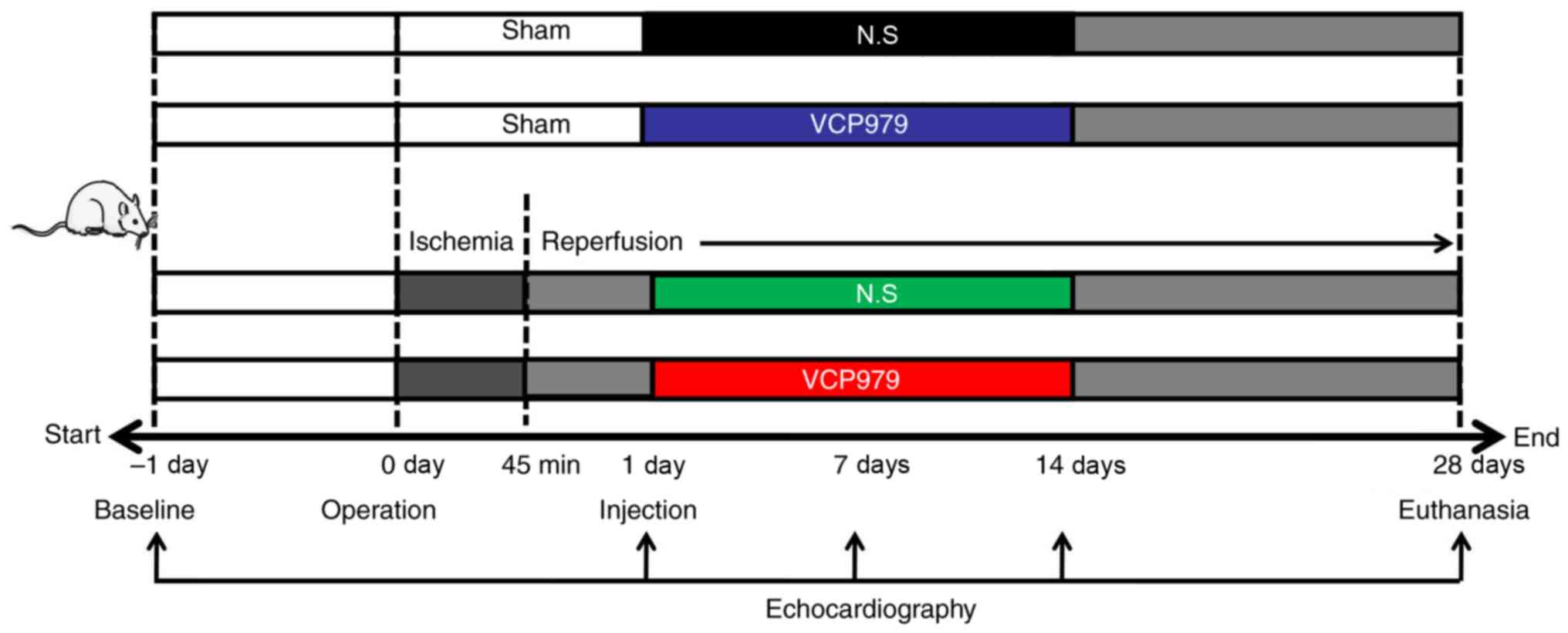
Introduction
Ischemic heart disease is one of the most common
causes of morbidity and disability worldwide (1). Timely and effective myocardial
reperfusion by percutaneous coronary intervention is essential to
salvage reversible injuries in the ischemic myocardium (2). However, reperfusion may
paradoxically aggravate the tissue damage by promoting myocardial
death, cardiac dysfunction, ventricular remodeling and the
inflammatory response, all of which fall under myocardial
ischemia/reperfusion (MI/R) injury (2,3).
Short-term hypertrophic and fibrotic response is adaptive and
protective, yet persistent pathological hypertrophy and excessive
fibrosis accelerates the progression of ventricular remodeling
following MI/R injury (4).
Currently, the cardioprotective medicines available for the
treatment of patients with heart failure, including vasodilators,
loop diuretics and inotropic agents, can alleviate congestion and
improve hemodynamics short-term. However, few drugs are capable of
preventing end-organ damage or improving long-term outcomes
(5). Therefore, it is of vital
importance to explore novel therapeutic options to alleviate
ventricular remodeling and prevent progression to heart
failure.
Previous preclinical and clinical studies have shown
that numerous small-molecule compounds can prevent myocardial cell
death during ischemia and subsequent reperfusion (6,7).
It has been reported that small molecule drugs with a low molecular
weight show good spatial dispersion in vivo and are easier
to absorb than multiple protein-based drugs, thus exerting obvious
effects on numerous pathological processes (8). Small-molecule compounds may
therefore lead to the development of new therapeutic agents.
Previous evidence has revealed that MI/R injury is
associated with the activation of the p38-mitogen-activated protein
kinase (MAPK) signaling pathway in murine models. The suppression
of p38-MAPK decreases the production of inflammatory cytokines,
including interleukin (IL)-1, IL-8 and tumor necrosis factor-α
(TNF-α), suggesting the therapeutic potential of p38-MAPK
inhibitors in ischemic heart disease (9,10)
However, the classic p38 inhibitor SB203580 has a low specificity
during other protein kinase functions (11). VCP979, a small-molecule compound
with novel chemical pharmacophores, was developed by the authors'
colleagues at the Monash University as a novel anti-fibrotic and
anti-inflammatory agent (12).
VCP979 was developed as a selective inhibitor of p38-MAPK. The
authors' preliminary studies also indicated that VCP979 possesses a
p38-MAPK inhibitory activity and has significant anti-fibrotic
effects in vitro and in vivo (11,13). In the diabetic murine model,
VCP979 can decrease the infarct volume and promote ischemic stroke
recovery and axonal/WM remodeling (11). However, whether its administration
can protect the heart from MI/R injury remains unclear. The aim of
the present study was to determine the role of the novel
small-molecule compound VCP979 in cardiac protection post MI/R
injury and explore the possible underlying mechanism.
Materials and methods
Animals
The 6-8 weeks old male C57BL/6 mice (22-26 g; n=60)
and neonatal Sprague-Dawley (SD) rats (6-7 g; n=35) were purchased
from Shanghai SLAC Laboratory Animal Co., Ltd. In the whole
experimental process, mice were bred at 20-25°C at 12-h light/dark
cycles and given food and water freely under specific pathogen-free
conditions. The experimental procedures were implemented following
the approval of the Institutional Animal Care and Use Committee of
Tongji University (Shanghai, China; approval no.
TJLAC-019-121).
MI/R injury model establishment
Briefly, mice were anesthetized with sodium
pentobarbital [60 mg/kg, intraperitoneal (i.p.) injection]. During
surgery, a heating pad was used to maintain the animal temperature
at 37.5-38.5°C. Next, mice were intubated and mechanically
ventilated using a Harvard rodent respirator. A left thoracotomy at
the 4-5th intercostal space was performed and the left anterior
descending coronary artery (LAD) was transiently ligated under 2 mm
of lower margin of the left auricle with an 8-0 suture, as
previously described (14). The
ligation was successful when the anterior wall of left ventricle
turned white. The MI/R groups were subjected to 45 min ischemia,
then reperfusion until day 28. The sham mice underwent the same
surgical procedure, except the LAD suture was not tightened. Then,
at 1 day after surgery, the cardiac function of each group was
detected respectively and mice with left-ventricular ejection
fraction (LVEF) below 50% were considered successful MI/R models.
Following surgery, mice were treated with normal saline (N.S) and
VCP979 (50 mg/kg/day) via an i.p. injection for 14 consecutive
days.
VCP979 administration protocol
The small-molecule compound VCP979 was stored as a
powder and dissolved in dimethyl sulfoxide initially, and then
further diluted in 0.9% N.S immediately before use.
All mice were randomly assigned to four groups (6
mice per group): i) Sham group injected with N.S; ii) sham group
injected with VCP979; iii) MI/R group injected with N.S; and iv)
MI/R group injected with VCP979. The treatment protocol is shown in
Fig. 1. Each experiment was
repeated at least three times.
Echocardiography
Transthoracic two-dimensional echocardiography using
a Vevo 2100 high-resolution imaging system with a 30-MHz linear
transducer (FUJIFILM Visual-Sonics, Inc.) was performed to assess
before and after MI/R injury with 2% isoflurane inhalation. The
cardiac function of all animals was evaluated through long-axis
scans using M-mode images including LVEF, left-ventricular
fractional shortening (LVFS), left-ventricular volume;
diastolic/systolic (LV Vol; d/s) and left-ventricular internal
dimension; diastolic/systolic (LVID; d/s) (15). Cardiac function in each group was
detected 1 day before and 1, 7, 14 and 28 days after surgery.
The safety detection of VCP979 in
mice
The blood (~1 ml) of sham groups was collected 14
days after N.S or VCP979 injection and the serum was isolated by
centrifugation (840 × g, 4°C, 10 min). Aspartate transaminase
(AST), alanine amino-transferase (ALT) and lactate dehydrogenase
(LDH) levels were measured using Beckmann AU680.
Cell culture and treatment
Neonatal cardiac myocytes (NCMs) and neonatal
cardiac fibroblasts (NCFs) were isolated from the hearts of
1-3-day-old SD rat pups as previously described (16). The non-adherent NCMs were
separated from the NCFs after 90 min of incubation. The NCMs and
NCFs were cultured in high-glucose DMEM (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.), BrdU (100 nM, only added in the
NCMs culture medium), 1% penicillin (100 U/ml) and 1% streptomycin
(100 µg/ml), and were incubated at 37°C in a humidified
atmosphere of 95% air and 5% CO2. In addition, NCMs and
NCFs were stimulated with Angiotensin II (Ang II; Sigma-Aldrich;
Merck KGaA) 200 nM for 24 h, and then treated with different
concentrations of VCP979 (0.1, 1, 3 and 9 µM) for another 24
h.
Cell supernatants LDH release assay
The LDH release to the medium was detected using an
LDH CytoTox 96 reagent kit (Promega Corporation), according to the
manufacturer's protocol. In a 96-well plate, 50 µl cell
supernatants were mixed with 50 µl CytoTox 96 reagents and
incubated at room temperature for 30 min. Finally, 50 µl
stop solutions was added and the absorbance value was measured at
490 nm using a microplate spectrophotometer (M200pro; Tecan Group,
Ltd.).
Measurement of cell proliferation
Cell proliferation was evaluated using a Cell
Counting Kit-8 (CCK-8) assay (Beijing Solarbio Science &
Technology Co., Ltd.). The NCFs were plated on 96-well plates at a
density of 1×104. Following stimulation with AngII (200
nM) for 24 h, the NCFs were treated with or without different
concentrations of VCP979 (0.1, 1, 3 and 9 µM) for another 24
h. According to the manufacturer's protocol, 10 µl CCK-8
solution was added to each well and the cells were incubated at
37°C for 1-2 h. A microplate spectrophotometer (M200pro) was used
to read the optical density of each well at 450 nm.
Immunofluorescence detection
Cardiomyocyte hypertrophy was determined using
TRITC-Phalloidin (Beijing Solarbio Science & Technology Co.,
Ltd.) staining, following the manufacturer's protocol. Briefly, the
cells (5×105 cells/well) were plated on 6-well plates
and fixed with 4% paraformaldehyde (PFA) for 10 min at room
temperature. Following 0.5% Triton X-100 permeability treatment,
the NCMs were stained with TRITC-Phalloidin (1:50) for 30 min at
room temperature in the dark, whereas the nucleus was stained with
4', 6-diamidino-2-phenylindole [room temperature (RT), 10 min].
Phalloidin can label the F-actin of the cytoskeleton with high
specificity and sensitivity. After staining with Phalloidin, the
ImageJ software 1.49p (National Institutes of Health) was used to
measure the cytoskeleton of each cell. A total of 5 different
fields were randomly selected and analyzed under an upright
fluorescent microscope (Leica DM2500; Leica Microsystems GmbH).
Histological analysis
The animals were euthanized on day 14 or 28 after
surgery. The tissues were dissected and fixed (4°C) in 4% PFA
overnight, embedded in paraffin, sectioned into 4-µm slices
and then stained (RT, 5 min) with hematoxylin and eosin (H&E)
and Masson's Trichrome. The relative myocyte cross-sectional areas
(CSAs) were detected by wheat germ agglutinin (WGA; Abcam)
conjugated to Alexa Fluor 488 (1:200; Thermo Fisher Scientific,
Inc.), as previously described (17) and imaged using a confocal
microscope (Leica SP8; Leica Microsystems, Inc.). In addition,
myocardial fibrosis was identified by immunohistochemistry (IHC)
using anti-collagen I (1:1,000; Abcam; cat. no. 34710). The
infiltration of inflammatory cells was detected by IHC with CD4
(1:1,000; Abcam; cat. no. 183685), CD8 [1:1,000; Cell Signaling
Technology, Inc. (CST); cat. no. 98941] and F4/80 (1:1,000; CST;
cat. no. 70076) antibodies. Next, 5 different fields were randomly
selected and imaged under an upright light microscope (Leica
DM750). The average areas of tissue fibrosis, relative size of
cells that are positive in IHC were measured using ImageJ software
(National Institutes of Health).
Western blotting
Mouse heart tissues and primary cells were collected
and protein concentrations were quantified using a bicinchoninic
acid protein assay kit (Thermo Fisher). Equivalent amounts of
proteins (30-50 µg) were loaded on SDS-polyacrylamide gels
(10-15%) and performed as previously described (18). Proteins were transferred onto
polyvinylidene fluoride membranes and blocked (RT, 30 min) with 5%
nonfat dry milk. The membranes were then incubated with the
following primary antibodies: Total p38-MAPK (CST; cat. no. 8690],
phospho-p38-MAPK (p-p38; CST; cat. no. 4511). Tubulin (CST; cat.
no. 2146) was used as the control. Following washing with PBS with
0.1% Tween-20, the membranes were incubated with the corresponding
secondary antibodies (1:5,000; Anti-rabbit IgG; CST; cat. no.
7074). The bands were visualized using an enhanced
chemiluminescence system (Minichemi™; Beijing Saiz Science &
Technology Co., Ltd.). The intensity of each protein band was
quantified using ImageJ software.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from mouse hearts or primary
cells using TRIzol reagent (Thermo Fisher Scientific, Inc.). For
mRNA detection in mouse hearts, infarcted tissues plus border zone
tissues in MI/R mice and the tissues of similar region in sham mice
were used. Then the RNA concentration was quantified using a
NanoDrop 2000 (Thermo Fisher Scientific, Inc.) and cDNA was
obtained using the PrimerScript RT reagent kit at 37°C for 15 min,
and 85°C for 5 sec (Takara Bio, Inc.). The mRNA levels were
detected by RT-qPCR with a SYBR Green Master Mix kit (Takara Bio,
Inc.) using real-time PCR systems (Thermo Fisher Scientific, Inc.).
The thermocycling conditions were as follows: 96°C for 30 sec, 57°C
for 30 sec and 72°C for 30 sec. The mRNA expression level was
calculated using the 2-ΔΔCq method (18). The β-actin and GAPDH genes were
used as the control. The primer sets used to identify the gene are
given in Table I.
 | Table IThe primer sequences information. |
Table I
The primer sequences information.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| m-β-actin |
GGCTGTATTCCCCTCCATCG |
CCAGTTGGTAACAATGCCATGT |
| m-ANP |
GCTTCCAGGCCATATTGGAG |
GGGGGCATGACCTCATCTT |
| m-BNP |
AGTCCTTCGGTCTCAAGGCA |
CCGATCCGGTCTATCTTGTGC |
| m-α-SMA |
GTCCCAGACATCAGGGAGTAA |
TCGGATACTTCAGCGTCAGGA |
| m-collagen-I |
GCTCCTCTTAGGGGCCACT |
ATTGGGGACCCTTAGGCCAT |
| m-IL-1β |
CGAGGCTAATAGGCTCATCT |
GTTTGGAAGCAGCCCTTCAT |
| m-IL-6 |
TGATGCACTTGCAGAAAACA |
ACCAGAGGAAATTTTCAATAGGC |
| m-Arg1 |
CAGAAGAATGGAAGAGTCAG |
CAGATATGCAGGGAGTCACC |
| m-CD206 |
AACAAGAATGGTGGGCAGTC |
AACTCCTCGTCCGTCTGTC |
| m-iNOS |
CACCAAGCTGAACTTGAGCGA |
CCATAGGAAAAGACTGCACCGA |
| m-TNF-α |
GCCAACGGCATGGATCTC |
GCAGCCTTGTCCCTTGAAGAG |
| m-Ang II |
CCAGGCCCGTTGTTCTTGAT |
GGAAGGGAGACTTGCTCATTC |
| r-β-actin |
CGTGCGTGACATTAAAGAG |
TTGCCGATAGTGATGACCT |
| r-GAPDH |
CGGCAAGTTCAACGGCACAG |
CGCCAGTAGACTCCACGACAT |
| r-collagen-I |
GGAGAGAGTGCCAACTCCAG |
GTGCTTTGGAAAATGGTGCT |
| r-TNF-α |
CTGTGCCTCAGCCTCTTCTC |
ACTGATGAGAGGGAGCCCAT |
| r-ANP |
GGTCCTTCGGATGCAGTATT |
CCACTTGAGCAGCATTGTGT |
| r-IL-1β |
ACGGGTTCCATGGTGAAGT |
CCTCTCAAGCAGAGCACAGA |
| r-IL-6 |
CCAGCCAGTTGCCTTCT |
GTCCTTAGCCACTCCTTCT |
| r-Ang II |
TTGGATAAAGAACCCGCCTC |
AAAGGGTAGACAGCTTGGC |
Statistical analysis
Results were analyzed using GraphPad Prism 5.0
analysis software (GraphPad Software, Inc.) and are expressed as
the mean ± standard error of the mean of 3 independent experiments.
In the present study, an unpaired two-tailed student's t-test was
used for comparisons between different groups. One-way analysis of
variance with a Bonferroni post hoc test was used for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference and P<0.01, a highly statistically
significant difference. The survival curve was estimated by the
Log-rank (Mantel-Cox) Test.
Results
Safety of VCP979 treatment in mice and
cultured cells
To ensure the safety of this newly developed
small-molecule compound, VCP979, the body weight of mice that
received VCP979 (50 mg/kg/day, i.p.) or control solvent for 14
consecutive days was first evaluated. As shown in Fig. 2A, there was no significant
difference in body weight between the two sham groups on days 0, 7,
14, 21 and 28 after surgery. H&E staining results showed that
the administration of VCP979 had no obvious impact on the pathology
of multiple murine organs, including the lung, liver, kidney,
spleen and heart (Fig. 2B).
Moreover, AST, ALT and LDH levels in serum were measured in sham
groups and no difference was observed after VCP979 injection
(Fig. 2C).
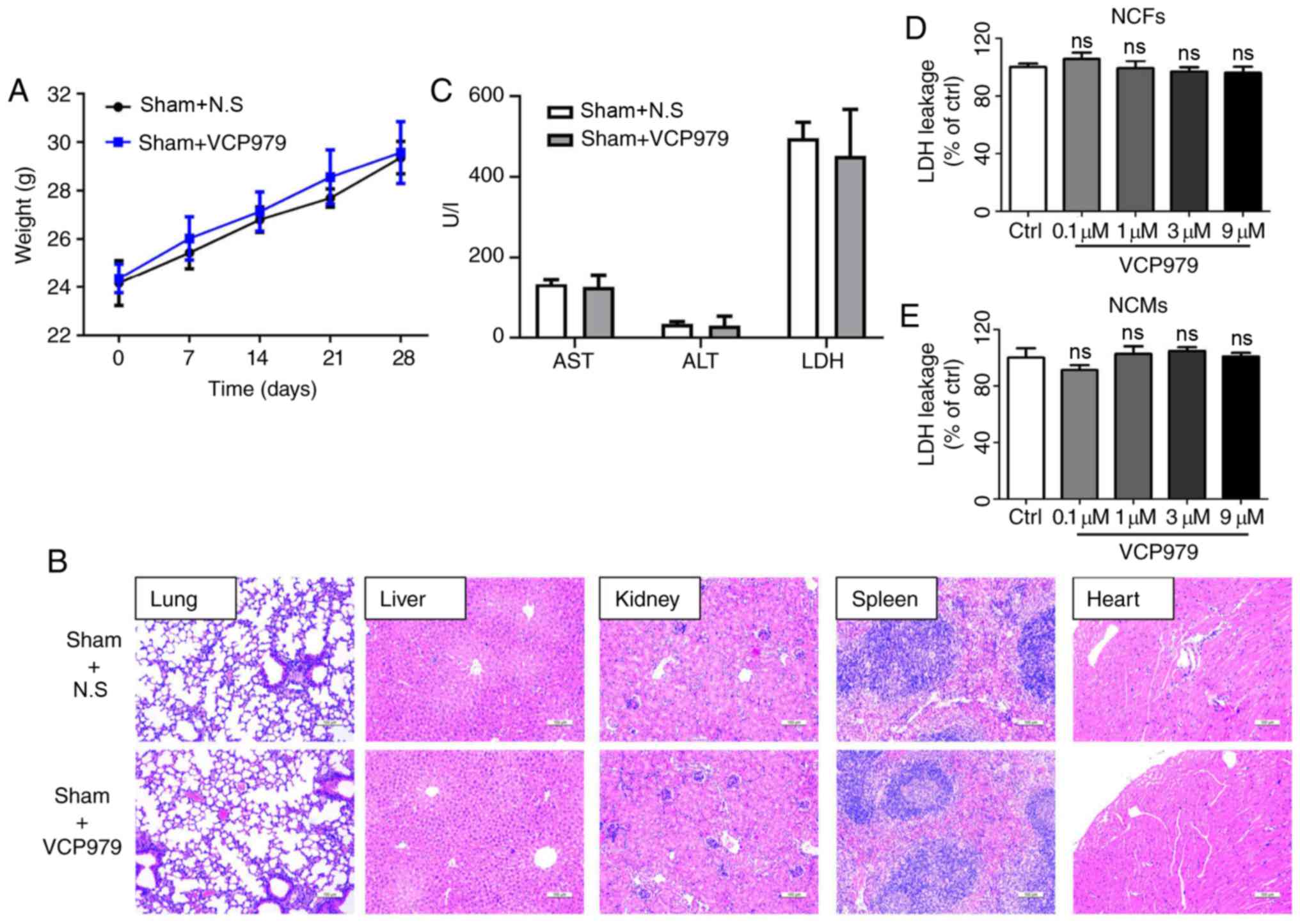 | Figure 2Safety of VCP979 treatment in mice
and cultured cells. (A) The body weight of mice in the sham groups
was recorded on days 0, 7, 14, 21 and 28 following surgery (n=6 per
group). (B) Representative hematoxylin and eosin-stained
histological sections of the lung, liver, kidney, spleen and heart
of mice after 28 days. Scale bar=100 µm. (C) The AST, ALT
and LDH levels in serum were measured in sham groups. LDH leakage
percent of the (D) NCFs and (E) NCMs following treatment with or
without different concentrations of VCP979 (0.1, 1, 3 and 9
µM) for 24 h. NCF, neonatal cardiac fibroblast; NCM,
neonatal cardiac myocyte; LDH, lactate dehydrogenase; AST,
aspartate transaminase; ALT, alanine aminotransferase. |
In addition, to clarify whether VCP979 could be
toxic to primary cells, the percentages of LDH leakage in the NCFs
and NCMs treated with or without VCP979 for 24 h were assessed.
Fig. 2D and E show that different
concentrations of VCP979 (0.1, 1, 3 and 9 µM) do not
significantly affect the percentages of LDH leakage in NCFs and
NCMs. These results indicated no significant toxic effects of
VCP979 at the present concentrations and duration.
VCP979 administration improves cardiac
function in murine MI/R injury models
Cardiac function was examined by M-mode
echocardiography on day 1 before and days 1, 7, 14 and 28 after
surgery (Fig. 3A). As compared
with the MI/R group, the administration of VCP979 significantly
increased both LVEF (28.1908±4.1065 vs. 40.5348±3.4986) and LVFS
(14.2999±2.1439 vs. 21.372±2.3741) on day 14 (P<0.05), but
decreased both LV Vol; d/s (99.3371±6.8049 vs.
69.3894±4.1022/51.8582±2.0042 vs. 37.1314±2.6628) and LVID; d/s
(4.6353±0.1338 vs. 3.8792±0.1285/3.8035±0.2779 vs. 2.8924±0.2081)
on day 28 (Fig. 3A). These
results demonstrated that VCP979 could improve ventricular regional
and global function (LVEF and LVFS) and inhibit the progression of
ventricular remodeling (LV Vol; d/s and LVID; d/s). In addition,
there was no significant difference in the survival rate between
the sham and MI/R groups injected with N.S or VCP979 during the 28
days of observation (Fig.
3C).
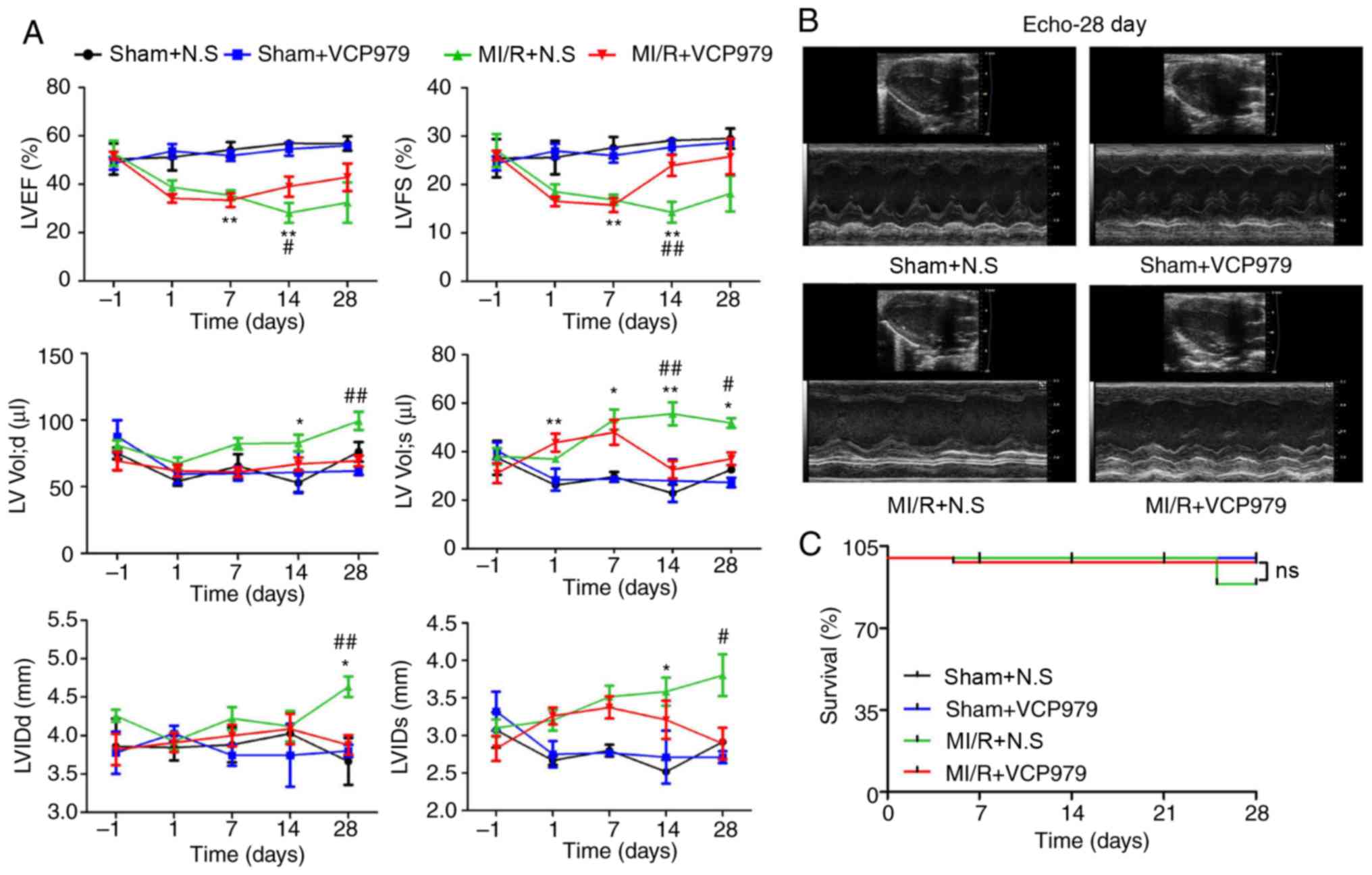 | Figure 3VCP979 administration improved
cardiac function in mice post-MI/R injury. (A) Cardiac function was
detected on days 1 before and 1, 7, 14 and 28 after surgery (n=6
per group). (B) Representative images of M-mode echocardiography
from the sham+N.S, sham+VCP979, MI/R+N.S and MI/R+VCP979 groups on
day 28. (C) The survival curves of the sham and MI/R groups
injected with N.S or VCP979 were observed for 28 days (n=12 per
group). Results are expressed as the mean ± standard error of the
mean. *P<0.05 and **P<0.01, sham+N.S
vs. MI/R+N.S group. #P<0.05 and
##P<0.01, MI/R+N.S vs. MI/R+VCP979. N.S, normal
saline; MI/R, myocardial ischemia/reperfusion; LVEF, left
ventricular ejection fraction; LV vol, left ventricular volume;
LVFS, left ventricular fractional shortening; LVIDd/s,
left-ventricular internal dimension; diastolic/systolic. |
VCP979 administration attenuates
myocardial fibrosis following MI/R injury
Next, myocardial fibrosis was analyzed by Masson's
Trichrome staining and IHC. Fig. 4A
and C showed that MI/R surgery significantly increased the
average area of myocardial fibrosis on day 28 (P<0.001),
compared with the sham mice (0.4283±0.1253 vs. 2.3253±0.1181) and
the administration of VCP979 greatly reduced the fibrosis area, as
compared with the N.S-treated MI/R group (2.3253±0.1181 vs.
1.245±0.0801). Furthermore, treatment with VCP979 attenuated
collagen I expression in MI/R hearts (12.247±0.5022 vs.
9.805±0.4045, Fig. 4B and D). As
shown by Fig. 4E, mRNA levels of
α-smooth muscle actin (α-SMA), collagen I and Ang II were
significantly decreased in the MI/R+VCP979 group, as compared with
the MI/R+N.S group on day 28 (1.5504±0.0923 vs. 1.257±0.0514,
2.9636±0.225 vs. 1.5357±0.2741 and 2.9806±0.1888 vs. 1.3883±0.0974,
respectively; P<0.05). These findings suggested that VCP979
plays an important role in attenuating myocardial fibrosis in
murine MI/R injury models.
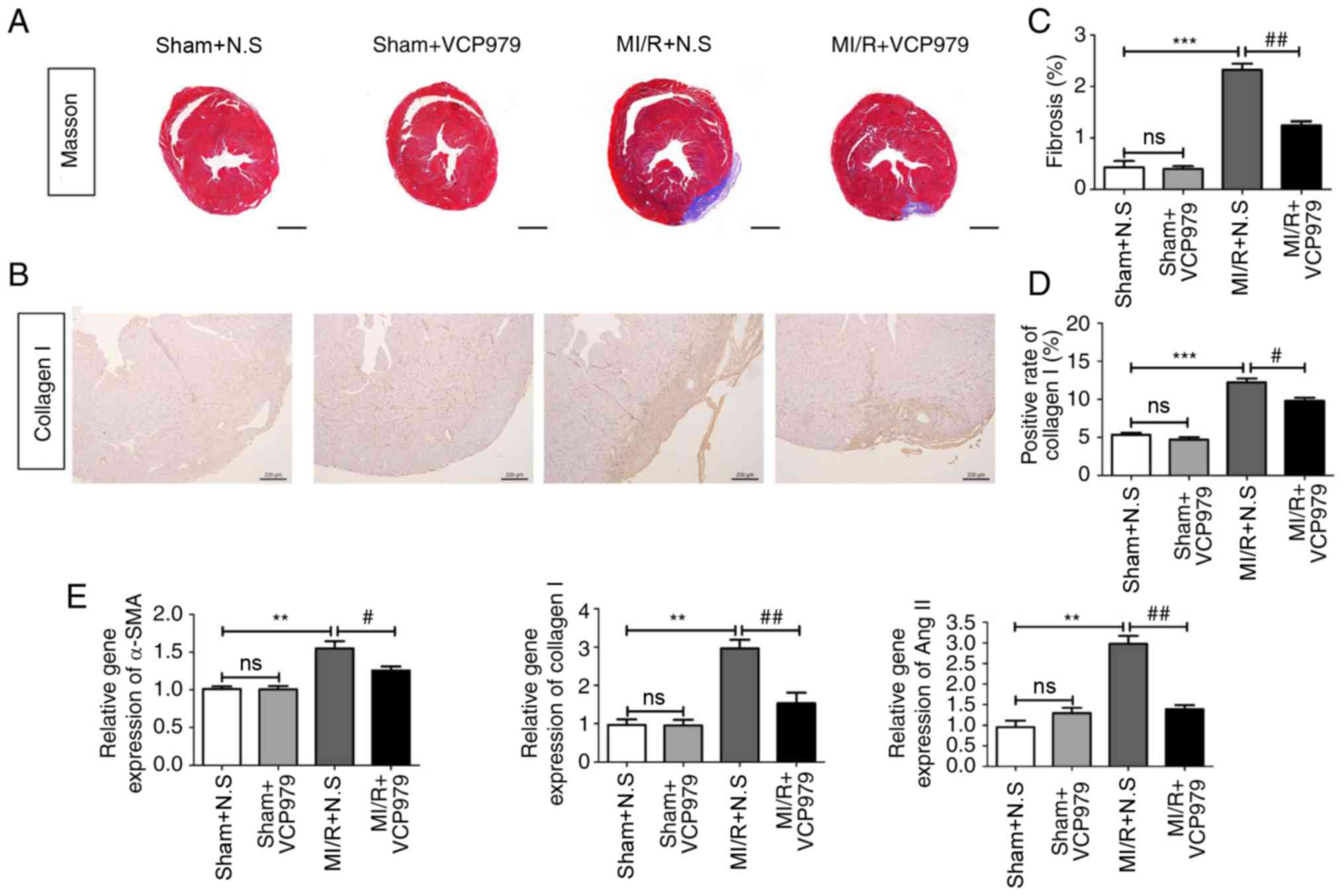 | Figure 4VCP979 administration of attenuated
myocardial fibrosis following MI/R injury. (A) Representative
Masson's Trichrome-stained histological sections of hearts from the
sham+N.S, sham+VCP979, MI/R+N.S and MI/R+VCP979 groups. Scale bar=1
mm. (B) Representative collagen I staining pictures. Scale bar=200
µm. (C) The fibrosis area of heart tissues and the (D)
positive rate of collagen I were measured using ImageJ software.
(E) mRNA levels of α-SMA, collagen I and Ang II in heart tissues of
different groups. Results are expressed as the mean ± standard
error of the mean. **P<0.01 and
***P<0.001, sham+N.S vs. MI/R+N.S.
#P<0.05 and ##P<0.01, MI/R+N.S vs.
MI/R+VCP979. SMA, smooth muscle actin; MI/R, myocardial
ischaemia/reperfusion; N.S, normal saline; Ang II, angiotensin II;
ns, not significant. |
VCP979 treatment alleviates pathological
hypertrophy of hearts subjected to MI/R injury
To evaluate the role of VCP979 on pathological
hypertrophy, the CSAs were detected by WGA staining. Fig. 5A and B showed that the average
cell size in the MI/R+VCP979 group was significantly decreased
compared with in the MI/R+N.S group (541.3636±54.7254 vs.
305.9167±28.6484; P<0.001). In addition, the continuous 2-weeks
VCP979 injection significantly reduced the ratio of heart weight
(g) to body weight (g), as compared with N.S injection in MI/R mice
(0.469±0.0037 vs. 0.4417±0.0064; P<0.05; Fig. 5C). Furthermore, RT-qPCR analysis
of the infarcted area and border zone of left ventricle
demonstrated that VCP979 treatment significantly decreased the mRNA
levels of atrial natriuretic peptide (ANP) and brain natriuretic
peptide (BNP) on day 28, compared with the N.S-treated MI/R group
(24.9505±1.2631 vs. 18.8687±0.3303 and 2.2903±0.0402 vs.
1.482±0.0653, respectively; P<0.01; Fig. 5D). This phenomenon suggested that
VCP979 plays a protective role in MI/R injury induced pathological
hypertrophy.
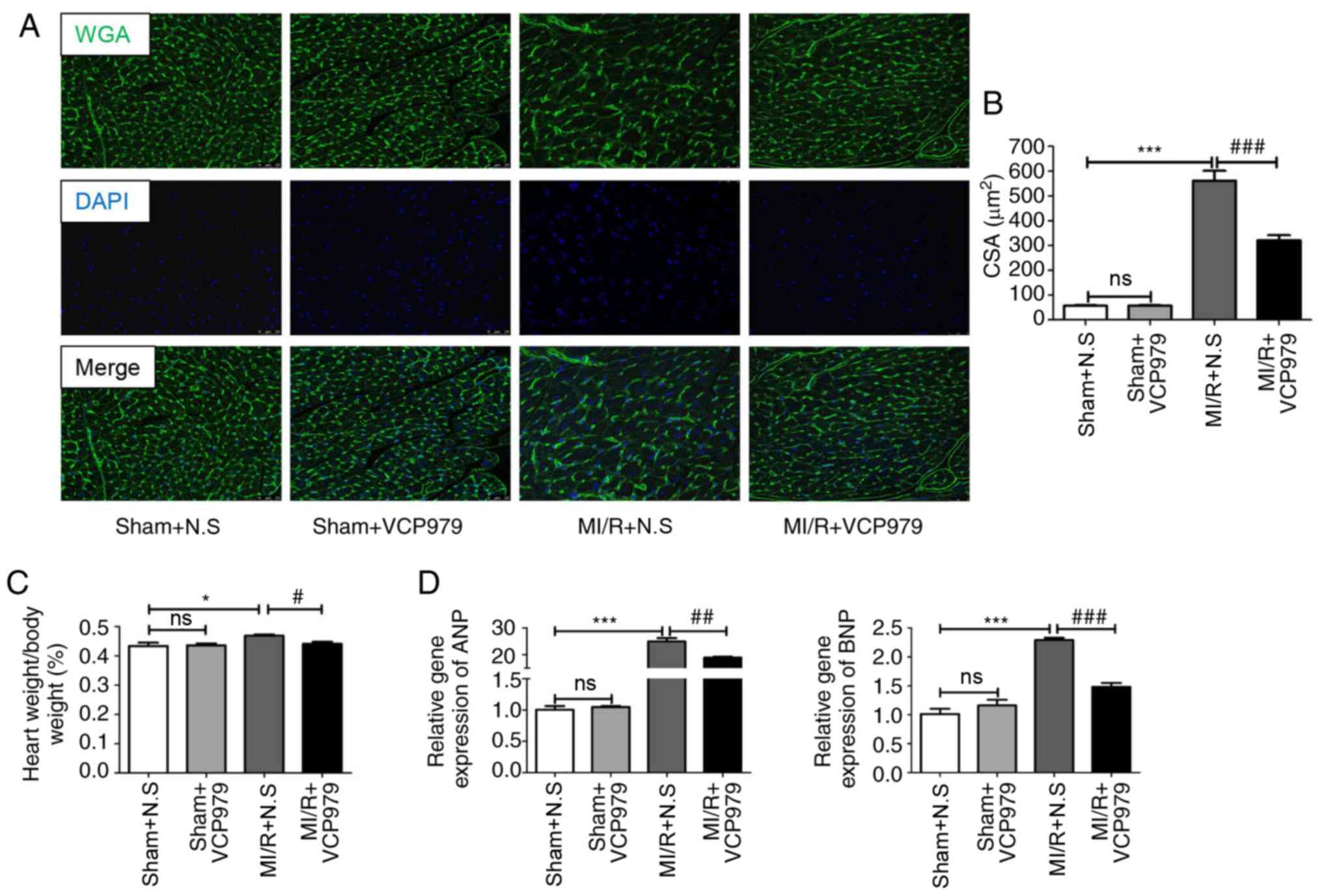 | Figure 5VCP979 treatment can alleviate
pathological hypertrophy of the heart after day 28 in vivo.
(A) Representative WGA immunofluorescence-stained sections. Green,
WGA; Blue, nucleus. Scale bar=25 µm. (B) The cross-sectional
areas of the heart sections were estimated using ImageJ software.
(C) The ratio of heart weight (g) to body weight (g) post-MI/R
injury on day 28. (D) mRNA expression of ANP and BNP in hearts.
Results are expressed as the mean ± standard error of the mean.
*P<0.05 and ***P<0.001, sham+N.S vs.
MI/R+N.S. #P<0.05, ##P<0.01 and
###P<0.001. MI/R+N.S vs. MI/R+VCP979. WGA, wheat germ
agglutinin; ANP, atrial natriuretic peptide; BNP, brain natriuretic
peptide; MI/R, myocardial ischemia/reperfusion; N.S, normal saline;
ns, not significant. |
VCP979 treatment prevents inflammatory
cell infiltration and suppresses the p38-MAPK signaling pathway in
MI/R hearts
Pro-inflammatory cell infiltration has been reported
to be closely associated with heart injury (14,19). In order to verify the potential
mechanism through which the administration of VCP979 protects heart
function following MI/R injury, the infiltration of inflammatory
cells was detected by IHC and the results revealed fewer numbers of
cluster of differentiation (CD)4+, CD8+ and
F4/80+ inflammatory cells in hearts of MI/R+VCP979 group
compared with those from the MI/R+N.S group on day 14 post-MI/R
injury (0.7508±0.0891 vs. 0.2296±0.0555, 0.5315±0.0819 vs.
0.2414±0.0198 and 2.8303±0.3082 vs. 1.7483±0.1626, respectively;
Fig. 6A and B). Next, the mRNA
expression of several inflammatory cytokines and macrophage
polarization-related factors in murine heart tissues was assessed
on day 14. Fig. 6C showed that,
as compared with the MI/R group, VCP979 treatment significantly
increased the levels of arginase 1 (Arg1) and CD206 (1.0092±0.0937
vs. 1.6763±0.0677 and 1.0021±0.0467 vs. 1.3567±0.0072,
respectively; P<0.01), while it decreased inducible nitric oxide
synthase (iNOS), IL-6 and TNF-α (1.0032±0.0565 vs. 0.4094±0.0534,
1.0127±0.1089 vs. 0.3444±0.0311 and 1.1481±0.0655 vs.
0.8184±0.0037, respectively) mRNA levels on day 14 after
surgery.
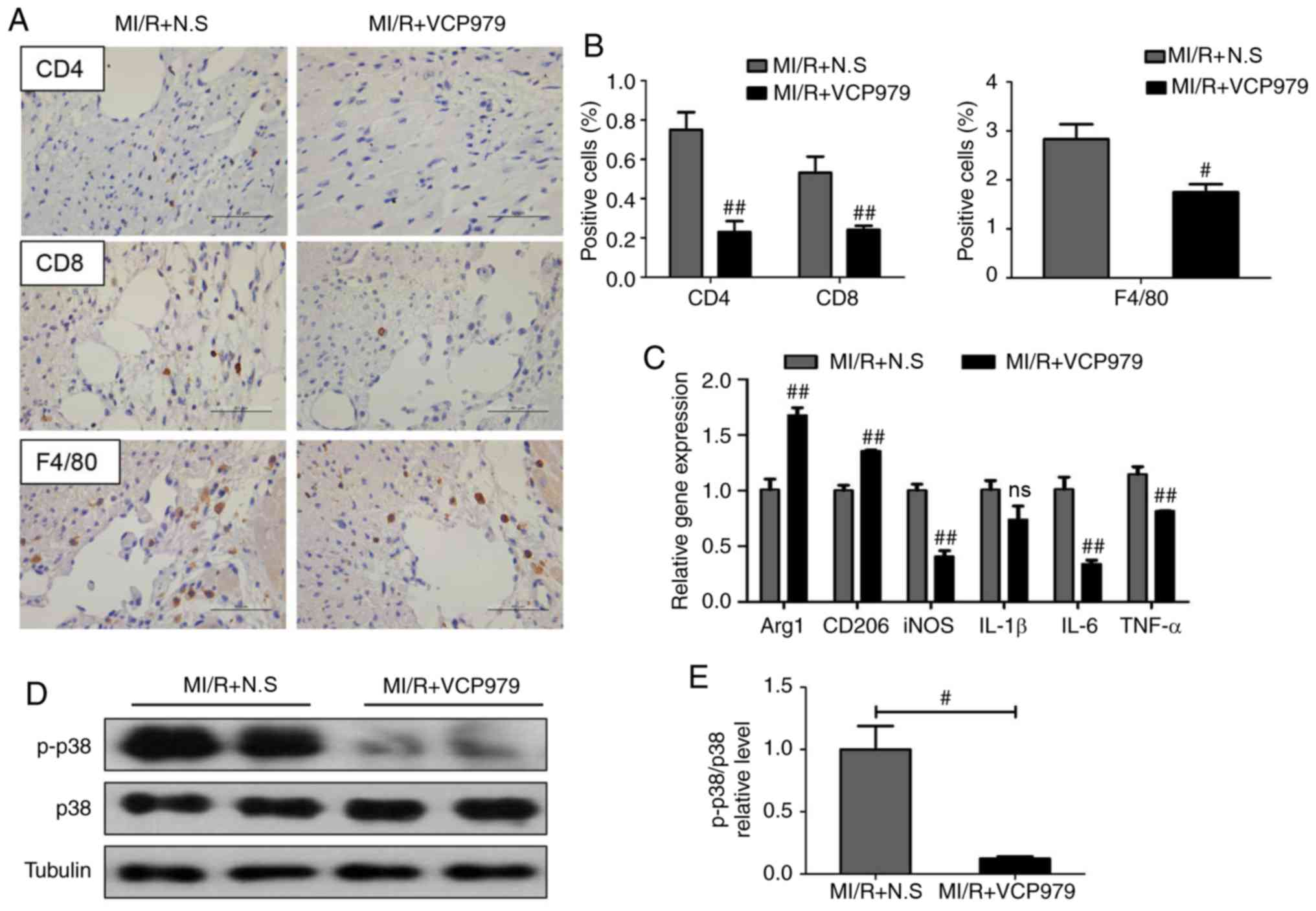 | Figure 6VCP979 treatment prevents
inflammatory cell infiltration and suppresses the p38-MAPK
signaling pathway on day 14. (A) Representative CD4, CD8 and F4/80
staining pictures. Scale bar=50 µm. (B) The relative
positive cells of the sections were determined by ImageJ software.
(C) Reverse transcription-quantitative PCR analysis of mRNA levels
of Arg1, CD206, iNOS, IL-1β, IL-6 and TNF-α in the heart tissues.
(D) Western blotting analysis and (E) quantitative analysis of
p-p38 and p38 protein levels in heart tissues from the MI/R+N.S and
MI/R+VCP979. Results are shown as mean ± standard error of the
mean. #P<0.05 and ##P<0.01, MI/R+N.S
vs. MI/R+VCP979 group. MAPK, mitogen-activated protein kinase;
iNOS, inducible nitric oxide synthase; p-, phosphorylated-; IL,
interleukin; TNF, tumor necrosis factor; CD, cluster of
differentiation; MI/R, myocardial ischemia/reperfusion; N.S, normal
saline; ns, not significant; Arg1, arginase 1. |
Next whether VCP979 could suppress the p38-MAPK
signaling pathway in vivo was explored. Fig. 6D and E showed that the
administration of VCP979 significantly decreased the ratio of
p-p38/p38 in MI/R hearts compared with the N.S treatment MI/R group
(1.0005±0.1884 vs. 0.1236±0.0164; P<0.05).
Addition of VCP979 inhibits the Ang
II-induced proliferation and collagen synthesis of NCFs and
hypertrophy of NCMs, and suppresses the p38-MAPK signaling
pathway
To explored whether VCP979 exerts its functions on
fibroblasts and cardiomyocytes directly, NCFs and NCMs were
isolated and the effect of VCP979 on cells exposed to Ang II (200
nM) was investigated. As shown in Fig. 7A, 24 h stimulation with Ang II
significantly increased the proliferation of NCFs (100.1229±1.2103
vs. 107.7961±2.902; P<0.05) and high concentrations (1, 3 and 9
µM) of VCP979 markedly reversed this elevation
(96.9753±0.8734, 93.0633±0.7293 and 91.28±0.4052, respectively;
Fig. 7A). Meanwhile, the collagen
I mRNA level in NCFs was significantly reduced following treatment
with high concentrations (1, 3 and 9 µM) of VCP979 for 24 h
(1.1763±0.013 vs. 1.0471±0.0051, 0.8838±0.0096, 0.8916±0.0028,
respectively; P<0.05; Fig.
7B). Next, VCP979 treatment was shown to significantly decrease
the ANP mRNA level in Ang II-induced NCMs at different
concentrations (0.1, 1, 3 and 9 µM; 38.8748±0.4596 vs.
26.2532±1.3328, 20.0562±0.8087, 19.568±1.0699 and 15.474±1.8371,
respectively; P<0.05; Fig.
7C). Phalloidin staining was performed to evaluate the role of
VCP979 against Ang II-stimulated cardiac hypertrophy. As shown in
Fig. 7D and E, Ang II notably
increased the average cell size of NCMs (4483.127±369.8357 vs.
7986.913±236.3841), which could be partially reversed by treatment
with VCP979 (9 µM, 24 h, 6680.613±385.5386). Furthermore,
the present results revealed that the addition of VCP979 (9
µM, 24 h) can decrease the ratio of p-p38/p38 in NCFs
exposed to Ang II in vitro (2.9061±0.1981 vs. 0.1473±0.0602,
Fig. 7F and G). In addition,
VCP979 treatment can significantly decreased the expression of
IL-1β, IL-6 and TNF-α mRNA levels in NCFs (3.4058±0.2912 vs.
1.8504±0.2208, 7.5461±0.9675 vs. 1.5955±0.2052 and 2.3879±0.1921
vs. 0.6044±0.0533, respectively; P<0.05; Fig. 7H) and NCMs (2.687±0.2218 vs.
2.4503±0.6911, 3.1266±0.3756 vs. 0.3447±0.0316 and 1.4729±0.0503
vs. 0.7569±0.0552, respectively; Fig.
7I), compared with the Ang II group.
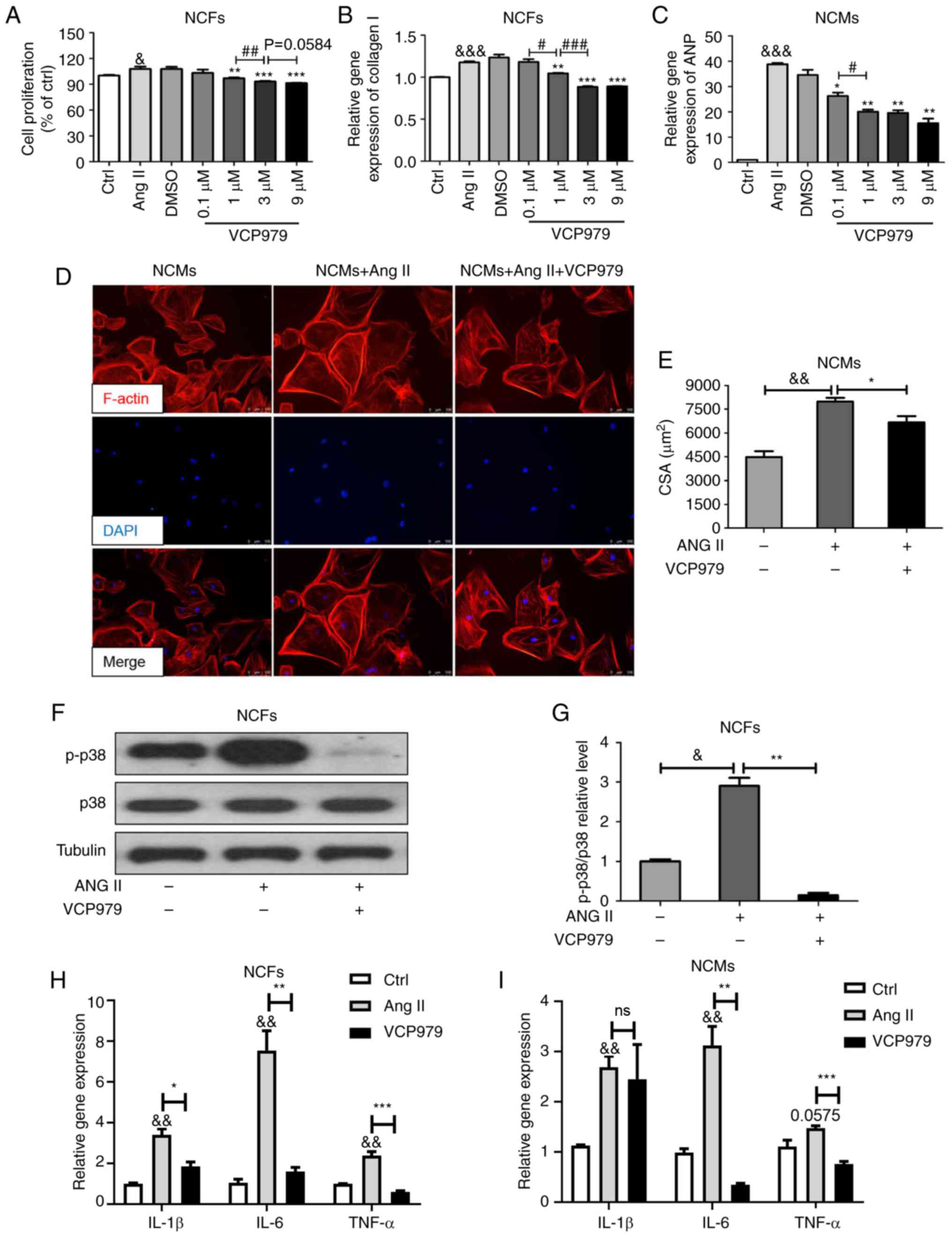 | Figure 7Addition of VCP979 inhibits the Ang
II-induced proliferation and collagen synthesis of NCFs and
hypertrophy of NCMs and suppresses the p38-MAPK signaling pathway
in vitro. (A) Ang II-induced proliferation percentage in
NCFs following treatment with different concentrations of VCP979.
(B) Collagen I mRNA level in NCFs and (C) ANP mRNA level in NCMs
following treatment with VCP979 for 24 h. (D) Representative
Phalloidin immunofluorescence staining pictures of NCMs. Phalloidin
is highly integrated with F-actin. Red, F-actin; Blue, nucleus.
Scale bar=100 µm. (E) The CSAs of the NCMs were measured
using ImageJ software. (F) Western blotting and (G) analysis of
p-p38 proteins in Ang II-induced NCFs, with or without the addition
of VCP979. The mRNA expression of IL-1β, IL-6 and TNF-α in (H) NCFs
and (I) NCMs. Results are presented as the mean ± standard error of
the mean. & P<0.05, &&P<0.01 and
&&&P<0.001, control vs. Ang II-induced
group. *P<0.05, **P<0.01 and
***P<0.001, Ang II-induced vs. VCP979 treatment
group. #P<0.05, ##P<0.01 and
###P<0.001, significant differences between various
concentrations of VCP979 in (A-C). NCFs, neonatal cardiac
fibroblasts; NCMs, neonatal cardiac myocytes; p-, phosphorylated-;
MAPK, mitogen-activated protein kinase; ANP, atrial natriuretic
peptide; CSAs, myocyte cross-sectional areas; IL, interleukin; TNF,
tumor necrosis factor; CD, cluster of differentiation; Ang II,
angiotensin. |
Discussion
Early coronary reperfusion can limit the infarct
size and improve prognosis, while persistent reperfusion can itself
induce cardiomyocyte damage. Existing pharmacological treatment is
not entirely capable of preventing the progression of ventricular
remodeling in MI/R injury and new drugs for ischemic heart disease
are still being developed (20).
In the present study, it was found that the administration of
VCP979 attenuated MI/R-induced cardiac dysfunction, markedly
alleviated myocardial fibrosis and pathological hypertrophy, and
had an anti-inflammatory effect in murine MI/R injury models. In
vitro, VCP979 treatment inhibited Ang II-induced proliferation
and collagen synthesis in NCFs and hypertrophy in NCMs. VCP979 is a
selective inhibitor for p38-MAPKα and is a much poor inhibitor of
p38-MAPKβ. It does not inhibit the p38-MAPKγ or δ isoform. A
previous study revealed that p38-MAPKα is most readily detected in
cardiac myocytes or whole hearts with little or no p38-MAPKβ
(21). Furthermore, VCP979
treatment inhibited the p38-MAPK signaling pathway in murine hearts
subjected to MI/R injury as well as Ang II-induced NCFs collagen
synthesis. Therefore, these findings provided the first evidence
that a novel small-molecule compound, VCP979, may be an effective
strategy for the protection of the heart from MI/R injury.
First, the safety of this newly developed
small-molecule compound VCP979 was verified in vivo and
in vitro. The results confirmed that different
concentrations of VCP979 (50 mg/kg/day in vivo and 0.01-9
µM in vitro, the dose chosen is based on preliminary
PK data) caused no pathological damage on murine organs and primary
cells, suggesting the safety of VCP979 treatment in murine models
and cultured cells. As it is in the early development process, the
pharmacokinetics of the compound in vivo is not fully clear
and needs further investigation.
Ventricular remodeling triggered by MI/R injury is a
progressive process that eventually leads to heart failure
(3). Cardiac fibroblasts play
vital and dynamic roles in regulating cardiac function (4). Under physiological conditions,
cardiac fibroblasts supply a scaffold for cardiomyocytes and ensure
cardiac pump function (4). By
contrast, cardiac fibroblasts expand following injury, (22) and participate in the pathogenesis
of adverse post-infarction remodeling (23). Cardiac hypertrophy is another key
pathological change that follows MI/R injury and contributes to
cardiac dysfunction (24).
Persistent pathological cardiac hypertrophy leads to increased
physical, oxidative and nitrosative stress at the cellular level
and further alters the shape, size and function of the myocardium
(25). Therefore, targeting
pathological myocardial fibrosis and hypertrophy may be a promising
therapeutic strategy for ventricular remodeling (25). However, few effective drugs can
prevent or reverse the process of ventricular remodeling (25). It has been previously reported
that the long-term daily treatment of rats with the p38-MAPK
signaling pathway inhibitor RWJ67657 can reduce pathological
myocardial fibrosis and hypertrophy and improve cardiac function
(LVFS) following MI (26).
Similarly, the present results showed a reduction in pathological
myocardial fibrosis and hypertrophy accompanied by improved cardiac
function following 2 weeks of continuous VCP979 administration,
which indicated that VCP979 may be useful for alleviating ventricle
remodeling following MI/R injury.
Reperfusion damage accelerates the infiltration of
inflammatory leukocytes into the injured myocardium (27). Inflammatory responses activated by
MI/R injury contribute to collagen deposition and cardiac
hypertrophy, but reduce left ventricular compliance (19). Cardiac inflammation is
characterized by the recruitment and activation of immune cells
mainly neutrophils, macrophages and lymphocytes at different stages
post MI/R injury (3). T
cell-mediated chronic inflammation has been confirmed as a major
player in the etiology and development of ventricular remodeling
post-MI/R injury (28).
Subsequent studies have shown the role of CD4+T-cells in
the pathology and healing of the heart following MI or MI/R injury
(29-31). Recent results showed an increase
in CD4+ T cells accompanied by high levels of
CD4+ T cell-associated inflammatory factors in MI/R
myocardia on days 7 and 14, as compared with the sham group
(14). The present study revealed
that VCP979 inhibited the infiltration of CD4+ cells in
injured hearts on day 14 following MI/R injury. As for
CD8+ T cells, little is known about their role in MI/R
injury. A subset of CD8+ cells expressing AT2R was shown
to have no cytotoxic activity, suggesting a potential
cardioprotective role in rat MI models (32). The addition of anti-CD8 antibodies
significantly reduced the number of cardiomyocytes destroyed by
lymphocytes post-MI (33). The
present data revealed that VCP979 treatment also markedly decreased
the infiltration of CD8+ T-cells in MI/R hearts on day
14.
Macrophages are equally pivotal for local infarct
zone remodeling, as they are involved in the initiation and
resolution phases of cardiac inflammatory cascades (34). Preclinical data has shown that
increased cardiac wall tension may stimulate local macrophage
proliferation following heart failure (35). There are two typical subtypes of
macrophages post-MI/R injury: Classically activated M1 macrophages
and alternatively activated M2 macrophages (36). M1 to M2 macrophage transition
improved heart repair and M2 macrophages promoted scar formation
and neo-vascularization (37).
Arg1, a M2 polarization marker, promoted an anti-inflammatory
reaction by downregulating M1 markers (iNOS) following MI (38). In the present study, VCP979
treatment markedly decreased the infiltration of F4/80+
macrophages in MI/R hearts. In addition, a down-regulation of M1
markers (iNOS) and an elevated expression of M2 polarization
markers (Arg1, CD206) were identified in reperfused myocardia on
day 14. Collectively, the present results indicated that VCP979 may
protect the heart from MI/R injury by inhibiting T cell- and
macrophage-medicated inflammatory response.
Upon infarction, specific inflammatory responses are
activated by cellular effectors, including immune cells,
cardiomyocytes, fibroblasts and vascular cells which secrete plenty
of pro/anti-inflammatory cytokines and chemokines (39). Pro-inflammatory cytokines and
chemokines are associated with the pathogenesis of heart repair and
ventricular remodeling in MI/R injury progression (25). Pro-inflammatory cytokines,
including IL-1β, IL-6, IL-8, TNF-α and mitochondrial pyruvate
carrier-1, are secreted and upregulated from adjacent
cardiomyocytes, resulting in leukocyte infiltration and infarct
area expansion (10). Other
cytokines, such as interferon-γ, IL-2 and IL-4, are secreted by T
cells and further stimulate the chemotaxis of monocytes and
neutrophils in MI/R injury (40).
Conversely, inhibitory cytokines, such as IL-10, are involved in
the suppression of extracellular matrix formation and
pro-inflammatory cytokine secretion (38). IL-4 and IL-13 may also repress
inflammatory responses and induce macrophage deactivation post-MI/R
injury (38). It was found that
VCP979 reduced the mRNA levels of pro-inflammatory cytokines in
reperfusion myocardia on day 14 following MI injury. Therefore,
these results indicated that VCP979 may contribute to cardiac
protection by inhibiting the inflammatory response.
Ang II is a key biologically active component of the
renin-Ang system and is produced by the hydrolysis of Ang I under
the action of Ang-converting enzyme (24). Previous studies have reported that
Ang II can modulate the proliferation and activity of cardiac
fibroblasts, and stimulate collagen synthesis (41,42). In addition, Ang II may directly
promote cell hypertrophy and accelerate protein synthesis and cell
size in vitro (41).
Clinical studies verified that the plasma Ang II level was elevated
in heart failure patients, which may be associated with cardiac
dysfunction and poor prognosis (43,44). Ang II can directly regulate
downstream inflammatory activities through several signaling
pathways, such as phospho-p38 (45). It was found that, in murine MI/R
injury models, the mRNA level of Ang II markedly increased in
hearts on day 28, but VCP979 treatment reversed this elevation.
Consistent with previous reports (41,46), the addition of Ang II induced
significant collagen synthesis in NCFs and hypertrophy in NCMs. The
present results further revealed that the addition of VCP979
prevented NCFs from Ang II-induced collagen synthesis and NCMs from
Ang II-induced hypertrophy in the cell culture system.
MAPKs signaling pathways, particularly p38-MAPK,
play crucial roles in the MI/R injury and consequent pathological
remodeling (9). Preclinical
evidence has shown that myocardial ischemia is a potent stimulant
of p38 activation, which might increase the production of
inflammatory cytokines and accelerate the rate of infarction/death
(9). A previous study has
demonstrated that the p38-MAPK signaling pathway inhibitor SB203580
significantly repressed MI/R injury through its anti-inflammatory,
anti-oxidative and anti-apoptosis effects (47). In the present experiments, the
small-molecule compound VCP979 inhibited the MI/R injury-triggered
activation of the p38-MAPK signaling pathway in murine models. In
addition, VCP979 markedly reduced NCFs collagen synthesis exposed
to Ang II by suppressing the p38-MAPK signaling pathway in
vitro. Although the present study demonstrated that VCP979
inhibited myocyte hypertrophy, whether VCP979 exerted its roles on
phosphorylated p38 MAPK in NCMs needs further investigation.
The present study, to the best of our knowledge was
the first to demonstrate that the novel small-molecule compound
VCP979 may improve ventricular remodeling in murine MI/R injury
models by ameliorating pathological myocardial fibrosis and
hypertrophy. The protective effects of VCP979 in murine MI/R injury
models were attributed to the inhibition of the inflammatory
response and the suppression of the p38-MAPK signaling pathway.
Therefore, VCP979 may serve as a small-molecule drug for the
clinical treatment of MI/R injury. Future clinical studies are
required to verify its efficacy and prospects.
Acknowledgments
The authors would like to thank Dr Liang Zheng from
the Research Center for Translational Medicine, Shanghai East
Hospital, Tongji University School of Medicine, for statistical
assistance.
Funding
The present study was supported by the National
Nature Science Foundation of China (NSFC; grant nos. 81670458,
81470393 and 81370434), Shanghai Municipal Health and Family
Planning Commission [grant nos. ZY(2018-2020)-FWTX-2007], Shanghai
Key Medical Discipline for Critical Care Medicine (grant nos.
2017zz02017), The National Key Research and Development Program of
China (grant nos. 2017YFA0105600), The Science and Technology
Commission of Shanghai Municipality (grant nos. 17431906600),
Three-year plan on TCM of Pudong Health Bureau of Shanghai (grant
nos. PDZY-2018-0603) and the National Health and Medical Research
Council of Australia (program grant no. 1092642 and development
grant no. 546243 to BHW).
Availability of data and materials
All data generated or analyzed during this study are
included in this paper, and more detailed data in the current study
are available from the corresponding author on reasonable
request.
Authors' contributions
ZL, XZ and BW contributed to the conception and
design of the study. JL, QM, XL, RZ, DY, XG, HC, FL, XG, HF
contributed to acquisition, analysis and interpretation of the
data. JL, XZ, ZL and BW wrote and revised the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The experimental procedures were implemented
following the approval of the Institutional Animal Care and Use
Committee of Tongji University (Shanghai, China; approval no.
TJLAC-019-121).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Moran AE, Forouzanfar MH, Roth GA, Mensah
GA, Ezzati M, Flaxman A, Murray CJ and Naghavi M: The global burden
of ischemic heart disease in 1990 and 2010: The global burden of
disease 2010 study. Circulation. 129:1493–1501. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miura T and Miki T: Limitation of
myocardial infarct size in the clinical setting: Current status and
challenges in translating animal experiments into clinical therapy.
Basic Res Cardiol. 103:501–513. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med. 17:1391–1401.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li Y, Li Z, Zhang C, Li P, Wu Y, Wang C,
Bond Lau W, Ma XL and Du J: Cardiac fibroblast-specific activating
transcription factor 3 protects against heart failure by
suppressing MAP2K3-p38 signaling. Circulation. 135:2041–2057. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Krum H and Teerlink JR: Medical therapy
for chronic heart failure. Lancet. 378:713–721. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kloner RA: Current state of clinical
translation of cardioprotective agents for acute myocardial
infarction. Circ Res. 113:451–463. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gerczuk PZ and Kloner RA: An update on
cardioprotection: A review of the latest adjunctive therapies to
limit myocardial infarction size in clinical trials. J Am Coll
Cardiol. 59:969–978. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arkin MR and Wells JA: Small-molecule
inhibitors of protein-protein interactions: Progressing towards the
dream. Nat Rev Drug Discov. 3:301–317. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kumphune S, Chattipakorn S and
Chattipakorn N: Role of p38 inhibition in cardiac
ischemia/reperfusion injury. Eur J Clin Pharmacol. 68:513–524.
2012. View Article : Google Scholar
|
|
10
|
Prompunt E, Sanit J, Barrère-Lemaire S,
Nargeot J, Noordali H, Madhani M and Kumphune S: The
cardioprotective effects of secretory leukocyte protease inhibitor
against myocardial ischemia/reperfusion injury. Exp Ther Med.
15:5231–5242. 2018.PubMed/NCBI
|
|
11
|
Cai Y, Lu C, Xu T, Ma Y, Min S, Scammells
P, Wang B and Ju S: Diffusion tensor imaging evaluation of
axonal/white matter remodeling in a mouse model of diabetic stroke
treated with novel p38 MAPK inhibitor, VCP979. J Biomed
Nanotechnol. 14:585–593. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vinh NB, Devine SM, Munoz L, Ryan RM, Wang
BH, Krum H, Chalmers DK, Simpson JS and Scammells PJ: Design,
synthesis, and biological evaluation of tetra-substituted
thiophenes as inhibitors of p38α MAPK. ChemistryOpen. 4:56–64.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu T, Ding J, Ge H, Xu X, Scammells P,
Wang B and Ju S: Effects of VCP979 novel p38 mitogen activated
protein kinase inhibitor on progression of pancreatic cancer in
mouse model with diabetic conditions. J Biomed Nanotechnol.
15:1325–1333. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yuan D, Tie J, Xu Z, Liu G, Ge X, Wang Z,
Zhang X, Gong S, Liu G, Meng Q, et al: Dynamic profile of CD4(+)
T-cell- associated cytokines/chemokines following murine myocardial
infarction/reperfusion. Mediators Inflamm. 2019:94836472019.
View Article : Google Scholar
|
|
15
|
Yang L, Wang B, Zhou Q, Wang Y, Liu X, Liu
Z and Zhan Z: MicroRNA-21 prevents excessive inflammation and
cardiac dysfunction after myocardial infarction through targeting
KBTBD7. Cell Death Dis. 9:7692018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu J, Ye Q, Xu S, Chang YX, Liu X, Ma Y,
Zhu Y and Hua S: Shengmai injection alleviates H2O2 induced
oxidative stress through activation of AKT and inhibition of ERK
pathways in neonatal rat cardiomyocytes. J Ethnopharmacol.
239:1116772019. View Article : Google Scholar
|
|
17
|
Dolber PC, Beyer EC, Junker JL and Spach
MS: Distribution of gap junctions in dog and rat ventricle studied
with a double-label technique. J Mol Cell Cardiol. 24:1443–1457.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhuang R, Wu J, Lin F, Han L, Liang X,
Meng Q, Jiang Y, Wang Z, Yue A, Gu Y, et al: Fasudil preserves lung
endothelial function and reduces pulmonary vascular remodeling in a
rat model of endstage pulmonary hypertension with left heart
disease. Int J Mol Med. 42:1341–1352. 2018.PubMed/NCBI
|
|
19
|
Dick SA and Epelman S: Chronic heart
failure and inflammation: What do we really know? Circ Res.
119:159–176. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tamargo J, Caballero R and Delpón E: New
drugs in preclinical and early stage clinical development in the
treatment of heart failure. Expert Opin Investig Drugs. 28:51–71.
2019. View Article : Google Scholar
|
|
21
|
Lemke LE, Bloem LJ, Fouts R, Esterman M,
Sandusky G and Vlahos CJ: Decreased p38 MAPK activity in end-stage
failing human myocardium: p38 MAPK alpha is the predominant isoform
expressed in human heart. J Mol Cell Cardiol. 33:1527–1540. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shinde AV and Frangogiannis NG:
Fibroblasts in myocardial infarction: A role in inflammation and
repair. J Mol Cell Cardiol. 70:74–82. 2014. View Article : Google Scholar
|
|
23
|
Kaur H, Takefuji M, Ngai CY, Carvalho J,
Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Möllmann H,
et al: Targeted ablation of periostin-expressing activated
fibroblasts prevents adverse cardiac remodeling in mice. Circ Res.
118:1906–1917. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ma Y, Hu Y, Wu J, Wen J, Li S, Zhang L,
Zhang J, Li Y and Li J: Epigallocatechin-3-gallate inhibits
angiotensin II-induced cardiomyocyte hypertrophy via regulating
Hippo signaling pathway in H9c2 rat cardiomyocytes. Acta Biochim
Biophys Sin (Shanghai). 51:422–430. 2019. View Article : Google Scholar
|
|
25
|
French BA and Kramer CM: Mechanisms of
post-infarct left ventricular remodeling. Drug Discov Today Dis
Mech. 4:185–196. 2007. View Article : Google Scholar
|
|
26
|
Kompa AR, See F, Lewis DA, Adrahtas A,
Cantwell DM, Wang BH and Krum H: Long-term but not short-term p38
mitogen-activated protein kinase inhibition improves cardiac
function and reduces cardiac remodeling post-myocardial infarction.
J Pharmacol Exp Ther. 325:741–750. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Di Napoli P, Taccardi AA, De Caterina R
and Barsotti A: Pathophysiology of ischemia-reperfusion injury:
Experimental data. Ital Heart J. 3(Suppl 4): 24s–28s.
2002.PubMed/NCBI
|
|
28
|
Hofmann U and Frantz S: Role of
lymphocytes in myocardial injury, healing, and remodeling after
myocardial infarction. Circ Res. 116:354–367. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hofmann U, Beyersdorf N, Weirather J,
Podolskaya A, Bauersachs J, Ertl G, Kerkau T and Frantz S:
Activation of CD4+ T lymphocytes improves wound healing and
survival after experimental myocardial infarction in mice.
Circulation. 125:1652–1663. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Boag SE, Andreano E and Spyridopoulos I:
Lymphocyte communication in myocardial ischemia/reperfusion injury.
Antioxid Redox Signal. 26:660–675. 2017. View Article : Google Scholar
|
|
31
|
Hofmann U and Frantz S: Role of T-cells in
myocardial infarction. Eur Heart J. 37:873–879. 2016. View Article : Google Scholar
|
|
32
|
Stabile E, Kinnaird T, la Sala A, Hanson
SK, Watkins C, Campia U, Shou M, Zbinden S, Fuchs S, Kornfeld H, et
al: CD8+ T lymphocytes regulate the arteriogenic response to
ischemia by infiltrating the site of collateral vessel development
and recruiting CD4+ mononuclear cells through the expression of
interleukin-16. Circulation. 113:118–124. 2006. View Article : Google Scholar
|
|
33
|
Varda-Bloom N, Leor J, Ohad DG, Hasin Y,
Amar M, Fixler R, Battler A, Eldar M and Hasin D: Cytotoxic T
lymphocytes are activated following myocardial infarction and can
recognize and kill healthy myocytes in vitro. J Mol Cell Cardiol.
32:2141–2149. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Panizzi P, Swirski FK, Figueiredo JL,
Waterman P, Sosnovik DE, Aikawa E, Libby P, Pittet M, Weissleder R
and Nahrendorf M: Impaired infarct healing in atherosclerotic mice
with Ly-6C(hi) monocytosis. J Am Coll Cardiol. 55:1629–1638. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sager HB, Hulsmans M, Lavine KJ, Moreira
MB, Heidt T, Courties G, Sun Y, Iwamoto Y, Tricot B, Khan OF, et
al: Proliferation and recruitment contribute to myocardial
macrophage expansion in chronic heart failure. Circ Res.
119:853–864. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao J, Li X, Hu J, Chen F, Qiao S, Sun X,
Gao L, Xie J and Xu B: Mesenchymal stromal cell-derived exosomes
attenuate myocardial ischemia-reperfusion injury through
miR-182-regulated macrophage polarization. Cardiovasc Res.
115:1205–1216. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Prabhu SD: It takes two to tango: Monocyte
and macrophage duality in the infarcted heart. Circ Res.
114:1558–1560. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Frangogiannis NG, Mendoza LH, Lindsey ML,
Ballantyne CM, Michael LH, Smith CW and Entman ML: IL-10 is induced
in the reperfused myocardium and may modulate the reaction to
injury. J Immunol. 165:2798–2808. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Prabhu SD and Frangogiannis NG: The
biological basis for cardiac repair after myocardial infarction:
From inflammation to fibrosis. Circ Res. 119:91–112. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang Z, Day YJ, Toufektsian MC, Xu Y,
Ramos SI, Marshall MA, French BA and Linden J: Myocardial
infarct-sparing effect of adenosine A2A receptor activation is due
to its action on CD4+ T lymphocytes. Circulation. 114:2056–2064.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Iwata M, Cowling RT, Yeo SJ and Greenberg
B: Targeting the ACE2-Ang-(1-7) pathway in cardiac fibroblasts to
treat cardiac remodeling and heart failure. J Mol Cell Cardiol.
51:542–547. 2011. View Article : Google Scholar
|
|
42
|
Song Q, Liu L, Yu J, Zhang J, Xu M, Sun L,
Luo H, Feng Z and Meng G: Dihydromyricetin attenuated Ang II
induced cardiac fibroblasts proliferation related to inhibitory of
oxidative stress. Eur J Pharmacol. 807:159–167. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Iwata M, Cowling RT, Gurantz D, Moore C,
Zhang S, Yuan JX and Greenberg BH: Angiotensin-(1-7) binds to
specific receptors on cardiac fibroblasts to initiate antifibrotic
and antitrophic effects. Am J Physiol Heart Circ Physiol.
289:H2356–H2363. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Basu R, Poglitsch M, Yogasundaram H,
Thomas J, Rowe BH and Oudit GY: Roles of angiotensin peptides and
recombinant human ACE2 in heart failure. J Am Coll Cardiol.
69:805–819. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shatanawi A, Romero MJ, Iddings JA,
Chandra S, Umapathy NS, Verin AD, Caldwell RB and Caldwell RW:
Angiotensin II-induced vascular endothelial dysfunction through
RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase
pathway. Am J Physiol Cell Physiol. 300:C1181–C1192. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Du M, Huang K, Gao L, Yang L, Wang WS,
Wang B, Huang K and Huang D: Nardosinone protects H9c2 cardiac
cells from angiotensin II-induced hypertrophy. J Huazhong Univ Sci
Technolog Med Sci. 33:822–826. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang X, Wang Y, Shen W, Ma S, Chen W and
Qi R: Rosa rugosa flavonoids alleviate myocardial ischemia
reperfusion injury in mice by suppressing JNK and p38 MAPK.
Microcirculation. 24:2017. View Article : Google Scholar
|