Introduction
Pulmonary hypertension (PH) is a progressive and
lethal pulmonary inflammatory disease characterized by sustained
increases in pulmonary arterial pressure (PAP) and pulmonary
arteriolar remodeling, which ultimately results in right heart
hypertrophy, failure or even premature mortality (1-4).
Development of PH is considered to involve the interaction between
multiple factors (5). There is
considerable circumstantial evidence that inflammatory responses
serve a key role in the pathogenesis or progression of PH (6,7).
Systemic or perivascular inflammation or immune dysregulation is a
primary feature observed in patients with PH and experimental
animal models of PH (7,8). Pulmonary inflammation initiates and
participates in the injury of pulmonary arterial endothelial cells
and aberrant proliferation of pulmonary vascular smooth muscle
cells, and subsequently accelerates the vascular remodeling and
progression of PH (4,9). This remodeling is supported by
histopathological data demonstrating infiltration of macrophages,
monocytes and lymphocytes in the vicinity of the remodeled
pulmonary arterioles and plexiform lesions of patients and animal
models [hypoxia and the monocrota-line (MCT) rat model] of PH
(10-12). In addition, increased levels of
both serum and tissue pro-inflammatory cytokines have been observed
in patients with PH and animal models of PH (7,12).
Despite an evident association between PH and dysregulation of the
adaptive immune response, the regulatory mechanisms that result
from alterations in lymphocyte subsets and the production of
inflammatory cytokines from injured lung tissues and immune cells
remain incompletely understood. Although immunosuppressive drugs
and several immunotherapeutic approaches may alleviate inflammation
and prevent vascular remodeling in animal models of PH (13-15), they lack therapeutic specificity
(13-15). Therefore, further elucidating the
molecular and cellular mechanism underlying PH-mediated
inflammation and examining new immunotherapeutic strategies or
agents is critical (14).
In our previous studies, it was demonstrated that
connexin (Cx)-mediated gap junction channels in T lymphocytes of
hypertensive animals and patients were involved in
hypertension-mediated inflammation (16-18), and that expressional or functional
inhibition of Cx (16-18), or treatment with various
anti-inflammatory drugs (β-estradiol and hydrogen sulfide)
(19,20) improved hypertension- and
pro-inflammatory stimuli-mediated inflammation by decreasing the
proportion of CD4+ T lymphocytes and levels of
pro-inflammatory cytokines (16-20). However, whether Cxs participate in
PH-mediated inflammation, and whether the inhibition of Cxs
alleviates inflammation in PH remain unknown. Carbenoxolone (CBX),
a semisynthetic derivative of glycyrrhetinic acid that inhibits gap
junction activity (21), has been
used to evaluate the role of Cx-mediated intercellular
communication in acute lung inflammation (22), and to treat pulmonary inflammatory
diseases (23,24). Inhalation of CBX has been
demonstrated to significantly decrease lung inflammation in
experimental asthma animal models, by decreasing the production of
interleukin (IL)-4 and -5 and by decreasing the infiltration of
inflammatory cells in perivascular regions (24). In addition, CBX has been
demonstrated to decrease differentiation of Th17 cells by
decreasing IL-23 production in antigen presenting cells (25). Based on our and other published
studies, it was hypothesized that CBX may alleviate PH-mediated
inflammation by regulating Cxs or gap junctions in T
lymphocytes.
Studies have demonstrated that the MCT-treated rat
model and PH exhibit similar pathological characteristics in
vascular remodeling, production of inflammatory cytokines and
infiltration of inflammatory cells (2,8);
therefore, this model was deemed to be well-suited for studying the
anti-inflammatory effects and corresponding molecular mechanisms of
CBX on MCT-induced pulmonary inflammation, and to examine the
association between Cxs function in T cells and MCT-induced
pulmonary inflammation. To investigate the therapeutic effects of
CBX on inflammation and pulmonary arteriolar remodeling caused by
MCT, physiological and echocardiographic parameters in the heart
were measured, histopathological changes in lung tissues were
assessed, and the levels of cytokines and distribution of
CD4+ and CD8+ T cells in lung tissues were
determined in CBX-treated MCT rats. Additionally, to the best of
our knowledge, the present study was the first to determine whether
Cx40 and Cx43 may serve as potential therapeutic targets in
PH-mediated inflammation, and whether CBX may improve the
pro-inflammatory microenvironment in a rat MCT model by regulating
Cx40 and Cx43 expression in T cell subsets. Although the precise
molecular mechanisms of CBX-mediated regulation of Cxs are
incompletely understood, the role of Cxs in T cells in the
progression of MCT-mediated pulmonary inflammation was
investigated, and the inhibition of Cxs or Cxs-based gap junctions
using CBX may be a potential therapeutic method for attenuating
inflammatory lung injury.
Materials and methods
Establishment of the animal model and
treatment schedules
Male Sprague-Dawley rats (age, 8-10 weeks; body
weight, 200-250 g) were obtained from Beijing Vital River
Laboratory Animal Technology Co., Ltd. All rats were housed in a
specific pathogen-free barrier facility with a 12: 12 h day: night
cycle at 22±2°C and 60-65% humidity. Rats had ad libitum
access to chow and water. All animal experimental protocols were
approved by the Institutional Animal Care and Use Committee of the
Medical College of Shihezi University (permit no. A2019-027-01) and
all experiments were performed in strict accordance with the
Guidelines on the Care and Use of Animals provided by the American
Physiological Society (NIH Publication no. 85-23, revised 1996)
(26).
A total of 24 rats were used, and were randomly
divided into the following four groups, with 6 rats/group: Control
group; CBX-treated rats (cat. no. C4790; Sigma-Aldrich; Merck
KGaA); MCT group; and MCT rats treated with CBX (MCT + CBX group).
Rats from the MCT and MCT + CBX groups received a single
intraperitoneal (i.p.) injection of MCT (cat. no. C2401; 60 mg/kg;
day 0; Sigma-Aldrich; Merck KGaA). The rats in the MCT + CBX group
received daily i.p. injections of CBX (20 mg/kg) for 28 days
following MCT administration, whereas the control rats received
daily i.p. administration of normal saline or CBX from day 0 to day
28. The CBX dose used was based on a previous study (27). On day 28, all animals underwent
echocardiography measurement. Rats were then sacrificed under deep
anesthesia by i.p. administration of sodium pentobarbital (100
mg/kg), and the lungs, hearts and blood samples were collected.
Doppler echocardiography measurement
The Doppler echo parameter 'pulmonary artery
acceleration time' (PAAT) is negatively correlated with the mean
pulmonary arterial pressure (PAP) measured invasively, namely
increased pulmonary hypertension or an increase in PAP as judged by
a decreased PAAT (28,29). Therefore, PAAT is considered as an
echocardiographic indicator of PH (30). PH was also assessed using Doppler
echocardiography at day 28 of the study. Transthoracic closed-chest
echocardiography was performed by an experienced doctor using a
Vivid E9 ultrasound system equipped with a 12-MHz transducer (GE
Healthcare). Rats were anesthetized by i.p. injection of 3% sodium
pentobarbital (40 mg/kg) and placed in a shallow left lateral
decubitus position, and an ultrasound gel was applied to the shaved
chest. Blood flow through the pulmonary artery and PAAT were
measured in the two-dimensional short-axis parasternal view by
M-mode and Pulsed-wave Doppler at the level of the pulmonary valve.
Papillary muscles were used as the reference point for
echocardiography measurements. PAAT was measured from the onset of
systolic flow to peak pulmonary outflow velocity according to the
American Society of Echocardiography guidelines (31). The acquisition of Echo images and
all the echocardiographic analyses were performed using Echopac
BT11 software (v.6.5; GE Healthcare).
Measurement of right-ventricular
hypertrophy
Rats were sacrificed under deep anesthesia by i.p.
administration of sodium pentobarbital (100 mg/kg), and the entire
heart was isolated, immediately dissected and weighed to assess
right-ventricular hypertrophy (RVH). The atria and extraneous blood
vessels were removed from the isolated heart in cooled 0.9% saline
solution. Subsequently, the two ventricles of the heart were
separated into the free right ventricle (RV) and the left ventricle
(LV) wall with the interventricular septum (S), and the 2 portions
were immediately blotted dry and weighed separately. Finally, a
weight ratio of RV to LV plus S [RV/(LV+S)] was calculated for
determination of the RVH index (RVHI).
Histopathological examination of lung
tissues
The left lung tissues obtained from sacrificed rats
were fixed in 4% paraformaldehyde for 48 h at 4°C, embedded in
paraffin and cut into 4 µm-thick sections using a microtome.
Sections were stained with hematoxylin and eosin or Masson trichome
staining according to the manufacturer's protocol (cat. no. 1345;
Beijing Solarbio Science & Technology Co., Ltd.), and the
histopathological changes in lung tissues and pulmonary arterioles
(small arterioles with an external diameter of 15-50 µm and
medium-sized arterioles with an external diameter of 50-150
µm) were examined under a light microscope and imaged using
a digital camera (BX51; Olympus Corporation) at a magnification,
×200 or ×400. For quantitative analysis, the microscopic images of
lung tissue sections were analyzed using Image-Pro Plus v.6.0
(Media Cybernetics, Inc.). Pulmonary vascular remodeling in the
arterioles was evaluated by the percentage of vascular wall
thickness (WT%) and the percentage of the vascular wall area (WA%).
The formula for WT% was: WT% = [2 × (external diameter of the
pulmonary arterioles - internal diameter of the pulmonary
arterioles)]/(external diameter of the pulmonary arterioles) ×100;
and the formula for WA% was: WA%=(external area - internal
area)/external area ×100. The formula for WT% and WA% was carried
out as described by Yang et al (8) with some minor modifications. A total
of 20 randomly selected pulmonary arterioles/rat (6 rats/group)
that were nearly round were analyzed and the average from each
group was calculated. Pulmonary vascular remodeling and lung
fibrosis was assessed using Image-Pro Plus v.6.0 by 2 professional
pathologists whom each assessed 20 different non-overlapping fields
of each section. The lung fibrosis index was analyzed by
calculating the ratio of the total area of collagen to the total
area of connective tissue in each visual field (32).
ELISA
Blood samples (5 ml) from the abdominal aorta of
anesthetized rats were allowed to clot for 15 min at 22-25°C,
centrifuged at 1,100 × g for 10 min at 4°C and the plasma was
collected and stored at -80°C. The right lung tissues (400 mg)
obtained from rats were minced extensively and homogenized for 30
sec using an ultrasonic homogenizer (Beijing HeDe Biotechnology
Co., Ltd.) in PBS (pH 7.2; lung tissue to PBS, 1:10) on ice.
Samples were then centrifuged at 12,000 × g for 15 min at 4°C, and
the lung tissue supernatant was obtained. Both the lung tissue
supernatant and previously prepared serum were used to measure the
concentrations of IL-1β (cat. no. 70-EK301B), IL-6 (cat. no.
70-EK3062/2), IL-10 (cat. no. 70-EK3102/2) and tumor necrosis
factor-α (TNF-α; cat. no. 70-EK382HS-96) using ELISA kits (Hangzhou
Multi-Sciences Biotech Co., Ltd.) according to the manufacturer's
protocols. Measurements of each sample were performed 3 times and
the inflammatory cytokine concentrations are expressed as pg/ml of
the supernatant.
Preparation of mononuclear cells from
lung tissues
Washed right lung tissues were dissociated into
single-cell suspensions as described previously, with certain
modifications (33-35). Lung tissues were minced with
sterilized scissors and forceps [RNase was inactivated by high
temperature (121°C)], and were enzymatically digested with
RPMI-1640 medium (cat. no. 11875085; Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 1% BSA (Beijing Solarbio
Science & Technology Co., Ltd.), 10 mmol/l HEPES, 60 units/ml
deoxyribonuclease I (cat. no. D4527), 1 mg/ml type XI collagenase
(cat. no. C7657), 1,000 U hyaluronidase (cat. no. H3631), and 0.1
mg/ml Kunitz-type soybean trypsin inhibitor (cat. no. 7659;
Sigma-Aldrich; Merck KGaA), and the tissues were agitated for 40
min in a 37°C incubator. Following digestion, the lung tissue and
supernatant were filtered through a 200-gauge stainless steel mesh
to remove cell clumps and undigested tissue. The filtered
suspension was centrifuged at 200 × g for 10 min at room
temperature, and lung cells were resuspended in FACS buffer (cat.
no. 00-4222-57; Thermo Fisher Scientific, Inc.) at a concentration
of ~2×106/ml. The number of live cells was measured
using light microscopy at low resolution (magnification, ×40) by
staining cells with 0.4% Trypan Blue for 10 min at 22-25°C.
Flow cytometry
All isolated mononuclear cells (>1×106
cells/ml) harvested from lung tissues in 500 µL FACS buffer
were blocked via incubation with 5% normal mouse serum (cat. no.
31880; Invitrogen; Thermo Fisher Scientific, Inc.) for 15 min at
4°C. Cells were subsequently stained with fluorescein
isothiocyanate (FITC)-conjugated anti-rat CD3 (cat. no. 201403),
allophycocyanin (APC)-conjugated anti-rat CD4 (cat. no. 201509) and
phycoerythrin (PE)-conjugated anti-rat CD8 (cat. no. 201705)
antibodies (all at 1:100; all from BioLegend, Inc.) at 4°C for 30
min. FITC-, APC-, and PE- labelled IgG1or IgM isotype were used as
negative controls (1:100; cat. no. 401607 for FITC-conjugated
isotype ctrl antibody; cat. no. 400111 for PE-conjugated mouse
isotype ctrl antibody; cat. no. 400119 for APC-conjugated isotype
ctrl antibody; Biolegend, Inc.). For the labeling of Cxs in
CD4+ or CD8+ T cells from lung tissues,
stained T cells with APC-labeled anti-CD4 and PE-labeled anti-CD8
antibodies were incubated with permeabilization solution
(Cytofix/Cytoperm kit; BD Biosciences) for 30 min at room
temperature. Following permeabilization, cells were incubated with
anti-Cx40 monoclonal antibody (1:100; cat. no. sc-365107; Santa
Cruz Biotechnology, Inc.) or anti-Cx43 antibody (1:100; cat. no.
sc-13558; Santa Cruz Biotechnology, Inc.) for 30 min at 4°C.
Following washing using FACS buffer, the T cells were then
incubated with a FITC-conjugated anti-mouse secondary antibody
(1:100; cat. no. ZF0312; OriGene Technologies, Inc.) for 30 min at
4°C in the dark. All stained cells were analyzed using a FACSort
flow cytometer (BD Pharmingen; BD Biosciences) together with BD
CellQuest Pro software (v.2.0, system OS2; Becton Dickinson and
Company). Double-color flow cytometry was performed to calculate
the percentages of CD4/CD8 positive T cells, and Cx40/Cx43
expression in different T lymphocyte subpopulations.
Statistical analysis
All experimental data are presented as the mean ±
standard error of mean of 3 independent experiments. GraphPad Prism
v.5.0 (GraphPad Software, Inc.) was used for statistical analysis.
Comparisons between two groups were performed using an unpaired
Student's t-test, and comparisons between multiple groups were
performed using a one-way analysis of variance followed by Tukey's
post hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Administration of CBX prevents
MCT-induced hemodynamic changes
A change in the shape of the pulmonary artery
outflow waveform in combination with PAAT have been demonstrated to
be a good echocardiographic indicator of PH (36); therefore, doppler echocardiography
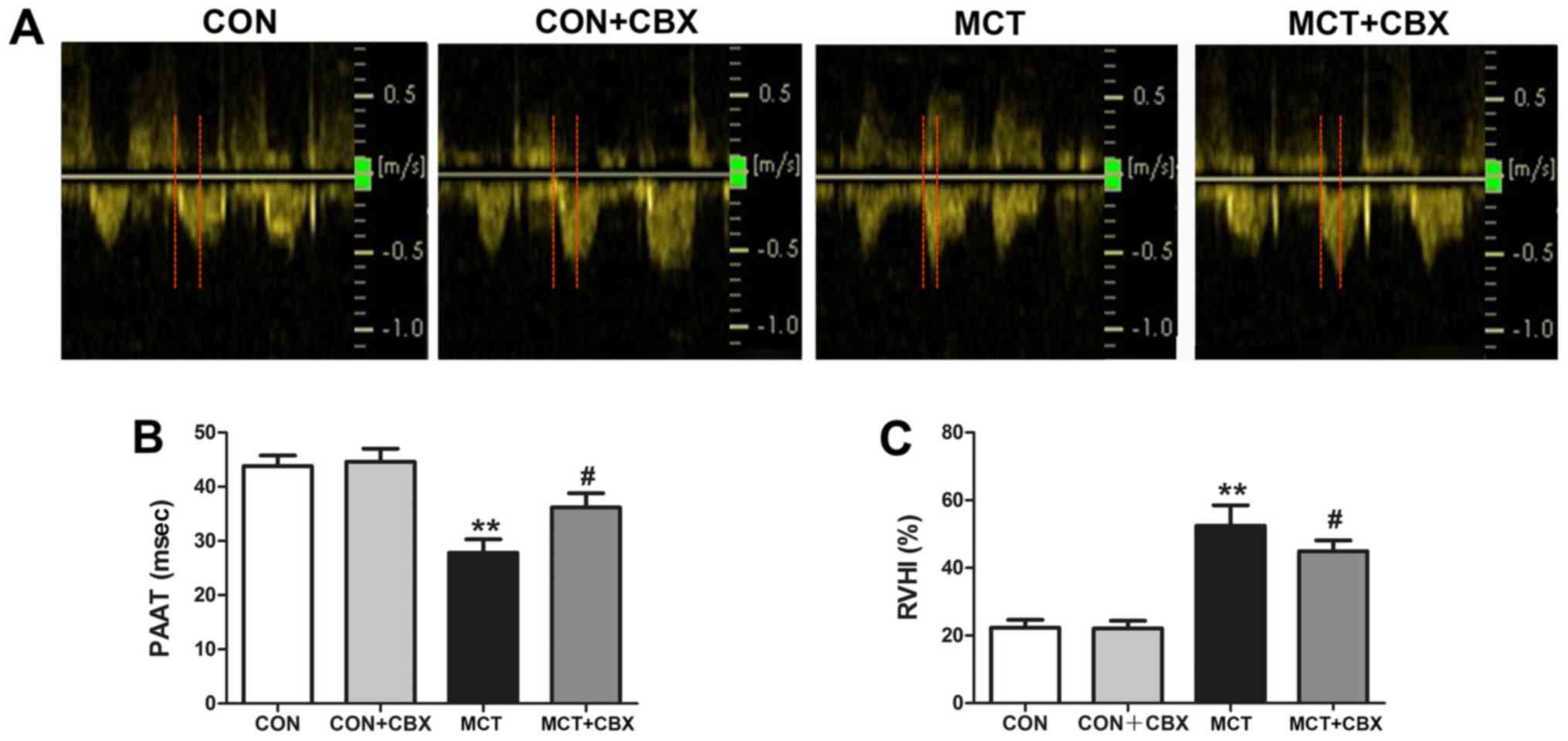
was used to confirm the establishment of MCT-induced PH (Fig. 1A and B). Fig. 1A shows representative images of
the pulmonary artery flow pattern among the four groups of rats.
Compared with the Control and CBX group, the MCT rats exhibited a
profile of pulmonary artery flow with more triangular,
dagger-shaped signals, indicative of high resistance in the
pulmonary artery (Fig. 1A). By
contrast, CBX administration attenuated the high resistance induced
by MCT (Fig. 1A). Doppler
echocardiography of the MCT group indicated that PAAT was
significantly decreased compared with the control group (Control,
43.80±1.99 ms; MCT, 27.80±2.52 ms; P<0.01; Fig. 1B), indicating that the rats in the
MCT group developed PH. The effect of CBX on pulmonary artery
function was investigated by measuring PAAT at the end of the
treatment period. Intraperitoneal administration of CBX
significantly increased PAAT compared with the MCT-treated rats
(MCT=27.80±2.52 ms; MCT + CBX=36.20 ± 2.60 ms; P<0.05; Fig. 1B). These results suggest that CBX
administration prevented the effect of MCT-mediated induction of
PH.
RVH is associated with PH, therefore right
ventricular hypertrophy was evaluated by measuring the RVHI. RVHI
in the MCT group was significantly increased compared with the
control group (Control=22.33±0.94%; MCT=52.46±2.48%; P<0.01;
Fig. 1C), suggesting that rats
exhibited RVH as a consequence of elevated pulmonary pressure when
treated with MCT. CBX administration of MCT-treated rats
ameliorated the deleterious effect of PH-induced RVH in rats, as
depicted by the decreased RVHI compared with the rats treated with
MCT alone (MCT=52.46±2.48%; MCT + CBX=44.90±1.32%; P<0.05;
Fig. 1C).
Administration of CBX attenuates
MCT-induced pulmonary vascular remodeling, lung fibrosis and
inflammatory cell infiltration
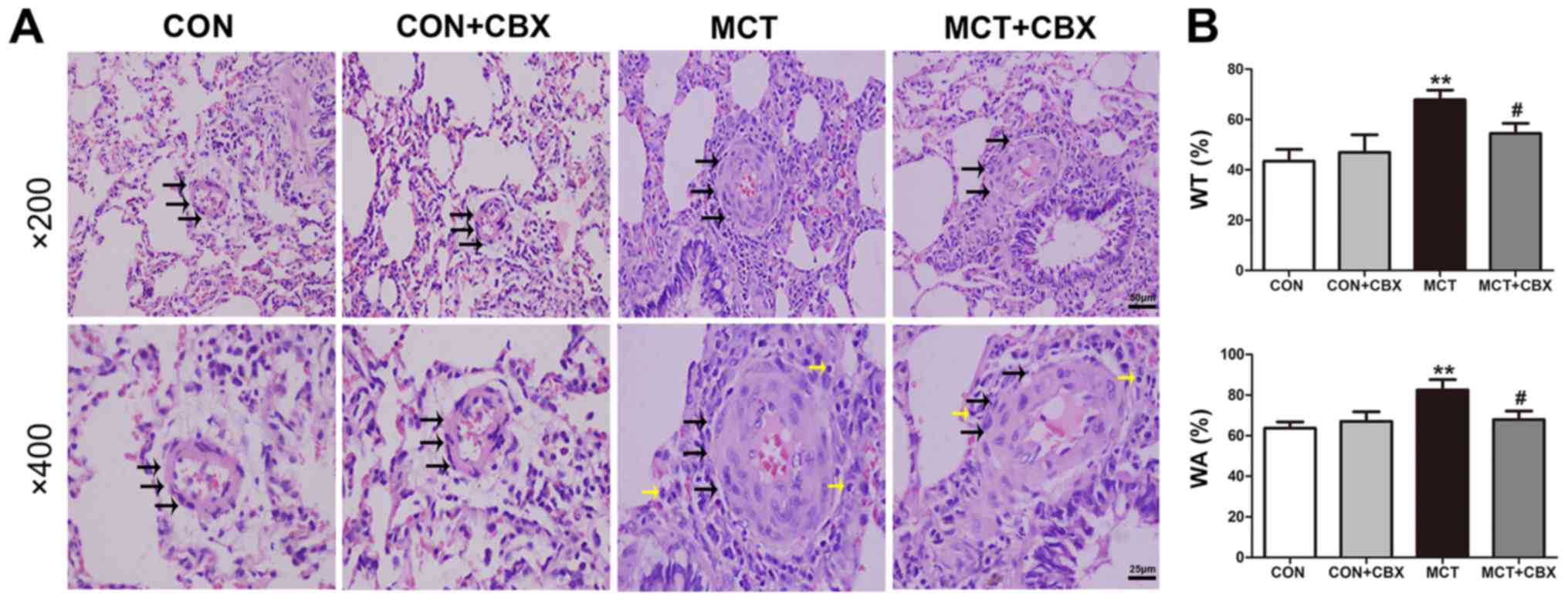
The effect of MCT on pulmonary vascular remodeling
in small arterioles and medium arterioles of lungs in the rat model
were determined, and the effect of CBX administration on
MCT-induced pulmonary arteriolar remodeling were evaluated
(Fig. 2 and 3). Compared with the control rats, MCT
treatment significantly induced muscularization, stenosis and
occlusion of the arterioles in the lungs, and this was accompanied
by inflammatory cell infiltration around the vessel wall and lung
tissues (Fig. 2A and 3A). The WT% in the small
(Control=20.45±2.24; MCT=64.21±4.47%) and medium pulmonary
arterioles (Control=43.51±4.62; MCT=67.82±3.88%), and the WA% in
the small (Control=58.74±3.15; MCT=91.24±3.34%) and medium
pulmonary arterioles (Control=63.71±3.07; MCT=82.56±5.04%) were
significantly increased in the MCT-treated rats compared with the
control (all P<0.01; Fig. 2B
and 3B). CBX administration
prevented medial hypertrophy, occlusion and muscularization of the
pulmonary arterioles, as well as inflammatory infiltration compared
with the MCT-treated rats (Fig.
2A and Fig. 3A). In
particular, CBX significantly decreased the MCT-induced increase in
WT% in the small (MCT=64.21±4.47%; MCT + CBX=50.41±3.80%) and in
medium pulmonary arterioles (MCT=67.82±3.88%; MCT +
CBX=54.53±3.98%), and the WA% in the small (MCT=91.24±3.34%; MCT +
CBX=77.62±5.04%) and medium pulmonary arterioles (MCT=82.56±5.04%;
MCT + CBX=67.93 ±4.20%) (all P<0.05; Fig. 2B and Fig. 3B).
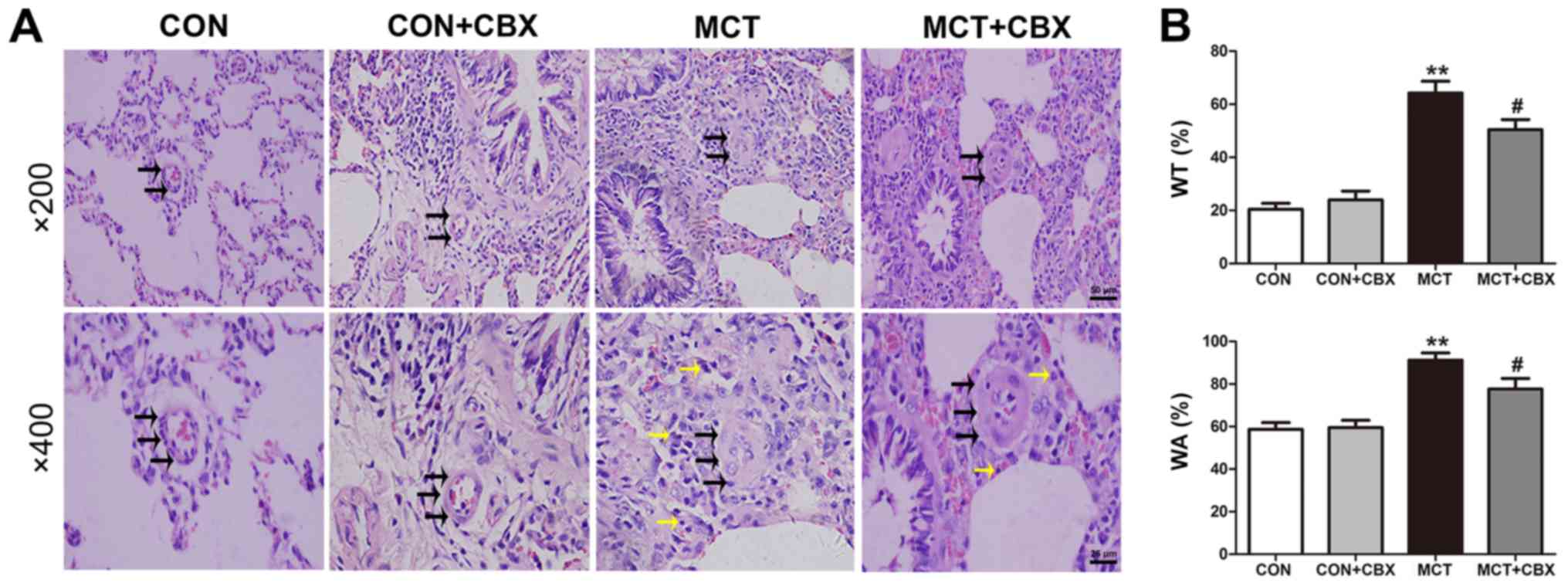 | Figure 2MCT-induced remodeling of small
pulmonary arterioles and inflammatory cell infiltration is
alleviated by CBX. (A) Representative hematoxylin and eosin
staining demonstrating histopathological changes in small pulmonary
arterioles at magnification, ×200 (scale bar=50 µm; upper
panel) and magnification, ×400 (scale bar=25 µm; lower
panel). Pulmonary arteriole walls in the control group were thin
and single layered. The intima, media and externa were difficult to
distinguish. Pulmonary vascular remodeling was observed in
MCT-treated animals, characterized by obliterated vessels (black
arrows); no obliteration was detected in the control rats and the
rats treated with a single intraperitoneal injection of CBX. CBX
prevented pulmonary arterial obliteration in small pulmonary
arterioles of MCT-treated animals. The black and yellow arrows
indicate pulmonary arterial obliteration and perivascular
infiltration of inflammatory cells in small pulmonary arterioles,
respectively. (B) WT% and WA% of the MCT-treated rats were
significantly increased, and CBX treatment of the MCT rats
significantly decreased WT% and WA%. Data are presented as the mean
± standard error of the mean of 6 rats/group.
**P<0.01 vs. control. #P<0.05 vs. MCT
group. CBX, carbenoxolone; MCT, monocrotaline; CON, control; WT%,
percentage of vascular wall thickness; WA%, percentage of the
vascular wall area. |
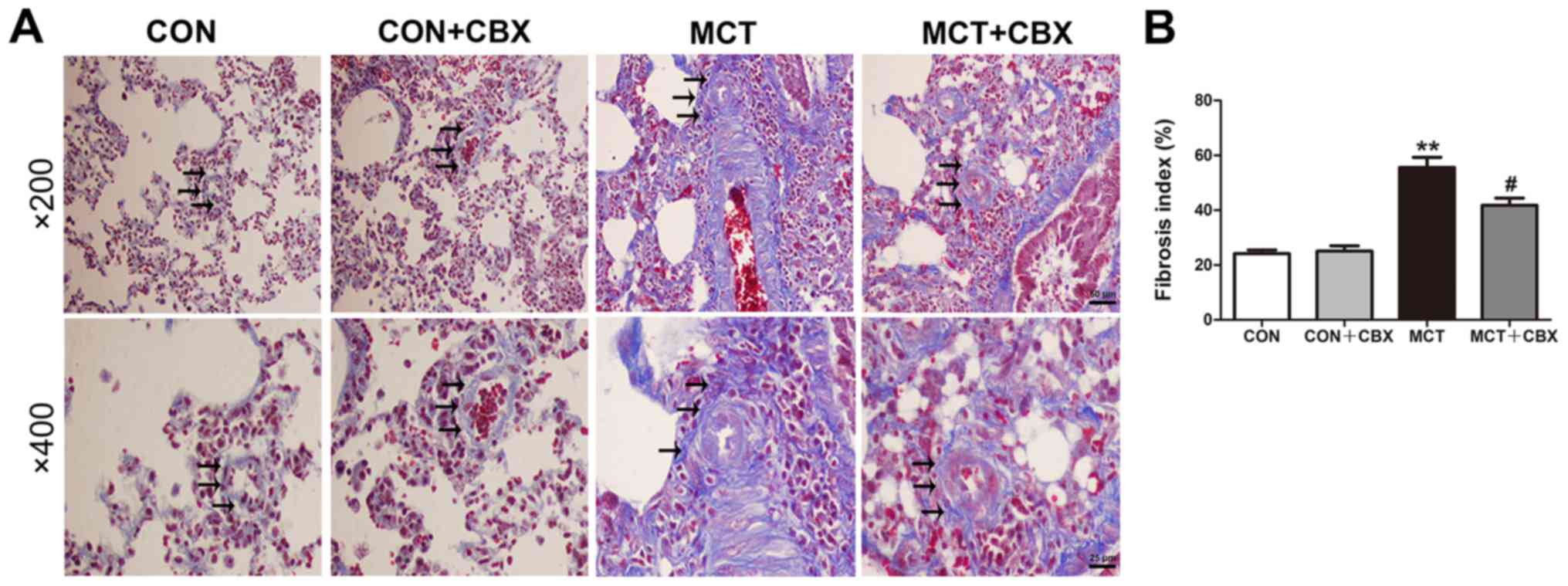
Lung fibrosis is a primary feature of MCT-induced
pulmonary inflammation (37).
Masson trichrome staining was used to demonstrate the presence of
fibrosis by collagen deposition. The results of Masson staining
indicated that the MCT-treated rats displayed notable collagen
deposition in the interstitium and arteries of the lungs compared
with the control rats (Control=24.18±1.31; MCT=55.55±3.76%;
P<0.01). CBX administration significantly decreased lung
interstitial fibrosis (MCT=55.55±3.76; MCT + CBX=41.78±2.62%;
P<0.05; Fig. 4A and B). The
histological results suggest that CBX exerted protective effects on
MCT-induced pulmonary vascular remodeling and lung fibrosis.
CBX administration prevents MCT-induced
changes in cytokine production in lung tissues and serum
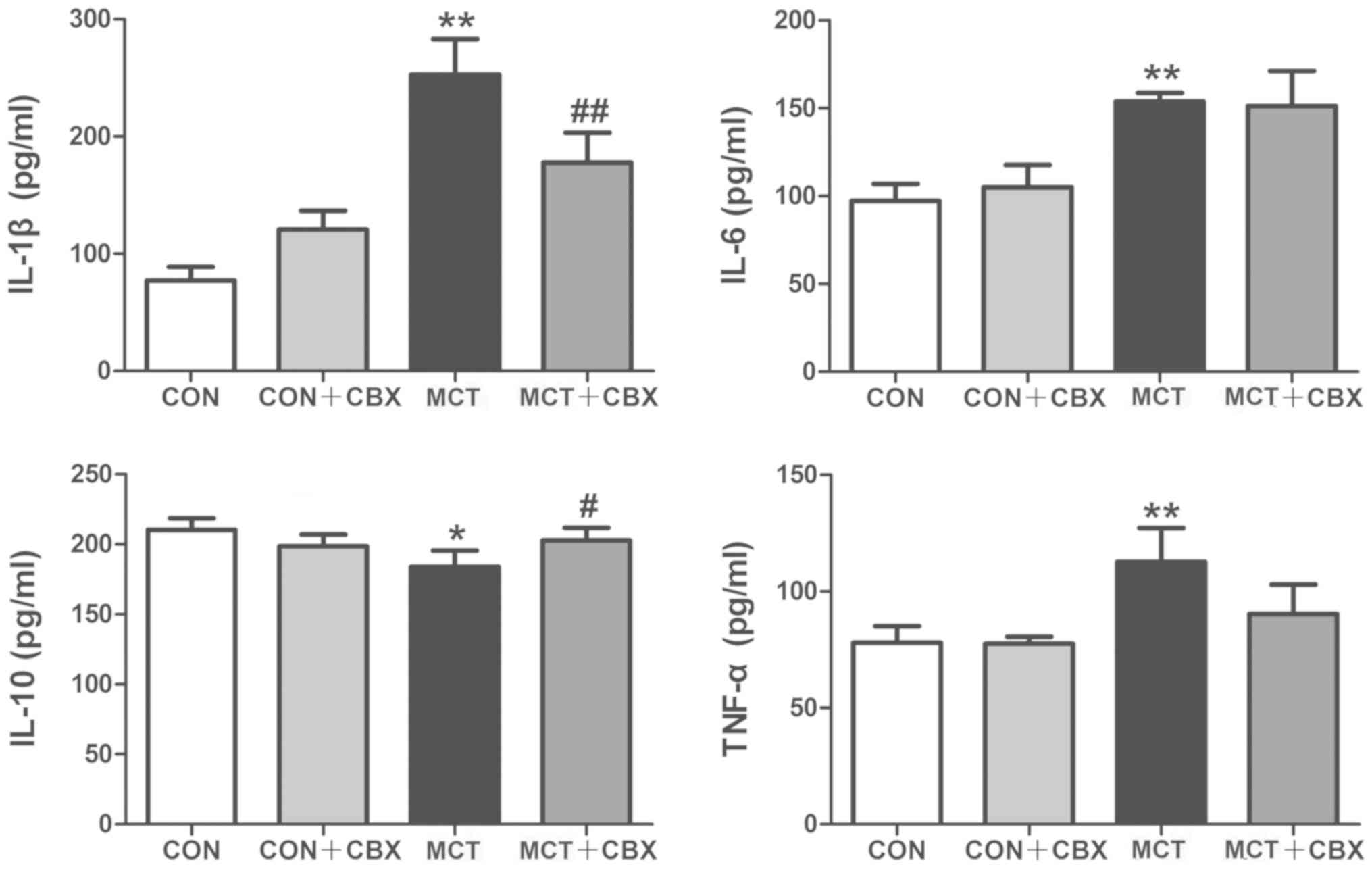
The inflammatory markers IL-1β, IL-6, IL-10 and
TNF-α serve pivotal roles in MCT-induced inflammation and pulmonary
vascular remodeling (38). To
identify the possible mechanisms involved in the beneficial effects
of CBX on MCT-induced systemic inflammation in rats, the levels of
these cytokines were measured in the lungs and serum using ELISAs.
Compared with the control rats, lung tissues and serum from the MCT
rats exhibited a significant increase in: IL-1β in the lung
(Control=130.0±6.23 pg/ml; MCT=702.50±27.86 pg/ml; P<0.01;
Fig. 5) and serum
(Control=77.16±5.80 pg/ml; MCT=253.0±15.13 pg/ml; P<0.01;
Fig. 6); IL-6 in the lung
(Control=93.24±2.87 pg/ml; MCT=232.3±15.05 pg/ml; P<0.01;
Fig. 5) and serum
(Control=97.20±4.85 pg/ml; MCT=153.90±2.44 pg/ml; P<0.01;
Fig. 6); and TNF-α concentration
in the lung (Control=130.10±7.52pg/ml; MCT=243.10±11.39 pg/ml;
P<0.01; Fig. 5) and serum
(Control=77.89±3.58 pg/ml; MCT=112.70±7.28 pg/ml; P<0.01;
Fig. 6), and a significant
decrease in IL-10 levels in the lung (Control=391.60±21.09 pg/ml;
MCT=227.80±15.30 pg/ml; P<0.01; Fig. 5) and serum (Control=210.20±4.20
pg/ml; MCT =184.20±5.69 pg/ml; P<0.05; Fig. 6). In contrast, CBX administration
significantly decreased the levels of pro-inflammatory cytokines:
IL-1β (MCT =702.50±27.86 pg/ml; MCT + CBX =461.40±21.84 pg/ml),
IL-6 (MCT =232.30±15.05 pg/ml; MCT + CBX =145.80±4.00 pg/ml) and
TNF-α (MCT =243.10±11.39 pg/ml; MCT + CBX =149.50±7.18 pg/ml) in
lung tissues (all P<0.01; Fig.
5) in MCT-treated rats, and significantly reduced the
MCT-induced increase in IL-1β (MCT =253.00±15.13 pg/ml; MCT + CBX
=177.50±12.78 pg/ml; P<0.01; Fig.
6) in the serum; however, the MCT-induced increases in the
serum levels of IL-6 and TNF-α were not attenuated by CBX when
compared with the MCT-treated group (P>0.05; Fig. 6). In addition, the levels of IL-10
in the lung (MCT =227.80±15.30 pg/ml; MCT + CBX =310.70±16.23
pg/ml; P<0.01; Fig. 5) and
serum (MCT =184.20±5.69 pg/ml; MCT + CBX =202.90±4.43 pg/ml;
P<0.05, Fig. 6) of MCT-treated
rats were significantly increased in the CBX treated rats. These
results demonstrate that CBX administration may alleviate pulmonary
inflammation induced by MCT in rats.
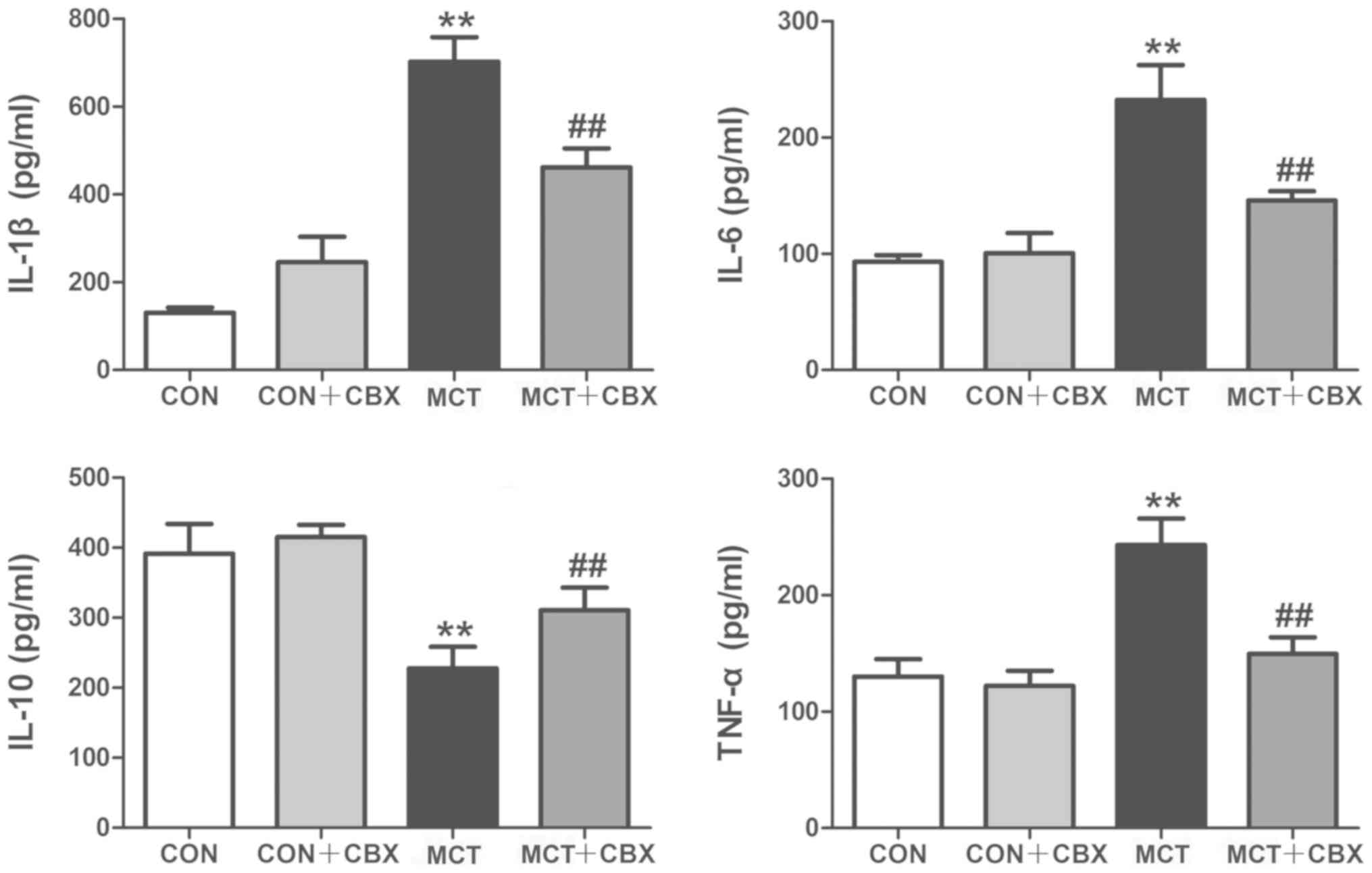 | Figure 5Effect of CBX on cytokine production
in rat lung tissues treated with MCT. Images indicate the protein
expression levels of IL-1β, IL-6, IL-10 and TNF-α in rat lung
tissues. Pulmonary levels of IL-1β, IL-6 and TNF-α were
significantly elevated in MCT-treated animals compared with the
control rats. Pulmonary levels of IL-10 were decreased in MCT
treated rats compared with the control group. Pro-inflammatory
cytokines IL-1β, IL-6 and TNF-α were increased in MCT-induced
inflammation, and CBX treatment significantly decreased the levels
of these cytokines. IL-10 levels were increased in the lung tissues
of the CBX-treated rats compared with the MCT-treated animals. Data
are presented as the mean ± standard error of the mean of 6
rats/group. **P<0.01 vs. control.
##P<0.01 vs. MCT treatment group. CBX, carbenoxolone;
IL, interleukin; MCT, monocrotaline; CON, control; TNF-α, tumor
necrosis factor-α. |
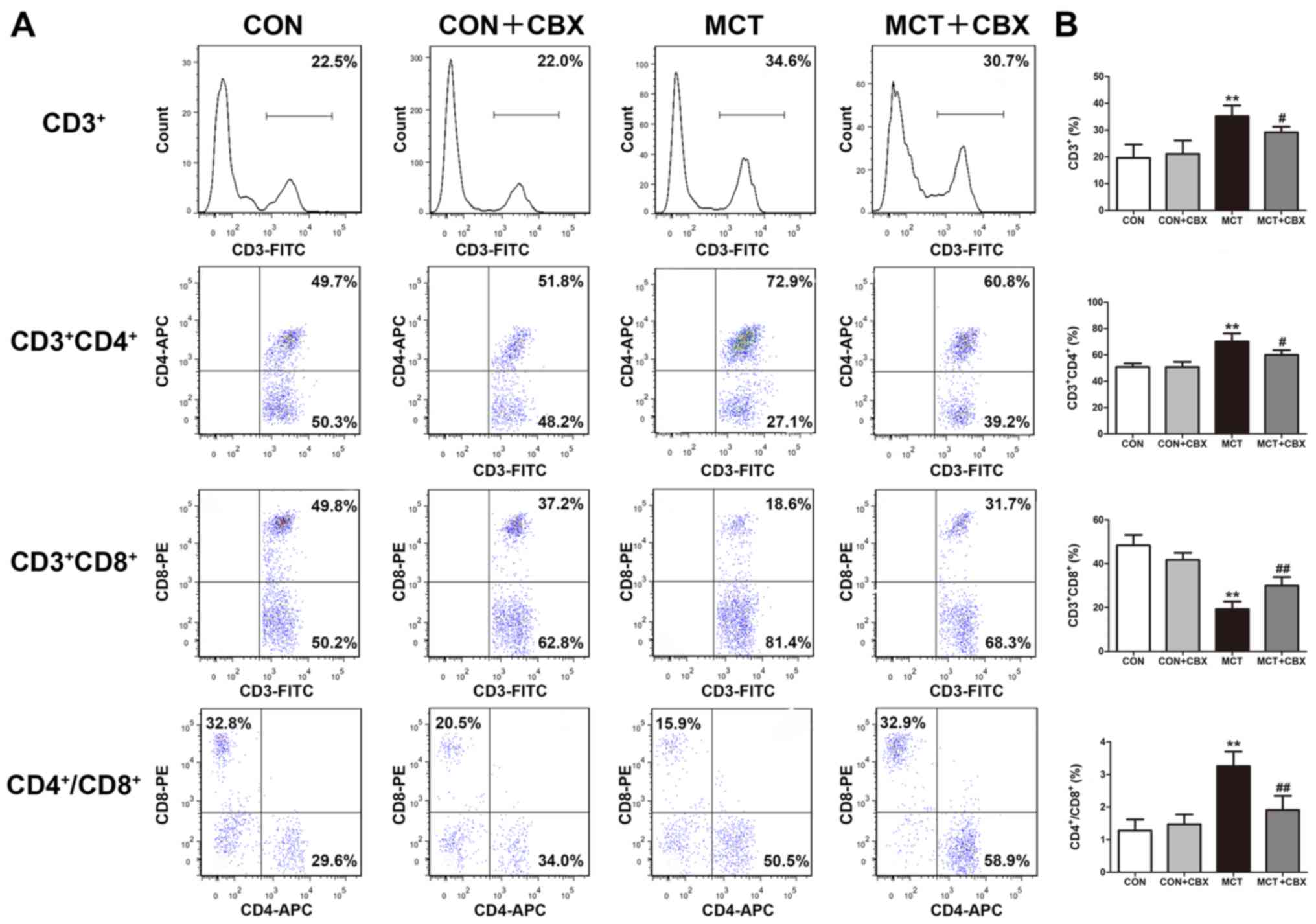
Administration of CBX alleviates
MCT-induced lung inflammation
It has previously been reported that T lymphocytes
serve a potential pathological role in MCT-induced pulmonary
inflammation (14,39) and in patients with PAH (39). To determine whether CBX prevented
MCT-induced changes to the adaptive immune system, T cell subset
analysis in lung tissues was performed. As demonstrated in Fig. 7, in the rats treated with MCT, the
percentages of the total CD3+ (Control=19.60±2.51%;
MCT=35.20±2.01%) and CD4+ (Control=50.78±1.45%;
MCT=70.23±3.08%) T cells in lung tissues were significantly
increased (both P<0.01; Fig.
7B), whereas CD8+ T cell counts were significantly
decreased in the lungs of MCT-treated rats (Control=48.40±2.39%;
MCT=19.25±1.74%; P<0.01; Fig.
7B) compared with the control rats. MCT treatment resulted in a
significant disruption of the CD4+/CD8+ T
cell subset ratios (Control=1.28±0.17; MCT=3.26±0.22; P<0.01;
Fig. 7B) in the lung tissues.
However, treatment with CBX resulted in significant decreases in
the percentages of CD3+ (MCT=35.20±2.01%; MCT +
CBX=29.13±1.06%; P<0.05), CD4+ T cells
(MCT=70.23±3.08%; MCT + CBX =59.93±1.89%; P<0.05) and
CD4+/CD8+T cell subset ratios (MCT=3.26±0.22;
MCT + CBX=1.91±0.22; P<0.01) as well as a significant increase
in the percentage of CD8+ T cells (MCT=19.25±1.74%; MCT
+ CBX=29.95±1.99%; P<0.01) compared with the MCT-treated rats
(Fig. 7B).
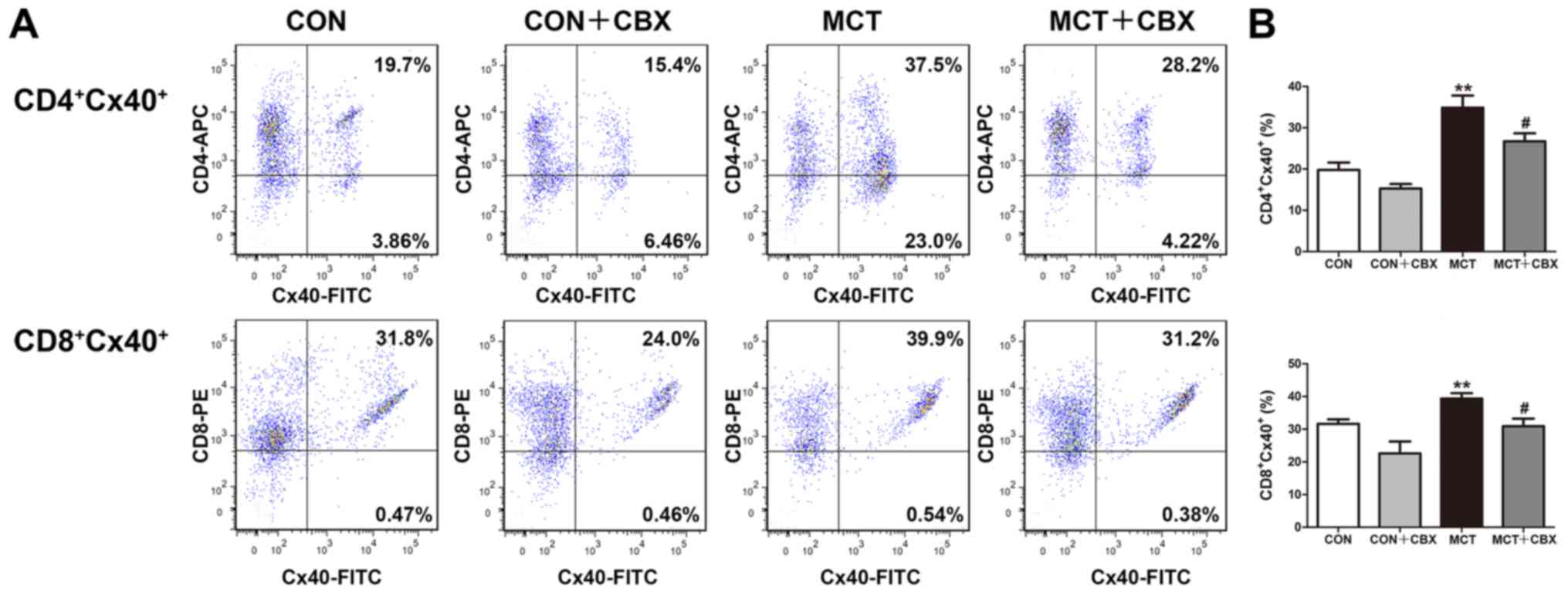
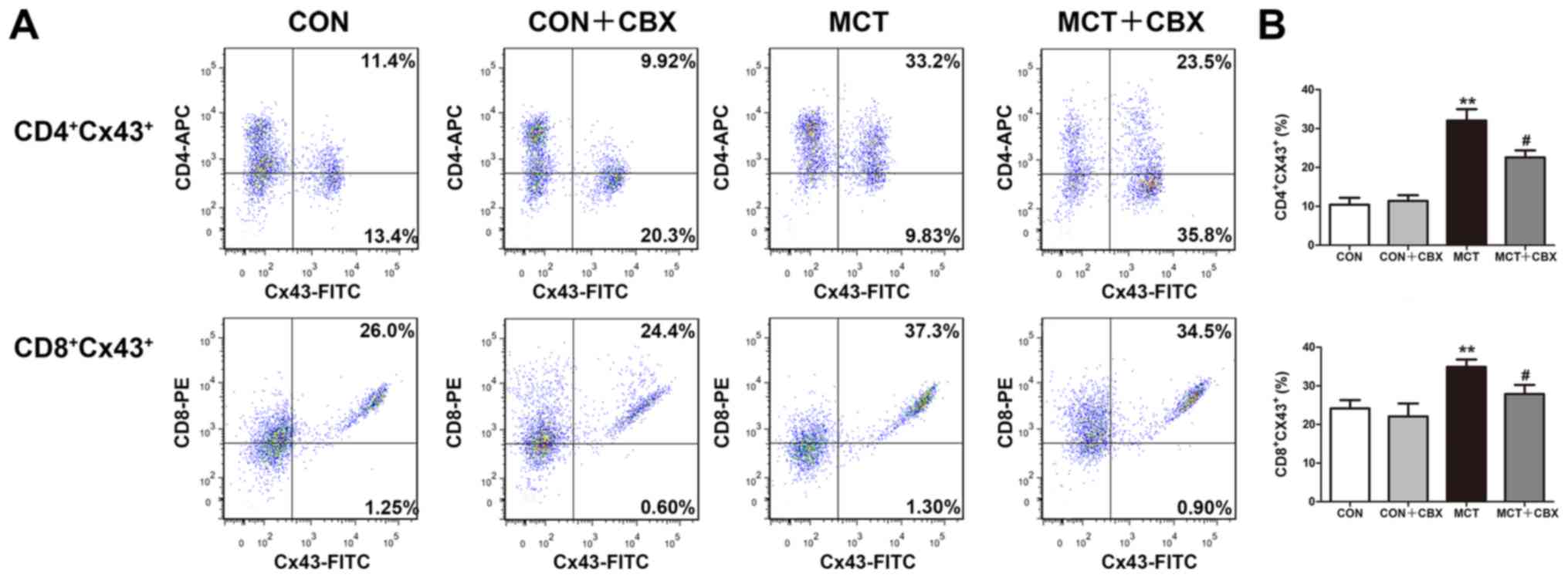
Treatment with CBX decreases Cx40 and
Cx43 protein expression levels in T lymphocyte subsets in the lungs
of MCT-treated rats
As the pro-inflammatory response is associated with
upregulation of Cx expression or the functionality of Cx-mediated
gap junction in T lymphocytes (16-18,40-42), the effect of MCT treatment on Cx
expression in T lymphocytes in lung tissues of MCT-treated rats was
studied, and in particular whether non-specific blocking of
channels formed of Cx decreased MCT-induced pulmonary or systemic
inflammation. Flow cytometry was used to detect Cx40 and Cx43
expression levels in CD4+ and CD8+ T
lymphocyte subsets from lung tissues. As indicated in Fig. 8 and 9, the percentages of
CD4+Cx40+ (Control=19.78±1.82%;
MCT=34.85±2.97%; P<0.01; Fig.
8B), CD8+Cx40+ (Control=31.67±1.34%;
MCT=39.37±1.67%; P<0.05; Fig.
8B), CD4+Cx43+ (Control=10.43±1.76%; MCT
=32.07±2.91%; P<0.01; Fig. 9B)
and CD8+Cx43+ (Control=24.17±2.15%; MCT
=34.90±1.90%; P<0.05; Fig. 9B)
double-positive T lymphocytes were significantly increased in the
lung tissues of MCT-treated rats compared with the control rats
treated with normal saline. Compared with the MCT-treated group,
CBX administration significantly decreased the MCT-induced
increases in the percentages of CD4+Cx40+
(MCT, 34.85±2.97%; MCT + CBX 26.70±1.97%; P<0.05; Fig. 8B), CD8+Cx40+
(MCT, 39.37±1.67%; MCT + CBX 30.92±2.30%; P<0.05; Fig. 8B), CD4+Cx43+
(MCT, 32.07±2.91%; MCT + CBX 22.58±1.79%; P<0.05; Fig. 9B) and
CD8+Cx43+ (MCT, 34.90±1.90%; MCT + CBX,
27.87±2.39%; P<0.05; Fig. 9B)
double-positive T lymphocytes in lung tissues.
Discussion
Elevated PAP, pulmonary vascular remodeling and RVH
are pathophysiological characteristics of PH (43) and MCT-induced lung damage
(11). In the present study, PAP
was assessed using the echo parameter, PAAT. A decrease in PAAT is
associated with an increase in PAP (30). In the present study, MCT-induced
lung damage was accompanied by a decrease in PAAT, an increase in
RVHI and thickening of the pulmonary arteriole walls in the
MCT-treated rats. A number of studies have demonstrated that
inflammation is an important initial step of pulmonary arteriolar
remodeling and lung damage in animal models of PH and patients with
PH (3,44), and these alterations are closely
associated with the development and outcome of PH (44). The histopathological data
corroborated these results, with notable inflammatory infiltration
in the lungs of rats treated with MCT, accompanied by accumulation
of the pro-inflammatory molecules, IL-1β, IL-6 and TNF-α, in the
lung tissues and serum. Inflammatory cells, in particular T
lymphocytes, have been observed in the lungs and in the walls of
resistance arteries in patients with PH (5). In the present study, the lung
tissues exhibited significantly increased numbers of
CD4+ T lymphocytes and significantly decreased numbers
of CD8+ T lymphocytes in the MCT-treated rats. The
elevation of CD4+ T cells and decrease in
CD8+ T cells in the lungs of MCT rats was consistent
with data from a previous study, which demonstrated the similar
changes to the T lymphocyte counts in the peripheral blood of
MCT-treated rats (2).
The presence of lung inflammation in pulmonary
inflammatory diseases indicates that drugs targeting the adaptive
immune component of the disease should be considered for treatment
(13). The structures mediating
direct cell-cell and cell extracellular environment interactions
are Cxs, which form channels including gap junction channels and
hemi-channels. Cxs participate in inflammatory responses and
regulate production of cytokines (40-42,45). Cx40 and Cx43 in T cells may serve
a pivotal role in mediating inflammatory changes (40-42,46) and the progression of
hypertension-mediated inflammatory response (16-20), as decreases in Cx expression or
blocking of Cx43 channels in T cells resulted in a decreased
production of inflammatory cytokines by T lymphocytes, decreased
activation and proliferation of T lymphocytes and attenuated
circulating inflammatory cell accumulation in spontaneously
hypertensive rats and hypertensive patients (16-20). Therefore, Cxs represent novel
potential targets for the treatment of cardiovascular disease
induced chronic low-grade inflammation. To further ascertain the
regulatory role of Cxs in T lymphocytes in MCT-induced PH mediated
lung inflammation, and whether Cxs were a suitable target for
therapeutic intervention in lung inflammation, CBX, a Cx inhibitor,
was used to investigate the effect of inhibiting Cx function in
lung inflammation and vascular remodeling in MCT-treated rats.
Previous studies have demonstrated the effectiveness of CBX in the
treatment of acute lung inflammation and pulmonary inflammatory
diseases (22-24). CBX inhalation significantly
alleviates asthma-induced lung inflammation by downregulating the
production of inflammatory cytokines, and by decreasing
inflammatory infiltration in perivascular areas (24). In addition, CBX has been suggested
to be effective in the treatment of Th17-mediated autoimmune
diseases by decreasing the number of Th17 cells and inhibiting the
synthesis of IL-23 in antigen presenting cells (25). The present study demonstrated that
CBX administration in the MCT-treated rats decreased PAAT, RVHI and
pulmonary vascular remodeling. The data from the collagen staining
assay also demonstrated a significant therapeutic effect of CBX in
decreasing collagen fiber proliferation and inflammatory
infiltration. Similarly, CBX administration decreased MCT-induced
lung inflammation by downregulating the levels of IL-1β, IL-6 and
TNF-α, and by decreasing the counts of
CD3+CD4+ T cells in lung tissues.
Furthermore, IL-10 levels, an anti-inflammatory cytokine, were
increased in the lungs of CBX-administered rats. These results are
similar to our previous studies (17,19,20) indicating that gap junction
blockers or anti-inflammatory agents (H2S donor and
β-estradiol) prevent hypertension-mediated inflammation by
decreasing inflammatory cytokine synthesis/secretion and preventing
an imbalance between T cell subsets in experimental rats and
patients with hypertension. The results suggest that CBX may
alleviate MCT-induced PAH and may protect against MCT-induced
inflammatory response by suppressing inflammation.
As aforementioned, our recent studies have
demonstrated the role of Cx-based gap junctions in
hypertension-mediated inflammatory responses (16-20), and various anti-inflammatory
agents and Cx blockers were observed to decrease inflammation by
inhibiting Cx expression or function in peripheral blood
lymphocytes of hypertensive animals and patients (16,17). In an attempt to clarify the
molecular mechanisms underlying the anti-inflammatory effects of
CBX in MCT-mediated pulmonary inflammation in rats, the expression
of Cx40 and Cx43 in T lymphocyte subsets was analyzed in lung
tissue homogenates. The results indicated that the percentages of
Cx40 and Cx43 expressing T lymphocytes were upregulated in lungs of
the MCT rats, indicating an association between Cxs in T
lymphocytes and MCT-mediated lung inflammation. This suggests a
functional significance of Cxs in controlling T lymphocyte-mediated
pulmonary inflammation and pulmonary vascular remodeling. CBX
administration decreased the percentages of Cx40- and
Cx43-expressing T lymphocytes in the lungs of MCT-treated rats.
Therefore, it may be assumed that CBX exerts its anti-inflammatory
and protective effects on lungs in MCT-treated by inhibiting Cx40
and Cx43 expression in T lymphocytes. These results highlight
mononuclear Cxs as a potential target for treating pulmonary
inflammation.
The present study has the following limitations.
Firstly, only one animal model of MCT-induced pulmonary
inflammation was established; other rodent models of PH or lung
inflammation and larger animals should be examined, to verify
long-term efficacy and safety of CBX therapy on PH or lung
inflammation. Secondly, the present study did not analyze the
expression levels of cytokines at the protein level by western blot
analysis. Although the analysis of cytokines protein expression
levels may provide a more convincing evidence for the therapeutic
effect of CBX on MCT induced pulmonary inflammation, a recent study
demonstrated that carbenoxolone may decrease pro-inflammatory
cytokine levels in lungs [IL-17, C-C motif chemokine 5, chemokine
(C-X-C motif) ligand 1, C-C motif chemokine 2, TNF-α, and IL-6] in
a murine model of lung ischemia-reperfusion injury (47). Therefore, carbenoxolone is
hypothesized to prevent the production of pro-inflammatory
cytokines from serum and lung in pulmonary inflammation. In
addition, only 1 experimental method was used to detect the
expression levels of Cxs. Future studies will be required to
further determine the alterations of Cxs expression at a protein
level in T lymphocytes of lung tissues and peripheral blood using
immunoblotting. Thirdly, CBX has not exhibited a clear selectivity
for any particular Cx subtypes; other mechanisms of action,
unrelated to Cxs inhibition, are considered to be involved in their
anti-inflammatory effects (43).
Thus, only the specific blockade of Cxs in T lymphocytes completely
explain the role of Cxs in MCT-induced pulmonary inflammation.
Although we have also tried to prevent MCT-induced pulmonary
inflammation in vivo by i.p. injection of Cx43 specific
mimetic peptide Gap27 (Gap27), both chronic (4 weeks) and
short-term (1 week) treatment schedules of Gap27 resulted in the
death of normal and MCT-treated animals, owing to the systemic
effect of Gap27 (Fan et al, unpublished data). Therefore, systemic
knockdown of Cx or systemic inhibition of Cxs systemically may
affect reproduction and life span or survival of experimental
animals, which is not beneficial to the study of chronic lung
inflammation. However, conditional T lymphocytes-specific knockdown
of Cxs using Cre/loxP-regulated RNA interference (RNAi) may be the
best choice for studying the role of Cxs in MCT or pulmonary
hypertension mediated inflammation, although it is not clear
whether RNAi knockdown of Cx43 will affect survival in hypertensive
animals. Further studies will be designed to determine whether Cxs
is involved in pulmonary inflammatory disease using conditional T
lymphocyte-specific knockdown of Cxs, or small interfering RNA
(siRNA) targeting of Cxs in T lymphocytes by siRNA strategies.
Finally, although having determined the involvement of Cxs in
pulmonary inflammation in the present study and previous studies
(43,48), future studies are required to
examine whether the changes of Cxs and the effect of CBX on Cxs are
present in different pulmonary inflammation-associated cell types
of the lung tissues, including pulmonary arterial endothelium,
smooth muscle and alveolar macrophages.
Despite these limitations, the present study
provided important evidence that Cxs and Cxs-based channels in T
lymphocytes may have an important role in pulmonary inflammatory
diseases, and inhibition of the Cxs-based channels using CBX
attenuated MCT-induced pulmonary inflammation and pulmonary
arteriolar thickening, as well as RVH by decreasing pulmonary
inflammatory monocyte infiltration and inhibiting pro-inflammatory
cytokine production in the lungs. The beneficial effects of CBX
were accompanied by attenuation of Cx40 and Cx43 expression in T
lymphocytes in lung tissues in the CBX-treated MCT rats. Together,
the present and previous studies from our study group support the
hypothesis that Cx and Cx-based channels may be novel therapeutic
targets for decreasing the T lymphocyte-mediated inflammatory
response in PH and other pulmonary inflammatory diseases.
Acknowledgments
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81660271 to
KM; grant no. 81460098 to XL; grant no. 81600325 to LZ; and grant
no. 81560081 to JS) and the International Cooperation Project of
Shihezi University (grant no. GJHZ201603 to KM).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
KM and JS conceived and designed the experiments.
ZF, LW, LLi and XL performed the experiments. LZ, ZF and LLiu
analyzed the data. LZ wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The protocol of the study was approved by
Institutional Animal Care and Use Committees (permit no.
A2019-027-01) of the Medical College of Shihezi University, and all
animal handling and experimental procedures were performed in
accordance with guidelines for the Care and Use of Laboratory
Animals published by the United States of America National
Institutes of Health (26).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sada Y, Dohi Y, Uga S, Higashi A,
Kinoshita H and Kihara Y: Non-suppressive regulatory T cell subset
expansion in pulmonary arterial hypertension. Heart Vessels.
31:1319–1326. 2016. View Article : Google Scholar
|
|
2
|
Gao L, Liu J, Hao Y, Zhao Z, Tan H, Zhang
J, Meng N, Zheng Q, Wang Z and Zhang Y: Chronic intermittent
hypobaric hypoxia attenuates monocrotaline-induced pulmonary
arterial hypertension via modulating inflammation and suppressing
NF-κB/p38 pathway. Iran J Basic Med Sci. 21:244–252.
2018.PubMed/NCBI
|
|
3
|
Deng Y, Guo SL, Wei B, Gao XC, Zhou YC and
Li JQ: Activation of nicotinic acetylcholine α7 receptor attenuates
progression of monocrotaline-induced pulmonary hypertension in rats
by downregulating the NLRP3 inflammasome. Front Pharmacol.
10:1282019. View Article : Google Scholar
|
|
4
|
Chen F, Wang H, Zhao J, Yan J, Meng H,
Zhan H, Chen L and Yuan L: Grape seed proanthocyanidin inhibits
monocro-taline-induced pulmonary arterial hypertension via
attenuating inflammation: In vivo and in vitro studies. J Nutr
Biochem. 67:72–77. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Austin ED, Rock MT, Mosse CA,
Vnencak-Jones CL, Yoder SM, Robbins IM, Loyd JE and Meyrick BO: T
lymphocyte subset abnormalities in the blood and lung in pulmonary
arterial hypertension. Respir Med. 104:454–462. 2010. View Article : Google Scholar :
|
|
6
|
Savai R, Pullamsetti SS, Kolbe J, Bieniek
E, Voswinckel R, Fink L, Scheed A, Ritter C, Dahal BK, Vater A, et
al: Immune and inflammatory cell involvement in the pathology of
idiopathic pulmonary arterial hypertension. Am J Respir Crit Care
Med. 186:897–908. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rabinovitch M, Guignabert C, Humbert M and
Nicolls MR: Inflammation and immunity in the pathogenesis of
pulmonary arterial hypertension. Circ Res. 115:165–175. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang JM, Zhou R, Zhang M, Tan HR and Yu
JQ: Betaine attenuates monocrotaline-induced pulmonary arterial
hypertension in rats via inhibiting inflammatory response.
Molecules. 23:E12742018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pang Y, Liang MT, Gong Y, Yang Y, Bu PL,
Zhang M and Yao HC: HGF reduces disease severity and inflammation
by attenuating the NF-κB signaling in a rat model of pulmonary
artery hypertension. Inflammation. 41:924–931. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li XQ, Wang HM, Yang CG, Zhang XH, Han DD
and Wang HL: Fluoxetine inhibited extracellular matrix of pulmonary
artery and inflammation of lungs in monocrotaline-treated rats.
Acta Pharmacol Sin. 32:217–222. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nogueira-Ferreira R, Vitorino R, Ferreira
R and Henriques-Coelho T: Exploring the monocrotaline animal model
for the study of pulmonary arterial hypertension: A network
approach. Pulm Pharmacol Ther. 35:8–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang B, Niu W, Xu D, Li Y, Liu M, Wang Y,
Luo Y, Zhao P, Liu Y, Dong M, et al: Oxymatrine prevents hypoxia-
and mono-crotaline-induced pulmonary hypertension in rats. Free
Radic Biol Med. 69:198–207. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cohen-Kaminsky S, Hautefort A, Price L,
Humbert M and Perros F: Inflammation in pulmonary hypertension:
What we know and what we could logically and safely target first.
Drug Discov Today. 19:1251–1256. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang W, Wang YL, Chen XY, Li YT, Hao W,
Jin YP and Han B: Dexamethasone attenuates development of
monocrotaline-induced pulmonary arterial hypertension. Mol Biol
Rep. 38:3277–3284. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao H, Xue Y, Guo Y, Sun Y, Liu D and
Wang X: Inhibition of endocan attenuates monocrotaline-induced
connective tissue disease related pulmonary arterial hypertension.
Int Immunopharmacol. 42:115–121. 2017. View Article : Google Scholar
|
|
16
|
Ni X, Li XZ, Fan ZR, Wang A, Zhang HC,
Zhang L, Li L, Si JQ and Ma KT: Increased expression and
functionality of the gap junction in peripheral blood lymphocytes
is associated with hypertension-mediated inflammation in
spontaneously hypertensive rats. Cell Mol Biol Lett. 23:402018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ni X, Wang A, Zhang L, Shan LY, Zhang HC,
Li L, Si JQ, Luo J, Li XZ and Ma KT: Up-regulation of gap junction
in peripheral blood T lymphocytes contributes to the inflammatory
response in essential hypertension. PLoS One. 12:e01847732017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang HC, Zhang ZS, Zhang L, Wang A, Zhu
H, Li L, Si JQ, Li XZ and Ma KT: Connexin 43 in splenic lymphocytes
is involved in the regulation of CD4+CD25+ T
lymphocyte proliferation and cytokine production in hypertensive
inflammation. Int J Mol Med. 41:13–24. 2018.
|
|
19
|
Ni X, Zhang L, Ma X, Shan LY, Li L, Si JQ,
Li XZ, Zhang YY and Ma KT: β-estradiol alleviates hypertension- and
concanavalin A-mediated inflammatory responses via modulation of
connexins in peripheral blood lymphocytes. Mol Med Rep.
19:3743–3755. 2019.PubMed/NCBI
|
|
20
|
Ni X, Zhang L, Peng M, Shen TW, Yu XS,
Shan LY, Li L, Si JQ, Li XZ and Ma KT: Hydrogen sulfide attenuates
hypertensive inflammation via regulating connexin expression in
spontaneously hypertensive rats. Med Sci Monit. 24:1205–1218. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Manjarrez-Marmolejo J and Franco-Pérez J:
Gap Junction Blockers: An overview of their effects on induced
seizures in animal models. Curr Neuropharmacol. 14:759–771. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
O'Donnell JJ III, Birukova AA, Beyer EC
and Birukov KG: Gap junction protein connexin43 exacerbates lung
vascular permeability. PLoS One. 9:e1009312014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Suzuki S, Matsuda Y, Sugawara T, Tabata T,
Ishibashi H, Hoshikawa Y, Kubo H and Kondo T: Effects of
carbenoxolone on alveolar fluid clearance and lung inflammation in
the rat. Crit Care Med. 32:1910–1915. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ram A, Singh SK, Singh VP, Kumar S and
Ghosh B: Inhaled carbenoxolone prevents allergic airway
inflammation and airway hyperreactivity in a mouse model of asthma.
Int Arch Allergy Immunol. 149:38–46. 2009. View Article : Google Scholar
|
|
25
|
Endong L, Shijie J, Sonobe Y, Di M, Hua L,
Kawanokuchi J, Mizuno T and Suzumura A: The gap-junction inhibitor
carben-oxolone suppresses the differentiation of Th17 cells through
inhibition of IL-23 expression in antigen presenting cells. J
Neuroimmunol. 240-241:58–64. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bayne K: Revised Guide for the Care and
Use of Laboratory Animals Available. American Physiological
Society. Physiologist. 39:199208–211. 1996.PubMed/NCBI
|
|
27
|
Sharma S, Sharma N, Saini A and Nehru B:
Carbenoxolone reverses the amyloid Beta 1-42 oligomer-induced
oxidative damage and anxiety-related behavior in rats. Neurotox
Res. 35:654–667. 2019. View Article : Google Scholar
|
|
28
|
Urboniene D, Haber I, Fang YH, Thenappan T
and Archer SL: Validation of high-resolution echocardiography and
magnetic resonance imaging vs. high-fidelity catheterization in
experimental pulmonary hypertension. Am J Physiol Lung Cell Mol
Physiol. 299:L401–L412. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Temple IP, Monfredi O, Quigley G,
Schneider H, Zi M, Cartwright EJ, Boyett MR, Mahadevan VS and Hart
G: Macitentan treatment retards the progression of established
pulmonary arterial hypertension in an animal model. Int J Cardiol.
177:423–428. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Flues K, Moraes-Silva IC, Mostarda C,
Souza PR, Diniz GP, Moreira ED, Piratello AC, Chaves ML, De Angelis
K, Salemi VM, et al: Cardiac and pulmonary arterial remodeling
after sinoaortic dener-vation in normotensive rats. Auton Neurosci.
166:47–53. 2012. View Article : Google Scholar
|
|
31
|
Lang RM, Bierig M, Devereux RB,
Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward
J, Shanewise JS, et al Chamber Quantification Writing Group;
American Society of Echocardiography's Guidelines and Standards
Committee; European Association of Echocardiography:
Recommendations for chamber quantification: A report from the
American Society of Echocardiography's Guidelines and Standards
Committee and the Chamber Quantification Writing Group, developed
in conjunction with the European Association of Echocardiography, a
branch of the European Society of Cardiology. J Am Soc
Echocardiogr. 18:1440–1463. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ji WJ, Ma YQ, Zhou X, Zhang YD, Lu RY, Guo
ZZ, Sun HY, Hu DC, Yang GH, Li YM, et al: Spironolactone attenuates
bleomycin-induced pulmonary injury partially via modulating
mononuclear phagocyte phenotype switching in circulating and
alveolar compartments. PLoS One. 8:e810902013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lei L, He ZY, Zhao C, Sun XJ and Zhong XN:
Elevated frequencies of CD4(+) IL-21(+) T, CD4(+) IL-21R(+) T and
IL-21(+) Th17 cells, and increased levels of IL-21 in
bleomycin-induced mice may be associated with dermal and pulmonary
inflammation and fibrosis. Int J Rheum Dis. 19:392–404. 2016.
View Article : Google Scholar
|
|
34
|
Barletta KE, Cagnina RE, Wallace KL, Ramos
SI, Mehrad B and Linden J: Leukocyte compartments in the mouse
lung: Distinguishing between marginated, interstitial, and alveolar
cells in response to injury. J Immunol Methods. 375:100–110. 2012.
View Article : Google Scholar :
|
|
35
|
Martinu T, Kinnier CV, Sun J, Kelly FL,
Nelson ME, Garantziotis S, Foster WM and Palmer SM: Allogeneic
sple-nocyte transfer and lipopolysaccharide inhalations induce
differential T cell expansion and lung injury: A novel model of
pulmonary graft-versus-host disease. PLoS One. 9:e979512014.
View Article : Google Scholar
|
|
36
|
Alencar AKN, Pimentel-Coelho PM, Montes
GC, da Silva MMC, Mendes LVP, Montagnoli TL, Silva AMS, Vasques JF,
Rosado-de-Castro PH, Gutfilen B, et al: Human mesenchymal stem cell
therapy reverses Su5416/hypoxia-induced pulmonary arterial
hypertension in mice. Front Pharmacol. 9:13952018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li L, Wei C, Kim IK, Janssen-Heininger Y
and Gupta S: Inhibition of nuclear factor-κB in the lungs prevents
monocrotaline-induced pulmonary hypertension in mice. Hypertension.
63:1260–1269. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yamazato Y, Yamazato M, Ishida A, Fujita J
and Ohya Y: Intratracheal administration of autologous bone
marrow-derived cells ameliorates monocrotaline-induced pulmonary
vessel remodeling and lung inflammation in rats. Lung. 196:147–155.
2018. View Article : Google Scholar
|
|
39
|
Marsh LM, Jandl K, Grünig G, Foris V,
Bashir M, Ghanim B, Klepetko W, Olschewski H, Olschewski A and
Kwapiszewska G: The inflammatory cell landscape in the lungs of
patients with idiopathic pulmonary arterial hypertension. Eur
Respir J. 51:17012142018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sáez PJ, Shoji KF, Aguirre A and Sáez JC:
Regulation of hemichannels and gap junction channels by cytokines
in antigen-presenting cells. Mediators Inflamm. 2014:7427342014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Oviedo-Orta E, Hoy T and Evans WH:
Intercellular communication in the immune system: Differential
expression of connexin40 and 43, and perturbation of gap junction
channel functions in peripheral blood and tonsil human lymphocyte
subpopulations. Immunology. 99:578–590. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mendoza-Naranjo A, Bouma G, Pereda C,
Ramírez M, Webb KF, Tittarelli A, López MN, Kalergis AM, Thrasher
AJ, Becker DL, et al: Functional gap junctions accumulate at the
immunological synapse and contribute to T cell activation. J
Immunol. 187:3121–3132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Freund-Michel V, Muller B, Marthan R,
Savineau JP and Guibert C: Expression and role of connexin-based
gap junctions in pulmonary inflammatory diseases. Pharmacol Ther.
164:105–119. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Feng S, Chen S, Yu W, Zhang D, Zhang C,
Tang C, Du J and Jin H: H2S inhibits pulmonary arterial endothelial
cell inflammation in rats with monocrotaline-induced pulmonary
hypertension. Lab Invest. 97:268–278. 2017. View Article : Google Scholar
|
|
45
|
Willebrords J, Crespo Yanguas S, Maes M,
Decrock E, Wang N, Leybaert L, Kwak BR, Green CR, Cogliati B and
Vinken M: Connexins and their channels in inflammation. Crit Rev
Biochem Mol Biol. 51:413–439. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Meloche J, Renard S, Provencher S and
Bonnet S: Anti-inflammatory and immunosuppressive agents in PAH.
Handb Exp Pharmacol. 218:437–476. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sharma AK, Charles EJ, Zhao Y, Narahari
AK, Baderdinni PK, Good ME, Lorenz UM, Kron IL, Bayliss DA,
Ravichandran KS, et al: Pannexin-1 channels on endothelial cells
mediate vascular inflammation during lung ischemia-reperfusion
injury. Am J Physiol Lung Cell Mol Physiol. 315:L301–L312. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Billaud M, Dahan D, Marthan R, Savineau JP
and Guibert C: Role of the gap junctions in the contractile
response to agonists in pulmonary artery from two rat models of
pulmonary hypertension. Respir Res. 12:302011. View Article : Google Scholar : PubMed/NCBI
|