Introduction
Neuropathic pain is a chronic pain condition that
may develop following damage, injury or dysfunction of nerves due
to trauma, surgery, disease or chemotherapy. The pain has been
described as a burning, shooting or crawling sensation, or similar
to electric shocks. Slight touch may induce severe pain in
patients, thereby seriously impairing quality of life. However,
currently prescribed therapeutics for neuropathic pain have been
insufficient for pain relief, and the development of new analgesics
is challenging. Accumulating evidence from animal models of
neuropathic pain indicate that microglia in the spinal dorsal horn
(SDH) have a critical role in the generation of neuropathic pain
following peripheral nerve injury (1-3).
Microglia immediately transform into an activated phenotype
following peripheral nerve injury and subsequently initiate
neuropathic pain. Activated microglia induce several
pain-associated factors including purinergic receptor P2X4 (P2RX4),
brain-derived neurotrophic factor (BDNF), interferon regulatory
factor 8 (IRF8), and pro-inflammatory cytokines (1,2,4,5),
which sustain higher levels of neuronal activity in the SDH.
Activation of microglia manifests as hypertrophy of
the cells' processes, and increased cell density and production of
cytokines. Profound activation of microglia is frequently observed
in the SDH after peripheral nerve injury (1-3).
We previously identified that ketamine, a classical painkiller,
inhibited not only N-methyl-D-aspartic acid receptors in neurons, a
canonical target of ketamine, but also large conductance
Ca2+-activated K+ (BK) channels in microglia
(3). Blockade of BK channel
function in microglia by selective inhibitors may attenuate the
generation of neuropathic pain (3). However, a previous investigation did
not rigorously address the specific role of microglial BK channels
in vivo as BK channel blockers, not microglia-selective gene
manipulation, were used to inhibit BK channels in microglia based
on their inhibitory kinetics (3).
BK channels are ubiquitously expressed in several tissues (6). Therefore, specific inhibition of
microglial BK channels is required for understanding the mechanisms
of neuropathic pain. The activation of microglia and subsequent
generation of pain is not restricted in neuropathic pain models.
Long-term opioid treatment in mice alters microglial phenotype,
which induces pain hypersensitivity (7). Neuropathic pain and opioid-induced
hyperalgesia share similar mechanisms that are mediated by
microglial activation including membrane translocation of P2RX4
from lysosomes and subsequent induction of BDNF (7). We previously demonstrated that BK
channels in spinal microglia are also activated following long-term
morphine treatment (8). In
particular, the Ca2+-activated K+ channel β3
auxiliary subunit (KCNMB3), one of a family of 4 auxiliary β
(β1-β4) subunits, was exclusively expressed in microglia in the
spinal cord according to single-cell polymerase chain reaction
(PCR) analysis (8). Furthermore,
morphine-induced hyperalgesia was markedly attenuated in spinal
cord specific-KCNMB3 knockdown mice (8). Therefore, the KCNMB3 knockdown
method enables the investigation of the specific role of BK
channels in spinal microglia during neuropathic pain.
BK channels contribute to an 'emergency brake' that
regulates excessive generation of action potentials and transmitter
release, and are broadly distributed throughout the body (6). BK channels are formed by a
pore-forming α subunit and 4 types of auxiliary β subunits, which
comprise a heterotetramer. The function of the BK channel is
distinct among tissues due to tissue-specific expression patterns
of β subunits (9). The β1 subunit
is distributed in smooth muscle cells, whereas the β2-β4 subunits
are expressed in the central nervous system (9,10).
In the spinal cord, we identified that KCNMB3 was exclusively
expressed in microglia, and that knockdown of KCNMB3 abrogated the
function of BK channels in microglia (8). Conversely, β2 and β4 subunits
expressed in astrocytes and neurons, respectively, were not
detected in spinal microglia (8).
Therefore, it is possible to evaluate the role of BK channels in
spinal microglia in vivo by manipulating the KCNMB3
gene. In the present study, the role of BK channels in microglia
during neuropathic pain were analyzed by in vivo knockdown
of KCNMB3, a microglia-specific subtype of BK channels in the
spinal cord.
Materials and methods
Animals
C57BL/6 mice (male, 8-10 weeks old; n=60) were
purchased from CLEA Japan, Inc. The mice were maintained in a 12 h
light:dark cycle (light beginning at 08:00) at 22-25°C ambient
temperature with food and water provided ad libitum. All
mice were handled daily for 5 days prior to the initiation of the
experiment to minimize their stress reactions to manipulation.
Surgical procedure and small interference
RNA (siRNA) injection
The mice were anesthetized with isoflurane (2%;
Pfizer, Inc.) in O2 (11). The back skin was additionally
anesthetized with 0.5% xylocaine (5 mg/kg), following removal of
the back hair by shaving. If no response was observed against a
noxious stimulus, including toe or tail pinch, the surgical
procedure was initiated. The lumber 5 (L5) transverse process was
identified and carefully removed with bone rongeurs. The L4 ventral
ramus was carefully isolated and freed from the adjacent nerve, and
then the L4 nerve was transected according to a previous method
(3). The incision was washed with
saline and closed. For the intrathecal injection of siRNA, KCNMB3
siRNA (#1: MSS224455; #2: MSS224454; #3: MSS284619) or control
siRNA (GC duplex negative control) were used. Each siRNA (20 pmol/5
µl) was suspended in Lipofectamine RNAiMAX, and then
injected intrathecally for 4 consecutive days according to a
previously described method (3,8).
All reagents were purchased from Thermo Fisher Scientific, Inc. The
knockdown efficacy of siRNA was examined by western blot analyses.
In certain experiments, the intrathecal injection of siRNA was
initiated at 5 days after nerve injury.
Western blot analysis
The L4 spinal dorsal horn was collected from mice
that were treated with siRNAs. The specimens were lysed in lysis
buffer [10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.5%
NP-40, phosphatase and protease inhibitor cocktail] and mixed with
Laemmli sample buffer. Protein concentration was determined using
the Pierce Bicinchoninic Acid Protein Assay kit (Thermo Fisher
Scientific, Inc.). Proteins (30 µg) were loaded into each
lane, separated by 12% SDS-PAGE and transferred to a PVDF membrane.
The blots were blocked with 0.2% Tween-20 in TBS (TBS-T) containing
5% Blocking One (Nacalai Tesque, Inc.) for 1 h at room temperature,
and were then incubated with primary antibodies, diluted in TBS-T
containing 5% Blocking One, at 4°C overnight. The primary
antibodies used were as follows: Anti-KCNMB3 mouse monoclonal
antibody (1:2,000; cat. no. NBP2-12916; Novus Biologicals, LLC) and
anti-β-actin mouse monoclonal antibody (1:5,000; cat. no. ab8226;
Abcam). After being washed with TBS-T, the membranes were incubated
with horseradish peroxidase (HRP)-conjugated secondary antibody
(1:1,000; cat. no. NA931; GE Healthcare) for 1 h at room
temperature. The membrane-bound HRP-labeled antibodies were
detected using Immobilon ECL Ultra Western HRP Substrate (Merck
KGaA) and an image analyzer (LAS-4000; Fujifilm). The bands that
were evaluated by apparent molecular size and were semi-quantified
using ImageJ 1.51j software (National Institutes of Health). The
band intensity was normalized to β-actin.
Behavioral testing
The mice were examined for mechanical
hypersensitivity of the hind paw following L4 nerve transection.
All mice were habituated to the testing environment for 3 days and
were examined for mechanical allodynia. The room temperature
remained stable at 22±1°C. The mice were placed in an acrylic
cylinder (6-cm diameter) with wire mesh floors and allowed to
habituate to the environment for 1 h. Calibrated von Frey filaments
(0.02-2.0 g; North Coast Medical, Inc.) were applied to the
midplantar surface of the hind paw (3,8).
The 50% paw withdrawal thresholds were calculated using the up-down
method (12). Behavioral tests
were performed for 3 or 10 days. Following behavioral analyses,
mice were euthanized with sodium pentobarbital [200 mg/kg,
intraperitoneal (i.p.) injection]. Animal death was confirmed by
cessation of respiration and heartbeat.
Patch clamp
For this protocol, six mice were euthanized with
sodium pentobarbital (200 mg/kg, i.p.) at 3 days after nerve
injury. Animal death was confirmed by cessation of respiration and
heartbeat. A block of the spinal cord from L3 to L5 was embedded in
1% agar. Transverse slices (200-µm thick) were cut from L4
segment with a vibratome (VT1000 S; Leica Biosystems Nussloch
GmbH). Ice-cold artificial cerebrospinal fluid (ACSF) saturated
with 95% O2 and 5% CO2 was used in slice
preparation. ACSF consisted of 124 mM NaCl, 2.5 mM KCl, 1.24 mM
KH2PO4, 1.3 mM MgSO4, 2.4 mM
CaCl2, 10 mM glucose, and 26 mM NaHCO3 (all
from Sigma-Aldrich; Merck KGaA). Spinal slices were stained with 25
µg/ml Alexa Fluor 488 conjugated isolectin GS-IB4
(IB4; cat. no. I21411; Thermo Fisher Scientific, Inc.)
for 30 min at room temperature in ACSF. A whole-cell patch clamp
recording was made from IB4-positive cells with marked
branched processes located at lamina II-III in L4 spinal cord in
the slice preparation. IB4-positive cells were visually
identified by the laser with a wavelength of 488 nm using an
upright microscope equipped with a ×40 water-immersion objective
(Zeiss GmbH). The external solution was ACSF. Patch electrodes were
fabricated using a Sutter P-97 (Sutter Instrument) from
borosilicate glass (1.5 mm outer diameter, 0.9 mm inner diameter;
G-1.5; Narishige Scientific Instrument Laboratories). Patch
pipettes were filled with internal solution containing 1% Lucifer
Yellow CH dilithium salt (Sigma-Aldrich; Merck KGaA). Internal
solution consisted of 120 mM KCl, 2 mM MgCl2, 10 mM
HEPES, 0.1 mM BAPTA adjusted to pH 7.3 with KOH (all from
Sigma-Aldrich; Merck KGaA). Voltage ramps from -120 to +30 mV were
applied for 300 msec to induce BK currents. The data were acquired
using Axopatch 200B amplifier, Digidata 1320 interface, and pClamp
9.0 software and analyzed with Clampfit 9 software (all from
Molecular Devices, LLC) according to the previous methods (3,8).
Immunohistochemistry
The mice were euthanized with sodium pentobarbital
(200 mg/kg, i.p.). Animal death was confirmed by cessation of
respiration and heartbeat. The mice were perfused transcardially
with 0.1 M phosphate buffer (PB), pH 7.4, followed by 4%
paraformaldehyde in 0.1 M PB, 3 and 10 days after nerve injury. The
L4 spinal segments were fixed with 4% paraformaldehyde (PFA;
Sigma-Aldrich; Merck KGaA) overnight at 4°C. The L4 segments were
further incubated with 30% sucrose (Sigma-Aldrich; Merck KGaA)
overnight to protect cryolesions. The spinal cord slices
(14-µm thick) were prepared by a CM1860 cryomicrotome (Leica
Microsystems, Inc.). In some experiments, the slices were fixed
with 4% PFA following the patch clamp recording, after which,
staining was performed according to the following method. Blocking
was performed by 1% normal donkey serum (Jackson ImmunoResearch
Laboratories, Inc.), 1% BSA (Sigma-Aldrich; Merck KGaA) and 0.1%
Triton X-100 (Sigma-Aldrich; Merck KGaA) in PBS for 1 h. The slices
were incubated with anti-Lucifer Yellow rabbit polyclonal antibody
(1:50,000; cat. no. A-5750; Thermo Fisher Scientific, Inc.) or
anti-ionized calcium-binding adapter molecule 1 (Iba1), a marker
for microglia, rabbit polyclonal antibody (1:2,000; cat. no.
019-19741; FUJIFILM Wako Pure Chemical Corporation) for 7 days at
4°C or overnight at 4°C, respectively. Following washing with PBS,
the slices were incubated with donkey anti-rabbit IgG conjugated
with Cy3 (cat. no. 711-165-152, 1:400; Jackson ImmunoResearch
Laboratories, Inc.) or Alexa488 (711-545-152, 1:400; Jackson
ImmunoResearch Laboratories, Inc.) for 2 h at 4°C, and then mounted
in Vectashield (Vector Laboratories, Inc.). The images were
acquired on a Nikon C2 scanning confocal microscope using a ×20
objective (NA 0.75; Nikon Corporation). Images were processed and
analyzed using ImageJ 1.51j software (National Institutes of
Health). The number of microglia in the SDH was counted. To analyze
the microglial morphology, Sholl analysis was performed manually
according to a previously described method (13): Concentric circles with 5 µm
increases in diameter were drawn around the soma and the number of
processes crossing each circles was counted. All measurements were
performed by an operator who was blinded to the identity of the
sections.
Reverse transcription quantitative PCR
(RT-qPCR) analyses
The total RNA was extracted from SDH samples with
TRIsure (Bioline; BiCat GmbH) according to the protocol of the
manufacturer. RT was performed using a QuantiTect Reverse
Transcription kit (Qiagen GmbH) with 500 ng extracted RNA,
according to the manufacturer's protocol (DNA elimination: 42°C for
2 min; RT: 42°C for 15 min; inactivation of QuantiTect Reverse
Transcriptase: 95°C for 3 min). RT-qPCR was performed using a
two-step protocol (initial denaturation: 95°C for 2 min and 45
cycles of denaturation: 95°C for 10 sec and annealing: 60°C for 10
sec) and processed in triplicate with a Corbett Rotor-Gene RG-3000A
Real-Time PCR System (Qiagen GmbH) using Thunderbird SYBR qPCR Mix
(10 µl) and 50X ROX reference dye (0.4 µl; both from
Toyobo Life Science) and cDNA template (0.2 µl). The primers
(600 nM) for each gene are listed below. The data were analyzed by
an RG-3000A software program (version Rotor-Gene 6.1.93; Corbett
Life Science; Qiagen GmbH.). All values were normalized to β-actin
expression. The 2−ΔΔCq method was used to calculate
relative mRNA expression levels (14). The primer sequences are described
as follows: P2RX4 forward, 5′-ACA ACG TGT CTC CTG GCT ACA AT-3′;
P2RX4 reverse, 5′-GTC AAA CTT GCC AGC CTT TCC-3′; BDNF forward,
5′-TAC CTG GAT GCC GCA AAC AT-3′; BDNF reverse, 5′-AGT TGG CCT TTG
GAT ACC GG-3′; IRF8 forward, 5′-GAG CTG CAG CAA TTC TAC GC-3′; IRF8
reverse, 5′-AAG GGT CTC TGG TGT GAG GT-3′; tumor necrosis factor-α
(TNF-α) forward, 5′-GTG GAA CTG GCA GAA GAG GC-3′; TNF-α reverse,
5′-AGA CAG AAG AGC GTG GTG GC-3′; interleukin-1β (IL-1β) forward,
5′-CTG TGT CTT TCC CGT GGA CC-3′; IL-1β reverse, 5′-CAG CTC ATA TGG
GTC CGA CA-3′; β-actin forward; 5′-AGA GGG AAA TCG TGC GTG AC-3′;
and β-actin reverse, 5′-CAA TAG TGA TGA CCT GGC CGT-3′.
Statistical analyses
The data are represented as the mean ± standard
error of the mean. Statistical analyses of the results were
performed with Student's unpaired t-test and one-way analysis of
variance (ANOVA) with post-hoc Dunnett's or Tukey's test, and
two-way ANOVA with post-hoc Tukey's test using the GraphPad Prism7
(GraphPad Software, Inc.) software package. P<0.05 was
considered to indicate a statistically significant difference.
Results
In vivo knockdown of KCNMB3 attenuates
the generation of neuropathic pain
To evaluate the role of BK channels in spinal
microglia on neuropathic pain, in vivo knockdown experiments
were performed by intrathecal injection of siRNA, as microglia in
the SDH, as well as several brain regions, are activated after
peripheral nerve injury (15). In
a previous study, we identified that the expression pattern of
KCNMB3 was restricted in microglia in the spinal cord (8). Therefore, KCNMB3 siRNA was used to
silence microglial BK channels in the spinal cord. KCNMB3 siRNA (20
pmol, 5 µl) was injected into the intrathecal space of naïve
mice for 4 consecutive days; the L4 segment of the SDH was then
collected. The knockdown efficacy of KCNMB3 siRNAs was examined
using western blot analyses. Among the siRNAs for KCNMB3, #3 siRNA
(MSS284619) resulted in almost complete depletion of KCNMB3 protein
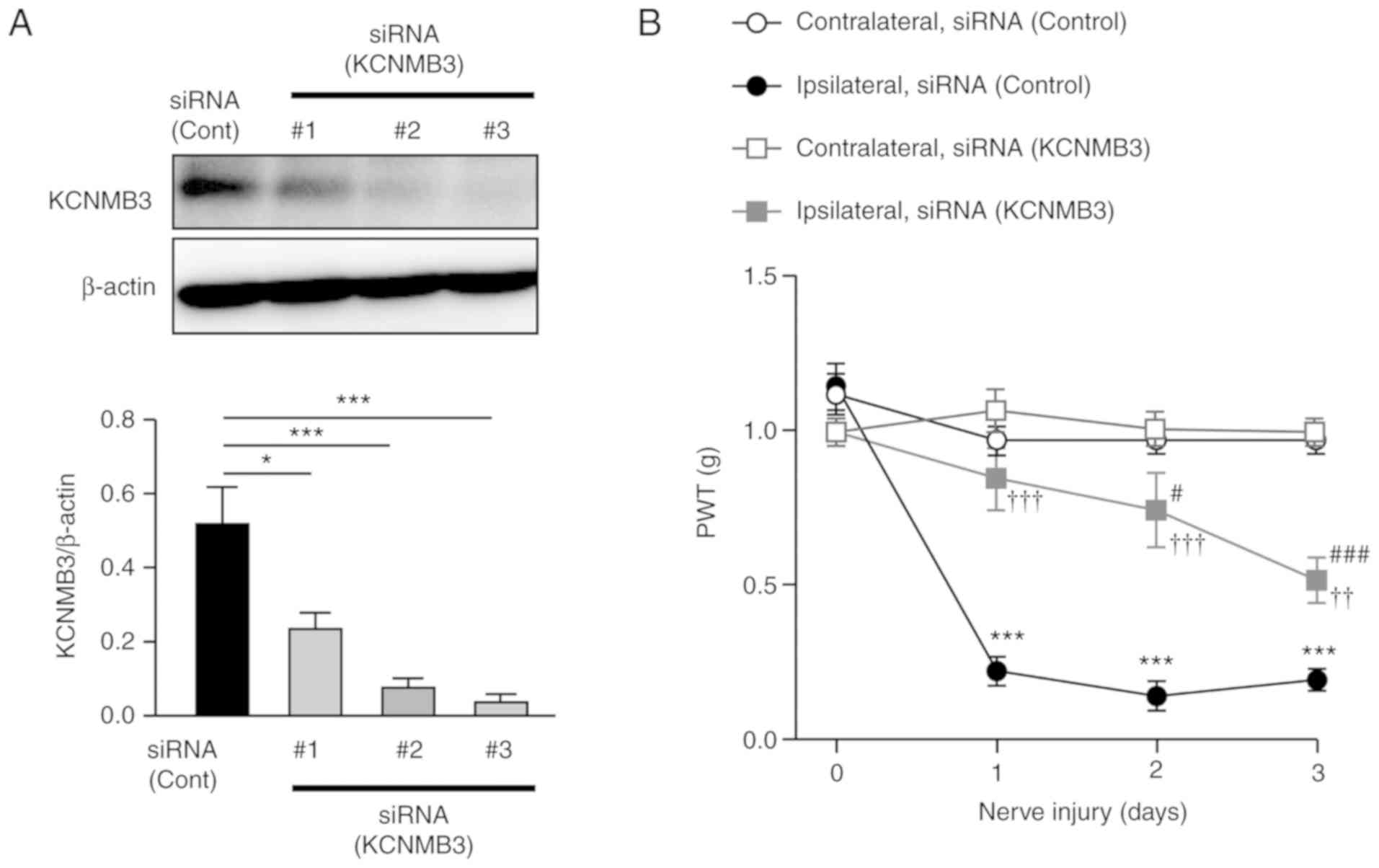
expression in the SDH (Fig. 1A).
Spinal microglia-specific knockdown of KCNMB3 was successful;
therefore, the #3 siRNA was used in subsequent experiments.
Nerve-injured mice exhibited a significant decreased in paw
withdrawal threshold (PWT) to mechanical stimulation applied to the
hind paw following nerve injury (Fig.
1B). Conversely, the decrease in PWT following nerve injury was
significantly attenuated in the KCNMB3 knockdown mice compared with
the control siRNA-treated group (Fig.
1B). To additionally evaluate the function of BK channels in
spinal microglia during neuropathic pain, whole-cell patch clamp
analyses from spinal microglia were performed 3 days after nerve
injury. Spinal cord slices from nerve-injured mice were stained
with IB4 to visualize the microglia. Although patch
pipette was applied to IB4+ cells, which have
branched processes (Fig. 2A),
IB4 preferentially labels nonpeptidergic primary
afferent fibers that terminate in laminas II of the SDH (16). Lucifer yellow was introduced
through the recording pipette to verify that the recorded cells
were microglia and not primary afferent fibers. Immunohistochemical
analyses using anti-Lucifer yellow antibodies revealed that the
recorded cells exhibited the characteristic structure of microglia
that have soma with branched processes, as demonstrated by the
Z-stack image in Fig. 2A,
indicating successful patch clamp recordings from microglia and not
primary afferent fibers in the SDH. Ipsilateral spinal microglia at
3 days after nerve injury exhibited large outward currents at +30
mV, in contrast to those in the sham-operated mice (Fig. 2B), similar to observations from
previous studies (3). These
current activations were significantly weakened in the spinal
microglia from the nerve-injured KCNMB3 knockdown mice (Fig. 2B). The Sholl analysis of the
microglial processes was performed to examine the morphological
changes of the microglia in the SDH. Morphological changes
associated with microglial activation, which were character-ized by
retraction of their processes, in nerve-injured mice were prevented
by KCNMB3 knockdown (Fig.
2C).
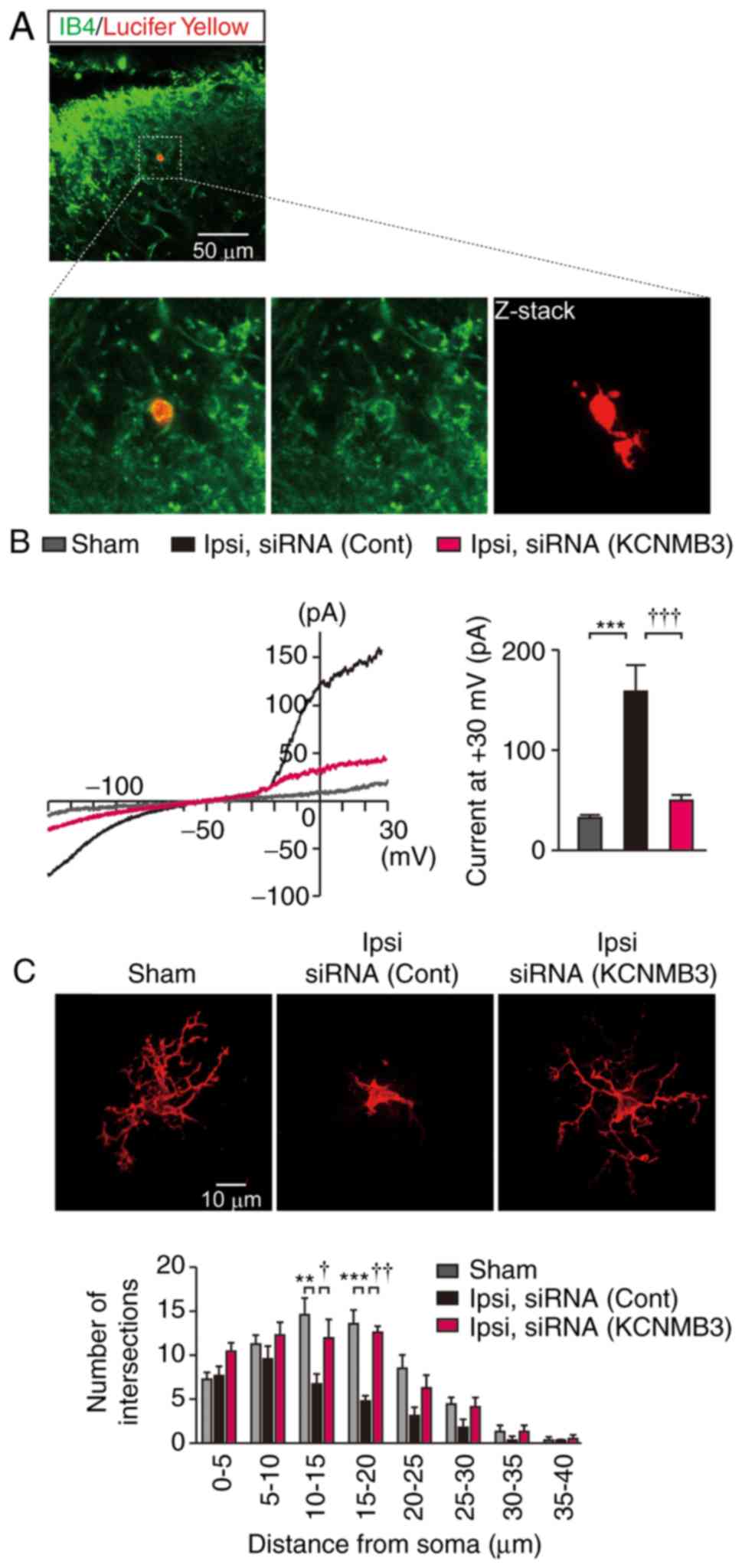 | Figure 2Patch clamp recording from
IB4-positive cells in the spinal dorsal horn. (A)
Lucifer yellow was introduced into IB4-positive cells
through patch pipette during recording, and then stained with
anti-Lucifer yellow antibody. Upper panel demonstrates merged image
of IB4 and anti-Lucifer yellow staining of the spinal
cord slices from nerve-injured mice on day 3. Lower panels
demonstrate enlarged image of the inset (square). The image on the
right-hand side in the lower panel demonstrates the Z-stack of
Lucifer yellow-positive cells. Scale bar, 50 µm. (B)
Representative BK currents in spinal microglia elicited by voltage
ramps. The bar chart represents statistical analysis of BK currents
at +30 mV. n=6 cells from 3 mice each. Data were analyzed by
one-way ANOVA followed by Tukey's post-hoc test.
***P<0.001. †††P<0.001. (C)
Representative Z-stack images of Lucifer yellow positive microglia
of sham-, siRNA (cont)-treated nerve-injured mice, and siRNA
(KCNMB3)-treated nerve-injured mice. Scale bar, 10 µm. Lower
panel indicated Sholl analysis of microglia in the SDH. n=6 cells
from 3 mice each. Data were analyzed by two-way ANOVA followed by
Tukey's post-hoc test. **P<0.01 and ***P<0.001.
†P<0.05 and ††P<0.01. Data are
presented as the mean ± standard error of the mean. IB4, isolectin
GS-IB4; ANOVA, analysis of variance; Ipsi, ipsilateral; siRNA,
small interfering RNA; Cont, control; KCNMB3,
Ca2+-activated K+ channel β3 auxiliary
subunit. |
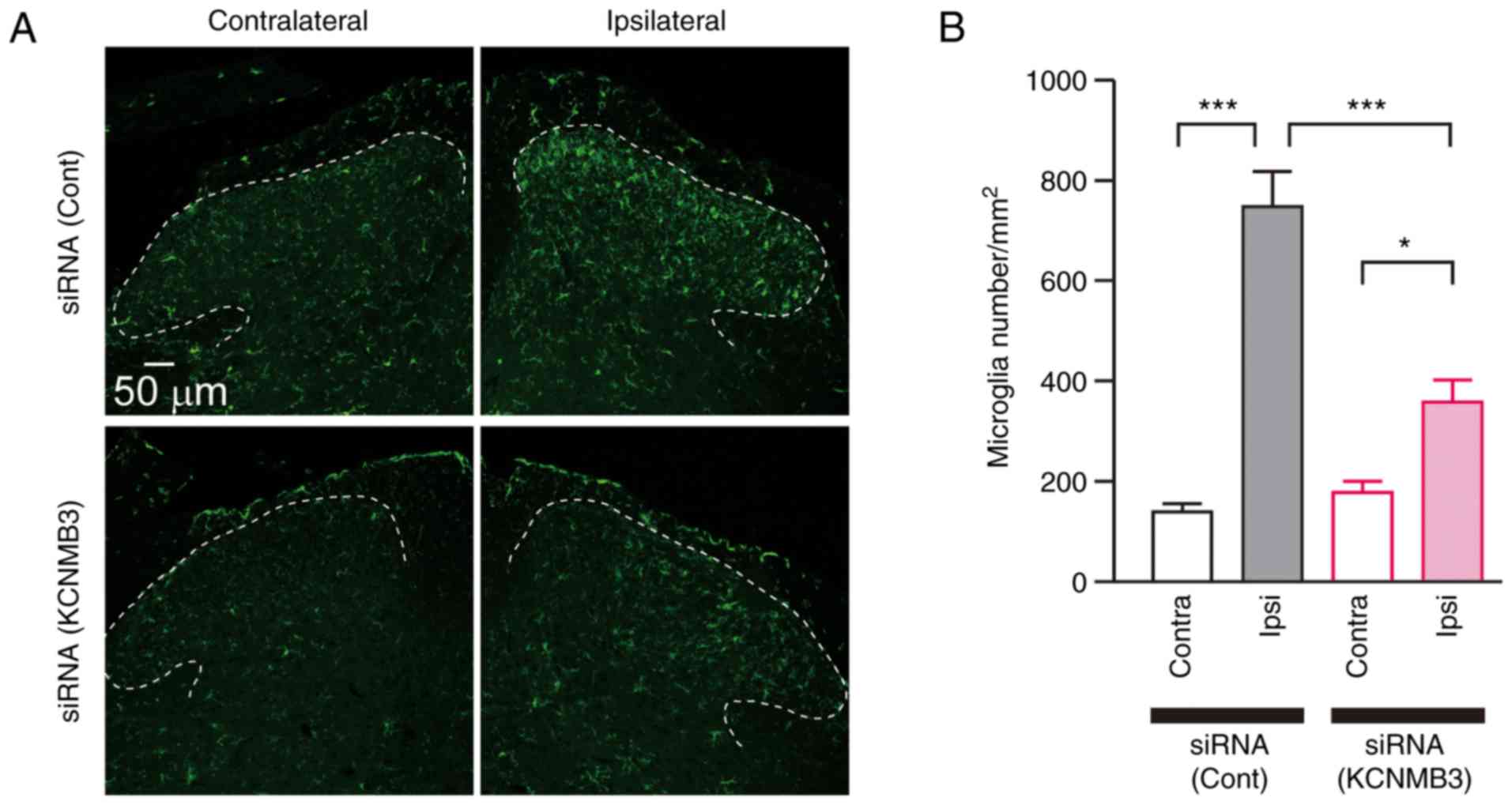
KCNMB3 knockdown prevents microglial
activation following nerve injury
The activation profile of microglia following nerve
injury was subsequently analyzed. The L4 spinal cord was collected
3 days after nerve injury and stained with anti-Iba1 antibody, a
microglia marker. The immunoreactivity of Iba1 in the ipsilateral
side of SDH was significantly increased compared with that of the
contralateral side (Fig. 3A).
Microglial density was also increased in the ipsilateral side of
SDH compared with the contralateral side (Fig. 3B). Conversely, KCNMB3 knockdown
markedly decreased Iba1 immunofluorescence and microglial density
in the ipsilateral side of SDH compared with that of control
siRNA-treated group (Fig. 3A and
B). The effects of KCNMB3 siRNA on pain-associated molecules
were additionally analyzed in the SDH. The messenger RNA (mRNA)
levels on the ipsilateral side of the SDH were normalized with
those on the contralateral side. Increased expression levels of
mRNAs for P2X4R, BDNF, IRF8, IL-1β and TNF-α in the SDH following
nerve injury were significantly attenuated by KCNMB3 knockdown
(Fig. 4). These results indicated
that KCNMB3 knockdown prevented activation of microglia.
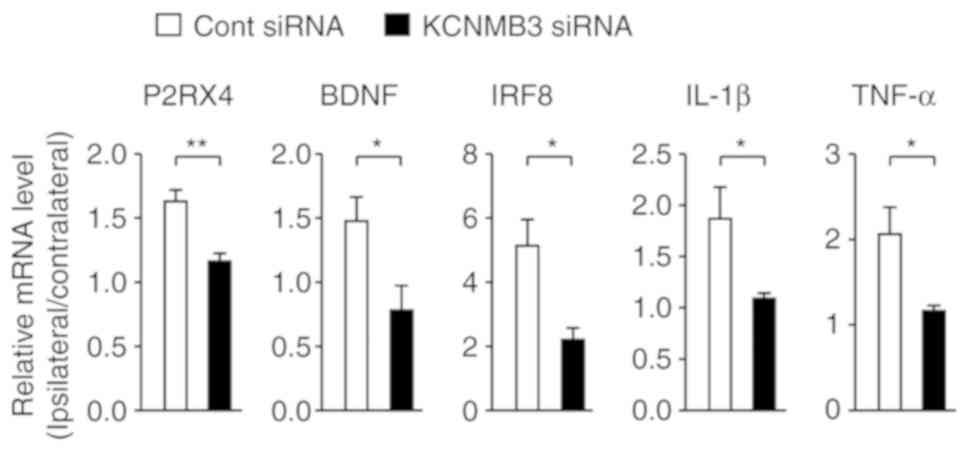 | Figure 4KCNMB3 knockdown prevents induction
of neuropathic pain-associated molecules. Relative expression
levels of mRNAs for P2RX4, BDNF, IRF8, IL-1β and TNF-α in the SDH 3
days after nerve injury. The mRNA levels in the ipsilateral side of
SDH were normalized to those in the contralateral side. Bars
represent mRNA levels. n=3-4 mice/group, unpaired t-test.
*P<0.05 and **P<0.01. Data are
presented as the mean ± standard error of the mean. KCNMB3,
Ca2+-activated K+ channel β3 auxiliary
subunit; P2RX4, purinergic receptor P2X4, BDNF, brain-derived
neuro-trophic factor; IRF8, interferon regulatory factor 8; IL-1β,
interleukin-1β; TNF-α, tumor necrosis factor-α; SDH, spinal dorsal
horn; cont, control; siRNA, small interfering RNA. |
Gene silencing of KCNMB3 in microglia
ameliorates neuropathic pain
To investigate the role of KCNMB3 in chronic pain,
intrathecal injection of KCNMB3 siRNA was initiated at day 5 after
nerve injury. Mechanical allodynia was gradually but significantly
alleviated by daily injection of KCNMB3 siRNA (Fig. 5A). The L4 spinal cord was
collected 10 days after nerve injury. Iba1 immunoreactivity and
microglia density in the SDH were significantly decreased by
intrathecal injection of KCNMB3 siRNA (Fig. 5B and C). In addition, increased
expression levels of mRNAs for pain-associated molecules, including
P2X4R, BDNF, IRF8, IL-1β and TNF-α, in the ipsilateral side of the
SDH were also suppressed by KCNMB3 siRNA treatment (Fig. 6). These results suggest that
KCNMB3 knockdown shifts microglia in a quiescent state.
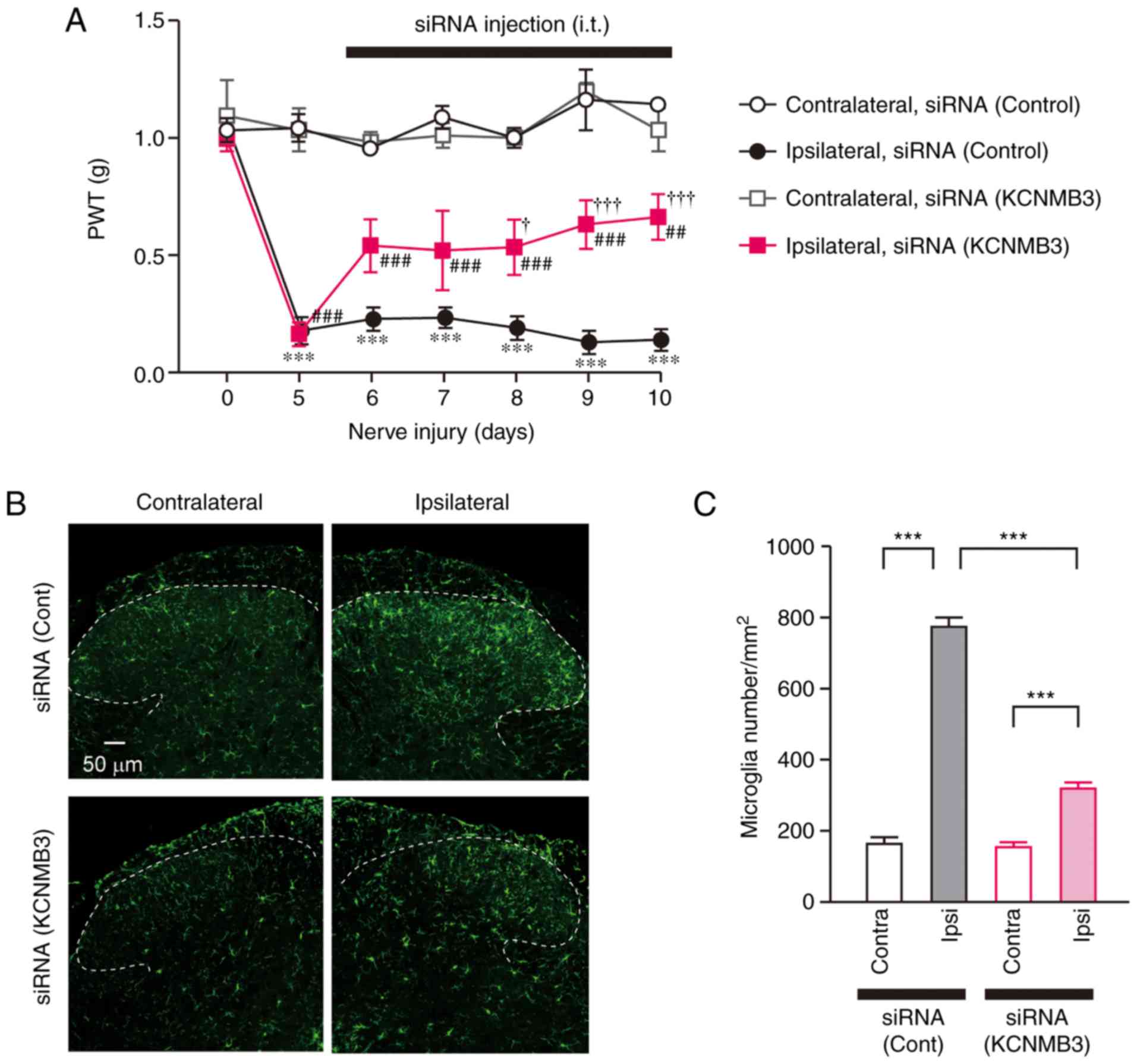 | Figure 5KCNMB3 knockdown ameliorates chronic
pain. (A) Time course of PWT following nerve injury. Intrathecal
injection of KCNMB3 siRNA was initiated at 5 days after nerve
injury. n=4 mice/group. Data were analyzed using two-way ANOVA
followed by Tukey's post-hoc test. ***P<0.001 vs.
Contralateral, siRNA (Control). †P<0.05 and
†††P<0.001 vs. Ipsilateral, siRNA (Control).
##P<0.01 and ###P<0.001 vs.
Contralateral, siRNA (KCNMB3). (B) Immunofluorescence of ionized
calcium-binding adapter molecule 1, a marker of microglia, 10 days
after nerve injury. KCNMB3 siRNA was injected intrathecally 5 days
after nerve injury. Dashed lines indicate the border of white and
gray matter of the spinal dorsal horn. Scale bar, 50 µm. (C)
The number of microglia in the spinal dorsal horn. Bars represent
microglia number. n=4 mice/group. Data were analyzed by two-way
ANOVA followed by Tukey's post-hoc test. ***P<0.001.
Data are presented as the mean ± standard error of the mean.
KCNMB3, Ca2+-activated K+ channel β3
auxiliary subunit; PWT, paw withdrawal threshold; siRNA, small
interfering RNA; cont, control; ANOVA, analysis of variance. |
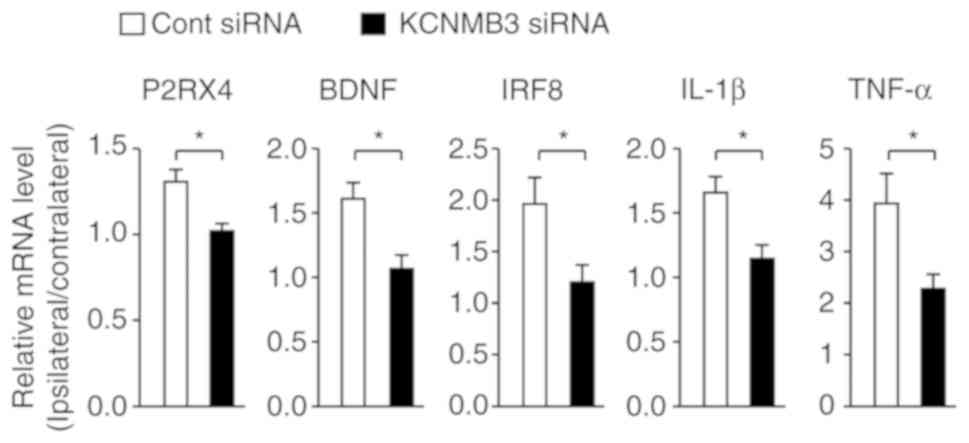 | Figure 6KCNMB3 knockdown inhibited the
expression levels of neuropathic pain-associated molecules.
Relative expression levels of mRNAs for P2RX4, BDNF, IRF8, IL-1β
and TNF-α in the SDH 10 days after nerve injury. The mRNA levels in
the ipsilateral side of the SDH were normalized with those in the
contralateral side. Bars represent mRNA levels. n=4 mice/group.
Data were analyzed using an unpaired Student's t-test.
*P<0.05. Data are presented as the mean ± standard
error of the mean. KCNMB3, Ca2+-activated K+
channel β3 auxiliary subunit; P2RX4, purinergic receptor P2X4,
BDNF, brain-derived neurotrophic factor; IRF8, interferon
regulatory factor 8; IL-1β, interleukin-1β; TNF-α, tumor necrosis
factor-α; SDH, spinal dorsal horn; cont, control; siRNA, small
interfering RNA. |
Discussion
Spinal microglia are known to be involved in pain,
including nerve injury-, cancer- and diabetic-induced neuropathic
pain and opioid-induced hyperalgesia (17). Microglia-derived molecules,
including IL-1β and TNF-α, sustain the pain state by the
facilitation of excitatory neurotransmission and decrease of
inhibitory neurotransmission in lamina II neurons in the spinal
cord (4). In addition,
microglia-derived BDNF leads to excitatory action of γ-aminobutyric
acid via a depolarizing shift in the anion reversal potential
(2). Due to amelioration of
allodynia by neutralizing IL-1β or quenching BDNF (2,4),
the functional inhibition of reactive microglia has significance in
pain relief. The major results from the present study revealed that
microglia-specific inhibition of BK channels resulted in a
decreased production of pain-associated molecules and attenuation
of neuropathic pain.
The family of Ca2+-activated
K+ channels is comprised of small conductance,
intermediate conductance, and large conductance (i.e. BK) channels.
Among the 3 types of Ca2+-activated K+
channels, BK channels are solely expressed in microglia (18). BK channels are involved in the
regulation of physiological processes, including neuronal
excitability and neurotransmitter release. However, the role of BK
channels in microglia remains to be completely elucidated.
Concordant with previous observations that the activation of
K+ channels are observed in reactive microglia (19,20), BK channels in microglia are
activated during neuropathic pain, which is involved in the
production of IL-1β and BDNF (3,8).
Previous studies have revealed that BK channel inhibitors impede
the secretion process of IL-1β from both lipopolysaccharide
[Toll-like receptor (TLR) 4 agonist]- and Pam3CSK4 (TLR2
agonist)-stimulated human monocytes (20). K+ efflux via BK
channels is involved in the activation of the NACHT, LRR and PYD
domain-containing protein 3 inflammasome, which is independent of
pannexin-1 or P2X purinoceptor 7 receptor pathways (21). Similarly, activation of BK
channels contributes to the inflammatory response in human
macrophages (22). Therefore, BK
channels contribute to the production of pro-inflammatory molecules
in microglia. BK currents are observed in resting microglia of
juvenile mice (20); however,
their expression levels are decreased in the microglia of young
adult mice (23). Regardless of
silent of BK channels in resting microglia, an increase in
intracellular Ca2+ concentration through patch pipette
caused robust activation of BK channels (8). Microglia in the developmental stage
are highly active, and contribute to synaptic pruning (24) and survival of cortical neurons
(25). Considering the
aforementioned data, activation of BK channels may contribute to
sustaining the active state of microglia.
Single-cell PCR analysis demonstrated that only
KCNMB3 and not KCNMB1, KCNMB2 or KCNMB4, is exclusively expressed
in spinal microglia and primary cultured microglia (8). However, it cannot be concluded that
the current activation of BK channels in spinal microglia following
nerve injury is due to an increase in their numbers. Microgliosis
in the SDH following nerve injury impedes the amount of KCNMB3
protein in a microglia. Furthermore, there is a requirement for the
evaluation of KCNMB3 protein levels in a microglial cell isolated
from the SDH via fluorescence-activated cell sorting in future
studies. Single-channel recording demonstrated that opening of the
BK channel coupled with KCNMB3 was observed at negative potentials
(26). By contrast, KCNMB1,
KCNMB2 and KCNMB4 are not activated at negative potentials
(9,27). In the present study, it was
observed that spinal microglia following nerve injury exhibited
large inward currents at negative potentials. Considering the
gating properties of the KCNMB protein family, it is conceivable
that functional KCNMB1, KCNMB2 and KCNMB4 are not expressed in
spinal microglia following nerve injury.
Microgliosis in the SDH was observed at 2 months
after nerve injury when pain was completely recovered (28). We hypothesized that KCNMB3 siRNA
injection during the maintenance phase of neuropathic pain would
not affect microgliosis in the SDH. Contrary to our hypothesis, the
number of spinal microglia observed following nerve injury was
decreased due to KCNMB3 siRNA treatment. The mechanisms responsible
for the decrease in the number of spinal microglia via KCNMB3
knockdown remain unknown. Microgliosis in the SDH following nerve
injury has previously been hypothesized to occur as a result of
microglial proliferation. BDNF is known to regulate the
proliferation and survival of cultured microglia (29). Therefore, BDNF decrease following
KCNMB siRNA injection may potentially affect the total number of
microglia. However, microglial proliferation in the SDH peaks 3
days after nerve injury (13).
Microglial proliferation during the induction phase of neuropathic
pain may be attenuated by KCNMB3 siRNA injection. Alternatively, it
is unlikely that KCNMB3 siRNA suppresses microglial proliferation
during the maintenance phase of neuropathic pain. An additional
mechanism for a decrease in the microglial density via KCNMB3 siRNA
may be the reestablishment of the microglial network. Tay et
al (30) demonstrated that
microglial egress and apoptosis were observed during the resolution
of microglial clusters. KCNMB3 siRNA may facilitate these processes
and shift the stable microglial network. Its role in the microglial
network should be assessed in future studies.
Changes in spinal density are associated with
synaptic connectivity and behavior. Microglia-derived BDNF is one
of the representative modulators of synaptic structure through the
formation of glutamatergic synapses (31). Liu et al (32) demonstrated that BDNF changed
synaptic connectivity in the SDH following peripheral nerve injury.
Additionally, BDNF expression is regulated by TNF-α (32). Therefore, inhibition of
inflammatory molecules and BDNF in spinal microglia by KCNMB3 siRNA
may indirectly modulate synaptic connectivity culminating in
attenuation of neuropathic pain.
Several molecules cause activation of microglia
during neuropathic pain (16,33). LPA (1-acyl-glycerol 3-phosphate)
is one of the simplest phospholipids that serves as an
extracellular signaling molecule. Following nerve injury, LPA is
produced by activated neurons and facilitates its production in
spinal microglia (33). LPA
stimulation causes increased expression levels of BDNF in microglia
(34). We previously identified
that LPA induced robust activation of BK currents in spinal
microglia, and BK channel inhibitor significantly attenuated
allodynia-like behaviors, which was caused by intrathecal injection
of LPA (3). A recent clinical
study demonstrated that LPA concentration in the cerebrospinal
fluid was correlated with pain symptoms in patients (35). Therefore, inhibition of BK channel
function in microglia is potentially mediated through the LPA
pathway in the spinal cord. In the present study, inhibition of
microglia-specific BK channels did not completely abrogate
neuropathic pain. This may be due to the involvement of factors
other than LPA in activation of microglia and the generation of
neuropathic pain (17,33).
The effectiveness of ultra-low doses of paxilline, a
BK channel-specific inhibitor, for epilepsy has been described
(36). We also observed the
effectiveness of paxilline in a model of morphine-induced
hyperalgesia (8). Paxilline may
cause side effects, although it effectively attenuated pathology.
Paxilline selectively binds to the BK channel pore-gate domain and
blocks channel activity. Previous evidence suggested that mice
lacking KCNMA1, which encodes the pore-forming subunit of the BK
channel, exhibit moderate ataxia (37); therefore, selective inhibition of
BK channels in microglia is required for therapy. Given this
evidence, KCNMB3, which is specifically expressed in microglia and
regulates pain-associated molecules, is a promising therapeutic
target for neuropathic pain.
In conclusion, the results of the present study
demonstrated that a microglia-specific subtype of BK channels
contributed to the induction of pro-inflammatory molecules and led
to the generation of neuropathic pain. The identification of the
role of BK channels improved understanding of microglial function
and neuropathic pain.
Abbreviations:
|
BDNF
|
brain-derived neurotrophic factor
|
|
BK
|
large conductance
Ca2+-activated K+ channels
|
|
IL
|
interleukin
|
|
IRF
|
interferon regulatory factor
|
|
LPA
|
lysophosphatidic acid
|
|
mRNA
|
messenger RNA
|
|
PWT
|
paw withdrawal threshold
|
|
SDH
|
spinal dorsal horn
|
|
siRNA
|
small interfering RNA
|
|
TLR
|
toll-like receptor
|
|
TNF
|
tumor necrosis factor
|
Acknowledgments
Not applicable.
Funding
The present study was supported by a Grant-in-Aid
for Scientific Research from the Japan for the Promotion of Science
(JSPS) (JP16K11477 to YHay).
Availability of data and materials
All the data generated and analyzed the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
YHay designed and supervised the study. KI, YHar, JZ
and YHay performed the experiments. KI, YHar, JZ, YM and YHay
analyzed the data. YHay wrote the manuscript. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were performed using approved
protocol (no., A27-187-0) and in accordance with the guidelines of
Kyushu University. Mice were housed in accordance with the
Institutional Animal Care and Use Committee regulations at Kyushu
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tsuda M, Shigemoto-Mogami Y, Koizumi S,
Mizokoshi A, Kohsaka S, Salter MW and Inoue K: P2X4 receptors
induced in spinal microglia gate tactile allodynia after nerve
injury. Nature. 424:778–783. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Coull JA, Beggs S, Boudreau D, Boivin D,
Tsuda M, Inoue K, Gravel C, Salter MW and De Koninck Y: BDNF from
microglia causes the shift in neuronal anion gradient underlying
neuropathic pain. Nature. 483:1017–1021. 2005. View Article : Google Scholar
|
|
3
|
Hayashi Y, Kawaji K, Sun L, Zhang X,
Koyano K, Yokoyama T, Kohsaka S, Inoue K and Nakanishi H:
Microglial Ca(2+)-activated K(+) channels are possible molecular
targets for the analgesic effects of S-ketamine on neuropathic
pain. J Neurosci. 31:17370–17382. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kawasaki Y, Zhang L, Cheng JK and Ji RR:
Cytokine mechanisms of central sensitization: Distinct and
overlapping role of interleukin-1beta, interleukin-6, and tumor
necrosis factor-alpha in regulating synaptic and neuronal activity
in the superficial spinal cord. J Neurosci. 28:5189–5194. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Masuda T, Tsuda M, Yoshinaga R,
Tozaki-Saitoh H, Ozato K, Tamura T and Inoue K: IRF8 is a critical
transcription factor for transforming microglia into a reactive
phenotype. Cell Rep. 1:334–340. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Salkoff L, Butler A, Ferreira G, Santi C
and Wei A: High-conductance potassium channels of the SLO family.
Nat Rev Neurosci. 7:921–931. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ferrini F, Trang T, Mattioli TA, Laffray
S, Del'Guidice T, Lorenzo LE, Castonguay A, Doyon N, Zhang W, Godin
AG, et al: Morphine hyperalgesia gated through microglia-mediated
disruption of neuronal Cl(-) homeostasis. Nat Neurosci. 16:183–192.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hayashi Y, Morinaga S, Zhang J, Satoh Y,
Meredith AL, Nakata T, Wu Z, Kohsaka S, Inoue K and Nakanishi H: BK
channels in microglia are required for morphine-induced
hyperalgesia. Nat Commun. 7:116972016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Contreras GF, Neely A, Alvarez O, Gonzalez
C and Latorre R: Modulation of BK channel voltage gating by
different auxiliary β subunits. Proc Natl Acad Sci USA.
109:18991–18996. 2012. View Article : Google Scholar
|
|
10
|
Brenner R, Chen QH, Vilaythong A, Toney
GM, Noebels JL and Aldrich RW: BK channel beta4 subunit reduces
dentate gyrus excitability and protects against temporal lobe
seizures. Nat Neurosci. 8:1752–1759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gargiulo S, Greco A, Gramanzini M,
Esposito S, Affuso A, Brunetti A and Vesce G: Mice anesthesia,
analgesia, and care, Part I: Anesthetic considerations in
preclinical research. ILAR J. 53:E55–E69. 2012. View Article : Google Scholar
|
|
12
|
Chaplan SR, Bach FW, Pogrel JW, Chung JM
and Yaksh TL: Quantitative assessment of tactile allodynia in the
rat paw. J Neurosci Methods. 53:55–63. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kohno K, Kitano J, Kohro Y, Tozaki-Saitoh
H, Inoue K and Tsuda M: Temporal kinetics of microgliosis in the
spinal dorsal horn after peripheral nerve injury in rodents. Biol
Pharm Bull. 41:1096–1102. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
15
|
Taylor AM, Mehrabani S, Liu S, Taylor AJ
and Cahill CM: Topography of microglial activation in sensory- and
affect-related brain regions in chronic pain. J Neurosci Res.
95:1330–335. 2017. View Article : Google Scholar
|
|
16
|
Griffin RS, Costigan M, Brenner GJ, Ma CH,
Scholz J, Moss A, Allchorne AJ, Stahl GL and Woolf CJ: Complement
induction in spinal cord microglia results in anaphylatoxin
C5a-mediated pain hypersensitivity. J Neurosci. 27:8699–8708. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Inoue K and Tsuda M: Microglia in
neuropathic pain: Cellular and molecular mechanisms and therapeutic
potential. Nat Rev Neurosci. 19:138–152. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schilling T and Eder C: Microglial K(+)
channel expression in young adult and aged mice. Glia. 63:664–672.
2015. View Article : Google Scholar
|
|
19
|
Bordey A and Spencer DD: Chemokine
modulation of high-conductance Ca(2+)-sensitive K(+) currents in
microglia from human hippocampi. Eur J Neurosci. 18:2893–2898.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schilling T and Eder C: Ion channel
expression in resting and activated microglia of hippocampal slices
from juvenile mice. Brain Res. 1186:21–28. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Parzych K, Zetterqvist AV, Wright WR,
Kirkby NS, Mitchell JA and Paul-Clark MJ: Differential role of
pannexin-1/ATP/P2X7 axis in IL-1β release by human monocytes. FASEB
J. 31:2439–2445. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Scheel O, Papavlassopoulos M, Blunck R,
Gebert A, Hartung T, Zähringer U, Seydel U and Schromm AB: Cell
activation by ligands of the toll-like receptor and interleukin-1
receptor family depends on the function of the large-conductance
potassium channel MaxiK in human macrophages. Infect Immun.
74:4354–4356. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Boucsein C, Zacharias R, Färber K,
Pavlovic S, Hanisch UK and Kettenmann H: Purinergic receptors on
microglial cells: Functional expression in acute brain slices and
modulation of microglial activation in vitro. Eur J Neurosci.
17:2267–2276. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Paolicelli RC, Bolasco G, Pagani F, Maggi
L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E,
Dumas L, et al: Synaptic pruning by microglia is necessary for
normal brain development. Science. 333:1456–1458. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ueno M, Fujita Y, Tanaka T, Nakamura Y,
Kikuta J, Ishii M and Yamashita T: Layer V cortical neurons require
microglial support for survival during postnatal development. Nat
Neurosci. 16:543–551. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zeng X, Xia XM and Lingle CJ:
Species-specific differences among KCNMB3 BK beta3 auxiliary
subunits: Some beta3 N-terminal variants may be primate-specific
subunits. J Gen Physiol. 132:115–129. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee US and Cui J: {beta} subunit-specific
modulations of BK channel function by a mutation associated with
epilepsy and dyskinesia. J Physiol. 587:1481–1498. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Masuda T, Ozono Y, Mikuriya S, Kohro Y,
Tozaki-Saitoh H, Iwatsuki K, Uneyama H, Ichikawa R, Salter MW,
Tsuda M and Inoue K: Dorsal horn neurons release extracellular ATP
in a VNUT-dependent manner that underlies neuropathic pain. Nat
Commun. 7:125292016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mizoguchi Y, Kato TA, Seki Y, Ohgidani M,
Sagata N, Horikawa H, Yamauchi Y, Sato-Kasai M, Hayakawa K, Inoue
R, et al: Brain-derived neurotrophic factor (BDNF) induces
sustained intracellular Ca2+ elevation through the up-regulation of
surface transient receptor potential 3 (TRPC3) channels in rodent
microglia. J Biol Chem. 289:18549–18555. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tay TL, Mai D, Dautzenberg J,
Fernández-Klett F, Lin G, Sagar, Datta M, Drougard A, Stempfl T,
Ardura-Fabregat A, et al: A new fate mapping system reveals
context-dependent random or clonal expansion of microglia. Nat
Neurosci. 20:793–803. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Parkhurst CN, Yang G, Ninan I, Savas JN,
Yates JR III, Lafaille JJ, Hempstead BL, Littman DR and Gan WB:
Microglia promote learning-dependent synapse formation through
brain-derived neurotrophic factor. Cell. 155:1596–1609. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu Y, Zhou LJ, Wang J, Li D, Ren WJ, Peng
J, Wei X, Xu T, Xin WJ, Pang RP, et al: TNF-α differentially
regulates synaptic plasticity in the hippocampus and spinal cord by
microglia-dependent mechanisms after peripheral nerve injury. J
Neurosci. 37:871–881. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ueda H, Matsunaga H, Olaposi OI and Nagai
J: Lysophosphatidic acid: Chemical signature of neuropathic pain.
Biochim Biophys Acta. 1831:61–73. 2013. View Article : Google Scholar
|
|
34
|
Fujita R, Ma Y and Ueda H:
Lysophosphatidic acid-induced membrane ruffling and brain-derived
neurotrophic factor gene expression are mediated by ATP release in
primary microglia. J Neurochem. 107:152–160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kuwajima K, Sumitani M, Kurano M, Kano K,
Nishikawa M, Uranbileg B, Tsuchida R, Ogata T, Aoki J, Yatomi Y and
Yamada Y: Lysophosphatidic acid is associated with neuropathic pain
intensity in humans: An exploratory study. PLoS One.
13:e02073102018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu J, Ye J, Zou X, Xu Z, Feng Y, Zou X,
Chen Z, Li Y and Cang Y: CRL4A(CRBN) E3 ubiquitin ligase restricts
BK channel activity and prevents epileptogenesis. Nat Commun.
5:39242014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Meredith AL, Thorneloe KS, Werner ME,
Nelson MT and Aldrich RW: Overactive bladder and incontinence in
the absence of the BK large conductance Ca2+-activated K+ channel.
J Biol Chem. 279:36746–36752. 2004. View Article : Google Scholar : PubMed/NCBI
|