Introduction
It is widely accepted that oxidative stress may
represent one characteristic feature in the pathological progress
of cardiac damage by affecting DNA, lipids and proteins, in
addition to activating a redox-regulated signalling cascade that
ultimately leads to cell death (1). Since mitochondria are the main
target of reactive oxygen species (ROS) production and damage
(2), the modulation of
mitochondrial dysfunction could assume clinical relevance in the
prevention of heart disease.
Nitric oxide (NO) has been reported to play either a
beneficial or harmful role in the control of cardiovascular system,
depending on the dose and duration of exposure (3). On the one hand, the blocking of NO
production via endothelial NO synthase (eNOS) would abolish the
cardioprotection against ischemia/reperfusion injury (4). On the other hand, inducible NOS
(iNOS)-dependent NO production could be detrimental for cardiac
function (5,6) through augmented formation of
peroxinitrites.
Thus, the modulation of mitochondria activity and NO
release could represent a therapeutic tool for the management of
cardiac disease. Genistein, the main soy-derived isoflavone with a
multitude of health benefits (7),
has been reported to exert protective effects on the cardiovascular
system through its antioxidant and anti-inflammatory function
(8), which would be related to
both mitochondria and NO. Hence, in human aortic and porcine
coronary endothelial cells, genistein enhanced eNOS expression and
augmented NO synthesis (9-11).
In addition, the opposite effects of genistein on iNOS/eNOS-related
NO release were found to be associated with its beneficial role
against isoproterenol-induced cardiac hypertrophy (12). It is also notable that in
different cell types, genistein was able to counteract the damage
caused by peroxidation by preserving mitochondrial function
(13,14). Mechanisms related to extracellular
signal-regulated kinases 1/2 (ERK1/2), mitogen-activated protein
kinase (MAPK) and Akt intracellular pathways have been reported to
be involved in the above effects (15).
Although there is extensive literature about the
cardiovascular effects elicited by genistein, data concerning its
function on cardiac cells remain scarce. Thus, in this study we
examined the effects of genistein on cardiomyoblast viability,
proliferation/migration and mitochondrial function in both
physiological and peroxidative conditions and analyzed the
involvement of eNOS/iNOS-dependent NO release and ERK1/2, p38MAPK
and Akt pathways. Since genistein has gained clinical consideration
for the management of postmenopausal symptoms, due to its molecular
structure that resembles that of estrogens, its effects were
compared with those elicited by 17β-estradiol.
Materials and methods
Culture of H9C2
Rat cardiomyoblasts (H9C2) were obtained from the
American Type Culture Collection (ATCC; cat. no. CRL-1446) and
cultured in Dulbecco's modified Eagle's medium (DMEM;
Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS;
Euroclone), 2 mM L-glutamine (Sigma-Aldrich) and 1%
penicillin-streptomycin (Sigma-Aldrich) at 37°C with 5%
CO2 in an incubator. For NO measurement, 7,500
cells/well were plated in 96-well plates with DMEM 0% FBS
supplemented with 1% penicillin-streptomycin-glutamine and without
phenol red (starvation medium, Sigma-Aldrich) for 4-6 h.
Mitochondrial membrane potential and cell viability were also
measured, following the same procedure used for NO measurement, but
with plating of 1×104 cells/well. For ROS
quantification, 2.5×104 cells/well were plated in
96-wells. For Trypan Blue test and western blot analysis,
4×105 cells/well were plated in 6-well plates in
complete medium, and at 80% confluency at least, they were
incubated with starvation medium overnight. Each experimental
protocol was repeated in five different cell samples.
NO release
The NO production was measured in H9C2 culture
supernatants using the Griess method (Promega; cat. no. G2930) as
previously performed in the same or similar cellular models
(16-20). H9C2 cardiomyoblasts were treated
for 30 min with genistein (10 pM, 100 pM, 10 nM, 100 nM and 1
µM; Sigma-Aldrich; cat. no. G6649) and 17β-estradiol (10 pM,
100 pM, 10 nM, 100 nM and 1 µM; Sigma-Aldrich; cat. no.
E8875). In addition, some samples were stimulated with the NOS
blocker, Nω-nitro-L-arginine methylester (L-NAME; 10 mM;
Sigma-Aldrich), the p38 MAPK inhibitor, SB203580 (100 nM;
Sigma-Aldrich), the phosphatidylinositol 3'-kinase (PI3K)
inhibitor, wortmannin (100 nM; Sigma-Aldrich), the MAPK/ERK
inhibitor, UO126 (100 nM; Sigma-Aldrich), the associated estrogen
receptors (ERs) inhibitor, fulvestrant (100 nM; Sigma-Aldrich), and
the G protein-coupled estrogenic receptors (GPER) inhibitor, G15
(100 nM; Sigma-Aldrich), which were administered for 30 min prior
to genistein (100 nM) and 17β-estradiol (100 nM). Those inhibitors
were also administered alone at the above reported concentrations,
to H9C2 for 30 min. In addition, in some experiments the effects of
genistein and 17β-estradiol were examined in peroxidative
conditions obtained by using hydrogen peroxide. In particular, 200
µM hydrogen peroxide was administered for 30 min after the
30 min-pretreatment of H9C2 with genistein (100 nM) and
17β-estradiol (100 nM) and the effects on NO release were examined.
At the end of the stimulations, NO production in the sample's
supernatants was examined by adding an equal volume of Griess
reagent following the manufacturer's instructions. At the end of
incubation, absorbance at 570 nm was measured by a spectrometer
(VICTOR™ X Multilabel Plate Reader; PerkinElmer) and the role of NO
production in this step was examined. The value of each sample was
quantified in respect to nitrite standard curve and expressed as
nitrite production (µM).
Cell viability
Oxidative stress was generated in H9C2 through 200
µM hydrogen peroxide for 30 min in starvation medium. Cell
viability was examined using the 1%
3-[4,5-dimeth-ylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide
(MTT; Life Technologies Italia; cat. no. CT02) dye, as previously
described (16-20). H9C2 cardiomyoblasts were treated
in physiological and peroxidative conditions with genistein (10 pM,
100 pM, 10 nM, 100 nM and 1 µM, for 30 min; Sigma-Aldrich)
and 17β-estradiol (10 pM, 100 pM, 10 nM, 100 nM and 1 µM,
for 30 min; Sigma-Aldrich), in the presence or absence of the same
inhibitors used in Griess assay. The same experimental protocol
described for NO release, with regard to estrogen/phytoestrogens
alone or in the presence/absence of inhibitors in physiologic and
peroxidative conditions, was followed. After each treatment, the
medium was removed, and fresh culture medium without red phenol and
FBS and with MTT dye was added in 96-well plates containing the
cells and incubated for 2 h at 37°C. Thereafter, the fresh culture
medium without red phenol and FBS was removed, and an MTT
solubilization solution (dimethyl sulfoxide; DMSO; Sigma-Aldrich)
in equal volume to the original culture medium was added and mixed
in a gyratory shaker until the complete dissolution of formazan
crystals. Cell viability was determined by measuring the absorbance
through a spectrometer (VICTOR™ X Multilabel Plate Reader;
PerkinElmer) with a wavelength of 570 nm and cell viability was
calculated by setting control cells as 100%.
Mitochondrial membrane potential
measurement
Mitochondrial membrane potential measurement in H9C2
was performed with JC-1 assay. Cells were stimulated as described
for cell viability. After stimulations, the medium of cells plated
in starvation medium was removed and incubated with
5,51,6,61-tetrachloro-1,11,3,31 tetraethylbenzimidazolyl
carbocyanine iodide (JC-1) 1X diluted in 1X Assay Buffer for 15 min
at 37°C in an incubator, following the manufacturer's instructions
(Cayman Chemical; cat. no. 10009172) and as previously performed
(13,16,18). After incubation, the cells were
washed twice with 1X Assay Buffer and then the mitochondrial
membrane potential was determined by measuring the red (excitation
550 nm/emission 600 nm) and green (excitation 485 nm/emission 535
nm) fluorescence through a spectrometer (VICTOR™ X Multilabel Plate
Reader; PerkinElmer). To identify cells undergoing apoptosis, the
ratio of fluorescent intensity of J-aggregates to fluorescent
intensity of monomers was used as an indicator of cell health.
Wound-healing migration assay
Cell migration was evaluated by the in vitro
scratch assay in serum-starved cells. Briefly, cell monolayers were
mechanically scratched with a sterile yellow tip (diameter=2 mm)
along the center of the plate and cell debris was removed by gentle
washing with PBS. Some samples were treated with genistein (10 pM,
100 nM) and 17β-estradiol (10 pM, 100 nM) for 24 h and 48 h. Images
of cell monolayers were taken using an optical microscope (Leica
ICC50HD) with a digital camera to evaluate wound closure. Migration
was quantified by calculating the area of wound at time points T0
(time of wound), T24 (24 h after wound) and T48 (48 h after wound)
by using ImageJ software (National Institutes of Health). For each
condition, the percentage of wound closure at several time points
throughout the course of the assay, was obtained through the
formula: % wound closure: [WA0-WA/WA0]*100,
where WA is the wound area and WA0 is the original size
of the wound area. Experiments were conducted in triplicate and
repeated at least five times.
Trypan blue proliferation assay
Cell proliferation was evaluated with Trypan blue
exclusion method. Cells were stimulated as described for cell
viability and mitochondrial membrane potential but with 24 h of
stimulation and without blockers. At the end of the stimulation,
the cells were detached and 50 µl cell suspension was
diluted 1:2 with Trypan blue and mixed by pipetting up and down and
then, 10 µl was put in the Burker chamber for cell counting.
Blue cells were the non-viable cells.
The percentage of viable cells was calculated by
dividing the number of viable cells by the number of total cells
and multiplying by 100%.
Glutathione (GSH) quantification
GSH measurement was performed with a specific kit
(Cayman Chemical; cat. no. 703002) as previously described
(13,16,20). H9C2 cardiomyoblasts were treated
in peroxidative conditions with genistein (10 pM, 100 nM) and
17β-estradiol (10 pM, 100 nM) for 30 min, in the presence of 200
µM hydrogen peroxide. The antioxidant 200 µM N
acetyl-cysteine (NAC; Sigma-Aldrich), administered for 30 min, was
used as the positive control. After treatments, the cells were
lysed using the 50 mM 2-(N-morpholino) ethanesulphonic acid (GSH
MES Buffer) and a rubber policeman. The cell pellet was centrifuged
at 10,000 × g for 15 min at 4°C. After centrifugation, the
supernatant was treated with an equal volume of metaphosphoric acid
(final concentration 5%; Sigma-Aldrich) for 5 min and centrifuged
at 2,000 × g for at ≥2 min. The supernatant was collected and
supplemented with 50 µl per ml of 4 M solution of
triethanolamine (Sigma-Aldrich). Then, 50 µl of the samples
was transferred to a 96-well plate where GSH was detected following
the manufacturer's instructions through a spectrometer (VICTOR™ X
Multilabel Plate Reader; PerkinElmer) at excitation/emission
wavelengths of 405-414 nM. GSH was expressed as nanomoles in
samples with 1.5 mg of protein/ml.
ROS quantification
The oxidation of 2,7-dichlo-rodihydrof luorescein
diacetate (H2DCFDA) into 2,7-dichlorodihydrofluorescein (DCF) was
used to assess ROS generation, following the manufacturer's
instructions (Abcam; cat. no. ab113851). Briefly, the cells in
96-well plates were stimulated with 30 min 200 µM hydrogen
peroxide alone or in the presence of genistein (10 pM, 100 pM, 10
nM, 100 nM and 1 µM; for 30 min) and 17β-estradiol (10 pM,
100 pM, 10 nM, 100 nM and 1 µM; for 30 min). The antioxidant
200 µM NAC was used as the positive control. After
treatments, the reactions were stopped by removing the medium and
washing with phosphate-buffered saline (PBS) followed by staining
with 10 µM H2DCFDA for 20 min at 37°C. The fluorescence
intensity of DCF was measured at an excitation and emission
wavelength of 485 and 530 nm by using a spectrophotometer (VICTOR™
X Multilabel Plate Reader; PerkinElmer) (13,16,18).
Oxygen consumption rate (OCR) assay
Oxygen consumption rate assay kit (MitoXpress-Xtra
HS Method) (Cayman Chemical; cat. no. 600800) was used to assess
the OCR, which is considered a parameter to study mitochondrial
function. Briefly, cells in the 96-well plates were stimulated with
30 min 200 µM hydrogen peroxide alone or in the presence of
genistein (10 pM and 100 nM; for 30 min) and 17β-estradiol (10 pM
and 100 nM; for 30 min). At the end of stimulation, 10 µM of
the probe (MitoXpress-Xtra Solution) was added in each well. The
plate was read at 380 nm with a spectrophotometer (VICTOR™ X
Multilabel Plate Reader; PerkinElmer). The results correspond to
the lifetime of the probe expressed in µs, calculated using
the formula: Lifetime (µs): (70-30)/ln (W1/W2) in which W1
and W2 represent window 1 and 2, respectively, for the measured
intensity readings at each time point; 70 and 30 represent the
delay time of W1 and W2.
Cell cycle analysis, by using propidium
iodide stain
Flow cytometry was used for cell cycle analysis.
This technique is widely used for measuring all phases of cell
cycle including apoptosis (21).
H9C2 cells (400,000 cells/well in 6-well plate), were stimulated
with genistein (10 pM and 100 nM; for 24 h) and 17β-estradiol (10
pM and 100 nM; for 24 h) alone, or in the presence of 200 µM
hydrogen peroxide. The effects of 200 µM hydrogen peroxide
for 30 min alone were also examined. At the end of each
stimulation, the culture medium was collected from each well and
transferred in a 15 ml tube in order to collect the cells that
eventually detached. Cells were detached with trypsin-EDTA
thereafter, an appropriate volume of culture medium was added, and
cell suspension was transferred to a tube and centrifuged at 900 ×
g for 5 min at room temperature. The supernatant was discarded, and
cells were fixed in 1 ml 70% ethanol for 1 h at -20°C. After 1 h,
the cells were centrifuged at 900 × g for 5 min at room
temperature, and ethanol as well as the supernatant were discarded.
Cells were washed with PBS and centrifuged again 900 × g for 5 min
at room temperature. Each pellet of cells was resuspended in 200
µl propidium iodide buffer (3.4 mM trisodium citrate, 9.65
mM sodium chloride, 0.003% tergitol), 25 µl RNase A (10
ng/ml; Cabru), and 10 µl propidium iodide (1 mg/ml;
Cabru).
Then, 50 µl of each sample was transferred in
a 96-well plate in triplicate, and after 15 min at 37°C in the
dark, the analysis was performed using Attune NxT (Life
Technologies).
Cell lysates
The H9C2 at confluence were plated in starvation
medium overnight at 37°C with 5% CO2. Western blot
analysis was performed in H9C2 treated with genistein (10 pM and
100 nM for 30 min) and 17β-estradiol (10 pM and 100 nM for 30 min)
in the presence or absence of specific inhibitors, as described
previously, for NO release and cell viability. In some samples, the
effects of 200 µM hydrogen peroxide, administered for 30 min
after the 30 min-pretreatment with 100 nM phytoestrogens/estrogens,
were also examined. Some samples of 200 µM hydrogen peroxide
were administered 30 min after phytoestrogens/estrogens. At the end
of stimulation, H9C2 cardiomyoblasts were lysed in iced Ripa buffer
supplemented with sodium orthovanadate (2 mM; Sigma-Aldrich) and
protease inhibitors cocktail (1 mM; Thermo Fisher Scientific) and
phenylmethanesulfonyl fluoride (1 mM; Sigma-Aldrich). The extracted
proteins were quantified through bicinchoninic acid protein (BCA;
Pierce) and used for electrophoresis and immunoblotting
studies.
Western blot analysis
Cell lysates (30 µg protein each sample) were
dissolved in 5X Laemmli buffer, boiled for 5 min and resolved in
10% sodium dodecyl sulfate poly-acrylamide gel electrophoresis gels
(Bio-Rad Laboratories). After electrophoresis they were transferred
to polyvinylidene fluoride membranes (Bio-Rad Laboratories), which
were incubated overnight at 4°C with specific primary antibodies:
Anti phospho-eNOS (1:1,000; Ser1177, Cell Signaling Technologies;
cat. no. 9570), anti eNOS (1:1,000; Cell Signaling Technologies,
cat. no. 32027), anti iNOS (1:500; Santa Cruz Biotechnology, cat.
no. sc-7271), anti phospho-Akt (1:1,000; Ser473, Santa Cruz
Biotechnology, cat. no. sc-33437), anti Akt (1:1,000; Santa Cruz
Biotechnology, cat. no. sc-81434), anti phospho-ERK1/2 (1:1,000;
Thr202/Tyr204, Santa Cruz Biotechnology, cat. no. sc-16982), anti
ERK1/2 (1:1,000; Cell Signaling Technologies, cat. no. 9102), anti
phospho-p38 MAP Kinase (1:1,000; Thr180/Tyr182, Cell Signaling
Technologies, cat. no. 9211), and anti p38 MAP Kinase (1:1,000;
Cell Signaling Technologies, cat. no. 9212). The membranes were
washed and then incubated with horseradish peroxidase-coupled goat
anti-rabbit IgG (Sigma-Aldrich), peroxidase-coupled rabbit
anti-goat IgG and horseradish peroxidase-coupled goat anti-mouse
IgG (Sigma-Aldrich) for 45 min and were developed through a
non-radioactive method using Western Lightning Chemiluminescence
(PerkinElmer Life and Analytical Sciences). Phosphorylated protein
expression was calculated as a ratio towards specific total protein
expression or β-actin (1:5,000; Santa Cruz Biotechnology; cat. no.
sc-47778) detection.
Statistical analysis
All data were recorded using the Institution's
database. Statistical analysis was performed using STATVIEW version
5.0.1 for Microsoft Windows (SAS Institute Inc.) and GraphPad Prism
6 (San Diego). Data were checked for normality prior to statistical
analysis. All the results obtained were examined through one-way
ANOVA followed by Tukey's test, by comparing preselected pairs of
column means. Pearson's coefficient was calculated for linear
correlation analysis in dose-response studies. Data are presented
as means ± standard deviation (SD) of five independent experiments
for each experimental protocol. P<0.05 was considered
statistically significant.
Results
Effects of genistein and 17β-estradiol on
cell viability, proliferation/migration and mitochondrial membrane
potential in the presence/absence of various inhibitors
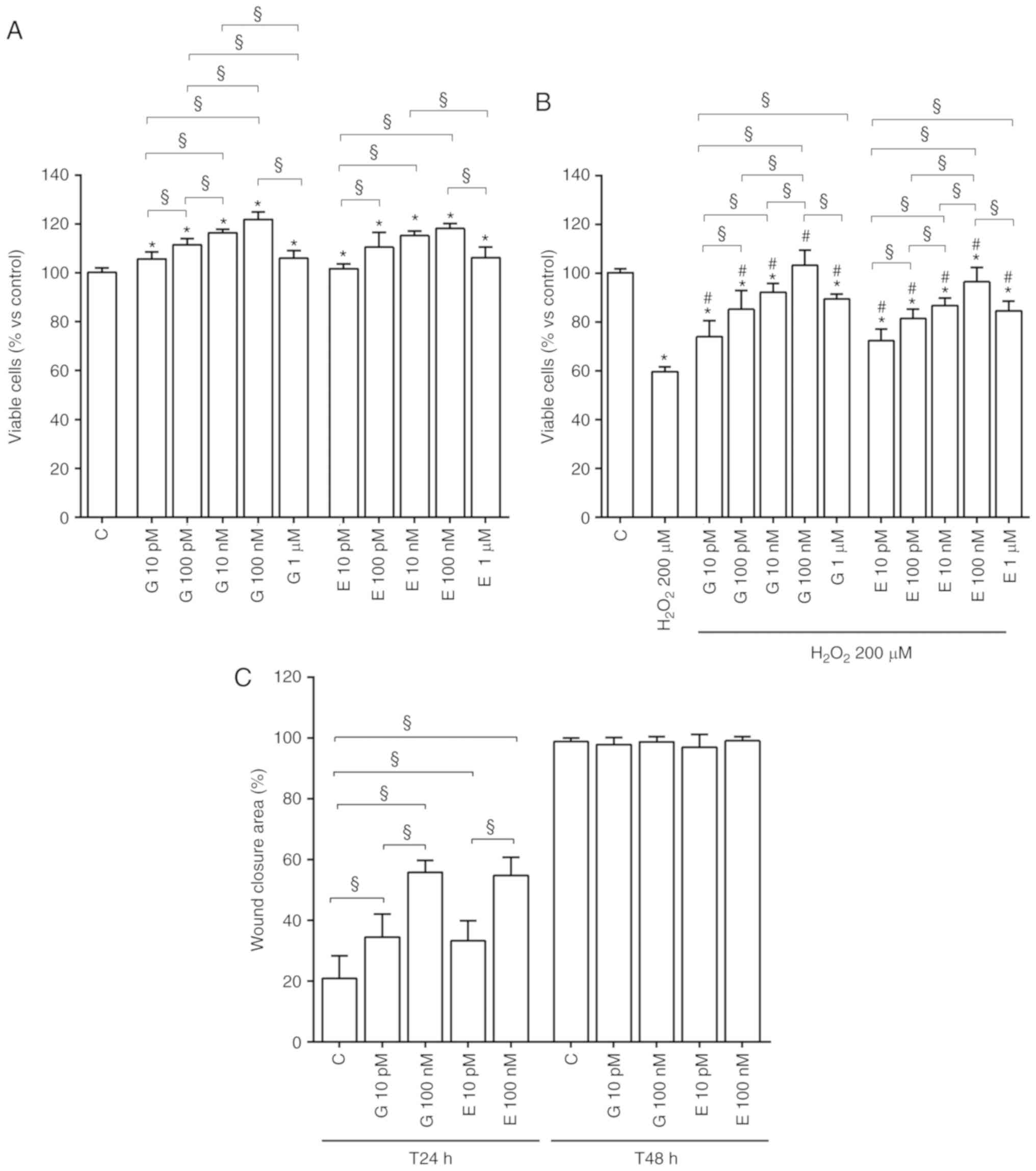
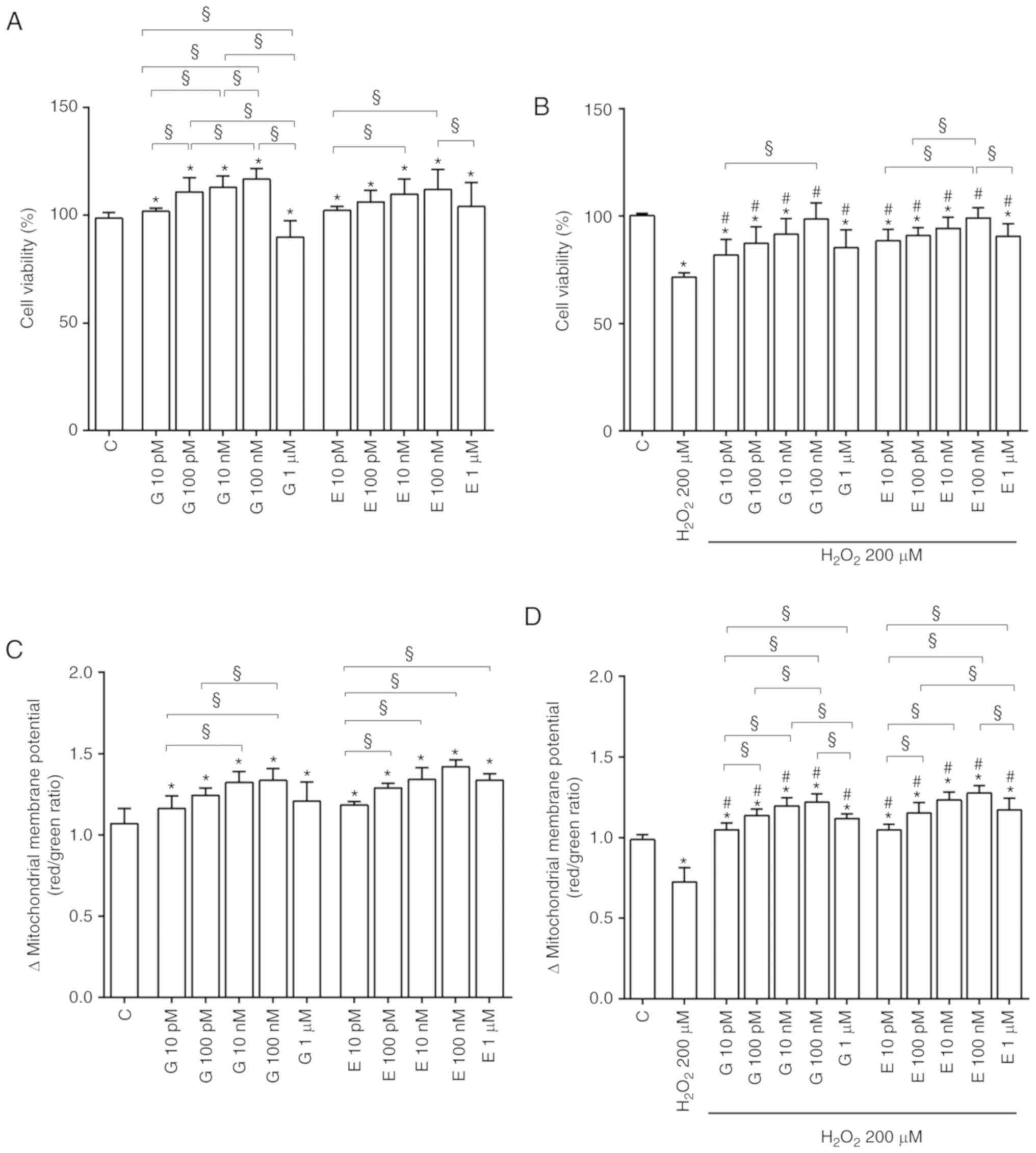
As shown in Fig.
1A, in physiological conditions genistein and 17β-estradiol
increased cell viability in a dose-dependent manner up to 100 nM.
At all doses genistein and 17β-estradiol improved the mitochondrial
membrane potential (Fig. 1C).
Furthermore, genistein and 17β-estradiol counteracted the effects
of 200 µM hydrogen peroxide on cell viability (Fig. 1B) and mitochondrial membrane
potential (Fig. 1D). Moreover,
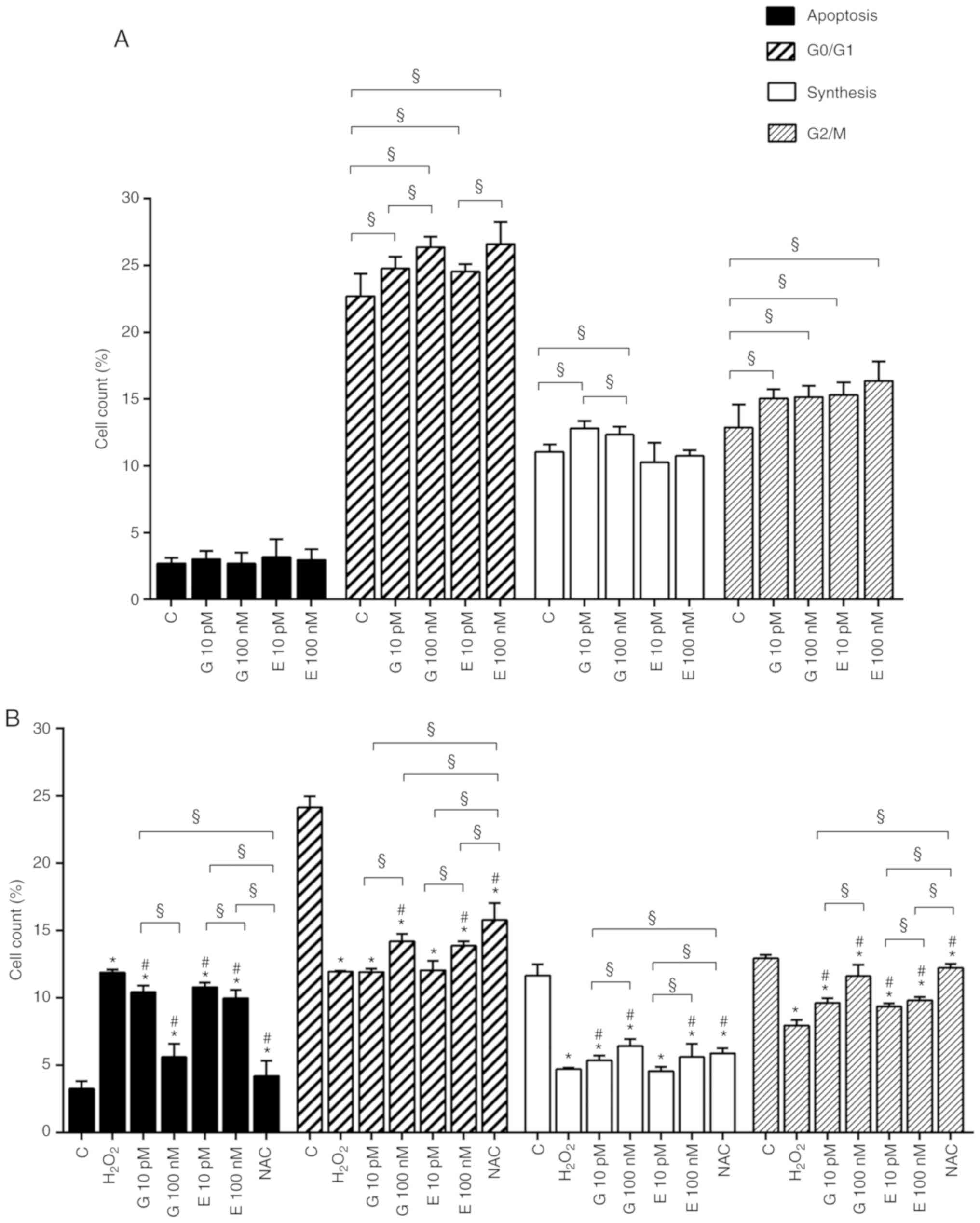
both agents were able to increase cell proliferation in
physiological (Fig. 2A) and
peroxidative (Fig. 2B) conditions
and increased cell migration (Fig.
2C and Fig. S1) up to 24 h
from the beginning of the stimulation. The above effects were
partly confirmed by the cell cycle analysis. Thus, in physiological
conditions, genistein increased the percentage of H9C2 in G0/G1, S
and G2/M phase (Figs. 3A and
S2A). As regarding estradiol,
both 10 pM and 100 nM were able to improve G0/G1 and G2/M phases
(Figs. 3A and S2A). In the peroxidative conditions,
both genistein and 17β-estradiol reduced apoptosis at all doses. In
comparison with hydrogen peroxide, 100 pM and 100 nM genistein and
10 pM and 100 nM estradiol were able to increase the percentage of
H9C2 in G0/G1. Doses of genistein and 17β-estradiol increased the
percentage of H9C2 in S and G2/M phase, as well (Figs. 3B and S2B).
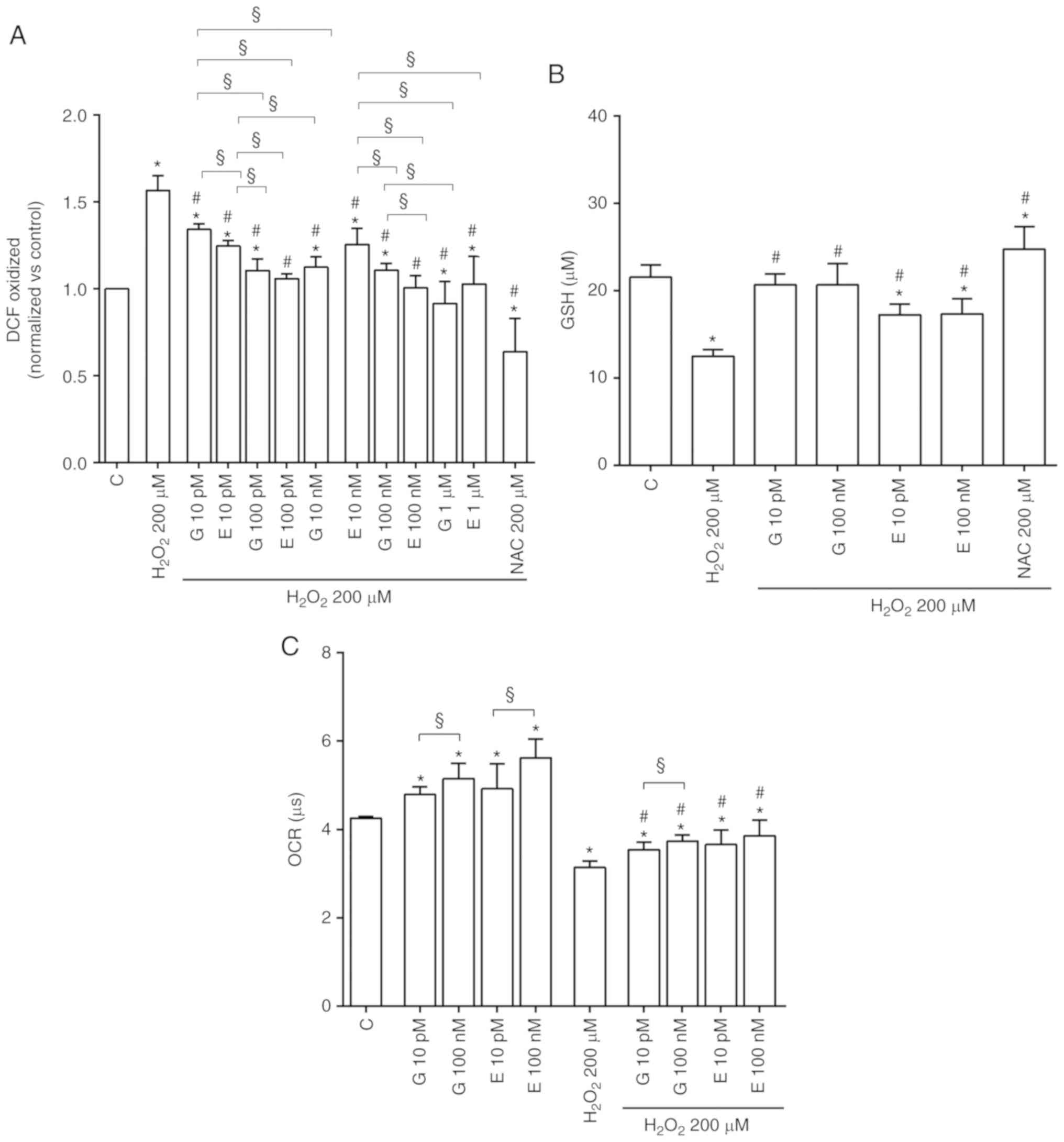
 | Figure 1Effects of genistein and
17β-estradiol on cell viability and mitochondrial membrane
potential of H9C2 cultured in physiological and peroxidative
conditions. Effects of genistein (G) 10 pM, 100 pM, 10 nM, 100 nM,
1 µM for 30 min and 17β-estradiol (E) 10 pM, 100 pM, 10 nM,
100 nM, 1 µM for 30 min, on cell viability and mitochondrial
membrane potential, are show in physiological (A) and (C) and
peroxidative (B) and (D) conditions. C=Control. Reported data are
means ± SD of five independent experiments. *P<0.05
vs. C; #P<0.05 vs. H2O2 200
µM. Square brackets indicate significance between groups
(§P<0.05). |
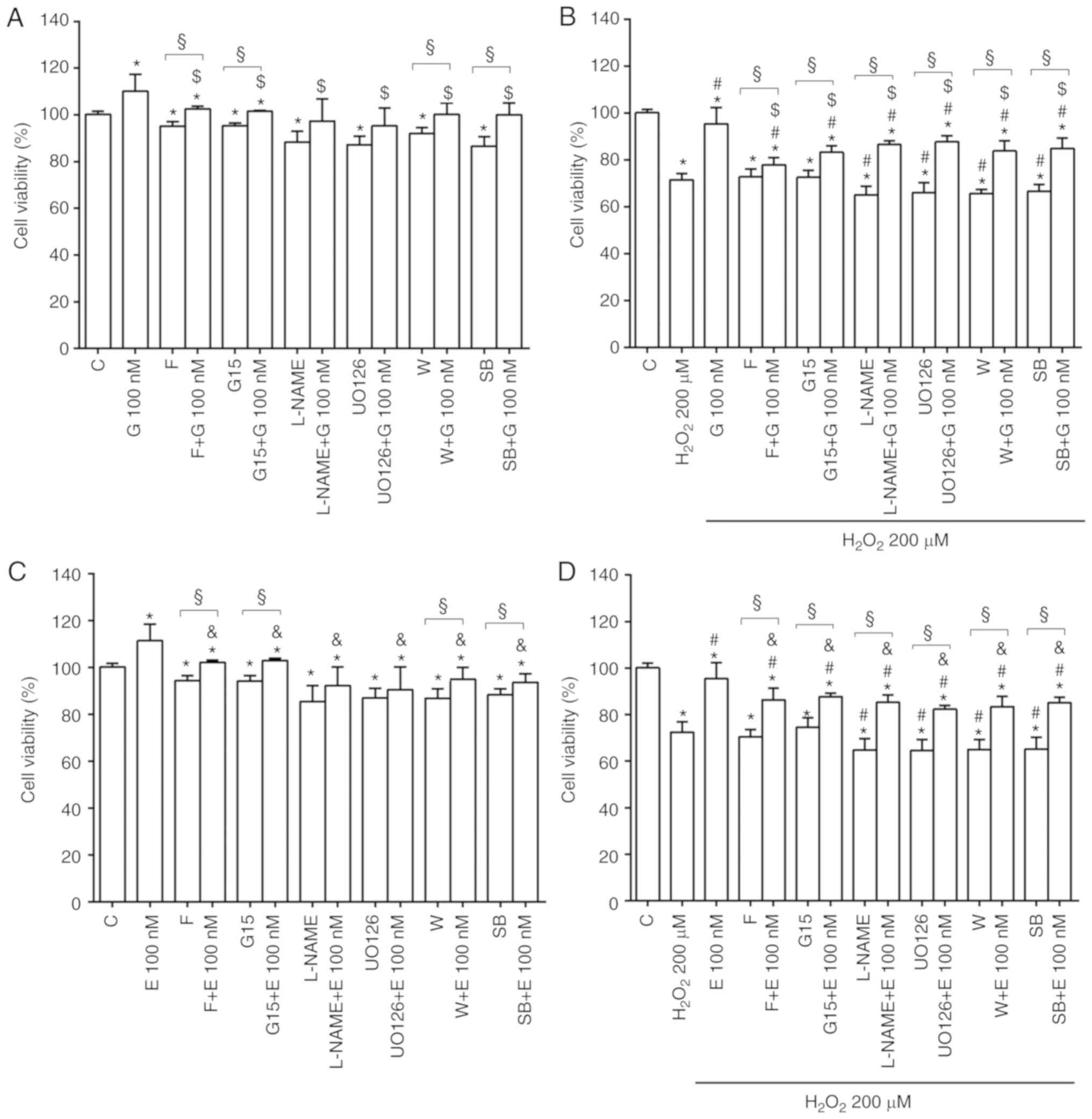
Of note is that, the effects of genistein and
17β-estradiol on cell viability (Fig.
4) and mitochondrial membrane potential (Fig. S3) were reduced or abolished by
fulvestrant, G15, L-NAME, UO126, wortmannin and SB203580. The
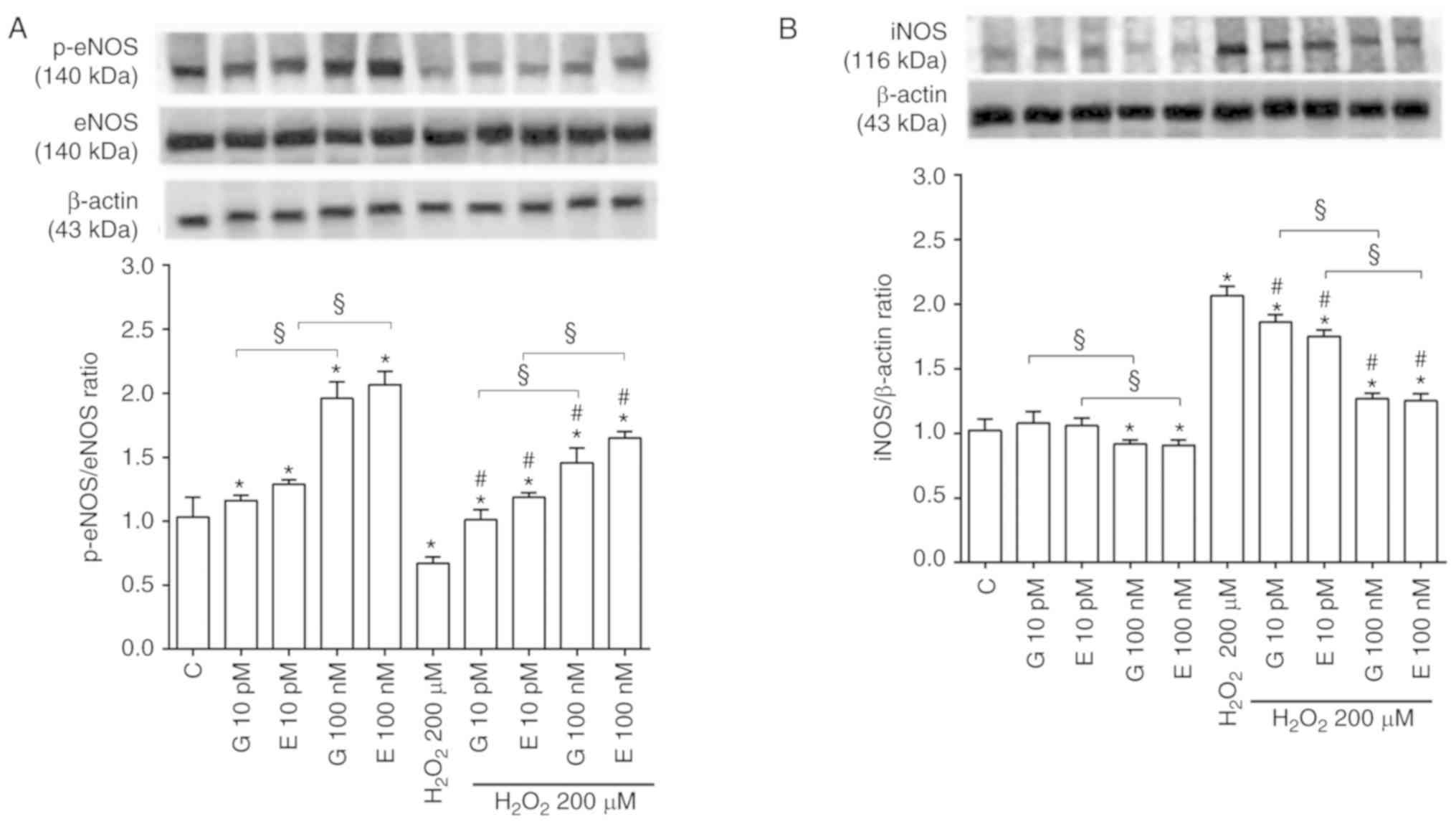
involvement of eNOS, iNOS, Akt, ERK1/2 and p38 MAPK was confirmed
by western blot analysis of their expression or activation. Thus,
in the physiological conditions, both genistein and 17β-estradiol
increased eNOS, Akt, ERK1/2 and p38 MAPK activation (Figs. 5A, S4A and S4C, S5A and S5C, S6A and S6C, S7A
and S7C), while causing either no effects or a slight decrease
of iNOS expression at 100 nM (Figs.
5B, S8A and S8C).
In H9C2 stimulated with hydrogen peroxide, eNOS,
Akt, ERK1/2 and p38 MAPK activation was reduced (Figs. 5A, S4B and S4D, S5B and S5D, S6B and S6D, S7B
and S7D) whereas iNOS expression was increased in comparison
with control (Figs. 5B, S8B and S8D). Notably, the pretreatment
with genistein and 17β-estradiol counteracted the inhibition of
eNOS (Figs. 5A, S4B and S4D) and the increased
expression of iNOS (Figs. 5B,
S8B and S8D). All inhibitors
were able to prevent the effects of genistein and 17β-estradiol
both in physiological and peroxidative conditions (Figs. S4-S8).
The protective effects elicited by genistein and
17β-estradiol against oxidative stress were confirmed by the
finding of a reduction of intracellular ROS release (Fig. 6A) and an increase of GSH
production (Fig. 6B).
Effects of genistein and 17β-estradiol on
mitochondrial oxygen consumption
As shown in Fig.
6C, both genistein and 17β-estradiol increased mitochondrial
oxygen consumption in H9C2-cultured physiological conditions at any
doses. Moreover, they were able to counteract the effects of
hydrogen peroxide.
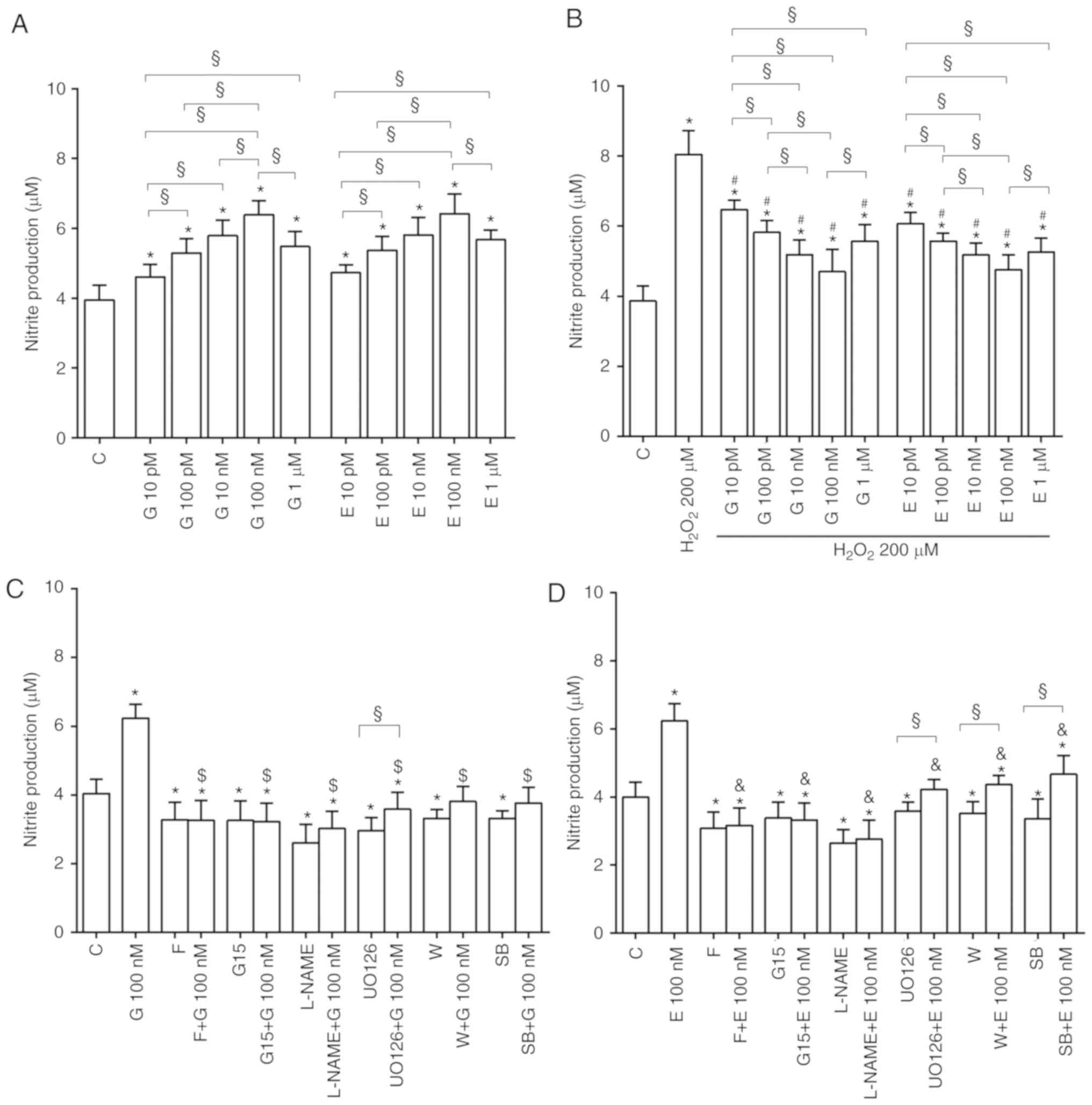
Effects of genistein and 17β-estradiol on
NO release
As shown in Fig.
7A, in physiological conditions genistein and 17β-estradiol
increased NO release with a maximum effect at 100 nM. Both agents
were able to counteract the effects of hydrogen peroxide on NO
release (Fig. 7B).
Fig. 7C and D
shows that, the presence of various inhibitors, the effects of
genistein and 17β-estradiol on NO release by H9C2 cultured in
physiological and peroxidative conditions were abolished.
Discussion
The cardiovascular benefits exerted by estrogens
have been widely described. For instance, in postmenopausal women
coronary heart disease has been linked to loss of estrogen
protection, and estrogen administration has been reported to be
associated with reduced cardiovascular risk factors (10). Furthermore, in anesthetized pig,
the administration of 17β-estradiol was shown to induce
vasodilation through NO release (10).
Nevertheless, estrogen therapy has been associated
with increased incidence of complications. For this reason,
alternative agents related to estrogens, among which
phytoestrogens, have gained wide attention (22). Thus, epidemiological studies have
revealed low rates of cardiovascular diseases (CVD) among Asian
populations whose diet is rich in phytoestrogens. This finding
would suggest a plausible causal inverse relationship between
phytoestrogens and CVD (13,23-26). However, up to-date, information
about the cellular mechanisms underlying the cardiovascular effects
of phytoestrogens is scarce.
The results obtained with our study have shown, to
the best of our knowledge, for the first time that genistein plays
an important role in the modulation of cell viability,
proliferation/migration, mitochondrial membrane potential and
oxygen consumption in H9C2, cultured either in physiological or
pathological conditions. In addition, cell cycle analysis revealed
an increase in the percentage of H9C2 cultured in physiological
conditions, in S phase in response to genistein. Moreover, in
peroxidative conditions, a reduction of apoptosis and an increase
of S and G2/M phases was observed in H9C2 treated with genistein.
Similar effects were observed in the presence of estradiol. Our
findings regarding cell viability would not confirm those obtained
by Gutiérrez-Venegas et al (27) who showed an absence of effects of
genistein in H9C2. In those experiments, however, genistein was
administered with lipopolysaccharide and for longer periods. By
contrast, our data confirm previous results obtained in H9C2
cultured in an experimental setup, the chemical-induced hypoxic
condition (28), which would be
more similar to our experimental model.
The doses of genistein were comparable to the ones
previously used and were in the range of nutritional concentrations
(29,30). Moreover, the effects of genistein
were similar to those elicited by 17β-estradiol, which was
administered in a concentration range that can commonly be found in
menstrual or menopausal women (31,32). In addition, similar doses of
estradiol affected calcium movements and modulated hypertrophic
signalling in the same cellular model (33,34).
In our study, the use of specific inhibitors,
allowed us to highlight the involvement of pathways related to Akt,
ERK1/2, and p38MAPK in the mechanisms of action of both genistein
and 17β-estradiol on cell viability and mitochondrial membrane
potential. Their involvement was also confirmed by western blot
analysis. Those findings are in agreement with previous ones
showing the role of the above kinases in the protective effects
elicited by genistein and 17β-estradiol in H9C2 (30,35).
The observation of an improvement of mitochondrial
membrane potential and oxygen consumption in response to genistein
and 17β-estradiol is of particular relevance and could be involved
in the mechanism of protection exerted by estrogens and
phytoestrogens against peroxidation. Thus, mitochondrial membrane
potential has been considered a good indicator of the energy status
of the mitochondria and, above all, of cellular homeostasis.
Interestingly, changes in mitochondrial membrane potential are
reportedly correlated with cell survival or death through apoptosis
(36-38).
Furthermore, the improvement of mitochondrial
function could be at the basis of increased cell
proliferation/migration observed in response to both genistein and
estradiol up to 100 nM. Concerning this issue, data related to the
phytoestrogens are quite new. Hence, previous findings collected in
vascular smooth muscle cells or cardiac fibroblasts showed
inhibitory effects elicited by genistein (39-41). Regarding estra-diol, our results
are in agreement with previous ones showing either an increase or
decrease of cardiomyoblast proliferation at low and high estrogenic
concentrations, respectively (42).
In H9C2 cultured in physiological conditions,
estrogens and phytoestrogens were shown, for the first time, to
acutely increase NO release. As observed for cell viability and
mitochondrial membrane potential, those effects were related to the
involvement of ERK1/2, p38MAPK and Ak-related pathways, which are
well known to play a key role in eNOS activation (43-45). By contrast, in peroxidative
conditions, the two agents reduced the excessive release of NO
caused by hydrogen peroxide. These results are particularly
relevant if we consider the different role played by NO. NO is
synthe-sized from L-arginine by three isoforms of NOS (46). These are the inducible and
calcium-independent NOS (iNOS), the constitutive and
calcium-dependent, neuronal NOS (nNOS) and the constitutive and
calcium-dependent, endothelial NOS (eNOS) (47). It is notable that, while NO
produced in low concentration, as in the case of the eNOS
activation, would act as a messenger and cytoprotective factor via
direct interactions with transition metals and other free radicals
(48), the stimulation of iNOS
and NO overproduction could increase reactive nitrous species (RNS)
formation and cause cellular death.
In the present study, eNOS phosphorylation was found
to be increased by genistein and 17β-estradiol in H9C2 cultured in
physiological conditions. By contrast, when H9C2 cardio-myoblasts
were subjected to peroxidation, eNOS activation was slightly
decreased in comparison with the strong iNOS expression; this could
explain the findings regarding NO production. Since these effects
were counteracted by estro-gens and phytoestrogens, the modulation
of the imbalance of the various isoforms of NOS and NO release
could be hypothesized to be at basis of the protective effects
elicited by genistein and 17β-estradiol against peroxidation and
increased cell death. Our findings are similar to those recently
reported by Ma et al who showed a role for NO in the
antiapoptotic effects of bioactive organosulfur compounds of
garlic, such as allicin, in H9C2 (49). In addition, the biological actions
of the polyphenol agent, licochalcone C, against oxidative stress
injuries in H9C2 have been found to be related to the maintenance
of the balance between the constitutive and inducible isoforms of
NOS (50). Finally, recent data
by Zuo et al, have shown that Panax ginseng was able to
protect cardiomyocytes against ischemic/reperfusion damage by
triggering the cascade of the so-called reperfusion injury salvage
kinase (RISK) pathways, which involves ERK1/2 and Akt, and the
subsequent eNOS-related NO release modulation (51). Interestingly, the pretreatment of
H9C2 with L-NAME, the non-selective NOS inhibitor, was able, not
only to reduce the effects of the two agents on NO release, but
also on cell viability and mitochondrial membrane potential.
It could therefore be hypothesized that a key role
is played by NO as modulator of mitochondrial function in mediating
the protective effects elicited by phytoestrogens and estrogens in
H9C2. Our speculations are supported by previous findings showing
an increased oxidative phosphorylation efficiency by NO and
beneficial effects on mitochondrial matrix pH and mitochondrial
membrane potential (52).
The protective effect of genistein and 17β-estradiol
on the cardiomyoblast response to oxidative stress, has been
confirmed by the analysis of intracellular ROS release and GSH
production (53-56). Oxidative stress, which is caused
by the accumulation of intracellular ROS or RNS, is one of the
leading factors triggering cardiomyocyte apoptosis by affecting
mitochondrial function. Moreover, ROS may have direct detrimental
effects on cellular structure and function by modulating myocardial
remodeling, which represents a risk factor for CVD. For this
reason, inhibiting ROS production or the enhancement of ROS
scavenging could be used as a therapeutic strategy for treating
CVD. Those species are generated constantly in vivo, and can
cause oxidative damage to DNA, proteins and lipids resulting in
cellular apoptotic death. Activation of the PI3K/Akt pathway in
H9C2 cells can suppress apoptosis and promote cell survival
(56). Therefore, the results we
have obtained would suggest that both genistein and 17β-estradiol
can protect H9C2 from peroxidation through the modulation of
mitochondrial function by the involvement of Akt pathway. Our data
are in agreement with those obtained by Zuo et al regarding
the role of ginseng in H9C2 (51)
and are similar to what was found in other cell types (30,25,57-61).
The involvement of ERs and GPER receptors in the
mechanisms of action of genistein and 17β-estradiol in H9C2 has
been confirmed by using fulvestrant and G15. Our findings are in
line with previous observations on the role of both estrogenic
receptors in the cardiovascular effects of estrogens and
phytoestrogens. In particular, although ERs are involved in the
cardio-protection exerted by 17β-estradiol, its protective effects
have been also described in the absence of ERs (62). Those findings lead to speculations
about the existence of alternative receptors such as GPER and
signalling pathways involved in 17β-estradiol-mediated regulation
of cardiovascular function. In particular, GPER activation leads to
the downstream enhancement of signalling molecules, such as ERK1/2
and Akt (63). Concerning
genistein, our data regarding the role of GPER in the cardiac
protective effects are quite new, since previous findings mainly
involved the reproductive system.
In conclusion, this study has shown, to the best of
our knowledge, for the first time that, genistein improves
viability/proliferation and mitochondrial function of H9C2 through
mechanisms not so different from those suggested by the effects of
estradiol. In particular, ERs and GPER receptors, the RISK pathway
and the modulation of NO release by eNOS/iNOS could play a role in
exerting their physiological effects and protection against
peroxidation. These findings could be of clinical relevance for the
management of cardiovascular disease in postmenopausal women, in
which the use of phytoestrogens may be an alternative hormonal
therapy for the amelioration of postmenopausal CVD.
Supplementary Data
Acknowledgments
We would like to thank Azienda Ospedaliera
Universitaria della Carità, Novara, for their assistance in
purchasing materials.
Abbreviations:
|
DCF
|
2,7-dichlorodihydrofluorescein
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
eNOS
|
endothelial NOS
|
|
ERK1/2
|
extracellular- signal-regulated
kinases 1/2
|
|
ERs
|
estrogen receptors
|
|
FBS
|
fetal bovine serum
|
|
GPER
|
G protein-
coupled-estrogenic-receptors
|
|
H2DCFDA
|
2,7-dichlorodihydrofluorescein
diacetate
|
|
iNOS
|
inducible NOS
|
|
L-NAME
|
Nω-nitro-L-arginine methylester
|
|
MTT
|
1%
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide
|
|
NO
|
nitric oxide
|
|
NOS
|
nitric oxide synthase
|
|
PBS
|
phosphate-buffered saline
|
|
p38MAPK
|
p38 mitogen activated protein
kinase
|
|
ROS
|
reactive oxygen species
|
|
JC-1
|
5,51,6,61-tetrachloro-1,11,3,31
tetraethylbenzimidazolyl carbocyanine iodide
|
Funding
This study was (partially) funded by the AGING
Project-Department of Excellence: DIMET, Università del Piemonte
Orientale.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
EG and SG contributed to the conception and design
of the study as well as the drafting and revising of the
manuscript. SF, GR, GC and CL contributed to performing of the
experiments, the analysis and interpretation of data, and the
drafting and revising of the manuscript. DM was involved in the
design, writing and revising of the manuscript. All authors have
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors have no competing insterests to
disclose.
References
|
1
|
Radak Z, Zhao Z, Goto S and Koltai E:
Age-associated neuro-degeneration and oxidative damage to lipids,
proteins and DNA. Mol Aspects Med. 32:305–315. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Murphy E and Steenbergen C: Mechanisms
underlying acute protection from cardiac ischemia-reperfusion
injury. Physiol Rev. 88:581–609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guan LY, Fu PY, Li PD, Li ZN, Liu HY, Xin
MG and Li W: Mechanisms of hepatic ischemia-reperfusion injury and
protective effects of nitric oxide. World J Gastrointest Surg.
6:122–128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu L, Zhou L, Wu X, Liu C, Fan Y and Li Q:
Hypoxic preconditioning protects cardiomyocytes against
hypoxia/reoxygenation injury through AMPK/eNOS/PGC-1α signaling
pathway. Int J Clin Exp Pathol. 7:7378–7388. 2014.
|
|
5
|
Gealekman O, Abassi Z, Rubinstein I,
Winaver J and Binah O: Role of myocardial inducible nitric oxide
synthase in contractile dysfunction and beta-adrenergic
hyporesponsiveness in rats with experimental volume-overload heart
failure. Circulation. 105:236–243. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang B, Larson DF and Watson RR:
Modulation of iNOS activity in age-related cardiac dysfunction.
Life Sci. 75:655–667. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ganai AA and Farooqi H: Bioactivity of
genistein: A review of in vitro and in vivo studies. Biomed
Pharmacother. 76:30–38. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Si H and Liu D: Phytochemical genistein in
the regulation of vascular function: New insights. Curr Med Chem.
14:2581–2589. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Si H, Yu J, Jiang H, Lum H and Liu D:
Phytoestrogen genistein up-regulates endothelial nitric oxide
synthase expression via activation of cAMP response element-binding
protein in human aortic endothelial cells. Endocrinology.
153:3190–3198. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grossini E, Molinari C, Mary DA, Uberti F,
Caimmi PP, Surico N and Vacca G: Intracoronary genistein acutely
increases coronary blood flow in anesthetized pigs through
beta-adrenergic mediated nitric oxide release and estrogenic
receptors. Endocrinology. 149:2678–2687. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schmitt CA and Dirsch VM: Modulation of
endothelial nitric oxide by plant-derived products. Nitric Oxide.
21:77–91. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meng Y, Zhang Y, Ma Z, Zhou H, Ni J, Liao
H and Tang Q: Genistein attenuates pathological cardiac hypertrophy
in vivo and in vitro. Herz. 44:247–256. 2019. View Article : Google Scholar
|
|
13
|
Surico D, Ercoli A, Farruggio S, Raina G,
Filippini D, Mary D, Minisini R, Surico N, Pirisi M and Grossini E:
Modulation of oxidative stress by 17 β-estradiol and genistein in
human hepatic cell lines in vitro. Cell Physiol Biochem.
42:1051–1062. 2017. View Article : Google Scholar
|
|
14
|
Liu F, Cao JG, Li C, Tan JS and Fu XH:
Protective effects of 7-difluoromethyl-5,4'-imethoxygenistein
against human aorta endothelial injury caused by lysophosphatidyl
choline. Mol Cell Biochem. 363:147–155. 2012. View Article : Google Scholar
|
|
15
|
Yang Y, Nie W, Yuan J, Zhang B, Wang Z, Wu
Z and Guo Y: Genistein activates endothelial nitric oxide synthase
in broiler pulmonary arterial endothelial cells by an Akt-dependent
mechanism. Exp Mol Med. 42:768–776. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
De Cillà S, Farruggio S, Vujosevic S,
Raina G, Filippini D, Gatti V, Clemente N, Mary D, Vezzola D,
Casini G, et al: Anti-vascular endothelial growth factors protect
retinal pigment epithelium cells against oxidation by modulating
nitric oxide release and autophagy. Cell Physiol Biochem.
42:1725–1738. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Grossini E, Farruggio S, Qoqaiche F, Raina
G, Camillo L, Sigaudo L, Mary D, Surico N and Surico D: Monomeric
adipo-nectin modulates nitric oxide release and calcium movements
in porcine aortic endothelial cells in normal/high glucose
conditions. Life Sci. 161:1–9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Surico D, Farruggio S, Marotta P, Raina G,
Mary D, Surico N, Vacca G and Grossini E: Human chorionic
gonadotropin protects vascular endothelial cells from oxidative
stress by apoptosis inhibition, cell survival signalling activation
and mitochondrial function protection. Cell Physiol Biochem.
36:2108–2120. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Grossini E, Bellofatto K, Farruggio S,
Sigaudo L, Marotta P, Raina G, De Giuli V, Mary D, Pollesello P,
Minisini R, et al: Levosimendan inhibits peroxidation in
hepatocytes by modulating apoptosis/autophagy interplay. PLoS One.
10:e01247422015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Grossini E, Gramaglia C, Farruggio S,
Bellofatto K, Anchisi C, Mary D, Vacca G and Zeppegno P: Asenapine
increases nitric oxide release and protects porcine coronary artery
endothelial cells against peroxidation. Vascul Pharmacol.
60:127–141. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Riccardi C and Nicoletti I: Analysis of
apoptosis by propidium iodide staining and flow cytometry. Nat
Protoc. 1:1458–1461. 2006. View Article : Google Scholar
|
|
22
|
Gencel VB, Benjamin MM, Bahou SN and
Khalil RA: Vascular effects of phytoestrogens and alternative
menopausal hormone therapy in cardiovascular disease. Mini Rev Med
Chem. 12:149–174. 2012. View Article : Google Scholar :
|
|
23
|
Lissin LW and Cooke JP: Phytoestrogens and
cardiovascular health. J Am Coll Cardiol. 35:1403–1410. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Behl C, Skutella T, Lezoualc'h F, Post A,
Widmann M, Newton CJ and Holsboer F: Neuroprotection against
oxidative stress by estrogens: Structure-activity relationship. Mol
Pharmacol. 51:535–541. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xi YD, Yu HL, Ma WW, Ding BJ, Ding J, Yuan
LH, Feng JF and Xiao R: Genistein inhibits mitochondrial-targeted
oxidative damage induced by beta-amyloid peptide, 25-35 in PC12
cells. J Bioenerg Biomembr. 43:399–407. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nuedling S, Kahlert S, Loebbert K,
Doevendans PA, Meyer R, Vetter H and Grohé C: 17 Beta-estradiol
stimulates expression of endothelial and inducible NO synthase in
rat myocardium in-vitro and in-vivo. Cardiovasc Res. 43:666–674.
1999. View Article : Google Scholar
|
|
27
|
Gutiérrez-Venegas G, Torras-Ceballos A,
Gómez-Mora JA and Fernández-Rojas B: Luteolin, quercetin, genistein
and quer-cetagetin inhibit the effects of lipopolysaccharide
obtained from Porphyromonas gingivalis in H9c2 cardiomyoblasts.
Cell Mol Biol Lett. 22:192017. View Article : Google Scholar
|
|
28
|
Shi YN, Zhang XQ, Hu ZY, Zhang CJ, Liao
DF, Huang HL and Qin L: Genistein protects H9c2 cardiomyocytes
against chemical hypoxia-induced injury via inhibition of
apoptosis. Pharmacology. 103:282–290. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sienkiewicz P, Surazyński A, Pałka J and
Miltyk W: Nutritional concentration of genistein protects human
dermal fibroblasts from oxidative stress-induced collagen
biosynthesis inhibition through IGF-I receptor-mediated signaling.
Acta Pol Pharm. 65:203–211. 2008.PubMed/NCBI
|
|
30
|
Hu WS, Lin YM, Ho TJ, Chen RJ, Li YH, Tsai
FJ, Tsai CH, Day CH, Chen TS and Huang CY: Genistein suppresses the
isoproterenol-treated H9c2 cardiomyoblast cell apoptosis associated
with P-38, Erk1/2, JNK, and NFκB signaling protein activation. Am J
Chin Med. 41:1125–1136. 2013. View Article : Google Scholar
|
|
31
|
Itagaki T, Shimizu I, Cheng X, Yuan Y,
Oshio A, Tamaki K, Fukuno H, Honda H, Okamura Y and Ito S: Opposing
effects of oestradiol and progesterone on intracellular pathways
and activation processes in the oxidative stress induced activation
of cultured rat hepatic stellate cells. Gut. 54:1782–1789. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
White MM, Zamudio S, Stevens T, Tyler R,
Lindenfeld J, Leslie K and Moore LG: Estrogen, progesterone, and
vascular reactivity: Potential cellular mechanisms. Endocr Rev.
16:739–751. 1995.PubMed/NCBI
|
|
33
|
Yang X, Mao X, Xu G, Xing S, Chattopadhyay
A, Jin S and Salama G: Estradiol up-regulates L-type
Ca2+ channels via membrane-bound estrogen
receptor/phosphoinositide-3-kinase/Akt/cAMP response
element-binding protein signaling pathway. Heart Rhythm.
15:741–749. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pai P, Velmurugan BK, Kuo CH, Yen CY, Ho
TJ, Lin YM, Chen YF, Lai CH, Day CH and Huang CY: 17β-Estradiol
and/or estrogen receptor alpha blocks isoproterenol-induced calcium
accumulation and hypertrophy via GSK3β/PP2A/NFAT3/ANP pathway. Mol
Cell Biochem. 434:181–195. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Urata Y, Ihara Y, Murata H, Goto S, Koji
T, Yodoi J, Inoue S and Kondo T: 17Beta-estradiol protects against
oxidative stress-induced cell death through the
glutathione/glutaredoxin-dependent redox regulation of Akt in
myocardiac H9c2 cells. J Biol Chem. 281:13092–13102. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Javadov S and Karmazyn M: Mitochondrial
permeability transition pore opening as an endpoint to initiate
cell death and as a putative target for cardioprotection. Cell
Physiol Biochem. 20:1–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ormel J and De Jonge P: Unipolar
depression and the progression of coronary artery disease: Toward
an integrative model. Psychother Psychosom. 80:264–274. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Russell JW, Golovoy D, Vincent AM,
Mahendru P, Olzmann JA, Mentzer A and Feldman EL: High
glucose-induced oxidative stress and mitochondrial dysfunction in
neurons. FASEB J. 16:1738–1748. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu JY, Lee JJ, Lim Y, Kim TJ, Jin YR,
Sheen YY and Yun YP: Genistein inhibits rat aortic smooth muscle
cell proliferation through the induction of p27kip1. J Pharmacol
Sci. 107:90–98. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tsai YC, Leu SY, Peng YJ, Lee YM, Hsu CH,
Chou SC, Yen MH and Cheng PY: Genistein suppresses leptin-induced
proliferation and migration of vascular smooth muscle cells and
neointima formation. J Mol Med. 21:422–431. 2017.
|
|
41
|
Pillai MS and Shivakumar K: Genistein
abolishes nucleoside uptake by cardiac fibroblasts. Mol Cell
Biochem. 332:121–125. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kilić A, Javadov S and Karmazyn M:
Estrogen exerts concentration-dependent pro-and anti-hypertrophic
effects on adult cultured ventricular myocytes. Role of NHE-1 in
estrogen-induced hypertrophy. J Mol Cell Cardiol. 46:360–369. 2009.
View Article : Google Scholar
|
|
43
|
Ciccarelli M, Cipolletta E, Santulli G,
Campanile A, Pumiglia K, Cervero P, Pastore L, Astone D, Trimarco B
and Iaccarino G: Endothelial beta2 adrenergic signaling to AKT:
Role of Gi and SRC. Cell Signal. 19:1949–1955. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ferro A, Queen LR, Priest RM, Xu B, Ritter
JM, Poston L and Ward JP: Activation of nitric oxide synthase by
beta 2-adre-noceptors in human umbilical vein endothelium in vitro.
Br J Pharmacol. 126:1872–1880. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Grossini E, Caimmi P, Molinari C, Uberti
F, Mary D and Vacca G: CCK receptors-related signaling involved in
nitric oxide production caused by gastrin 17 in porcine coronary
endo-thelial cells. Mol Cell Endocrinol. 350:20–30. 2012.
View Article : Google Scholar
|
|
46
|
Alderton WK, Cooper CE and Knowles RG:
Nitric oxide synthases: Structure, function and inhibition. Biochem
J. 357:593–615. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bhutto IA, Baba T, Merges C, McLeod DS and
Lutty GA: Low nitric oxide synthases (NOSs) in eyes with
age-related macular degeneration (AMD). Exp Eye Res. 90:155–167.
2010. View Article : Google Scholar
|
|
48
|
Liaudet L, Soriano FG and Szabó C: Biology
of nitric oxide signaling. Crit Care Med. 28(Suppl 4): N37–N52.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ma L, Chen S, Li L, Deng L, Li Y and Li H:
Effect of allicin against ischemia/hypoxia-induced H9C2 myoblast
apoptosis via eNOS/NO pathway-mediated antioxidant activity. Evid
Based Complement Alternat Med. 2018:32079732018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Franceschelli S, Pesce M, Ferrone A, Gatta
DM, Patruno A, Lutiis MA, Quiles JL, Grilli A, Felaco M and
Speranza L: Biological effect of Licochalcone C on the regulation
of PI3K/Akt/eNOS and Nf-κb/iNOS/NO signaling pathways in H9C2 cells
in response to LPS stimulation. Int J Mol Sci. 18:E6902017.
View Article : Google Scholar
|
|
51
|
Zuo YH, Han QB, Dong GT, Yue RQ, Ren XC,
Liu JX, Liu L, Luo P and Zhou H: Panax ginseng polysaccharide
protected H9c2 cardiomyocyte from hypoxia/reoxygenation injury
through regulating mitochondrial metabolism and RISK pathway. Front
Physiol. 9:6992018. View Article : Google Scholar :
|
|
52
|
Zaobornyj T and Ghafourifar P: Strategic
localization of heart mitochondrial NOS: A review of the evidence.
Am J Physiol Heart Circ Physiol. 303:H1283–H1293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Celojevic D, Petersen A, Karlsson JO,
Behndig A and Zetterberg M: Effects of 17β-estradiol on
proliferation, cell viability and intracellular redox status in
native human lens epithelial cells. Mol Vis. 17:1987–1996.
2011.
|
|
54
|
Chetty CS, Vemuri MC, Reddy GR and Suresh
C: Protective effect of 17-beta-estradiol in human neurocellular
models of lead exposure. Neurotoxicology. 28:396–401. 2007.
View Article : Google Scholar
|
|
55
|
Sandoval M, Cutini P, Rauschemberger M and
Massheimer V: The soyabean isoflavone genistein modulates
endothelial cell behavior. Br J Nutr. 104:171–179. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Uttara B, Singh AV, Zamboni P and Mahajan
RT: Oxidative stress and neurodegenerative diseases: A review of
upstream and downstream antioxidant therapeutic options. Curr
Neuropharmacol. 7:65–74. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yu HL, Zhang XH, Xiao R, Li L, Xiang L,
Feng JF, Yuan LH and Ma WW: Effects of genistein and folic acid on
neuronal membrane and mitochondrial membrane damaged by β-amyloid
peptides 31-35. Zhonghua Yu Fang Yi Xue Za Zhi. 44:607–611. 2010.In
Chinese. PubMed/NCBI
|
|
58
|
Borrás C, Gambini J, López-Grueso R,
Pallardó FV and Viña J: Direct antioxidant and protective effect of
estradiol on isolated mitochondria. Biochim Biophys Acta.
1802:205–211. 2010. View Article : Google Scholar
|
|
59
|
Richardson TE, Yu AE, Wen Y, Yang SH and
Simpkins JW: Estrogen prevents oxidative damage to the mitochondria
in Friedreich's ataxia skin fibroblasts. PLoS One. 7:e346002012.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Asokan Shibu M, Kuo WW, Kuo CH, Day CH,
Shen CY, Chung LC, Lai CH, Pan LF, Vijaya Padma V and Huang CY:
Potential phytoestrogen alternatives exert cardio-protective
mechanisms via estrogen receptors. Biomedicine (Taipei). 7:112017.
View Article : Google Scholar
|
|
61
|
Patten RD, Pourati I, Aronovitz MJ, Baur
J, Celestin F, Chen X, Michael A, Haq S, Nuedling S, Grohe C, et
al: 17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in
vitro via activation of phospho-inositide-3 kinase/Akt signaling.
Circ Res. 95:692–699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Feldman RD and Limbird LE: GPER (GPR30): A
nongenomic receptor (GPCR) for steroid hormones with implications
for cardiovascular disease and cancer. Annu Rev Pharmacol Toxicol.
57:567–584. 2017. View Article : Google Scholar
|
|
63
|
Prossnitz ER and Barton M: The G
protein-coupled estrogen receptor GPER in health and disease. Nat
Rev Endocrinol. 7:715–726. 2011. View Article : Google Scholar : PubMed/NCBI
|