Introduction
Viral myocarditis (VMC) is the most common type of
myocarditis, and it is mainly caused by Coxsackievirus B (CVB)
infection (1-3). In total, ~20% of sudden mortalities
in infants are due to viral myocarditis or fatal ventricular
arrhythmias caused by viral myocarditis (4,5).
The prognosis of patients with persistent viral myocarditis is
poor, and the incidence of viral myocarditis has increased in the
past years (6). Viral myocarditis
is a serious threat to pediatric health (7). Chronic inflammation of myocardial
cells can cause myocardial cell growth inhibition, hypertrophy,
apoptosis and myocardial fibrosis, thus causing dilated
cardiomyopathy and heart failure (8,9).
The currently available treatments for viral myocarditis remain
unsatisfactory. Therefore, it is important to study the molecular
mechanism of the pathogenesis of viral myocarditis and to identify
new effective diagnostic and therapeutic targets.
MicroRNAs (miRNAs) are a class of small endogenous
non-coding RNAs that can post-transcriptionally regulate gene
expression by binding to the 3′-untranslated region (UTR) of target
mRNAs (10-12). miRNAs have been identified to play
important roles in the regulation of various biological processes,
including cell proliferation, differentiation and apoptosis
(13-15). In addition, miRNAs are considered
to be involved in the regulation of vascular proliferation, cardiac
development, heart failure and cardiac hypertrophy (16-18). Several miRNAs, including miR-155,
miR-21, miR-146a, miR-208b, miR-499-5p and miR-148a, have been
reported to play important roles in the pathogenesis of viral
myocarditis (19-21).
Sirtuin 1 (SIRT1) is a NAD+-dependent
deacetylase and it is important for cell apoptosis (22). Previous studies have indicated
that SIRT1 plays an important role in the regulation of oxidative
stress and inflammatory response (23-26). SIRT1 overexpression and SIRT1
activator SRT1720 treatment could attenuate renal lipid content and
expression of lipogenesis, oxidative stress and inflammatory
markers in mice (27). SIRT1
controls acetaminophen hepatotoxicity by regulating inflammation
and oxidative stress (24). SIRT1
activation could attenuate inflammation and premature senescence
involved in chronic lung diseases (28). In addition, a previous study
indicated that SIRT1 attenuates endoplasmic reticulum
stress-induced cardiomyocyte apoptosis by regulating PERK/eIF2α,
ATF6/CHOP, and IRE1α/JNK pathways (29). A number of other studies have also
demonstrated the important regulatory role of SIRT1 in
cardiomyocyte apoptosis (30-32). In addition, SIRT1 has been found
to be a target of miR-543 (33,34) and miR-217 (35). Collectively, the present results
suggested that miR-543 and miR-217 may have a role in viral
myocarditis development by regulating the inflammatory response and
cardiomyocyte apoptosis via SIRT1. To the best of our knowledge,
the expression levels and roles of miR-543 and miR-217 in viral
myocarditis, and the relationship between miR-543, miR-217 and
SIRT1 in cardiomyocytes remain unclear.
Therefore, the aim of the present study was to
investigate the expression levels and role of miR-217 and miR-543
in viral myocarditis, and to explore their underlying
mechanism.
Materials and methods
Clinical samples
In total, 30 children (female to male ratio, 13:17;
age range, between 9 months and 12 years) with viral myocarditis
and healthy volunteers were enrolled at Wuhan Children's Hospital
between May 2015 and May 2017. Blood samples (500 µl) were
collected from each participant. Informed consent was obtained from
each patient and the parents or the legal guardians of the
patients. The present study was approved by The Ethics Committee of
Wuhan Children's Hospital.
Animal experiments
In total, 40 male BALB/C mice (weight, 18-22 g; age,
4 weeks) were obtained from Vital River Laboratories Co., Ltd. Mice
were housed at 25±5°C, with 50-70% humidity, with a 12-h dark/light
cycle and free access to food and water. The in vivo animal
experiments were carried out according to the to the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals (36). This study was
approved by The Animal Ethics Committee of Tongji Medical College,
Huazhong University of Science and Technology.
The mice were divided into two groups: i) Control
group (mice were not infected and were intraperitoneally injected
with 0.2 ml PBS); and ii) CVB3 infection group (1 h after
intraperitoneal injection with 103 TCID50 of CVB3 virus,
the mice were injected with 0.2 ml PBS). The CVB3 strain used in
the present study was obtained from The Center for Endemic Disease
Control of China. The CVB3 was amplified in HeLa cells (American
Type Culture Collection) according to a previous study (37). Mice in the CVB3 infection group
were injected with CVB3 for three days, and sacrificed on day 21.
At day 21, the peripheral blood was collected and stored at −20°C.
After the mice were sacrificed by decapitation, myocardial tissues
were harvested and stored in liquid nitrogen until use.
Cell culture and infection
Embryonic rat heart-derived H9C2 cells and HeLa
cells were obtained from The American Type Culture Collection. All
cells were grown in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
containing 10% FBS (Gibco; Thermo Fisher Scientific, Inc.), 1%
streptomycin-penicillin solution and maintained at 37°C with 5%
CO2.
CVB3 infection in H9C2 cells was performed by
treating H9C2 cells with 104 PFU/ml CVB3 virus. H9C2
cells were randomly divided into the following groups: i) Control
group, normal H9c2 cells without any treatment; ii) CVB3 group,
H9C2 cells treated with 104 PFU/ml CVB3 virus, as
previously described (38); iii)
CVB3 + inhibitor control-1 group, H9C2 cells were transfected with
the inhibitor control of miR-217 for 48 h and then treated with
104 PFU/ml CVB3 virus; iv) CVB3+ miR-217 inhibitor group
(CVB3 + miR-217 inhibitor), H9C2 cells were transfected with
miR-217 inhibitor for 48 h and then treated with 104
PFU/ml CVB3 virus; v) CVB3 + miR-217 inhibitor + SIRT1-small
interfering (si) RNA group (CVB3 + miR-217 inhibitor +
SIRT1-siRNA), H9C2 cells were co-transfected with miR-217 inhibitor
and SIRT1-siRNA for 48 h and then treated with 104
PFU/ml CVB3 virus; vi) CVB3 + inhibitor control-2 group, H9C2 cells
were transfected with the inhibitor control of miR-543 for 48 h and
then treated with 104 PFU/ml CVB3 virus; vii) CVB3+
miR-543 inhibitor group (CVB3 + miR-543 inhibitor), H9C2 cells were
transfected with the miR-543 inhibitor for 48 h and then treated
with 104 PFU/ml CVB3 virus; viii) CVB3 + miR-543
inhibitor + SIRT1-siRNA group (CVB3 + miR-543 inhibitor +
SIRT1-siRNA), H9C2 cells were co-transfected with miR-543 inhibitor
and SIRT1-siRNA for 48 h and then treated with 104
PFU/ml CVB3 virus for 48 h.
Cell transfection
H9C2 cells were seeded into six-well plates
(1×106 cells/well) and cultured at 37°C for 24 h. Then,
H9C2 cells were transfected with the 100 nM inhibitor control of
miR-217 inhibitor (inhibitor control-1: 5′-GCC UCC GGC UUC GCA CCU
CU-3′), 100 nM inhibitor control of miR-543 inhibitor (inhibitor
control-2: 5′-CAG UAC UUU UGU GUA GUA CAA-3′), 100 nM miR-217
inhibitor (5′-UAC UGC AUC AGG AAC UGA UUG GA-3′), 100 nM miR-543
inhibitor (5′-AAG AAG UGC ACC GCG AAU GUU U-3′), 1 µM
control-siRNA (cat. no. sc-36869; Santa Cruz Biotechnology, Inc.),
1 µM SIRT1-siRNA (cat. no. sc-40986; Santa Cruz
Biotechnology, Inc.), 100 nM miR-217 inhibitor + 1 µM
SIRT1-siRNA or 100 nM miR-543 inhibitor + 1 µM SIRT1-siRNA
by using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. After
48 h of cell transfection, transfection efficiency was detected
using reverse transcription-quantitative (RT-q)PCR.
RT-qPCR
Total RNA from blood samples and cells was collected
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. Total
RNA was reverse transcribed into cDNA using the PrimeScript RT
Reagent Kit (Takara Bio, Inc.) according to the manufacturer's
protocol. The temperature protocol for the reverse transcription
reaction was as follows: 25°C for 5 min, 42°C for 60 min and 80°C
for 2 min. cDNAs were analyzed by RT-qPCR using SYBR Premix Ex Taq
(Takara Bio, Inc.) according to the manufacturer's instructions.
The amplification conditions were as following: Initial
denaturation at 95°C for 5 min, followed by 40 cycles of 95°C for
15 sec and 60°C for 30 sec. The primer sequences used for the
RT-qPCR were as follows: miR-543 forward 5′-CAG TGC TAA AAC ATT CGC
GG-3′ and reverse 5′-TAT GGT TGT TCA CGA CTC CTT CAC-3′; miR-217
forward 5′-TAC TGC ATC AGG AAC TGA CTG GA-3′; and reverse 5′-GTG
CAG GGT CCG A GG T-3′; U6 forward 5′-GCT TCG GCA GCA CAT ATA CTA
AAA T-3′ and reverse 5′-CGC TTC ACG AAT TTG CGT GT C AT-3′; SIRT1
forward 5′-AAT CCA GTC ATT AAA GGT CTA CAA-3′ and reverse
5′-TAGGACCATTACTGCCAGAGG-3′; GAPDH forward 5′-CTT TGG TAT CGT GGA
AGG ACT C-3′ and reverse 5′-GTA GAG GCA GGG ATG ATG TTC T-3′. U6
and GAPDH were used as the internal controls to normalize the
expression level of miRNA and mRNA, respectively. The relative gene
expression was calculated using the 2−ΔΔCq method
(39).
Western blotting
Total protein from cells was harvested using a RIPA
tissue/cell lysis buffer (cat. no. R0010; Beijing Solarbio Science
& Technology Co., Ltd.). Bicinchoninic acid assay (Thermo
Fisher Scientific, Inc.) was performed to determine the protein
concentrations. Equal amount of protein (25 µg/lane) was
separated by 12% SDS-PAGE and transferred to PVDF (Roche). Then,
after blocking with 5% non-fat milk for 1 h at room temperature,
the membranes were incubated with the following primary antibodies:
SIRT1 (1:1,000; cat. no. 9475; Cell Signaling Technology, Inc.),
phosphorylated (p)-AMP-activated protein kinase-α (p-AMPK-α; 1:
1,000; cat. no. 50081; Cell Signaling Technology, Inc.), AMPK-α
(1:1,000; cat. no. 5831; Cell Signaling Technology, Inc.), p65
(1:1,000; cat. no. 8242; Cell Signaling Technology, Inc.), p-NF-κB
p65 (p-p65; 1:1,000; cat. no. 3033; Cell Signaling Technology,
Inc.), Bax (1:1,000; cat. no. 14796; Cell Signaling Technology,
Inc.), Bcl-2 (1:1,000; cat. no. ab196495; Abcam), and β-actin
(1:1,000; cat. no. 4970; Cell Signaling Technology, Inc.),
overnight at 4°C, followed by incubation with horseradish
peroxidase-conjugated goat anti-rabbit immunoglobulin G secondary
antibody (1:2,000; cat. no. 7074; Cell Signaling Technology, Inc.)
at room temperature for 1 h. An ECL system (Pierce; Thermo Fisher
Scientific, Inc.) was used to visualize protein bands according to
the manufacturer's instructions and proteins were quantified using
Quantity One Image software (version 4.6; Bio-Rad Laboratories,
Inc.).
CCK-8 assay
After treatment, CCK-8 assay was used to analyze
cell viability. Sells were seeded in 96-well plates (Corning Inc.)
and incubated for 48 h at 37°. Subsequently, 10 µg/ml CCK-8
solution was added to each well, and the cells were then incubated
at 37° for 4 h. The optical density was detected at a wavelength of
450 nm using a micro-plate reader (Thermo Fisher Scientific,
Inc.).
Cell apoptosis assay
The Annexin V-FITC/propidium iodide (PI) apoptosis
detection kit (cat. no. 70-AP101-100; MultiSciences) was used to
analyze cell apoptosis. Following indicated treatments, cells were
collected with 0.25% trypsin, washed with PBS, and then stained
with 5 µl Annexin V-FITC and 5 µl PI for 30 min at
room temperature in the dark. Finally, flow cytometry (BD
Biosciences) was performed to analyze cell apoptosis, and data were
analyzed using WinMDI software (version 2.5; Purdue University
Cytometry Laboratories; www.cyto.purdue.edu/flowcyt/software/Catalog.htm).
ELISA
To measure the production of inflammatory cytokines
interleukin (IL)-6 and IL-1β in cell culture medium, the culture
supernatants of H9C2 cells were collected, and the level of IL-6
(cat. no. ab100772), and IL-1β (cat. no. ab100768) was detected by
using ELISA kits (Abcam) following the manufacturer's
instructions.
MDA and SOD detection
The SOD activity (cat. no. ab118970) and the level
of MDA (cat. no. Ab118970) were determined by using commercially
available kits (Abcam) following the manufacturer's protocols.
Automatic biochemical analysis
The contents of creatine kinase (CK-MB) and lactate
dehydrogenase (LDH) in the peripheral blood of mice in different
groups were detected according to the manufacturer's protocols
(Beckman Coulter, Inc.).
Dual-luciferase reporter assay
TargetScan software (version 7.1; www.targetscan.org/vert_71) was used to predict the
putative target genes of miR-217 and miR-543. SIRT1 was found to be
a potential target of both miR-217 and miR-543. To confirm the
targets and the putative binding sites, the wild-type (WT) 3′UTR of
SIRT1 or the mutant (MUT) 3′UTR SIRT1 were cloned into the
dual-luciferase reporter vector pmiR-RB-REPORT (Guangzhou RiboBio
Co., Ltd.) following the manufacturer's instructions. The
QuikChange Site-Directed Mutagenesis kit (Agilent Technologies,
Inc.) was used to make a point mutation in the miR-217 and miR-543
binding domain on the 3′UTR of SIRT1. H9C2 cells were
co-transfected with 100 ng WT-SIRT1 or 100 ng MUT-SIRT1 and 50 nM
miR-217 and miR-543 mimic or 50 nM mimic control using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) following the manufacturer's protocol. After 48
h, luciferase activity was determined by using a
Dual-luciferase® reporter assay system (Promega
Corporation) according to the manufacturer's protocol. Firefly
luciferase activity was normalized to Renilla luciferase
activity.
Statistical analysis
All data are presented as the mean ± SD from >3
independent experiments. All statistical analyses were performed
using SPSS 17.0 software (SPSS, Inc.). Two groups were compared
using Student's t-test. One-way ANOVA followed by Tukey's post hoc
test was used to compare multiple groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
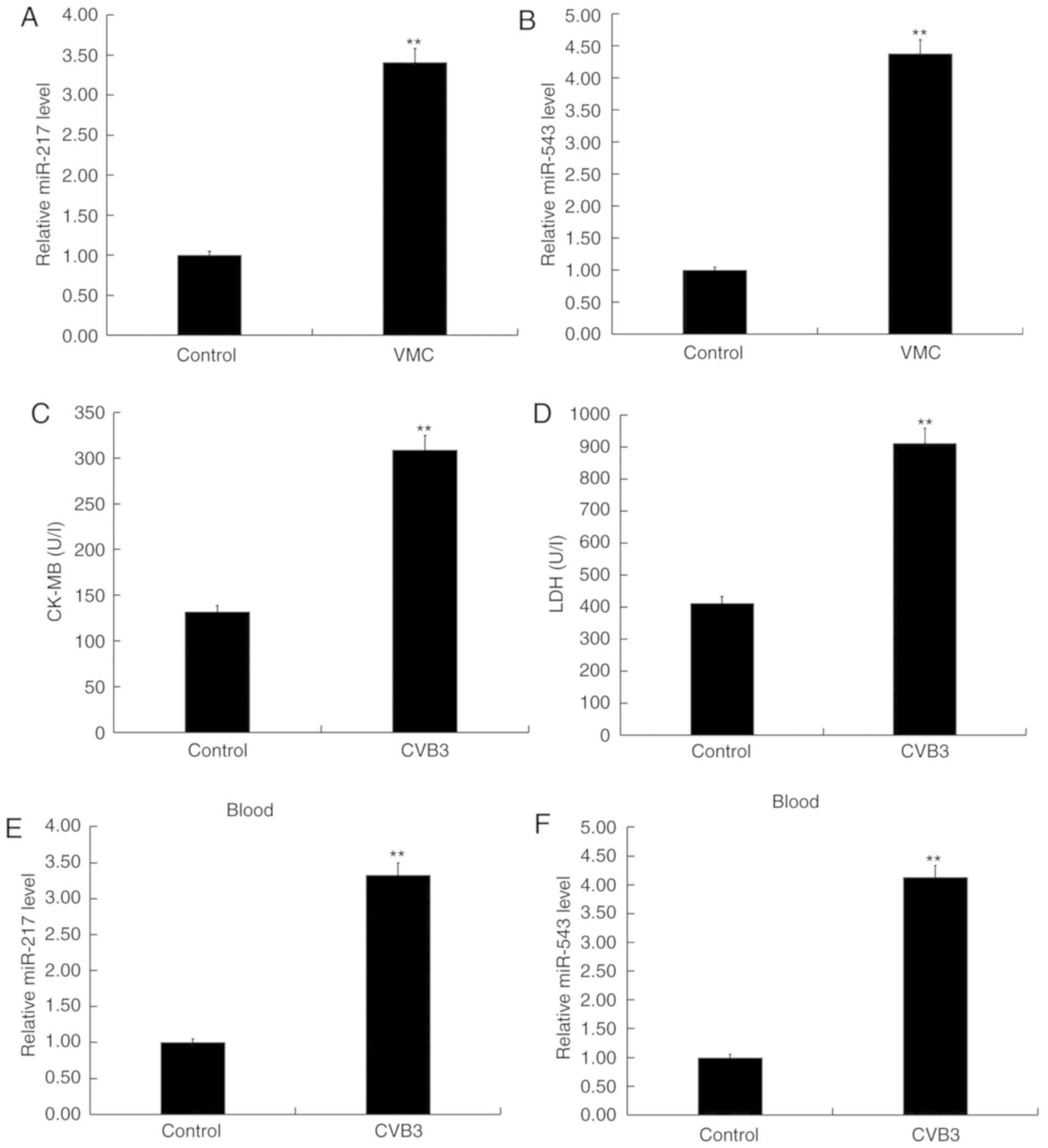
miR-217 and miR-543 are upregulated in
viral myocarditis
The levels of miR-217 and miR-543 were detected in
the blood samples collected from children with viral myocarditis
and in the blood samples from healthy volunteers using RT-qPCR, and
the results indicated that the levels of miR-217 and miR-543 in the
blood samples from children with viral myocarditis was
significantly higher than that in the blood samples from the
healthy volunteers (P<0.01; Fig.
1A and B). In addition, the levels of miR-217 and miR-543 in
the blood and myocardial tissues of CVB3-infected mice were
detected. As shown in Fig. 1C and
D, CVB3 infection significantly increased the levels of CK-MB
and LDH in the blood of CVB3 infected mice (both P<0.01;
Fig. 1C and D), indicating the
successful establishment of an in vivo model of viral
myocarditis (37). As expected,
the levels of miR-217 and miR-543 in the blood and myocardial
tissues of CVB3-infected mice were significantly increased compared
with the control group (all P<0.01; Fig. 1E-H). In addition, an in
vitro model of viral myocarditis was also established in the
present study, and, compared with the control group, the levels of
miR-217 and miR-543 were significantly increased in H9C2 cells
infected with CVB3 (both P<0.01; Fig. 1I and J). Collectively, the present
results suggested that miR-217 and miR-543 may play an important
role in the development of viral myocarditis.
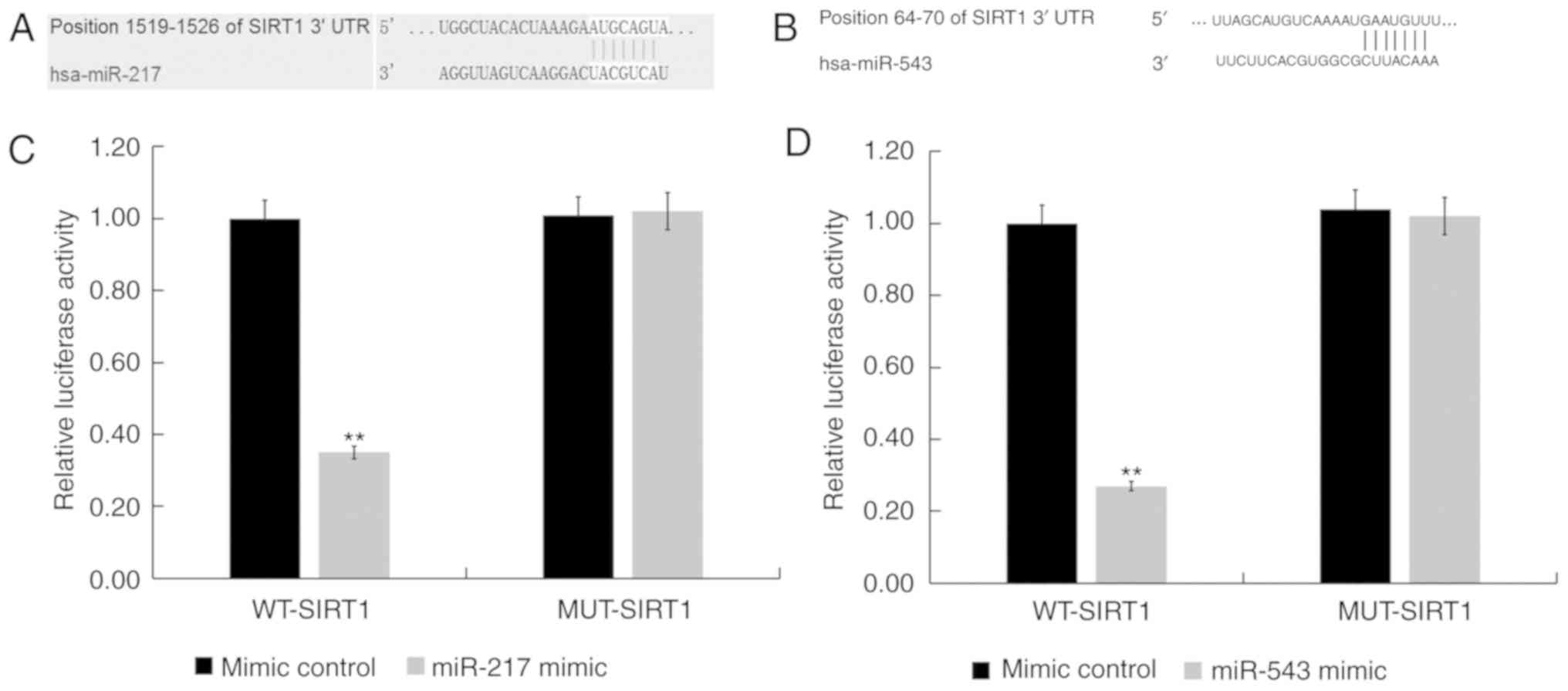
SIRT1 is a target of miR-217 and
miR-543
To examine the role of miR-217 and miR-543 in viral
myocarditis, the putative targets of these two miRNAs were
investigated. TargetScan was used to predict the potential targets
of miR-217 and miR-543, and SIRT1 was found to be a potential
target of both miR-217 and miR-543 (Fig. 2A and B). Results from the
luciferase reporter assay indicated that the luciferase activity
was significantly reduced in H9C2 cells co-transfected with miR-217
and miR-543 mimics and SIRT1-WT reporter plasmids, but
co-transfection with miR-217 and miR-543 mimic and SIRT1-MUT
reporter plasmids did not alter the luciferase activity (both
P<0.01; Fig. 2C and D).
Collectively, the present results indicated that SIRT1 was a target
of both miR-217 and miR-543.
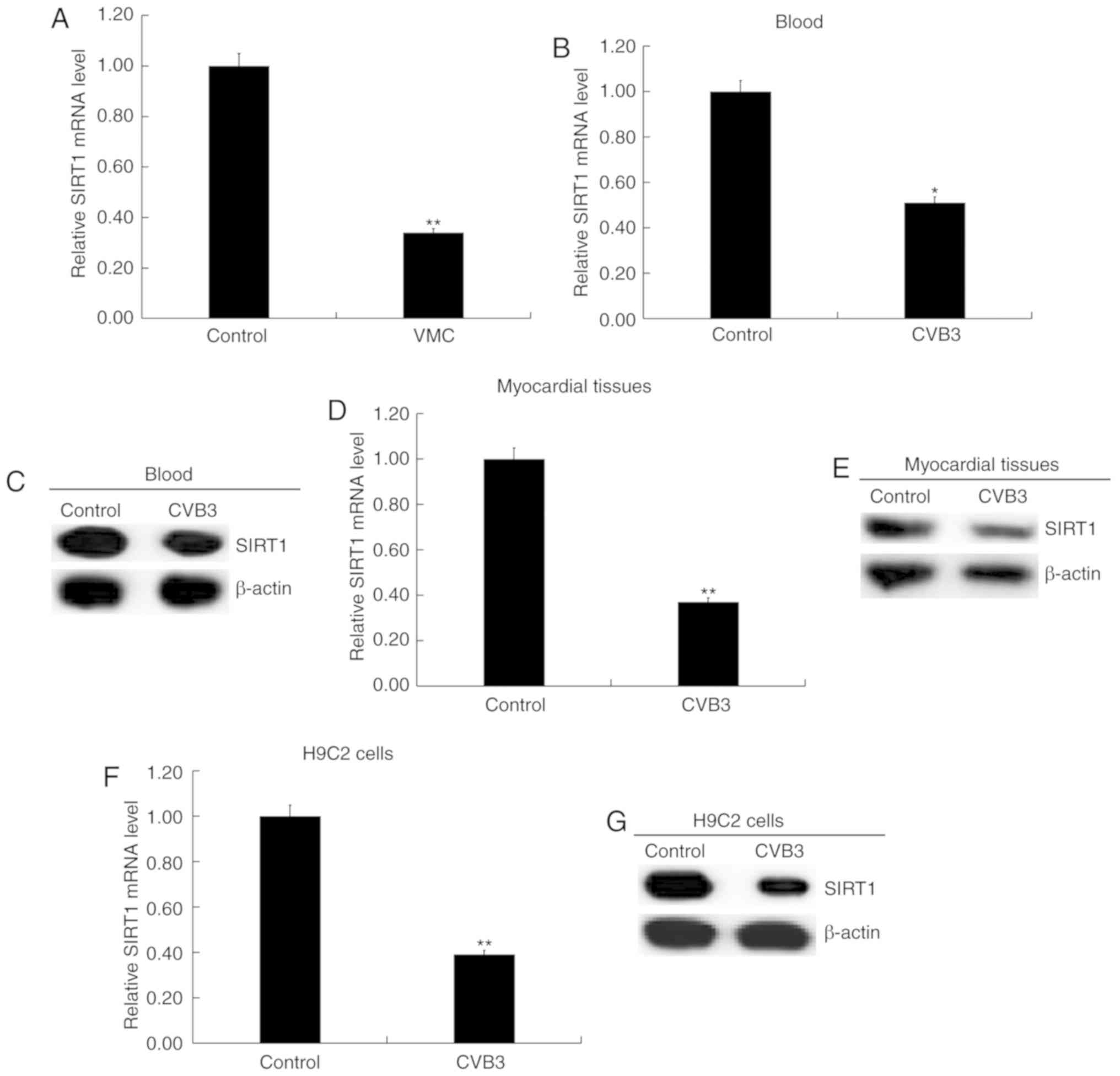
SIRT1 is downregulated in viral
myocarditis
As shown in Fig.
3A, the mRNA level of SIRT1 in the blood samples from children
with viral myocarditis was significantly lower compared with
healthy volunteers (P<0.01; Fig.
3A). Compared with the control group, the mRNA levels of SIRT1
in the blood (P<0.01; Fig. 3B)
and myocardial tissues (P<0.01; Fig. 3D) of CVB3-infected mice were
significantly decreased. The protein levels of SIRT1 in the blood
(Fig. 3C) and myocardial tissues
(Fig. 3E) of CVB3-infected mice
were also markedly decreased. In addition, SIRT1 mRNA (P<0.01;
Fig. 3F) and protein (Fig. 3G) expression levels were also
reduced in H9C2 cells infected with CVB3. The present results
further suggested that miR-217 and miR-543 may play critical roles
in the development of viral myocarditis.
miR-217 and miR-543 downregulation
increases cell proliferation and suppresses cell apoptosis in
CVB3-infected H9C2 cells
To investigate the effect of miR-217 and miR-543 on
myocarditis, H9C2 cells were transfected with the inhibitor control
of miR-217 inhibitor, inhibitor control of miR-543 inhibitor,
miR-217 inhibitor, miR-543 inhibitor, miR-217 inhibitor +
SIRT1-siRNA or miR-543 inhibitor + SIRT1-siRNA for 48 h.
Subsequently, H9C2 cells were infected with CVB3 virus. miR-217
inhibitor and miR-543 inhibitor significantly reduced the
expression levels of miR-217 and miR-543 in H9C2 cells,
respectively (P<0.01; Fig. 4A and
B). SIRT1-siRNA significantly inhibited the mRNA (P<0.01;
Fig. 4C) and protein (Fig. 4D) expression of SIRT1 in H9C2
cells. miR-217 inhibitor (P<0.01; Fig. 4E) and miR-543 inhibitor
(P<0.01; Fig. 4G)
significantly increased the mRNA expression of SIRT1 in H9C2 cells,
and these effects were reversed by SIRT1-siRNA (both P<0.01;
Fig. 4E and G). The protein
expression levels of SIRT1 increased after transfection with miR
inhibitors and decreased following SIRT1-siRNA transfection
(Fig. 4F and H).
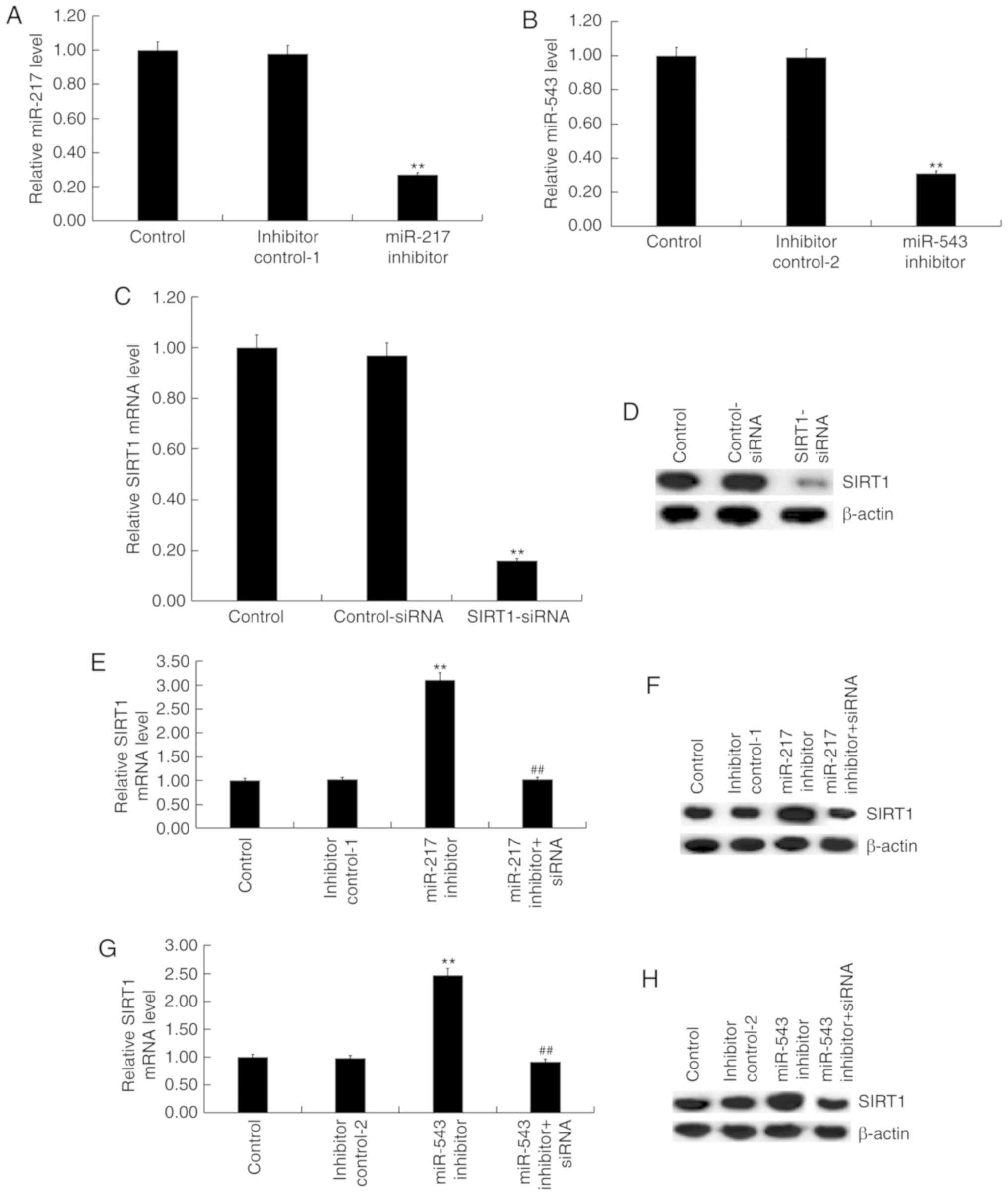 | Figure 4miR-217 and miR-543 negatively
regulate the expression of SIRT1 in H9C2 cells. H9C2 cells were
transfected with (A) miR-217 or (B) miR-543 inhibitors, and the
levels of miR-217 and miR-543 in H9C2 cells were detected using
RT-qPCR analysis. H9C2 cells were transfected with control-siRNA or
SIRT1-siRNA, and the (C) mRNA and (D) protein expression levels of
SIRT1 were detected using RT-qPCR and western blotting. H9C2 cells
were transfected with inhibitor control-1, miR-217 inhibitor or
miR-217 inhibitor + SIRT1-siRNA, and the (E) mRNA and (F) protein
expression levels of SIRT1 were detected using RT-qPCR and western
blotting. H9C2 cells were transfected with inhibitor control-1,
miR-543 inhibitor or miR-543 inhibitor + SIRT1-siRNA, and the (G)
mRNA and (H) protein expression levels of SIRT1 were detected using
RT-qPCR and western blotting. **P<0.01 vs.
corresponding control; ##P<0.01 vs. corresponding miR
inhibitor. siRNA, small interfering RNA; miR, microRNA; SIRT1,
sirtuin 1; RT-qPCR, reverse transcription-quantitative PCR;
inhibitor control-1, miR-217 inhibitor control; inhibitor
control-2, miR-543 inhibitor control. |
In addition, the results of CCK-8 assay suggested
that the decreased cell viability caused by CVB3 infection was
significantly reversed following transfection with miR-217
inhibitor (P<0.05; Fig. 5A)
and miR-543 inhibitor (P<0.05; Fig. 5B), and these changes were
significantly eliminated by SIRT1-siRNA (both P<0.05; Fig. 5A and B). Cell apoptosis assay
indicated that the increased cell apoptosis caused by CVB3
infection was significantly decreased by miR-217 inhibitor
(P<0.01; Fig. 5C) and miR-543
inhibitor (P<0.01; Fig. 5D),
and these changes were significantly suppressed by SIRT1-siRNA
(both P<0.01; Fig. 5C and D).
In addition, the decrease in the protein expression levels of Bcl-2
and Bax caused by CVB3 infection was significantly reversed by
miR-217 inhibitor (Fig. 5E) and
miR-543 inhibitor (Fig. 5F), and
these changes were suppressed by SIRT1-siRNA.
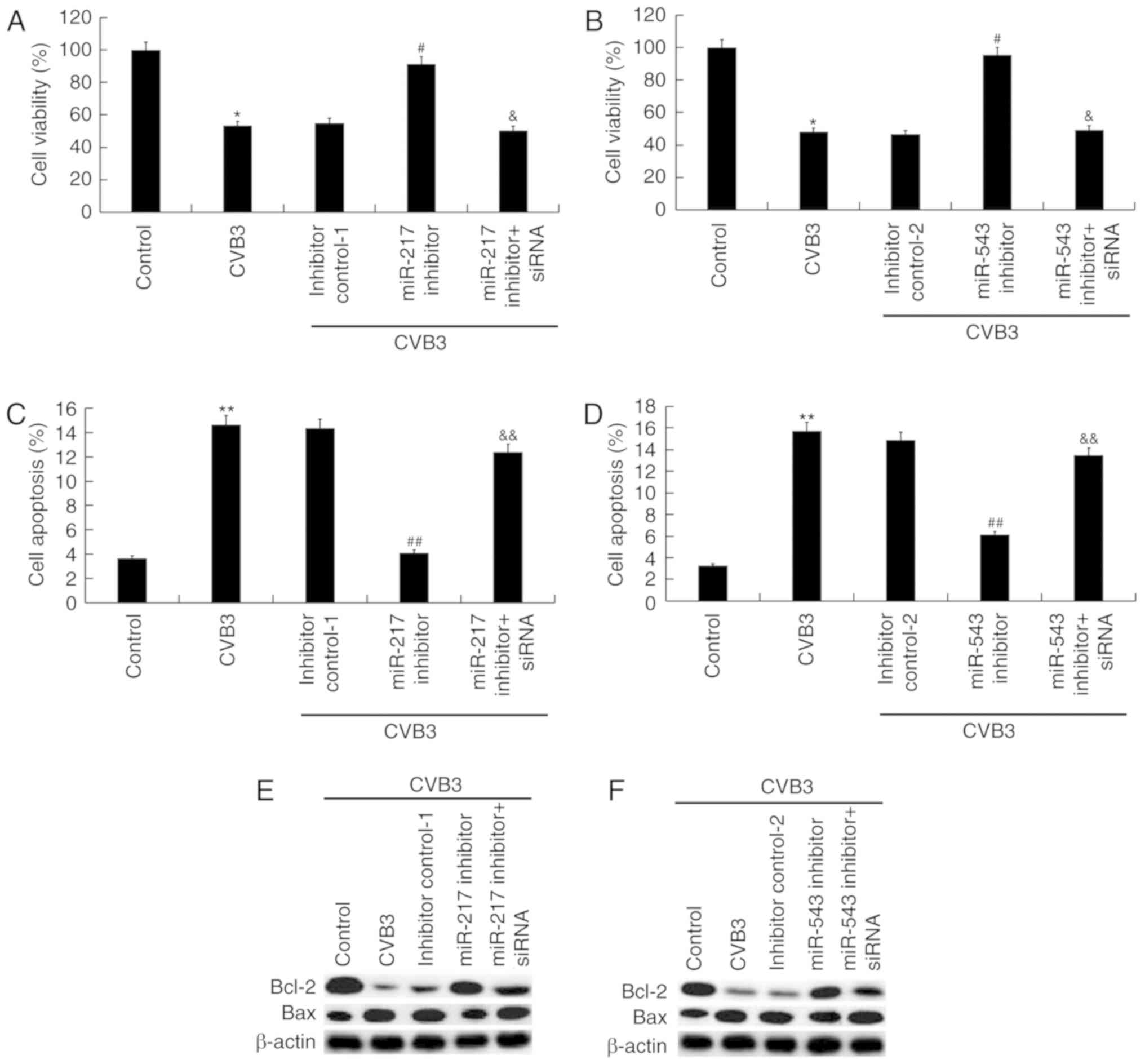 | Figure 5Role of miR-217 and miR-543 on
CVB3-infected H9C2 cells. H9C2 cells were transfected with miR-217
or miR-543 inhibitors, miR-217 or miR-543 inhibitor controls,
miR-217 inhibitor + SIRT1-siRNA, or miR-543 inhibitor + SIRT1-siRNA
for 48 h and then treated with 104 PFU/ml CVB3 virus for
48 h. Effects of (A) miR-217 and (B) miR-543 on cell viability of
CVB3-infected H9C2 cells. Effect of (C) miR-217 and (D) miR-543 on
cell apoptosis of CVB3-infected H9C2 cells. Effects of (E) miR-217
and (F) miR-543 on Bax and Bcl-2 protein expression levels in
CVB3-infected H9C2 cells. *P<0.05,
**P<0.01 vs. corresponding control;
&P<0.05, &&P<0.01 vs.
corresponding miR inhibitor. #P<0.05,
##P<0.01 vs. CVB3. CVB3, Coxsackievirus B3; miR,
microRNA; SIRT1, sirtuin 1; siRNA, small interfering RNA; inhibitor
control-1, miR-217 inhibitor control; inhibitor control-2, miR-543
inhibitor control. |
miR-217 and miR-543 downregulation
decreases oxidative stress in CVB3-infected H9C2 cells
To assess the oxidative stress, the level of MDA (an
indicator of lipid peroxidation) (40) and the activity of SOD (an enzyme
involved in the removal of free radicals) (41) were determined. The presented
findings suggested that compared with the control group, CVB3
infection significantly enhanced the level of MDA (P<0.01) and
inhibited SOD activity (P<0.01) in H9C2 cells. Compared with the
CVB3 infection group, miR-217 inhibitor significantly decreased MDA
levels (P<0.05; Fig. 6A) and
increased SOD activity (P<0.01; Fig. 6C), and these changes were
suppressed by SIRT1-siRNA. miR-543 served a similar role in
regulating MDA level and SOD activity (Fig. 6B and D).
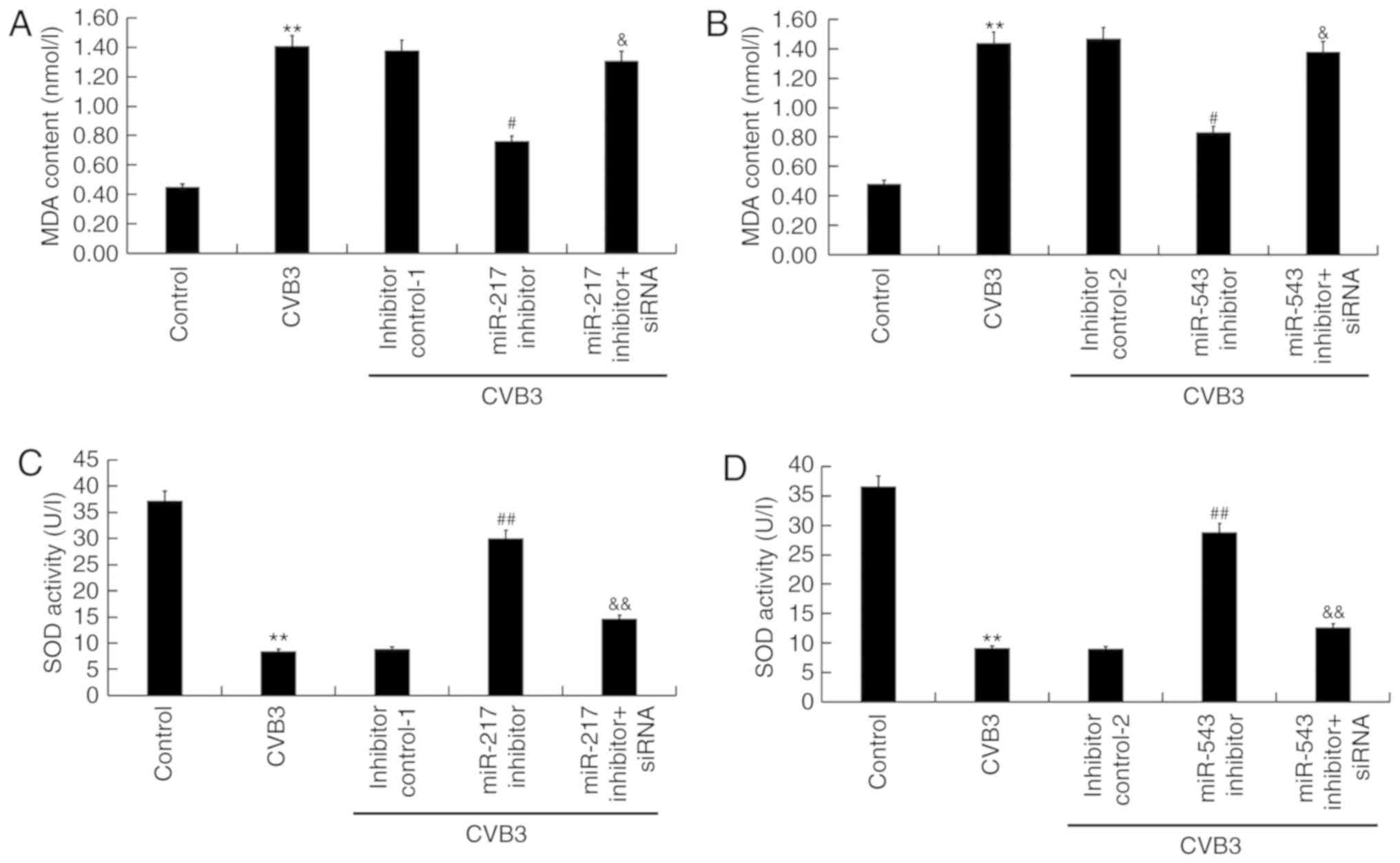 | Figure 6Effects of miR-217 and miR-543 on
oxidative stress in CVB3-infected H9C2 cells. H9C2 cells were
transfected with miR-217 or miR-543 inhibitors, miR-217 or miR-543
inhibitor controls, miR-217 inhibitor + SIRT1-siRNA, or miR-543
inhibitor + SIRT1-siRNA for 48 h and then treated with
104 PFU/ml CVB3 virus for 48 h. Effects of (A) miR-217
and (B) miR-543 on MDA levels in CVB3-infected H9C2 cells. Effect
of (C) miR-217 and (D) miR-543 on SOD activity in CVB3-infected
H9C2 cells. **P<0.01 vs. corresponding control;
&P<0.05, &&P<0.01 vs.
corresponding miR inhibitor; #P<0.05,
##P<0.01 vs. CVB3. CVB3, Coxsackievirus B3; miR,
microRNA; SIRT1, sirtuin 1; siRNA, small interfering RNA; SOD,
superoxide dismutase; MDA, malondialdehyde; inhibitor control-1,
miR-217 inhibitor control; inhibitor control-2, miR-543 inhibitor
control. |
miR-217 and miR-543 downregulation
reduces inflammatory response in CVB3-infected H9C2 cells
To study the effect of miR-217 inhibitor and miR-543
inhibitor on inflammatory response in CVB3-infected H9C2 cells, the
levels of IL-1β and IL-6 were measured by ELISA. It was found that,
compared with the control group, the levels of IL-1β and IL-6 in
CVB3-infected H9C2 cells increased significantly (P<0.01). The
increased levels of IL-1β and IL-6 in CVB3-infected H9C2 cells were
significantly reduced by miR-217 inhibitor (both P<0.01;
Fig. 7A and C) and miR-543
inhibitor (both P<0.01; Fig. 7B
and D). Notably, the effects of miR-217 and miR-543 inhibitors
were suppressed by SIRT1-siRNA.
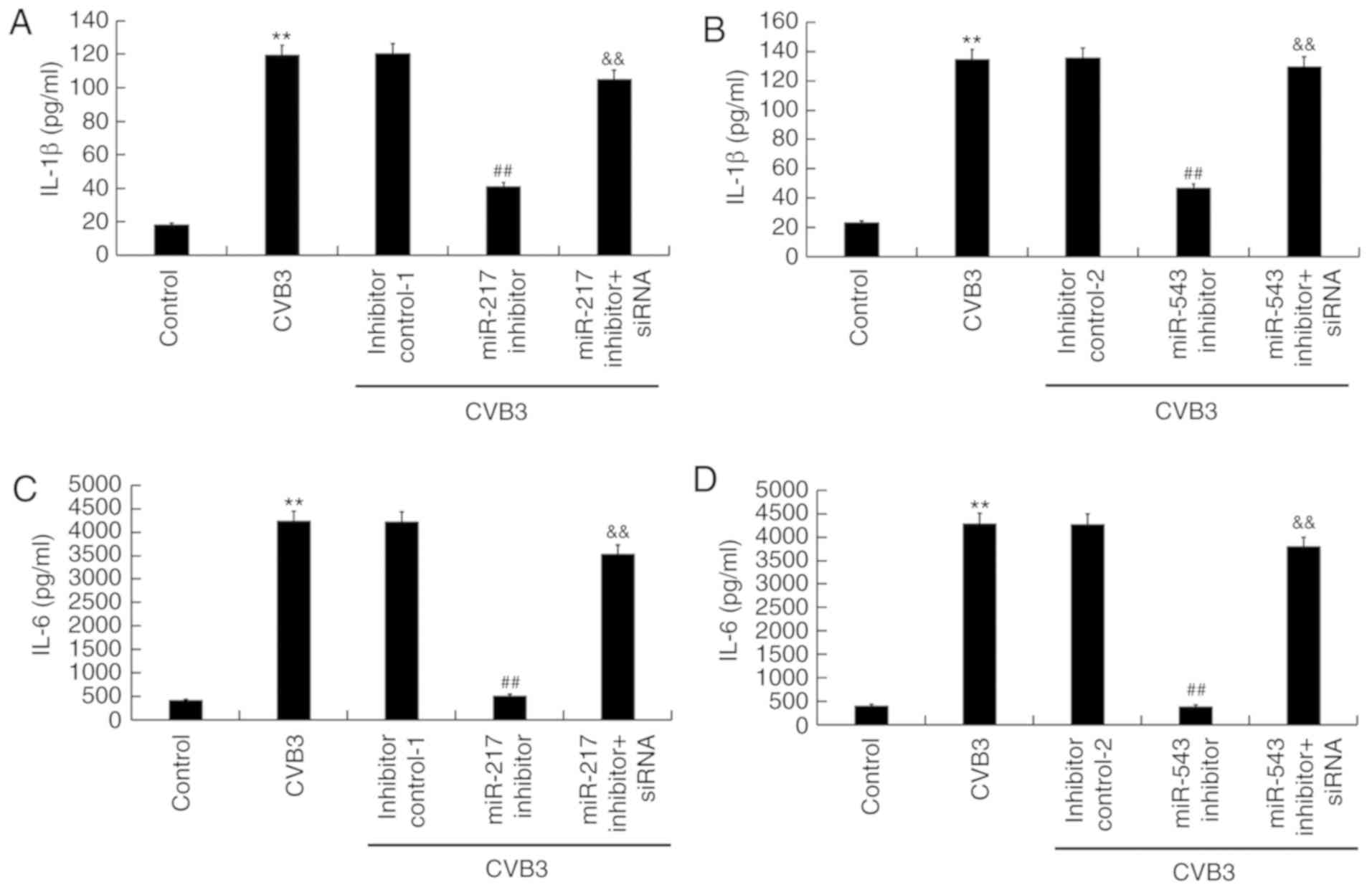 | Figure 7Effects of miR-217 and miR-543 on
inflammatory response in CVB3-infected H9C2 cells. H9C2 cells were
transfected with miR-217 or miR-543 inhibitors, miR-217 or miR-543
inhibitor controls, miR-217 inhibitor + SIRT1-siRNA, or miR-543
inhibitor + SIRT1-siRNA for 48 h and then treated with
104 PFU/ml CVB3 virus for 48 h. Effects of (A) miR-217
and (B) miR-543 on IL-1β levels in CVB3-infected H9C2 cells. Effect
of (C) miR-217 and (D) miR-543 on IL-6 levels in CVB3-infected H9C2
cells. **P<0.01 vs. corresponding control;
&&P<0.01 vs. corresponding miR inhibitor;
##P<0.01 vs. CVB3. CVB3, Coxsackievirus B3; miR,
microRNA; SIRT1, sirtuin 1; siRNA, small interfering RNA; IL,
interleukin; inhibitor control-1, miR-217 inhibitor control;
inhibitor control-2, miR-543 inhibitor control. |
Effect of miR-217 and miR-543
downregulation on the SIRT1/AMPK-α/NF-κB pathway in CVB3-infected
H9C2 cells
The activity of the SIRT1/AMPK-α/NF-κB pathway in
CVB3-infected H9C2 cells was then analyzed. As shown in Fig. 8, CVB3 infection markedly inhibited
SIRT1 protein (Fig. 8A and E)
expression and significantly reduced SIRT1 mRNA (P<0.01;
Fig. 8B and F) levels, decreased
p-AMPK-α protein level and p-AMPK-α/AMPK-α ratio (Fig. 8A, C, E and G), and promoted the
phosphorylation level of p65 protein and p-p65/p65 ratio (Fig. 8A, D, E and H) in H9C2 cells.
Compared with the CVB3 group, miR-217 (Fig. 8A-D) and miR-543 (Fig. 8E-H) inhibition notably increased
SIRT1 and p-AMPK-α expression levels, and p-AMPK-α/AMPK-α ratio,
and decreased p-p65 protein level and p-p65/p65 ratio. SIRT1 siRNA
significantly reversed the effects of miR-217 and miR-543
inhibition on the activity of the SIRT1/AMPK-α/NF-κB pathway
(P<0.01; Fig. 8).
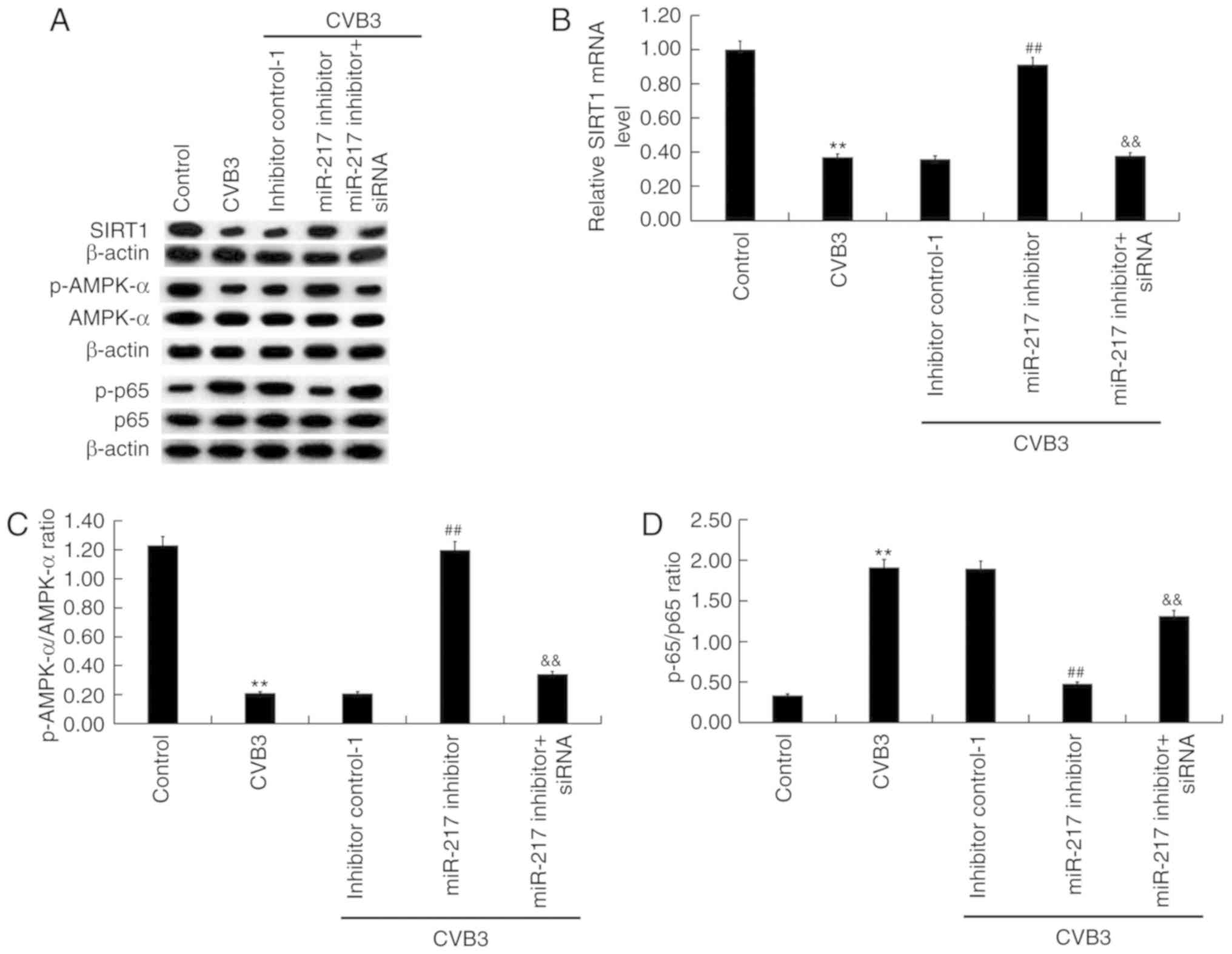 | Figure 8Effects of miR-217 and miR-543 on the
SIRT1/AMPK-α/NF-κB pathway in CVB3-infected H9C2 cells. H9C2 cells
were transfected with miR-217 or miR-543 inhibitors, miR-217 or
miR-543 inhibitor controls, miR-217 inhibitor + SIRT1-siRNA, or
miR-543 inhibitor + SIRT1-siRNA for 48 h and then treated with
104 PFU/ml CVB3 virus for 48 h. Effects of miR-217 on
the (A) protein and (B) mRNA expression levels of SIRT1, (C)
p-AMPK-α/AMPK-α ratio and (D) p-p65/p65 ratio in CVB3-infected H9C2
cells. Effects of miR-217 on the (E) protein and (F) mRNA
expression levels of SIRT1, (G) p-AMPK-α/AMPK-α ratio and (H)
p-p65/p65 ratio in CVB3-infected H9C2 cells. **P<0.01
vs. corresponding control; &&P<0.01 vs.
corresponding miR inhibitor; ##P<0.01 vs. CVB3. p-,
phosphorylated; AMPK-α, AMP-activated protein kinase-α; CVB3,
Coxsackievirus B3; miR, microRNA; SIRT1, sirtuin 1; siRNA, small
interfering RNA; inhibitor control-1, miR-217 inhibitor control;
inhibitor control-2, miR-543 inhibitor control. |
Discussion
The present study suggested that miR-217 and miR-543
were significantly upregulated in viral myocarditis. SIRT1, a
target of miR-217 and miR-543, was significantly downregulated in
viral myocarditis. CVB3 infection significantly reduced H9C2 cell
proliferation, induced H9C2 cell apoptosis, upregulated the
production of IL-6, IL-1β, and MDA content, and inhibited SOD
activity. Compared with the CVB3 infection group, downregulation of
miR-217 and miR-543 significantly increased cell proliferation,
reduced cell apoptosis, decreased the synthesis of IL-6, IL-1β,
decreased the level of MDA, and increased SOD activity. The effects
of miR-217 and miR-543 inhibition on CVB3-infected cells were
reversed by SIRT1 knockdown. The present results suggested that
miR-217 and miR-543 may be novel therapeutic targets for treating
viral myocarditis.
Viral myocarditis is a clinically common disease in
children and its incidence is increasing (42). The clinical manifestations of
viral myocarditis are not typical, and if the diagnosis and
treatment are not timely, the prognosis of pediatric patients can
be seriously affected (42).
Therefore, it is urgent to find new effective diagnostic markers
and therapeutic targets for viral myocarditis. Numerous previous
studies have shown that miRNAs play important roles in the
occurrence and development of viral myocarditis (19-21). In the present study, it was found
that the levels of miR-217 and miR-543 in blood samples from
children with viral myocarditis were significantly higher than that
in blood samples from healthy volunteers. It was also found that
miR-217 and miR-543 were upregulated in both in vivo and
in vitro model of viral myocarditis induced by CVB3
infection (37,38). The present findings suggested a
potential role for miR-217 and miR-543 in viral myocarditis. Then,
to examine the role of miR-217 and miR-543 in viral myocarditis,
the relationship between SIRT1, miR-217 and miR-543 was
investigated in cardiomyocytes using H9C2 cells, and the present
results indicated that SIRT1 was a direct target of both miR-217
and miR-543 in H9C2 cells. Additionally, SIRT1 was found to be
downregulated in patients with viral myocarditis, and in mice and
H9C2 cells infected with CVB3.
Pathological changes of cardiomyocytes are the main
pathological alterations occurring in viral myocarditis, and these
changes are due to direct viral and immune-mediated damages to
cardiomyocytes (43). In
addition, the present study investigated the effects of miR-217 and
miR-543 on CVB3-infected H9C2 cells. In line with a previous study
(38), it was found that CVB3
infection significantly inhibited H9C2 cell viability and induced
cell apoptosis. miR-217 and miR-543 inhibition significantly
promoted H9C2 cell viability and inhibited H9C2 cell apoptosis.
Inflammatory response and oxidative stress play a key role in the
pathological development of viral myocarditis. Continuous chronic
inflammation can induce myocardial cell necrosis, cardiac
hypertrophy and cardiomyocyte apoptosis, which can lead to heart
failure (44). In the present
study, the effects of miR-217 and miR-543 inhibition on
inflammatory response and oxidative stress were examined in
CVB3-infected H9C2 cells. The present findings suggested that
inhibition of miR-217 and miR-543 significantly decreased
CVB3-mediated upregulation of IL-6, IL-1β and MDA, and reversed
CVB3-mediated inhibition of SOD activity, indicating the potential
of miR-217 and miR-543 inhibition in suppressing the inflammatory
response and oxidative stress in CVB3-infected H9C2 cells. A
previous study demonstrated that the activation of the AMPKα-SIRT1
pathway prevents NF-κB inflammation (45). Therefore, to examine the molecular
mechanism of miR-217 and miR-543 in CVB3-infected H9C2 cells, the
SIRT1/AMPK-α/NF-κB pathway was investigated in the present study.
The present results showed that CVB3 infection significantly
inhibited SIRT1 protein and mRNA levels, decreased p-AMPK-α protein
level and p-AMPK-α/AMPK-α ratio, and promoted p-p65 protein level
and p-p65/p65 ratio in H9C2 cells, and these alterations were
significantly reversed by miR-217 and miR-543 inhibition. Notably,
the effects of miR-217 and miR-543 inhibition on CVB3-infected H9C2
cells were suppressed following SIRT1 silencing.
Collectively, the present study suggested that
miR-217 and miR-543 were significantly upregulated in viral
myocarditis, and miR-217 and miR-543 inhibition may attenuate viral
myocarditis by inhibiting the apoptosis of cardiomyocytes and
preventing the inflammatory response and oxidative stress by
targeting SIRT1. The present results suggested that miR-217 and
miR-543 may be novel potential therapeutic targets for the
treatment of viral myocarditis.
Funding
No funding was received.
Availability of data and materials
The datasets generated and analyzed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
KX designed the study, and collected and interpreted
the data. YZ contributed to the acquisition of the data, manuscript
preparation and literature search. DS performed the statistical
analysis. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Informed consent was obtained from each patient and
the parents or the legal guardians of the patients. The use of
human samples was approved by The Ethics Committee of Wuhan
Children's Hospital. Animal experiments were approved by The Animal
Ethics Committee of Tongji Medical College, Huazhong University of
Science and Technology.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Yajima T: Viral myocarditis: Potential
defense mechanisms within the cardiomyocyte against virus
infection. Future Microbiol. 6:551–566. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gaaloul I, Riabi S, Harrath R, Evans M,
Salem NH, Mlayeh S, Huber S and Aouni M: Sudden unexpected death
related to enterovirus myocarditis: Histopathology,
immunohistochemistry and molecular pathology diagnosis at
post-mortem. BMC Infect Dis. 12:2122012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang T, Zhang J, Xiao A, Liu W, Shang Y
and An J: Melittin ameliorates CVB3-induced myocarditis via
activation of the HDAC2-mediated GSK-3β/Nrf2/ARE signaling pathway.
Biochem Biophys Res Commun. 480:126–131. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Márquez-González H, López-Gallegos D and
González- Espi nosa AM: Effect of immune therapy in the prognosis
of viral myocarditis in pediatric patients. Rev Med Inst Mex Seguro
Soc. 54(Suppl 3): S296–S301. 2016.In Spanish.
|
|
5
|
Yu M, Long Q, Li HH, Liang W, Liao YH,
Yuan J and Cheng X: IL-9 inhibits viral replication in
coxsackievirus B3-induced myocarditis. Front Immunol. 7:4092016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang C, Dong C and Xiong S: IL-33 enhances
macrophage M2 polarization and protects mice from CVB3-induced
viral myocarditis. J Mol Cell Cardiol. 103:22–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pollack A, Kontorovich AR, Fuster V and
Dec GW: Viral myocarditis-diagnosis, treatment options, and current
controversies. Nat Rev Cardiol. 12:670–680. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rienks M, Papageorgiou A, Wouters K,
Verhesen W, Leeuwen RV, Carai P, Summer G, Westermann D and Heymans
SA: novel 72-kDa leukocyte-derived osteoglycin enhances the
activation of toll-like receptor 4 and exacerbates cardiac
inflammation during viral myocarditis. Cell Mol Life Sci.
74:1511–1525. 2017. View Article : Google Scholar
|
|
9
|
Yue-Chun L, Guang-Yi C, Li-Sha G, Chao X,
Xinqiao T, Cong L, Xiao-Ya D and Xiangjun Y: The protective effects
of ivabradine in preventing progression from viral myocarditis to
dilated cardiomyopathy. Front Pharmacol. 7:4082016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hammond SM: An overview of microRNAs. Adv
Drug Deliv Rev. 87:3–14. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ghildiyal M and Zamore PD: Small silencing
RNAs: An expanding universe. Nat Rev Genet. 10:94–108. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Soifer HS, Rossi JJ and Saetrom P:
MicroRNAs in disease and potential therapeutic applications. Mol
Ther. 15:2070–2079. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Krol J, Loedige I and Filipowicz W: The
widespread regulation of microRNA biogenesis, function and decay.
Nat Rev Genet. 11:597–610. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
O'Connell RM, Rao DS, Chaudhuri AA and
Baltimore D: Physiological and pathological roles for microRNAs in
the immune system. Nat Rev Immunol. 10:111–122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bardooli F, McAlindon E, Littlejohns B,
Suleiman MS, Bucciarelli-Ducci C and Baumbach A: TCT-184 Early
changes in circulating miRNA 133a are indicative of cardiac
remodelling after 3 months in patients presenting with acute ST
elevation myocardial infarction. J Am Coll Cardiol. 68:B75–B76.
2016. View Article : Google Scholar
|
|
17
|
Wang Y, Ouyang M, Wang Q and Jian Z:
MicroRNA-142-3p inhibits hypoxia/reoxygenation-induced apoptosis
and fibrosis of cardiomyocytes by targeting high mobility group box
1. Int J Mol Med. 38:1377–1386. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Singh GB, Raut SK, Khanna S, Kumar A,
Sharma S, Prasad R and Khullar M: MicroRNA-200c modulates DUSP-1
expression in diabetes-induced cardiac hypertrophy. Mol Cell
Biochem. 424:1–11. 2017. View Article : Google Scholar
|
|
19
|
Corsten MF, Papageorgiou A, Verhesen W,
Carai P, Lindow M, Obad S, Summer G, Coort SL, Hazebroek M, van
Leeuwen R, et al: MicroRNA profiling identifies microRNA-155 as an
adverse mediator of cardiac injury and dysfunction during acute
viral myocarditis. Circ Res. 111:415–425. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Corsten MF, Dennert R, Jochems S,
Kuznetsova T, Devaux Y, Hofstra L, Wagner DR, Staessen JA, Heymans
S and Schroen B: Circulating MicroRNA-208b and MicroRNA-499 reflect
myocardial damage in cardiovascular disease. Circ Cardiovasc Genet.
3:499–506. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bao JL and Lin L: MiR-155 and miR-148a
reduce cardiac injury by inhibiting NF-κB pathway during acute
viral myocarditis. Eur Rev Med Pharmacol Sci. 18:2349–2356.
2014.PubMed/NCBI
|
|
22
|
Nogueiras R, Habegger KM, Chaudhary N,
Finan B, Banks AS, Dietrich MO, Horvath TL, Sinclair DA, Pfluger PT
and Tschöp MH: Sirtuin 1 and sirtuin 3: Physiological modulators of
metabolism. Physiol Rev. 92:1479–1514. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
da Cunha MSB and Arruda SF:
Tucum-do-Cerrado (Bactris setosa Mart.) may promote anti-aging
effect by upregulating SIRT1-Nrf2 pathway and attenuating oxidative
stress and inflammation. Nutrients. 9:pii: E1243. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rada P, Pardo V, Mobasher MA,
García-Martínez I, Ruiz L, González-Rodríguez Á, Sanchez-Ramos C,
Muntané J, Alemany S, James LP, et al: SIRT1 controls acetaminophen
hepatotoxicity by modulating inflammation and oxidative stress.
Antioxid Redox Signal. 28:1187–1208. 2018. View Article : Google Scholar
|
|
25
|
Chan SH, Hung CH, Shih JY, Chu PM, Cheng
YH, Lin HC and Tsai KL: SIRT1 inhibition causes oxidative stress
and inflammation in patients with coronary artery disease. Redox
Biol. 13:301–309. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cheng YY, Kao CL, Ma HI, Hung CH, Wang CT,
Liu DH, Chen PY and Tsai KL: SIRT1-related inhibition of
pro-inflammatory responses and oxidative stress are involved in the
mechanism of nonspecific low back pain relief after exercise
through modulation of Toll-like receptor 4. J Biochem. 158:299–308.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nguyen LT, Mak CH, Chen H, Zaky AA, Wong
MG, Pollock CA and Saad S: SIRT1 attenuates kidney disorders in
male offspring due to maternal high-fat diet. Nutrient. 11:pii:
E146. 2019. View Article : Google Scholar
|
|
28
|
Hwang JW, Yao H, Caito S, Sundar IK and
Rahman I: Redox regulation of SIRT1 in inflammation and cellular
senescence. Free Radic Biol Med. 61:95–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo R, Liu W, Liu B, Zhang B, Li W and Xu
Y: SIRT1 suppresses cardiomyocyte apoptosis in diabetic
cardiomyopathy: An insight into endoplasmic reticulum stress
response mechanism. Int J Cardiol. 191:36–45. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mao Q, Liang X, Wu Y and Lu Y: Resveratrol
attenuates cardiomyocyte apoptosis in rats induced by coronary
micro-embolization through SIRT1-mediated deacetylation of p53. J
Cardiovasc Pharmacol Ther. 24:551–558. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang WX, He BM, Wu Y, Qiao JF and Peng
ZY: Melatonin protects against sepsis-induced cardiac dysfunction
by regulating apoptosis and autophagy via activation of SIRT1 in
mice. Life Sci. 217:8–15. 2019. View Article : Google Scholar
|
|
32
|
Ben Salem I, Boussabbeh M, Pires Da Silva
J, Guilbert A, Bacha H, Abid-Essefi S and Lemaire C: SIRT1 protects
cardiac cells against apoptosis induced by zearalenone or its
metabolites α- and β-zearalenol through an autophagy-dependent
pathway. Toxicol Appl Pharmacol. 314:82–90. 2017. View Article : Google Scholar
|
|
33
|
Li J, Dong G, Wang B, Gao W and Yang Q:
miR-543 promotes gastric cancer cell proliferation by targeting
SIRT1. Biochem Biophys Res Commun. 469:15–21. 2016. View Article : Google Scholar
|
|
34
|
Hu X, Chi L, Zhang W, Bai T, Zhao W, Feng
Z and Tian H: Down-regulation of the miR-543 alleviates insulin
resistance through targeting the SIRT1. Biochem Biophys Res Commun.
468:781–787. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yin H, Liang X, Jogasuria A, Davidson NO
and You M: miR-217 regulates ethanol-induced hepatic inflammation
by disrupting sirtuin 1-lipin-1 signaling. Am J Pathol.
185:1286–1296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bayne K: Revised guide for the care and
use of laboratory animals available American physiological society.
Physiologist 39. 199:208–211. 1996.
|
|
37
|
Xin L, Ma X, Xiao Z, Yao H and Liu Z:
Coxsackievirus B3 induces autophagy in HeLa cells via the
AMPK/MEK/ERK and Ras/Raf/MEK/ERK signaling pathways. Infect Genet
Evol. 36:46–54. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Qi L, Xin Q and Wenjun J: Inhibition of
iNOS protects cardiomyocytes against coxsackievirus B3-induced cell
injury by suppressing autophagy. Biomed Pharmacother. 91:673–679.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
40
|
Tsikas D: Assessment of lipid peroxidation
by measuring malondialdehyde (MDA) and relatives in biological
samples: Analytical and biological challenges. Anal Biochem.
524:13–30. 2017. View Article : Google Scholar
|
|
41
|
Wu JQ, Kosten TR and Zhang XY: Free
radicals, antioxidant defense systems, and schizophrenia. Prog
Neuropsychopharmacol Biol Psychiatry. 46:200–206. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Casadonte JR, Mazwi ML, Gambetta KE, Palac
HL, McBride ME, Eltayeb OM, Monge MC, Backer CL and Costello JM:
Risk factors for cardiac arrest or mechanical circulatory support
in children with fulminant myocarditis. Pediatr Cardiol.
38:128–134. 2017. View Article : Google Scholar
|
|
43
|
Simpson KE, Storch GA, Lee CK, Ward KE,
Danon S, Simon CM, Delaney JW, Tong A and Canter CE: High frequency
of detection by PCR of viral nucleic acid in the blood of infants
presenting with clinical myocarditis. Pediatr Cardiol. 37:399–404.
2016. View Article : Google Scholar :
|
|
44
|
Tse G, Yeo JM, Chan YW, Lai ET and Yan BP:
What is the arrhythmic substrate in viral myocarditis? Insights
from clinical and animal studies. Front Physiol. 7:3082016.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tian Y, Ma J, Wang W, Zhang L, Xu J, Wang
K and Li D: Resveratrol supplement inhibited the NF-κB inflammation
pathway through activating AMPKα-SIRT1 pathway in mice with fatty
liver. Mol Cell Biochem. 422:75–84. 2016. View Article : Google Scholar : PubMed/NCBI
|