Introduction
Skin cancer is the most common cancer in western
countries, and it was one of the ten leading cancer types when
estimating new cases and deaths in the United States in 2017
(1). The main cause of all types
of skin cancer is exposure to ultraviolet radiation from the sun
and other sources (2). Skin
cancer is divided into two categories: Melanoma and non-melanoma.
Non-melanoma skin cancer includes basal cell carcinoma and squamous
cell carcinoma. Basal cell carcinoma (BCC) accounts for ~75% of
skin cancer cases, squamous cell carcinoma (SCC) accounts for ~20%
of cases, and melanoma accounts for only ~5% of cases (3). In comparison with other skin
cancers, the incidence rate of melanoma is the lowest, but it is
the deadliest.
Mitogen-activated protein kinase (MAPK) signal
transduction pathways are highly conserved among eukaryotes during
evolution and can regulate different cellular functions, such as
proliferation, differentiation, apoptosis, cell-cell adhesion,
inflammation, migration and invasion, in response to various
environmental signals (4). There
are four types of P38 MAPK isoforms in mammalian cells: p38α (also
known as MAPK14), p38β (also known as MAPK 11), p38γ (also known as
MAPK 12), and p38δ (also known as MAPK13) (5). Regarding structural similarity, p38α
and p38β are 75% similar, while p38γ and p38δ are 70% similar
(5). P38α and p38β are
universally expressed, while p38γ and p38δ are more tissue-specific
(6). Regarding kinase inhibition,
p38α and p38β can be targeted by a class of pyridinyl imidazole
drugs, but these drugs cannot inhibit p38γ and p38δ (7). However, this type of chemical
inhibition does not allow us to distinguish whether functions are
mediated by p38α or p38β. In the present study, specific short
hairpin (sh)RNA or small interfering (si)RNA were used in order to
knockdown p38α or p38β and to clearly differentiate their
biological functions.
P38α is an essential protein during embryonic
development, and it can regulate various cellular functions.
Notably, multiple proteins can be directly phosphorylated by p38α.
Additionally, the p38α pathway can control the production of
different extracellular signaling molecules, such as growth
factors, cytokines and chemokines (8). The p38α protein can regulate
different cellular functions during tumor formation at different
stages of development and for different types of cancers. P38α is
reported to act as a tumor suppressor in the initial stages
(9) but promotes tumor activity
in the later stages of tumorigenesis (10,11). In the initial stages, p38α can
regulate cell homeostasis by balancing cell apoptosis,
proliferation and differentiation (12). In the later stages, it can
facilitate tumor cell survival and dissemination (13). Therefore, p38α can have different
roles in different types of cancers.
Epithelial-mesenchymal transition (EMT) is a
biological process that allows a polarized epithelial cell to
undergo certain biological changes, such as polarity stimulation or
loss, thus shifting the cell to a more aggressive phenotype; these
altered cells resemble mesenchymal cells (14). Mesenchymal cells gain migratory
and invasion abilities, develop apoptosis resistance and have
increased extracellular matrix (ECM) component production (15,16). Once mesenchymal cell formation
occurs, the cells can migrate away from their origin. There are
several biomarkers for evaluating EMT, such as twist family bHLH
transcription factor 1 (Twist), snail family transcriptional
repressor 1 (Snai1, also known as Snail) and vimentin. By contrast,
E-cadherin, zonula occludens-1 (ZO-1) and cytokeratin serve as
mesenchymal-epithelial transition (MET) markers (17). Snail and Twist can
transcriptionally activate the downstream targets of various
signaling pathways, thus regulating EMT (18). A previous study has shown that
certain transcription factors, such as Snail, Slug, Twist, zinc
finger E-box binding homeobox (ZEB) 1, ZEB2 and transcription
factor 3 (TCF3), can regulate EMT by transcriptionally inhibiting
E-cadherin expression via suppressing its promoter activity
(19). In addition, vimentin,
which serves as a cytoskeleton marker of EMT, is extremely abundant
in various cancer cells, and its expression is highly correlated
with cancer invasion and poor prognosis (20). Furthermore, matrix
metallopeptidases (MMPs) are involved in the development and
progression of different cancers. MMPs, such as the gelatinases
MMP2 and MMP9, can degrade type IV collagen, a major component of
the ECM, to regulate cancer cell metastasis (21). Whether p38α or p38β could be
involved in EMT or MET in A375 melanoma cells remains unknown.
A previous study indicated that p38α may act as a
tumor promoter and enhance melanoma cell metastasis under cytokine
stimulation (22,23). Therefore, p38α can regulate the
migration abilities of tumor cells, as well as EMT processes
(24). In other types of cancer,
p38α has been reported to regulate cell proliferation,
differentiation, apoptosis and numerous cellular processes, but in
melanoma, its precise role remains unknown. In addition, p38β has
been demonstrated to regulate cell apoptosis, differentiation and
metabolism (25-27). However, the functions of p38β,
which shares 75% structural similarity with p38α, have not been
investigated in melanoma. Therefore, the goal of the present study
was to identify the biological functions of p38α and p38β.
The present study investigated the roles of p38α and
p38β by ablating their gene functions using specific shRNA in A375
melanoma cells. Based on proliferation, apoptosis, migration and
aging assay analyses, the data identified that p38α, but not p38β,
may regulate melanoma cell proliferation and migration. Thus, p38α
might be a valuable therapeutic target in patients with
melanoma.
Materials and methods
Cell culture and treatment
A375 human melanoma cells and B16F0 mouse melanoma
cells were obtained from the Bioresource Collection & Research
Center (Hsinchu, Taiwan) and 293T cells were purchased from the
American Type Culture Collection; cat. no. CRL-3216). Both cell
lines were maintained in Dulbecco's modified Eagle's medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (FBS; HyClone; GE Healthcare Life Sciences), 2 mM
glutamine, 2 mM sodium pyruvate, 100 µg/ml penicillin, and
100 µg/ml streptomycin in humidified air (5% CO2)
at 37°C.
Gene silencing using shRNA
shRNA-encoding plasmids were purchased from the
National RNAi Core Facility (Academia Sinica, Taipei, Taiwan). 293T
cells were co-transfected with three plasmids: pMD. G (0.5
µg), pCMVΔr89.1 (4.5 µg) and p38α (5 µg) or
p38β (5 µg) shRNA plasmid (cloned in the pLKO.1 vector). The
plasmids were transfected using the PureFection transfection
reagent (cat. no. LV750A-1), according to the manufacturer's
instructions (System Biosciences LLC). After transfection for 24 h,
the medium was replaced with fresh medium. The lentiviral
particle-containing medium was then harvested after 24 and 48 h,
and used to transduce the A375 cells. After 48 h of infection, 2
µg/ml puromycin was added to the media for 48 h to remove
the non-transfected cells. The stable cells were then routinely
cultured in the presence of 1 µg/ml puromycin.
The shRNA oligoribonucleotide sequences were as
follows: p38α #1, GTT CAG TTC CTT ATC TAC CAA; p38α #2, CCA TGA GGC
AAG AAA CTA TAT; p38α, #3 GCC GTA TAG GAT GTC AGA CAA; p38β #1, GCC
ACG TCC ATC GAG GAC TTC; p38β #2 CCT GTC CTC TTC TGG CTA CTG; and
p38β, #3, CGG CTC CGT CTG TTC GGC CTA.
siRNA silencing
B16F0 cells were cultured to reach 80% confluence on
the day of transfection. siRNAs (10 nM) were transfected using the
PureFection transfection reagent, according to the manufacturer's
instructions (System Biosciences). p38α siRNA (cat. no.
MAPK14_Hs01_00018467), p38β siRNA (cat. no. MAPK11_Hs01_00071113)
and siRNA universal negative control #1 (cat. no. SIC001) were
purchased from Sigma-Aldrich (Merck KGaA). At 48 h
post-transfection, subsequent experiments were performed.
Western blot analysis
Cultured A375 human melanoma cells were scraped,
washed twice with PBS and centrifuged at 23,000 x g (28). The cell pellets were lysed in
buffer containing 50 mM Tris (pH 7.5), 0.5 M NaCl, 1.0 mM EDTA (pH
7.5), 10% glycerol, 1 mM BME, 1% IGEPAL-630 and protease inhibitor
cocktail (Roche Molecular Diagnostics). After incubation for 30 min
on ice, the supernatants were collected by centrifugation at 23,000
x g for 10 min at 4°C. The Bradford method was performed to
determine the protein concentrations. Samples containing the same
amounts of protein (10 µg) were analyzed by western
blotting. Proteins were separated on 10% or 12% SDS-PAGE gels
(depending on the molecular weight) and transferred onto PVDF
membranes (EMD Millipore). Non-specific protein binding was
prevented by incubating the membranes with blocking buffer (5%
non-fat dry milk, 20 mM Tris-HCl pH 7.6, 150 mM NaCl and 0.1%
Tween-20) for at least 1 h at room temperature. Then, the membranes
were incubated with the following specific primary antibodies:
Anti-p38α (clone A1F7; cat. no. sc-33688; Santa Cruz Biotechnology,
Inc.), anti-p38β (clone N-14; cat. no. sc-15918; Santa Cruz
Biotechnology, Inc.), anti-E-cadherin (cat. no. sc-8426; Santa Cruz
Biotechnology, Inc.), anti-Twist (cat. no. sc-81417; Santa Cruz
Biotechnology, Inc.), anti-vimentin (clone RV202; cat. no.
sc-32322; Santa Cruz Biotechnology, Inc.), anti-Snai1 (clone T-18;
cat. no. sc-10433; Santa Cruz Biotechnology, Inc.), anti-MMP2
(clone 8B4; cat. no. sc-13595; Santa Cruz Biotechnology, Inc.),
anti-MMP9 (clone M-17; cat. no. sc-6841; Santa Cruz Biotechnology,
Inc.), anti-vascular endothelial growth factor (VEGF; clone 147;
cat. no. sc-507; Santa Cruz Biotechnology, Inc.), anti-p53 (cat.
no. sc-1311; Santa Cruz Biotechnology, Inc.), anti-p21 (cat. no.
sc-6246; Santa Cruz Biotechnology, Inc.), anti-p16 (cat. no.
10883-1-AP; ProteinTech Group, Inc.) and anti-β-actin (clone C4;
cat. no. sc-47778; Santa Cruz Biotechnology, Inc.). The dilution of
primary antibodies was 1:1,000. All membranes with primary
antibodies were placed on an orbital shaker at 4°C overnight. The
membranes were then incubated with horseradish
peroxidase-conjugated secondary antibodies (1:3,000; all from Santa
Cruz Biotechnology, Inc.) mouse anti-rabbit immunoglobulin (Ig) G
(cat. no. sc-2357), mouse-IgGκ light chain binding protein (cat.
no. sc-516102) and mouse anti-goat IgG (cat. no. sc-2354) for 2 h
at room temperature. Densitometric analysis of the immunoblots was
performed using a Fuji LAS 3000 imaging system and the Image Lab
Software (version 5.2.1, Bio-Rad Laboratories, Inc.).
Cell viability assay
A375 human melanoma cells (shp38α #1 and shp38β #2
stable clones; 2x104 cells/ml) were inoculated in
96-well plates in 200 µl of medium containing 10% FBS. After
12, 24, 36, 48 and 72 h, 20 µl of MTT solution (5 mg/ml) was
added to each well for 3 h. The MTT solution was removed, and
dimethyl sulfoxide (DMSO) was added. Cell viability was measured at
570 nm using a spectrophotometer.
Senescence-associated β-galactosidase
(SA-β-gal) staining
The assay was performed using the Senescence
β-Galactosidase Staining kit (cat. no. 9680; Cell Signaling
Technology, Inc.), according to the manufacturer's instructions
(29). SA-β-gal staining was
performed to determine the percentage of SA-Gal-positive cells.
A375 human melanoma cells (shp38α #1 and shp38β#2 stable clones;
1x105 cells/ml) were seeded on 6-well plates for 24 h.
The cells were then fixed with 4% para-formaldehyde at room
temperature for 30 min. After fixation, the cells were washed with
PBS three times and incubated with X-Gal at pH 6.0 overnight at
37°C. After washing, the cells were counted using bright-field
light microscopy (DP73; Olympus Corporation) at x100
magnification.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
A375 non-infected control, shp38α and shp38β cell
lines were collected by trypsinization. RNA was then extracted
using the Directzol™ RNA MiniPrep kit (Zymo Research Corporation).
RNA samples (1 µg) were reverse transcribed into cDNA. An
aliquot of RNA was incubated with 0.5 µg of oligo dT (MD
Bio.). Following incubation at 70°C for 15 min, 0.25 mM dNTPs (MD
Bio.), 20 U of RNasin I Plus RNase Inhibitor (Promega Corporation)
and 20 U of M-MLV Reverse Transcriptase (Promega Corporation) were
added and incubated at 42°C for 90 min for cDNA synthesis. Then, a
GeneAmp PCR system 2400 (PerkinElmer, Inc.) was used to amplify the
cDNA. qPCR analysis was performed using SYBR-Green I Master
Mix (Bio-Rad Laboratories, Inc.). The total reaction volume used
for PCR was 10 µl, and included 2 µl of cDNA, 5
µl of SYBR-Green, 0.5 µl of 10 µM forward
primer and reverse primer, and 2 µl of ddH2O. The
reactions were incubated in Applied Biosystems MicroAmp Optical
8-Tube Strips with 8-Cap Strips (0.1 ml) at 95°C for 10 min,
followed by 40 cycles of 95°C for 15 sec, 55°C for 15 sec and 72°C
for 30 sec, and all data were collected in triplicate. An Applied
Biosystems QuantStudio™ 3 Real-Time PCR System was used to detect
mRNA expression. All reactions were run in triplicate. The cycle
number at which the reaction crossed the threshold cycle (Cq) was
determined for each gene and the relative amount of each gene to
GAPDH was described using the equation 2ΔCq where
ΔCq=Cqinterest gene-CqGAPDH) (30). The primers were as follows: p38α,
forward ACC TGT CTC CAG TGG GCT CT and reverse CAC GTA ACC CCG TTT
TTG TG; p38β, forward CAT CTT CCG TGG AGC CAA C and reverse CAC TGT
CCA GCA CCA GCA T; and GAPDH, forward CCA GCC GAG CCA CAT CGC TC
and reverse ATG AGC CCC AGC CTT CTC CAT.
Migration assay
To determine the cell migration ability, A375 human
melanoma cells (shp38α #1 and shp38β #2 stable clones) were seeded
at a density of 3x104 cells/well in a 48-well Boyden
chamber (Neuro Probe Inc.) plate; the polycarbonate membrane
filters had 8 µm pores. The lower compartment was
supplemented with DMEM and 10% FBS. Cells were seeded in the upper
part of the Boyden chamber in serum-free medium and incubated for
24 h. Cotton swabs were used to remove the cells on the upper
surface (non-migrated cells). Next, the cells on the lower surface
of the membrane filters were fixed with methanol and stained with a
0.05% Giemsa solution for 1 h. The filters were then rinsed twice
with distilled water until no additional stain remained, and
air-dried for 20 min. The cells that migrated to the lower side of
the filter were observed under a light microscope at x100
magnification. Four fields were selected randomly and counted for
each filter. Three independent experiments were performed.
Cell cycle analysis
For flow cytometry analysis of cell cycle phase
distributions, cells were seeded at 80% confluency in 10 cm dishes,
and after 24 h, the cells were trypsinized and washed twice with
PBS. Next, 70% ethanol was used to fix the cells at 4°C for 1 h.
After that, the cells were stained with a propidium iodide
(Sigma-Aldrich; Merck KGaA) solution (20 µg/ml PI, 0.1%
Triton-X, 0.2 mg/ml RNase in ice-cold PBS) and incubated at room
temperature for 15 min. Data analysis was conducted using flow
cytometry (BD Biosciences). MODFIT™ LT 3.0 software (Verity
Software House, Inc.) was used to analyze the cell cycle
contribution. Three independent experiments were performed.
Wound healing assay
To examine whether p38α or p38β knockdown can reduce
the migration ability of A375 melanoma cells, A375 cells were
plated in a six-well plate and grown for 24 h. Cells were scratched
with a pipette tip and washed with PBS to remove the non-adhered
floating cells. Mitomycin C (5 µg/ml) was added to inhibit
cell proliferation. Cells were maintained in 10% FBS medium. The
scratches were photographed at 0 and 24 h, and the relative
migration was analyzed by ImageJ software with 64-bit Java
1.8.0_112 (National Institutes of Health).
Statistical analysis
Each experiment was repeated at least three times.
Statistical analyses were performed using GraphPad Prism version 5
(GraphPad Software, Inc.). The data were analyzed by one-way ANOVA.
Significance between the individual means was determined by Tukey's
test. Imaging results were quantified by ImageJ and processed with
Adobe Photoshop (Adobe Systems, Inc). P<0.05 was considered to
indicate a statistically significant difference.
Results
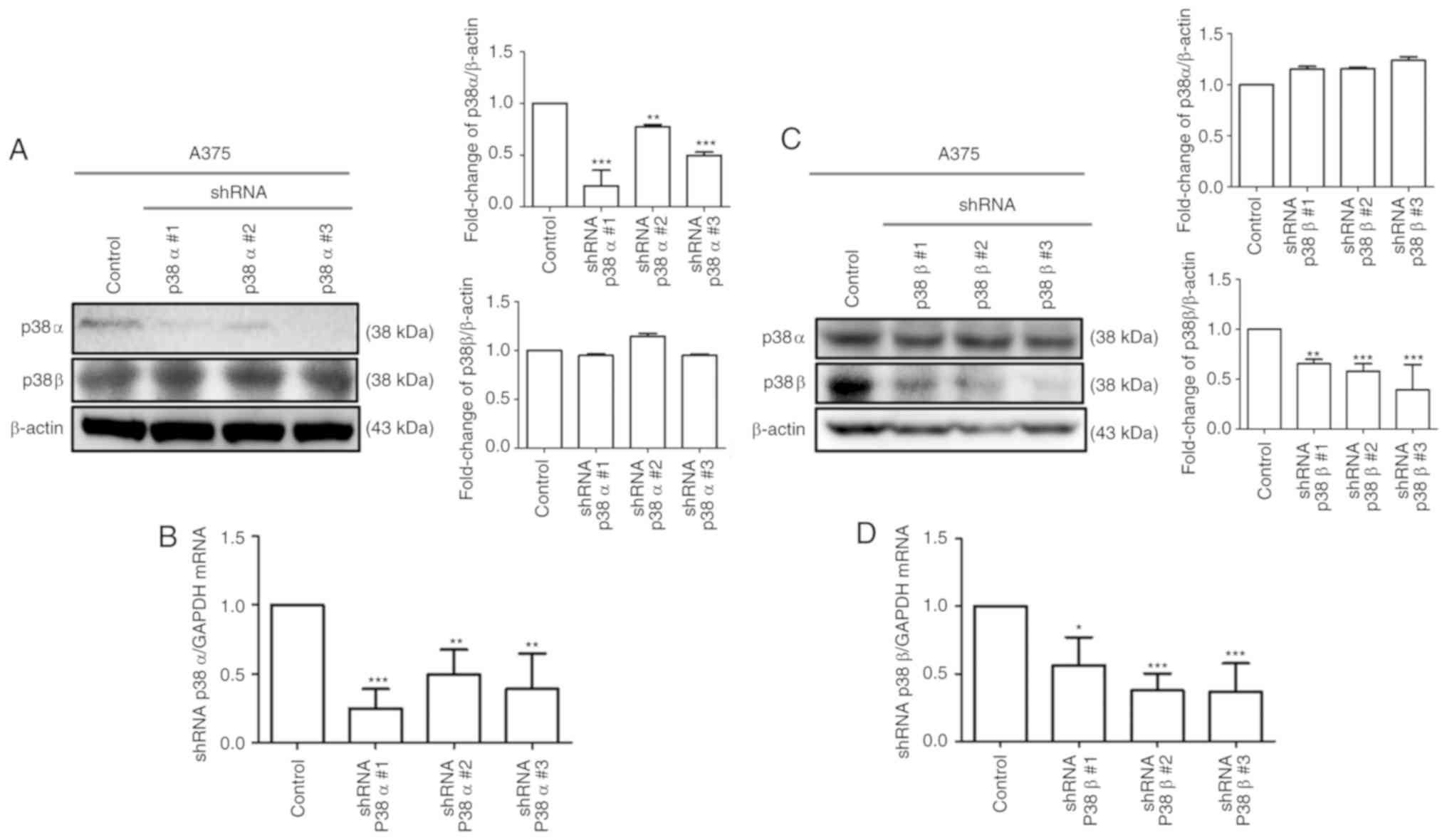
MAPK p38α and p38β silencing efficiency
in the A375 melanoma cell line
To determine the silencing efficiency, the mRNA and
protein expression levels of p38α and p38β were measured following
knockdown using shRNA (Fig. 1).
The silencing specificity of the shRNA against p38α and p38β was
evaluated by RT-qPCR to determine mRNA levels and by western
blotting to determine protein levels. As shown in Fig. 1A and C, p38α and p38β protein
levels were significantly decreased in p38α and p38β shRNA stable
clone cells. Similar results were obtained for the mRNA expression
levels (Fig. 1B and D). The
clonal lines shp38α #1 and shp38β#2 had the highest knockdown
efficiencies, and therefore these were selected for subsequent
experiments. A shRNA scrambled control was also used in A375
melanoma cells to confirm the transduction and knockdown efficiency
(Fig. S1). Notably, the results
demonstrated that the shRNA sequences were specific, with the p38α
shRNA not affecting the p38β mRNA and protein levels, and the p38β
shRNA not affecting the p38α levels Fig. 1).
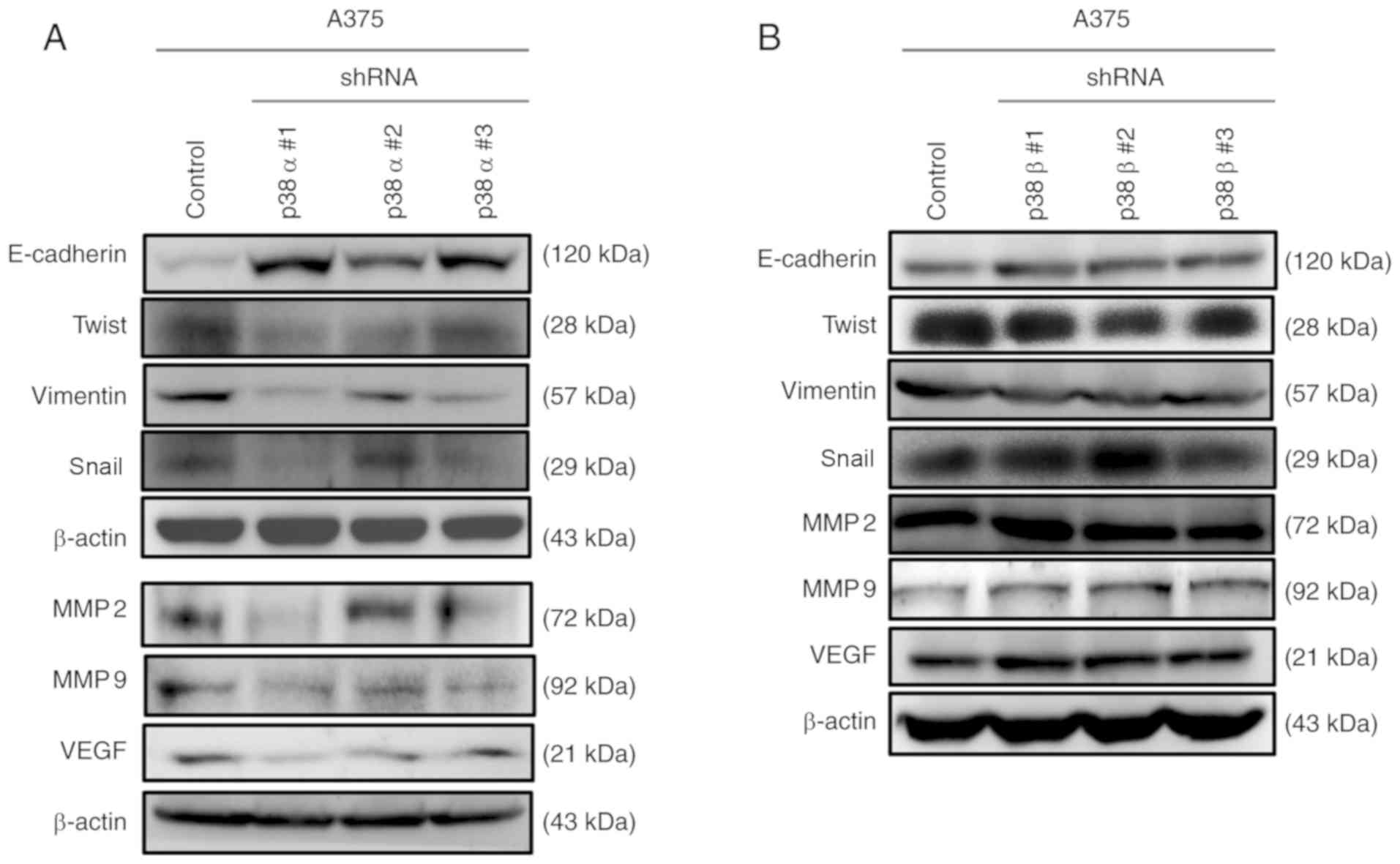
Knockdown of p38α, but not p38β, reverses
EMT and impairs VEGF expression in A375 melanoma cells
It has been reported that p38α regulates the
production of different extracellular signaling molecules, such as
growth factors, cytokines and chemokines. These signaling molecules
promote EMT and increase cell migration and invasion abilities
(17). To examine EMT and MET
markers, western blot analysis was performed. The results
demonstrated that the protein expression levels of the MET marker
E-cadherin were significantly increased after p38α silencing.
Furthermore, the protein levels of EMT markers, including Twist,
Snai1, vimentin, MMP2 and MMP9, were decreased after p38α
silencing. Of note, VEGF, which acts as an angiogenesis and
metastasis mediator, was downregulated after p38α shRNA
transduction (Fig. 2A). By
contrast, there were no obvious changes in MET, EMT or angiogenesis
markers in cells transduced with p38β shRNA compared with those in
control cells (Fig. 2B).
Furthermore, the EMT markers MMP2 and MMP9 were downregulated in
p38α, but not p38β, shRNA stable clone cells when compared with the
scrambled control in A375 melanoma cells (Fig. S1). Taken together, these results
demonstrated that the knockdown of p38α triggered a molecular
switch from a mesenchymal phenotype to an epithelial-like phenotype
in A375 melanoma cells. However, p38β was not involved in the
regulation of EMT and angiogenesis-related gene expression.
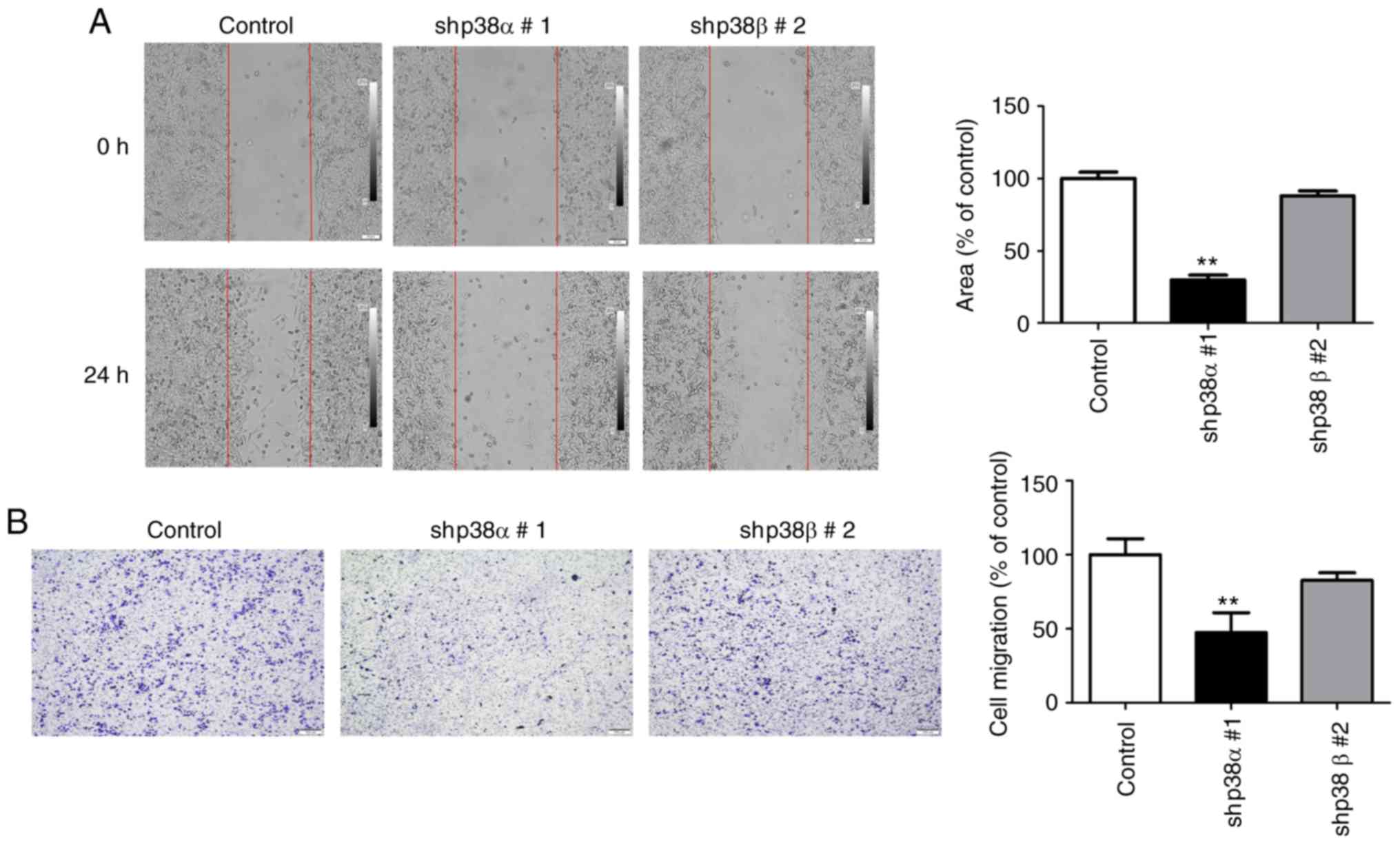
Effects of silencing p38α or p38β on A375
cell migration
p38α silencing has been shown to inhibit melanoma
cell motility under IL-6 stimulation (22,31); however, no study has investigated
the migration ability after p38α silencing and without cytokine
stimulation. The present study examined the migration ability of
p38α shRNA and p38β shRNA A375 stable clonal cell lines by wound
healing and Transwell assays. As presented in Fig. 3A and B, silencing p38α via shRNA
significantly reduced the migration ability of A375 cells. By
contrast, in the p38β shRNA stable clone, the migratory ability did
not change significantly compared with the control cells (Fig. 3A and B). To further confirm these
results, transient transfection was performed with p38α and
p38β-specific siRNAs into B16F0 melanoma cells, and the knockdown
efficiencies were analyzed by western blotting (Fig. S2A). Knockdown of p38α, but not
p38β, inhibited B16F0 melanoma cell migration (Fig. S2B). These findings indicated that
p38α, but not p38β, was crucial for the migration ability of A375
human melanoma cells.
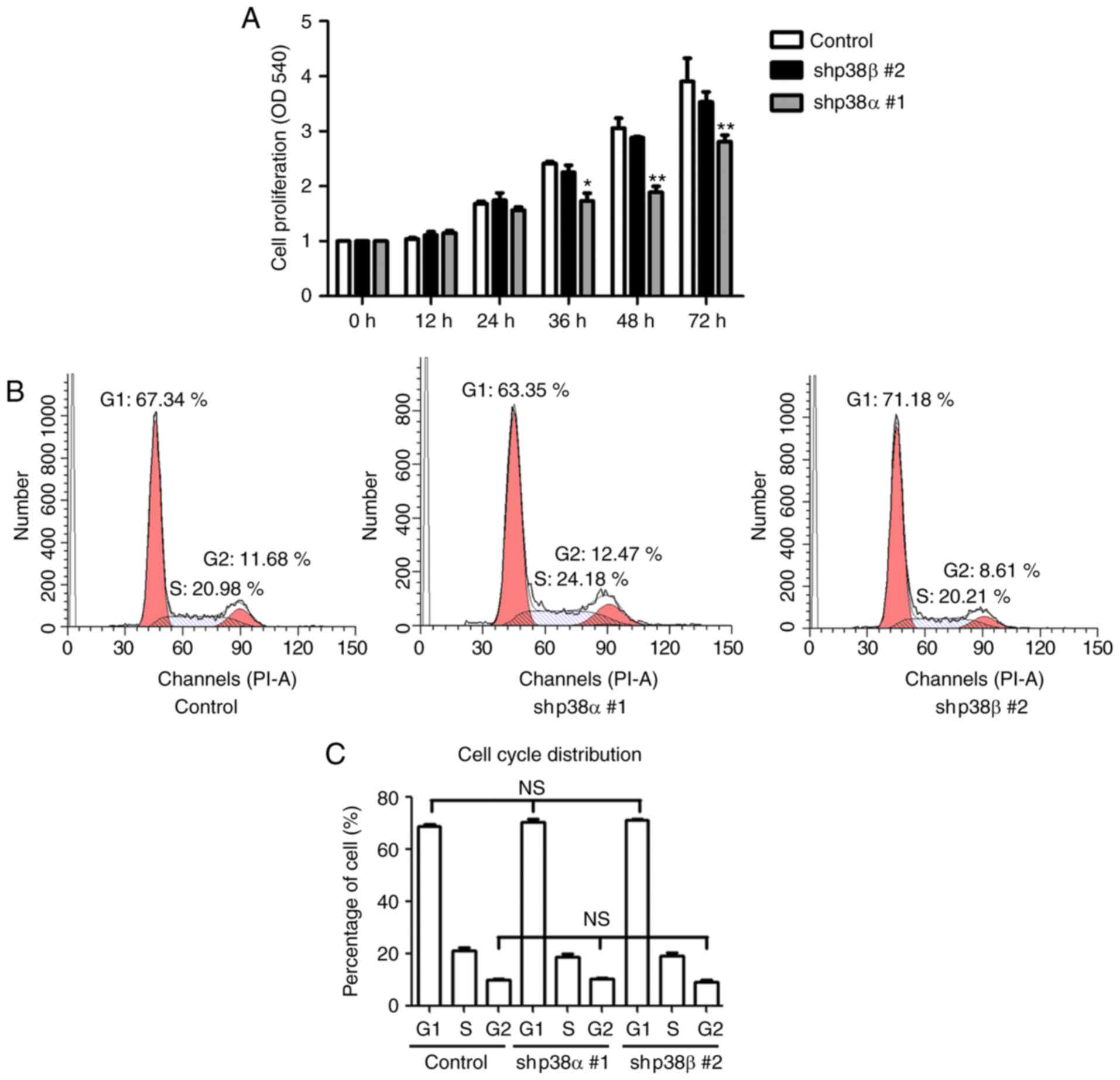
Knockdown of p38α inhibits cell
proliferation but not cell cycle progression
To analyze cell proliferation, an MTT assay was
performed. The results demonstrated that the p38α shRNA stable
clone had a lower proliferation rate compared with control cells
(Fig. 4A). By contrast, no
significant changes in the p38β shRNA stable clone proliferation
were observed (Fig. 4A). To
understand how p38α regulates cell growth, the cell cycle phase
distribution was analyzed by flow cytometry. There were no
significant changes in the cell cycle phase distribution between
control, p38α shRNA and p38β shRNA stable clones (Fig. 4B). Taken together, these data
suggested that knockdown of p38α partially reduced the
proliferation of A375 melanoma cells, while p38β was not involved
in regulating cell proliferation.
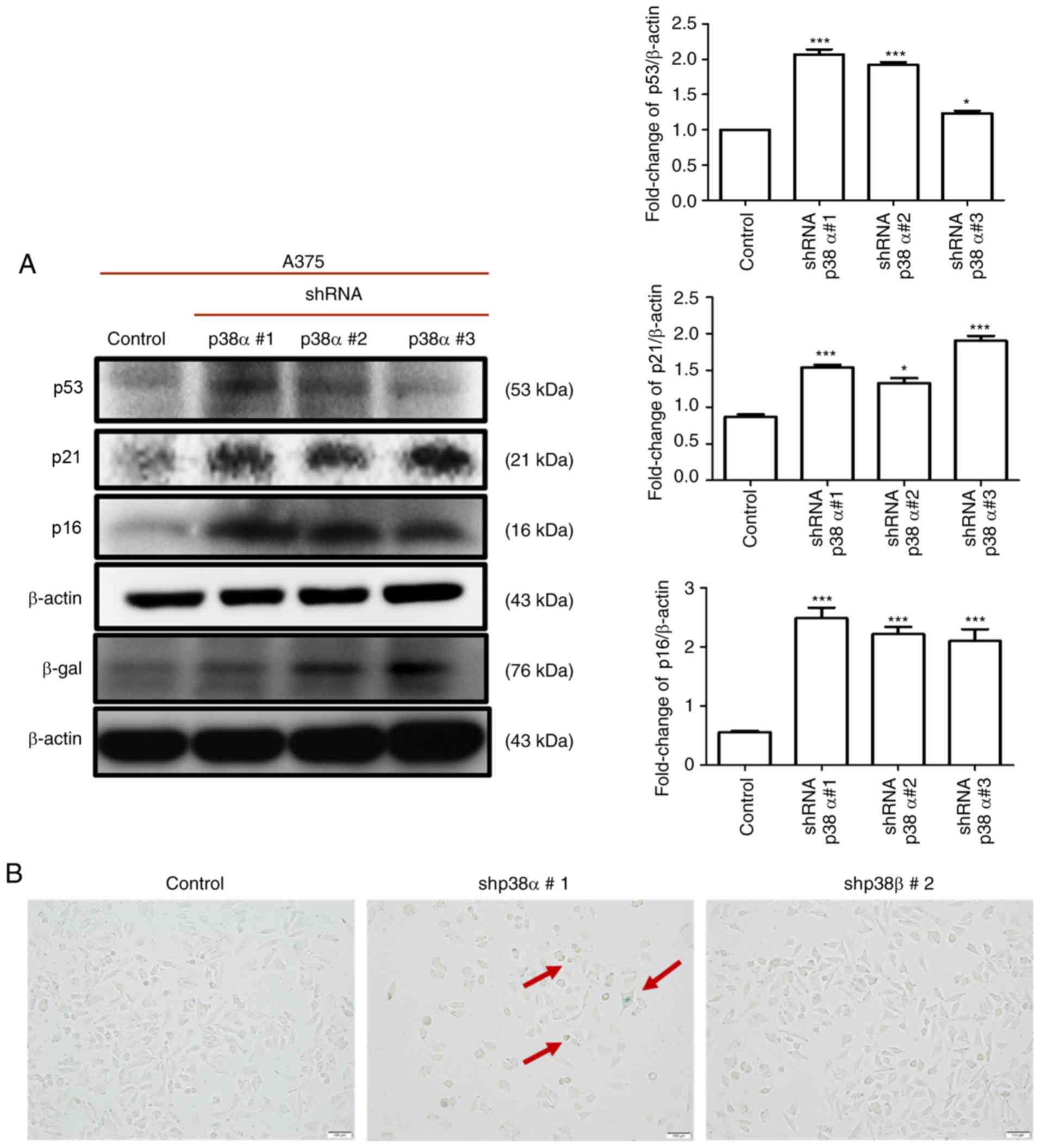
Knockdown of p38α induces senescence-like
features but not cell cycle arrest to reduce growth in A375
cells
To understand whether the knockdown of p38α or p38β
could induce cell senescence, a SA-β-gal assay was performed and
the protein expression of senescence-related markers p16, p21, p53
and β-gal was detected by western blotting. In the p38α shRNA
stable clones, a significant increase was observed in the numbers
of SA-β-gal-positive cells (Fig.
5B) and in the expression levels of p16, p21, p53 and β-gal
(Fig. 5A). By contrast, no
positive staining for SA-β-gal was observed in the p38β shRNA
stable clone (Fig. 5B). These
data suggested that p38α knockdown induced cellular senescence,
while p38β was not involved in regulating cell senescence.
Discussion
The current study revealed that, between p38α and
p38β, only p38α was significantly associated with EMT marker and
VEGF expression, cellular migration and proliferation in A375
melanoma cells. This finding is important because high p38α
expression is associated with cancer development in melanoma
(32). Therefore, targeting p38α
may be a potential treatment approach for patients with
melanoma.
While there have been reports about the
contradictory roles of p38 MAPK in cancers, the significance of the
p38 MAPK signaling pathway in regulating various cancers is widely
recognized (5,33-35). In cancer cells, the activation of
p38 MAPK leads to growth inhibition, but it stimulates cell growth
after activation in some other types of cancer (36,37). Furthermore, p38 MAPK activation
has also been shown to be associated with metastasis and
tumorigenesis. Conversely, several studies have reported that p38
MAPK may be a negative regulator of metastasis and tumorigenesis
(38-40). In addition, p38 MAPK has opposite
roles in regulating cell death, and it can mediate either cell
survival or cell death depending on, not only the type of stimulus,
but also the type of cell. The important question that remains to
be answered is, therefore, whether the p38 MAPK isoforms have a
pro-oncogenic or tumor-suppressive role in different cancer cells.
To this end, the present study aimed to identify the specific roles
of p38α and p38β in melanoma.
A crucial step in melanoma progression is EMT, a
process that regulates melanoma migration, invasion and metastasis
(41,42). The present study demonstrated that
p38α regulated melanoma EMT. Previous studies have reported that
high expression of p38α in melanoma enhances metastasis (22,23); the current findings, therefore,
together with previous literature, support a tumor-promoting role
of p38α in melanoma. However, contradictory results have been
reported in other types of cancer, such as breast cancer (9,43,44), lung cancer (11,45) and liver cancer (46,47), where p38α has been found to act as
a tumor suppressor in the initial step of tumor formation and to
act as a tumor promoter in the later stages of cancer to enhance
metastasis. The present study demonstrated that p38α, but not p38β,
regulated melanoma migration by upregulating the protein expression
of EMT markers, including Twist, Snail, vimentin, MMP2 and MMP9,
and by downregulating the protein expression of the MET marker
E-cadherin. Furthermore, the results of wound healing and Transwell
assays showed that the migration ability was impaired in shp38α,
but not in shp38β, stable clones. Taken together, these results
indicated that p38α enhanced melanoma EMT and migration
ability.
Angiogenesis contributes to providing adequate blood
supply to the tumor and subsequently enhances tumor growth and
progression (48). VEGF is a
pro-angiogenic stimulator that can bind specifically to different
receptor tyrosine kinases, such as VEGFR1/2/3, and send angiogenic
signals (49). A previous study
has shown that p38 activation can enhance endothelial cell
migration and tumor formation (50). However, the precise effect of
different p38 isoforms on VEGF expression in melanoma remained
unknown to date. The present study demonstrated that the knockdown
of p38α, but not p38β, decreased the expression levels of VEGF. In
addition to promoting angiogenesis, p38α has been shown to
participate in regulating pro-inflammatory cytokines and chemokines
(22,31). Whether p38α enhances angiogenesis
by upregulating pro-inflammatory cytokines or chemokines requires
further study.
Uncontrolled proliferation is a characteristic of
cancer (51). Therefore, cell
cycle arrest or cellular senescence, which are considered a barrier
to tumorigenesis, may inhibit proliferation (52,53). Having discovered that silencing
p38α, but not p38β, reduced cell proliferation, it was hypothesized
that the inhibition of p38α or p38β could affect the cell cycle. To
examine this hypothesis, flow cytometry was used for cell cycle
phase distribution analysis. However, the results demonstrated that
neither the shp38α nor the shp38β stable clonal cells had any
changes in their percentages of cells in the G1/S and G2/M phases,
indicating that cell cycle arrest was not the primary mechanism of
growth reduction caused by p38α knockdown. Thus, other mechanisms,
that may be involved in proliferation reduction, need to be
examined. In a previous study, overexpres-sion of p16 and p14,
which are markers of senescence, inhibited melanoma A375 cell
proliferation, migration and invasion, and promoted apoptosis
(54). Therefore, it was
hypothesized that senescence activation in shp38α stable clonal
cells may be another mechanism that caused proliferation and EMT
inhibition. The present results demonstrated that p38α knockdown,
but not p38β, induced cellular senescence. Together, these findings
suggested that p38α served a crucial role in regulating cell
proliferation and cellular senescence.
In conclusion, when comparing the roles of p38α and
p38β, only p38α was identified to be significantly associated with
regulating EMT and senescence in melanoma cells. In the present
in vitro study, shRNA was used to specifically knockdown
p38α or p38β in the A375 melanoma cell line and the results
revealed that only p38α was a crucial factor in regulating cell
proliferation and migration, suggesting that p38α may have an
oncogenic-maintaining role. The present study highlighted the
distinct and often opposing functions of the individual p38 MAPK
isoforms in melanoma. These novel findings indicated that targeting
p38α may provide a potential strategy in treating melanoma.
Supplementary Data
Acknowledgements
Not applicable.
Funding
This study was supported by China Medical University
Hospital (grant no. DMR-108-137).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
SYW and SCN conceived and designed the study. CJC,
CYH and WWK performed the experiments. SCN wrote the manuscript.
All authors have read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy and integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fogel AL, Jaju PD, Li S, Halpern-Felsher
B, Tang JY and Sarin KY: Factors influencing and modifying the
decision to pursue genetic testing for skin cancer risk. J Am Acad
Dermatol. 76:829–835. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bradford PT: Skin cancer in skin of color.
Dermatol Nurs. 21:170–177. 2009.PubMed/NCBI
|
|
4
|
Koul HK, Pal M and Koul S: Role of p38 MAP
kinase signal transduction in solid tumors. Genes Cancer.
4:342–359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cuenda A and Rousseau S: P38 MAP-kinases
pathway regulation, function and role in human diseases. Biochim
Biophys Acta. 1773:1358–1375. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cuadrado A and Nebreda AR: Mechanisms and
functions of p38 MAPK signalling. Biochem J. 429:403–417. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Patel SB, Cameron PM, O'Keefe SJ,
Frantz-Wattley B, Thompson J, O'Neill EA, Tennis T, Liu L, Becker
JW and Scapin G: The three-dimensional structure of MAP kinase
p38beta: Different features of the ATP-binding site in p38beta
compared with p38alpha. Acta Crystallogr D Biol Crystallogr.
65:777–785. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Katz M, Amit I and Yarden Y: Regulation of
MAPKs by growth factors and receptor tyrosine kinases. Biochim
Biophys Acta. 1773:1161–1176. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bulavin DV and Fornace AJ Jr: P38 MAP
kinase's emerging role as a tumor suppressor. Adv Cancer Res.
92:95–118. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Igea A and Nebreda AR: The stress kinase
p38α as a target for cancer therapy. Cancer Res. 75:3997–4002.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gupta J, del Barco Barrantes I, Igea A,
Sakellariou S, Pateras IS, Gorgoulis VG and Nebreda AR: Dual
function of p38α MAPK in colon cancer: Suppression of
colitis-associated tumor initiation but requirement for cancer cell
survival. Cancer Cell. 25:484–500. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chiacchiera F, Matrone A, Ferrari E,
Ingravallo G, Lo Sasso G, Murzilli S, Petruzzelli M, Salvatore L,
Moschetta A and Simone C: P38alpha blockade inhibits colorectal
cancer growth in vivo by inducing a switch from HIF1alpha- to
FoxO-dependent transcription. Cell Death Differ. 16:1203–1214.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kalluri R and Neilson EG:
Epithelial-mesenchymal transition and its implications for
fibrosis. J Clin Invest. 112:1776–1784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hay ED: An overview of
epithelio-mesenchymal transformation. Acta Anat (Basel). 154:8–20.
1995. View Article : Google Scholar
|
|
17
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Voutsadakis IA: The network of
pluripotency, epithelial-mesenchymal transition, and prognosis of
breast cancer. Breast Cancer (Dove Med Press). 7:303–319. 2015.
|
|
20
|
Liu CY, Lin HH, Tang MJ and Wang YK:
Vimentin contributes to epithelial-mesenchymal transition cancer
cell mechanics by mediating cytoskeletal organization and focal
adhesion maturation. Oncotarget. 6:15966–15983. 2015.PubMed/NCBI
|
|
21
|
Bauvois B: New facets of matrix
metalloproteinases MMP-2 and MMP-9 as cell surface transducers:
Outside-in signaling and relationship to tumor progression. Biochim
Biophys Acta. 1825:29–36. 2012.
|
|
22
|
Linnskog R, Jonsson G, Axelsson L, Prasad
CP and Andersson T: Interleukin-6 drives melanoma cell motility
through p38α-MAPK-dependent up-regulation of WNT5A expression. Mol
Oncol. 8:1365–1378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yan Q, Bach DQ, Gatla N, Sun P, Liu JW, Lu
JY, Paller AS and Wang XQ: Deacetylated GM3 promotes
uPAR-associated membrane molecular complex to activate p38 MAPK in
metastatic melanoma. Mol Cancer Res. 11:665–675. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
del Barco Barrantes I and Nebreda AR:
Roles of p38 MAPKs in invasion and metastasis. Biochem Soc Trans.
40:79–84. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee WH, Liu FH, Lee YL and Huang HM:
Interferon-alpha induces the growth inhibition of human T-cell
leukaemia line jurkat through p38alpha and p38beta. J Biochem.
147:645–650. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hale KK, Trollinger D, Rihanek M and
Manthey CL: Differential expression and activation of p38
mitogen-activated protein kinase alpha, beta, gamma, and delta in
inflammatory cell lineages. J Immunol. 162:4246–4252.
1999.PubMed/NCBI
|
|
27
|
Kuma Y, Campbell DG and Cuenda A:
Identification of glycogen synthase as a new substrate for
stress-activated protein kinase 2b/p38beta. Biochem J. 379:133–139.
2004. View Article : Google Scholar
|
|
28
|
Lee MR, Lin C, Lu CC, Kuo SC, Tsao JW,
Juan YN, Chiu HY, Lee FY, Yang JS and Tsai FJ: YC-1 induces G0/G1
phase arrest and mitochondria-dependent apoptosis in
cisplatin-resistant human oral cancer CAR cells. Biomedicine
(Taipei). 7:122017. View Article : Google Scholar
|
|
29
|
Su D, Zhu S, Han X, Feng Y, Huang H, Ren
G, Pan L, Zhang Y, Lu J and Huang B: BMP4-Smad signaling pathway
mediates adriamycin-induced premature senescence in lung cancer
cells. J Biol Chem. 284:12153–12164. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Piazza VG, Bartke A, Miquet JG and Sotelo
AI: Analysis of different approaches for the selection of reference
genes in RT-qPCR experiments: A case study in skeletal muscle of
growing mice. Int J Mol Sci. 18:E10602017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Linnskog R, Mohapatra P, Moradi F, Prasad
CP and Andersson T: Demonstration of a WNT5A-IL-6 positive feedback
loop in melanoma cells: Dual interference of this loop more
effectively impairs melanoma cell invasion. Oncotarget.
7:37790–37802. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu K, Yu D, Cho YY, Bode AM, Ma W, Yao K,
Li S, Li J, Bowden GT and Dong Z and Dong Z: Sunlight UV-induced
skin cancer relies upon activation of the p38α signaling pathway.
Cancer Res. 73:2181–2188. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu H, He J and Yang J: Tumor cell p38
MAPK: A trigger of cancer bone osteolysis. Cancer Cell
Microenviron. 2:e4642015.PubMed/NCBI
|
|
34
|
Gonzalez-Villasana V, Fuentes-Mattei E,
Ivan C, Dalton HJ, Rodriguez-Aguayo C, Fernandez-de Thomas RJ,
Aslan B, Del C, Monroig P, Velazquez-Torres G, Previs RA, et al:
Rac1/Pak1/p38/MMP-2 axis regulates angiogenesis in ovarian cancer.
Clin Cancer Res. 21:2127–2137. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Loesch M and Chen G: The p38 MAPK stress
pathway as a tumor suppressor or more? Front Biosci. 13:3581–3593.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ding XZ and Adrian TE: MEK/ERK-mediated
proliferation is negatively regulated by P38 map kinase in the
human pancreatic cancer cell line, PANC-1. Biochem Biophys Res
Commun. 282:447–453. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cocolakis E, Lemay S, Ali S and Lebrun JJ:
The p38 MAPK pathway is required for cell growth inhibition of
human breast cancer cells in response to activin. J Biol Chem.
276:18430–18436. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Grossi V, Peserico A, Tezil T and Simone
C: p38α MAPK pathway: A key factor in colorectal cancer therapy and
chemoresistance. World J Gastroenterol. 20:9744–9758. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tai TW, Su FC, Chen CY, Jou IM and Lin CF:
Activation of p38 MAPK-regulated Bcl-xL signaling increases
survival against zoledronic acid-induced apoptosis in osteoclast
precursors. Bone. 67:166–174. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dreissigacker U, Mueller MS, Unger M,
Siegert P, Genze F, Gierschik P and Giehl K: Oncogenic K-Ras
down-regulates Rac1 and RhoA activity and enhances migration and
invasion of pancreatic carcinoma cells through activation of p38.
Cell Signal. 18:1156–1168. 2006. View Article : Google Scholar
|
|
41
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nakamura M and Tokura Y:
Epithelial-mesenchymal transition in the skin. J Dermatol Sci.
61:7–13. 2011. View Article : Google Scholar
|
|
43
|
Campbell RM, Anderson BD, Brooks NA,
Brooks HB, Chan EM, De Dios A, Gilmour R, Graff JR, Jambrina E,
Mader M, et al: Characterization of LY2228820 dimesylate, a potent
and selective inhibitor of p38 MAPK with antitumor activity. Mol
Cancer Ther. 13:364–374. 2014. View Article : Google Scholar
|
|
44
|
Pereira L, Igea A, Canovas B, Dolado I and
Nebreda AR: Inhibition of p38 MAPK sensitizes tumour cells to
cisplatin-induced apoptosis mediated by reactive oxygen species and
JNK. EMBO Mol Med. 5:1759–1774. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ventura JJ, Tenbaum S, Perdiguero E, Huth
M, Guerra C, Barbacid M, Pasparakis M and Nebreda AR: p38alpha MAP
kinase is essential in lung stem and progenitor cell proliferation
and differentiation. Nat Genet. 39:750–758. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hui L, Bakiri L, Mairhorfer A, Schweifer
N, Haslinger C, Kenner L, Komnenovic V, Scheuch H, Beug H and
Wagner EF: p38alpha suppresses normal and cancer cell proliferation
by antagonizing the JNK-c-Jun pathway. Nat Genet. 39:741–749. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sakurai T, He G, Matsuzawa A, Yu GY, Maeda
S, Hardiman G and Karin M: Hepatocyte necrosis induced by oxidative
stress and IL-1 alpha release mediate carcinogen-induced
compensatory proliferation and liver tumorigenesis. Cancer Cell.
14:156–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rajabi M and Mousa SA: The role of
angiogenesis in cancer treatment. Biomedicines. 5:E342017.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mehrad B, Keane MP and Strieter RM:
Chemokines as mediators of angiogenesis. Thromb Haemost.
97:755–762. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yoshizuka N, Chen RM, Xu Z, Liao R, Hong
L, Hu WY, Yu G, Han J, Chen L and Sun P: A novel function of
p38-regulated/activated kinase in endothelial cell migration and
tumor angiogenesis. Mol Cell Biol. 32:606–618. 2012. View Article : Google Scholar :
|
|
51
|
Fouad YA and Aanei C: Revisiting the
hallmarks of cancer. Am J Cancer Res. 7:1016–1036. 2017.PubMed/NCBI
|
|
52
|
Cairney CJ, Bilsland AE, Evans TR, Roffey
J, Bennett DC, Narita M, Torrance CJ and Keith WN: Cancer cell
senescence: A new frontier in drug development. Drug Discov Today.
17:269–276. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Prieur A and Peeper DS: Cellular
senescence in vivo: A barrier to tumorigenesis. Curr Opin Cell
Biol. 20:150–155. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bai M, Yu NZ, Long F, Feng C and Wang XJ:
Effects of CDKN2A (p16INK4A/p14ARF) over-expression on
proliferation and migration of human melanoma A375 Cells. Cell
Physiol Biochem. 40:1367–1376. 2016. View Article : Google Scholar : PubMed/NCBI
|