Introduction
Sepsis is a heterogeneous syndrome that is caused by
a dysregulated host response to infection (1). It was reported that there were
391,544 cases of severe sepsis and the crude case mortality rate
for severe sepsis was 37.7% in 2003 (2). Due to population aging, individuals
live longer with chronic diseases; furthermore, due to the spread
of antibiotic-resistant organisms and the broader use of
immunosuppressive and chemotherapeutic agents, the number of cases
of sepsis is increasing (3,4).
Sepsis imposes a substantial global burden in terms of morbidity
and mortality, and there is currently no satisfactory therapeutic
method, as sepsis is characterized by a complicated
pathophysiological process leading from infection, sepsis and
severe sepsis to septic shock. The disproportionate inflammatory
response to invasive infection is a triggering event, which is
closely associated with sustained excessive inflammation and immune
suppression and leads to a failure in restoring normal homeostasis.
Pro-inflammatory cytokines, such as tumor necrosis factor (TNF),
interleukin (IL)-1β, IL-12 and IL-18, serve a vital role in the
pathogenesis of sepsis, and the inhibition of these cytokines is
associated with reduced bacteremia and systemic inflammatory
response (5).
Inflammasomes are cytosolic multimeric protein
complexes that facilitate the maturation and secretion primarily of
IL-1β and IL-18 against invading pathogens or danger signals in
immune cells (6). The activation
of inflammasomes during sepsis can amplify inflammatory responses.
The nucleotide-binding domain and leucinerich repeat pyrin 3 domain
(NLRP3), one of the most well-characterized sensor proteins and
inflammasomes, initiates innate immune defense responses in
response to different danger or cell stress signals triggered by
exogenous and endogenous pathogens, reactive oxygen species (ROS),
mitochondrial (mt) DNA and extracellular ATP (7). Activated NLRP3 recruits the adaptor
protein apoptosis-associated speck-like protein containing a CARD
(ASC) and procaspase-1 to form a complex, resulting in the
autocatalytic activation of caspase-1, which cleaves the pro-forms
of inflammatory cytokines IL-1β and IL-18 into their mature forms
which act as potent cytokines secreted from the cell to become
involved in the cell immune response. Notably, the NLRP3
inflammasome has been reported to be closely associated with the
innate immune defense response in sepsis (7) and other infectious diseases in
animal models (8-10), indicating that the lack of NLRP3
contributes to the low production of inflammatory cytokines,
reduces susceptibility to inflammation and facilitates the recovery
of inflammatory response and organ damage. Mice with NLRP3-, ASC-
or caspase-1 knockdown exhibit a major defect in the maturation of
IL-1β and IL-18 in response to lipopolysaccharide (LPS) and ATP
stimulation, or are resistant to LPS-induced lethality (11,12). In vitro, macrophages serve
an important role in mediating the inflammatory response during
infection. Bacterial LPS and/or ATP may activate NLRP3 and promote
the maturation of IL-1β and IL-18 in macrophages (13,14).
Autophagy serves different roles in terms of
protection and impairment in a variety of models, however, its role
in human diseases has not been clearly defined. Autophagy is a
dynamic process from autophagosomal induction and formation to
autophagosome-lysosome fusion, which enables the regeneration of
damaged proteins and organelles, including mitochondria (15). A previous study provided evidence
that damaged mitochondria contribute to NLRP3 inflammasome-related
sepsis (16). Mitophagy, a
primary mitochondrial autophagy system, can selectively clear
impaired mitochondria and inhibit mitochondrial dysfunction. Danger
signals, such as the release of mtROS and mtDNA released from
damaged mitochondria, can activate the NLRP3 inflammasome and
further initiate cell and tissue injury (13,17).
Hydrogen (H2) serves a protective role in
inflammation-related diseases. In our previous investigations,
H2 alleviated the excessive release of pro-inflammatory
cytokines and oxidative stress factors and protected against cell,
tissue and organ damage (18-20). In addition, H2 was
shown to regulate the process of autophagy to mitigate the
inflammatory response and lung injury in sepsis (19). Therefore, it was hypothesized that
NLRP3 activation and autophagy, particularly mitophagy, may
regulate the inflammatory response and mitochondrial dysfunction in
the pathogenesis of sepsis, which is reversed by H2.
Materials and methods
Cell culture and treatment
Primary alveolar macrophages (PAMs) were obtained
from 60 male C57BL/6 adult mice (weight 20-25 g; age, 6-8 weeks;
Laboratory Animal Center of the Academy of Military Medical
Sciences). All the mice had free access to standard animal chow and
water, and were housed at room temperature (20-22°C) with 30-70%
humidity on a 12-h light/dark cycle. Following sacrifice of the
mice, the lungs were removed and washed in warm sterile
phosphate-buffered saline (PBS) supplemented with 5 mM EDTA five
times; the bronchoalveolar lavage (BAL) fluid was collected and
centrifuged at 500 × g 4°C for 5 min. The cells were resuspended in
DMEM supplemented with 10% (v/v) FBS, 1% (v/v) penicillin and
streptomycin to adjust the cell density to 1.5×106/ml.
The cells were seeded into 6-well plates and incubated at 37°C/5%
CO2 to allow the cells to adhere for 2 h. The adherent
PAMs were then collected for the following experiments. The cells
were randomly divided into two for two experiment parts: The first
part of the experiment included five groups, as follows: Control,
LPS (cells were incubated with 1 µg/ml LPS for 6 h with
normal medium), LPS+ATP (cells were incubated with 1 µg/ml
LPS for 5 h followed by treatment with 5 mM ATP for 1 h with normal
medium), LPS+H2 (cells were incubated with 1
µg/ml LPS for 6 h with H2-rich medium), and
LPS+ATP+H2 (cells were incubated with 1 µg/ml LPS
for 5 h followed by treatment with 5 mM ATP for 1 h with
H2-rich medium). The second part of the experiment
included six groups, as follows: LPS, LPS+H2, cecal
ligation and puncture (CLP)+H2+rapamycin (Rap;
BioVision, Inc., 10 nM Rap was added to the H2-rich
medium 1 h before LPS treatment), CLP+H2+3-MA
(BioVision, Inc.; 1.5 mM 3-MA was added to the H2-rich
medium 1 h before LPS treatment), CLP+H2+NLRP3 inhibitor
(20 µM NLRP3 inhibitor MCC950, Sigma Aldrich; Merck KGaA,
was added into the H2-rich medium 1 h before LPS
treatment), and CLP+H2+NLRP3 inhibitor+3-MA [20
µM NLRP3 inhibitor and 10 mg/kg 3-MA, (BioVision, Inc.)]
were added to the H2-rich medium 1 h before LPS
treatment). The culture supernatants of the first and the second
parts of the experiment were collected to detect IL-1β, IL-18,
TNF-α and IL-6 by ELISA. The cells were collected to evaluate
mitochondrial function through mitochondrial membrane potential
(MMP), the respiratory control ratio (RCR), ATP content and mtDNA,
to analyze the protein expression of caspase-1 P10, NLRP3, ASC,
microtubule-associated protein 1 light chain 3 (LC3), Beclin 1,
PINK1, Parkin and voltage-dependent anion channel (VDAC) by western
blotting, and to examine the mRNA expression of LC3, Beclin 1,
PINK1 and Parkin by reverse transcription-quantitative polymerase
chain reaction (RT-qPCR) analysis.
Animals, CLP procedure and treatment
Another 130 male C57BL/6 mice (weighing 20-25 g and
aged 6-8 weeks) were obtained from the Laboratory Animal Center of
the Academy of Military Medical Sciences (Beijing, China). All mice
had free access to standard animal chow and water and were housed
at room temperature (20-22°C) with 30-70% humidity on a 12/12-h
light/dark cycle. All experimental procedures were approved by the
Institutional Animal Care and Use Committee of Tianjin Medical
University (Tianjin, China) and were performed in accordance with
the National Institutes of Health Guide for the Care and Use of
Laboratory Animals. All efforts were made to minimize the suffering
of the 130 mice (n=23/group) used. A sepsis model was established
by CLP as previously described (18). The mice were randomly divided into
seven groups as follows: Control, CLP, CLP+H2
[H2-rich saline (5 ml/kg) was injected i.p. 1 and 6 h
following the sham and CLP procedures], CLP+H2+Rap (Rap,
an inducer of autophagy, was administered by intraperitoneal
injection at 10 mg/kg BW 1 h before CLP; H2 treatment
was as in the CLP+H2 group), CLP+H2+3-MA
(3-MA, an inhibitor of autophagy, was administered by
intraperitoneal injection at 15 mg/kg BW 1 h before CLP;
H2 treatment was as for the CLP+H2 group).
Subsequently, the mice were anesthetized with sodium pentobarbital
(50 mg/kg, intraperitoneally) and sacrificed via decollation; lung
tissues were collected to determine the tissue pathological scores
by hematoxylin and eosin (H&E) staining, wet-to-dry (W/D)
weight ratio and myeloperoxidase (MPO) activity. BAL fluid was
collected to analyze the total proteins in the lung. Liver and
kidney tissues were collected to investigate pathological scores.
Blood and tissue homogenates of the lung, liver and kidney were
obtained to detect the biochemical parameters of the liver and
kidney, and the levels of IL-1β, IL-18 and TNF-α by ELISA. Other
parts of the lung, liver and kidney were collected and proteins
were extracted for the detection of procaspase-1, caspase-1 P10,
NLRP3 and ASC by western blotting. The survival rate was analyzed
1, 2, 3, 5 and 7 days after surgery.
ELISA
The supernatants and tissues of the lung, liver and
kidney were collected. The tissue samples were homogenized and
centrifuged at 10,000 g for 10 min at 4°C, and the supernatants
were used to analyze IL-1β (cat. no. RLB00; R&D Systems, Inc.),
IL-18 (cat. no. 7625; R&D Systems, Inc.), TNF-α (cat. no.
RTA00; R&D Systems, Inc.) and IL-6 (cat. no. R6000B; R&D
Systems, Inc.) using ELISA commercial kits according to the
manufacturer's instructions.
Transmission electron microscopy
(TEM)
The lung tissues were harvested, cut into small
pieces and immediately fixed in 0.25% glutaraldehyde at 4°C
overnight. The macrophages were isolated, concentrated and fixed in
0.25% glutaraldehyde at 4°C overnight. Following fixation and
dehydration, 50-nm sections were prepared with using an
ultramicrotome (Ultracut UCT; Leica Microsystems, Inc.). The
sections were stained with 2% uranyl acetate for 30 min at 4°C,
followed by staining in lead citrate solution 30 min at room
temperature. The sections were observed using a transmission
electron microscope (Hitachi, Ltd., Tokyo, Japan).
Western blot assay
Macrophages and the lung, liver and kidney tissues
were harvested and lysed in RIPA lysis buffer. Protein quantitation
was performed with the BCA assay kit (Thermo Fisher Scientific,
Inc.). Protein (15 µg) was loaded onto 10% SDA-PAGE gels and
electrophoretically transferred onto PVDF membranes (EMD Millipore)
by conventional wet blotting. The membranes were blocked with
blocking buffer (5% non-fat dry milk) for 1 h, and then incubated
with primary antibodies against procaspase-1, caspase-1 P10 (cat.
no. sc-56036, 1:200, Santa Cruz Biotechnology, Inc.), NLRP3 (cat.
no. ab214185; 1:500, Abcam), ASC (cat. no. sc-514414, 1:200, Santa
Cruz Biotechnology, Inc.), LC3 (cat. no. ab48394; 1:500, Abcam),
Beclin 1 (cat. no. ab62557; 1:500, Abcam), PINK1 (cat. no. ab23707;
1:500, Abcam), Parkin (cat. no. ab77924; 1:500, Abcam), VDAC (cat.
no. ab14734; 1:200, Abcam) and β-actin (cat. no. ab8227; 1:2,000,
Abcam) in blocking buffer at 4°C overnight. Subsequently, the
membranes were washed with TBST three times, followed by incubation
with corresponding horseradish peroxidase-conjugated goat IgG
secondary antibodies (cat. nos. ab6721 and ab6728; 1:5,000, Abcam)
at 37°C for 1.5 h. Following washing with TBST three times, the
blots were analyzed with ECL reagent (EMD Millipore). The membranes
were visualized using the UVP Bio-Imaging System. The blot
densities were analyzed with Quantity One (v4.6) software (Bio-Rad
Laboratories, Inc.). Protein expression was normalized to
β-actin.
RT-qPCR analysis
RNA was isolated from cells using the TRIzol reagent
(cat. no. 15596026; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. cDNA was obtained using a RevertAid
First Strand cDNA Synthesis kit (cat. no. K1621; Thermo Fisher
Scientific, Inc.). relative mRNA expression was detected by qPCR
using SYBR PCR Master Mix (Thermo Fisher Scientific, Inc.) on a
7500 Real-time PCR system (Thermo Fisher Scientific, Inc.). The
amplification conditions were as follows: Pre-degeneration at 95°C
for 10 min, then 40 cycles of denaturing at 95°C for 15 sec,
annealing at 60°C for 30 sec and extension at 72°C for 30 sec, and
a final extension at 72°C for 5 min. The gene-specific primer
sequences are listed in Table I.
Relative fold expressions were normalized to GAPDH and calculated
using the 2−∆∆Cq method (21).
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene | Primer | Sequence
('5-3') |
|---|
| LC3 | Forward |
TCCACTCCCATCTCCGAAGT |
| Reverse |
TTGCTGTCCCGAATGTCTCC |
| Beclin 1 | Forward |
ACCAGCGGGAGTATAGTGAGT |
| Reverse |
CAGCTGGATCTGGGCGTAG |
| PINK1 | Forward |
CCATCGGGATCTCAAGTCCG |
| Reverse |
GATCACTAGCCAGGGACAGC |
| Parkin | Forward |
TTCCCGTTCAGCTCTGGG |
| Reverse |
CCCTGCATCCACTGGTGC |
| GAPDH | Forward |
GTGTTTCCTCGTCCCGTAGA |
| Reverse |
AATCTCCACTTTGCCACTGC |
Mitochondrial function
MMP measurement
MMP was identified using JC-1 dye (Beyotime
Institute of Biotechnology) according to the manufacturer's
instructions. Following the experiment, the cells were dyed with
JC-1 staining solution at 37°C for 30 min, and the fluorescent
properties changed from green to red when the level of MMP was
high. Red fluorescent JC-1 aggregates form in hyperpolarized
membranes, whereas green fluorescent monomeric forms indicate
membrane depolarization. The higher the ratio of red to green
fluorescence, the more intact the mitochondrial membrane.
RCR analysis
The isolation of intact mitochondria from cells was
performed according to the method described by Iglesias-González
et al (22). The cells
were centrifuged at 1,000 × g for 10 min at 4°C, following which
the supernatant was collected and centrifuged again at 10,000 × g
for 10 min at 4°C. The resulting mitochondrial pellet was gently
washed twice, resuspended and diluted in a minimal volume of
isolation buffer. All steps of mitochondrial isolation were
performed on ice at 4°C. The protein concentration was determined
using the Pierce BCA Protein Assay kit (Thermo Fisher Scientific,
Inc.). The isolated mitochondria (1 mg/ml) were incubated with 2 ml
respiration buffer (70 mmol/l sucrose, 1 mmol/l EDTA, 225 mmol/l
mannitol, 200 mmol/l KH2PO4, 200 mmol/l
K2HPO4 and 0.1% BSA; pH 7.4) at 30°C in a
temperature-controlled water bath with continuous stirring. The
mitochondrial oxygen consumption was evaluated with a Clark-type
electrode (Hansatech-Instruments, Ltd.). The state 3 respiration
rate reflects the oxygen consumption rate in the presence of ADP,
whereas the state 4 respiration rate reflects the oxygen
consumption rate when ADP is exhausted. The RCRs were estimated as
state 3/state 4.
ATP detection
The ATP content was determined with the ATP
Bioluminescence Assay kit (no. 11699709001, Roche Diagnostics)
according to the manufacturer's instructions.
mtDNA detection
Total DNA was isolated from cells with a DNeasy
Blood & Tissue kit according to the manufacturer's instructions
(Qiagen, Inc.). The quality of the extracted DNA was evaluated by
qPCR analysis. The cycling program for amplification was 95°C for 2
min, followed by 95°C for 10 sec, 60°C for 30 sec, and 95°C for 10
sec for 40 cycles. The copy number of mtDNA was normalized to that
of nuclear DNA (cytochrome c oxidase I/18S ribosomal RNA).
The mitochondrial primer sequences were selected and were as
follows: Mouse cytochrome c: Forward 5′-TTT GGG TCC CTT CTA
GGA GTC-3′ and reverse 5′-CCG ACA TGA AGG AAT AAG CAA-3′; murine
β2-microglobulin: Forward 5′-ATG GGA AGC CGA ACA TAC TG-3′ and
reverse 5′-CAG TCT CAG TGG GGG TGA AT-3′. The relative gene
expression was analyzed, and the ratio of mtDNA/nDNA was 100% in
the control or LPS groups, according to CFX Manager 2.1 software
(Bio-Rad Laboratories, Inc.).
Determination of mtROS
mtROS production was measured using the ROS-specific
fluorescent probe, dichloro-dihydro-fluorescein diacetate
(DCFH-DA). The fluorescence intensity was measured in a fluorescent
microplate reader (BioTek Instruments, Inc.). The fluorescence
intensity was normalized to that of the control group.
Tissue pathological scores
Following the experiment, the lung, liver and kidney
tissues were perfused with 10% formalin and fixed for 24 h at room
temperature. After embedding in paraffin, 5 µm sections of
lung tissues were cut, dewaxed and stained with H&E.
Histopathological analysis of the tissues was performed following
staining with H&E. The sections were observed and photographed
under a microscope (Biorevo BZ-9000; Keyence Corporation), and
evaluated by at least two pathologists who were blinded to the
study protocol. The scoring standard is shown in Table SI.
Lung MPO
The lung tissues were harvested and collected to
measure the lung MPO activity using a commercial kit (cat. no.
K747; BioVision, Inc.) following the manufacturer's
instructions.
BAL total protein
The mice were sacrificed, and a small catheter was
placed in the trachea and secured. Isotonic saline (500 µl)
was slowly instilled and then gently withdrawn. This fluid was
instilled and withdrawn three times in total. The BALF was
centrifuged at 1,500 g for 10 min, at 4°C and the supernatant was
used to assess the total protein in the BAL using a protein assay
commercial kit (Bio-Rad Laboratories, Inc.).
Lung W/D ratio
The harvested wet lung was weighed and then placed
in an oven for 24 h at 80°C. The lung was then weighed when it had
dried.
Biochemical parameters of the liver and
kidney
Blood was collected for the measurement of
biochemical parameters [alanine aminotransferase (ALT), aspartate
aminotransferase (AST), blood urea nitrogen (BUN) and creatinine
(Cr)] with commercial ELISA kits (R&D Systems, Inc.) according
to the manufacturer's instructions.
Statistical analysis
The survival rates are expressed as percentages and
analysis was performed with the log-rank test. All other results of
experiments are expressed as the mean ± standard deviation and were
analyzed by one-way ANOVA with the Tukey's multiple comparisons
test. Statistical analyses were performed using GraphPad Prism
(v.5.0; GraphPad Software, Inc.). In all tests, P<0.05 was
considered to indicate a statistically significant difference.
Results
H2 downregulates
NLRP3-mediated cytokine release following LPS and ATP stimulation
in PAMs
The ATP-driven activation of caspase-1 is a common
model for NLRP3 inflammasome-mediated caspase-1 activation in
LPS-induced macrophages (11,12). To investigate the effect of
H2 on cytokines and inflammasome pathways, LPS, ATP and
H2-rich medium were used to treat macrophages. As shown
in Fig. 1, the expression levels
of IL-1β (Fig. 1A), IL-18
(Fig. 1B), TNF-α (Fig. 1C), IL-6 (Fig. 1D) and caspase-1 P 10 (Fig. 1E) were increased in the LPS and
LPS+ATP groups compared with those in the Con group (P<0.05);
however, no significant differences in TNF-α (Fig. 1C) or IL-6 (Fig. 1D) were detected between the LPS
and LPS+ATP groups (P>0.05). H2 treatment alleviated
the increased expression of IL-1β (Fig. 1A), IL-18 (Fig. 1B) and caspase-1 P 10 (Fig. 1E and H) compared with expression
in the LPS and LPS+ATP groups (P<0.05).
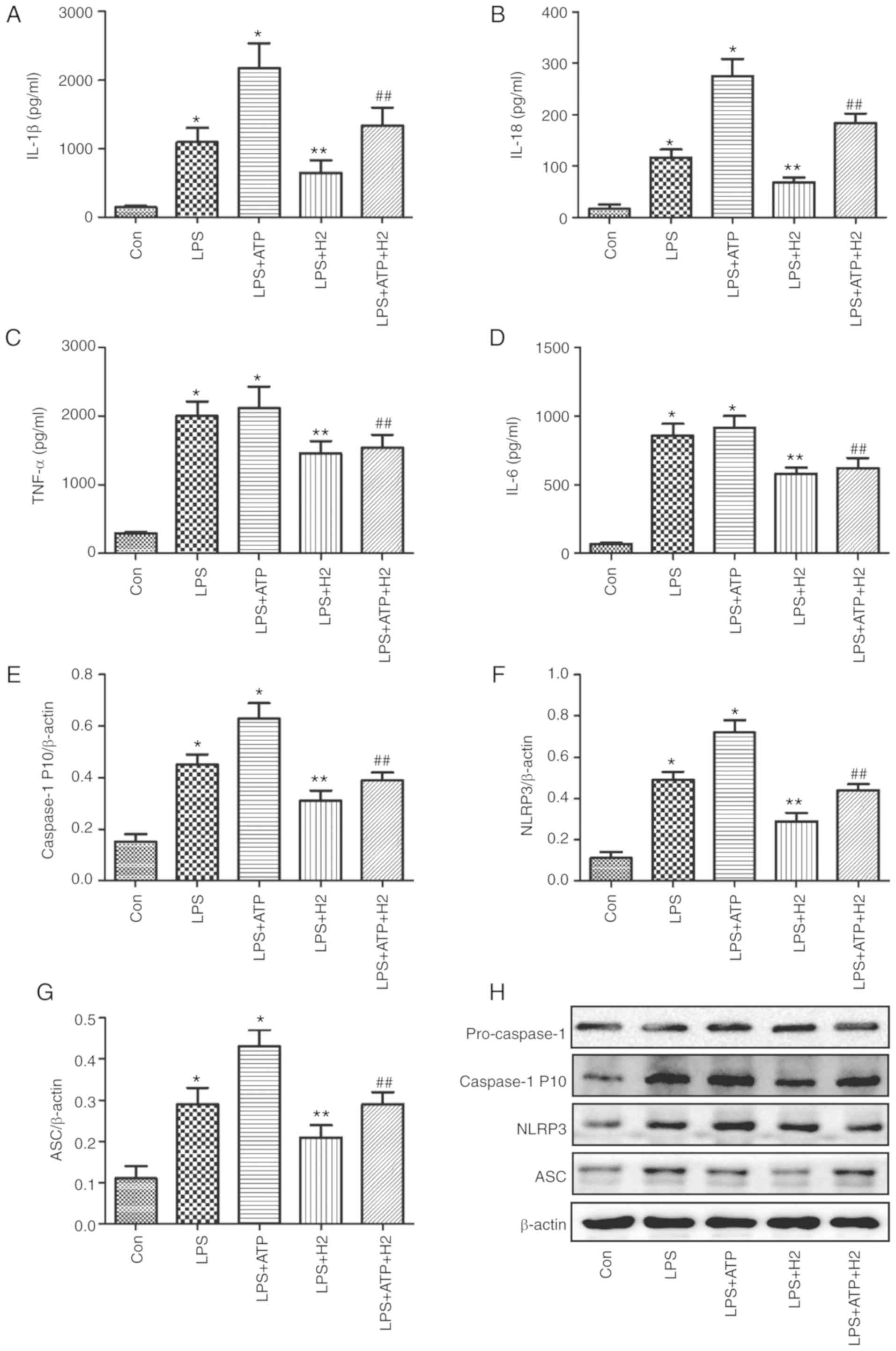 | Figure 1Cytokine and inflammasome protein
expression in macrophages treated with LPS, ATP and H2.
Macrophages were exposed to by LPS, ATP and H2. The
culture supernatants were collected to detect cytokines (A) IL-1β,
(B) IL-18, (C) TNF-α and (D) IL-6 by ELISA, and cells were
harvested to measure the expression of (E) caspase-1, (F) NLRP3 and
(G) ASC by western blotting. (H) Blots of caspase-1, NLRP3 and ASC
in cells. Data are expressed as the mean ± standard deviation
(n=6). *P<0.05 vs. Con group, **P<0.05
vs. LPS group, ##P<0.05 vs. LPS+ATP group. LPS,
lipopolysaccharide; IL, interleukin; TNF, tumor necrosis factor;
ASC, apoptosis-associated speck-like protein containing a CARD;
NLRP3, NACHT, LRR and PYD domains-containing protein 3;
H2, hydrogen; Con, control. |
Macrophages can express intracellular Nod-like
receptors (NLRs) when induced by detrimental stimulation. The NLRP3
inflammasome is a critical member of the NLR family, as it
interacts with ASC and sequentially regulates the expression of
mature IL-1β, IL-18 and caspase-1. The NLRP3 inflammasome pathway
protein was investigated in the present study. Consistent with the
changes in IL-1β, IL-18 and caspase-1 P10, LPS and ATP activated
the inflammasome, NLRP3 (Fig. 1F and
H) and increased the expression of ASC (Fig. 1G and H) (P<0.05), and there was
a significant difference between groups LPS and LPS+ATP
(P<0.05). The expression levels of NLRP3 (Fig. 1F and H) and ASC (Fig. 1G and H) were reduced in the
LPS+H2 and LPS+ATP+H2 groups compared with
those in the LPS and LPS+ATP groups, respectively (P<0.05).
These data indicated that LPS and ATP activated the inflammasome
and promoted the maturation and release of cytokines, whereas
H2 alleviated inflammasome activation and the release of
cytokines in response to LPS and ATP.
H2 improves the mitochondrial
dysfunction induced by LPS and ATP in PAMs
Mitochondrial function was assessed through MMP,
RCR, ATP content and ROS. mtDNA is a damage-associated molecular
pattern. When mitochondria are injured, mtDNA is released into the
cytosol. LPS and ATP treatment reduced MMP (the
monomer/J-aggregates value was upregulated), mtDNA copy number, RCR
and ATP content, and increased ROS release in macrophages (Fig. 2A-E, P<0.05); however, there was
no significant difference between the LPS and LPS+ATP groups
(Fig. 2A, P>0.05).
H2-rich medium markedly increased MMP (the
monomer/J-aggregates value was downregulated), mtDNA copy number,
RCR and ATP content, and reduced ROS release in the LPS and
ATP-treated cells compared with results in the LPS group and
LPS+ATP group, respectively (Fig.
2A-E, P<0.05). These data suggest that H2-rich
medium treatment alleviated the mitochondrial dysfunction and
injury in macrophages induced by LPS and ATP.
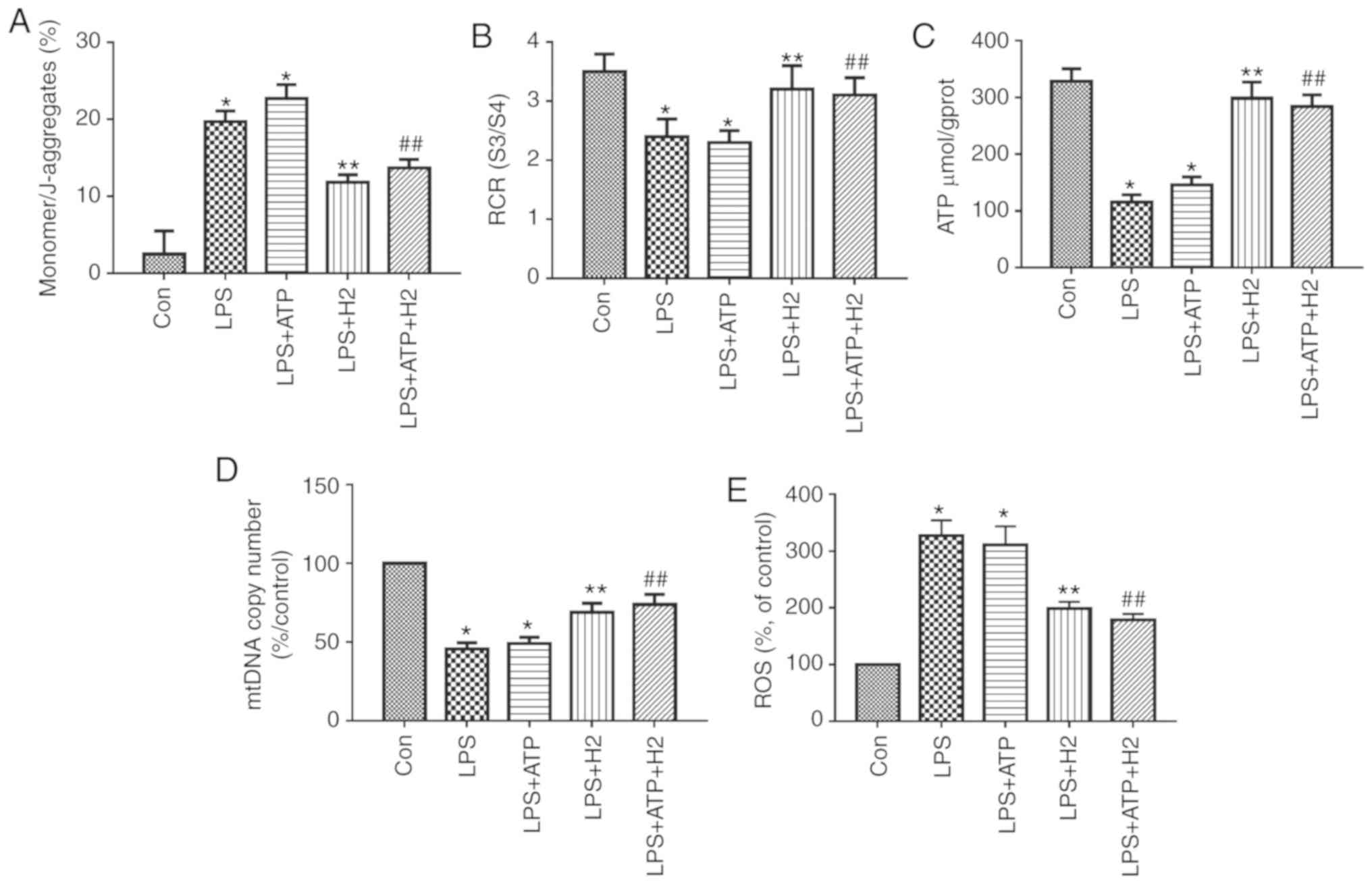 | Figure 2Mitochondria and mtDNA in macrophages
treated with LPS, ATP and H2. Macrophages were treated
by LPS, ATP and H2. Cells were harvested to measure (A)
MMP, (B) RCR, (C) ATP, (D) mtDNA and (E) ROS release. Data are
expressed as the mean ± standard deviation (n=6).
*P<0.05 vs. Con group, **P<0.05 vs. LPS group,
##P<0.05 vs. LPS+ATP group. mtDNA, mitochondrial DNA;
LPS, lipopolysaccharide; MMP, mitochondrial membrane potential;
RCR, respiratory control ratio; ROS, reactive oxygen species;
H2, hydrogen; Con, control. |
Effect of H2 on
autophagy-related proteins in alveolar macrophages during LPS and
ATP treatment
Autophagy is a vital process responsible for
removing senescent or damaged organelles. LC3I, a cytosolic form of
LC3, is conjugated to phosphatidylethanolamine to form
LC3-phosphatidylethanolamine conjugate (LC3II), which reflects the
autophagic activity. Beclin 1 is a core component of the autophagy
machinery. LPS or LPS+ATP induced the increasing of number of
autophagosomes compared with that in the Con group (Fig. 3A, P<0.05), H2
treatment further promoted the number of autophagosomes in the
LPS+H2 group and LPS+ATP+H2 group compared
with that in the LPS group or LPS+ATP group, respectively (Fig. 3A, P<0.05). As shown in Fig. 3B, C and G, LPS and ATP enhanced
autophagic activity, as indicated by the alternation of LC3I into
LC3II and the increased expression of Beclin 1 in macrophages
(P<0.05), whereas H2 further promoted the process of
autophagy. The expression levels of LC3II and Beclin 1 were further
elevated in the LPS+H2 and LPS+ATP+H2 groups
compared with those in the LPS or LPS+ATP groups, respectively
(P<0.05).
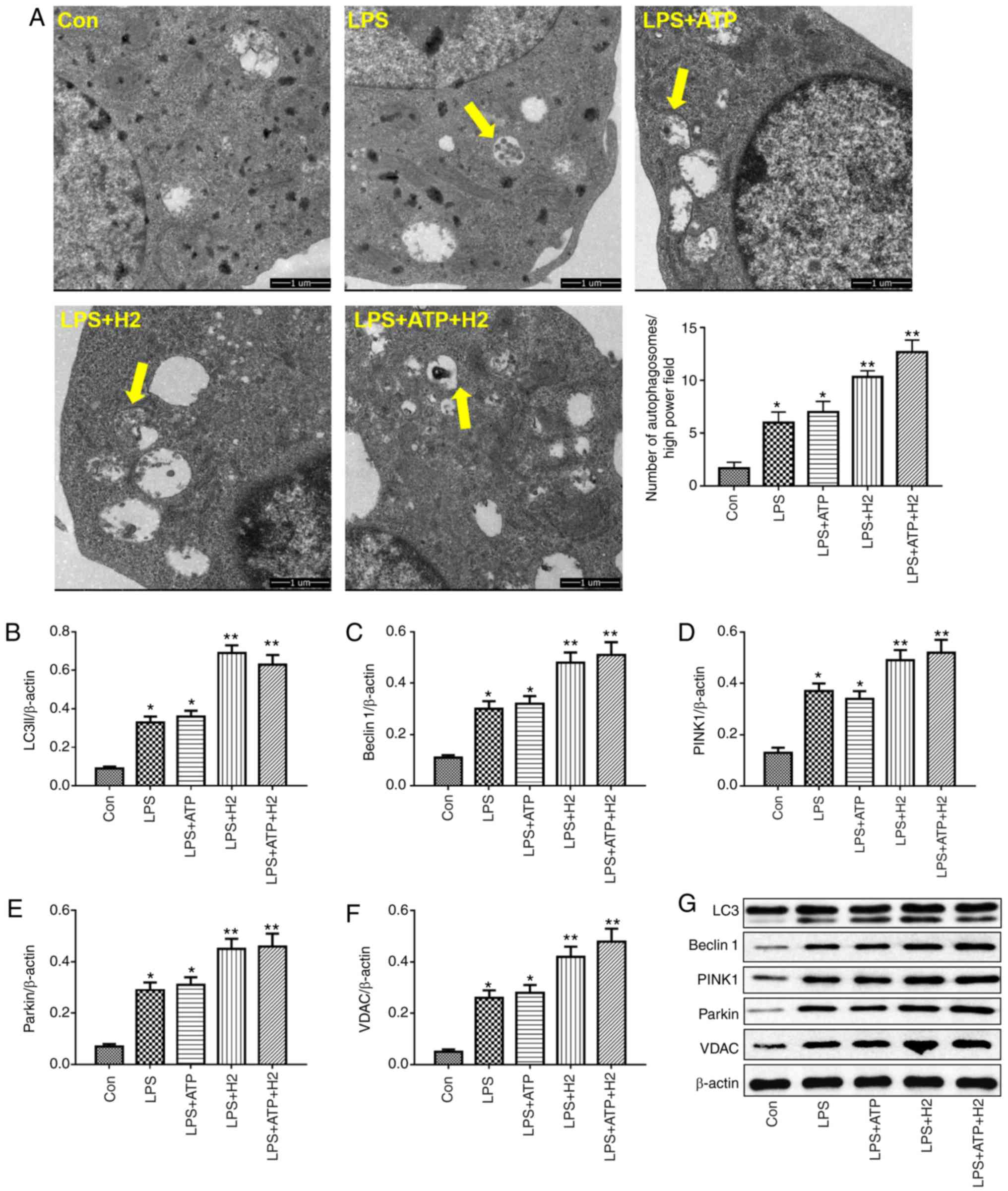 | Figure 3Autophagy-related protein expression
in macrophages treated with LPS, ATP and H2. Macrophages
were treated by LPS, ATP and H2. Cells were harvested to
measure the (A) number of autophagosomes (indicated by yellow
arrows; magnification, ×10,000), and the expression of (B) LC3, (C)
Beclin 1, (D) PINK1, (E) Parkin and (F) VDAC by western blotting.
(G) Representative blots of LC3, Beclin 1, Parkin and VDAC in
cells. Data are expressed as the mean ± standard deviation (n=6).
*P<0.05 vs. Con group, **P<0.05 vs. LPS
group. LPS, lipopolysaccharide; LC3, microtubule-associated protein
1 light chain 3; PINK1, PTEN-induced putative kinase 1; VDAC,
voltage-dependent anion channel; H2, hydrogen; Con,
control. |
Parkin is the most well-known E3 ubiquitin ligase
involved in mitophagy. The PINK-Parkin pathway serves a critical
role in mitophagy (23). VDACs
are necessary for efficient recruitment of Parkin to the
mitochondria during the process of mitophagy. Consistent with the
variations in LC3 and Beclin 1, LPS and ATP also induced mitophagic
activity, with increased expression of PINK1, Parkin and VDAC
(Fig. 3D-G, P<0.05). Compared
with the LPS and LPS+ATP groups, H2 treatment further
increased the expression of PINK1, Parkin and VDAC in the
LPS+H2 and LPS+ATP+H2 groups (Fig. 3D-G, P<0.05). This
autophagy-related protein increase, particularly mitophagic
protein, indicated that H2 was able to increase
mitophagic activity in macrophages stimulated by LPS and ATP.
H2 regulates NLRP3-mediated
cytokine release via autophagy in LPS-treated macrophages
Accumulated evidence has verified that autophagy
regulates the NLRP3 inflammasome activation as an upstream pathway
(13). To examine the induction
and inhibition of autophagy, autophagy-related gene expression was
detected following treatment with autophagy inducer (Rap) or
inhibitor (3-MA). It was observed that the mRNA expression levels
of LC3, Beclin 1, PINK1 and Parkin were increased in the
LPS+H2+Rap group and reduced in the
LPS+H2+3-MA group compared with those in the
LPS+H2 group (Fig.
S1, P<0.05). To further investigate the effect of
H2 on NLRP3 inflammasome activation via autophagy, the
cells were treated with autophagy inducer Rap and inhibitor 3-MA,
and NLRP3 inhibitor MCC950. Compared with the levels in the LPS
group, the expression levels of IL-1β (Fig. 4A), IL-18 (Fig. 4B), TNF-α (Fig. 4C), IL-6 (Fig. 4D) and caspase-1 P10 (Fig. 4E) were considerably reduced in the
LPS+H2, LPS+H2+Rap and
LPS+H2+NLRP3 inhibitor groups (P<0.05). Compared with
the LPS+H2 group, the downregulation of IL-1β (Fig. 4A), IL-18 (Fig. 4B), TNF-α (Fig. 4C), IL-6 (Fig. 4D) and caspase-1 P10 (Fig. 4E) was significant in the
LPS+H2+Rap group (P<0.05). IL-1β (Fig. 4A), IL-18 (Fig. 4B) and caspase-1 P10 (Fig. 4E) were also significantly
decreased in the LPS+H2+NLRP3 inhibitor group
(P<0.05); however, TNF-α (Fig.
4C) and IL-6 (Fig. 4D) did
not differ significantly between the LPS+H2 and
LPS+H2+NLRP3 inhibitor groups (P>0.05); Compared with
expression in the LPS+H2 group, IL-1β (Fig. 4A), IL-18 (Fig. 4B), TNF-α (Fig. 4C), IL-6 (Fig. 4D) and caspase-1 P10 (Fig. 4E) were increased in the
LPS+H2+3-MA group (P<0.05). To investigate whether
H2 reduced the activation of NLRP3 via autophagy, 3-MA
was used to inhibit autophagy in the LPS+H2+NLRP3
inhibitor group; the result demonstrated that, compared with the
LPS+H2+NLRP3 inhibitor group, all the above-mentioned
indicators (Fig. 4A-E) were
markedly upregulated in the LPS+H2+3-MA+NLRP3 inhibitor
group (P<0.05).
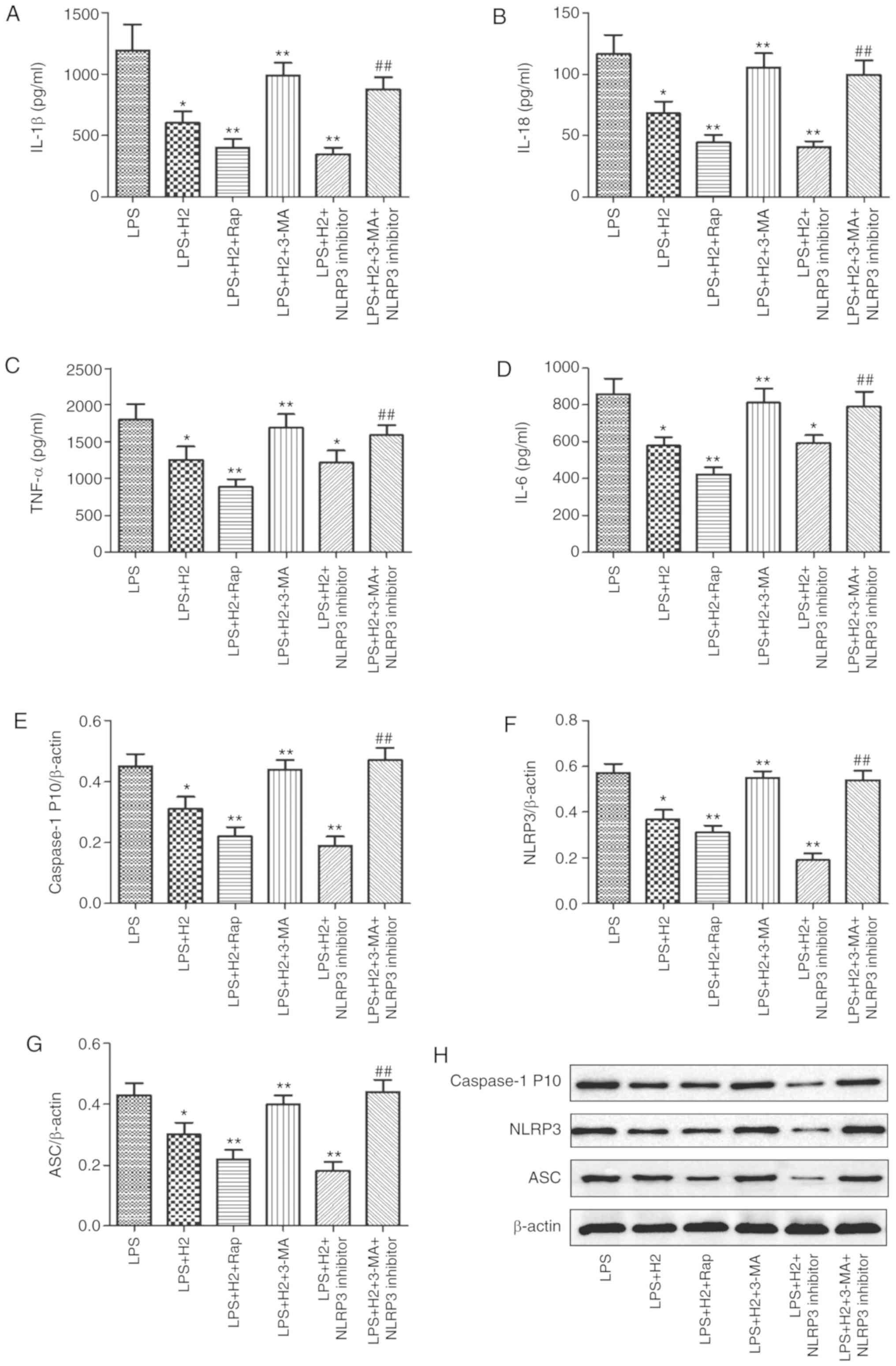 | Figure 4Effect of autophagy on inflammasome
activation in macrophages induced by LPS and treated with
H2. Macrophages were treated by LPS, ATP and
H2, autophagy inducer Rap, and autophagy inhibitor 3-MA.
The culture supernatants were collected to detect cytokines (A)
IL-1β, (B) IL-18, (C) TNF-α and (D) IL-6 by ELISA, and cells were
harvested to measure the expression of (E) caspase-1, (F) NLRP3 and
(G) ASC by western blotting. (H) Representative blots of caspase-1,
NLRP3 and ASC in cells. Data are expressed as the mean ± standard
deviation (n=6). *P<0.05 vs. LPS group,
**P<0.05 vs. LPS+H2 group,
##P<0.05 vs. LPS+H2+NLRP3 inhibitor group.
LPS, lipopolysaccharide; Rap, rapamycin; 3-MA, 3-methyladenine; IL,
interleukin; TNF, tumor necrosis factor; NLRP3, NACHT, LRR and PYD
domains-containing protein 3; ASC, apoptosis-associated speck-like
protein containing a CARD; H2, hydrogen. |
Consistent with the results in cytokines, NLRP3 and
ASC were reduced in the LPS+H2 group compared with
levels in the LPS group; the autophagy inducer and NLRP3 inhibitor
further reduced the expression of NLRP3 and ASC in macrophages
treated with LPS (Fig. 4F and G,
P<0.05). Compared with those in the LPS+H2 group, the
expression levels of NLRP3 and ASC were increased in the
LPS+H2+3-MA group. Compared with expression in the
LPS+H2+NLRP3 inhibitor group, NLRP3 and ASC were
significantly aggravated in the LPS+H2+3-MA+NLRP3
inhibitor group (Fig. 4F and G,
P<0.05). These results suggest that H2 and Rap
synergistically induced autophagy and inhibited NLRP3 inflammasome
activation and that H2 reversed this inhibition of the
NLRP3 inflammasome by the autophagy inhibitor 3-MA.
H2 improves mitochondrial dysfunction via
the autophagy-mediated NLRP3 inflammasome pathway in LPS-treated
macrophages
To investigate the effect of H2 on
mitochondrial dysfunction and damage via the autophagy-mediated
NLRP3 inflammasome pathway, the cells were treated with autophagy
inducer Rap and inhibitor 3-MA, and with the NLRP3 inhibitor
MCC950. MMP, RCR, ATP content, mtDNA and ROS were assessed to
verify this hypothesis. H2 treatment increased MMP (the
monomer/J-aggregates value was downregulated), RCR, ATP content and
mtDNA copy number and reduced ROS release in the LPS+H2
group compared with results in the LPS group (Fig. 5A-E, P<0.05). The autophagy
inducer Rap and NLRP3 inhibitor further increased MMP (the
monomer/J-aggregates value was down-regulated), RCR, ATP content
and mtDNA copy number and reduced ROS release in the
LPS+H2+Rap and LPS+H2+NLRP3 inhibitor groups
compared with results in the LPS+H2 group (Fig. 5A-D, P<0.05). The autophagy
inhibitor 3-MA markedly reduced MMP (the monomer/J-aggregates value
was increased), RCR, ATP content and mtDNA copy number and
increased ROS release in the LPS+H2+3-MA group compared
with results in the LPS+H2 group (Fig. 5A-E, P<0.05). Compared with the
LPS+H2+NLRP3 inhibitor group, MMP (the
monomer/J-aggregates value was increased), RCR, ATP content and
mtDNA copy number were significantly reduced and ROS release was
increased in the LPS+H2+3-MA+NLRP3 inhibitor group
(Fig. 5A-E, P<0.05).
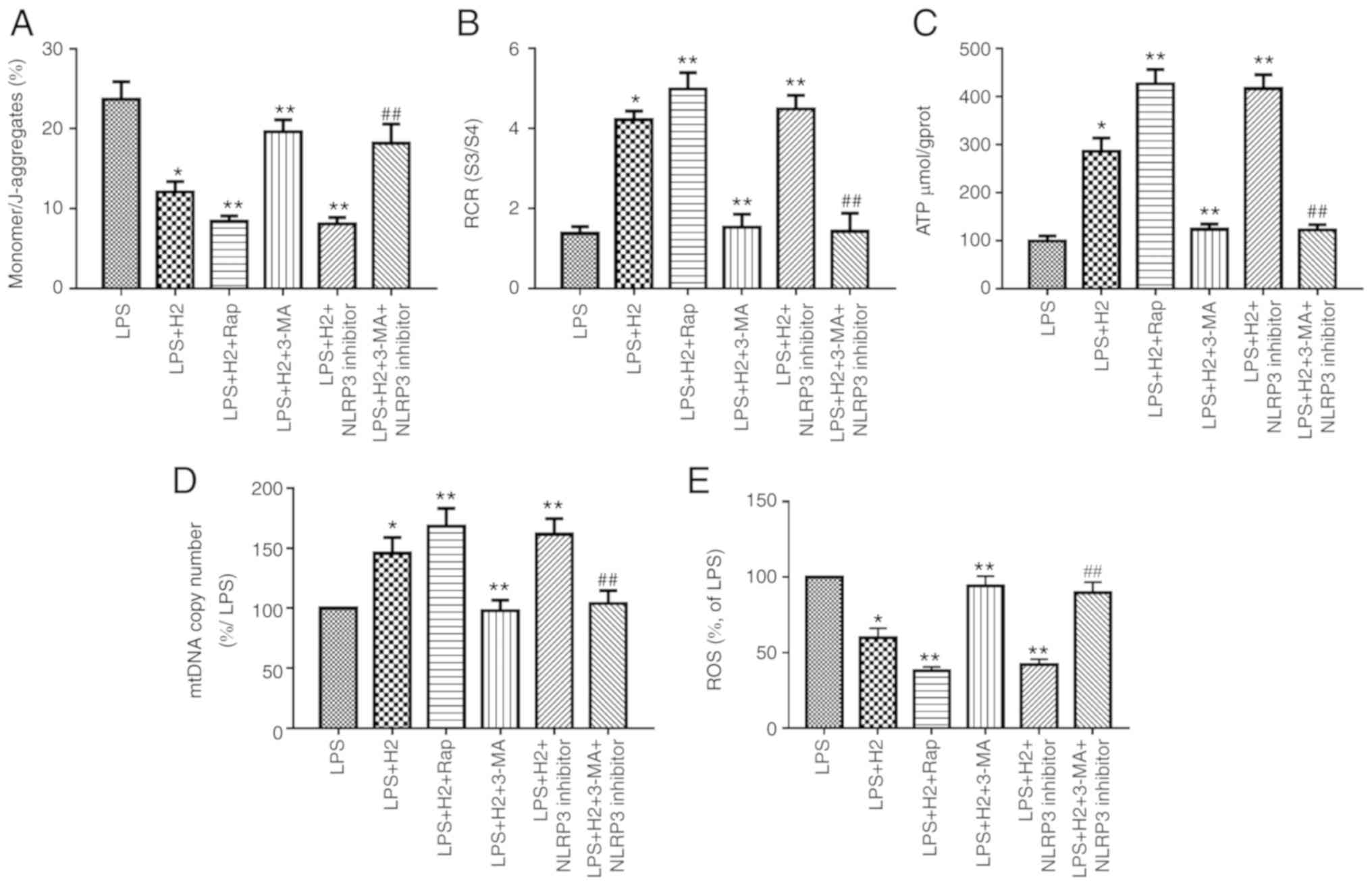 | Figure 5H2 alleviates
mitochondrial dysfunction in macrophages induced by LPS via
autophagy-mediated NLRP3 inactivation. Macrophages were treated
with LPS, ATP and H2, autophagy inducer Rap and
autophagy inhibitor 3-MA. Cells were harvested to measure (A) MMP,
(B) RCR, (C) ATP, (D) mtDNA and (E) ROS release. Data are expressed
as the mean ± standard deviation (n=6). *P<0.05 vs.
LPS group, **P<0.05 vs. LPS+H2 group,
##P<0.05 vs. LPS+H2+NLRP3 inhibitor group.
LPS, lipopolysaccharide; Rap, rapamycin; 3-MA, 3-methyladenine;
MMP, mitochondrial membrane potential; RCR, respiratory control
ratio; mtDNA, mitochondrial DNA; ROS, reactive oxygen species;
H2, hydrogen. |
H2 improves survival rate via
the autophagy-mediated NLRP3 inflammasome pathway in mice following
CLP
To investigate the effect of the autophagy-mediated
NLRP3 inflammasome pathway on H2 promoting the survival
rate in CLP mice, the mice were treated with autophagy inducer Rap
and inhibitor 3-MA following CLP, and the survival rate at 1, 2, 3,
5 and 7 days was recorded. The results (Fig. 6K) revealed that the survival rate
was higher in the CLP+H2 and CLP+H2+Rap
groups compared with that in the CLP group (P<0.05). Compared
with that in the CLP+H2, group, the survival rate was
lower in the CLP+H2+3-MA group (P<0.05).
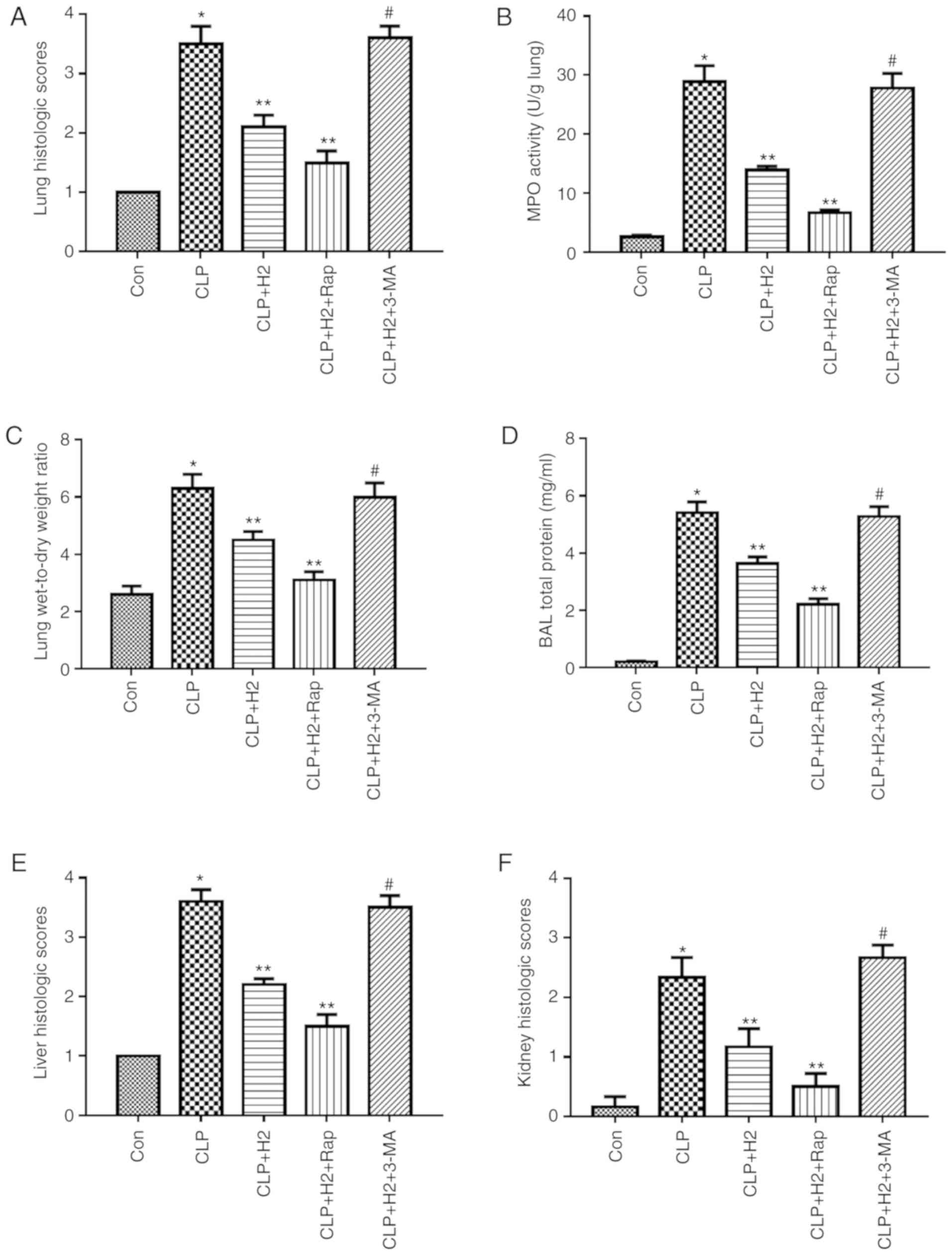 | Figure 6Effect of autophagy-mediated NACHT,
LRR and PYD domains-containing protein 3 inactivation on lung
injury, biochemical parameters of liver and kidney and survival
rate in LPS-induced macrophages treated with H2. Sepsis
was produced by CLP. Septic mice were treated with H2,
autophagy inducer Rap and autophagy inhibitor 3-MA. After 24 h,
lung tissues were collected to detect (A) pathological tissue
changes by hematoxylin and eosin staining, (B) MPO activity and (C)
W/D weight ratio; (D) bronchoalveolar lavage fluid was collected to
analyze total proteins. (E) Liver and (F) kidney tissues were
collected to investigate pathological scores. Blood was obtained to
measure the biochemical parameters (G) ALT, (H) AST, (I) Cr and (J)
BUN. (K) Survival rate was analyzed at 1, 2, 3, 5 and 7 days
post-CLP (n=20). Data are expressed as the mean ± standard
deviation (n=6). *P<0.05 vs. Con group,
**P<0.05 vs. CLP group, #P<0.05 vs.
CLP+H2 group. LPS, lipopolysaccharide; CLP, cecal
ligation and puncture; Rap, rapamycin; 3-MA, 3-methyladenine; MPO,
myeloperoxidase, W/D, wet/dry; ALT, alanine transaminase; AST,
aspartate transaminase; BUN, blood urea nitrogen; Cr, creatinine;
H2, hydrogen; Con, control. |
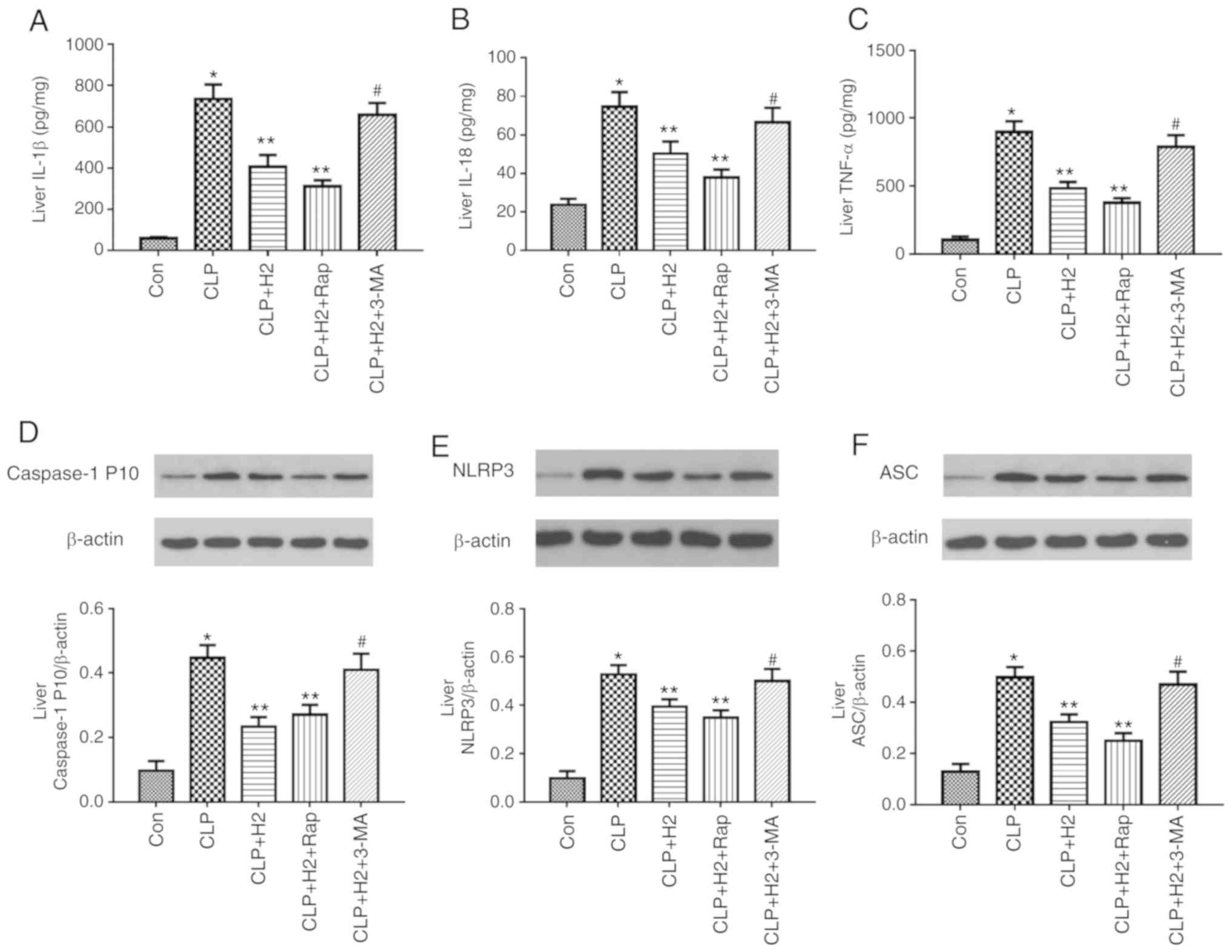
H2 improves lung, kidney and
liver dysfunction via the autophagy-mediated NLRP3 inflammasome
pathway in mice following CLP
Consistent with our previous research, CLP induced
severe acute lung injury (ALI) at 24 h post-CLP, as shown by lung
histopathology, lung MPO activity, protein concentration in BAL
fluid and lung W/D ratio (Fig.
6A-D and Fig. S2,
P<0.05). H2 ameliorated ALI via decreasing the levels
of these indicators (Fig. 6A-D,
P<0.05). To evaluate the effect of the autophagy-mediated NLRP3
inflammasome pathway on H2 improving organ dysfunction
in vivo, ALI was assessed through inducing or inhibiting
autophagy or NLRP3 inflammasome activation. The autophagy inducer
and NLRP3 inhibitor ameliorated lung histopathology, lung MPO
activity, protein concentration in BAL fluid and lung W/D ratio
following CLP+H2 treatment compared results in the
CLP+H2 group. However, 3-MA treatment reversed the
beneficial effect of H2 and the NLRP3 inhibitor on lung
histopathology, lung MPO activity, protein concentration in BAL
fluid and lung W/D ratio in mice following CLP and H2
treatment (Fig. 6A-D,
P<0.05)
Liver and kidney injury exhibited the same trend as
ALI in the present study. H2 significantly improved the
histopathological changes of the liver and kidney, and the serum
levels of ALT, AST, Cr and BUN in the CLP mice (Fig. 6E-J, Fig. S2, P<0.05). The autophagy
inducer ameliorated liver and kidney injury in mice following
CLP+H2 treatment. 3-MA treatment reversed the beneficial
effect of H2 on liver and kidney injury in the mice
following CLP+H2 treatment (Fig. 6A-D, P<0.05).
These data suggest that H2 and the
autophagy inducer Rap exerted a protective effect against acute
lung, liver and kidney injury induced by CLP and that H2
improved the acute changes in the lung, liver and kidney induced
via autophagy-mediated NLRP3 inflammasome inactivation in
sepsis.
Lack of autophagy reverses the inhibitory
effect of H2 on the NLRP3 inflammasome and cytokines in
sepsis
To examine the role of autophagic proteins during
NLRP3 inflammasome activation following H2 treatment in
septic mice, the mice were treated with autophagy inducer and
autophagy inhibitor. H2 and the autophagy inducer Rap
attenuated the release of IL-β, IL-18, TNF-α and caspase-1 P10 and
the expression of NLRP3 and ASC in the lungs of the septic mice.
The autophagy inhibitor 3-MA exacerbated the release of IL-1β,
IL-18, TNF-α and caspase-1 P10 and the expression of NLRP3 and ASC
in mice treated by CLP and H2 (Fig. 7A-F, P<0.05). Consistent with
the changes in the lungs of septic mice, the abnormal variation of
cytokines and NLRP3 inflammasome exhibited the same trend in the
liver (Fig. 8A-F, P<0.05) and
kidney (Fig. 9A-F, P<0.05).
These results indicate that autophagic activity increases and acts
synergistically with H2 treatment to improve excessive
cytokine release in septic mice. The lack of autophagy partly
reversed the inhibitory effect of H2 on the NLRP3
inflammasome and cytokines in sepsis.
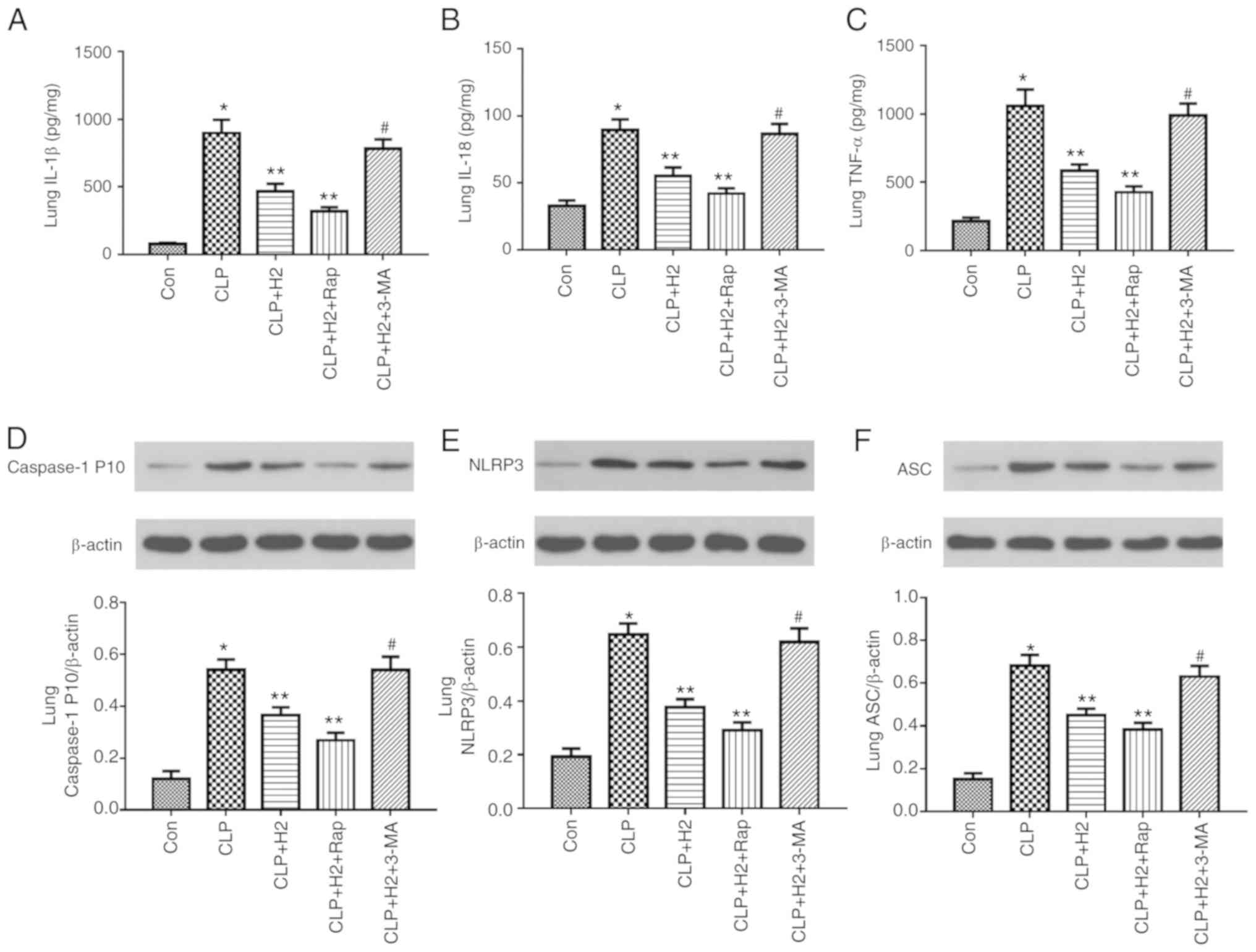 | Figure 7H2 alleviates cytokine
release and NLRP3 inflammasome activation in the lungs of septic
mice via increasing autophagy. Septic mice were treated with
H2, autophagy inducer Rap and autophagy inhibitor 3-MA.
After 24 h, lung tissues were collected to detect cytokines (A)
IL-1β, (B) IL-18 and (C) TNF-α by ELISA and the expression of (D)
caspase-1, (E) NLRP3 and (F) ASC by western blotting. Data are
expressed as the mean ± standard deviation (n=6).
*P<0.05 vs. Con group, **P<0.05 vs. the
CLP group, #P<0.05 vs. CLP+H2 group. Rap,
rapamycin; 3-MA, 3-methyladenine; IL, interleukin; TNF, tumor
necrosis factor; NLRP3, NACHT, LRR and PYD domains-containing
protein 3; ASC, apoptosis-associated speck-like protein containing
a CARD; CLP, cecal ligation and puncture; H2, hydrogen;
Con, control. |
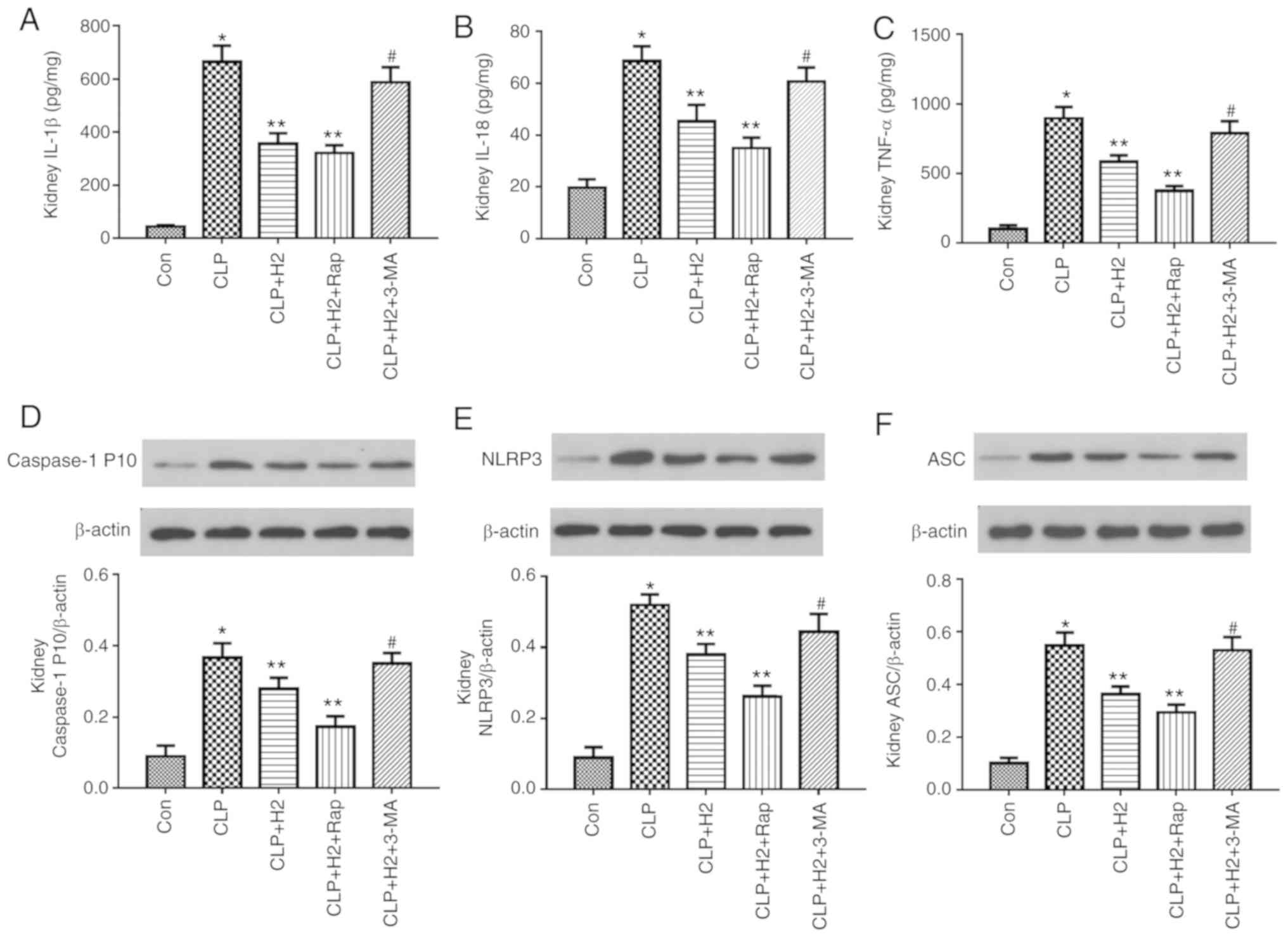 | Figure 8H2 alleviates cytokine
release and NLRP3 inflammasome activation in the livers of septic
mice via increasing autophagy. Septic mice were treated with
H2, autophagy inducer Rap, and autophagy inhibitor 3-MA.
After 24 h, liver tissues were collected to detect cytokines (A)
IL-1β, (B) IL-18 and (C) TNF-α by ELISA and the expression of (D)
caspase-1, (E) NLRP3 and (F) ASC by western blotting. Data are
expressed as mean ± standard deviation (n=6). *P<0.05
vs. Con group, **P<0.05 vs. CLP group,
#P<0.05 vs. CLP+H2 group. Rap, rapamycin;
3-MA, 3-methyladenine; IL, interleukin; TNF, tumor necrosis factor;
NLRP3, NACHT, LRR and PYD domains-containing protein 3; ASC,
apoptosis-associated speck-like protein containing a CARD; CLP,
cecal ligation and puncture; H2, hydrogen; Con,
control. |
 | Figure 9H2 alleviates cytokine
release and NLRP3 inflammasome activation in the kidneys of septic
mice via increasing autophagy. Septic mice were treated with
H2, autophagy inducer Rap and autophagy inhibitor 3-MA.
After 24 h, kidney tissues were collected to detect cytokines (A)
IL-1β, (B) IL-18 and (C) TNF-α by ELISA and the expression of (D)
caspase-1, (E) NLRP3 and (F) ASC by western blotting. Data are
expressed as the mean ± standard deviation (n=6).
*P<0.05 vs. Con group, **P<0.05 vs. CLP
group, #P<0.05 vs. CLP+H2 group. Rap,
rapamycin; 3-MA, 3-methyladenine; IL, interleukin; TNF, tumor
necrosis factor; NLRP3, NACHT, LRR and PYD domains-containing
protein 3; ASC, apoptosis-associated speck-like protein containing
a CARD; CLP, cecal ligation and puncture; H2, hydrogen;
Con, control. |
Discussion
Our previous and present research verified that
H2 exerts a protective effect during sepsis; however,
the mechanisms underlying the role of H2 in suppressing
the pathological development of septic organ injury remain to be
fully elucidated. Mitochondrial dysfunction and structural damage
have been identified as key physiopathological factors in sepsis
and are correlated with the severity of organ dysfunction and
outcome of sepsis (24).
Macrophages are the first line of defense against pathogens in
sepsis (25). Macrophage
mitochondrial dysfunction is involved in the pathogenesis of sepsis
through several mechanisms, including systemic inflammatory
responses, oxidative stress, energy metabolism and the intrinsic
apoptotic pathway (26). It was
previously reported that mitochondrial dysfunction induced by LPS
is associated with mtDNA depletion and results in a lack of
mitochondrial transcription (27,28). Mitochondria are the main source of
ROS, the production of which is a consequence of primary oxidative
injury of mitochondrial respiratory complex proteins and mtDNA
following exposure to LPS. The present study demonstrated that LPS
and ATP treatment led to the accumulation of physiologically
abnormal mitochondria and promoted mitochondrial dysfunction, as
shown by the reduced MMP, mitochondrial RCR, ATP content and mtDNA
copy number and increased ROS in macrophages. H2
alleviated the mitochondrial dysfunction in macrophages induced by
LPS and ATP, and improved lung, liver and kidney injury. Therefore,
H2 protected organ function in septic mice and enhanced
the survival rate of sepsis.
Accumulating evidence indicates that cellular
damage induced by acute inflammatory responses leads to the
progression of systemic inflammatory response syndrome. Macrophage
activation contributes to the release of various pro-inflammatory
cytokines, including IL-1β, IL-6 and TNF-α, and leads to excessive
ROS production, exacerbating the inflammatory cascade in sepsis.
The inflammasome is a complex of cytoplasmic proteins formed in
response to endogenous or exogenous pathogen-associated molecular
patterns or danger-associated molecular patterns, which leads to an
inflammatory response by modulating the cleavage and mature of
pro-inflammatory cytokines, including pro-IL-1β and pro-IL18
(29). NLRP3, one of the most
well-characterized inflammasomes, has been shown to be involved in
conditions including sepsis and infectious diseases (30,31). Excessive activation of the NLRP3
inflammasome is considered to be an important factor in the
pathological development of septic injury (32). Once activated, NLRP3 recruits ASC
and procas-pase-1 to form a complex, which results in caspase-1
activation and the maturation of pro-inflammatory cytokines IL-1β
and IL-18. Results reported by Wu et al (33), Ganz et al (34) and Li et al (35) indicate that activation of the
NLRP3 inflammasome increases the expression of ASC in septic liver
tissue. NLRP3 inflammasome activation and excessive cytokine
expression were also observed in LPS and ATP-induced macrophages.
Consistent with the results of previous studies, LPS and ATP
treatment contributed to inflammasome pathway activation in the
present study, including the increased expression of NLRP3 and
caspase-1 and the maturation of IL-β and IL-18 in macrophages. The
release of TNF-α and IL-6 exhibited the same trend as IL-β and
IL-18 in macrophages stimulated by LPS and ATP. In addition,
inflammasome activation in sepsis was detected in vivo. The
findings were consistent with the changes of macrophages in
vitro: The NLRP3 inflammasome, ASC and procaspase-1 were
markedly increased, with excessive release of cytokines IL-1β,
IL-18, TNF-α and IL-6. In line with our previous findings,
H2 improved organ damage via alleviating the excessive
release of pro-inflammatory cytokines. Furthermore, H2
inactivated the inflammasome via reducing the expression of NLRP3
and procaspase-1 in macrophages induced by LPS and ATP, and in the
lung, kidney and liver of septic mice. Whether H2
mitigated the inflammatory response via regulating the inflammasome
activation was also investigated. The results demonstrated that
H2 alleviated the inflammatory response and organ damage
via inhibiting NLRP3 inflammasome pathway activation.
Autophagy is a self-degradation process for
recycling organelles released by damaged cells. Under certain
conditions, mitophagy selectively removes specific proteins and
injured organelles, such as mitochondria (36). It has been reported that cardiac
dysfunction and liver injury may be alleviated by the activation of
autophagy in septic mice (37-39). Of note, it was reported that the
activation of mitophagy was necessary to prevent organ injury in
sepsis (40). LC3 and Beclin 1
are two key molecules involved in the initiation and progression of
autophagy. LC3 is prone to the degradation of damaged mitochondria
by binding p62 to the autophagosome (41). Nakahira et al reported that
LPS and ATP treatment produced a greater abundance of swollen
mitochondria with severely disrupted cristae in LC3B and Beclin
1-knockdown macrophages, and macrophages were prone to exhibit
severe mitochondrial derangement and dysfunction following LPS and
ATP treatment (13). The
PINK1-Parkin pathway is part of the main mitophagic process
(42). The lack of PINK1 and
Parkin prevented the activation of mitophagy. In Pink1 and
Parkin-knockout mice, sepsis was more likely to induce tissue
injury, increasing mortality (43). The present study examined the
activation and effect of autophagy on mitochondrial dysfunction,
organ injury, organ function and survival rate following
H2 treatment in vitro and in vivo. It was
observed that the activation of autophagy, particularly mitophagy,
was enhanced in macrophages induced by LPS and ATP, and that LC3I
conversion to LC3II and the expression levels of Beclin 1, PINK1,
Parkin and VDAC were increased in macrophages induced by LPS and
ATP. H2 treatment increased PINK1/Parkin-mediated
mitophagy via increasing the expression of LC3II, Beclin 1, PINK1,
Parkin and VDAC in macrophages. The mechanisms of LPS inducing
autophagy may be associated with mitochondrial damage, ROS
generation and the excessive production of inflammatory cytokines,
which promote autophagy under LPS stimulation (44); in addition, autophagy, as a
protective mechanism, increases in stress conditions, such as LPS
exposure. H2 exerts a protective effect during LPS
challenge, which improves the autophagic process to inhibit
mitochondrial damage, ROS generation and the excessive production
of inflammatory cytokines.
Autophagy is closely associated with NLRP3
inflammasome activation. Autophagy acts to restore the balance of
inflammatory responses, as uncontrolled and detrimental
inflammation is inhibited via inflammasome inactivation and
proinflammatory cytokine clearance (45). The lack of autophagy increased
NLRP3 activation and the secretion of pro-inflammatory cytokines
following LPS stimulation (46),
whereas the induction of autophagy contributed to the degradation
of NLRP3 and reduced the level of IL-1β (47). Mitophagy can recycle damaged
mitochondria selectively, which activates the NLRP3 inflammasome
(16). Furthermore, the immune
cross-talk between autophagy and the inflammasome pathways in
bacterial infection and sepsis have been investigated (48). The interplay between inflammasomes
and autophagy varies markedly in sepsis and is affected by factors
including pathogens, infection conditions, time duration, host
cells and animal models (49). To
further investigate the effect of autophagy and the inflammasome on
H2 alleviating mitochondrial dysfunction and organ
damage in sepsis, an autophagy inducer and inhibitor were used in
the present study to examine the NLRP3 pathway, cytokines, organ
dysfunction and damage indicators. As mentioned above, Rap
attenuated NLRP3 activation, the expression of ASC, cleavage of
caspase-1 and the maturation of IL-1β and IL-18, and also improved
mitochondrial dysfunction, tissue damage in the lung, liver and
kidney, alleviated lung injury and liver and kidney dysfunction and
improved survival rates in septic mice treated with H2.
The autophagy inhibitor reversed the effect of inflammasome
inactivation on these indicators in septic mice receiving
H2 treatment. These results suggest that H2
alleviated the sepsis-induced mitochondrial dysfunction and
inflammatory response via autophagy-mediated NLRP3 inflammasome
inactivation.
In conclusion, the results of the present study
demonstrate that H2 alleviated the mitochondrial
dysfunction induced by LPS and ATP in macrophages and improved
tissue injury in the lung, liver and kidney in septic mice.
H2 was shown to exert a protective effect in sepsis via
suppressing autophagy/mitophagy-mediated NLRP3 inflammasome
activation under LPS stimulation or in an animal sepsis model. The
contribution of autophagy and inflammasome inactivation to this
protective process may uncover a mechanistic link between them when
H2 is administered to protect against sepsis. However,
the mechanisms underlying the role of H2 treatment in
sepsis requires further investigation.
Supplementary Data
Abbreviations:
|
3-MA
|
3-methyladenine
|
|
ASC
|
apoptosis-associated speck-like
protein containing a CARD
|
|
ATP
|
adenosine triphosphate
|
|
BALF
|
bronchoalveolar lavage fluid
|
|
BCA
|
bicinchoninic acid
|
|
CLP
|
cecal ligation and puncture
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
H2
|
hydrogen
|
|
IL
|
interleukin
|
|
JC-1
|
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl benzimidalyl
carbocyanine iodide
|
|
LPS
|
lipopolysaccharide
|
|
LC3
|
microtubule-associated protein 1
light chain 3
|
|
MMP
|
mitochondrial membrane potential
|
|
MPO
|
myeloperoxidase
|
|
mtDNA
|
mitochondrial DNA
|
|
NLRP3
|
NACHT, LRR and PYD domains-containing
protein 3
|
|
PAM
|
primary alveolar macrophage
|
|
PINK1
|
PTEN-induced putative kinase 1
|
|
Rap
|
rapamycin
|
|
RCR
|
respiratory control ratio
|
|
ROS
|
reactive oxygen species
|
|
SDS-PAGE
|
sodium dodecyl sulfate-polyacrylamide
gel electrophoresis
|
|
TNF
|
tumor necrosis factor
|
|
VDAC
|
voltage-dependent anion channel
|
Acknowledgments
Not applicable.
Funding
This study was supported by grants from the
National Natural Science Foundation of China (grant. nos. 81601667
to HC, 81671888 to YY, 81772043 to KX and 81801889 to YL) and the
Natural Science Foundation of Tianjin City (grant. no.
18JCYBJC93700).
Availability of data and materials
The datasets used and/or analyzed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
YY and KX designed the study and helped to edit and
revise the manuscript; HC and XMa were involved in animal model
establishment and sample collection, and drafted the manuscript;
XMe, YW and YL were involved in the in vitro experiments;
JF, LZ and YZ performed the detection of indicators. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Institutional Animal Care and Use Committee of Tianjin Medical
University and were performed in accordance with the National
Institutes of Health Guide for Care and Use of Laboratory Animals.
All efforts were made to minimize animal suffering and the number
of animals used.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
van der Poll T, van de Veerdonk FL,
Scicluna BP and Netea MG: The immunopathology of sepsis and
potential therapeutic targets. Nat Rev Immunol. 17:407–420. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dombrovskiy VY, Martin AA, Sunderram J and
Paz HL: Rapid increase in hospitalization and mortality rates for
severe sepsis in the United States: A trend analysis from 1993 to
2003. Crit Care Med. 35:1244–1250. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dellinger RP, Levy MM, Rhodes A, Annane D,
Gerlach H, Opal SM, Sevransky JE, Sprung CL, Douglas IS, Jaeschke
R, et al: Surviving sepsis campaign: International guidelines for
management of severe sepsis and septic shock, 2012. Intensive Care
Med. 39:165–228. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wiersinga WJ, Leopold SJ, Cranendonk DR
and van der Poll T: Host innate immune responses to sepsis.
Virulence. 5:36–44. 2014. View Article : Google Scholar :
|
|
6
|
Lamkanfi M and Dixit VM: Mechanisms and
functions of inflammasomes. Cell. 157:1013–1022. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rahim I, Djerdjouri B, Sayed RK,
Fernández-Ortiz M, Fernández-Gil B, Hidalgo-Gutiérrez A, López LC,
Escames G, Reiter RJ and Acuña-Castroviejo D: Melatonin
administration to wild-type mice and nontreated NLRP3 mutant mice
share similar inhibition of the inflammatory response during
sepsis. J Pineal Res. 63:2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Feng Z, Qi S, Zhang Y, Qi Z, Yan L, Zhou
J, He F, Li Q, Yang Y, Chen Q, et al: Ly6G+ neutrophil-derived
miR-223 inhibits the NLRP3 inflammasome in mitochondrial
DAMP-induced acute lung injury. Cell Death Dis. 8:e31702017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu Q, Ci X, Wen Z and Peng L: Diosmetin
alleviates lipopoly-saccharide-induced acute lung injury through
activating the Nrf2 pathway and inhibiting the NLRP3 inflammasome.
Biomol Ther (Seoul). 26:157–166. 2018. View Article : Google Scholar
|
|
10
|
Zhang Y and Li X, Grailer JJ, Wang N, Wang
M, Yao J, Zhong R, Gao GF, Ward PA, Tan DX and Li X: Melatonin
alleviates acute lung injury through inhibiting the NLRP3
inflammasome. J Pineal Res. 60:405–414. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Allen Li P, Banerjee H, Franklin S, Herzog
S, Johnston L, McDowell C, Paskind J, Rodman M, Salfeld LJ, et al:
Mice deficient in IL-1 beta-converting enzyme are defective in
production of mature IL-1 beta and resistant to endotoxic shock.
Cell. 80:401–411. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sutterwala FS, Ogura Y, Szczepanik M,
Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ,
Galán JE, Askenase PW and Flavell RA: Critical role for
NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its
regulation of caspase-1. Immunity. 24:317–327. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nakahira K, Haspel JA, Rathinam VA, Lee
SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim
HP, et al: Autophagy proteins regulate innate immune responses by
inhibiting the release of mitochondrial DNA mediated by the NALP3
inflammasome. Nat Immunol. 12:222–230. 2011. View Article : Google Scholar
|
|
14
|
Zhang B, Wei W and Qiu J: ALK is required
for NLRP3 inflammasome activation in macrophages. Biochem Biophys
Res Commun. 501:246–252. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim MJ, Yoon JH and Ryu JH: Mitophagy: A
balance regulator of NLRP3 inflammasome activation. BMB Rep.
49:529–535. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shimada K, Crother TR, Karlin J, Dagvadorj
J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et
al: Oxidized mitochondrial DNA activates the NLRP3 inflammasome
during apoptosis. Immunity. 36:401–414. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie K, Yu Y, Pei Y, Hou L, Chen S, Xiong L
and Wang G: Protective effects of hydrogen gas on murine
polymicrobial sepsis via reducing oxidative stress and HMGB1
release. Shock. 34:90–97. 2010. View Article : Google Scholar
|
|
19
|
Dong A, Wang L, Wang YY, Bian YX, Yu YH
and Xie KL: Role of autophagy in hydrogen-induced reduction of lung
injury in septic mice. Chin J Anesthesiol. 37:632–636. 2017.
|
|
20
|
Chen HG, Xie KL, Han HZ, Wang WN, Liu DQ,
Wang GL and Yu YH: Heme oxygenase-1 mediates the anti-inflammatory
effect of molecular hydrogen in LPS-stimulated RAW 264.7
macrophages. Int J Surg. 11:1060–1066. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Iglesias-González J, Sánchez-Iglesias S,
Beiras-Iglesias A, Soto-Otero R and Méndez-Álvarez E: A simple
method for isolating rat brain mitochondria with high metabolic
activity: Effects of EDTA and EGTA. J Neurosci Methods. 213:39–42.
2013. View Article : Google Scholar
|
|
23
|
Narendra DP, Jin SM, Tanaka A, Suen DF,
Gautier CA, Shen J, Cookson MR and Youle RJ: PINK1 is selectively
stabilized on impaired mitochondria to activate Parkin. PLoS Biol.
8:e10002982010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Azevedo LC: Mitochondrial dysfunction
during sepsis. Endocr Metab Immune Disord Drug Targets. 10:214–223.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mège JL, Mehraj V and Capo C: Macrophage
polarization and bacterial infections. Curr Opin Infect Dis.
24:230–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Deng SY, Zhang LM, Ai YH, Pan PH, Zhao SP,
Su XL, Wu DD, Tan HY, Zhang LN and Tsung A: Role of interferon
regulatory factor-1 in lipopolysaccharide-induced mitochondrial
damage and oxidative stress responses in macrophages. Int J Mol
Med. 40:1261–1269. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yamada H, Arai T, Endo N, Yamashita K,
Fukuda K, Sasada M and Uchiyama T: LPS-induced ROS generation and
changes in glutathione level and their relation to the maturation
of human monocyte-derived dendritic cells. Life Sci. 78:926–933.
2006. View Article : Google Scholar
|
|
28
|
Suliman HB, Welty-Wolf KE, Carraway M,
Tatro L and Piantadosi CA: Lipopolysaccharide induces oxidative
cardiac mitochondrial damage and biogenesis. Cardiovasc Res.
64:279–288. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mariathasan S, Newton K, Monack DM, Vucic
D, French DM, Lee WP, Roose-Girma M, Erickson S and Dixit VM:
Differential activation of the inflammasome by caspase-1 adaptors
ASC and Ipaf. Nature. 430:213–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reboldi A, Dang EV, McDonald JG, Liang G,
Russell DW and Cyster JG: Inflammation. 25-Hydroxycholesterol
suppresses interleukin-1-driven inflammation downstream of type I
interferon. Science. 345:679–684. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wirnsberger G, Zwolanek F, Asaoka T,
Kozieradzki I, Tortola L, Wimmer RA, Kavirayani A, Fresser F, Baier
G, Langdon WY, et al: Inhibition of CBLB protects from lethal
Candida albicans sepsis. Nat Med. 22:915–923. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hao H, Cao L, Jiang C, Che Y, Zhang S,
Takahashi S, Wang G and Gonzalez FJ: Farnesoid X receptor
regulation of the NLRP3 inflammasome underlies
cholestasis-associated sepsis. Cell Metab. 25:856–867.e5. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu Y, Ren J, Zhou B, Ding C, Chen J, Wang
G, Gu G, Wu X, Liu S, Hu D and Li J: Gene silencing of non-obese
diabetic receptor family (NLRP3) protects against the
sepsis-induced hyper-bile acidaemia in a rat model. Clin Exp
Immunol. 179:277–293. 2015. View Article : Google Scholar :
|
|
34
|
Ganz M, Csak T, Nath B and Szabo G:
Lipopolysaccharide induces and activates the Nalp3 inflammasome in
the liver. World J Gastroenterol. 17:4772–4778. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li S, Wu H, Han D, Ma S, Fan W, Wang Y,
Zhang R, Fan M, Huang Y, Fu X and Cao F: A novel mechanism of
mesenchymal stromal cell-mediated protection against sepsis:
Restricting inflammasome activation in macrophages by increasing
mitophagy and decreasing mitochondrial ROS. Oxid Med Cell Longev.
2018:35376092018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Saito T and Sadoshima J: Molecular
mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ
Res. 116:1477–1490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hsieh CH, Pai PY, Hsueh HW, Yuan SS and
Hsieh YC: Complete induction of autophagy is essential for
cardioprotection in sepsis. Ann Surg. 253:1190–1200. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yen YT, Yang HR, Lo HC, Hsieh YC, Tsai SC,
Hong CW and Hsieh CH: Enhancing autophagy with activated protein C
and rapamycin protects against sepsis-induced acute lung injury.
Surgery. 153:689–698. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin CW, Lo S, Perng DS, Wu DB, Lee PH,
Chang YF, Kuo PL, Yu ML, Yuan SS and Hsieh YC: Complete activation
of autophagic process attenuates liver injury and improves survival
in septic mice. Shock. 41:241–249. 2014. View Article : Google Scholar
|
|
40
|
Carchman E and Zuckerbraun B:
Mitophagy/mitochondrial biogenesis is necessary to prevent organ
injury in sepsis and is dependent on TLR9 signaling. J Am Coll
Surg. 213(Suppl): S592011. View Article : Google Scholar
|
|
41
|
Lazarou M: Keeping the immune system in
check: A role for mitophagy. Immunol Cell Biol. 93:3–10. 2015.
View Article : Google Scholar
|
|
42
|
Vives-Bauza C, Zhou C, Huang Y, Cui M, de
Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al:
PINK1-dependent recruitment of Parkin to mitochondria in mitophagy.
Proc Natl Acad Sci USA. 107:378–383. 2010. View Article : Google Scholar
|
|
43
|
Kang R, Zeng L, Xie Y, Yan Z, Zhou B, Cao
L, Klionsky DJ, Tracey KJ, Li J, Wang H, et al: A novel PINK1- and
PARK2-dependent protective neuroimmune pathway in lethal sepsis.
Autophagy. 12:2374–2385. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang D, Zhou J, Ye LC, Li J, Wu Z, Li Y
and Li C: Autophagy maintains the integrity of endothelial barrier
in LPS-induced lung injury. J Cell Physiol. 233:688–698. 2018.
View Article : Google Scholar
|
|
45
|
Cadwell K: Crosstalk between autophagy and
inflammatory signalling pathways: Balancing defence and
homeostasis. Nat Rev Immunol. 16:661–675. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yin JJ, Xie G, Zhang N and Li Y:
Inhibiting autophagy promotes endoplasmic reticulum stress and the
ROS-induced nod-like receptor 3-dependent proinflammatory response
in HepG2 cells. Mol Med Rep. 14:3999–4007. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Giegerich AK, Kuchler L, Sha LK, Knape T,
Heide H, Wittig I, Behrends C, Brüne B and von Knethen A:
Autophagy-dependent PELI3 degradation inhibits proinflammatory IL1B
expression. Autophagy. 10:1937–1952. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Suzuki T, Franchi L, Toma C, Ashida H,
Ogawa M, Yoshikawa Y, Mimuro H, Inohara N, Sasakawa C and Nuñez G:
Differential regulation of caspase-1 activation, pyroptosis, and
autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS
Pathog. 3:e1112007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gan Pu Q, Li C, Li R, Tan Y, Li S, Wei X,
Lan Y, Deng L, Liang XH, et al: Atg7 deficiency intensifies
inflammasome activation and pyroptosis in pseudomonas sepsis. J
Immunol. 198:3205–3213. 2017. View Article : Google Scholar :
|














