Introduction
Non-alcoholic fatty liver disease (NAFLD) is a
condition in which fat accumulates in the livers of patients
without a history of alcohol consumption (1). The disease occurs in close
association with obesity, visceral adiposity, type 2 diabetes and
the spectrum of metabolic syndrome (2).
The more advanced form of the disease is designated
as non-alcoholic steatohepatitis (NASH) and manifests, apart from
steatosis, as intra-lobular inflammation, hepatocellular ballooning
and progressive fibrosis (3).
Long-lasting NASH may progress to liver cirrhosis and
hepatocellular carcinoma (HCC) (4). The higher incidence of co-morbidity
risk factors in patients with NASH-derived HCC leads to a lower
overall survival compared to patients with HCC which has developed
due to other etiologies. Over the past decades, the prevalence of
NAFLD/NASH has markedly increased, more than doubling from
1999-2002 to 2009-2012. Among the west-ernized countries, 20% of
the general adult population presents with hepatic steatosis, with
2-3% suffering from NASH (2).
Due to the multifactorial nature of NAFLD/NASH, a
variety of drug candidates aiming at different molecular targets,
involved with disease etiology and pathogenesis, have been tested
to reverse or ameliorate the disease. These include insulin
sensitizers (such as thiazolidinediones and metformin);
hepatoprotective agents, including those with antioxidant
properties (such as ursodeoxycholic acid and vitamin E); bile acid
analogues (such as obeticholic acid), metabolically active agents
(such as weight loss drugs and hypouricemic agents); and drugs that
affect fat metabolism (such as statins and other hypolipidemic
agents, fibrates, acetyl-CoA carboxylase inhibitors and aramchol)
(5-7). More drug candidates which target
specific molecular pathways, such as the peroxisome
proliferator-activated receptor (PPAR), farnesoid X receptor (FXR),
glucagon-like peptide-1 and thyroid hormone receptor β (TRβ), are
under development; however, to date, to the best of our knowledge,
the US FDA has not approved any drug for the specific treatment of
NAFLD/NASH (8).
Namodenoson is a synthetic adenosine derivative that
binds with high selectivity to the A3 adenosine receptor (A3AR) in
pathological liver cells, inducing robust anti-inflammatory and
anti-cancer effects. It has been well established that
extracellular adenosine signaling is mediated via 4 receptors:
A1, A2A, A2B and A3.
The latter has the lowest affinity to adenosine and is
overexpressed in liver cells derived from inflammatory and tumor
tissues, whereas a low expression is found in normal liver cells
(9-11).
It has been well documented that A3AR is upregulated
in tumor and inflammatory liver tissues compared to normal liver
cells, suggesting a role for A3AR in human liver diseases (9). Selective agonists which bind to the
A3AR with high affinity induce a marked anti-inflammatory and
anti-cancer effect in experimental animal models of autoimmune
hepatitis and in orthotopic and xenograft liver cancer models
(9,10).
Data from a phase I/II trial in patients with
advanced HCC treated with Namodenoson demonstrated anti-tumor
activity and the prolongation of the overall survival time with an
excellent safety profile (12).
The molecular mechanisms of action involved with the
anti-inflammatory and anticancer effects of A3AR agonists, entail
inhibition of cyclic adenosine mono-phosphate (c-AMP), protein
kinase A (PKA), phosphoinositide 3-kinase (PI3K) and p-Akt
(9,13,14). These effects give rise to the
de-regulation of the nuclear factor κ-light-chain-enhancer of
activated B cells (NF-κB) and the Wnt/β-catenin pathways, known to
mediate anti-inflammatory, anti-steatotic, anti-fibrotic and
anticancer effects (9,15-17).
Of note, Namodenoson also acts as a protective agent
against liver damage in ischemia and partial hepatectomy. It has
been found to exert a protective effect on normal liver tissues in
an experimental model of liver inflammation (9,11),
suggesting its use as a therapeutic agent in subjects with hepatic
impairment. Taken together, these findings suggest a differential
effect of Namodenoson on pathological cells manifested via its
anti-inflammatory/anticancer effect, and at the same time, a
hepatoprotective effect towards normal liver cells.
Based on the unique characteristics of Namodenoson,
in this study, we aimed to determine the utility of the drug in 3
experimental models of NAFLD/NASH disease. The findings of this
study demonstrate that Namodenoson exhibits a triple-mechanism of
action in the liver, exerting anti-steatotic, anti-inflammatory and
anti-fibrotic effects. The upstream targeting of these pathways is
likely an effective and symbiotic combination for a promising
candidate for the treatment of NASH.
Materials and methods
Reagents
The A3AR agonist, Namodenoson, is a compound known
generically as 2-chloro-N6-(3-iodobenzyl)-adenos
ine-5′-N-methyl-uronamide, was synthesized for Can-Fite BioPharma
by Albany Molecular Research Inc. A stock solution of 5.44 mg/ml
was prepared in DMSO and further diluted in PBS for the in
vivo and in vitro experiments.
STAM mouse model
C57BL/6 mice (14-day-pregnant, female) were obtained
from Japan SLC, Inc. All animals used in this study were housed and
cared for in accordance with the Japanese Pharmacological Society
Guidelines for Animal Use. The animals were maintained in a SPF
facility under controlled conditions of temperature (23±2°C),
humidity (45±10%), lighting (12-h artificial light and dark cycle;
light from 08:00 to 20:00) and air exchange. A high pressure was
maintained in the experimental room to prevent the contamination of
the facility. The animals were housed in TPX cages (CLEA Japan,
Inc.) with a maximum of 4 mice per cage. Sterilized Paper-Clean
(Japan SLC) was used for bedding and was replaced once a week. A
sterilized solid high-fat diet (HFD) was provided ad
libitum. NASH was induced in 24 male mice by a single
subcutaneous injection of 200 µg streptozotocin (STZ,
Sigma-Aldrich Co. LLC.) solution 2 days after birth. At 4 weeks of
age, feeding with a HFD (57 kcal% fat, cat. no. HFD32, CLEA Japan,
Inc.) was initiated till sacrifice. Namodenoson at 200 µg/kg
(n=8) or equal amounts of DMSO (n=16) treatments were administered
p.o., thrice daily between weeks 6-9.
Mouse model of carbon tetrachloride
(CCl4)-induced liver fibrosis
Eleven-week-old male C57bl/6J male mice (n=21), from
Harlan Laboratories, and housed in a barrier facility and received
care according to the NIH guidelines. All procedures involving
laboratory animals were evaluated and approved by The Hebrew
University Institutional Animal Care and Use Committee and followed
the guidelines for laboratory animal welfare.
The mice (n=16) received an intraperitoneal (i.p.)
injection of CCl4 at 0.5 ml/kg body weight (1:10 v/v in corn oil)
(Sigma). Namodenoson at the dose of 100 µg/kg body was
administered by i.p. injection twice a week, commencing one day
after the first dose of CCl4 (n=8) or the vehicle (n=8) (corn oil)
twice a week for 6 weeks, as previously described (18). In addition, 5 naïve mice with no
treatments have been included as well. The mice were sacrificed at
72 h after the final CCl4 injection. Whole livers and serum were
collected for histological, cytological, biochemical and molecular
analyses.
The animals of both models were sacrificed by
exsangui-nation through direct cardiac puncture under ether
anesthesia.
Serum alanine aminotransferase (ALT)
measurement
Blood samples were collected from C57bl/6J male
mouse hearts at a volume of 0.5 ml. The samples were centrifuged
for 5 min at 3,584 × g, at 4°C, and the supernatants (volume of 600
µl) were transferred to Eppendorf tubes. Serum samples of 32
µl were dropped onto the strip of the ALT (Reflotron) and
analyzed by Reflovet® plus (Roche).
Histopathological analyses
Livers from the STAM and CCl4 model mice were
harvested and fixed in 4% formaldehyde for 48 h. Paraffin slides of
3-5 µ thickness were stained as described below. The
evaluation of the slides was performed using a microscope (Olympus
BX60, serial no. 7D04032 at a magnification of ×10) and microscope
Camera (Olympus DP73, serial no. OH05504). Ten random fields were
observed for each slide. For the STAM model slides, hematoxylin and
eosin (H&E) staining was used to analyze inflammation,
steatosis and ballooning, combined as the NAFLD activity score
(NAS) and calculated according to the Kleiner criteria (19) (Table
I). To assess the adiponectin quantity, immunofluorescence
mouse anti-adiponectin (1:50, cat. no. ab22554, Abcam) was used.
The stained sections were examined under a fluorescence microscope
(E600, Nikon) equipped with a Plan Fluor objective connected to a
CCD camera (DMX1200F, Nikon). Digital images were collected and
analyzed using Image-Pro Plus software 6.3. Images were assembled
using Adobe Photoshop (Adobe Systems). In addition,
immunohistological analysis utilizing rabbit
anti-adiponectin/Acrp30 antibody (cat. no. NB100-6581; Novus
Biologicals) at a dilution of 1:10 was performed to reproduce
images. For the CCl4 model slides, the following analyses were
performed: i) Sirius Red staining for fibrosis assessment; and ii)
immunohistological analysis utilizing rabbit
anti-adiponectin/Acrp30 antibody and rabbit anti leptin/OB antibody
(cat. no. NBP1-59324; Novus Biologicals) at a 1:10 dilution,
respectively, for the assessment of adiponectin and leptin.
 | Table INAS calculation by Kleiner
criteria. |
Table I
NAS calculation by Kleiner
criteria.
| Item | Score | Extent |
|---|
| Steatosis | 0 | <5% |
| 1 | 5-33% |
| 2 | >33-66% |
| 3 | >66% |
| Hepatocyte
ballooning | 0 | None |
| 1 | Few balloon
cells |
| 2 | Many
cells/prominent ballooning |
| Lobular
inflammation | 0 | No foci |
| 1 | <2
foci/200x |
| 2 | 2-4 foci/200x |
| 3 | >4
foci/200x |
Inflammation assessment (H&E
staining)
For the assessment of inflammation, H&E staining
was carried out. The sections were graded as follows: Grade 0, no
inflammation; grade 1, mild/few inflammatory cells, 10-20 per X20
high-power field (HPF); grade 2, moderate/more inflammatory cells,
20-50 per X20 HPF; and grade 3, severe/many inflammatory cells,
>50 per X20 HPF.
Fibrosis assessment (Sirius Red
staining)
The liver sections were stained with Picro-Sirius
Red solution (Waldeck). Morphometric analysis was performed using
'Image Pro Plus; version 6.3 (Media Cybernetics)'.
Adiponectin and leptin assessment
For adiponectin and leptin assessment, the sections
were graded as follows: Grade 0, no positive reaction of IHC at
all; grade 1, only few cells are immunopositive to the stain (<5
cells per a ×10 field); grade 2, very mild immune reaction (>5
and <15 cells per a ×10 field); grade 3, mild immune reaction
(>15 and <25 cells per a ×10 field); grade 4, moderate immune
reaction (>25 and <50 cells per a ×10 field); and grade 5,
high immune reaction (>50 cells per a ×10 field).
RT-qPCR. Total cellular RNA was isolated from liver
tissue, using 2 ml TRI Reagent (Bio LAB; Jerusalem, Israel) for
each 3 cm of tissue. The samples were homogenized for 5 min at room
temperature of 25°C. 0.2 ml of Chloroform (Bio LAB; Jerusalem,
Israel) were added to each sample, incubated for 15 min at room
temperature and centrifuged (1,400 rpm) for 15 min at 4°C. The
supernatant in each sample was transferred to new Eppendorf, 0.5 ml
of isopro-panol (Bio-Lab Ltd.) were added to the sample for 10 min
at 25°C and centrifuged (32,256 × g) for 10 min at 4°C. The
supernatants were removed and 1 ml of 75% ethanol was added to the
pellet and centrifuged (12,600 × g) for 5 min at 4°C. The pellets
were dried in air at room temperature for 15 min, 50 µl of
DEPC were added, and the samples were heated for 10 min at 55°C.
The preparation of cDNA was performed using High Capacity cDNA
Isolation kit (R&D Systems). RT-qPCR was performed for the
quantification of the expression of the gene that encoded α-SMA
(Rhenium Assay ID: Mm00725412s1), compared to GAPDH (Rhenium Assay
ID: Mm99999915g1) as a housekeeping gene using TaqMan Fast Advanced
Master Mix (Applied Biosystems). Relative gene expression data were
analyzed using the 2−ΔΔCq method (20).
Semi-quantitative PCR
For the LX2 cells, total RNA was extracted using
TRI-Reagent (Sigma). RNA (500 ng) was reverse transcribed using the
high-capacity cDNA reverse transcription kit (Thermo Fisher
Scientific) according to the manufacturer's instructions. RT-qPCR
was performed for the quantification gene expression of the encoded
α-SMA, compared to β-actin as a housekeeping gene using TaqMan Fast
Advanced Master Mix (Applied Biosystems). For α-SMA, the primers
forward. 5′-ACT GGG ACG ACA TGG AAA AG-3′ and reverse. 5′-TAC ATG
GCT GGG ACA TTG AA-3′ were added. The PCR reaction was performed as
follows: Heating to 94°C for 2 min, 25 cycles of 94°C for 15 sec,
56°C for 45 sec, and 68°C for 2 min. For the amplification of human
β-actin, the primers forward. 5′-TGG GAA TGG GTC AGA AGG ACT-3′ and
reverse. 5′-TTT CAC GGT TGG CCT TAG GGT-3′ were used. The PCR
condition included heating to 94°C for 2 min, 25 cycles of 94°C for
30 sec, 56°C for 1 min and 30 sec, and 72°C for 45 sec. The PCR
products were electrophoresed on 2% agarose gels, stained with
ethidium bromide and visualized with UV illumination. Total Lab
v1.11 software was used for quantification.
Cell culture
LX2 human hepatic stellate cells (HSCs; ATCC) were
grown in DMEM containing penicillin (10 units/ml), streptomycin (10
µg/ml), L-glutamine (2 mM) and 10% fetal bovine serum (FBS).
The cells were maintained in T-75 flasks at 37°C in a 5%
CO2 incubator and transferred to a freshly prepared
medium twice weekly. For all experiments, serum-starved cells were
used. FBS was omitted from the cultures for 18 h and the experiment
was carried out on monolayers of cells in DMEM supplemented with 1%
FBS in a 37°C, 5% in a CO2 incubator.
3H-thymidine incorporation
assay
3H-thymidine incorporation assay was used
to evaluate cell growth. LX2 cells (5,000 cells/well) were
incubated (37°C) with 10 nM Namodenoson in the absence or presence
of the antagonist, MRS 1523 (5 nM) (cat. no. M1809; Sigma-Aldrich),
in a 96-well plate for 48 h. Each well was pulsed with 1 µ
Ci 3H-thymidine for the last 24 h. The cells were
harvested and the 3H-thymidine uptake was determined in
an LKB liquid scintillation counter (LKB). These experiments were
repeated at least 4 times.
Western blot analysis
Protein extracts from liver tissue or the LX-2 cell
line were utilized. The liver tissue was homogenized with an
ice-cold RIPA lysis buffer (Sigma) with protease phosphatase
inhibitor cocktail (Roche). The LX2 cells were rinsed with ice-cold
PBS and transferred to ice-cold lysis buffer (TNN buffer, 50 mM
Tris buffer pH 7.5, 150 mM NaCl, NP 40 0.5% for 20 min). Cell
debris was removed by centrifugation for 10 min, at 7,500 × g, 4°C.
The supernatant was utilized for western blot analysis. Protein
concentrations were determined using the NanoDrop spectrophotometer
(Thermo Fisher Scientific). Equal amounts of the sample (50
µg) were separated by SDS-PAGE, using 4-12% polyacrylamide
gels. The resolved proteins were then electroblotted onto
nitrocellulose membranes (Pall Corp.). The membranes were then
blocked with 5% bovine serum albumin and incubated with the desired
primary antibody [A3AR, sc-13938; PI3K, sc-1637; glycogen synthase
kinase 3β (GSK-3β), sc-9166; β-catenin, sc-7963; cyclin D1,
sc-8396; NF-κB, sc-372; lymphoid enhancer-binding factor 1 (LEF-1),
sc-374522; α-SMA, sc-32251; and β-actin, sc-47778; dilution
1:1,000; all from Santa Cruz Biotechnology] for 24 h at 4°C. The
blots were then washed and incubated with a secondary antibody
(Abcam; mouse ab97020, rabbit ab97048) for 1 h at room temperature.
Bands were recorded using the BCIP/NBT color development kit
(Promega). Densitometry of protein expression was normalized
against β-actin and expressed as a percentage of the control.
Statistical analysis
Statistical analyses for the comparison between 2
groups were performed using the Student's t-test. Significant
differences between multiple groups were determined using one-way
ANOVA followed by Tukey's post-hoc analysis. P-values <0.05 were
considered to indicate statistically significant differences. Data
are presented as the means ± SD. The Chi-squared test was used for
the assessment of ascites. For both analyses, P-values <0.05
were considered indicate statistically significant differences.
Results
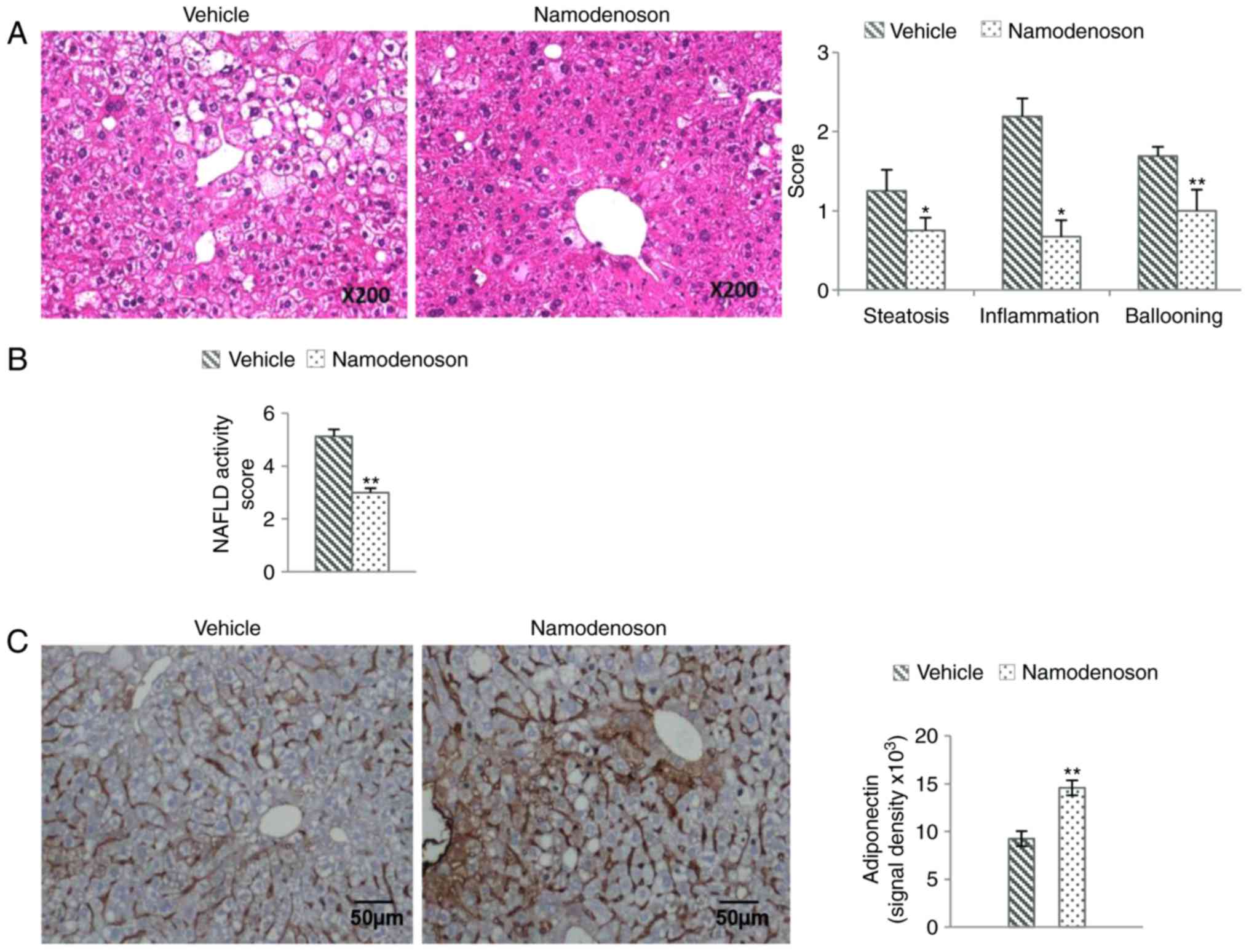
Namodenoson improves NAS in the STAM
model
A representative photomicrograph of H&E-stained
liver sections is depicted in Fig.
1A, demonstrating that in the vehicle-treated group,
inflammatory cell infiltration, severe micro- and macrovesicular
fat deposition and hepatocellular ballooning had occurred.
Namodenoson treatment between weeks 6 to 9 resulted in a
significant reduction in steatosis and an improvement in
inflammation and ballooning, as demonstrated by morphometric
analysis (P<0.05, P<0.05 and P<0.01, respectively),
summing up to a significant decrease in the NAS score compared to
the vehicle-treated group (Fig.
1B). Moreover, intense adiponectin staining was observed in
liver sections derived from the Namodenoson-treated group compared
to the vehicle treated one (Fig.
1C, left panel) and quantification is depicted in the right
panel of Fig. 1C, utilizing
immunofluorescence antibody (P<0.01).
Anti-NAFLD/NASH effects of Namodenoson in
the CCl4 model
In the CCl4 model, the serum ALT levels increased
from 63.65±2.36 to 294.75±69.24 (P<0.01). Namodenoson
significantly decreased the serum ALT levels, returning them to
near normal levels (69.35±17.64; P<0.01; Fig. 2A). Namodenoson also reversed the
ascites induced by CCl4 compared to the vehicle-treated group
(P<0.05; Fig. 2B).
Morphometric analysis of the liver sections derived from the
CCL4-treated mice revealed an upregulation of inflammation,
fibrosis, adiponectin and leptin, whereas a significant reduction
was noted in the samples derived from the Namodenoson-treated mice
(P<0.01; Fig. 2C).
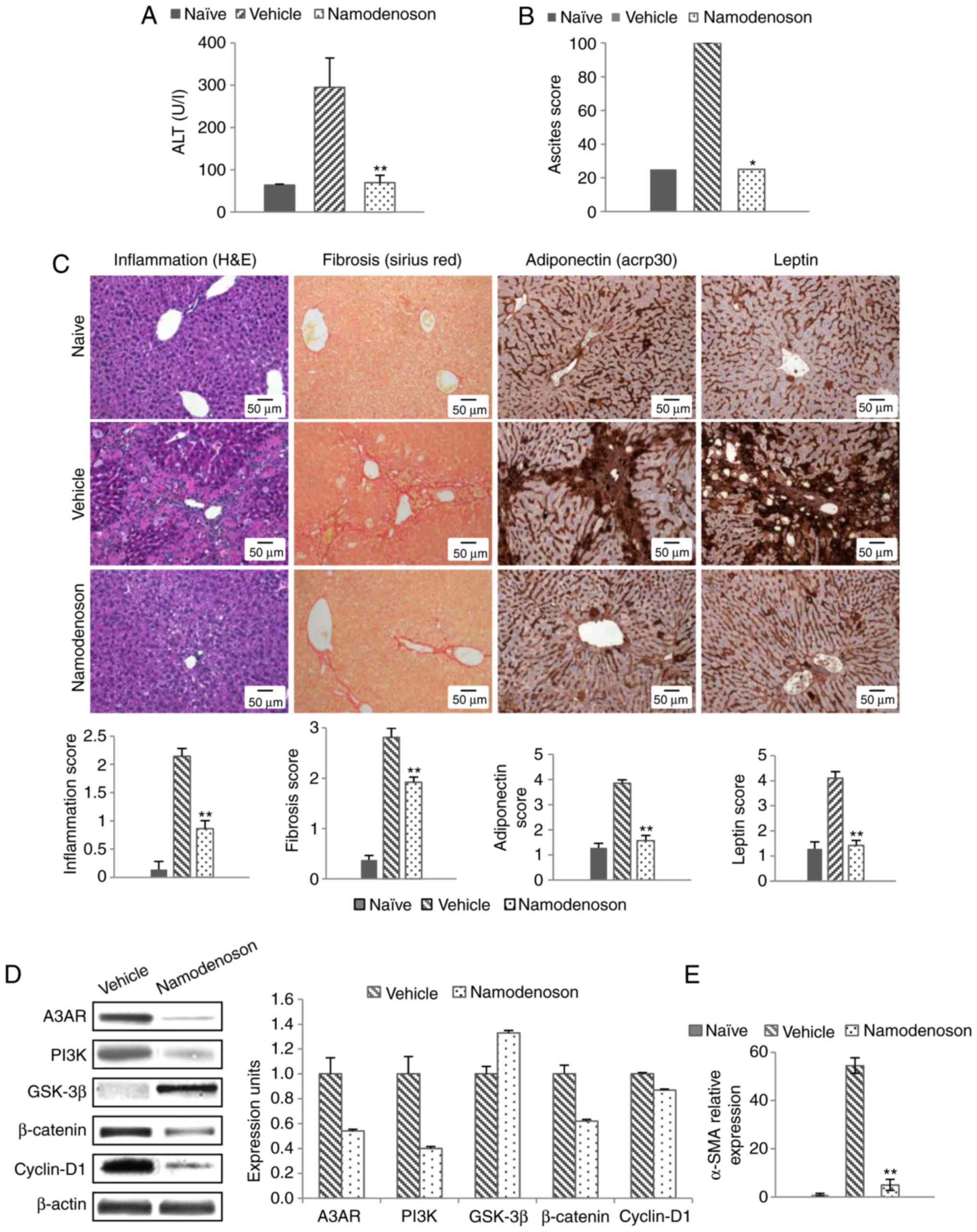 | Figure 2Namodenoson ameliorates liver
functions and pathology via the deregulation of the Wnt/β-catenin
pathway in the CCl4 model. CCL4-injected mice were treated twice
weekly via i.p. injection with Namodenoson (100 µg/kg) or
the vehicle (DMSO). The levels of (A) serum ALT and (B) ascites
were reversed to those of the naïve group following treatment with
Namodenoson. (C) Representative stained liver sections
demonstrating an improvement in inflammation (H&E staining),
fibrosis (Sirius Red staining), adiponectin and leptin
(immunohistochemistry); original magnification, ×40 (D)
Representative western blots of the protein expression of A3AR,
PI3K, GSK-3β, β-catenin and cyclin-D1 in mouse liver extracts.
β-Actin served as an internal control. (E) mRNA expression of
α-SMA. The data represent the means ± SEM. Statistical analysis was
carried out by one-way ANOVA followed by Tukey's post-hoc analysis
in panels A, C and E. The Chi-squared test was used for ascite
assessment in panel B; *P<0.05,
**P<0.01, vehicle vs. naïve and Namodenoson vs.
vehicle; n=8 animals/group. A3AR, A3 adenosine receptor; PI3K,
phosphoinositide 3-kinase; GSK-3β, glycogen synthase kinase 3β;
α-SMA, α-smooth muscle actin. |
Western blot analysis of the liver extracts derived
from the animals in the CCl4 model revealed that Namodenoson
treatment induced a decrease in the A3AR and PI3K expression levels
followed by an increase in the GSK-3β expression level, with a
subsequent reduction in the levels of β-catenin and cyclin D1, key
proteins of the Wnt signaling pathway (Fig. 2D). The mRNA expression level of
α-SMA was markedly decreased in the Namodenoson-treated group
compared with the vehicle-treated group (P<0.01; Fig. 2E).
Namodenoson inhibits LX2 cell
Proliferation and modulates key growth regulatory proteins
Namodenoson inhibited LX2 cell proliferation as was
measured by 3H-thymidine incorporation. The highly
specific A3AR antagonist, MRS 1523, counteracted the inhibitory
effect of Namodenoson (P<0.01; Fig. 3A). Western blot analysis revealed
that upstream, Namodenoson treatment induced the downregulation of
A3AR, PI3K, NF-κB and α-SMA expression; these effects were reversed
by MRS 1523, demonstrating the specificity of the response to the
agonist (Fig. 3B). Downstream,
GSK-3β was up-regulated whereas β-catenin, Lef-1 and cyclin D1 were
down-regulated (Fig. 3C).
Moreover, a decrease in α-SMA mRNA expression was noted (Fig. 3D). As can been seen, the protein
profile was very similar to that of the liver extracts derived from
the Namodenoson-treated CCL4 animals, supporting the notion that
Namodenoson deregulates the Wnt/β-catenin pathway.
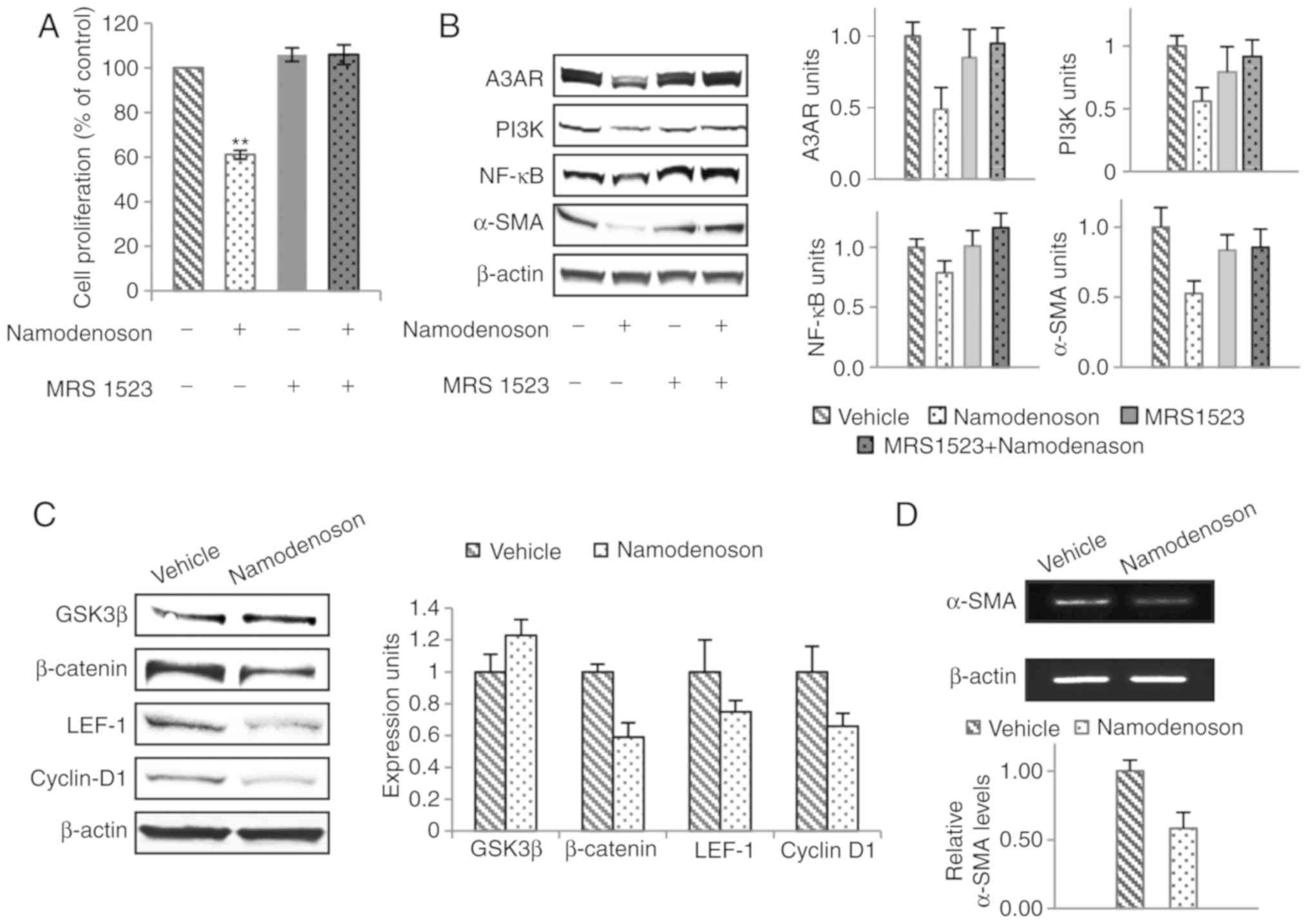 | Figure 3Namodenoson induces the proliferation
inhibition and modulation of cell growth regulatory proteins in
LX-2 HSCs. Cells were incubated for 48 h with the vehicle,
Namodenoson (10 nM), the A3AR antagonist, MRS 1523 (5 nM), and a
combination of the agonist and the antagonist. (A) Cell
proliferation. The data represent the means ± SEM. Statistical
analysis was carried out by one-way ANOVA followed by Tukey's
post-hoc analysis. **P<0.01, vehicle vs. Namodenoson;
4 independent experiments. (B) Western blot analyses of A3AR, PI3K,
NF-κB and α-SMA. (C) Western blot analyses of β-catenin, GSK-3β,
LEF-1 and cyclin D1; β-actin was used as a loading control. (D)
α-SMA mRNA expression examined by RT-qPCR. A3AR, A3 adenosine
receptor; PI3K, phosphoinositide 3-kinase; GSK-3β, glycogen
synthase kinase 3β; LEF-1, lymphoid enhancer-binding factor 1;
α-SMA, α-smooth muscle actin. |
Discussion
In this study, we demonstrate that the A3AR agonist,
Namodenoson, exerts significant anti-inflammatory, anti-steatotic
and anti-fibrotic effects on the STAM and the CCl4 animal models,
as well as on human LX2 HSCs. Namodenoson is an agonist at the Gi
protein-associated A3AR, and is over-expressed in inflammatory and
cancer cells; however, its expression is very low even absent in
normal body cells (9,13). It has been previously demonstrated
by us and others that A3AR activation with a specific agonist
induces the apoptosis of both inflammatory and cancer cells,
whereas normal cells are refractory to the effect of the agonist.
The effect towards the pathological cells was mediated via the
deregulation of both the NF-κB and the Wnt/β-catenin signaling
pathways and led to the inhibition of inflammatory cytokine
production, resulting in the apoptosis of inflammatory and cancer
cells (10,13,21).
These unique characteristics of the A3AR agonists
served as the rational to try and utilize Namodenoson as an
anti-NAFLD/NASH agent. Indeed, in the STAM model, a decrease in
steatosis, lobular inflammation and ballooning was recorded,
altogether yielding a significant reduction in NAS, pointing
towards the anti-NAFLD/NASH effects of Namodenoson. In the CCl4
model, Namodenoson also induced a robust anti-inflammatory effect
manifested by its capability to reverse the H&E inflammation
morphometric score and ALT serum levels to normal values. Similar
data were previously observed in a mouse model of Concanavalin
A-induced hepatitis upon treatment with A3AR ligands, which
reversed the increase in liver enzymes to normal values and
inhibited inflammatory cytokine production and additional
manifestations of liver inflammation (10,21).
In the current study, a decrease in NF-κB expression
levels, both ex vivo in the CCl4 model, and in the LX-2
hepatic stellate cells in vitro, supports the
anti-inflammatory effects of Namodenoson. Moreover, morphometric
analysis of adiponectin, usually negatively associated with
parameters of the metabolic syndrome, revealed a significant
increase in adiponectin staining in the Namodenoson-treated mice,
supporting the anti-inflammatory and anti-metabolic effects of the
drug. Of note, adiponectin is known to be overexpressed in the CCl4
model (22) and in the current
study Namodenoson treatment reversed the adiponectin levels to
those of the naïve mice, demonstrating its beneficial effect.
The following molecular chain of events took place
both in the liver extracts derived from the CCl4 model and in the
LX-2 HSCs. Upstream, the downregulation of A3AR and PI3K occurred
immediately following treatment with Namodenoson, followed by a
decrease in the expression levels of NF-κB and α-SMA. The response
was highly specific to Namodenoson treatment since the introduction
of an antagonist (MRS 1523), to the LX-2 cell cultures, reversed
the decrease in the levels of A3AR, PI3K, NF-κB and α-SMA. A3AR
downregulation upon treatment with a specific agonist has been
previously described and is a result of receptor internalization
and degradation in the cytoplasm. The receptor is then
re-synthesized and re-cycle to the cell surface (23).
A plethora of mechanistic pathways occur downstream
to PI3K deregulation and play a role in mediating the effects of
Namodenoson recorded in the present study. Both the Wnt/β-catenin
and the NF-κB pathways are controlled upstream by PI3K and were
deregulated upon treatment with Namodenoson. Moreover, the
Wnt/β-catenin pathway is also known to be involved in the
pathogenesis of fibrosis and steatosis in hepatic stellate cells
(15,24,25). In the CCl4 liver extracts and the
LX2 HSCs, GSK-3β was found to be upregulated with a subsequent
decrease in the expression levels of β-catenin, Lef-1 and cyclin
D1, the latter being responsible for proliferation of HSCs in the
liver and also essential for the evolution of liver fibrosis and
steatosis (26). Additional
support for the anti-fibrotic effect of Namodenoson came from its
ability to decrease mRNA and protein expression levels of α-SMA,
known directly to be controlled by PI3K, and serve as biomarker for
fibrosis (16). A decrease in
PI3K most probably led to a subsequent decrease in α-SMA,
contributing to the anti-fibrotic effect of Namodenoson. Moreover,
adiponectin directly inhibits the activation of HSCs, thereby
playing a role in the prevention of fibrosis (27).
It thus seems that the Wnt/β-catenin pathway acts as
the 'policeman' at the crossroads of steatosis, inflammation and
fibrosis. A cartoon summarizing the molecular events taking place
in pathological liver cells upon treatment with Namodenoson is
presented in Fig. 4.
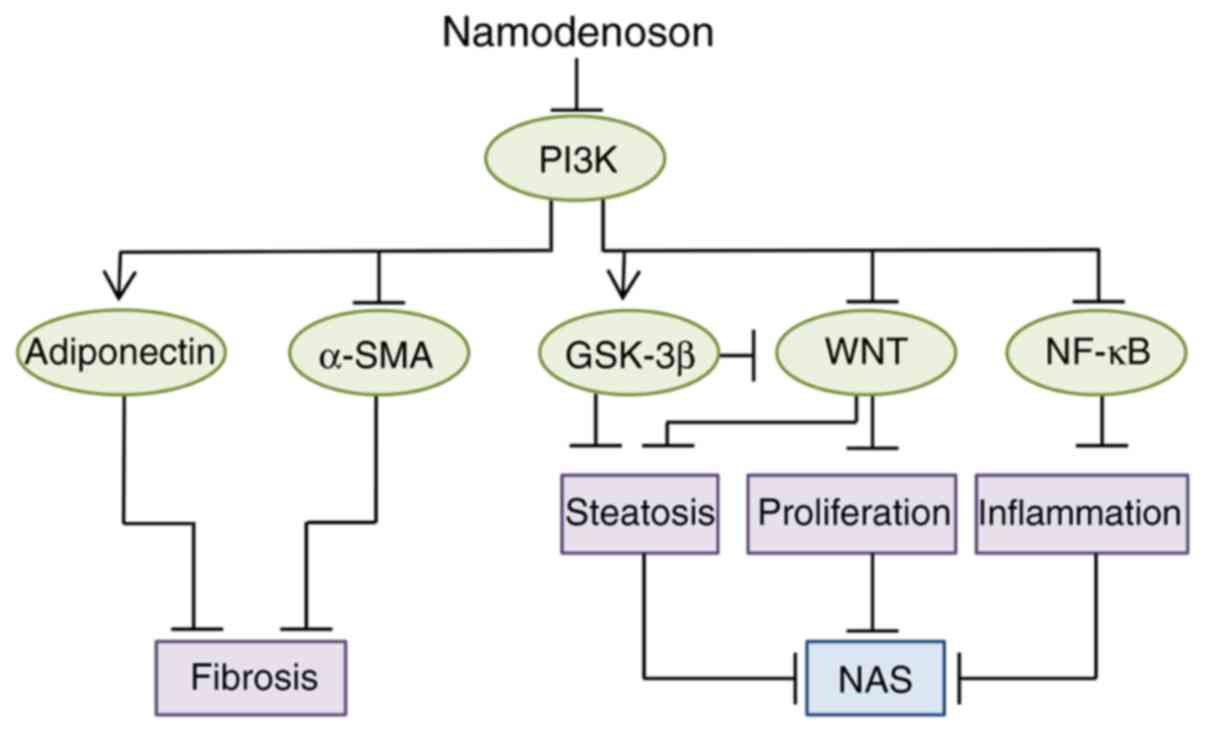 | Figure 4Proposed mechanism confers the
anti-NASH effect of Namodenoson in the liver. Namodenoson via A3AR,
downregulates the key protein PI3K, deregulates the NF-κB and the
WNT signaling pathways, as well as α-SMA, leading to the inhibition
of proliferation, inflammation, steatosis and fibrosis. The latter
is also inhibited by adiponectin which is upregulated upon the
inhibition of PI3K. The GSK3-β expression level is also increased,
affecting as well the decrease of steatosis, altogether resulting
in NAS inhibition. A3AR, A3 adenosine receptor; PI3K,
phosphoinositide 3-kinase; GSK-3β, glycogen synthase kinase 3β;
LEF-1, lymphoid enhancer-binding factor 1; α-SMA, α-smooth muscle
actin; NAS, non-alcoholic fatty liver disease (NAFLD) activity
score. |
A side benefit of Namodenoson can be the
cardio-protective and neuroprotective characteristic of the drug,
extensively published over the past decade and may compensate for
the cardiovascular and diabetic diseases which are part of the
pathogenesis of NAFLD/NASH (28,29).
Taken together, this study demonstrates that
Namodenoson exhibits a triple-mechanism of action in the liver,
exerting anti-steatosis, anti-inflammatory and anti-fibrotic
effects through the activation of the A3AR and deregulation of the
NF-κB and the Wnt/β-catenin pathways. Thus, the upstream targeting
of these pathways is likely an effective and symbiotic combination
for a promising candidate for the treatment of NASH.
Abbreviations:
|
NASH
|
non-alcoholic steatohepatitis
|
|
A3AR
|
A3 adenosine receptor
|
|
HSC
|
hepatic stellate cell line
|
|
NAFLD
|
non-alcoholic fatty liver disease
|
|
NAS
|
NAFLD activity score
|
|
ALT
|
alanine aminotransferase
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
NF-κB
|
nuclear factor κ-light-chain-enhancer
of activated B cells
|
|
LEF-1
|
lymphoid enhancer-binding factor 1
|
|
GSK-3β
|
glycogen synthase kinase 3β
|
|
α-SMA
|
α-smooth muscle actin
|
|
HCC
|
hepatocellular carcinoma
|
|
PPAR
|
peroxisome proliferator-activated
receptor
|
|
FXR
|
farnesoid X receptor
|
|
TRβ
|
thyroid hormone receptor β
|
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
We confirm that the compound described in the
manuscript is in the purity of 99.9%. the source is Albany
Molecular Research Inc USA. All data generated or analyzed during
this study are included in this published article or are available
from the corresponding author on reasonable request.
Authors' contributions
PF, RS and SC developed the study concept and
designed the study. SC, II, FB, JA and AS performed the experiments
and procedures. PF, SC and II wrote the manuscript. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures involving laboratory animals were
evaluated and approved by The Hebrew University Institutional
Animal Care and Use Committee and followed the guidelines for
laboratory animal welfare.
Patient consent for publication
Not applicable.
Competing interests
The authors with potential competing interests are
detailed as follows: PF is an employment and stock options holder;
SC is a consultant and stock options holder; FB is an employment
and stock options holder; II is an employment and RS a stock
options holder. The authors with no competing interests are
detailed as follows: JA and AS.
References
|
1
|
Ratziu V, Bellentanib S, Cortez-Pintoc H,
Dayd C and Marchesinie G: A position statement on NAFLD/NASH based
on the EASL 2009, special conference. J Hepatol. 53:372–384. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Williams CD, Stengel J, Asike MI, Torres
DM, Shaw J, Contreras M, Landt CL and Harrison SA: Prevalence of
nonalcoholic fatty liver disease and nonalcoholic steatohepatitis
among a largely middle-aged population utilizing ultrasound and
liver biopsy: A prospective study. Gastroenterology. 140:124–131.
2011. View Article : Google Scholar
|
|
3
|
Chalasani N, Younossi Z, Lavine JE, Diehl
AM, Brunt EM, Cusi K, Charlton M and Sanyal AJ: The diagnosis and
management of non-alcoholic fatty liver disease: Practice guideline
by the American association for the study of liver diseases,
American college of gastroenterology, and the American
gastro-enterological association. Hepatology. 55:2005–2023. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dhamija E, Paul SB and Kedia S:
Non-alcoholic fatty liver disease associated with hepatocellular
carcinoma: An increasing concern. Indian J Med Res. 149:9–17. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ratziu V, Goodman Z and Sanyal A: Current
efforts and trends in the treatment of NASH. J Hepatol. 62(Suppl
1): S65–S75. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Takahashi Y, Sugimoto K, Inui H and
Fukusato T: Current pharmacological therapies for nonalcoholic
fatty liver disease/nonalcoholic steatohepatitis. World J
Gastroenterol. 21:3777–3785. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guillaume M and Ratziu V: Pharmacological
agents for nonalcoholic steatohepatitis. Hepatol Int. 7(Suppl 2):
833–841. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cave MC, Clair HB, Hardesty JE, Falkner
KC, Feng W, Clark BJ, Sidey J, Shi H, Aqel BA, McClain CJ and
Prough RA: Nuclear receptors and nonalcoholic fatty liver disease.
Biochim Biophys Acta. 1859:1083–1099. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bar-Yehuda S, Stemmer SM, Madi L, Castel
D, Ochaion A, Cohen S, Barer F, Zabutti A, Perez-Liz G, Del Valle L
and Fishman P: The A3 adenosine receptor agonist CF102 induces
apoptosis of hepatocellular carcinoma via de-regulation of the Wnt
and NF-kappaB signal transduction pathways. Int J Oncol.
33:287–295. 2008.PubMed/NCBI
|
|
10
|
Cohen S, Stemmer SM, Zozulya G, Ochaion A,
Patoka R, Barer F, Bar-Yehuda S, Rath-Wolfson L, Jacobson KA and
Fishman P: CF102 an A3 adenosine receptor agonist mediates
anti-tumor and anti-inflammatory effects in the liver. J Cell
Physiol. 226:2438–2447. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ohana G, Cohen S, Rath-Wolfson L and
Fishman P: A3 adenosine receptor agonist, CF102, protects against
hepatic ischemia/reper-fusion injury following partial hepatectomy.
Mol Med Rep. 14:4335–4341. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stemmer SM, Benjaminov O, Medalia G,
Ciuraru NB, Silverman MH, Bar-Yehuda S, Fishman S, Harpaz Z,
Farbstein M, Cohen S, et al: CF102 for the treatment of
hepatocellular carcinoma: A phase I/II, open-label, dose-escalation
study. The Oncologist. 18:25–26. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ochaion A, Bar-Yehuda S, Cohen S, Amital
H, Jacobson KA, Joshi BV, Gao ZG, Barer F, Patoka R, Del Valle L,
et al: The A3 adenosine receptor agonist CF502 inhibits the PI3K,
PKB/Akt and NF-kappaB signaling pathway in synoviocytes from
rheumatoid arthritis patients and in adjuvant-induced arthritis
rats. Biochem Pharmacol. 76:482–494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fishman P, Bar-Yehuda S, Madi L,
Rath-Wolfson L, Ochaion A, Cohen S and Baharav E: The PI3K-NF-κB
signal transduction pathway is involved in mediating the
anti-inflammatory effect of IB-MECA in adjuvant induced arthritis.
Arthritis Res Ther. 8:R332006. View
Article : Google Scholar
|
|
15
|
Miao CG, Yang YY, He X, Huang C, Huang Y,
Zhang L, Lv XW, Jin Y and Li J: Wnt signaling in liver fibrosis:
Progress challenges and potential directions. Biochimie.
95:2326–2335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang C, Liu XQ, Sun HN, Meng XM, Bao YW,
Zhang HP, Pan FM and Zhang C: Octreotide attenuates hepatic
fibrosis and hepatic stellate cells proliferation and activation by
inhibiting Wnt/β-catenin signaling pathway, c-Myc and cyclin D1.
Int Immunopharmacol. 63:183–190. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matsuda S, Kobayashi M and Kitagishi Y:
Roles for PI3K/akt/PTEN pathway in cell signaling of nonalcoholic
fatty liver disease. ISRN Endocrinol. 2013:4724322013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Abu-Tair L, Axelrod JH, Doron S, Ovadya Y,
Krizhanovsky V, Galun E, Amer J and Safadi R: Natural killer
cell-dependent anti-fibrotic pathway in liver injury via toll-like
receptor-9. PLoS One. 8:e825712013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kleiner DE, Brunt EM, Van Natta M, Behling
C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS,
Unalp-Arida A, et al: Design and validation of a histological
scoring system for nonalcoholic fatty liver disease. Hepatology.
41:1313–1321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Gomez G and Sitkovsky MV: Differential
requirement for A2a and A3 adenosine receptors for the protective
effect of inosine in vivo. Blood. 102:4472–4478. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yoda-Murakami M, Taniguchi M, Takahashi K,
Kawamata S, Saito K, Choi-Miura NH and Tomita M: Change in
expression of GBP28/adiponectin in carbon
tetrachloride-administrated mouse liver. Biochem Biophys Res
Commun. 285:372–377. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Madi L, Bar-Yehuda S, Barer F, Ardon E,
Ochaion A and Fishman P: A3 adenosine receptor activation in
melanoma cells: Association between receptor fate and tumor growth
inhibition. J Biol Chem. 278:42121–42130. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ge WS, Wang YJ, Wu JX, Fan JG, Chen YW and
Zhu L: β-catenin is overexpressed in hepatic fibrosis and blockage
of Wnt/β-catenin signaling inhibits hepatic stellate cell
activation. Mol Med Rep. 9:2145–2151. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang JN, Li L, Li LY, Yan Q, Li J and Xu
T: Emerging role and therapeutic implication of Wnt signaling
pathway in liver fibrosis. Gene. 674:57–69. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Núñez KG, Gonzalez-Rosario J, Thevenot PT
and Cohen AJ: Cyclin D1 in the liver: Role of noncanonical
signaling in liver steatosis and hormone regulation. Ochsner J.
17:56–65. 2017.PubMed/NCBI
|
|
27
|
Udomsinprasert W, Honsawek S and
Poovorawan Y: Adiponectin as a novel biomarker for liver fibrosis.
World J Hepatol. 10:708–718. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen GJ, Harvey BK, Shen H, Chou J, Victor
A and Wang Y: Activation of adenosine A3 receptors reduces ischemic
brain injury in rodents. J Neurosci Res. 84:1848–1855. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cross HR, Murphy E, Black RG, Auchampach J
and Steenbergen C: Overexpression of A(3) adenosine receptors
decreases heart rate, preserves energetics, and protects ischemic
hearts. Am J Physiol Heart Circ Physiol. 283:H1562–H1568. 2002.
View Article : Google Scholar : PubMed/NCBI
|