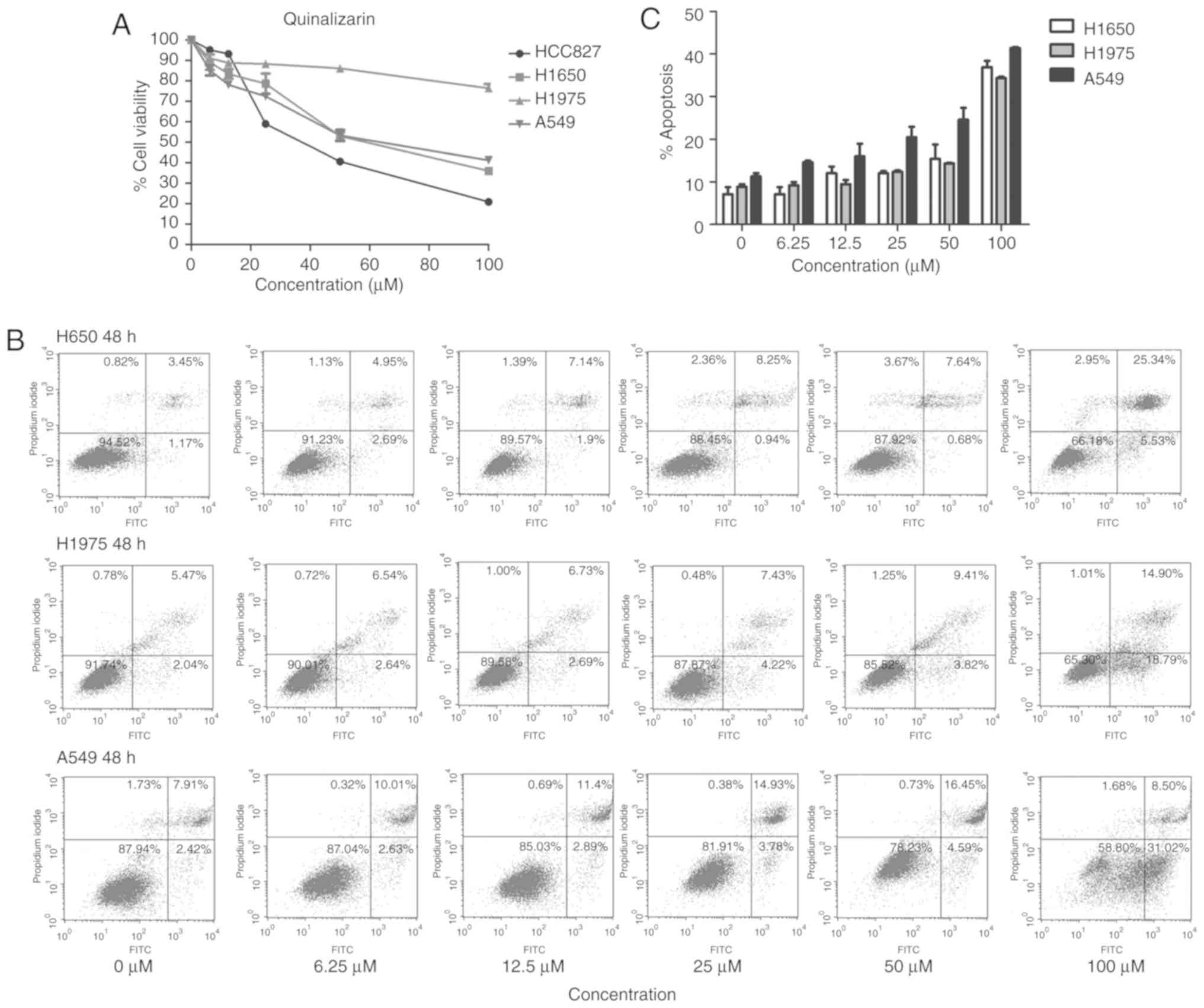
The MTT assay was also used to explore the influence
of quinalizarin on these four cell lines; the results showed that
quinalizarin suppressed cell viability. Additionally, CK2α
expression was negatively associated with cell viability (Figs. 2 and 4A). The highest inhibition rate was
found in the HCC827 cells, which are known to bear an
EGFR-sensitive mutation. Thereafter, the total apoptosis rates of
the resistant cell lines were tested by flow cytometry after
treatment with quinalizarin for 48 h (Fig. 4B). The result showed that
quinalizarin promoted apoptosis in H1650, H1975 and A549 cells,
with the strongest effect being shown in the A549 cells bearing the
EGFR wild-type genotype (Fig.
4C).
Quinalizarin enhances the suppression of
cell viability mediated by icotinib in both primary and secondary
drug resistant cell lines
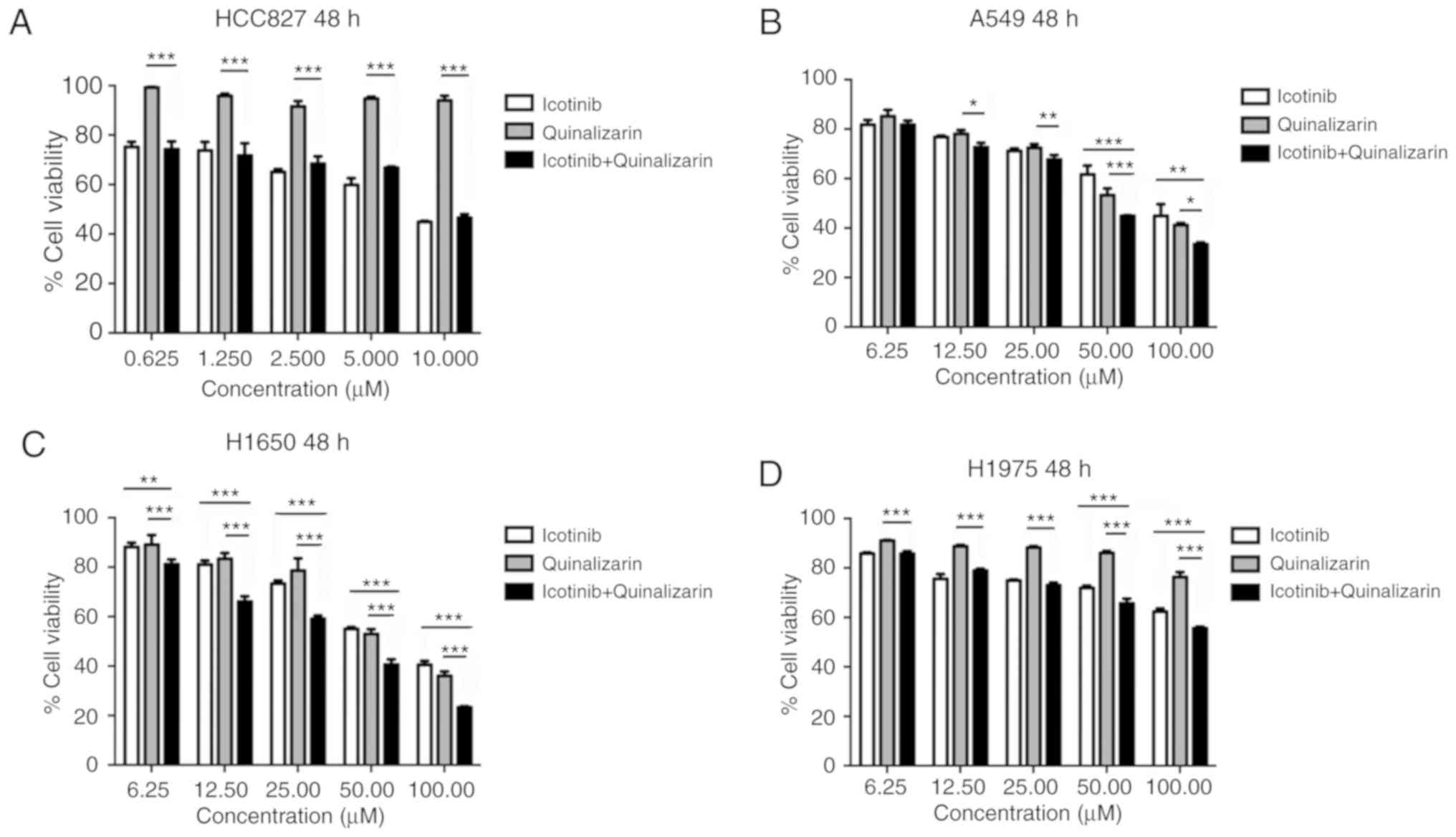
From the above experiments, the authors of the
current study found that quinalizarin and icotinib, individually,
suppressed the viability of four different lung adenocarcinoma
cells. The cell viability after the cells were treated with a
combination of quinalizarin and icotinib was then evaluated
(Fig. 5). Fig. 5A shows that quinalizarin did not
enhance the reduction of cell viability mediated by icotinib in the
HCC827 cells. However, quinalizarin enhanced the suppression of
cell viability mediated by icotinib in A549, H1650 and H1975 cell
lines, which were shown to be primary or secondary EGFR-TKI
resistant cell lines. When treated with 100 µM quinalizarin
and icotinib, the viability of A549 cells were significantly lower
than that of cells treated with quinalizarin (P<0.05) or
icotinib (P<0.01) alone (Fig.
5B). When treated with 100 µM quinalizarin and icotinib,
the viability of H1650 and H1975 cells were significantly lower
than that of cells treated with quinalizarin or icotinib alone (all
P<0.001; Fig. 5C and D).
Moreover, the suppression effect was more prominent in the H1650
cells.
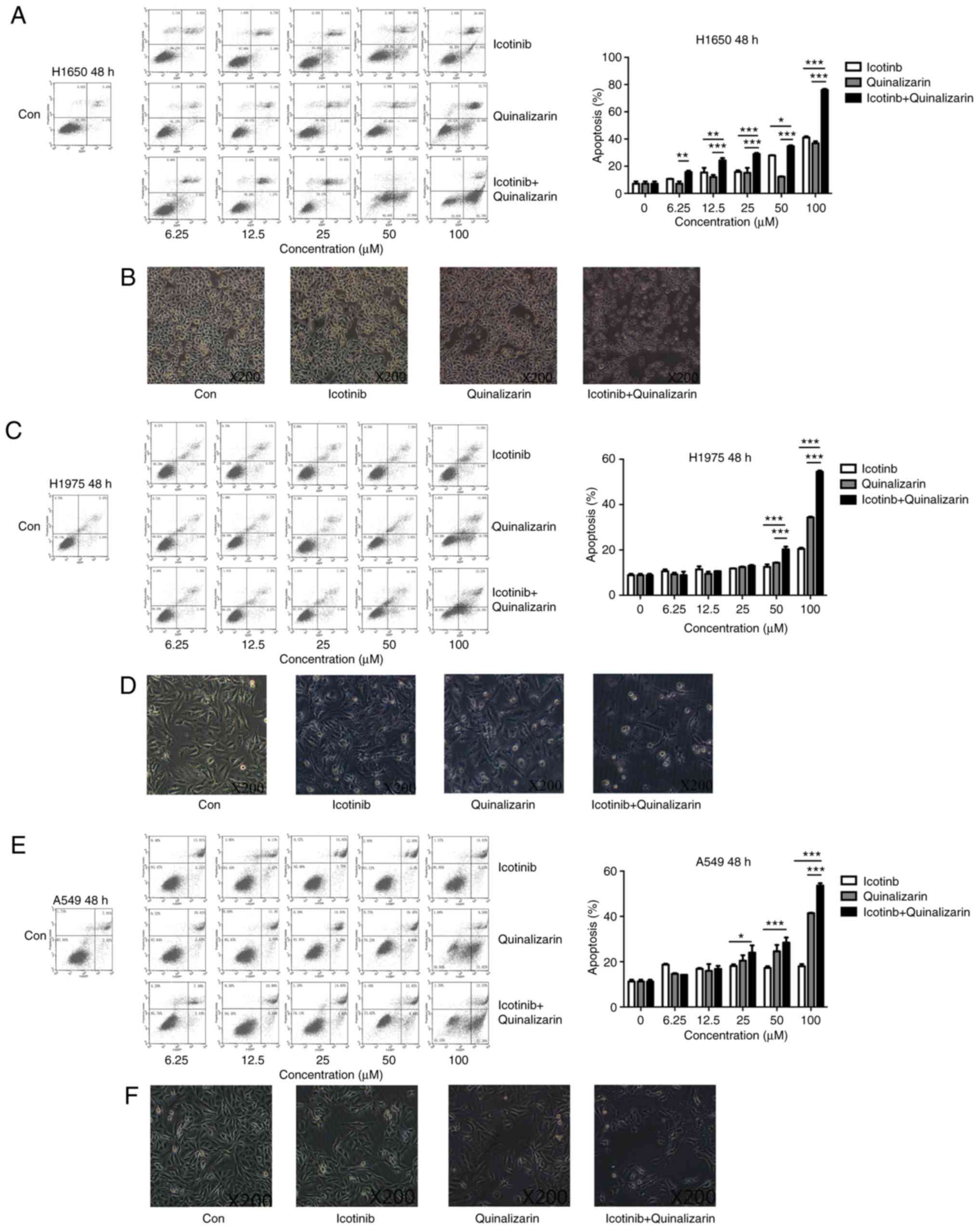
Quinalizarin increases the apoptosis rate
of EGFR-resistant cells when treated together with icotinib
As described previously, quinalizarin enhanced the
suppression of cell proliferation mediated by icotinib in both
primary and secondary drug resistant cell lines. To determine how
these effects occur, flow cytometry was performed to assess the
apoptosis rates after the cells were treated with quinalizarin and
icotinib. Fig. 6A shows that the
combination of 100 µM quinalizarin and icotinib
significantly increased the total apoptosis rate of the H1650 cells
compared with quinalizarin or icotinib alone (both P<0.001). In
addition, Fig. 6B shows that
H1650 cell morphology changed after the cells were treated with
both quinalizarin and icotinib. The cells treated with the
combination of the drugs were small and round, and some of them
showed obvious vacuolization. These changes are considered signs of
cell apoptosis. Thus, the authors of the current study deduced that
quinalizarin, together with icotinib, mediated cell apoptosis and
increased the apoptosis rate in the H1650 cells. Similar results
were also observed in H1975 (Fig. 6C
and D) and A549 (Fig. 6E and
F).
The total apoptosis rate and cell morphology in
H1650, H1975 and A549 were shown in Fig. 6. Data showed that the combination
treatment enhanced the total apoptosis rate compared with
quinalizarin or icotinib alone in all cell lines (P<0.0001 at
100 µM). The results also suggested that the largest
increase in the total apoptosis rate was in the H1650 cells after
they were treated with both drugs.
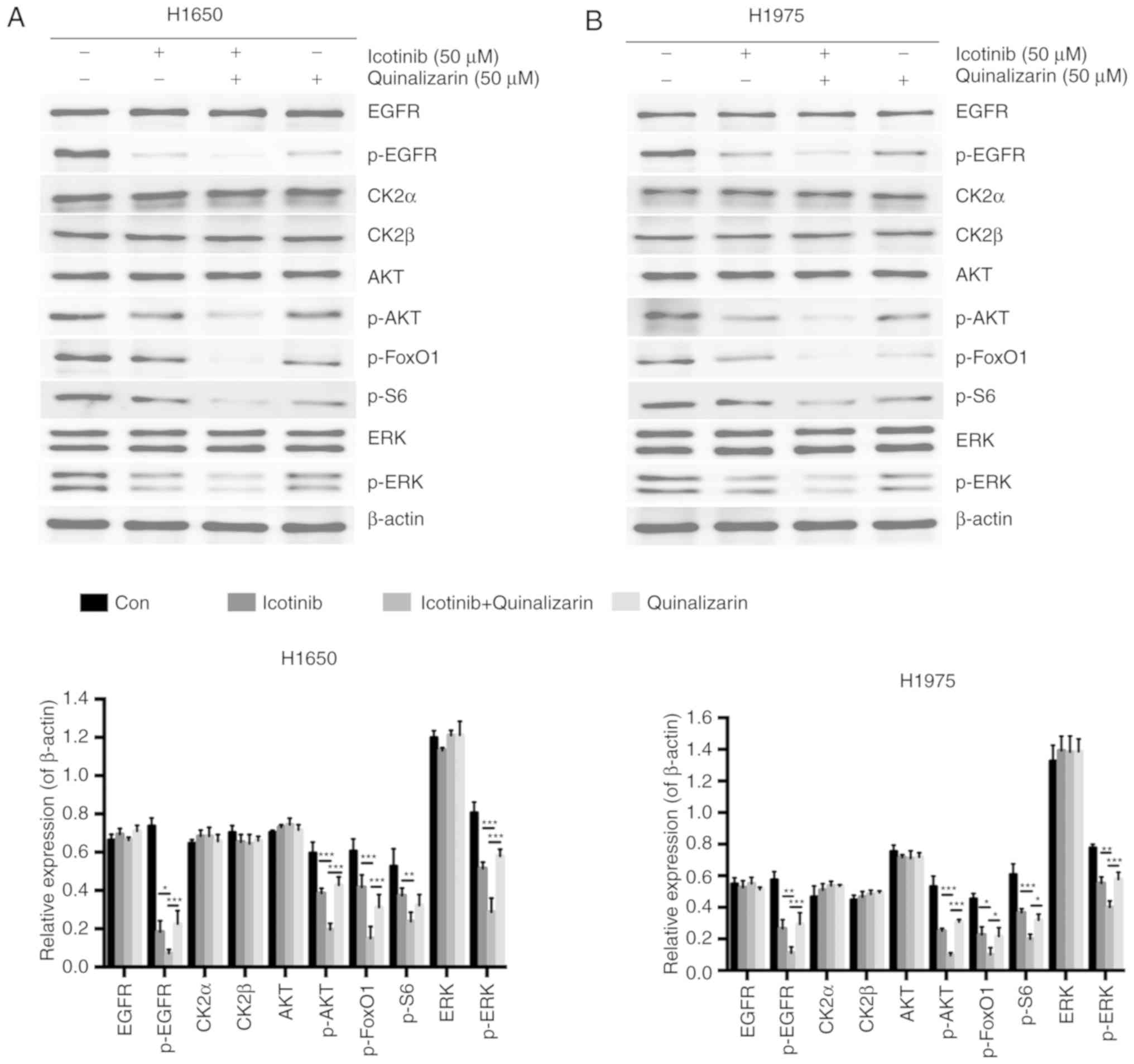
Quinalizarin and icotinib together
decrease AKT and ERK protein expression in EGFR-resistant cell
lines
It was shown earlier that quinalizarin may sensitize
lung adenocar-cinoma cancer cells to icotinib by inhibiting cell
viability and promoting apoptosis. To further explore the
underlying mechanisms, a western blot analysis was performed to
assess whether the combination of these two drugs resulted in a
reduction of the EGFR signaling pathways. A previous study showed
that the efficacy of EGFR-TKIs was partly due to the modulation of
the PI3K-AKT-mTOR and the Ras-Raf-MEK-ERK signaling pathways
(12). Several specific CK2
inhibitors have also already been shown to work with these two
pathways (24,25). Since AKT and ERK are regarded as
key members of these pathways, the authors of the current study
mainly evaluated the levels of EGFR, CK2α, CK2β, AKT and ERK, and
the corresponding phosphorylated forms of EGFR, AKT, ERK, FoxO1 and
S6 (Fig. 7); FoxO1 and S6 are
downstream of AKT (26).
The results showed that p-EGFR was significantly
down-regulated by combining 50 µM quinalizarin and 50
µM icotinib compared with quinalizarin alone (P<0.05 in
H1650 and P<0.01 in H1975) and icotinib alone (both P<0.001).
The combination treatment also significantly decreased the
expression of p-AKT in both cells lines (all P<0.001), p-ERK in
H1650 cells (both P<0.001) and in H1975 cells (P<0.01 for
icotinib alone and P<0.001 for quinalizarin alone), p-FoxO1 in
H1650 cells (both P<0.001) and in H1975 cells (both P<0.05),
and p-S6 in H1650 (P<0.01 for icotinib alone) and in H1975 cells
(P<0.05 for quinalizarin alone and P<0.001 for icotinib
alone).
Discussion
EGFR-TKIs provide a way to improve the treatment
outcomes for lethal NSCLC (4-6).
However, the critical problems related to EGFR-TKIs is unavoidable
drug resistance, which prevents patients from further benefits
(22). The authors of the current
study illustrated several ways in which EGFR-TKIs work and
discussed the possible mechanisms devoted to the drug
resistance.
In accordance with a great body of literature, the
current study found that the protein kinase CK2 was almost
connected, either indirect or directly, to all analysed pathways,
including PTEN, S6 and AKT within the PI3K-AKT-mTOR signaling
pathway (17,18) as well as RAF within the Ras-Raf
MEK-ERK downstream pathway (27).
CK2 has recently emerged as a promising target for lung cancer
therapy (28). It is a ubiquitous
serine/threonine protein kinase that has been demonstrated to be
involved in cell growth and proliferation, as well as in inhibiting
apoptosis (16,25). Other studies have revealed that
the activity of CK2 is up to 2-3-fold higher in lung cancer cells
compared with normal lung tissues (20,29). Hung et al (30) revealed that the CK2 inhibitor
suppressed lung cancer growth in a murine xenograft model. Li et
al (31) found that the CK2
inhibitor quinalizarin reduced cell proliferation and induced
apoptosis in NSCLC cells. Kim and Kim (32) reported that the CK2 inhibitor
CX-4945 suppressed the proliferative activity of human cancer
cells. Ku et al (33)
showed that CX-4945 induced cell migration and suppressed
metastasis in A549 human lung cancer cells. Di Maira et al
(34) demonstrated that the
inhibition of CK2 reversed the multidrug resistance in a CEM cell
line with a high CK2 level. Thus, the authors of the current study
deduced that CK2 inhibitors might not only have antineoplastic
effects but also partly reverse the EGFR-TKI-resistance of human
lung adenocarcinoma cell lines.
The current study showed that the sensitivity of
lung adenocarcinoma cells to icotinib was different between the
cell lines and was associated with various EGFR mutation
types. The cells with an EGFR-sensitive mutation (HCC827) were more
sensitive to icotinib than those that possessed wild-type
EGFR (A549) and those with an EGFR-resistant mutation (H1650
and H1975). The reason that icotinib prevented the tumor from
growing is because it competed for EGFR binding and inhibited the
downstream signaling pathways, such as PI3K-AKT-mTOR,
Ras-Raf-Mek-ERK and STAT. The current study demonstrated that the
downstream signaling pathways were normally overexpressed or
activated in the EGFR-sensitive mutant cell line (HCC827) and these
pathways play a prominent role in cell growth (35). The inhibition of these pathways
showed a more remarkable anti-tumor effect and HCC827 cells were
more sensitive to icotinib. The A549, which possesses wild-type
EGFR, has fewer activated basal pathways and is less
sensitive to EGFR-TKIs (36,37). The H1975 cell line possesses a
T790M mutation that changes the binding site on EGFR and weakens
the competition of EGFR-TKIs (23). The H1650 cell line does not have
PTEN expression, which negatively regulates the PI3K-AKT-mTOR
signaling pathway (38). In the
current study, it was not surprising to find that H1975 and H1650
cells were resistant to icotinib, an EGFR-TKI.
In the current study, quinalizarin inhibited cell
viabilty in the cell lines that possessed wild-type EGFR and
were EGFR-resistant, as well as in the EGFR-sensitive cells.
Quinalizarin also promoted apoptosis in the cell lines that
possessed wild-type EGFR and were EGFR-resistant. CK2 plays
an important role in the activation of the Ras-Raf-MEK-ERK
signaling pathway (39,40). CK2 inhibitors suppress the
PI3K-AKT-mTOR signaling pathway and promote apoptosis (24,41). The Ras-Raf-MEK-ERK and
PI3K-AKT-mTOR signaling pathways are important for proliferation
and apoptosis (42-44), which is why quinalizarin inhibited
cell proliferation and promote apoptosis in these cells.
Additionally, in the current study, quinalizarin
synergized with anti-tumor effects mediated by icotinib. The
combination of quinalizarin with icotinib had a greater inhibitory
effect on cell viability and further promoted apoptosis in the lung
adenocarcinoma cells that were EGFR-resistant. The results of the
current study were in line with the previous literature, which
indicated that CK2 inhibition suppresses the PI3K-AKT-mTOR
signaling pathway and promotes apoptosis by itself (41). Bliesath et al (45) insisted that the combination of
EGFR-TKIs and CX-4945 shows better antitumor effects by suppressing
the PI3K-AKT-mTOR signaling pathway compared with EGFR-TKI alone.
In line with the study, the current results showed again that
combining a CK2 inhibitor and icotinib together had better
antitumor activity than icotinib alone, which might be explained by
the fact that the combination treatment inhibited the activity of
the PI3K-AKT-mTOR signaling pathway to a greater degree. So et
al (46) reported that the
CK2 inhibitor CX-4945 synergized with EGFR-TKIs on EGFR-mutant lung
cancer cells (exon 19del and T790M), which were established as
secondary resistant cell lines to EGFR-TKIs. In accordance with the
aforementioned study, the current study demonstrated the potential
ways in which quinalizarin was able to make the H1975 cells
sensitive to icotinib. PTEN loss (H1650) has been shown to
contribute to EGFR-TKI resistance (38) and CK2 further induces the
stability of PTEN (18), which
may be another way that quinalizarin acts to sensitize H1650 cells
to icotinib.
Further experiments on the exact mechanism of the
combination treatment were also conducted. The result showed that
p-AKT and p-ERK were obviously downregulated when the cells were
treated with both quinalizarin and icotinib. The Ras-Raf-MEK-ERK
and PI3K-AKT-mTOR signaling pathways are main pathways that
regulate cell survival and proliferation, and AKT and ERK are the
key members of these two pathways. A number of studies insist that
EGFR-TKIs play a pivotal role in anti-cancer therapy mainly by
acting on these two signaling pathways (47,48). Similarly, in the current study,
CK2 inhibition by quinalizarin also had an impact on these two
signaling pathways. Thus, it is reasonable to conclude that
quinalizarin may sensitize cells to icotinib by inhibiting the
proliferation and promoting the apoptosis mediated by AKT and ERK
in EGFR-TKI-resistant human adenocarcinoma cell lines.
Acknowledgments
The authors sincerely thank Professor Mathias
Montenarh from Medical Biochemistry and Molecular Biology, Saarland
University (Homburg, Germany) for his time, support and valuable
suggestions throughout the development of this paper.
Funding
This study was supported by The National Natural
Science Foundation of China (grant no. 81101690 to RM and grant no.
81672979 to GW), The Foundation for Fostering Key Talents from
Middle-aged and Young Medical Personnel in Wuhan (2016), Natural
Science Foundation of Hubei Province of China (grant no.
2016cfb426).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GW and RM conceived and designed the experiment. KL
and FZ performed the experiments. KL and YZ analyzed the data. QL
and ZL interpreted the results and wrote the draft of the
manuscript. SZ performed the MTT assay and LL performed part of the
western blot analysis. SZ and LL wrote the final vision of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bareschino MA, Schettino C, Rossi A,
Maione P, Sacco PC, Zeppa R and Gridelli C: Treatment of advanced
non small cell lung cancer. J Thorac Dis. 3:122–133. 2011.
|
|
2
|
Shi Y, Au JS, Thongprasert S, Srinivasan
S, Tsai CM, Khoa MT, Heeroma K, Itoh Y, Cornelio G and Yang PC: A
prospective, molecular epidemiology study of EGFR mutations in
asian patients with advanced non-small-cell lung cancer of
adeno-carcinoma histology (PIONEER). J Thorac Oncol. 9:154–162.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu YL, Chu DT, Han B, Liu X, Zhang L, Zhou
C, Liao M, Mok T, Jiang H, Duffield E and Fukuoka M: Phase III,
randomized, open-label, first-line study in asia of gefitinib
versus carbo-platin/paclitaxel in clinically selected patients with
advanced non-small-cell lung cancer: Evaluation of patients
recruited from mainland China. Asia-Pac J Clin Oncol. 8:232–243.
2012. View Article : Google Scholar
|
|
4
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et
al: Gefitinib or carboplatin-paclitaxel in pulmonary
adenocarcinoma. N Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thongprasert S, Duffield E, Saijo N, Wu
YL, Yang JC, Chu DT, Liao M, Chen YM, Kuo HP, Negoro S, et al:
Health-related quality-of-life in a randomized phase III first-line
study of gefi-tinib versus carboplatin/paclitaxel in clinically
selected patients from Asia with advanced NSCLC (IPASS). J Thorac
Oncol. 6:1872–1880. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ellis PM, Coakley N, Feld R, Kuruvilla S
and Ung YC: Use of the epidermal growth factor receptor inhibitors
gefitinib, erlotinib, afatinib, dacomitinib, and icotinib in the
treatment of non-small-cell lung cancer: A systematic review. Curr
Oncol. 22:e183–e215. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jackman D, Pao W, Riely GJ, Engelman JA,
Kris MG, Janne PA, Lynch T, Johnson BE and Miller VA: Clinical
definition of acquired resistance to epidermal growth factor
receptor tyrosine kinase inhibitors in non-small-cell lung cancer.
J Clin Oncol. 28:357–360. 2010. View Article : Google Scholar
|
|
8
|
Gao Z, Chen W, Zhang X, Cai P, Fang X, Xu
Q, Sun Y and Gu Y: Icotinib, a potent and specific EGFR tyrosine
kinase inhibitor, inhibits growth of squamous cell carcinoma cell
line A431 through negatively regulating AKT signaling. Biomed
Pharmacother. 67:351–356. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen X, Zhu Q, Liu Y, Liu P, Yin Y, Guo R,
Lu K, Gu Y, Liu L, Wang J, et al: Icotinib is an active treatment
of non-small-cell lung cancer: A retrospective study. PLoS One.
9:e958972014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liang W, Wu X, Fang W, Zhao Y, Yang Y, Hu
Z, Xue C, Zhang J, Zhang J, Ma Y, et al: Network meta-analysis of
erlotinib, gefitinib, afatinib and icotinib in patients with
advanced non-small-cell lung cancer harboring EGFR mutations. PLoS
One. 9:e852452014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi Y, Zhang L, Liu X, Zhou C, Zhang L,
Zhang S, Wang D, Li Q, Qin S, Hu C, et al: Icotinib versus
gefitinib in previously treated advanced non-small-cell lung cancer
(ICOGEN): A randomised, double-blind phase 3 non-inferiority trial.
Lancet Oncol. 14:953–961. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Antonicelli A, Cafarotti S, Indini A,
Galli A, Russo A, Cesario A, Lococo FM, Russo P, Mainini AF,
Bonifati LG, et al: EGFR-targeted therapy for non-small cell lung
cancer: Focus on EGFR oncogenic mutation. Int J Med Sci.
10:320–330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou C and Yao LD: Strategies to improve
outcomes of patients with EGRF-mutant non-small cell lung cancer:
Review of the literature. J Thorac Oncol. 11:174–186. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yun CH, Mengwasser KE, Toms AV, Woo MS,
Greulich H, Wong KK, Meyerson M and Eck MJ: The T790M mutation in
EGFR kinase causes drug resistance by increasing the affinity for
ATP. Proc Natl Acad Sci USA. 105:2070–2075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Meggio F and Pinna LA:
One-thousand-and-one substrates of protein kinase CK2? FASEB J.
17:349–368. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ortega CE, Seidner Y and Dominguez I:
Mining CK2 in cancer. PLoS One. 9:e1156092014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Torres J and Pulido R: The tumor
suppressor PTEN is phosphorylated by the protein kinase CK2 at its
C terminus. implications for PTEN stability to proteasome-mediated
degradation. J Biol Chem. 276:993–998. 2001. View Article : Google Scholar
|
|
19
|
Cozza G, Mazzorana M, Papinutto E, Bain J,
Elliott M, di Maira G, Gianoncelli A, Pagano MA, Sarno S, Ruzzene
M, et al: Quinalizarin as a potent, selective and cell-permeable
inhibitor of protein kinase CK2. Biochem J. 421:387–395. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Daya-Makin M, Sanghera JS, Mogentale TL,
Lipp M, Parchomchuk J, Hogg JC and Pelech SL: Activation of a
tumor-associated protein kinase (p40TAK) and casein kinase 2 in
human squamous cell carcinomas and adenocarcinomas of the lung.
Cancer Res. 54:2262–2268. 1994.PubMed/NCBI
|
|
21
|
Faust M, Schuster N and Montenarh M:
Specific binding of protein kinase CK2 catalytic subunits to
tubulin. FEBS Lett. 462:51–56. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee CK, Kim S, Lee JS, Lee JE, Kim SM,
Yang IS, Kim HR, Lee JH, Kim S and Cho B: Next-generation
sequencing reveals novel resistance mechanisms and molecular
heterogeneity in EGFR-mutant non-small cell lung cancer with
acquired resistance to EGFR-TKIs. Lung Cancer. 113:106–114. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cross DA, Ashton SE, Ghiorghiu S, Eberlein
C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ,
et al: AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated
resistance to EGFR inhibitors in lung cancer. Cancer Discov.
4:1046–1061. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Di Maira G, Salvi M, Arrigoni G, Marin O,
Sarno S, Brustolon F, Pinna LA and Ruzzene M: Protein kinase CK2
phosphorylates and upregulates Akt/PKB. Cell Death Differ.
12:668–677. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Duncan JS and Litchfield DW: Too much of a
good thing: The role of protein kinase CK2 in tumorigenesis and
prospects for therapeutic inhibition of CK2. Biochim Biophys Acta.
1784:33–47. 2008. View Article : Google Scholar
|
|
26
|
Hay N: Interplay between FOXO, TOR, and
Akt. Biochim Biophys Acta. 1813:1965–1970. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu SG and Shih JY: Management of acquired
resistance to EGFR TKI-targeted therapy in advanced non-small cell
lung cancer. Mol Cancer. 17:382018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chua MM, Ortega CE, Sheikh A, Lee M,
Abdul-Rassoul H, Hartshorn KL and Dominguez I: CK2 in cancer:
Cellular and biochemical mechanisms and potential therapeutic
target. Pharmaceuticals (Basel). 10. pp. E182017, View Article : Google Scholar
|
|
29
|
Yaylim I and Isbir T: Enhanced casein
kinase II (CK II) activity in human lung tumours. Anticancer Res.
22:215–218. 2002.PubMed/NCBI
|
|
30
|
Hung MS, Xu Z, Chen Y, Smith E, Mao JH,
Hsieh D, Lin YC, Yang CT, Jablons DM and You L: Hematein, a casein
kinase II inhibitor, inhibits lung cancer tumor growth in a murine
xenograft model. Int j Oncol. 43:1517–1522. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li Q, Li K, Yang T, Zhang S, Zhou Y, Li Z,
Xiong J, Zhou F, Zhou X, Liu L, et al: Association of protein
kinase CK2 inhibition with cellular radiosensitivity of non-small
cell lung cancer. Sci Rep. 7:161342017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim J and Kim SH: Druggability of the CK2
inhibitor CX-4945 as an anticancer drug and beyond. Arch Pharm Res.
35:1293–1296. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ku MJ, Park JW, Ryu BJ, Son YJ, Kim SH and
Lee SY: CK2 inhibitor CX4945 induces sequential inactivation of
proteins in the signaling pathways related with cell migration and
suppresses metastasis of A549 human lung cancer cells. Bioorg Med
Chem Lett. 23:5609–5613. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Di Maira G, Brustolon F, Bertacchini J,
Tosoni K, Marmiroli S, Pinna LA and Ruzzene M: Pharmacological
inhibition of protein kinase CK2 reverts the multidrug resistance
phenotype of a CEM cell line characterized by high CK2 level.
Oncogene. 26:6915–6926. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Asati V, Mahapatra DK and Bharti SK:
PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as
anticancer agents: Structural and pharmacological perspectives. Eur
J Med Chem. 109:314–341. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lieber M, Smith B, Szakal A, Nelson-Rees W
and Todaro G: A continuous tumor-cell line from a human lung
carcinoma with properties of type II alveolar epithelial cells. Int
J Cancer. 17:62–70. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Greve G, Schiffmann I, Pfeifer D, Pantic
M, Schuler J and Lubbert M: The pan-HDAC inhibitor panobinostat
acts as a sensitizer for erlotinib activity in EGFR-mutated and
-wildtype non-small cell lung cancer cells. BMC Cancer. 15:9472015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sos ML, Koker M, Weir BA, Heynck S,
Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P,
et al: PTEN loss contributes to erlotinib resistance in EGFR-mutant
lung cancer by activation of Akt and EGFR. Cancer Res.
69:3256–3261. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ritt DA, Zhou M, Conrads TP, Veenstra TD,
Copeland TD and Morrison DK: CK2 is a component of the KSR1
scaffold complex that contributes to raf kinase activation. Curr
Biol. 17:179–184. 2007. View Article : Google Scholar
|
|
40
|
Parker R, Clifton-Bligh R and Molloy MP:
Phosphoproteomics of MAPK inhibition in BRAF-mutated cells and a
role for the lethal synergism of dual BRAF and CK2 inhibition. Mol
Cancer Ther. 13:1894–1906. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
So KS, Rho JK, Choi YJ, Kim SY, Choi CM,
Chun YJ and Lee JC: AKT/mTOR down-regulation by CX-4945, a CK2
inhibitor, promotes apoptosis in chemorefractory non-small cell
lung cancer cells. Anticancer Res. 35:1537–1542. 2015.PubMed/NCBI
|
|
42
|
Yip PY: Phosphatidylinositol
3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR)
signaling pathway in non-small cell lung cancer. Transl Lung Cancer
Res. 4:165–176. 2015.PubMed/NCBI
|
|
43
|
Reungwetwattana T and Dy GK: Targeted
therapies in development for non-small cell lung cancer. J
Carcinog. 12:222013. View Article : Google Scholar
|
|
44
|
Zer A and Leighl N: Promising targets and
current clinical trials in metastatic non-squamous NSCLC. Front
Oncol. 4:3292014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bliesath J, Huser N, Omori M, Bunag D,
Proffitt C, Streiner N, Ho C, Siddiqui-Jain A, O'Brien SE, Lim JK,
et al: Combined inhibition of EGFR and CK2 augments the attenuation
of PI3K-Akt-mTOR signaling and the killing of cancer cells. Cancer
Lett. 322:113–118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
So KS, Kim CH, Rho JK, Kim SY, Choi YJ,
Song JS, Kim WS, Choi CM, Chun YJ and Lee JC:
Autophagosome-mediated EGFR down-regulation induced by the CK2
inhibitor enhances the efficacy of EGFR-TKI on EGFR-mutant lung
cancer cells with resistance by T790M. PLoS One. 9:e1140002014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ke EE and Wu YL: EGFR as a pharmacological
target in EGFR-mutant non-small-cell lung cancer: Where do we stand
now? Trends Pharmacol Sci. 37:887–903. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Morgillo F, Della Corte CM, Fasano M and
Ciardiello F: Mechanisms of resistance to EGFR-targeted drugs: Lung
cancer. ESMO Open. 1:e0000602016. View Article : Google Scholar : PubMed/NCBI
|