Introduction
Colorectal cancer (CRC) is one of the most common
malignant tumors worldwide, with the second highest incidence in
men, the third in women, and the third and fourth in cancer-related
deaths among men and women (1).
Despite recent improvements in screening strategies and the
development of more effective CRC treatments, the prognosis for
advanced CRC remains poor (2).
Many risk factors, including viral and bacterial infections,
alcohol, tobacco smoke, aging, ulcerative colitis, and a sedentary
lifestyle, as well as genetic mutations may be associated with CRC
(3). There are three types of
genetic aberrations, namely chromosomal instability (CIN),
microsatellite instability (MSI), and CpG island methylator
phenotype (CIMP), involved in the pathogenesis of CRC (4).
Myeloid differentiation factor 88 (MyD88) is
an essential adaptor molecule for IL-1 and Toll-like
receptor (TLR) signaling (5). TLRs are a family of pattern
recognition receptors that identify various pathogen-associated
molecular patterns (PAMPs), molecules derived from various
pathogens, and activate host innate immune defense against pathogen
invasion. MyD88 signaling plays a predominant role in
mediating systemic and cardiac cytokine responses in the survival
of activated CD4+ T cells to promote tumor cell
proliferation, invasion, metastasis and are correlated with the
prognosis of HCC patient-mediated inflammatory pathway injury and
neurodegenerative tissue injury (6-9).
MyD88 expression is an adverse prognostic factor in ovarian
cancer and is essential in adenovirus keratitis (10,11). MyD88 is a therapeutic
target for inflammatory lung diseases (12). It also contributes to ocular
surface homeostasis (13).
However, the role of MyD88 in CRC and the mode of action
following its expression remain unknown.
Previous findings showed MyD88 expression in
cancer tissue and adjacent normal colorectal tissues of patients
with CRC; however, the expression levels were significantly higher
in the cancer tissues than in the adjacent tissues (14). The MyD88 expression level
was correlated with the clinical stage, T stage, M stage and lymph
node metastasis, and the survival rate of patients with CRC and
higher MyD88 expression was significantly lower than that of
the patients with CRC and lower MyD88 expression.
The aim of the present study was to determine the
role of MyD88 in CRC. The MyD88 gene was knocked down
to dissect its functional role in CRC cells. In addition, the
mechanism of MyD88 knockdown, which causes change in the
related signal pathway, was explored. The findings showed that
MyD88 is a crucial factor affecting CRC progression.
Materials and methods
MyD88 siRNA synthesis and
transfection
siRNA target sequences were identified on the human
MyD88 sequence. According to the siRNA design guidelines,
DNA template oligonucleotides corresponding to three different
siRNA sequences (siRNA-1, siRNA-2 and siRNA-3) were designed as
follows: siRNA-1: GCC TAT CGC TGT TCT TGA A, siRNA-2: GAC TGA TTC
CTA TTA AAT A, siRNA-3: CAGCGAGCTAATTGAGAAA. These siRNA and
negative control (NC) sequences were produced by Genechem Co. Ltd..
The SW480 and HCT116 cells were cultured in a medium with 10% FBS.
When these CRC cells were at approximately 90% confluency, the NC,
siRNA-1, siRNA-2 and siRNA-3 sequences were transfected into the
cells, using Lipofectamine 3000 (Invitrogen), according to the
manufacturer's instructions.
RNA preparation and quantitative PCR amplification.
RT-qPCR was used to test mRNA expression of the MyD88 gene.
Total RNA was extracted from CRC cell lines, using the Qiagen
RNeasy kit (Qiagen Bioinformatics) according to the manufacturer's
instructions, and quantified using UV260/280 nm to an absorption
ratio of z1.8. An RT Reagent kit (Takara Bio Lnc.) was used for
reverse transcription of RNA into cDNA. The primers (BioSune
Biotechnology Co., Ltd.) MyD88 F: GGC TGC TCT CAA CAT GCG A,
R: CTG TGT CCG CAC GTT CAA GA and GAPDH F: GAA GGT GAA GGT
CGG AGT C, R: GAA GAT GGT GAT GGG ATT TC, were designed to amplify
cDNA with SYBR Premix EX Taq kit (Takara Bio Lnc.). PCR conditions
were 95°C for 2 min, 95°C for 15 sec, and 60°C for 30 sec for 40
cycles. The relative amount of MyD88 mRNA was normalized to that of
GAPDH. The 2−ΔΔCq method was used to calculate
this as Livak and Schmittgen had reported (15).
Construction of lentiviral vectors
In order to establish better stable transfection for
later experiments, we constructed lentiviral-based siRNA targeting
MyD88 vector. The primers, designed by BioSune Biotechnology
Co., Ltd. contained two restriction sites, AgeI (underlined)
and EcoRI (in bold) (siRNA1 F: CCGGGC CTA TCG CTG TTC TTG AAT
TCA AGA GAT TCAA GAA CAG CGA TAG GCT TTT TTG GTA CC, R:
AATTGG TAC CAA AAA AGC CTA TCG CTG TTC TTG AAT CTC TTG AAT
TCA AGA ACA GCG ATA GGC; siRNA2 F: CCGGGA CTG ATT CCT ATT AAA TAT
TCA AGA GAT ATT TAA TAG GAA TCA GTC TTT TTT GGT ACC, R: AAT
TGG TAC CAA AAA AGA CTG ATT CCT ATT AAA TAT CTC TTG AAT ATT TAA
TAG GAA TCA GTC; and siRNA3 F: CCGGCA GCG AGC TAA TTG AGA AAT
TCA AGA GAT TTC TCA ATT AGC TCG CTG TTT TTT GGT ACC, R: AAT
TGG TAC CAA AAA ACA GCG AGC TAA TTG AGA AAT CTC TTG AAT TTC TCA
ATT AGC TCG CTG). The primers were designed to anneal and form
double-stranded DNA. pLKO.1-EGFP-Puro (Fenghui Bio) was used as an
empty vector for purification and recovery (Tiangen) after double
digestion with AgeI and EcoRI (Thermo Fisher
Scientific, Inc.). The purified linear empty vector and the
double-stranded DNA formed by annealing were then ligated with T4
DNA Ligase (Invitrogen) and transformed, using stabl3, cloned,
sequenced, and then subjected to plasmid extraction (Tiangen) for
the next experiment.
Lentiviral infection and stable cell line
screening
The SW480 and HCT116 cell lines were obtained from
the cell bank of the Chinese Academy of Sciences (Shanghai, China).
The two cell lines were cultured and maintained in DMEM (Gibco BRL)
with 10% fetal bovine serum (FBS), and incubated at 37°C and 5%
CO2. The validated MyD88 knockdown plasmid and
empty vector were co-transfected with pMD2.G and psPAX2 (Fenghui
Bio) into 293T cells, and the supernatant was filtered after 2 days
of culture. HCT116 and SW480 cells were infected with the
supernatant, and after 2 weeks of screening with puromycin, the
protein was extracted for verification. Stably expressed cells were
selected for subsequent experiments.
Western blot analysis
Western and IP cell lysis buffer (Beyotime),
containing 1% PMSF (Amresco), was used to cleave proteins for 30
min on ice. After 30 min, the lysed cells were centrifuged at
12,000 × g for 10 min at 4°C to extract the supernatant for protein
quantification using the BCA Protein Assay kit (Thermo Fisher
Scientific, Inc.), and boiled for 10 min by adding 5X SDS. The
total amount of protein (50 µg) was added to the prepared
12% SDS-PAGE gels for electrophoretic separation and transferred to
0.45 µm PVDF membranes (Amersham Hybond, GE Healthcare). The
membranes were then blocked with 1% albumin from bovine serum
(Amresco) for 2 h. Next, the membranes were incubated overnight
with diluted MyD88 (1:1,000, AF5195; Affinity), GAPDH
(1:1,000, ab181602; Abcam), NF-κB p65 (1:1,000, AF5006;
Affinity), p-NF-κB p65 (1:1,000, #3033; Cell Signaling),
c-jun (1:1,000, bs-0670R; Bioss), and p-c-jun
(1:1,000, bs-3172R; Bioss) antibodies on a shaker at 4°C. The
membranes were washed for 10 min three times with TBS-T (0.1%
Tween-20) at room temperature, incubated in goat anti-rabbit IgG
H&L (HRP) (1:2,000, ab7090; Abcam) for 1 h, and then washed.
Subsequently the membranes were exposed to enhanced
chemiluminescence substrate detection solution (Lulong
Biotech).
Colony formation assay
Cells (500/well) were seeded in a 6-cm plate, and
cultured for 14 days in a culture medium, containing 10% FBS. The
culture solution was discarded and cells were infiltrated with
methanol for 10 min, stained with crystal violet, washed with
water, dried and counted.
Cell proliferation assay
CCK-8 (Dojindo) was used to detect cell
proliferation. Cells were seeded, at densities of 1,000 cells/well,
in a 96-well plate, then incubated at 37°C, 5% CO2.
After 24, 48, 72, and 96 h, the culture solution was discarded and
100 µl of 10% CCK-8 serum-free medium was added. Cell
proliferation was estimated using a microplate reader (Bio-Tek)
after 1 h of incubation.
Cell migration and invasion assay
In the migration experiment, 4×104 CRC
cells, in serum-free medium, were seeded into the upper chamber of
a Transwell insert (8-mm pore size; Corning Inc.), and a medium
with 20% FBS was added in the lower chamber as a chemoattractant.
In the invasion experiment, Matrigel (BD Bioscience) was coated on
the upper chamber seeded with 9×104 cells, and the lower
chamber contained 20% FBS medium. After incubation at 37°C, 5%
CO2 for 48 h, the Transwell chamber was taken out and
the medium in the well was discarded and washed with calcium-free
PBS. The cells were, then, fixed with methanol for 30 min and
stained with 0.1% crystal violet for 20 min. The upper unmigrated
cells were gently wiped off with a cotton swab, and counted under
microscope.
Wound healing assay for CRC cell
migration
The cells were seeded in a 6-well plate. After the
cells had grown to 100% confluency, the cell layer was scratched
with a 20 µl pipette tip and the medium containing 10% FBS
was replaced with a serum-free medium. Images of the cells were
captured at 0, 24, 48, and 72 h.
Animal experiments
The experiment performed with animals was approved
by the Institutional Animal Care and Use Committee at the Fujian
Medical University. Ten five-week-old BALB/c nude mice raised at
the Animal Experimental Center of Fujian Medical University were
used in this study. HCT116-pLKO.1 or HCT116-pLKO.1-sh3 cells
(2×106), in serum-free DMEM medium, were injected into
the bilateral subcutaneous (HCT116-pLKO.1 cells on one side and
HCT116-pLKO.1-sh3 cells on the other side) of 10 mice. The
manipulation was stable, rapid and gentle to reduce discomfort in
mice. During tumor growth, the tumor volume was measured every four
days using the formula: length x width 2/2. and the
correlation function graph was plotted. After 6 weeks of tumor
growth the nude mice were euthanized by carbon dioxide (100%
CO2 gas replacement rate at 10-30% container
volume/min), and the tumors were removed after the heartbeat and
respiratory arrest of nude mice, photographed and immediately
weighed. The tumors were soaked in formalin for immunohistochemical
analysis.
Immunohistochemistry
Immunohistochemistry was performed on
formalin-fixed, paraffin-embedded tumor tissue specimens from the
mice. After dewaxing, hydration, and antigen retrieval, the
remaining experimental procedures were performed in accordance with
the UltraSensitiveTM SP (mouse/rabbit) IHC kit
instructions. Finally, the degree of staining of the slices was
observed under the microscope after DAB staining, hematoxylin
counterstaining and neutral resin sealing.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 5 software. The data were expressed as the means ± standard
deviation (SD) and analyzed by one-way ANOVA or Student's t-test.
Differences with P-values <0.05 were considered to be
statistically significant.
Results
Expression of MyD88 mRNA and protein
after siRNA transfection
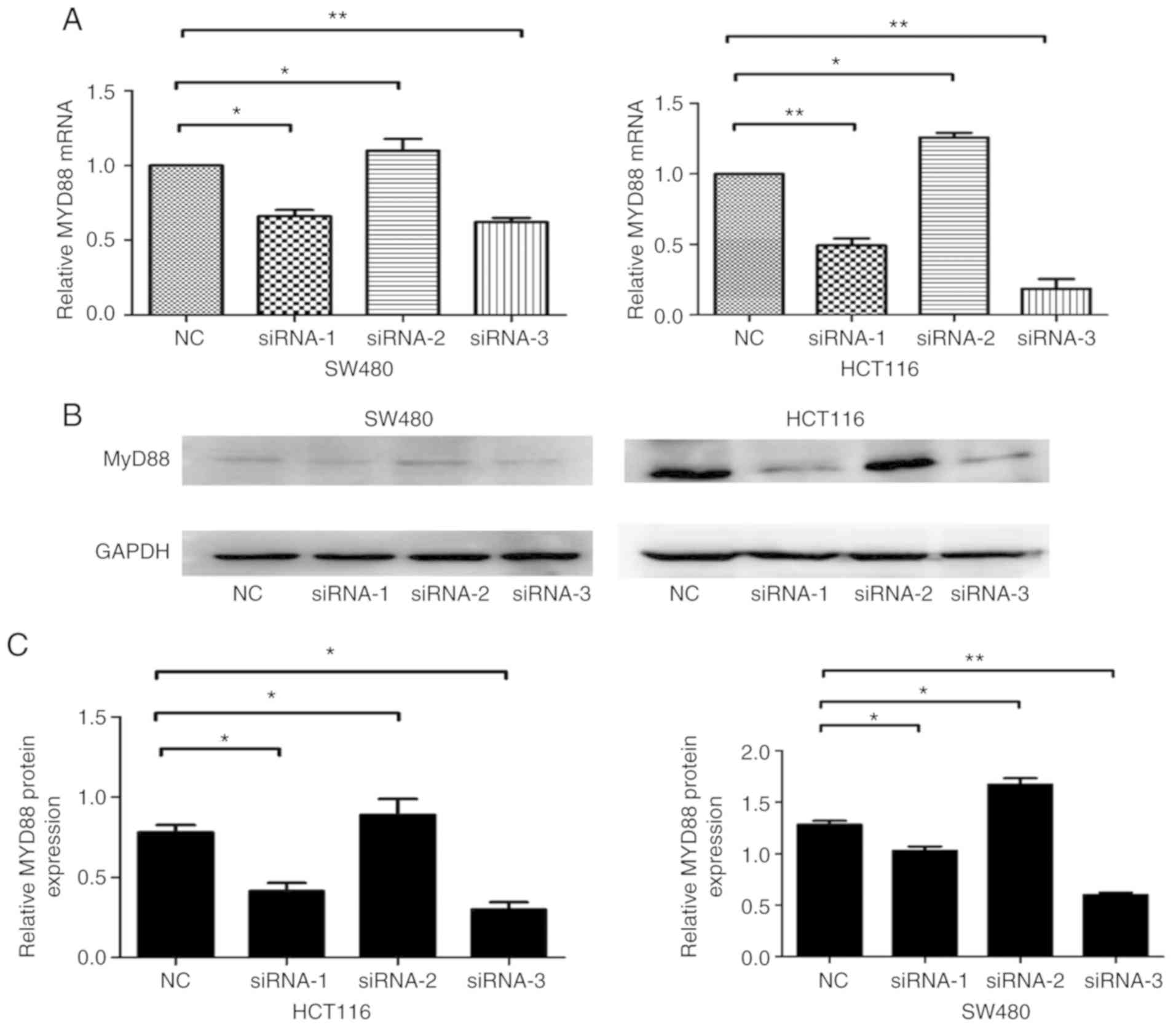
An RNAi-based method was employed to silence
MyD88 and detect the effects of knocking down the gene. NC,
siRNA-1, siRNA-2 and siRNA-3 sequences were transfected into SW480
and HCT116 cells. The MyD88 mRNA and protein levels,
determined 48 h after transfection using RT-qPCR and western blot
analysis, were shown to decrease in the siRNA-transfected SW480 and
HCT116 cells. Compared with NCsiRNA and siRNA-2, which had no
effect on MyD88 mRNA and protein expression levels, siRNA-1
and siRNA-3 sequences inhibited MyD88 mRNA and protein
expression levels (Fig. 1A-C).
Since siRNA-1 and siRNA-3 were the most effective siRNA, and these
sequences were selected to silence MyD88 in this study.
Expression of MyD88 mRNA and protein
after infection with pLKO.1-sh1 and pLKO.1-sh3 lentiviral
vectors
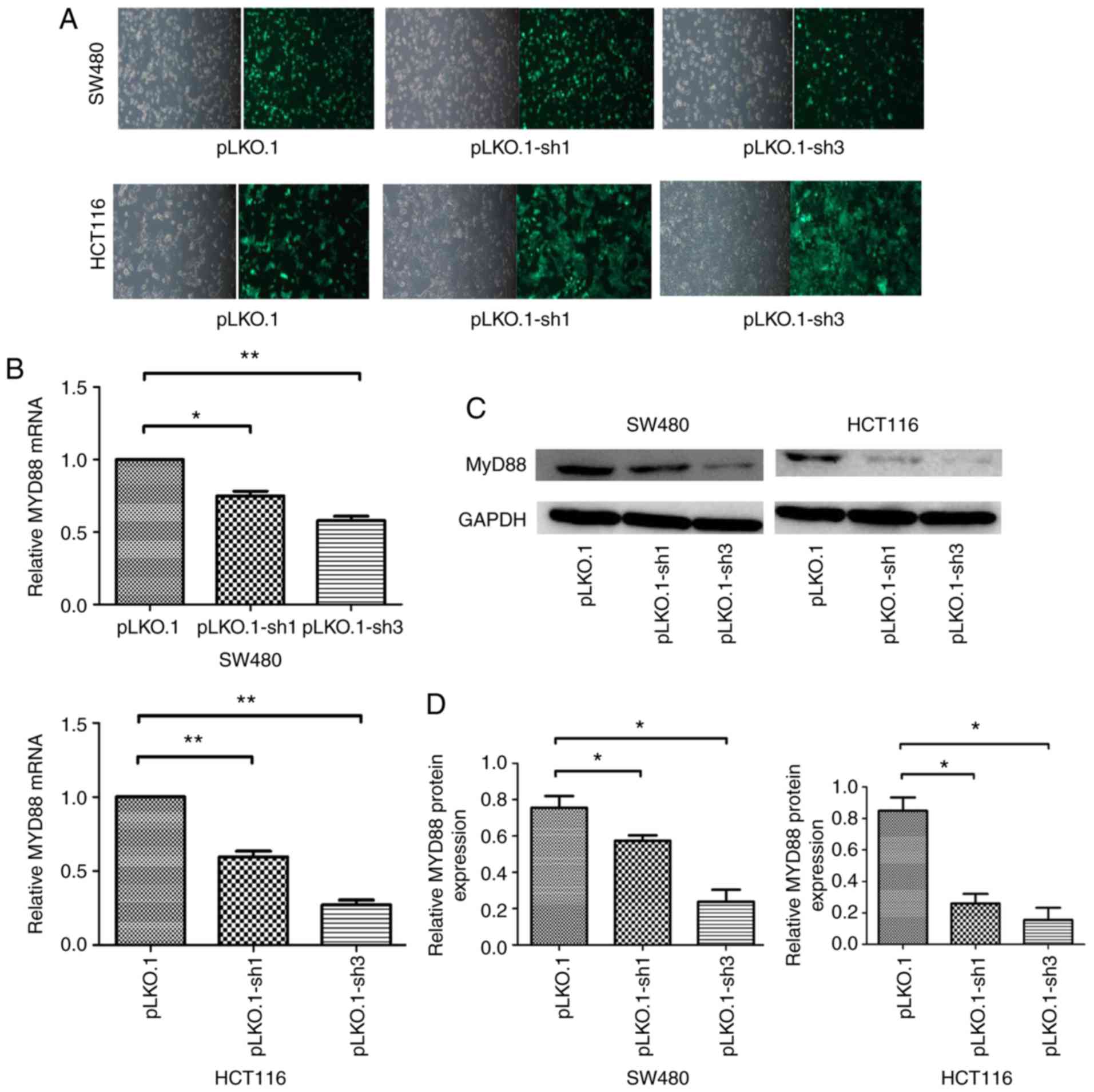
The NC, siRNA-1 and siRNA-3 sequences were
constructed into lentiviral vectors and packaged as lentiviruses
carrying NC, siRNA-1 and siRNA-2 sequences named pLKO.1, pLKO.1-sh1
and pLKO.1-sh3, respectively. Infection efficiency was quantified
by counting cells under a fluorescence microscope 48 h after
infection, because the pLKO.1 vector can express green fluorescent
protein in the SW480 and HCT116 cells, which had >95% efficiency
(Fig. 2A). Quantitative PCR and
western blot analyses showed that MyD88 mRNA and protein
levels were markedly inhibited after infection in SW480 and HCT116
cells. Compared with the pLKO.1 group, the pLKO.1-sh1 and
pLKO.1-sh3 groups revealed relatively lower MyD88 mRNA
expression levels in SW480 and HCT116 cells (Fig. 2B). Consistent with mRNA
expression, western blotting showed inhibition of MyD88
protein expression in the pLKO.1-sh1 and pLKO.1-sh3 groups compared
with pLKO.1 group in SW480 and HCT116 cells (Fig. 2C and D). To generate stably
transfected pLKO.1, the pLKO.1-sh1 and pLKO.1-sh3 cells, the
culture medium was replaced with a selection medium containing 2
µg/ml puromycin (Sigma) for two weeks. Stably transfected
CRC cells were cultured using medium containing 0.5 µg/ml
puromycin. These cells were then used for subsequent
experiments.
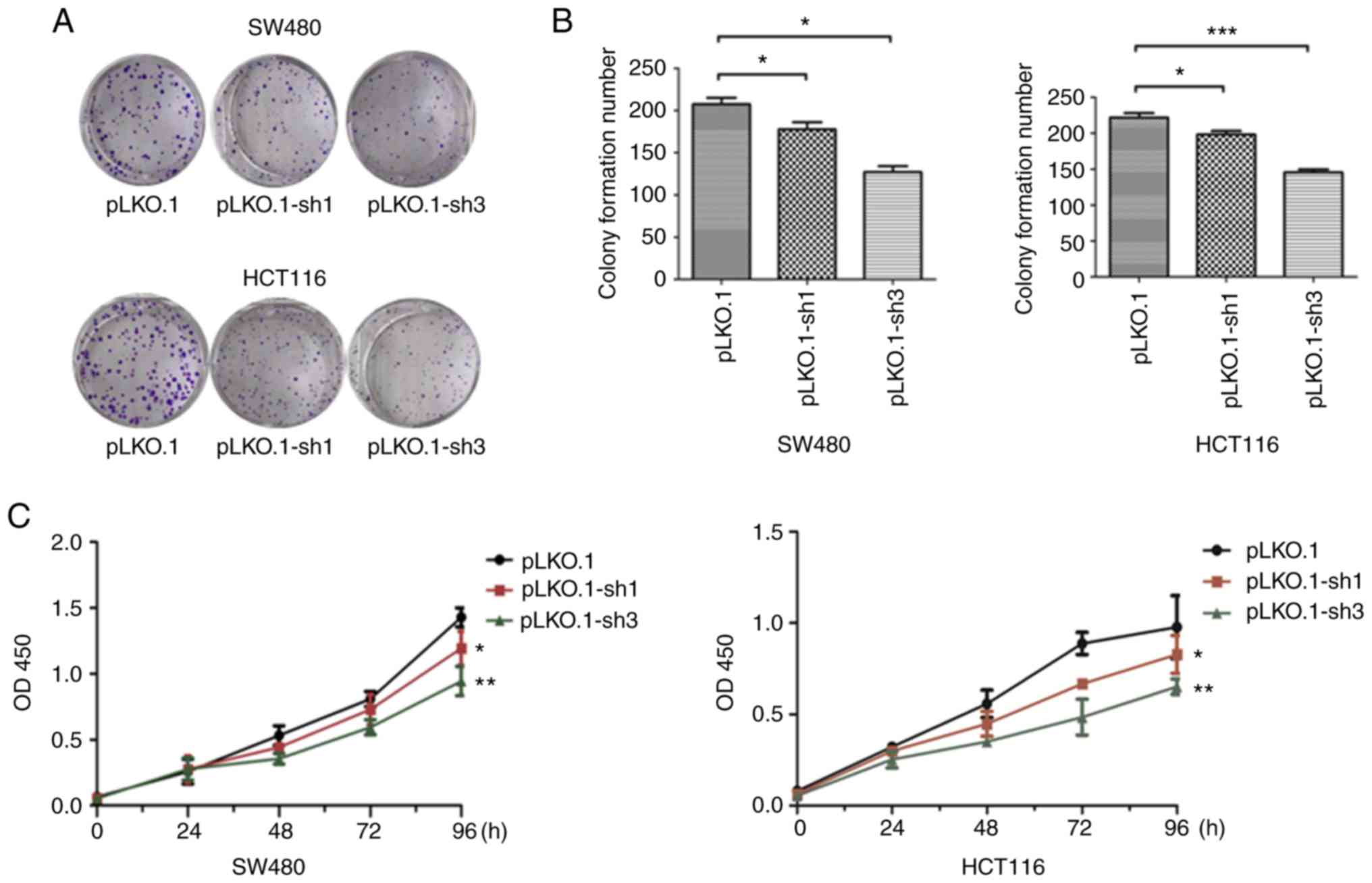
Knockdown of MyD88 reduces the
proliferation of SW480 and HCT116 cells
To verify whether the expression of MyD88 can
affect tumor proliferation, stably transfected pLKO.1, the
pLKO.1-sh1 and pLKO.1-sh3 CRC cells were used. Cell proliferation
was estimated using colony formation and CCK-8 assays. The colony
formation assay showed a decrease (P<0.05) in the number of
colonies in the pLKO.1-sh1 and pLKO.1-sh3 groups, while there was
no significant difference in the colony numbers in the pLKO.1-sh1
and pLKO.1-sh3 groups (P>0.05), when compared to the pLKO.1
group in the SW480 and HCT116 cells (Fig. 3A and B). CCK-8 experiment results
showed that compared to the LKO.1 group, cell proliferation of the
pLKO.1-sh1 and pLKO.1-sh3 groups, which were similar, was lower in
the SW480 and HCT116 cells (P<0.05) (Fig. 3C). The cell viabilities of the
pLKO.1-sh1 and pLKO.1-sh3 groups were decreased when compared with
the pLKO.1 group in both colorectal carcinoma cell lines. These
results indicate that silencing MyD88 contributes to the
inhibition of CRC cell proliferation.
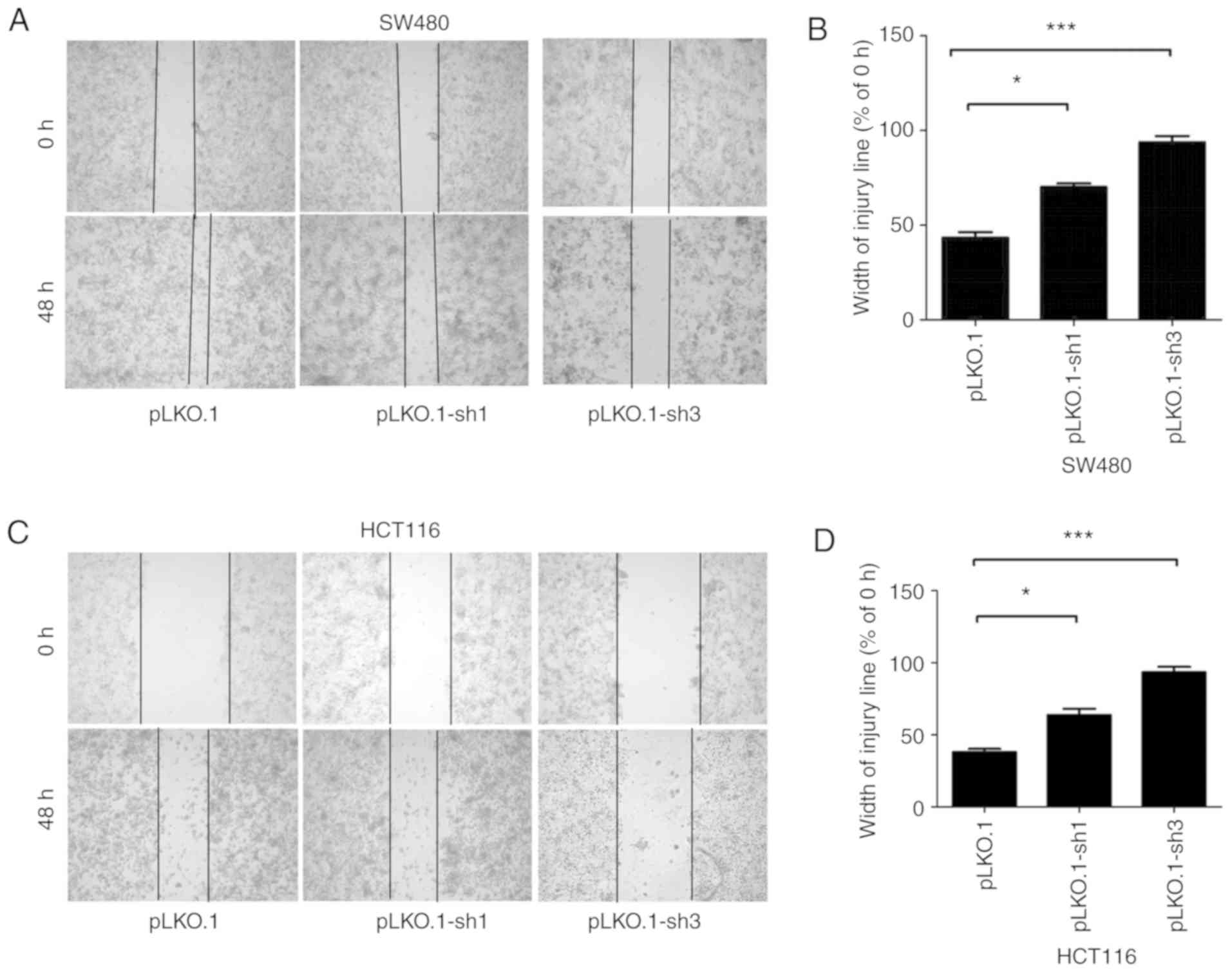
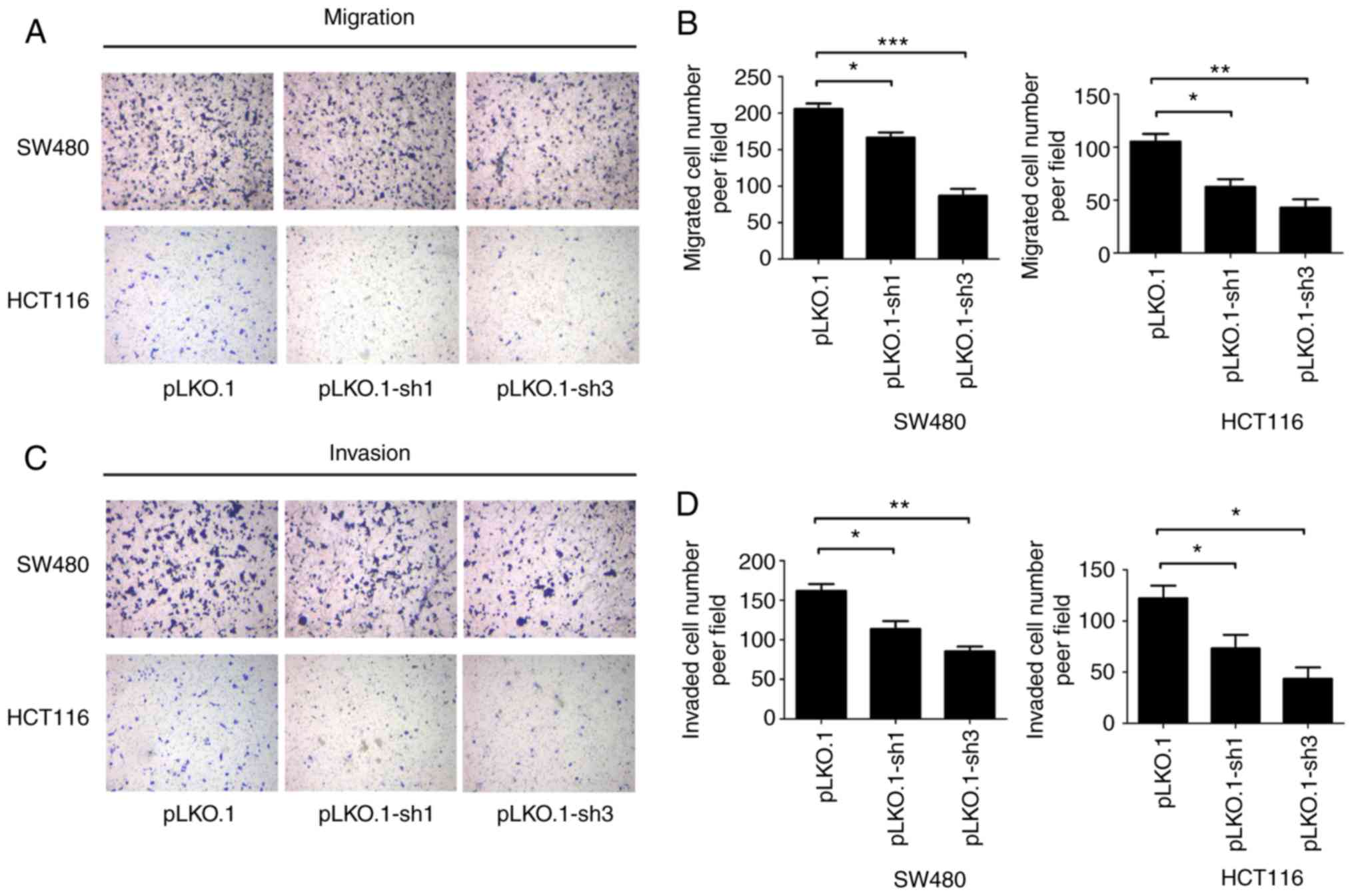
MyD88 knockdown attenuates migration and
invasion of colorectal carcinoma cells
To determine the ability of MyD88 knockdown
in colorectal carcinoma cells to suppress migration, the Transwell
assay and scratch test were used to verify the migration ability of
SW480 and HCT116 cells harboring MyD88 knockdown. Similar to
migration, the invasiveness of the SW480 and HCT116 cells was
tested using the Transwell assay. Wound healing/scratch was used to
confirm changes in cell migration by observing the extent of wound
closure. In the pLKO.1-sh1 and pLKO.1-sh3 groups (P>0.05), the
healing speed of the scratches were lower than in the pLKO.1 group
in SW480 and HCT116 cells (P<0.05) (Fig. 4A-D). To further verify the results
of the migration, we used the Transwell experiment. The number of
colorectal cells in the pLKO.1 group that migrated through the
Transwell polycarbonate filter was markedly higher than that of
cells in the pLKO.1-sh1 group (P<0.05), which was similar to the
number of cells in the pLKO.1-sh3 group in the SW480 and HCT116
cells (P>0.05) (Fig. 5A and
B). The results showed that knockdown of MyD88
significantly suppressed cell migration ability compared with the
NC group. In vitro cell invasion showed that the number of
cells penetrating the basement membrane was significantly higher in
the pLKO.1 group than in the pLKO.1-sh1 and pLKO.1-sh3 groups
(P>0.05) in the SW480 and HCT116 cells (Fig. 5C and D).
MyD88 affects NF-κB and AP-1 signaling
pathways
In the above experiments, we confirmed that
MyD88 can affect the biological behavior of CRC cells, but
the underlying mechanisms remain unknown. To investigate the
mechanism by which MyD88 silencing leads to a decrease in
proliferation and invasion, we evaluated NF-κB (p65),
p-NF-κB (p-p65), AP-1 (c-jun) and p-AP-1
(pc-Jun) proteins; these proteins are critical for the growth
and invasion of cancer cells, and are very important for
NF-κB and AP-1 signaling pathways. Western blot
analysis showed that NF-κB (p65), p-NF-κB (p-p65),
AP-1 (c-jun) and p-AP-1 (p-c-jun) protein levels in
the pLKO.1 group cells increased compared with those in the
pLKO.1-sh1 and pLKO.1-sh3 groups (P<0.05) in the SW480 and
HCT116 cells (Fig. 6A-D). The
results indicated that MYD88 suppressed the expression of
related signaling pathway proteins.
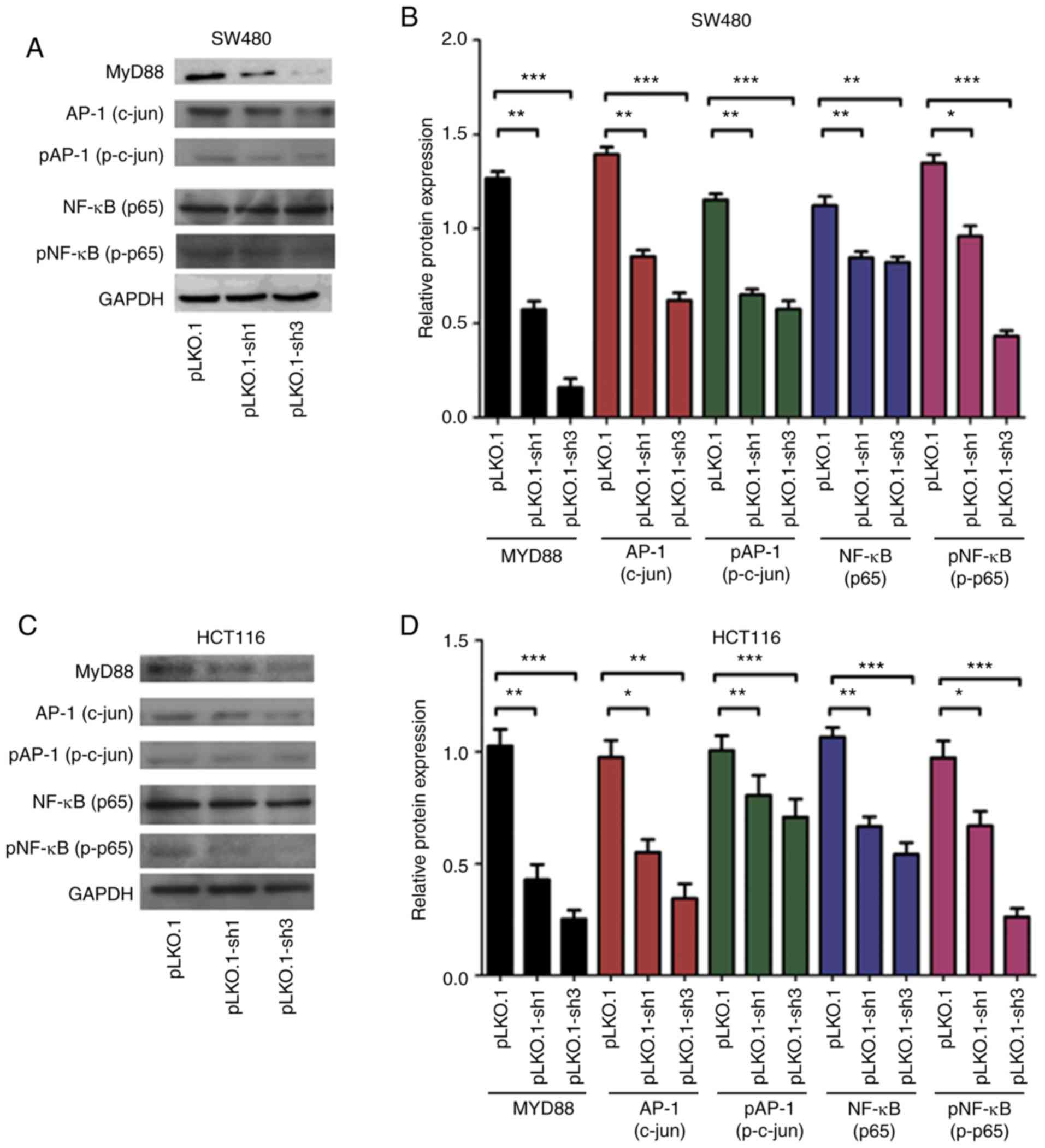 | Figure 6Western blot analysis revealed that
the silencing of MyD88 markedly inhibited the expression
NF-κB (p65), p-NF-κB (p-p65), AP-1 (c-jun) and
p-AP-1 (p-c-jun) protein in the SW480 and HCT116 cells. (A)
The protein electropherogram showed the protein expression
situation of NF-κB (p65), p-NF-κB (p-p65), AP-1
(c-jun), p-AP-1 (p-c-jun) and MyD88 in pLKO.1,
pLKO.1-sh1 and pLKO.1-sh3 groups in the SW480 cells. (B) The
protein expression of the pLKO.1, pLKO.1-sh1 and pLKO.1-sh3 groups
was analyzed using a histogram in the SW480 cells. (C) An
electropherogram was used to show the protein expression of
NF-κB (p65), p-NF-κB (p-p65), AP-1 (c-jun),
p-AP-1 (p-c-jun) and MyD88 in pLKO.1, pLKO.1-sh1 and
pLKO.1-sh3 groups in the HCT116 cells. (D) The protein expression
of the pLKO.1, pLKO.1-sh1 and pLKO.1-sh3 groups was analyzed using
a histogram in the HCT116 cells. Error bars represent mean ± SEM,
representative of three experiments. *P<0.05,
**P<0.01, ***P<0.001. |
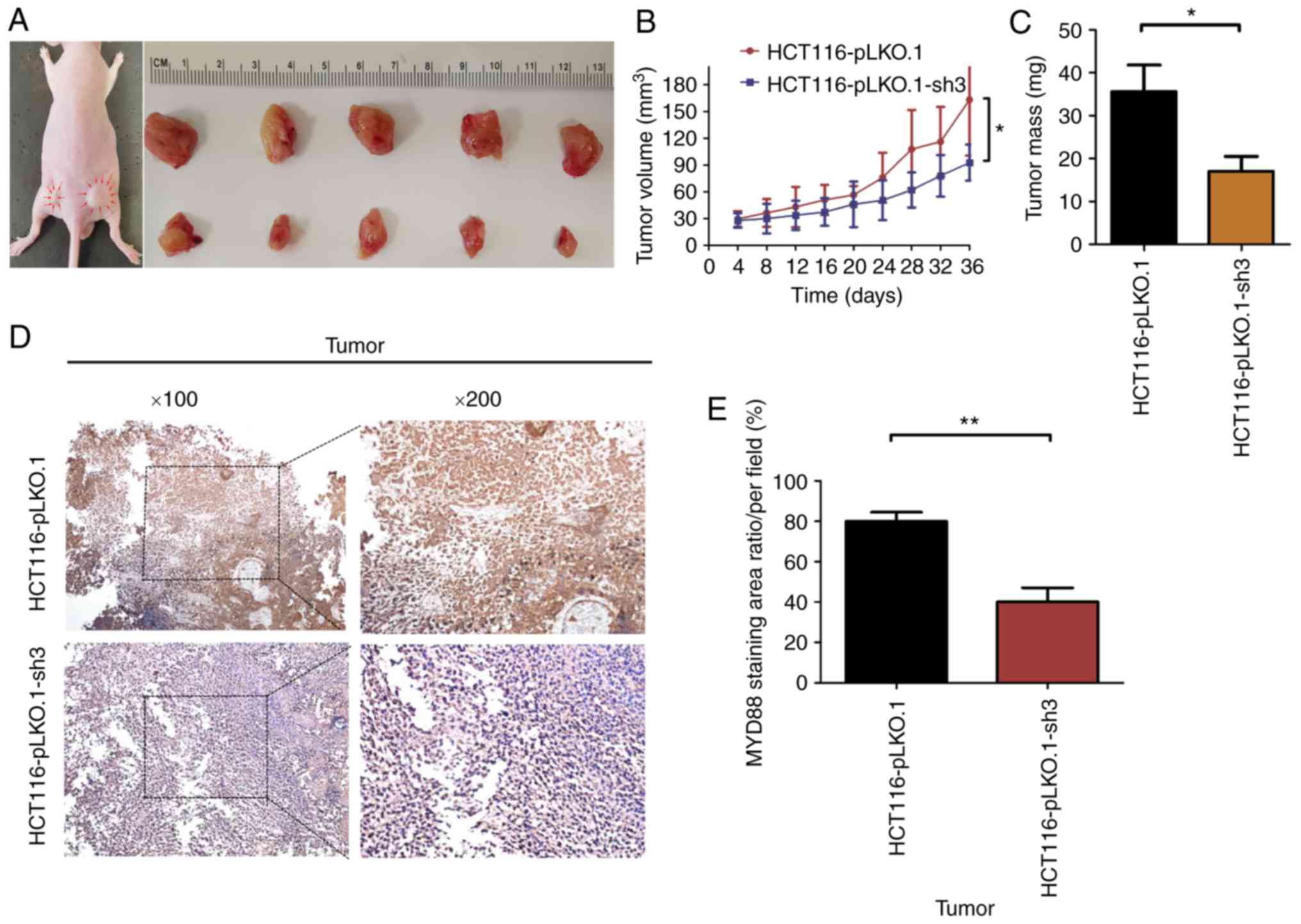
Knockdown of MyD88 suppresses tumor
growth in BALB/c nude mice with subcutaneous xenograft tumors
To verify whether tumor growth in vivo was
the same as in vitro, the HCT116-pLKO.1 or HCT116-pLKO.1-sh3
cells were subcutaneously xenografted into 5-week-old BALB/c nude
mice (5 mice/group). At 6 weeks of subcutaneous tumor growth, nude
mice were all successfully established in a subcutaneous mouse
model (Fig. 7A). At autopsy, none
of the mice had any ascites or liver, lung or lymph node
metastasis. The results showed that the growth rate of subcutaneous
tumors in the HCT116-pLKO.1-sh3 group was significantly lower than
that in the HCT116-pLKO.1 group (P<0.05) (Fig. 7B). In addition, the tumor weight
of the HCT116-pLKO.1-sh3 group was markedly lower than that of the
HCT116-pLKO.1 group (P<0.05) (Fig.
7C). Since MyD88 protein expression was inhibited in
HCT116-pLKO.1-sh3 cells, immunohistochemistry was used to analyze
the expression levels of MyD88 in subcutaneous xenograft
tumors. Compared with the HCT116-pLKO.1 group, MyD88
expression levels were markedly inhibited in the HCT116-pLKO.1-sh3
group (P<0.05) (Fig. 7D and
E).
Discussion
MyD88 plays a central role in innate immune
response. It regulates NF-κB signaling activity (16) and the MAPKs-c-jun (AP-1)
signaling pathway (17).
MyD88 and MyD88-related signaling have been shown to
be involved in the progression and carcinogenesis of
cancer-associated cells, with both intrinsic and extrinsic
inflammation. In addition, detection of abnormal MyD88
expression is used to predict the prognosis of various human
cancers (e.g., lymph, liver) (18). Hence, MyD88 can be
considered a potential carcinogenic marker for related research. It
will have a positive impact on the pathogenic factors and treatment
of CRC.
In the current study, we successfully knocked down
the MyD88 gene and verified the knockdown effect of the
protein using cell fluorescence, RT-qPCR and western blot analysis.
We determined whether MyD88 gene was able to affect the
biological behavior of CRC cells, and subsequently confirmed the
ability of MyD88 knockdown to inhibit cell proliferation and
invasion in vivo using relevant cell function tests. The
results suggest that knockdown MyD88 can suppress
proliferation, invasion, and migration of CRC. These findings agree
with those of previous studies which showed that, the SNP of
MyD88 was associated with poor survival of Caucasian
patients with CRC (19). Ikebe
et al (20) reported that
siMyD88 can decrease the invasiveness and migration ability
of pancreatic cancer cells. Wang et al (21) indicated that a high MyD88
expression is associated with liver metastasis and poor prognosis
in CRC patients. We also found that MyD88 expression was
high in CRC tissues and was associated with poor survival in
colorectal carcinoma patients (14).
Similarly, we explored whether knockdown of
MyD88 gene had the same effect in vitro. We used
subcutaneous injection of colon cancer cells to conduct
subcutaneous tumorigenesis experiments, confirming that knockdown
of MyD88 gene in mice can also inhibit tumor growth. This
finding is in agreement with previous studies, which considered
that MyD88 increased the risk of developing colorectal
neoplasm (22,23). Our findings indicate that
MyD88 can promote the growth of CRC. Thus, knockdown of
MyD88 gene can inhibit tumor growth, invasion, migration and
other related behavioral changes.
However, the specific mechanism by which
MyD88 induces biological behavior in CRC is unclear.
Previous findings showed that MyD88 can protect the
intestine from tumorigenesis via IL-18/MyD88 signaling as
compared to colitis-associated cancer animal models of
IL-18−/−, IL-18R1−/−, and
MyD88−/− mice, as well as wild-type mice
(17). By contrast, MyD88
can promote the malignant transformation of colitis to colorectal
cancer through the TLR/MyD88 pathway in the
MyD88-knockout mice after LPS treatment (24). To date, it remains unclear how and
why MyD88 possesses these dual functions in CRC, and the
underlying mechanisms require further investigation.
As previously shown, MyD88 signaling plays a
role in regulating the growth of normal epithelial and cancer cells
in the gut. The activated form of NF-kB is upregulated and
functionally correlated with many tumors, modulating proliferation
and invasion. AP-1 and, specifically, c-Jun, affect
tumor cell proliferation, migration and invasion (25). Recent studies have shown that
mitogen-activated protein kinases (MAPKs) and nuclear
factor-κB (NF-κB) signaling pathways are two key factors
leading to the development of lipopolysaccharide (LPS)-induced
acute lung injury (ALI) (26),
osteoclastogenesis (27),
neuroinflammation (28),
atherosclerosis (29),
tumorigenesis in prostate carcinoma and glioblastoma (25), and activation of human dendritic
cells (30). Under the premise of
MyD88-induced tumor bioethology, we investigated whether
knockdown MyD88 induced changes in NF-κB (p65) and
AP-1 (MAPKs-c-jun) proteins. In knockdown of MyD88
stably transfected cell lines, we found that p65,
phosphorylated p65, c-jun, and phosphorylated
c-jun protein expression resulted in different degrees of
decline. This demonstrates that knockdown of MyD88 gene can
affect MyD88-mediated activation of the NF-κB (p65)
and AP-1 (MAPKs-c-jun) pathways, thereby effectively
retarding the aggressive transformation of the tumor.
In summary, MyD88 could be an independent
factor to explore its role in colon cancer, which can be used as a
factor in the pathogenesis of colon cancer, and deserves further
development as a potential diagnostic and prognostic biomarker. We
found that knocking down the MyD88 gene affects
proliferation, invasion, and migration of CRC cells, and can reduce
the activity of NF-κB and AP-1 pathways. These
results show that the MyD88 gene plays an important role in
promoting CRC and may be exploited as a diagnostic and prognostic
biomarker for CRC.
Acknowledgments
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81702424 and 81872364), the
Fujian Provincial Health Department Young and Middle-aged Talents
Training Project (grant no. 2018-ZQN-46), the Joint Funds for the
Innovation of Science and Technology, Fujian Province (grant no.
2017Y9092), the Project of Science and Technology Research Program
in Fujian Province (grant no. 2016B044), the Fujian Provincial
Natural Science Foundation (grant no. 2018J05127), and the National
Clinical Key Specialty Construction Project (General Surgery) of
China.
Availability of data and materials
The datasets used or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JY, GZ, YH and WZ conceived and designed the study.
GZ, ZC and CL performed the experiments. SY and JY interpreted the
results. GZ and ZC wrote the paper, and all authors contributed to
writing. All authors read and approved the final manuscript.
Ethics approval and consent to
pariticipate
The experiment performed with animals was approved
by the Institutional Animal Care and Use Committee at the Fujian
Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Testa U, Pelosi E and Castelli G:
Colorectal cancer: Genetic abnormalities, tumor progression, tumor
heterogeneity, clonal evolution and tumor-initiating cells. Med Sci
(Basel). 6:E312018.
|
|
2
|
Okugawa Y, Grady WM and Goel A: Epigenetic
alterations in colorectal cancer: Emerging biomarkers.
Gastroenterology. 149:1204–1225.e12. 2015. View Article : Google Scholar :
|
|
3
|
Yaghoubi N, Soltani A, Ghazvini K,
Hassanian SM and Hashemy SI: PD-1/PD-L1 blockade as a novel
treatment for colorectal cancer. Biomed Pharmacother. 110:312–318.
2019. View Article : Google Scholar
|
|
4
|
Marmol I, Sánchez-de-Diego C, Pradilla
Dieste A, Cerrada E and Rodriguez Yoldi MJ: Colorectal carcinoma: A
general overview and future perspectives in colorectal cancer. Int
J Mol Sci. 18:E1972017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lingel A, Ehlers E, Wang Q, Cao M, Wood C,
Lin R and Zhang L: Kaposi's sarcoma-associated herpesvirus reduces
cellular myeloid differentiation primary-response gene 88 (MyD88)
expression via modulation of its RNA. J Virol. 90:180–188. 2016.
View Article : Google Scholar :
|
|
6
|
Feng Y, Zou L, Chen C, Li D and Chao W:
Role of cardiac- and myeloid-MyD88 signaling in endotoxin shock: A
study with tissue-specific deletion models. Anesthesiology.
121:1258–1269. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang H, Li M, Hung CY, Sinha M, Lee LM,
Wiesner DL, LeBert V, Lerksuthirat T, Galles K, Suresh M, et al:
MyD88 shapes vaccine immunity by extrinsically regulating survival
of CD4+ T cells during the contraction phase. PLoS
Pathog. 12:e10057872016. View Article : Google Scholar
|
|
8
|
Xu X, Yin Y, Tang J, Xie Y, Han Z, Zhang
X, Liu Q, Qin X, Huang X and Sun B: Long non-coding RNA Myd88
promotes growth and metastasis in hepatocellular carcinoma via
regulating Myd88 expression through H3K27 modification. Cell Death
Dis. 8:e31242017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Syeda S, Patel AK, Lee T and Hackam AS:
Reduced photoreceptor death and improved retinal function during
retinal degeneration in mice lacking innate immunity adaptor
protein MyD88. Exp Neurol. 267:1–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
d'Adhemar CJ, Spillane CD, Gallagher MF,
Bates M, Costello KM, Barry-O'Crowley J, Haley K, Kernan N, Murphy
C, Smyth PC, et al: The MyD88+ phenotype is an adverse prognostic
factor in epithelial ovarian cancer. PLoS One. 9:e1008162014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou X, Ramke M, Chintakuntlawar AV, Lee
JY, Rajaiya J and Chodosh J: Role of MyD88 in adenovirus keratitis.
Immunol Cell Biol. 95:108–116. 2017. View Article : Google Scholar
|
|
12
|
Di Padova F, Quesniaux VFJ and Ryffel B:
MyD88 as a therapeutic target for inflammatory lung diseases.
Expert Opin Ther Targets. 22:401–408. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reins RY, Courson J, Lema C and Redfern
RL: MyD88 contribution to ocular surface homeostasis. PLoS One.
12:e01821532017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu G, Zheng W, Huang Y, Hua J, Yang S and
Ye J: Expression of MyD88 in cancer tissue of patients with
colorectal cancer and clinical significancer. Journal of Jilin
University (Medicine Edition). 44:1047–1051. 2018.
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
16
|
Yan F, Guan J, Peng Y and Zheng X: MyD88
NEDDylation negatively regulates MyD88-dependent NF-κB signaling
through antagonizing its ubiquitination. Biochem Biophys Res
Commun. 482:632–637. 2017. View Article : Google Scholar
|
|
17
|
Tanishima M, Takashima S, Honda A, Yasuda
D, Tanikawa T, Ishii S and MaruYama T: Identification of optineurin
as an interleukin-1 receptor-associated kinase 1-binding protein
and its role in regulation of MyD88-dependent signaling. J Biol
Chem. 292:17250–17257. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang L, Yu K, Zhang X and Yu S: Dual
functional roles of the MyD88 signaling in colorectal cancer
development. Biomed Pharmacother. 107:177–184. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Klimosch SN, Försti A, Eckert J, Knezevic
J, Bevier M, von Schönfels W, Heits N, Walter J, Hinz S, Lascorz J,
et al: Functional TLR5 genetic variants affect human colorectal
cancer survival. Cancer Res. 73:7232–7242. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ikebe M, Kitaura Y, Nakamura M, Tanaka H,
Yamasaki A, Nagai S, Wada J, Yanai K, Koga K, Sato N, et al:
Lipopolysaccharide (LPS) increases the invasive ability of
pancreatic cancer cells through the TLR4/MyD88 signaling pathway. J
Surg Oncol. 100:725–731. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang EL, Qian ZR, Nakasono M, Tanahashi T,
Yoshimoto K, Bando Y, Kudo E, Shimada M and Sano T: High expression
of toll-like receptor 4/myeloid differentiation factor 88 signals
correlates with poor prognosis in colorectal cancer. Br J Cancer.
102:908–915. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rakoff-Nahoum S and Medzhitov R:
Regulation of spontaneous intestinal tumorigenesis through the
adaptor protein MyD88. Science. 317:124–127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chang CM, Chia VM, Gunter MJ, Zanetti KA,
Ryan BM, Goodman JE, Harris CC, Weissfeld J, Huang WY, Chanock S,
et al: Innate immunity gene polymorphisms and the risk of
colorectal neoplasia. Carcinogenesis. 34:2512–2520. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schiechl G, Bauer B, Fuss I, Lang SA,
Moser C, Ruemmele P, Rose-John S, Neurath MF, Geissler EK, Schlitt
HJ, et al: Tumor development in murine ulcerative colitis depends
on MyD88 signaling of colonic F4/80+CD11b(high)Gr1(low)
macrophages. J Clin Invest. 121:1692–1708. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Galardi S, Mercatelli N, Farace MG and
Ciafre SA: NF-κB and c-Jun induce the expression of the oncogenic
miR-221 and miR-222 in prostate carcinoma and glioblastoma cells.
Nucleic Acids Res. 39:3892–3902. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen X, Yang X, Liu T, Guan M, Feng X,
Dong W, Chu X, Liu J, Tian X, Ci X, et al: Kaempferol regulates
MAPKs and NF-κB signaling pathways to attenuate LPS-induced acute
lung injury in mice. Int Immunopharmacol. 14:209–216. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park JW, Yoon HJ, Kang WY, Cho S, Seong
SJ, Lee HW, Yoon YR and Kim HJ: G protein-coupled receptor 84
controls osteoclastogenesis through inhibition of NF-κB and MAPK
signaling pathways. J Cell Physiol. 233:1481–1489. 2018. View Article : Google Scholar
|
|
28
|
Zheng Y, Fang W, Fan S, Liao W, Xiong Y,
Liao S, Li Y, Xiao S and Liu J: Neurotropin inhibits
neuroinflammation via suppressing NF-κB and MAPKs signaling
pathways in lipopoly-saccharide-stimulated BV2 cells. J Pharmacol
Sci. 136:242–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pan JX: LncRNA H19 promotes
atherosclerosis by regulating MAPK and NF-κB signaling pathway. Eur
Rev Med Pharmacol Sci. 21:322–328. 2017.PubMed/NCBI
|
|
30
|
Parola C, Salogni L, Vaira X, Scutera S,
Somma P, Salvi V, Musso T, Tabbia G, Bardessono M, Pasquali C, et
al: Selective activation of human dendritic cells by OM-85 through
a NF-κB and MAPK dependent pathway. PLoS One. 8:e828672013.
View Article : Google Scholar
|