Introduction
Colorectal cancer, which is one of the most common
types of malignant tumor, has the third highest incidence in men
and the second highest incidence in women globally; as a result,
colorectal cancer is responsible for a high number of mortalities
(1). Radical surgical resection
is currently the only method for treating colorectal cancer
(2). The primary cause of failure
in colorectal cancer surgery is metastasis and tumor recurrence. To
date, no effective medication has been identified to inhibit the
progression of colorectal cancer. Therefore, the development of
protective therapies to suppress the progression of colorectal
cancer is necessary in treatment of colorectal cancer.
Epithelial mesenchymal transition (EMT) is a key
mechanism in tumor metastasis (3,4).
In tumor cells, it is a biological process in which tumor cells
allow epithelial cells to transform into cells with mesenchymal
phenotype characteristics. During the process, tumor epithelial
cells lose their polarity and adhesion among cells is decreased.
Through this, interstitial cell characteristics including high
migration and invasion ability, anti-apoptosis and degradation of
extracellular matrix are obtained (5,6).
Therefore, EMT, which serves an important role in the process of
tumor metastasis is also a critical condition for invasion and
metastasis in colorectal cancer, (7).
As a class of serine/threonine protein kinases in
eukaryotic cells, the primary function of adenosine
monophosphate-activated protein kinases (AMPK) is to serve as a
cellular energy sensor to modulate cellular responses to a low
nutrient environment (8). A
previous study has indicated that AMPK may regulate cell
proliferation, growth and autophagy (9). The tumor suppressor gene
serine/threonine kinase 11 may activate AMPK, while tumor
suppressor gene TSC complex subunit 2 (TSC2) is a downstream
effector of AMPK (10). These
studies provide evidence that AMPK may be a potential target for
cancer therapy. Mammalian target of rapamycin (mTOR) is also a
serine/threonine protein kinase that regulates cell growth by
integrating nutrient and growth factor signaling (11). Numerous studies have indicated
that rapamycin may inhibit the proliferation of colorectal cancer
cells in vivo and in vitro, and it has been suggested
that the mTOR pathway is activated in patients with colorectal
cancer (12,13). Therefore, targeting AMPK and mTOR
signal pathways may be a promising protective strategy for
management of colorectal cancer.
As a class of serine proteases, human
kallikrein-related peptidases (KLKs) are encoded by 15 different
genes that localize to human chromosome 19q13.4 and have a highly
conserved set of gene structures and protein sequences (14). KLKs have been demonstrated to
serve important roles in tumor growth and metastasis and are
considered as markers for various types of cancer (15). A previous study indicated that
KLKs were closely associated with colorectal cancer, as it
participated in tumor cell proliferation, apoptosis and prognosis
(16). An additional study also
identified that KLK promoted the invasion and metastasis of cancer
cells by hydrolyzing certain macromolecular substances including
the extracellular matrix (ECM), cell adhesion molecules (CAM), and
importantly, KLK may degrade almost all ECM molecules (17). It was also demonstrated that the
AMPK/TSC2/mTOR signaling pathway was involved in the process of
tissue kallikrein to protecting the SH-SY5Y neuronal cell line
against oxygen and glucose deprivation‑induced injury (18). KLK12 is known as kallikrein 5, and
a previous study has demonstrated that KLK12 mRNA levels were
upregulated in cancer tissues including gastric, breast and
prostate cancer (19). It has
been suggested that KLK12 may become a novel tumor biomarker, as
data from a recent study demonstrated that the mRNA expression of
KLK12 was upregulated in phase III colorectal cancer tissues
(20), which also suggested that
KLK12 may be involved in the pathogenesis of cancer and malignant
changes in tissues (19).
However, the mechanisms underlying the effect of KLK12 in human
colorectal cancer remain unclear.
The present study was performed to investigate the
role of KLK12 in cellular and animal colorectal cancer models, and
to identify the potential underlying mechanisms. The results from
the study increased understanding on the role of KLK12 in
colorectal cancer.
Materials and methods
Human tissue samples
From May 2016 to May 2017, 45 patients who received
radical colorectal cancer resection in The First Affiliated
Hospital of Zhejiang Chinese Medical University (Hangzhou, China),
were recruited to the study. Specimens were obtained immediately
following surgical resection and stored at −80°C for subsequent
analysis. All patients had a negative preoperative history of
chemotherapy or radiotherapy and no other types of cancer were
diagnosed concomitantly. The present study was approved by the
Ethical Committee of The First Affiliated Hospital of Zhejiang
Chinese Medical University and written informed consent was
obtained from each patient.
Immunohistochemistry (IHC)
Tissues were fixed with 4% paraformaldehyde for 24 h
at room temperature and embedded in paraffin. The paraffin-embedded
block tissues were cut into 4 μm sections. The samples were
dehydrated in a graded ethanol (100,−95, 90, 80, 70 and 0%), and
for antigen retrieval, deparaffinized sections were incubated in a
microwave oven at 95°C for 5 min and cooled for 1 h at room
temperature. Following washing with PBS for two times, the sections
were treated with methanol containing 3% hydrogen peroxide for 10
min at room temperature and blocked with 5% goat serum (Beijing
Solarbio Science & Technology Co., Ltd.) at room temperature
for 20 min. Subsequently, anti-KLK12 antibody (cat. no. AF3095;
R&D systems, Inc.) was added to the sections at 4°C and
incubated for 24 h. Following washing with PBS three times,
biotin-labeled goat anti-rabbit IgG secondary antibody (1:5,000,
cat. no. ab97049, Abcam) was added to the sections and incubated
for 10 min at room temperature. The sections were then stained by
0.05% DAB staining solution (Leica Microsystems, Inc.) at room
temperature for 15 min and stained with 2 g/l hematoxylin (Beijing
Solarbio Science & Technology Co., Ltd.) for 3 min at room
temperature. The sections were then observed using an MF43
fluorescence microscope (Guangzhou Micro-shot Technology Co.,
Ltd.), with magnification of ×100 and 200.
Cell culture
Colorectal cancer HT-29, LoVo, SW-480, SW-620,
Caco2, HCT-116, HCT-15, RKO, LS174T and DLD-1 cell lines, and the
normal colon CCD-18Co cell line were purchased from American Type
Culture Collection. RPMI-1640 medium containing 2 mM L-glutamine,
10% fetal bovine serum (FBS), penicillin G (100 U/ml) and
streptomycin (0.1 mg/ml) was used as cell culture medium and
cultured with the cells at 37°C with 5% CO2. All culture
reagents were purchased from Gibco; Thermo Fisher Scientific, Inc.
HT-29 cells were inoculated and grown to 80% confluency prior to
treatment with adequate concentrations of rapamycin (10 nM; cat.
no. V900930; Sigma-Aldrich; Merck KGaA) and
5-amino-imidazole-4-carboxamide ribonucleotide (AICAR; 2 mM; cat.
no. A9978; Sigma-Aldrich; Merck KGaA) for 90 min, and the cells
were then transfected with KLK12 plasmid for 12, 24 and 48 h to
detect cell viability.
Transfection
50 nmol/l small interfering RNA targeted KLK12
(siKLK12) and negative siRNA (Shanghai GenePharma Co., Ltd.) were
inserted into pLKO.1-TRC vector (Shanghai Institute of Biochemistry
and Cell Biology, Chinese Academy of Sciences) to produce the
recombinant plasmid. siRNA was used to transfect HT-29 cells using
Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.).
The KLK12 coding region was inserted into the pLKO.1-TRC vector,
and the HT-29 cells were transfected with KLK12 plasmid or empty
vector. The cells that did not undergo transfection were used as a
control. Sequences of the siRNAs used in the present study were as
follows: KLK12 siRNA sense, 5′-AAACAG UGA CAG CCA CGU ATT-3′; KLK12
siRNA antisense, 5′-UAC GUG GCU GUC ACU GUU UGG-3′; negative siRNA
sense, 5′-UUC UCC GAA CGU GUC ACG UTT-3′; negative siRNA antisense,
5′-ACG UGA CAC GUU CGG AGA ATT-3′. Transfected cells were cultured
in complete medium for 48 h prior to use in the invasion, migration
and apoptosis experiments.
MTT assay
HT-29 cells were transfected with or without
exposure to AICAR/rapamycin. The transfected cells were seeded in
96-well plates at a density of 5×103/l. Following
incubation at 37°C for 12, 24 and 48 h, 10 μl MTT solution
(5 mg/ml) was added into the wells and the plate was incubated in
an atmosphere containing 5% CO2 at 37°C for 4 h. Next, a
microplate reader was used to measure the absorbance at 450 nm.
Cell viability was detected by MTT assay (Thermo Fisher Scientific,
Inc.) according the manufacturer's protocol.
Wound healing assay
After 48 h of cell culture transfection, the cell
density of each group was ~90%. The cells were serum-starved for 24
h, and 200 μl sterile tip was used to scratch the cells in
the well plate. The medium was discarded and the surface was gently
washed 3 times with PBS to rinse off the exfoliated cells. After 0
and 24 h, the cell culture plate was placed under a 40-fold
inverted microscope (Olympus Corporation) to be observed, and
images at the same position were captured twice.
Transwell assay
Transwell chambers (8 μm; Corning
Incorporated) was placed on a 24-well plate with a layer of 50
μl Matrigel (BD Biosciences) coated onto the Transwell
chamber 37°C for 30 min. Then, 6×104 transfected cells
were cultured in serum-free medium for 12 h to eliminate the
effects of the serum and then resuspended in RPMI-1640 medium
without FBS. A total of 100 μl suspended cells were added to
the Transwell chamber, while 400 μl RPMI-1640 medium
containing 20% FBS was added to the lower chambers. The cells were
cultured with 5% CO2 for 24 h at 37°C in an incubator.
The Transwell chamber was then removed, the culture solution in the
Transwell plate was discarded and washed with calcium-free PBS
twice, and the chamber was fixed in 100% methanol solution at 4°C
for 30 min and stained with 0.1% crystal violet for 20 min at room
temperature. Next, PBS was used to wash the chamber, and the upper
chamber liquid was aspirated. The unmigrated cells in the upper
layer were gently wiped off using a cotton swab. The microporous
membrane was removed using a small tweezers carefully and dried
with the bottom side up and then transferred to a glass slide,
sealed with a neutral gum. Images were observed and captured using
an inverted light microscope (Olympus Corporation), with the
magnification of ×200.
Apoptosis assay
An Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) apoptosis detection kit (BD
Biosciences Medical Devices Shanghai Co., Ltd.) was used to detect
HT-29 cells apoptosis. Colorectal cancer HT-29 cells
(5×105 cells per well) were seeded in 6-well plates
until cell density reached 85%. siKLK12, KLK12 plasmids and the
corresponding control plasmids were used to transfect HT-29 cells,
which were then cultured in serum-free medium at 37°C for 5 h.
After 48 h of culture, the cells were centrifuged at 4°C for 10
min, at the speed of 5,000 × g, and washed twice with PBS, and 100
μl 1X Annexin V Binding Buffer and 5 μl FITC-labeled
Annexin V (20 μg/ml) were added to the cells and incubated
at room temperature for 20 min. Next, the cells were added with 5
μl PI (50 μg/ml) for 5 min and 400 μl binding
buffer (BD Biosciences). A flow cytometer (BD Biosciences Medical
Devices Shanghai Co., Ltd.) was used to analyze cell apoptosis;
among those cells, FITC−/PI− cells
represented healthy living cells, FITC−/PI+
cells represented necrotic cells, FITC+/PI−
cells indicated early apoptotic cells,
FITC+/PI+ cells represented late apoptotic
cells.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Following transfection, HT-29 cells were incubated
in an incubator for 48 h, and total RNA from HT-29 cells
(2×104 cells/well in 6-well plates) and 50 mg of
colorectal cancer and normal tissues were extracted using
TRIzol® reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. NanoDrop™ 2000
spectrophotometer (NanoDrop Technologies; Thermo Fisher Scientific,
Inc.) was conducted to measure RNA quality; an A260/A280 ratio
between 1.8-2.0 was required for the generation of cDNA. The
oligo-dT or stem-loop reverse transcriptase primers (Takara Bio,
Inc.) were used to obtain cDNA, and the RT thermocycler conditions
were set at 42°C for 60 min, 70°C for 5 min and maintenance at 4°C.
qPCR was performed with SYBR® Premix Ex Taq™ II (Takara
Bio Inc.) using real-time PCR Detection System (ABI 7500; Thermo
Fisher Scientific, Inc.). PCR reaction conditions were as follows:
Pretreatment at 95°C for 10 min, followed by 40 cycles at 94°C for
15 sec, 60°C for 1 min and 60°C for 1 min, and preservation at 4°C.
The 2−ΔΔCq method was used to quantify the data
(21). The primers used in this
assay are listed in Table I.
 | Table IPrimers for reverse transcription
quantitative polymerase chain reaction. |
Table I
Primers for reverse transcription
quantitative polymerase chain reaction.
| Genes | Forward | Reverse |
|---|
| KLK12 |
GCCTCAACCTCTCCATCGTC |
CTTGAAGGACTCCCCCACAC |
| E-cadherin C |
GAGAGCTACACGTTCACGG |
GGGTGTCGAGGGAAAAATAGG |
| Vimentin |
TGCCGTTGAAGCTGCTAACTA |
CCAGAGGGAGTGAATCCAGATTA |
| Snail |
TCGGAAGCCTAACTACAGCGA |
AGATGAGCATTGGCAGCGAG |
| MMP-2 |
TTGATGGCATCGCTCAGATC |
TTGTCACGTGGCGTCACAGT |
| MMP-9 |
GACGCAGACATCGTCATCCA |
CACAACTCGTCATCGTCGAAA |
| GAPDH |
AACGTGTCAGTGGTGGACCTG |
AGTGGGTGTCGCTGTTGAAGT |
Western blot analysis
The HT-29 cells were treated with siKLK12, KLK12 and
the corresponding control plasmid and then cultured in an incubated
for 48 h. Total proteins were extracted from HT-29 cells
(2×104 cells/well in 6-well plates) and tissues (50 mg)
by radioimmunoprecipitation assay lysis buffer (Cell Signaling
Technology, Inc.). A BCA Protein Assay kit (Pierce; Thermo Fisher
Scientific, Inc.) was applied to measure the concentrations of
proteins, which were adjusted to a concentration of 6
μg/μl using 1X loading and DEPC water. The samples (5
μl) were separated by 10% SDS-PAGE gels and then transferred
to polyvinylidene fluoride membrane (PVDF; EMD Millipore).
Following blocking in 5% nonfat milk in PBST (0.1% Tween-20 in PBS)
for 1 h at 37°C, the PVDF membranes were probed with the primary
antibodies overnight at 4°C, followed washing 3 times with PBST.
Next, the membrane was incubated with the secondary antibody
(horseradish peroxidase-conjugated goat anti-mouse/rabbit IgG,
1:2,000; cat. nos. sc-516102/sc-2357; Santa Cruz Biotechnology,
Inc.) at room temperature for 2 h and washed with PBST 3 times. An
EZ-ECL kit (Biological Industries) was used to develop the
membranes, and the gray values of the bands were analyzed and
quantified using ImageJ v5.0 software (Bio-Rad Laboratories, Inc.).
The antibodies used in the study were as follows: Anti-GAPDH
(mouse; 1:1,000; cat. no. LS-B1625; LifeSpan BioSciences, Inc.);
anti-epithelial (E)-cadherin (mouse; 1:1,000; cat. no. ab1416;
Abcam); anti-vimentin (rabbit; 1:1,000; cat. no. ab92547; Abcam);
anti-matrix metalloproteinase (MMP)-2 (rabbit; 1:1,000; cat. no.
ab37150; Abcam); anti-MMP-9 (rabbit; 1:1,000; cat. no. ab73734;
Abcam); anti-zinc finger protein SNAI1 (Snail; rabbit; 1:1,000;
cat. no. 3879; Cell Signaling Technology, Inc.); anti-cleaved
caspase-3 (rabbit; 1:1,000; cat. no. ab13847; Abcam); anti-Bax
(rabbit; 1:1,000; cat. no. ab32503; Abcam); anti-Bcl-2 (rabbit;
1:1,000; cat. no. ab32124; Abcam); anti-AMPK (mouse; 1:1,000; cat.
no. ab32047; Abcam); anti-phosphorylated (p)-AMPK (rabbit; 1:1,000;
cat. no. ab133448; Abcam); anti-mTOR (rabbit; 1:1,000; cat. no.
ab32028; Abcam); and anti-p-mTOR (rabbit; 1:1,000; cat. no.
ab84400; Abcam).
Statistical analysis
All the experimental data are presented as the mean
± standard deviation. Statistical analyses were performed using
GraphPad Prism 6 (GraphPad Software, Inc.) statistical software. A
one-way analysis of variance followed by Turkey's post hoc was used
to analyze differences among the experimental groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
KLK12 is highly expressed in colorectal
cancer tissues and cell lines
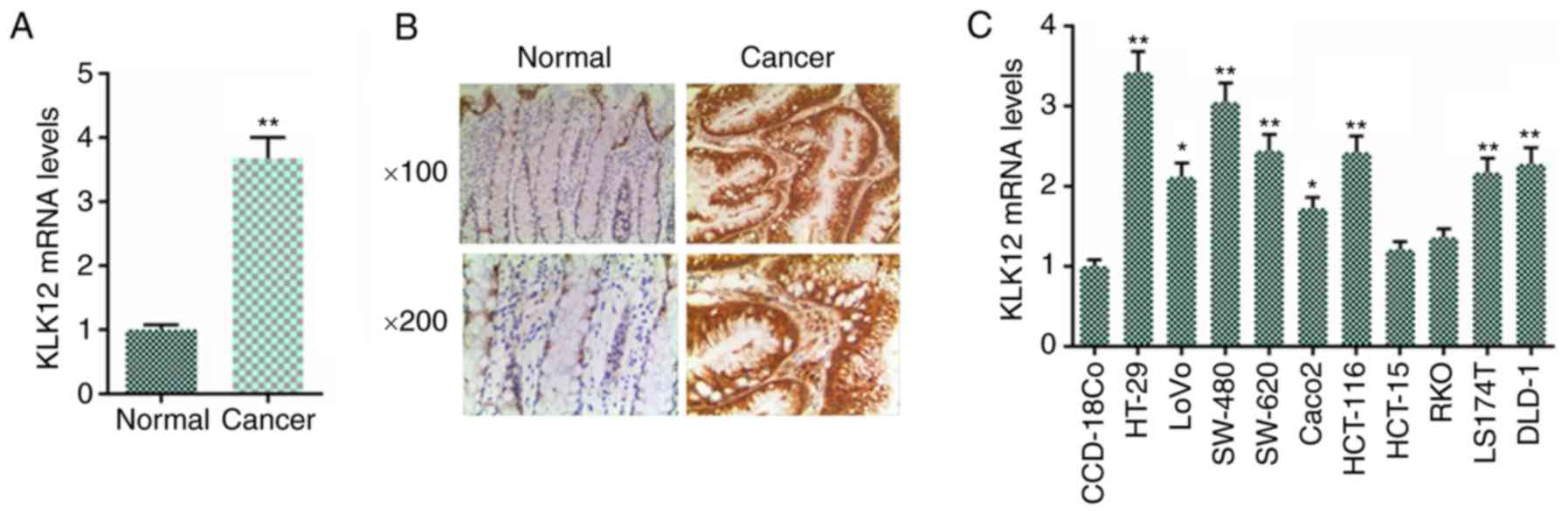
In the present study, the expression levels of KLK12
in colorectal cancer tissues and adjacent normal tissues of cancer
were initially explored using RT-qPCR. The results indicated that
the KLK12 mRNA level was increased in the cancer tissues compared
with the adjacent normal tissues (Fig. 1A). The IHC results indicated a
large number of brown particles in the cancer tissues (Fig. 1B). Next, the KLK12 levels in the
colorectal cancer cell lines and normal cells were determined using
RT-qPCR; the results in the cell lines were consistent with those
from the cancer tissues, in that the KLK12 mRNA levels were
identified to be increased at different degrees in the majority of
the colorectal cancer cell lines, with the exception of HCT-15 and
RKO cells, compared with normal colorectal cells CCD-18Co (Fig. 1C). HT-29 cells exhibited the
highest expression of KLK12; therefore, HT-29 cells were selected
to be used in the following experiments.
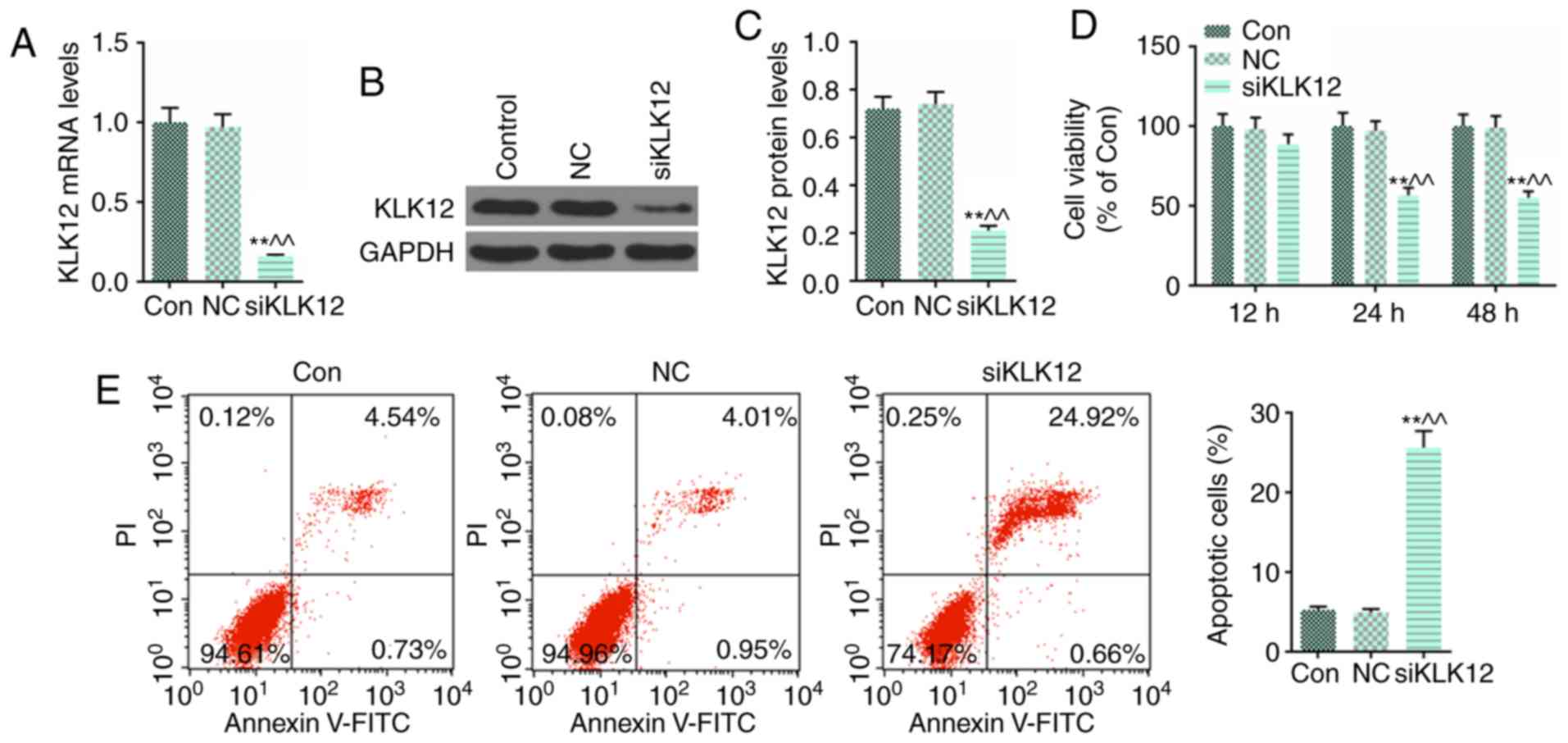
Knockdown of KLK12 inhibits HT-29 cells
viability and promotes the cell apoptosis
To further investigate the functional effects of
KLK12 on colorectal cancer HT-29 cells, KLK12 expression was
silenced by siRNA using Lipofectamine® 2000 transfection
reagent, and RT-qPCR (Fig. 2A)
and western blot analysis (Fig.
2B and C) were performed to confirm that the transfection was
successful. Subsequently, HT-29 cells were seeded in a 96-well
plate and transfected with small interfering RNA of KLK12 for 12,
24 and 48 h. The viability of HT-29 cells was analyzed using the
MTT assay. It was identified that knockdown of KLK12 significantly
decreased the viability of HT-29 cells at 24 and 48 h (Fig. 2D). Furthermore, cell apoptosis
levels were quantified by flow cytometry using Annexin V and PI
double staining, and it was demonstrated that KLK12 silencing
noticeably increased the proportion of apoptotic HT-29 cells
compared with the control and NC groups (Fig. 2E).
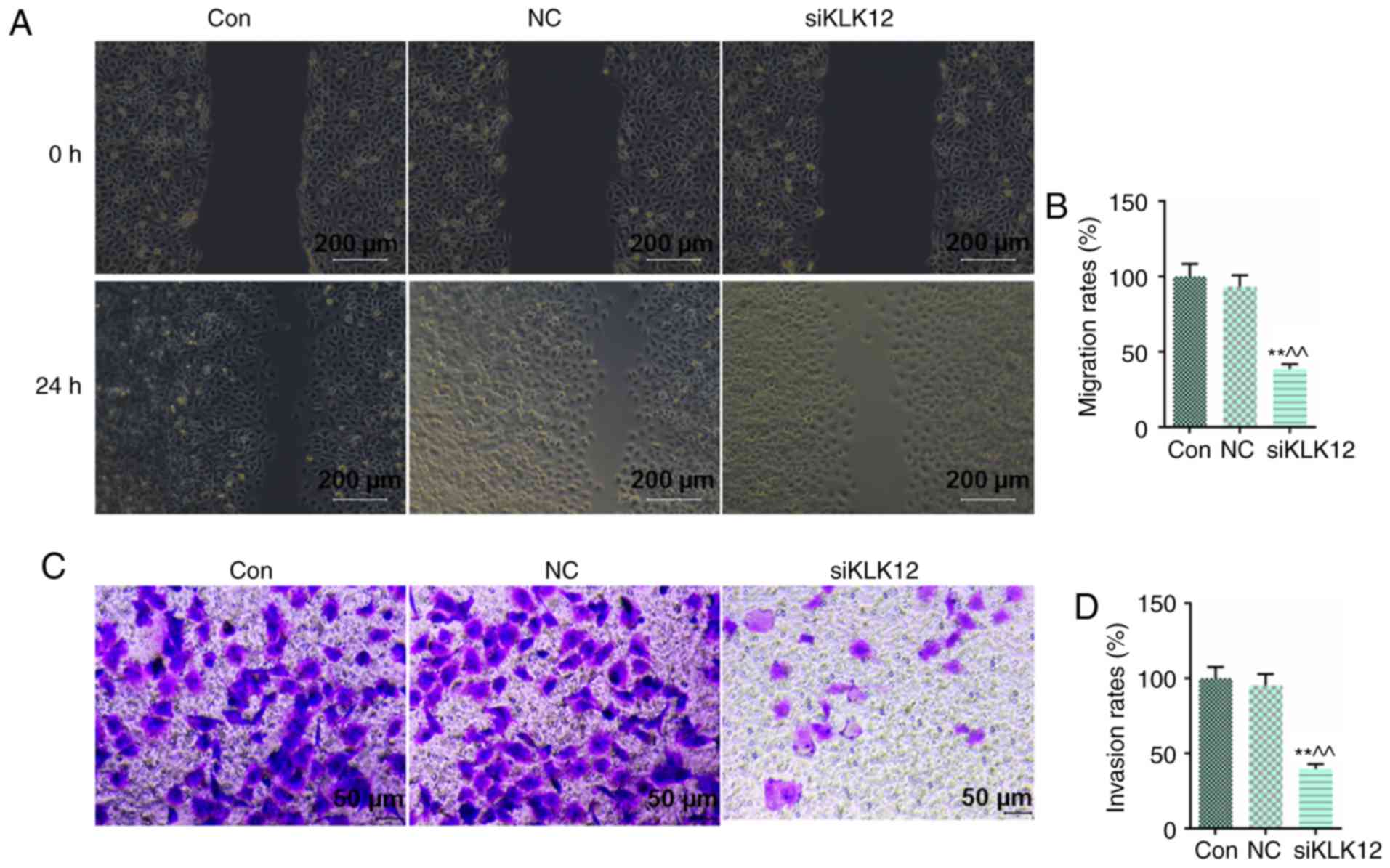
Knockdown of KLK12 inhibits HT-29 cells
migration and invasion
Migration and invasion are typical biological
characteristics of malignant tumors; therefore, invasion and
migration were determined by wound healing and Transwell invasion
assays, respectively. It was observed that KLK12 silencing for 24 h
significantly decreased the migration rate in HT-29 cells (Fig. 3A and B). Concomitantly, cell
invasion was inhibited following 24 h downregulation of KLK12 in
HT-29 cells (Fig. 3C and D).
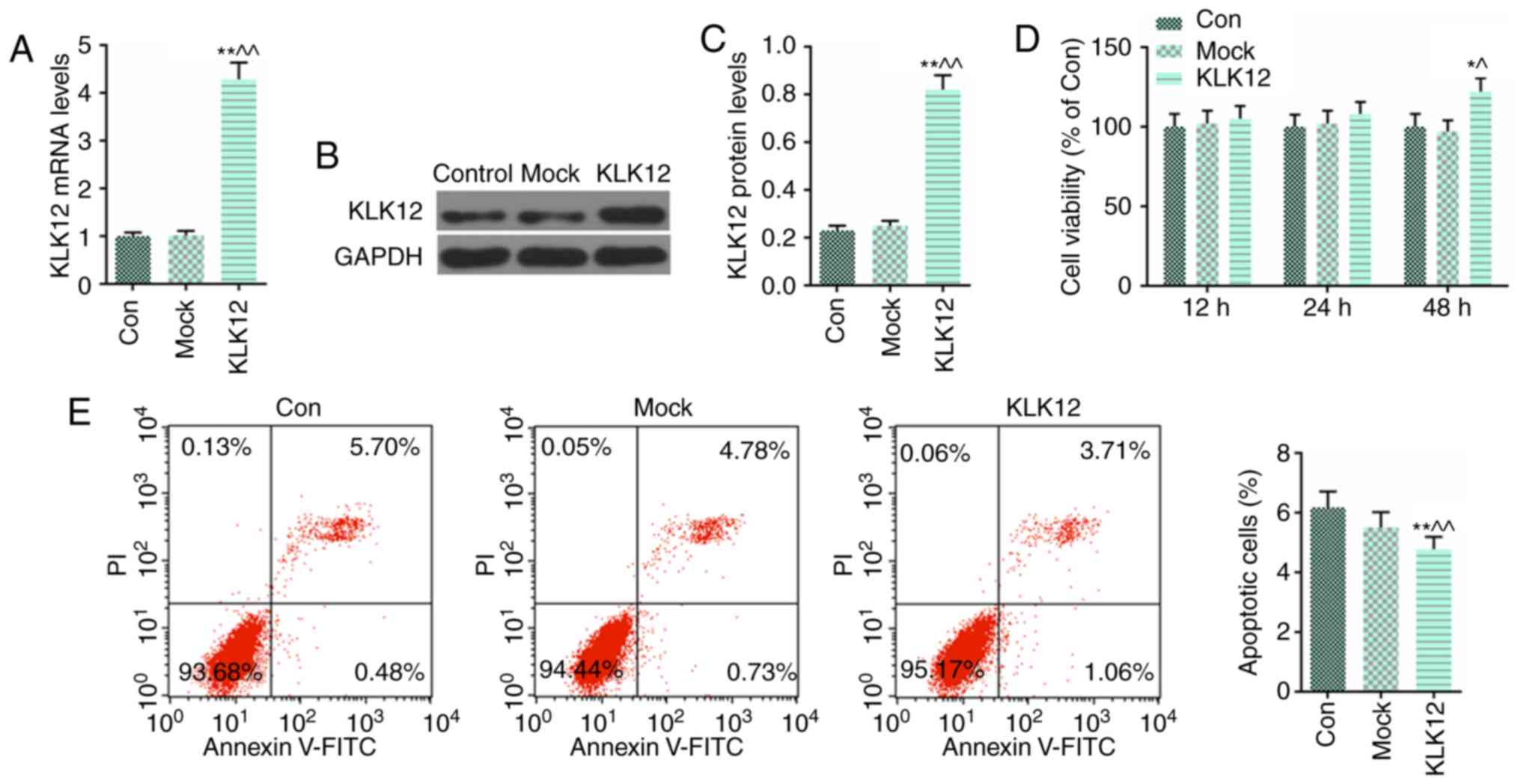
Overexpression of KLK12 suppresses
apoptosis in HT-29 cells
The expression level of KLK12 was overexpressed by a
KLK12 plasmid using Lipofectamine® 2000 transfection
reagent, and RT-qPCR (Fig. 4A)
and western blot analysis (Fig.
4B and C) were performed to confirm that the transfection was
successful. Subsequently, HT-29 cells were seeded in a 96-well
plate and transfected with the KLK12 plasmid for 12, 24 and 48 h.
The viability of the HT-29 cells was analyzed using the MTT assay.
KLK12 overexpression significantly increased the viability of HT-29
cells at 48 h (Fig. 4D).
Furthermore, cell apoptosis levels were quantified by flow
cytometry with Annexin V and PI double staining, and it was
identified that KLK12 overexpression significantly decreased the
proportion of apoptotic HT-29 cells compared with the control and
mock groups (Fig. 4E).
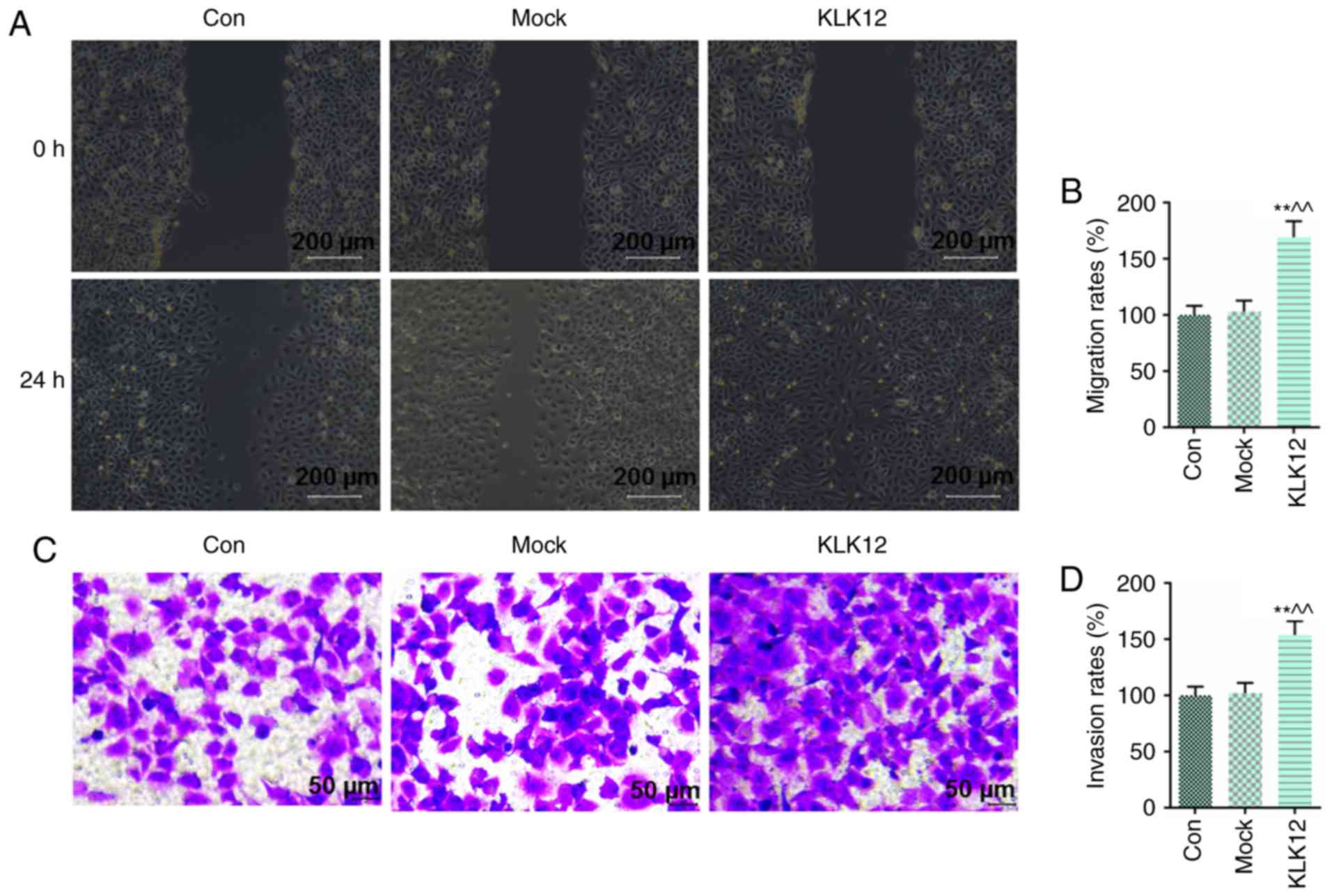
Upregulation of KLK12 promotes migration
and invasion in HT-29 cells
In the present study, invasion and migration were
determined by wound healing and Transwell invasion assays,
respectively. The results demonstrated that overexpressing KLK12
for 24 h significantly increased the migration rate of HT-29 cells
(Fig. 5A and B), and that cell
invasion was also increased following KLK12 upregulation for 24 h
in the HT-29 cells (Fig. 5C and
D).
EMT-associated and apoptosis-associated
proteins are altered in HT-29 cells treated with siKLK12/KLK12
The effects of siKLK12/KLK12 on the EMT-associated
proteins including E-cadherin, Vimentin, Snail, MMP-2 and MMP-9
were determined in HT-29 cells by performing RT-qPCR and western
blot analysis. It was identified that treatment with KLK12 siRNA
significantly upregulated the expression of E-cadherin and
downregulated the expression levels of vimentin, Snail, MMP-2 and
MMP-9 at mRNA levels (Fig. 6A)
and protein levels (Fig. 6B and
C). However, overexpression of KLK12 produced the opposite effect
in these proteins, in comparison with the results from siKLK12
treatment (Fig. 6A-C). It was
also identified that apoptosis was induced by siKLK12 treatment and
inhibited by KLK12 overexpression from the results of flow
cytometry; however, alterations to the apoptosis-associated
proteins remained unclear. Therefore, the effects of KLK12
silencing and over-expression on the expression of
apoptosis-association proteins including Bcl-2, Bax and cleaved
caspase-3 was examined in the HT-29 cells by western blot analysis.
The results indicated that KLK12 silencing induced apoptosis, as
the expression of anti-apoptosis protein Bcl2 was significantly
downregulated and the expression levels of pro-apoptosis proteins
Bax and cleaved caspase-3 were upregulated, when compared with the
control group (Fig. 6D and E),
while overexpression of KLK12 elicited the opposite effect in these
proteins, in comparison with the effects of siKLK12 treatment
(Fig. 6D and E).
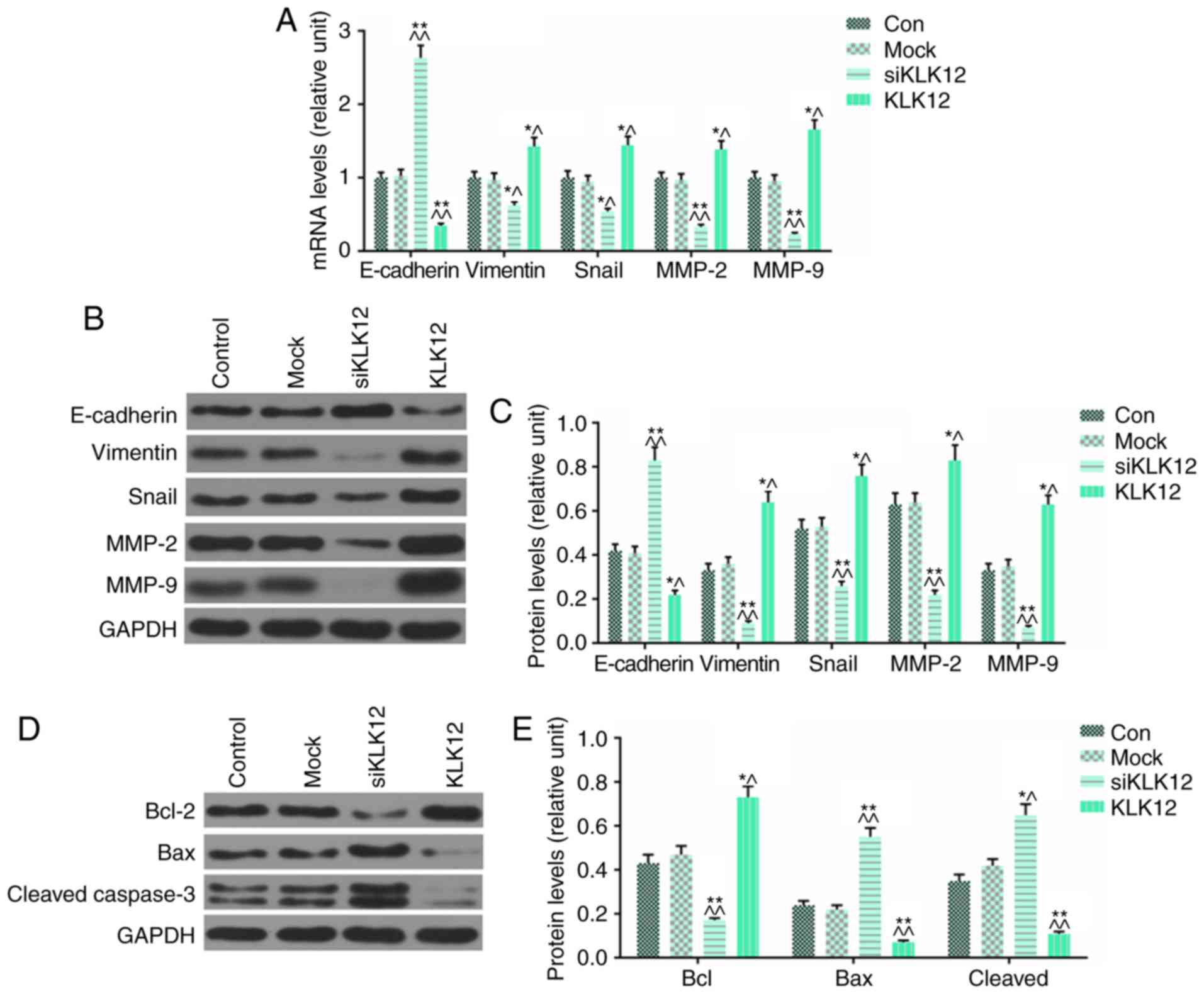 | Figure 6Expression of epithelial-mesenchymal
transition-associated and apoptosis-associated proteins is altered
in HT-29 cells treated with siKLK12 or KLK12 overexpression
plasmids. (A) The mRNA levels of E-cadherin, vimentin, Snail, MMP-2
and MMP-9 were analyzed by reverse transcription quantitative
polymerase chain reaction. (B and C) The protein levels of
E-cadherin, vimentin, Snail, MMP-2 and MMP-9 were analyzed by
western blot analysis. (D and E) The protein levels of Bcl-2, Bax
protein and cleaved caspase-3 were analyzed by western blot
analysis. *P<0.05 and **P<0.01 vs. NC
group. ^P<0.05 and ^^P<0.01 vs. mock
group. The 'KLK12' group represents the KLK12 overexpression group.
KLK12, kallikrein-related peptidase 12; si, small interfering RNA;
E-cadherin, epithelial cadherin; Snail, zinc finger protein SNAI1;
MMP, matrix metalloproteinase; NC, negative control. |
AMPK and mTOR pathways are involved in
HT-29 cells treated with siKLK12/KLK12
The AMPK/mTOR signaling pathway serves an important
role in tumor progression and metastasis. To explore the potential
mechanism of KLK12 in the regulation of colorectal cancer cell
migration and invasion, the present study examined the effects of
KLK12 on AMPK/mTOR signaling. The levels of p-AMPK, AMPK, p-mTOR
and mTOR in the HT-29 cells, which were transfected with siKLK12
and the KLK12 plasmid, were determined by western blot analysis.
The results demonstrated that in HT-29 cells transfected with
siKLK12, p-AMPK levels were significantly upregulated compared with
control group; by contrast, the expression of p-mTOR was decreased
(Fig. 7A and B). However,
overexpression of KLK12 resulted in the opposite effect in those
proteins compared with siKLK12 treatment (Fig. 7A and B). In order to confirm the
role of AMPK/mTOR in the progression of HT-29 cells, an activator
of the AMPK signaling pathway, AICAR (2 mM), and a specific
inhibitor of mTOR signaling pathway, rapamycin (10 nM), were
incubated with HT-29 cells with or without the KLK12 plasmid for
12, 24 and 48 h. The viability of the HT-29 cells was analyzed
using MTT assay. The data demonstrated that AICAR reversed the
effect of KLK12 overexpression on cell viability, and that AICAR
had significant inhibitory effect on viability of HT-29 cells
(Fig. 7C). Surprisingly, the
results of rapamycin on the effect of KLK12 on HT-29 cell viability
were consistent with AICAR (Fig.
7D).
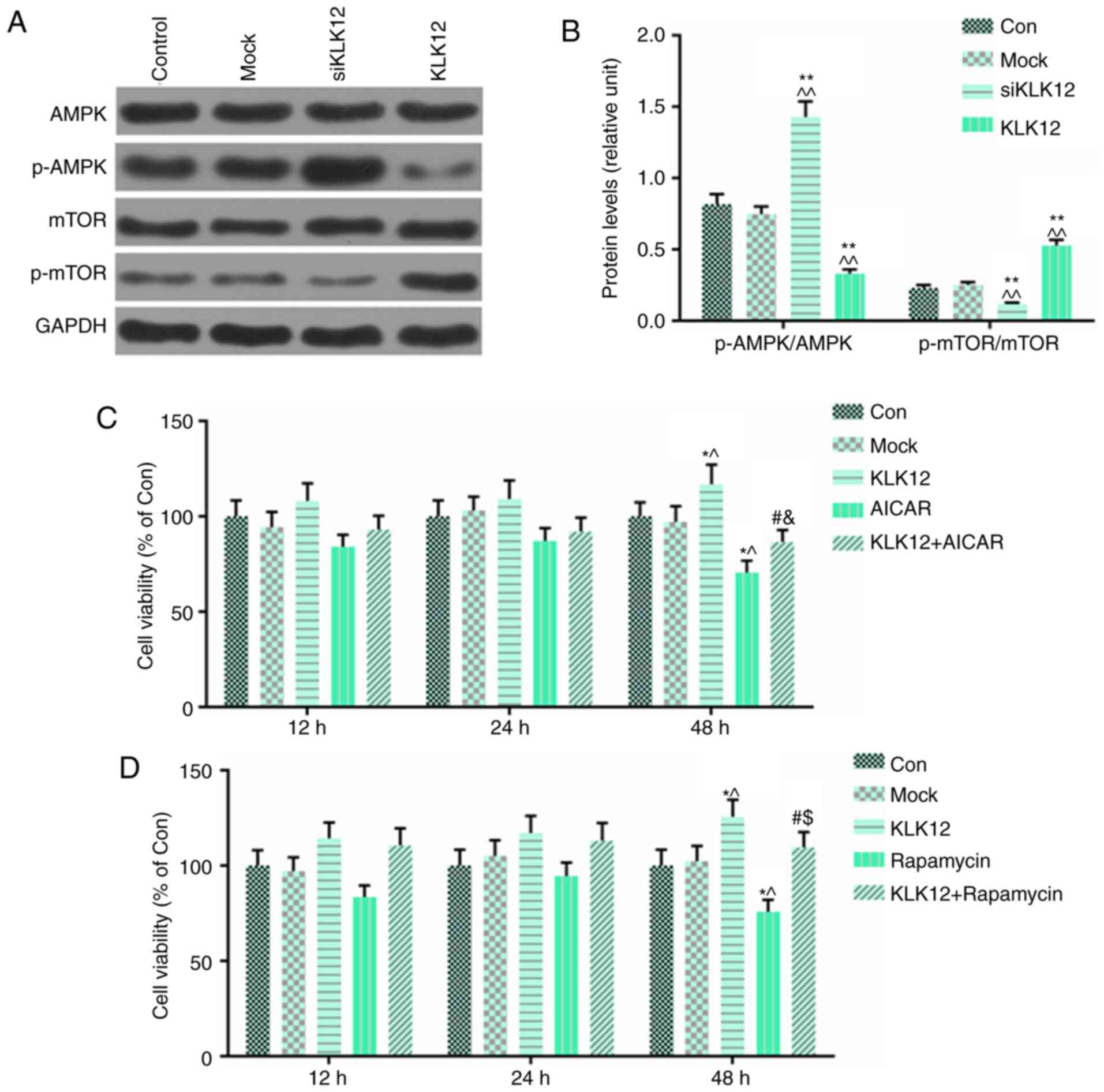 | Figure 7AMPK and mTOR pathways are involved
in HT-29 cells treated with siKLK12 or KLK12 overexpression
plasmids. (A and B) Protein levels of AMPK, p-AMPK, mTOR, p-mTOR
and GAPDH in HT-29 cells were (A) analyzed and (B) quantified by
western blot analysis. (C) Analysis of cell viability using the
CCK-8 assay in mock or KLK12-transfected HT-29 cells incubated with
or without AICAR treatment. (D) Analysis of cell viability using
the CCK-8 assay in mock or KLK12-transfected HT-29 cells incubated
with or without rapamycin treatment. *P<0.05 and
**P<0.01 vs. NC group. ^P<0.05 and
^^P<0.01 vs. mock group. #P<0.05 vs.
KLK12. &P<0.05 vs. AICAR. $P<0.05
vs. rapamycin. The 'KLK12' group represents the KLK12
overexpression group. KLK12, kallikrein-related peptidase 12; AMPK,
adenosine monophosphate-activated protein kinase; p-APMK,
phosphorylated adenosine monophosphate-activated protein kinase;
mTOR, mammalian target of rapamycin; p-mTOR, phosphorylated
mammalian target of rapamycin; CCK-8, Cell Counting Kit-8; AICAR,
5-aminoimidazole-4-carboxamide ribonucleotide. |
Discussion
Tissue or serum levels of KLK are examined
individually or in small groups as a diagnostic or prognostic
factor for determining different types of cancer (22,23). In the colorectal cancer, KLK has
been demonstrated to act as a diagnostic or prognostic factor
(16). However, KLK12 has not
been studied extensively in colorectal cancer. In the present
study, KLK12 overexpression was demonstrated promoting the
viability and inhibit apoptosis in the colorectal cancer-derived
HT29 cell line via the activation of the AMPK/mTOR signaling
pathways.
There have been multiple previous studies concerning
KLK12 in different types of cancer: Li et al identified that
KLK12 levels were significantly increased in gastric cancer cells
compared with in GES-1 cells (24). In addition, analysis of a combined
sample of 3,153 cases and 3,199 controls indicated that the KLK12
tag single nucleotide polymorphism rs3865443 was marginally and
statistically correlated with the risk of developing prostate
cancer (25). KLK12 expression at
mRNA level was markedly increased in MKN-45 gastric cancer cells
compared with normal mucosal cells and 2 other gastric cancer cell
lines (26); the mRNA level of
KLK12 observed in the present study was consistent with previous
studies. It was also identified that KLK12 expression levels were
increased in colorectal cancer tissues and colorectal
cancer-derived cell lines compared with their corresponding
controls. These data demonstrated that the oncogenic potential of
KLK12 in colorectal cancer was similar to that in other types of
cancer.
Metastasis is regarded as a major factor
contributing to the poor prognosis of patients with colorectal
cancer and is a typical feature of malignant tumors. Overexpression
of KLK12 protein was significantly associated with lymph node
metastasis, and the proliferation of gastric cancer MKN-45 cells
was markedly decreased by the knockdown of KLK12 protein (26).
Transfection of AGS cells with KLK12 siRNA led to
decreased cell proliferation (24). KLK12 efficiently cleaved human
extracellular matrix proteins fibronectin and tenascin, both of
which are involved in the regulation of endothelial cell adhesion
and migration (27). In the
present study, cell viability, migration and invasion were
inhibited when the HT-29 cells were transfected with siKLK12.
Concomitantly, E-cadherin expression was upregulated and vimentin,
Snail, MMP-2 and MMP-9 expression were downregulated in
siKLK12-trans-fected HT-29 cells. However, KLK12 overexpression
elicited the opposite effect on those factors. Therefore, it was
concluded that KLK12 may be involved in the process of EMT in
colorectal cancer, at least in HT-29 cells.
Previous data supports the hypothesis that targeting
apoptosis may be a promising and protective strategy for the
management of colorectal cancer. MLK7-AS1 knockdown promoted CRC
cell apoptosis in vitro (28). Lupeol-induced cellular apoptosis
of both colorectal cancer cell lines, which increased p53 and
decreased Bcl2 protein levels (29). In the present study, apoptosis
levels and the expression levels of apoptosis-associated proteins
were determined by flow cytometry and western blot analysis,
respectively. It was identified that cell apoptosis was induced by
the knockdown of KLK12 in HT-29 cells. In addition, the
anti-apoptosis protein (Bcl-2) was expressed at low levels and the
pro-apoptosis proteins (Bax and cleaved caspase-3) were expressed
at high levels in the siKLK12 group, compared with the control
group. However, KLK12 upregulation produced the opposite effect on
those factors. These results suggested that the function of KLK12
as a carcinogenic factor in colorectal cancer may be realized by
the inhibition of apoptosis, at least in HT-29 cells. However, the
precise mechanisms underlying KLK12 activity in colorectal cancer
required further investigation.
At present, the mechanism by which KLK12 induced
viability and metastasis of colorectal cancer is not fully
understood. AMPK, a ubiquitous serine/threonine protein kinase,
regulates tumorigenesis, development and chemical resistance
through negative regulation of mTOR (30). The phosphorylation of AMPK and
inhibition of downstream phosphorylation of mTOR in
cholangiocarcinoma cells were activated by FYN proto-oncogene, Src
family tyrosine kinase knockdown (31). In the present study, the functions
of KLK12 on AMPK/mTOR signaling were explored, and it was
identified that the phosphorylation of AMPK was activated and
downstream phosphorylation of mTOR was inhibited by KLK12
downregulation, while KLK12 overexpression inhibited the
phosphorylation levels of AMPK and promoted phosphorylation of
mTOR. Furthermore, the viability of KLK12 plasmid-transfected HT-29
cells incubated with AICAR or rapamycin was determined by MTT
assay. It was identified that cell viability was improved by HT-29
cells, which was reversed by AICAR or rapamycin. These data
demonstrated that the AMPK/mTOR signaling pathway was activated by
KLK12 in colorectal cancer. siKLK12 inhibited the viability and EMT
process of HT-29 cells via regulation of the AMPK/mTOR signaling
pathway. However, the method by which KLK12 functions in colorectal
cancer animal models or in patients with colorectal cancer remains
unclear. The present study also demonstrated that rapamycin may
produce limited effects on cell viability, compared with AICAR, and
this may be explained by a more marked inhibition of AICAR on mTOR
and its downstream genes.
Previous studies have indicated that KLK12 splice
variant KLK12sv3 may be used as a marker in predicting a desirable
prognosis in breast cancer (19).
Papachristopoulou et al (32) described the value of KLK12sv1/2
and KLK12sv3 in differentiating between benign and malignant breast
tumors and their potential prognostic value. We hypothesized that
KLK12 expression may also be associated with the survival and
prognosis of patients with colorectal cancer, however, whether it
may be used as a prognostic marker for colorectal cancer remains to
be explored. However, by referring to other studies (33,34), the effects of KLK12 on cell
proliferation and migration were verified. In the present study, it
was identified that silencing KLK12 inhibited the cell viability
and promoted apoptosis of human colorectal cancer HT-29 cells.
However, colorectal cancer is a highly heterogeneous disease, and
the role of KLK12 requires additional confirmation in multiple
different colorectal cancer cells.
In conclusion, KLK12 down-regulation inhibited cell
viability and metastasis via regulating AMPK/mTOR signaling pathway
in HT-29 cells. The present results provided that KLK12 was an
oncogene and could be used as a therapeutic target for treating
colorectal cancer.
Acknowledgements
Not applicable.
Abbreviations:
|
KLK12
|
kallikrein-related peptidase 12
|
|
RT-qPCR
|
reverse transcription quantitative
polymerase chain reaction
|
|
p-AMPK
|
phosphorylated adenosine
monophosphate-activated protein kinase
|
|
p-mTOR
|
phosphorylated mammalian target of
rapamycin
|
|
EMT
|
epithelial-mesenchymal transition
|
|
AMPK
|
adenosine monophosphate-activated
protein kinases
|
|
mTOR
|
mammalian target of rapamycin
|
|
KLK
|
kallikrein
|
|
PI
|
propidium iodide
|
Funding
This study was supported by the Zhejiang Provincial
Traditional Chinese Medicine Science Research Foundation (grant no.
2016ZB037); the Natural Science Foundation of Zhejiang Province
(grant no. LQ15H290004).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
QL and HZ made substantial contributions to the
conception and design of the study. XZ and ZF were responsible for
data acquisition, data analysis and interpretation. HZ, XZ and QL
were responsible for drafting the article and critically revising
it for important intellectual content. All authors provided final
approval of the version to be published. All authors are
accountable for all aspects of the study in ensuring that questions
related to the accuracy or integrity of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
All procedures performed in studies involving human
participants were in accordance with the ethical standards of the
institutional and/or national research committee and with the 1964
Helsinki declaration and its later amendments or comparable ethical
standards. The present study was approved by the Ethical Committee
of The First Affiliated Hospital of Zhejiang Chinese Medical
University and written informed consent was obtained from each
patient.
Patient consent for publication
Written informed consent was obtained from each
patient.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar
|
|
3
|
Das B, Sarkar N, Bishayee A and Sinha D:
Dietary phytochemicals in the regulation of epithelial to
mesenchymal transition and associated enzymes: A promising
anticancer therapeutic approach. Semin Cancer Biol. 56:196–218.
2019. View Article : Google Scholar
|
|
4
|
Iwatsuki M, Mimori K, Yokobori T, Ishi H,
Beppu T, Nakamori S, Baba H and Mori M: Epithelial-mesenchymal
transition in cancer development and its clinical significance.
Cancer Sci. 101:293–299. 2010. View Article : Google Scholar
|
|
5
|
Nieto MA: The ins and outs of the
epithelial to mesenchymal transition in health and disease. Annu
Rev Cell Dev Biol. 27:347–376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boesch M, Spizzo G and Seeber A: Concise
review: Aggressive colorectal cancer: Role of epithelial cell
adhesion molecule in cancer stem cells and
epithelial-to-mesenchymal transition. Stem Cells Transl Med.
7:495–501. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hardie DG and Sakamoto K: AMPK: A key
sensor of fuel and energy status in skeletal muscle. Physiology
(Bethesda). 21:48–60. 2006.
|
|
9
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lizcano JM, Göransson O, Toth R, Deak M,
Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG and
Alessi DR: LKB1 is a master kinase that activates 13 kinases of the
AMPK subfamily, including MARK/PAR-1. EMBO J. 23:833–843. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guertin DA and Sabatini DM: Defining the
role of mTOR in cancer. Cancer Cell. 12:9–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ji S, Tang S, Li K, Li Z, Liang W, Qiao X,
Wang Q, Yu S and Ye M: Licoricidin inhibits the growth of SW480
human colorectal adenocarcinoma cells in vitro and in vivo by
inducing cycle arrest, apoptosis and autophagy. Toxicol Appl
Pharmacol. 326:25–33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thent ZC, Zaidun NH, Azmi MF, Senin MI,
Haslan H and Salehuddin R: Is metformin a therapeutic paradigm for
colorectal cancer: Insight into the molecular pathway? . Curr Drug
Targets. 18:734–750. 2017. View Article : Google Scholar
|
|
14
|
Clements J, Hooper J, Dong Y and Harvey T:
The expanded human kallikrein (KLK) gene family: Genomic
organisation, tissue-specific expression and potential functions.
Biol Chem. 382:5–14. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Di Meo A, Wang C, Cheng Y, Diamandis EP
and Yousef GM: The miRNA-kallikrein interaction: A mosaic of
epigenetic regulation in cancer. Biol Chem. 399:973–982. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Talieri M, Li L, Zheng Y, Alexopoulou DK,
Soosaipillai A, Scorilas A, Xynopoulos D and Diamandis EP: The use
of kallikrein-related peptidases as adjuvant prognostic markers in
colorectal cancer. Br J Cancer. 100:1659–1665. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lawrence MG, Lai J and Clements JA:
Kallikreins on steroids: Structure, function, and hormonal
regulation of prostate-specific antigen and the extended kallikrein
locus. Endocr Rev. 31:407–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Y, Lu Z, Cui M, Yang Q, Tang Y and
Dong Q: Tissue kallikrein protects SH-SY5Y neuronal cells against
oxygen and glucose deprivation-induced injury through bradykinin B2
receptor-dependent regulation of autophagy induction. J Neurochem.
139:208–220. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Talieri M, Devetzi M, Scorilas A, Pappa E,
Tsapralis N, Missitzis I and Ardavanis A: Human kallikrein-related
peptidase 12 (KLK12) splice variants expression in breast cancer
and their clinical impact. Tumour Biol. 33:1075–1084. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kwon Y, Park M, Jang M, Yun S, Kim WK, Kim
S, Paik S, Lee HJ, Hong S, Kim TI, et al: Prognosis of stage III
colorectal carcinomas with FOLFOX adjuvant chemotherapy can be
predicted by molecular subtype. Oncotarget. 8:39367–39381. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Geng X, Liu Y, Dreyer T, Bronger H,
Drecoll E, Magdolen V and Dorn J: Elevated tumor tissue protein
expression levels of kallikrein-related peptidases KLK10 and KLK11
are associated with a better prognosis in advanced high-grade
serous ovarian cancer patients. Am J Cancer Res. 8:1856–1864.
2018.PubMed/NCBI
|
|
23
|
Tailor PD, Kodeboyina SK, Bai S, Patel N,
Sharma S, Ratnani A, Copland JA, She JX and Sharma A: Diagnostic
and prognostic biomarker potential of kallikrein family genes in
different cancer types. Oncotarget. 9:17876–17888. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li XS and He XL: Kallikrein 12
downregulation reduces AGS gastric cancer cell proliferation and
migration. Genet Mol Res. 15:2016.
|
|
25
|
Lose F, Batra J, O'Mara T, Fahey P,
Marquart L, Eeles RA, Easton DF, Al Olama AA, Kote-Jarai Z, Guy M,
et al: Common variation in Kallikrein genes KLK5, KLK6, KLK12, and
KLK13 and risk of prostate cancer and tumor aggressiveness. Urol
Oncol. 31:635–643. 2013. View Article : Google Scholar
|
|
26
|
Zhao EH, Shen ZY, Liu H, Jin X and Cao H:
Clinical significance of human kallikrein 12 gene expression in
gastric cancer. World. J Gastroenterol. 18:6597–6604. 2012.
|
|
27
|
Kryza T, Parent C, Pardessus J, Petit A,
Burlaud-Gaillard J, Reverdiau P, Iochmann S, Labas V, Courty Y and
Heuzé-Vourc'h N: Human kallikrein-related peptidase 12 stimulates
endothelial cell migration by remodeling the fibronectin matrix.
Sci Rep. 8:63312018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang R, Li J, Yan X, Jin K, Li W, Liu X,
Zhao J, Shang W and Zhao X: Long noncoding RNA MLK7AS-1 promotes
proliferation in human colorectal cancer via downregulation of p21
expression. Mol Med Rep. 19:1210–1221. 2019.
|
|
29
|
Wang Y, Hong D, Qian Y, Tu X, Wang K, Yang
X, Shao S, Kong X, Lou Z and Jin L: Lupeol inhibits growth and
migration in two human colorectal cancer cell lines by suppression
of Wnt-β-catenin pathway. Onco Targets Ther. 11:7987–7999. 2018.
View Article : Google Scholar :
|
|
30
|
Cork GK, Thompson J and Slawson C: Real
Talk: The inter-play between the mTOR, AMPK, and hexosamine
biosynthetic pathways in cell signaling. Front Endocrinol
(Lausanne). 9:pp. 5222018, View Article : Google Scholar
|
|
31
|
Lyu SC, Han DD, Li XL, Ma J, Wu Q, Dong
HM, Bai C and He Q: Fyn knockdown inhibits migration and invasion
in cholangiocarcinoma through the activated AMPK/mTOR signaling
pathway. Oncol Lett. 15:2085–2090. 2018.PubMed/NCBI
|
|
32
|
Papachristopoulou G, Tsapralis N,
Michaelidou K, Ardavanis-Loukeris G, Griniatsos I, Scorilas A and
Talieri M: Human kallikrein-related peptidase 12 (KLK12) splice
variants discriminate benign from cancerous breast tumors. Clin
Biochem. 58:78–85. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jia S, Qu T, Wang X, Feng M, Yang Y, Feng
X, Ma R, Li W, Hu Y, Feng Y, et al: KIAA-1199 promotes migration
and invasion by Wnt/β-catenin pathway and MMPs mediated EMT
progression and serves as a poor prognosis marker in gastric
cancer. PLoS One. 12:pp. e01750582017, View Article : Google Scholar
|
|
34
|
Yang D, Du G, Xu A, Xi X and Li D:
Expression of miR-149-3pinhibits proliferation, migration, and
invasion of bladder cancer by targeting S100A4. Am J Cancer Res.
7:2209–2219. 2017.
|