Introduction
Mechanical ventilation is a life-saving therapy for
patients with acute lung injury or acute respiratory distress
syndrome (ARDS), which are characterized by intractable hypoxemia
and bilateral lung infiltration (1). Moderate tidal volume does not cause
lung injury itself; however, several studies have demonstrated that
it may initiate or promote pre-existing lung injuries, causing
further damage to the endothelial barrier with subsequent
inflammation and pulmonary edema (2-4).
This syndrome has been termed as ventilator-associated lung injury
(VALI). Due to the high morbidity and mortality of patients with
ARDS, VALI remains a significant medical problem in intensive care
units (5). Therefore, further
investigation is required to develop effective strategies to
mitigate VALI.
Sphingosine 1-phosphate (S1P) is a pleiotropic
bioactive mediator that is involved in a variety of key biological
processes, including cellular proliferation, apoptosis, migration
and survival (6). The formation
of S1P is catalyzed by the phosphorylation of sphingosine by
sphingosine kinase (SPHK). Recent evidence has suggested a key role
for SPHK in several lung pathologies, including lung fibrosis
(7), pulmonary artery
hypertension (8), asthma
(9) and ischemia reperfusion
injury (10). Nishiuma et
al (11) reported that the
inhalation of SPHK1 inhibitor could attenuate airway inflammation
in a mouse model of asthma. A recent study revealed the expression
of SPHK to be dysregulated in VALI; however, whether this
contributes to the pathogenesis of VALI remains unclear.
The increased permeability of endothelial cells has
been shown to be the prominent feature of VALI (12,13). The disruption of the endothelial
barrier induces the transmigration of inflammatory cells, such as
neutrophils, and the formation of edema. Our previous study showed
that the activation of Ras homolog family member a (RhoA), and
subsequent phosphorylation of myosin light chain and contraction of
endothelial cells, may be involved in VALI (14). Furthermore, high tidal volume
ventilation-induced lung inflammation was found to be associated
with the upregulation of RhoA; treatment with RhoA inhibitor
suppressed the expression of Rho-associated coiled-coil forming
protein kinase (ROCK) and alleviated lung inflammation (15). A previous study showed that, in a
partial urethral obstruction model, upregulation of SPHK1 was
accompanied with the induction of RhoA expression, suggesting an
association between SPHK and RhoA in regulating endothelial barrier
function (16).
Through the comprehensive analysis of mouse VALI
genomics data, the present study found that the mRNA expression
levels of SPHK1, rather than SPHK2, were significantly upregulated
in mouse lung tissues following ventilation. Therefore, the present
results suggested that upregulation of SPHK1 may contribute to
endothelial hyperpermeability during the development of a two-hit
model of VALI by activating the RhoA signaling pathway. It is
well-known that bacteremia and/or circulating bacterial products,
such as lipopolysaccharide (LPS), are present in the circulation of
critically ill individuals (17,18). Therefore, in the present study, a
two-hit mouse model was established by systemic LPS (1 mg/kg)
followed by ventilation with a low tidal volume of 10 ml/kg. The
improvements identified in the present study were similar to the
reported clinical mechanical ventilation strategies (2,3).
Therefore, the present findings may have potential clinical
applications. In addition, in the present study, an in vitro
mechanical stretch system was used on primary cultured mouse lung
microvascular endothelial cells to evaluate the role of SPHK1 in
the mechanical stretch-induced activation of the RhoA signaling
pathway and endothelial hyperpermeability.
Materials and methods
Microarray data collection
The present study used the Gene Expression Omnibus
(GEO) database (http://www.ncbi.nlm.nih.gov/geo/) to retrieve
expression profile datasets. The search term used was: 'Ventilator
lung'.
Animal preparation
In total, 280 male ICR mice (aged 8-10 weeks,
weighting 25-30 g) were purchased from the Animal Experimentation
Center of the Second Military Medical University. All mice had free
access to water and food and were housed at room temperature
(20-22°C) with 30-70% humidity under a 12-h light/dark cycle. All
experimental protocols were approved by The Shanghai Jiaotong
University School of Medicine and the methods were conducted in
accordance with the institutional guidelines (ethical approval nos.
XHEC-C-2017-058 and XHEC-F-NSFC-2018-057).
Mechanical ventilation and drug
treatment
Mice underwent ventilation and were
intraperitoneally administered with 1 mg/kg LPS (LPS + ventilation
group) or saline (ventilation group) (2). After 12 h, the mice were
anesthetized with ketamine (70 mg/kg) and xylazine (10 mg/kg)
(19), and connected to a
ventilator (Inspira, Harvard Apparatus Ltd.) following tracheotomy
(10 ml/kg at 150 breaths/min) for 4 h. SPHK1 inhibitor SKI II (50
mg/kg) or ROCK1 inhibitor RKI-1447 (10 mg/kg) were injected
intraperitoneally 1 h prior to ventilation. All animals were
sacrificed following ventilation, and bronchoalveolar lavage (BAL)
fluid or lung tissues were collected. The blood samples were
centrifuged at 956 × g for 10 min at 4°C. Lung samples were fixed
in 4% paraformaldehyde for 1 day at room temperature, employed to
determine the lung wet-to-dry (W/D) ratio or stored at −80°C until
use.
Experimental groups
For the first set of experiments, the present study
aimed to investigate the effects of SPHK1 inhibition on VALI. The
mice were randomly assigned to five groups (n=7 per group): i)
Saline administration followed by spontaneous breathing
(non-ventilated group); ii) LPS administration followed by
spontaneous breathing (non-ventilated + LPS group); iii) saline
followed by ventilation (ventilated group); iv) LPS administration
followed by ventilation (ventilated + LPS group); and v) LPS and
subsequent SPHK1 inhibitor administration with ventilation
(ventilated + LPS + SPHK1 inhibitor group).
For the second set of experiments, the present study
aimed to investigate the effects of ROCK1 inhibition on VALI. The
mice were randomly allocated to the following five groups: i)
Saline administration followed by spontaneous breathing
(non-ventilated group); ii) LPS administration followed by
spontaneous breathing (non-ventilated + LPS group); iii) saline
administration followed by ventilation (ventilated group); iv) LPS
administration followed by ventilation (ventilated + LPS group);
and v) LPS and subsequent ROCK1 inhibitor administration with
ventilation (ventilated + LPS + ROCK1 inhibitor group).
BAL
BAL fluid collection was performed three times with
1 ml of saline as previously described (12). The total cell counts were
determined as previously described (20) and the protein concentration of BAL
fluid was analyzed using a commercially available bicinchoninic
protein assay kit.
Lung W/D weight ratio
The index of pulmonary edema formation was
calculated by obtaining the lung W/D weight ratio. The lung weights
prior to and following drying were used to calculate the lung W/D
ratio as previously described (13).
Evans blue dye extravasation assay
Evans blue dye was injected intravenously at 30
mg/kg 1 h prior to the termination of ventilation to assess
vascular leak, according to a previously described protocol
(13).
Examination of lung histopathology
The left lung tissues were fixed in 4%
paraformaldehyde at room temperature for 1 day, and then stained
with hematoxylin for 5-10 min at room temperature and eosin
(Beyotime Institute of Biotechnology) for 1-2 min at room
temperature. The pathogenesis of lung samples was graded by two
pathologists in a blinded manner as previously reported: i)
0=Normal tissue; ii) 1=minor inflammatory alterations; iii) 2=mild
to moderate inflammatory alterations without marked damage in the
lung architecture; iv) 3=moderate inflammatory injury with
thickening of the alveolar septa; v) 4=moderate to severe
inflammatory injury with the formation of nodules or areas of
pneumonitis; and vi) 5=severe inflammatory injury with total
obliteration of the field. The mean score was reported per
section.
Assay for S1P levels
The content of S1P in the serum was determined via
ELISA using a commercial kit, according to the manufacturer's
instructions (cat. no. abx585002; Abbexa Ltd.).
Assay for ROCK1 activity
The activities of ROCK1 in the lung tissue and in
mouse lung vascular endothelial cells (MLVECs) were analyzed via
ELISA using a commercial kit, according to the manufacturer's
instructions (cat. no. STA-416; Cell Biolabs, Inc.).
Isolation of MLVECs and cyclic
stretch
MLVECs isolation and culture were performed using a
modified method as previously described (9). Briefly, the anesthetized mice were
perfused with DMEM (Gibco; Thermo Fisher Scientific, Inc.) from the
right ventricle to remove blood from the lungs. Then, the
subpleural pulmonary tissue was cut into pieces and cultured in
DMEM with 20% FBS under 5% CO2 at 37°C for 60 h. The
tissue was removed and the adherent cells at passages three and
four were used. For cyclic stretch, MLVECs (1×106
cells/well) were seeded onto collagen I-coated Bioflex®
6-well culture plates (Flexcell International Corporation) and
grown to 80% confluence. MLVECs were then exposed to cyclic stretch
(8% linear elongation, sinusoidal wave, 30 cycles/min) for 4 h, as
previously described (21), by
using the Flexcell® FX-5000 Tension system (FX5K;
Flexcell International Corporation). The cells that did not receive
cyclic stretch were placed in the same incubator for 4 h at
37°C.
Analysis of MLVEC permeability
Endothelial cell permeability was analyzed according
to a previously published protocol (22). The assay was based on the high
affinity binding of an avidin-conjugated FITC-labeled tracer to
biotinylated extracellular matrix proteins immobilized on the
bottom of culture dishes covered with an MLVEC monolayer. The
BioFlex plates (Flexcell International Corporation) were coated
with biotinylated gelatin (Sigma-Aldrich; Merck KGaA), and MLVECs
were seeded in the wells (1×106 cells/well). After
cyclic stretch, cells were fixed with 3.7% formaldehyde at room
temperature for 10 min, and FITC-avidin (25 µg/ml) was added
to the culture medium for 3 min at room temperature. After washing,
elastic bottoms of plates were excised with a scalpel and
transferred onto a microslide. The fluorescence was then analyzed
under a microscope (magnification, ×20; Olympus Corporation).
Western blot analysis
For western blotting, the lung tissues and MLVECs
were homogenized in cold RIPA lysis buffer (Beyotime Institute of
Biotechnology) containing proteinase and phosphatase inhibitor
cocktail (Roche Diagnostics). Equal amounts of protein extract
(30-40 µg) were separated by 8-12% SDS-PAGE and transferred
onto PVDF membranes (EMD Millipore). After blocking in 5% non-fat
dry milk for 2 h at room temperature, the membranes were probed
overnight at 4°C with anti-SPHK1 (cat. no. ab71700; 1:500; Abcam),
anti-phosphorylated (p)-myosin phosphatase target subunit 1
(p-MYPT1; cat. no. 5163S; 1:1,000; Cell Signaling Technology,
Inc.), anti-MYPT1 (cat. no. 2634S; 1:1,000; Cell Signaling
Technology, Inc.), anti-RhoA (cat. no. ab187027; 1:3,000; Abcam)
and anti-β-actin (cat. no. sc-47778; 1:500; Santa Cruz
Biotechnology, Inc.) antibodies. After washing, the membranes were
incubated for 1 h at room temperature with horseradish
peroxidase-conjugated secondary antibodies (cat. nos. ab6721 and
ab6728; 1:5,000; Abcam). The antibody-reactive bands were
visualized via an enhanced chemiluminescence western blotting
detection system (EMD Millipore). The membranes were visualized
using the UVP Bio-Imaging system. Densitometric analysis was
performed using Quantity One software (version 4.6; Bio-Rad
Laboratories, Inc.).
Analysis of RhoA via pull-down assay
RhoA activity was measured via a pull-down assay
using glutathione S-transferase (GST) fusion of Rho-binding domain
(RBD) (Cell Signaling Technology, Inc.) according to the
manufacturer's instructions. Following treatment, lung tissues and
MLVECs were collected and lysed in cold RIPA lysis buffer.
Supernatants were incubated for 2 h at 4°C with GST-RBD coupled
beads. Centrifuge the samples at 3,824 × g for 10-30 sec and
precipitates were washed three times with RIPA lysis buffer and
suspended in 2X SDS sample buffer (Cell Signaling Technology,
Inc.). The activated RhoA bound to the beads or total RhoA in cell
extracts was detected using western blot analysis with a RhoA
antibody following the aforementioned protocol.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA isolated from mouse lungs tissue using
TRIzol® reagent (Thermo Fisher Scientific, Inc.) was
used for RT. Subsequently, qPCR was performed using
SYBR® Premix Ex Taq (2X; Takara Bio, Inc.) with a
StepOne Plus system (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The amplification conditions were as follows: Initial
denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for
15 sec, 60°C for 30 sec and 72°C for 30 sec, with a final extension
at 72°C for 5 min. Mouse β-actin served as an internal control,
using the 2−ΔΔCq method (23). The sequences of the primers used
are listed in Table I.
 | Table IPrimers used for qPCR. |
Table I
Primers used for qPCR.
| Gene symbol | Primer sequence
(5′-3′) |
|---|
| SPHK1 | F:
GAAGACCTGCTCATCAACTGC |
| R:
GGTGCCCACTGTGAAACG |
| β-actin | F:
CTGTATGCCTCTGGTCGTAC |
| R:
TGATGTCACGCACGATTTCC |
Statistical analysis
Data are presented as the mean ± SEM. Normal
distribution was assessed by Shapiro-Wilk test. Statistical
significance was measured according to sample distribution and
homogeneity of variance. Statistical comparison between two groups
were determined by Student's t-test for normally distributed data
and by Mann-Whitney U test for non-normally distributed data.
One-way ANOVA with Bonferroni's post hoc test was performed to
compare multiple groups using SPSS 19 (SPSS, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
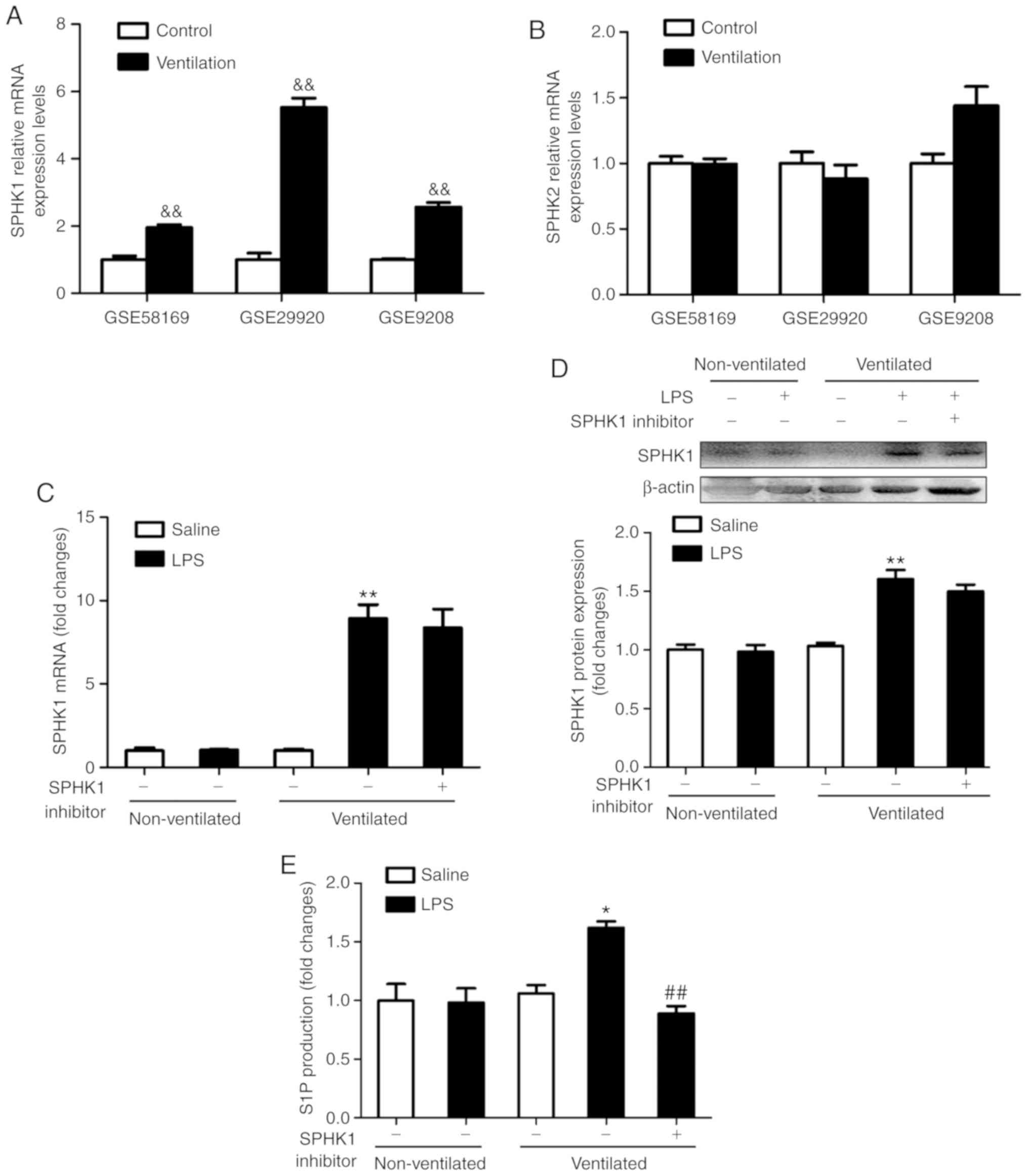
Pulmonary levels of SPHK1 and S1P are
upregulated in the two-hit model of VALI
To determine the expression levels of SPHK in
ventilated mouse lung tissues, the present study analyzed the gene
expression profiles of the GEO mouse VALI datasets GSE58169
(24), GSE29920 and GSE9208
(25). Collectively, the analysis
of these data revealed that the mRNA expression of SPHK1, rather
than SPHK2, was significantly upregulated in ventilated mouse lung
tissues (Fig. 1A and B). To
verify these findings, the expression of SPHK1 was investigated by
RT-qPCR and western blotting. As shown in Fig. 1C and D, there were no significant
differences in mRNA and protein concentration of SPHK1 in LPS alone
or ventilated alone group. However, in the ventilated + LPS group,
the expression of SPHK1 was significantly increased compared with
that in the non-ventilated group. SPHK1 inhibitor had no effects on
the mRNA and protein expression of SPHK1. Furthermore, consistent
with the increase in SPHK1 expression, the levels of pulmonary S1P
were significantly increased in the ventilated + LPS group, but
decreased following treatment with SPHK1 inhibitor (Fig. 1E).
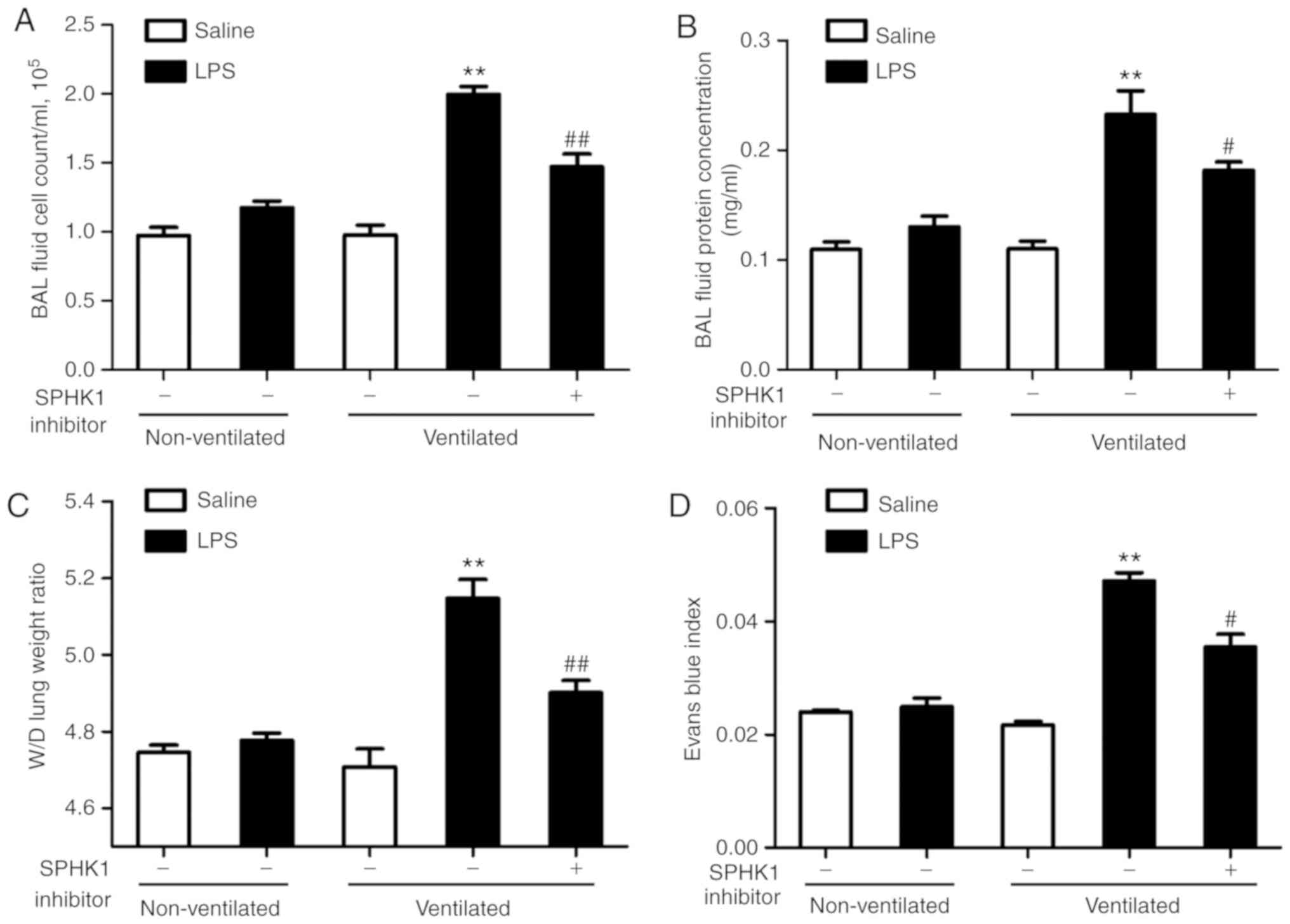
SPHK1 inhibitor attenuates lung vascular
hyperpermeability in the two-hit model of VALI
In the present study, it was investigated whether
SPHK1 inhibitor could exhibit a protective effect on the two-hit of
model of VALI. As shown in Fig. 2A
and B, LPS or ventilation alone induced minor non-significant
lung injury compared with the non-ventilated group. In line with
our previous study (13), low
tidal volume ventilation did not exert significant lung injury.
However, when combined with LPS, ventilation significantly induced
lung injury, indicating a synergistic effect of these two stimuli.
In addition, the degree of lung injury in the two-hit VALI models
was significantly attenuated following treatment with SPHK1
inhibitor. Similar results were observed in the lung W/D weight
ratio and the Evans blue assay (Fig.
2C and D). Ventilation + LPS led to increased lung W/D ratio
and Evans blue leakage from the vascular space into the lung
parenchyma, which were significantly alleviated following treatment
with SPHK1 inhibitor.
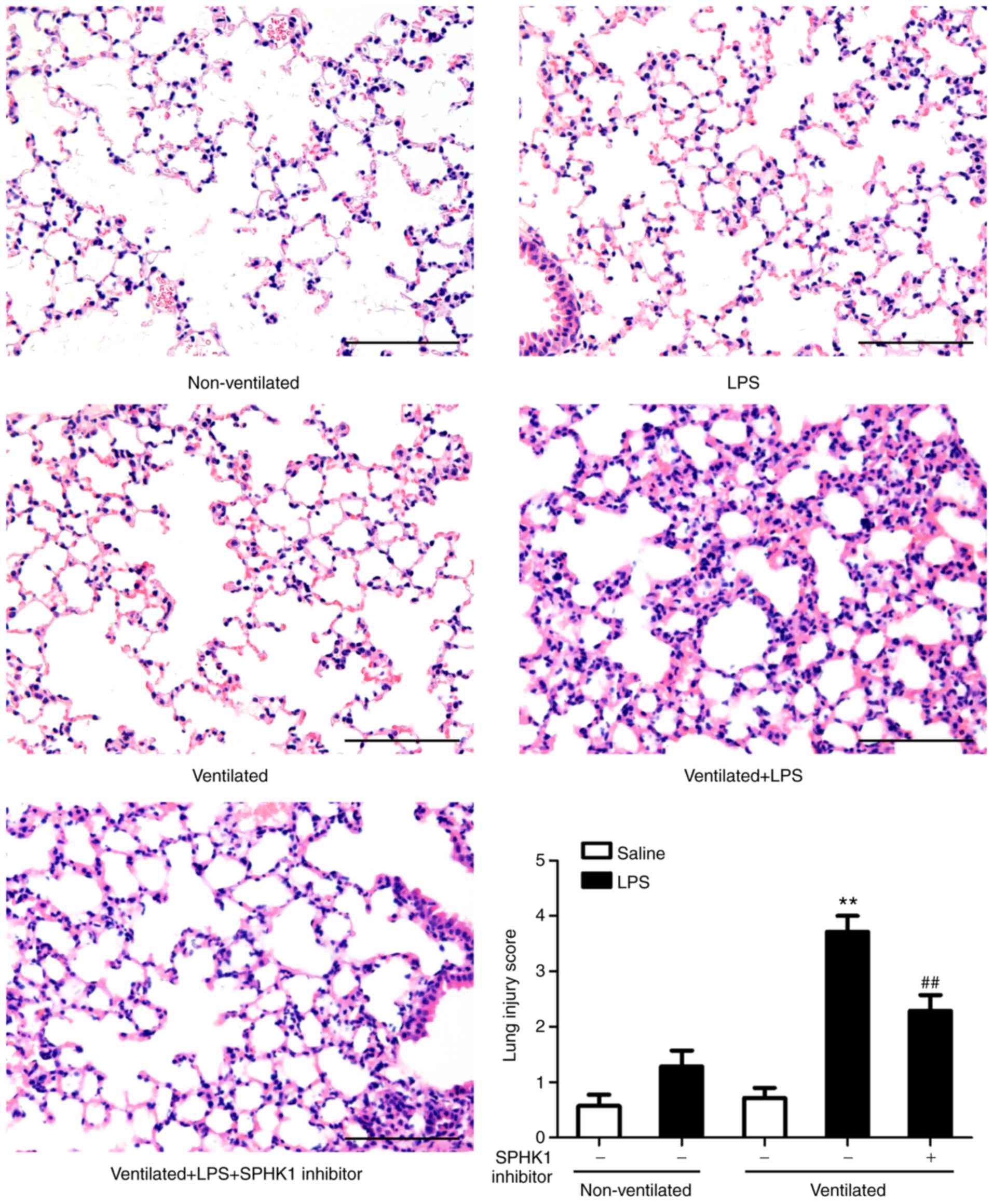
H&E staining was then performed to evaluate the
histology of tissues (Fig. 3).
Histopathological analysis revealed that mice in the two-hit VALI
group (ventilated + LPS) exhibited diffuse interstitial edema and
infiltration around the pulmonary vessels; alveolar and
interstitial edema were also observed compared with the LPS and
ventilated groups. Quantitative analysis demonstrated a significant
increase in the lung injury score in the ventilated + LPS group.
Following treatment with SPHK1 inhibitor, lung injury induced by
ventilation + LPS was significantly attenuated.
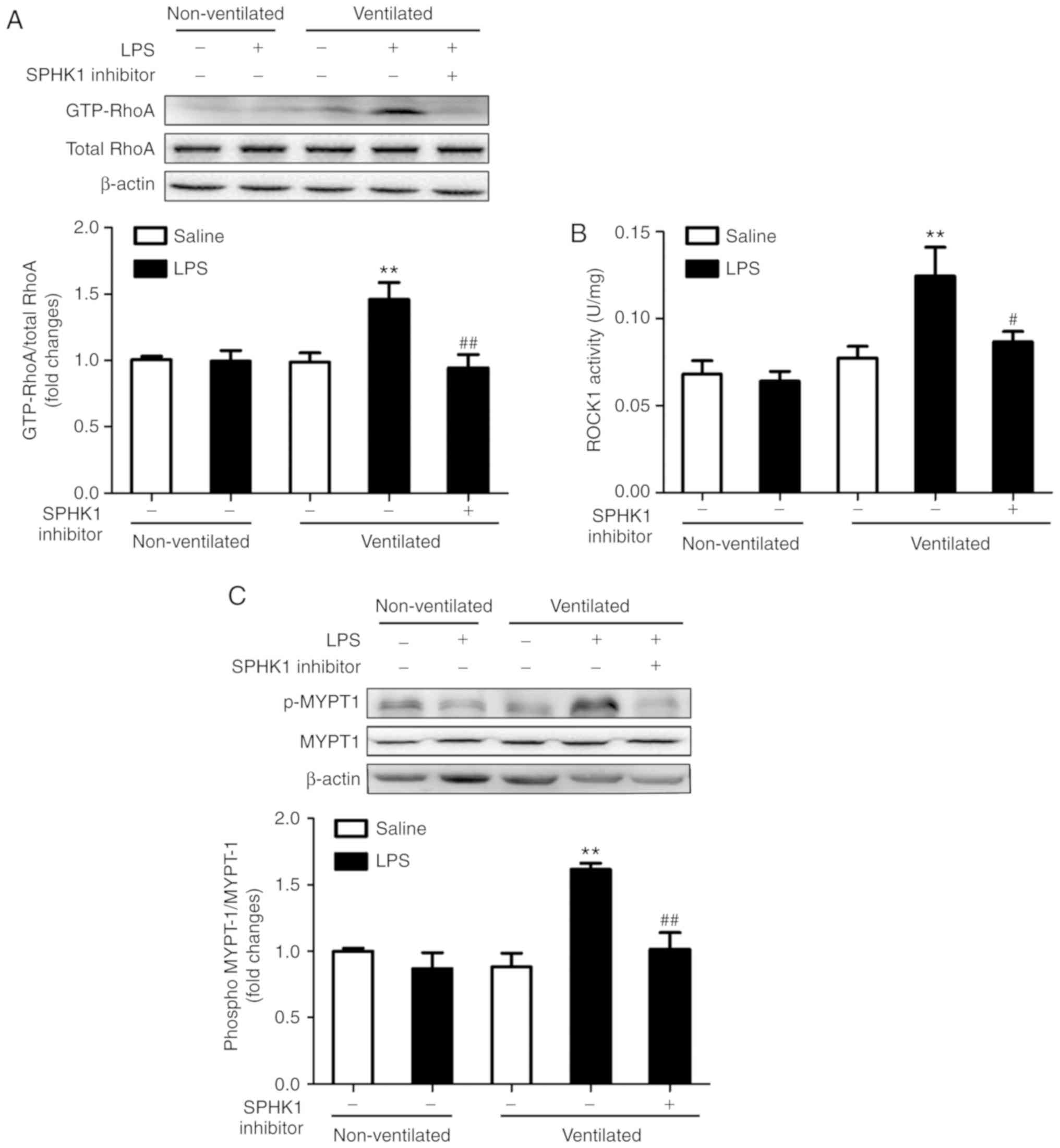
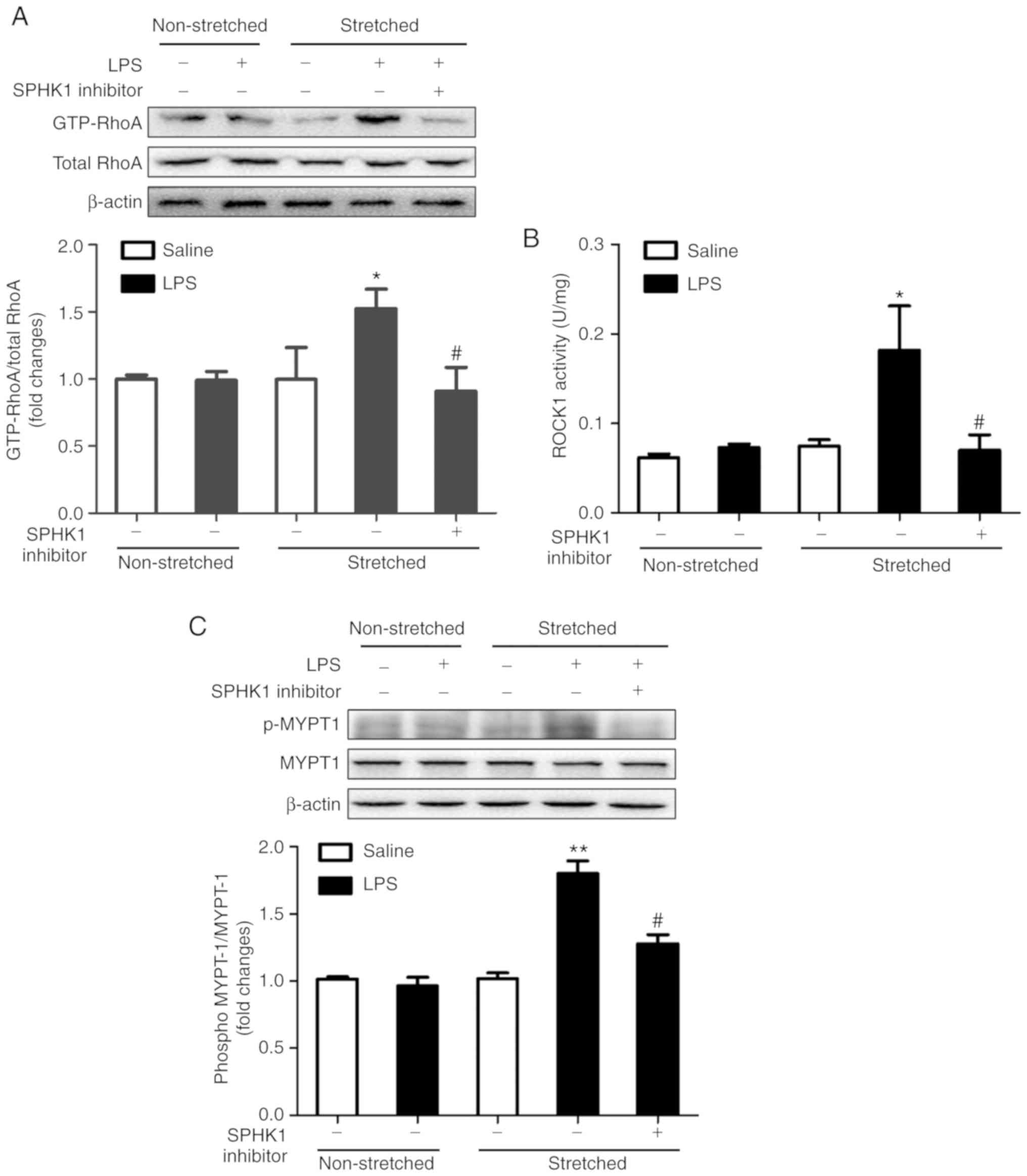
Effects of SPHK1 inhibitor on the
RhoA/ROCK pathway in a two-hit model of VALI
RhoA and its target protein, ROCK, are involved in
the calcium-sensitizing signaling pathway associated with the
cytoskeletal contractile response via the activity of myosin ATPase
(26). ROCK mediates the
phosphorylation of myosin light chain, ultimately resulting in the
reorganization of actin myosin, tension fiber formation and cell
contraction (27). MYPT-1 is the
regulatory subunit of myosin light chain phosphatase, which
functions as a downstream target of ROCK (28). The present study investigated the
activation of RhoA and ROCK1, and the phosphorylation of MYPT-1 in
the two-hit model of VALI. As presented in Fig. 4, LPS or ventilation alone had no
significant effect on RhoA and ROCK1 activity, and on the
phosphorylation levels of MYPT1. By contrast, mice in the two-hit
VALI group (ventilated + LPS) exhibited an increase in RhoA and
ROCK1 activity, and MYPT1 phosphorylation compared with the
non-ventilated + LPS or ventilated groups. Additionally, SPHK1
inhibitor treatment significantly attenuated the increases in RhoA
and ROCK1 activity, and the phosphorylation levels of MYPT1 in the
lung tissues from mice in the ventilated + LPS group. The present
results suggested that SPHK1 inhibitor suppressed the activity of
the RhoA/ROCK signaling pathway in animals with VALI.
Effects of SPHK1 inhibitor on the
RhoA/ROCK pathway in MLVECs
As lung endothelial cell hyperpermeability is caused
by endothelial overdistension during ventilation (1,29),
the effects of cyclic stretch and LPS were examined, and the effect
of SPHK1 inhibition on the activation of the RhoA/ROCK pathway was
investigated in primary cultured MLVECs. In the present study,
MLVECs were subjected to LPS treatment for 12 h and then exposed to
cyclic stretch (8% elongation) for 4 h. Consistent with the
findings in vivo, LPS or stretch alone led to minor changes
in the activation of RhoA and ROCK1, and the phosphorylation of
MYPT1. On the contrary, MLVECs subjected to the combination of
stretch and LPS treatment exhibited significant increases in the
activity of RhoA and ROCK1, and in the phosphorylation levels of
MYPT1. Treatment with SPHK1 inhibitor significantly inhibited the
activation of RhoA and ROCK1, and the phosphorylation of MYPT1
induced by cyclic stretch + LPS (Fig.
5).
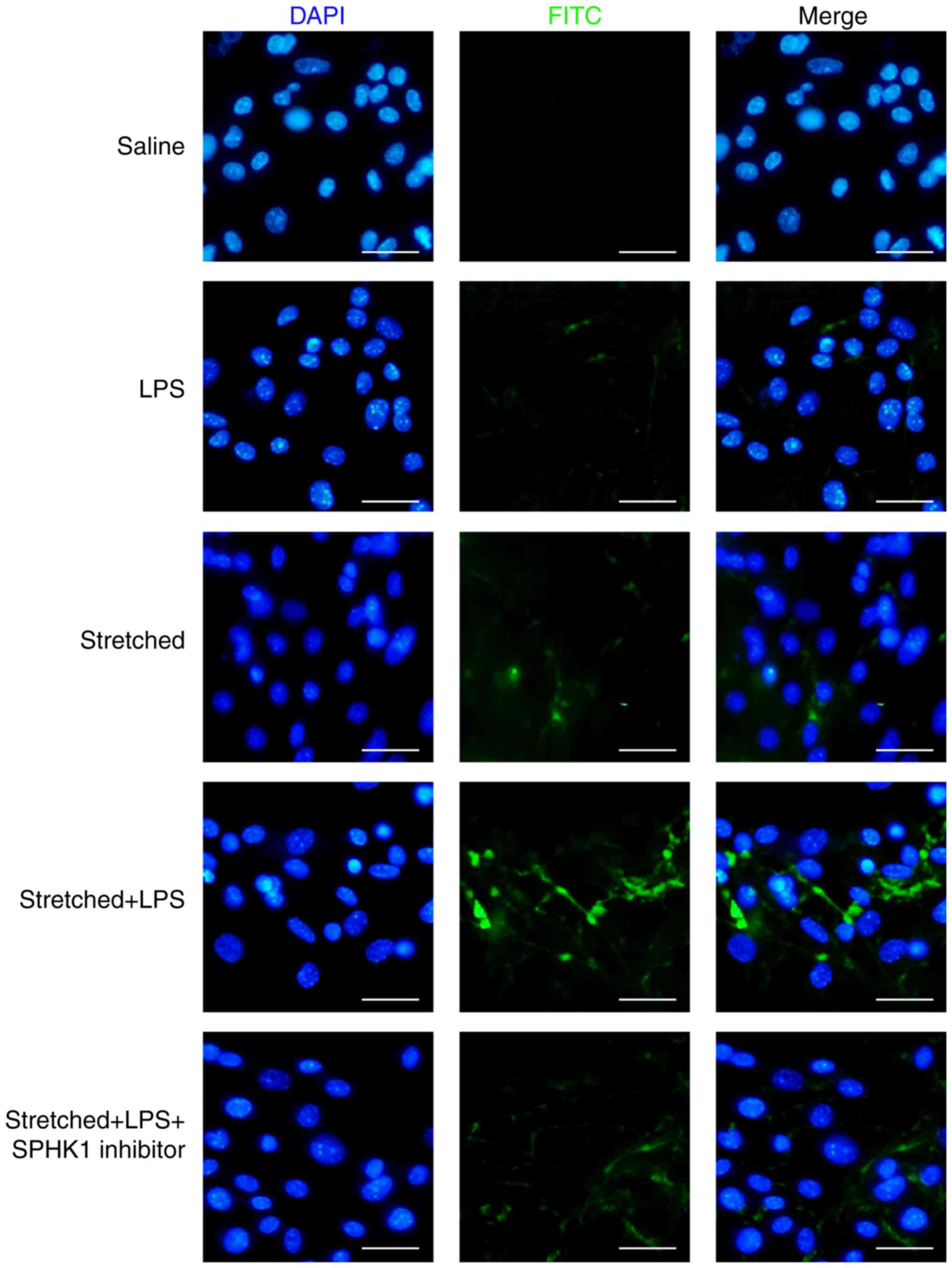
Effects of SPHK1 inhibitor on the
permeability of MLVECs
The protective effects of SPHK1 inhibitor against
stretch-associated endothelial hyperpermeability were further
assessed by a permeability assay. As presented in Fig. 6, LPS or mechanical stretch alone
had limited effects on endothelial permeability. By contrast,
analysis of the permeability sites in the LPS + stretch-challenged
endothelial monolayers revealed the presence of FITC-labeled
avidin, which entered cells via weakened cell junctions and
stretch-induced paracellular gaps; the combination of stretch and
LPS treatment markedly increased endothelial monolayer permeability
to FITC-labeled avidin. In addition, SPHK1 inhibitor treatment
markedly decreased the cellular entry of the fluorescent probe via
intercellular junctions, indicating that inhibition of SPHK1
attenuated the disruptive effects of LPS + cyclic stretch on the
permeability function of endothelial cells in vitro.
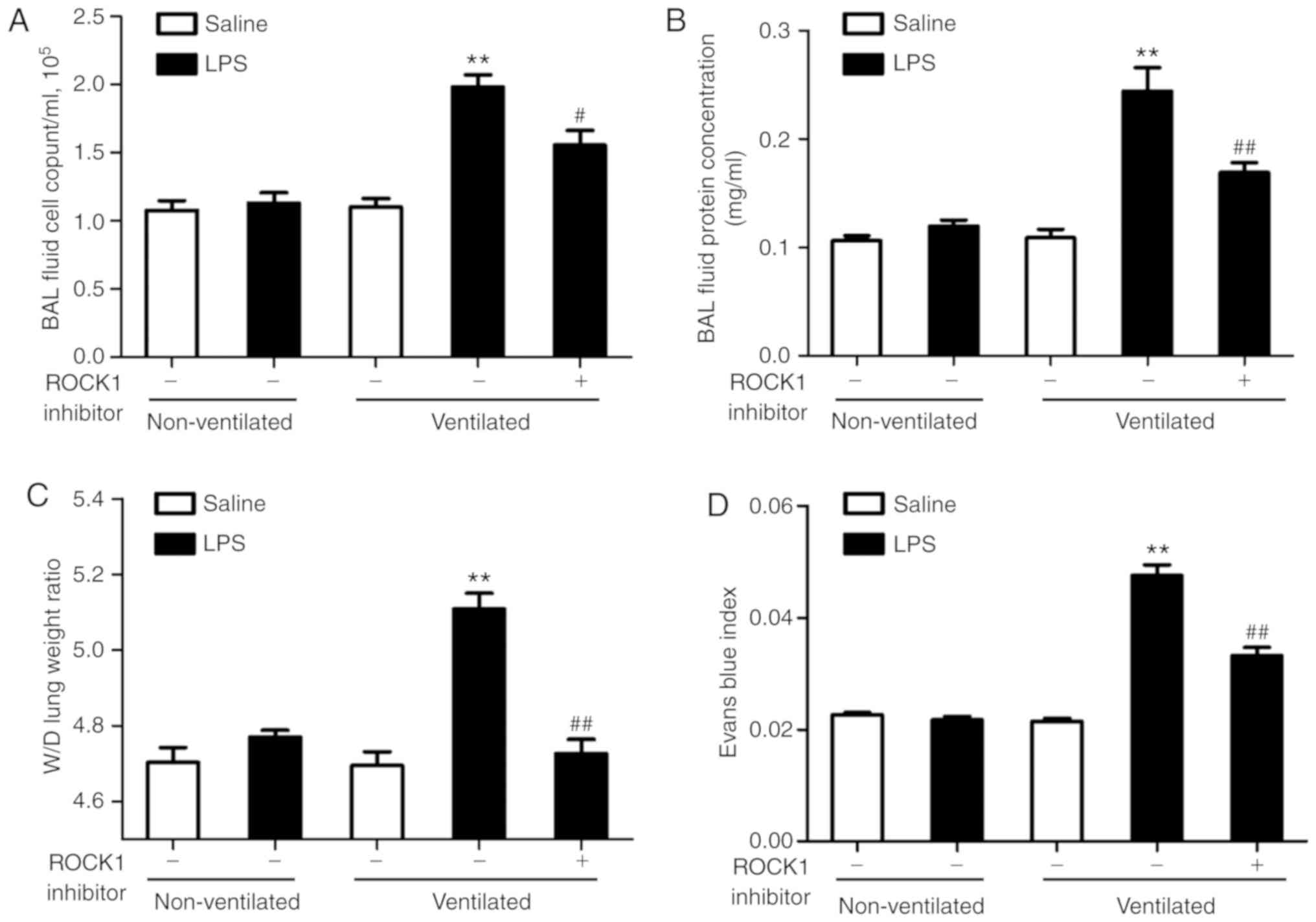
ROCK1 inhibitor attenuates lung vascular
hyperpermeability in the two-hit model of VALI
To assess the role of the RhoA/ROCK pathway in the
two-hit model of VALI, mice were treated with ROCK1 inhibitor
RKI-1447 (10 mg/kg) prior to ventilation. As presented in Fig. 7A and B, ROCK1 inhibitor exerted a
significant protective effect on the two-hit model of VALI as
assessed by measuring the cell count and the protein levels in the
BAL fluid. The results of the lung W/D ratio and Evans blue index
analyses also supported these findings (Fig. 7C and D). In addition, H&E
staining was performed to evaluate the effects of ROCK1 inhibitor
on lung histology (Fig. 8). The
degree of lung injury induced by LPS and ventilation was
significantly attenuated by ROCK1 inhibitor treatment.
Additionally, the present study investigated the
effects of ROCK1 inhibitor on the ROCK1/MYPT1 pathway in
vivo. As presented in Fig.
S1, ROCK activity was suppressed in response to ROCK1
inhibitor, and the levels of p-MYPT1 were also downregulated.
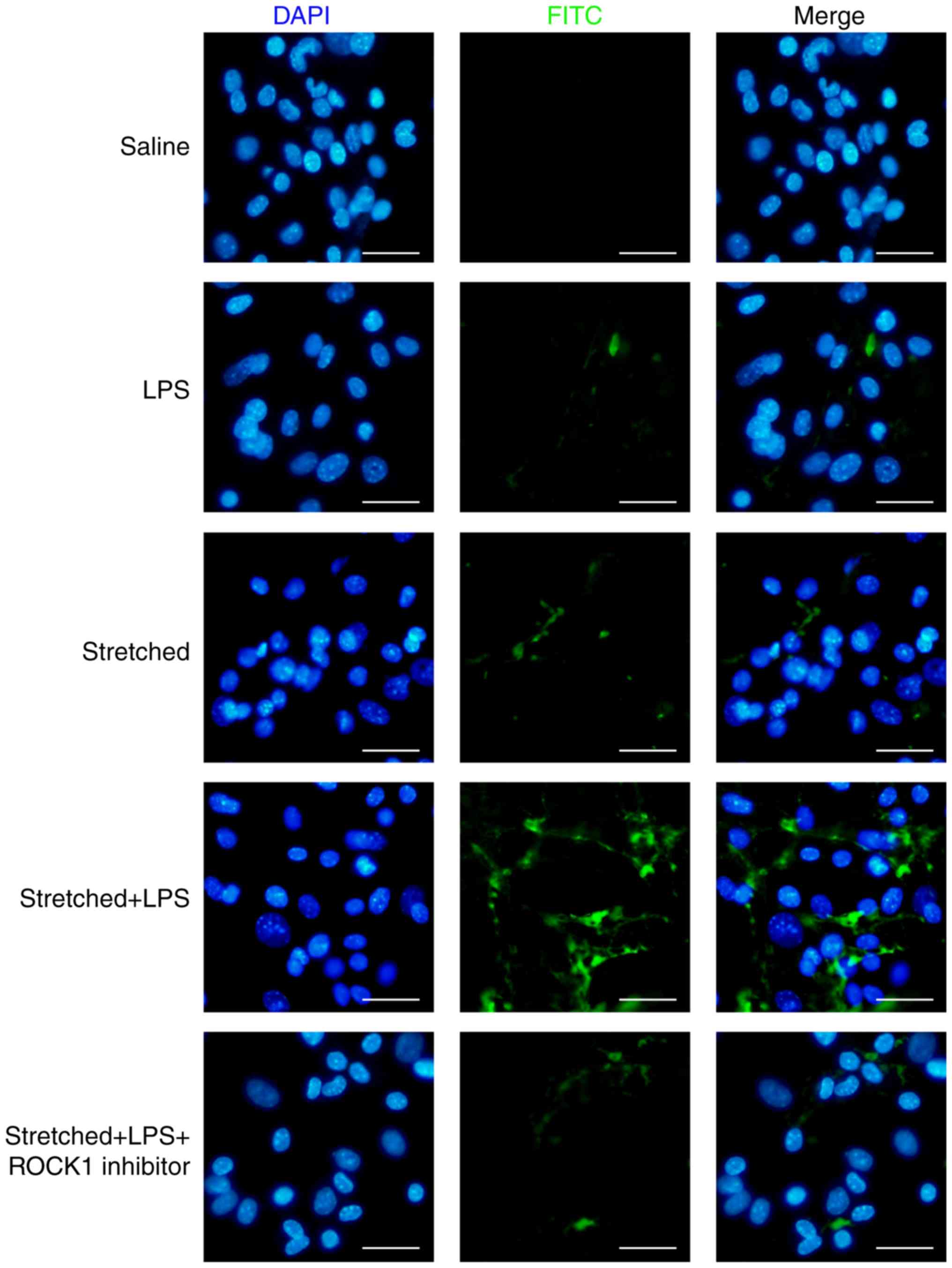
Effects of ROCK1 inhibitor on the
permeability of MLVECs
The present study investigated the effects of ROCK1
inhibitor on endothelial permeability in vitro. As presented
in Fig. 9, ROCK1 inhibition by
RKI-1447 (1 µM) could inhibit the entry of the fluorescent
probe in cells in the stretch + LPS group. The present results
suggested that inhibition of ROCK1 attenuated the
barrier-disruptive effects of LPS treatment and mechanical stretch
in vitro. In addition, in MLVECs, the activity of ROCK1 and
the phosphorylation levels of MYPT1 were reversed by treatment with
ROCK1 inhibitor (Fig. S2).
Discussion
In the present study, it was reported that SPHK1/S1P
signaling was induced in a two-hit model of VALI, and the present
results suggested that inhibition of SPHK1 activity may be a
potential therapeutic approach for treating VALI.
Previous studies have shown that positive-pressure
ventilation via moderate ventilation does not generally cause
extensive lung injury in animals with healthy lungs (3,13);
however, whether ventilation promotes injury in pre-injured lungs
requires further investigation. Investigating the effects of
ventilation is clinically important as patients receiving
ventilation therapy in intensive care with lung injury can exhibit
complications, such as sepsis (17,30,31). Brégeon et al (3) revealed that positive-pressure
ventilation using a tidal volume of 10 ml/kg was harmful under
conditions of endotoxemia, and may increase the susceptibility to
lung injury in the presence of this complication. In the present
study, a tidal volume of 10 ml/kg was selected and LPS (1 mg/kg)
was administered to mimic the ventilation strategy commonly used in
clinic. In addition, the concentration of LPS used reflected the
concentration range of circulating LPS associated with pre-injured
lungs (2,3). When establishing the two-hit model,
the present study intraperitoneally administrated 1 mg/kg LPS,
according to the study of O'Mahony et al (2); and no notable lung injury was
observed at this dose. Similarly, the present study reported that
ventilation or LPS alone could result in mild changes, not meeting
the criteria for ALI or ARDS (5,29).
By contrast, in the present study, the combined treatment of LPS
and ventilation resulted in molecular and histological alterations
associated with lung injury. These findings may support future
clinical application of the present findings. Furthermore,
Wadgaonkar et al (32)
revealed that the mRNA expression levels of SPHK1 increased ~5-fold
following the intratracheally administration of 2 mg/kg LPS; in
addition, significant upregulation of SPHK1 protein expression was
reported. The present study showed that intraperitoneal
administration of LPS at 1 mg/kg had no significant effect on
pulmonary SPHK1 expression. The discrepancy between our findings
and those of Wadgaonkar et al (32) may be due to an increased dose and
a different administration route of LPS.
Mammals have two isoforms of SPHK, including SPHK1
and SPHK2 (33). The expression
of these two isoenzymes has been identified in endothelial cells
and the lungs, and these enzymes were found to generate S1P
following activation (34). In a
previous study, SPHK1 rather than SPHK2, was found to be the major
isoenzyme mediating endothelial barrier function (35). Billich et al (36) reported that SPHK1 activity was
3-20 times increased compared with SPHK2 in mouse tissues. In
addition, the expression of SPHK1 was found to increase following
ventilation at 30 ml/kg for 4 h (24,25). During the development of pulmonary
arterial hypertension, unlike Sphk2−/− mice,
Sphk1−/− mice exhibit protection against
hypoxia-mediated pulmonary hypertension (8). In the present study, the gene
expression profiles from datasets generated in mice with VALI were
analyzed. In the present study, the mRNA expression levels of
SPHK1, rather than SPHK2, were significantly upregulated in
ventilated mouse lung tissues. Using an in vivo two-hit
model of VALI, it was observed that the mRNA and protein expression
levels of SPHK1 were increased following the combined treatment of
LPS and ventilation. In addition, the present study identified that
SPHK1 inhibitor treatment improved LPS + ventilation-induced lung
injury, suggesting that upregulation of SPHK1 may contribute to LPS
+ ventilation-induced lung injury.
Accumulating evidence demonstrated that mechanical
stretch disrupts the function of endothelial adherens junctions and
promotes vascular endothelial hyperpermeability (1,37).
However, conflicting findings regarding the effects of SPHK1 on the
regulation of endothelial function have been reported (38,39). For example, the activation of
SPHK1 was determined to mediate endothelial damage under high
glucose conditions or in streptozotocin-induced diabetic rats, and
inhibition of SPHK1 markedly protected endothelial cells and
suppressed the formation of vascular lesions (38). These previous findings were
consistent with the present study, as mechanical stretch resulted
in the upregulation of SPHK1, and SPHK1 inhibitor attenuated
mechanical stretch-induced lung edema and endothelial
hyperpermeability. In addition, SPHK1 has been reported to induce
endothelial cell proliferation, thus promoting hemangiogenesis and
lymphangiogenesis in breast cancer, both of which could be
suppressed by SPHK1 inhibitor (39). Collectively, these previous and
the present findings indicated that SPHK1 may modulate endothelial
function in a context-dependent manner.
The increased production of ROS (1), and the disorganized structure of the
actin and tubulin cytoskeleton (37), endothelial apoptosis (40) and disruptions in endothelial
interactions (13) are the main
causes of endothelial hyperpermeability in VALI. The present study
and several previous reports revealed that RhoA/ROCK1-mediated
phosphorylation of myosin light chain led to the contraction of
endothelial cells and breakdown of the pulmonary endothelial
barrier (14,27). In the present study, it was
identified that SPHK inhibitor attenuated the activity of RhoA and
ROCK1, and the phosphorylation of MYPT1 in vivo and in
vitro; consequently, the function of the pulmonary vascular
endothelial barrier was found to be improved following SPHK
inhibition. ROCK1 inhibitor could also attenuate lung injury and
the phosphorylation of MYPT1 in vivo and in vitro. In
addition, inhibition of SPHK1 was observed to notably protect
against high glucose-induced endothelial cell injury in a protein
kinase C (PKC)- and ERK1/2-dependent manner (38). SPHK1 has been reported as a
modulator of hypoxia inducible factor (HIF)-1α, which contributes
to hypoxia-induced endothelial dysfunction (41). Whether the PKC, ERK1/2 or HIF-1α
signaling pathways are involved in mediating the protective effects
of SPHK1 inhibitor against mechanical stretch-induced endothelial
hyperpermeability requires further investigation.
In conclusion, by using an LPS + ventilation model,
the present study suggested that upregulation of SPHK1 served an
essential role in the two-hit model of VALI. Additionally, SPHK1
inhibitor could exert protective effects via the suppression of
RhoA-mediated phosphorylation of MYPT1 and endothelial
hyperpermeability. Furthermore, lung injury induced by the combined
treatment of LPS and ventilation could be reversed by ROCK1
inhibitor.
However, the present study presents certain
limitations; the present findings were not confirmed using an SPHK1
activator or SPHK1 knockout mice, which may provide further insight
into the mechanisms underlying the protective effects of SPHK1
inhibitor in the two-hit VALI animal model. Nevertheless, although
the exact role of the SPHK1/S1P pathway in VALI remains to be
elucidated, the present findings suggested that SPHK1 inhibitor
could be a novel therapeutic agent for treating VALI.
Supplementary Data
Acknowledgments
The authors would like to thank Dr Yan-fei Mao for
excellent technical support. (Department of Anesthesiology and
Surgical Intensive Care Unit, Xinhua Hospital, Shanghai Jiaotong
University School of Medicine).
Funding
The present study was supported by grants from The
Shanghai Municipal Commission of Health and Family Planning to Dr
Jiang (grant no. 2017BR062), Shanghai Science and Technology
Commission to Dr Wang (grant no. 18YF1415500), and from The
National Natural Science Foundation of China to Dr Jiang (grant
nos. 81772117 and 81571929) and Dr Liu (grant no. 81672266).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YW analyzed the data and drafted the manuscript. TTG
performed the experiments and designed the study. DFX performed the
experiments and collected the data. XYZ performed the experiments
and drafted the manuscript. WWD performed the experiments. ZL
performed the experiments and analyzed the data. YJL performed the
experiments and supervised the study. LJ designed and supervised
the study. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved by The
Shanghai Jiaotong University School of Medicine and the methods
were conducted in accordance with the institutional guidelines
(ethical approval nos. XHEC-C-2017-058 and
XHEC-F-NSFC-2018-057).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lv Z, Wang Y, Liu YJ, Mao YF, Dong WW,
Ding ZN, Meng GX, Jiang L and Zhu XY: NLRP3 inflammasome activation
contributes to mechanical stretch-induced endothelial-mesenchymal
transition and pulmonary fibrosis. Crit Care Med. 46:e49–e58. 2018.
View Article : Google Scholar
|
|
2
|
O'Mahony DS, Liles WC, Altemeier WA,
Dhanireddy S, Frevert CW, Liggitt D, Martin TR and Matute-Bello G:
Mechanical ventilation interacts with endotoxemia to induce
extrapulmonary organ dysfunction. Crit Care. 10:R1362006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brégeon F, Delpierre S, Chetaille B,
Kajikawa O, Martin TR, Autillo-Touati A, Jammes Y and Pugin J:
Mechanical ventilation affects lung function and cytokine
production in an experimental model of endotoxemia. Anesthesiology.
102:331–339. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rizzo AN, Sammani S, Esquinca AE, Jacobson
JR, Garcia JG, Letsiou E and Dudek SM: Imatinib attenuates
inflammation and vascular leak in a clinically relevant two-hit
model of acute lung injury. Am J Physiol Lung Cell Mol Physiol.
309:L1294–L1304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Slutsky AS and Ranieri VM:
Ventilator-induced lung injury. N Engl J Med. 369:2126–2136. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Spiegel S and Milstien S: Sphingosine
1-phosphate, a key cell signaling molecule. J Biol Chem.
277:25851–25854. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang LS, Berdyshev E, Mathew B, Fu P,
Gorshkova IA, He D, Ma W, Noth I, Ma SF, Pendyala S, et al:
Targeting sphingosine kinase 1 attenuates bleomycin-induced
pulmonary fibrosis. FASEB J. 27:1749–1760. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen J, Tang H, Sysol JR, Moreno-Vinasco
L, Shioura KM, Chen T, Gorshkova I, Wang L, Huang LS, Usatyuk PV,
et al: The sphingosine kinase 1/sphingosine-1-phosphate pathway in
pulmonary arterial hypertension. Am J Respir Crit Care Med.
190:1032–1043. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Price MM, Oskeritzian CA, Falanga YT,
Harikumar KB, Allegood JC, Alvarez SE, Conrad D, Ryan JJ, Milstien
S and Spiegel S: A specific sphingosine kinase 1 inhibitor
attenuates airway hyperresponsiveness and inflammation in a mast
cell-dependent murine model of allergic asthma. J Allergy Clin
Immunol. 131:501–511.e1. 2013. View Article : Google Scholar
|
|
10
|
Wang L, Chen F, Pan Y, Lin L and Xiong X:
Effects of FTY720 on lung injury induced by hindlimb ischemia
reperfusion in rats. Mediators Inflamm. 2017:53013122017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nishiuma T, Nishimura Y, Okada T, Kuramoto
E, Kotani Y, Jahangeer S and Nakamura S: Inhalation of sphingosine
kinase inhibitor attenuates airway inflammation in asthmatic mouse
model. Am J Physiol Lung Cell Mol Physiol. 294:L1085–L1093. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Xu CF, Liu YJ, Mao YF, Lv Z, Li
SY, Zhu XY and Jiang L: Salidroside attenuates ventilation induced
lung injury via sirt1-dependent inhibition of NLRP3 inflammasome.
Cell Physiol Biochem. 42:34–43. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dong WW, Liu YJ, Lv Z, Mao YF, Wang YW,
Zhu XY and Jiang L: Lung endothelial barrier protection by
resveratrol involves inhibition of HMGB1 release and HMGB1-induced
mitochondrial oxidative damage via an Nrf2-dependent mechanism.
Free Radic Biol Med. 88:404–416. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu K, Mao YF, Zheng J, Peng ZY, Liu WW,
Liu Y, Xu WG, Sun XJ, Jiang CL and Jiang L: SC5b9-induced pulmonary
microvascular endothelial hyperpermeability participates in
ventilator-induced lung injury. Cell Biochem Biophys. 67:1421–1431.
2013. View Article : Google Scholar
|
|
15
|
Dai H, Zhang S, Du X, Zhang W, Jing R,
Wang X and Pan L: RhoA inhibitor suppresses the production of
microvesicles and rescues high ventilation induced lung injury. Int
Immunopharmacol. 72:74–81. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aydin M, Downing K, Villegas G, Zhang X,
Chua R, Melman A and Disanto ME: The sphingosine-1-phosphate
pathway is upregulated in response to partial urethral obstruction
in male rats and activates RhoA/Rho-kinase signalling. BJU Int.
106:562–571. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Parsons PE, Worthen GS, Moore EE, Tate RM
and Henson PM: The association of circulating endotoxin with the
development of the adult respiratory distress syndrome. Am Rev
Respir Dis. 140:294–301. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Martin TR, Rubenfeld GD, Ruzinski JT,
Goodman RB, Steinberg KP, Leturcq DJ, Moriarty AM, Raghu G,
Baughman RP and Hudson LD: Relationship between soluble CD14,
lipopolysaccharide binding protein, and the alveolar inflammatory
response in patients with acute respiratory distress syndrome. Am J
Respir Crit Care Med. 155:937–944. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Iwai Y, Honda S, Ozeki H, Hashimoto M and
Hirase H: A simple head-mountable LED device for chronic
stimulation of optogenetic molecules in freely moving mice.
Neurosci Res. 70:124–127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Birukova AA, Fu P, Chatchavalvanich S,
Burdette D, Oskolkova O, Bochkov VN and Birukov KG: Polar head
groups are important for barrier-protective effects of oxidized
phospholipids on pulmonary endothelium. Am J Physiol Lung Cell Mol
Physiol. 292:L924–L935. 2007. View Article : Google Scholar
|
|
21
|
Wu J, Yan Z, Schwartz DE, Yu J, Malik AB
and Hu G: Activation of NLRP3 inflammasome in alveolar macrophages
contributes to mechanical stretch-induced lung inflammation and
injury. J Immunol. 190:3590–3599. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dubrovskyi O, Birukova AA and Birukov KG:
Measurement of local permeability at subcellular level in cell
models of agonist- and ventilator-induced lung injury. Lab Invest.
93:254–263. 2013. View Article : Google Scholar :
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Spassov S, Pfeifer D, Strosing K, Ryter S,
Hummel M, Faller S and Hoetzel A: Genetic targets of hydrogen
sulfide in ventilator-induced lung injury-a microarray study. PLoS
One. 9:e1024012014. View Article : Google Scholar
|
|
25
|
Papaiahgari S, Yerrapureddy A, Reddy SR,
Reddy NM, Dodd OJ, Crow MT, Grigoryev DN, Barnes K, Tuder RM,
Yamamoto M, et al: Genetic and pharmacologic evidence links
oxidative stress to ventilator-induced lung injury in mice. Am J
Respir Crit Care Med. 176:1222–1235. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sari-Hassoun M, Clement MJ, Hamdi I,
Bollot G, Bauvais C, Joshi V, Toma F, Burgo A, Cailleret M,
Rosales-Hernández MC, et al: Cucurbitacin I elicits the formation
of actin/phosphomyosin II co-aggregates by stimulation of the
RhoA/ROCK pathway and inhibition of LIM-kinase. Biochem Pharmacol.
102:45–63. 2016. View Article : Google Scholar
|
|
27
|
Zhang M, Dong M, Liu W, Wang L, Luo Y, Li
Z and Jin F: 1α,25-dihydroxyvitamin D3 ameliorates seawater
aspiration-induced acute lung injury via NF-κB and RhoA/Rho kinase
pathways. PLoS One. 9:e1045072014. View Article : Google Scholar
|
|
28
|
MacKay CE, Shaifta Y, Snetkov VV, Francois
AA, Ward JPT and Knock GA: ROS-dependent activation of
RhoA/Rho-kinase in pulmonary artery: Role of Src-family kinases and
ARHGEF1. Free Radic Biol Med. 110:316–331. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xu CF, Liu YJ, Wang Y, Mao YF, Xu DF, Dong
WW, Zhu XY and Jiang L: Downregulation of R-Spondin1 contributes to
mechanical stretch-induced lung injury. Crit Care Med.
47:e587–e596. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Danner RL, Elin RJ, Hosseini JM, Wesley
RA, Reilly JM and Parillo JE: Endotoxemia in human septic shock.
Chest. 99:169–175. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Marshall JC, Foster D, Vincent JL, Cook
DJ, Cohen J, Dellinger RP, Opal S, Abraham E, Brett SJ, Smith T, et
al: Diagnostic and prognostic implications of endotoxemia in
critical illness: Results of the MEDIC study. J Infect Dis.
190:527–534. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wadgaonkar R, Patel V, Grinkina N, Romano
C, Liu J, Zhao Y, Sammani S, Garcia JG and Natarajan V:
Differential regulation of sphingosine kinases 1 and 2 in lung
injury. Am J Physiol Lung Cell Mol Physiol. 296:L603–L613. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Saba JD and Hla T: Point-counterpoint of
sphingosine 1-phosphate metabolism. Circ Res. 94:724–734. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao Y, Kalari SK, Usatyuk PV, Gorshkova
I, He D, Watkins T, Brindley DN, Sun C, Bittman R, Garcia JG, et
al: Intracellular generation of sphingosine 1-phosphate in human
lung endothelial cells: Role of lipid phosphate phosphatase-1 and
sphingosine kinase 1. J Biol Chem. 282:14165–14177. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tauseef M, Kini V, Knezevic N, Brannan M,
Ramchandaran R, Fyrst H, Saba J, Vogel SM, Malik AB and Mehta D:
Activation of sphingosine kinase-1 reverses the increase in lung
vascular permeability through sphingosine-1-phosphate receptor
signaling in endothelial cells. Circ Res. 103:1164–1172. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Billich A, Bornancin F, Dévay P,
Mechtcheriakova D, Urtz N and Baumruker T: Phosphorylation of the
immunomodulatory drug FTY720 by sphingosine kinases. J Biol Chem.
278:47408–47415. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Birukova AA, Fu P, Xing J, Yakubov B,
Cokic I and Birukov KG: Mechanotransduction by GEF-H1 as a novel
mechanism of ventilator-induced vascular endothelial permeability.
Am J Physiol Lung Cell Mol Physiol. 298:L837–L848. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang L, Xing XP, Holmes A, Wadham C,
Gamble JR, Vadas MA and Xia P: Activation of the sphingosine
kinase-signaling pathway by high glucose mediates the
proinflammatory phenotype of endothelial cells. Circ Res.
97:891–899. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nagahashi M, Ramachandran S, Kim EY,
Allegood JC, Rashid OM, Yamada A, Zhao R, Milstien S, Zhou H,
Spiegel S and Takabe K: Sphingosine-1-phosphate produced by
sphingosine kinase 1 promotes breast cancer progression by
stimulating angio-genesis and lymphangiogenesis. Cancer Res.
72:726–735. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Raaz U, Kuhn H, Wirtz H and Hammerschmidt
S: Rapamycin reduces high-amplitude, mechanical stretch-induced
apoptosis in pulmonary microvascular endothelial cells. Microvasc
Res. 77:297–303. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee SO, Kim JS, Lee MS and Lee HJ:
Anti-cancer effect of pristimerin by inhibition of HIF-1α involves
the SPHK-1 pathway in hypoxic prostate cancer cells. BMC Cancer.
16:7012016. View Article : Google Scholar
|