Introduction
Nonsense-mediated RNA decay (NMD) is a highly
conserved eukaryotic mRNA surveillance mechanism that eliminates
transcripts with a premature termination codon (PTC) located
>50-55 nts upstream of the last exon-exon junction (EJ),
resulting from mutations or aberrant splicing (1,2).
In addition to the targeting of PTC-harboring mRNAs for
surveillance, ~5-30% of cellular transcripts are finely degraded by
the NMD pathway as natural substrates for modulating biological
processes (3,4); therefore, a disrupted NMD may be
exploited to cause gene expression disruption without
quality-control and fine-tuning (5). In addition, NMD impacts the cellular
transcriptome in cell-type specific manners, indicating that NMD
perturbation has a selective effect in increasing the levels of
reported NMD substrates, such as NF-κ-B-inducing kinase
(NIK), transducing β like 2 (TBL2) and
N-acetyltransferase 9 (NAT9), in different cell lines
(6,7). The up-frameshift suppressor 1
homolog (UPF1) gene, encoding an ATP-dependent RNA helicase,
is the central regulator of the NMD pathway (8). A feedback loop between the NMD and
the UPF1 gene has been reported, in which UPF1 protein was
rate-limiting for NMD and responses to NMD perturbation (6). UPF1 mutations have been
deemed to play a causal role in malignant tumors, such as
pancreatic adenosquamous carcinomas and inflammatory
myofibroblastic tumors in our previous studies, suggesting that
perturbed NMD is involved in cell hyper-proliferation, abnormal
differentiation and stable activation of inflammation (7,9).
The modulation of the NMD pathway has been suggested to provide a
benefit in several disease therapies (5,10),
and therefore understanding the interactions between NMD and
diseases can optimize preventive and therapeutic approaches against
diseases. Herein, two somatic UPF1 mutations
(c.2935_2936insA and c.2030-2081del) were identified in two
patients with sporadic psoriasis scales, which perturbed the
ability of the UPF1-NMD pathway to degrade substrates in
keratinocyte cells. In the present study, to the best of our
knowledge, mutated UPF1 transcripts in psoriasis have been
discovered for the first time, which indicated a role of the NMD
pathway in skin disease.
A previous study hypothesized that there are
keratinocyte-specific targets of the NMD pathway, and the
amphiregulin (AREG) gene, which encodes the most abundant
epidermal growth factor receptor (EGFR) ligand in keratinocytes,
was suggested as one such substrate (11). AREG is overexpressed in a
wide spectrum of epithelial diseases, including squamous cell
carcinoma of the head and neck, colon cancer, lung cancer and
psoriasis (12-15). AREG transgenic animals show
epidermal acanthosis, keratinocyte hyperproliferation,
hyperkeratosis, cutaneous immune cell infiltration and angiogenesis
in the skin (16,17). However, it is unknown how
AREG is regulated post-transcriptionally by mRNA decay. In
the present study, to the best of our knowledge, it is the first
time that the potential mechanism of the AREG-NMD axis
involved in keratinocyte cells was explored. In addition, cell
activities such as differentiation, wound healing and inflammatory
response, which are highly relevant to the pathogenesis of numerous
skin diseases, were demonstrated to be regulated by UPF1 in
an AREG-dependent manner. This finding has created an
opportunity to better understand the NMD pathway in keratinocytes,
and indicates role of the NMD pathway in the development of
diseases associated with keratinocyte morbidities.
Materials and methods
Statistical analysis
The gene expression profiles of human paired
psoriasis tissues were retrieved from Gene Expression Omnibus (GEO)
database. After carefully screening the content, discarding the
datasets with incomplete information and those lacking control
patients, the 5 datasets with paired samples, GDS5392, GDS4602,
GDS2518, GDS3539, and GDS4600, were obtained (18-22). R packages (version 3.5.2) were
used to annotate the raw data and make the expression matrix
(23). The median of expression
level was chosen for genes matched by several probes, and paired t
test was used to compare UPF1 expression. Student's t-test
was used for the analysis of statistical significance between two
groups, and Bonferroni test was used for multiple comparisons after
the analysis of variance. Data for continuous variables are
expressed as the mean ± SD. All statistical analyses were performed
using SPSS 21.0 software (IBM Corp.). P<0.05 from a two-tailed
test was considered to indicate a statistically significant
difference.
Clinical samples
A total of 10 patients with a
dermatologist-confirmed diagnosis of vulgaris psoriasis, according
to the 'Consensus on diagnosis and treatment of Chinese integrative
medicine for psoriasis vulgaris', published in China (24,25), were recruited at the Affiliated
Hospital of Ningbo University (Ningbo, China) between January 2016
and July 2017. The patient information is shown in Table SI. The
lesion scales from 6 patients (P1-P6) were independent samples,
while the lesion scales and adjacent healthy samples from 4
patients (PA-PD) were paired samples. All the psoriasis patients
had no other autoimmune or systemic diseases, and required a
typical lesion of ≥1 cm in size that was suitable for biopsy, and
the target lesion and surrounding 5 cm area were not treated with
any therapeutic measures for at least 2 weeks before the sampling.
Skin scales or healthy cornified epidermal layer of patients with
psoriasis vulgaris were scraped by blunt scalpels with patients'
permission. The scales were promptly soaked in liquid-free nitrogen
RNA sample storage solution (Beijing Bomaide Gene Technology Co.,
Ltd.).
Imiquimod (IMQ)-induced psoriasis-like
mice
To create the well-established psoriasis-like skin
model (26), 8-11-weeks-old
Balb/c mice provided by the Experimental Animal Center of Hangzhou
Normal University were kept under standard laboratory conditions of
12-h light-dark cycles, 50% humidity and 24-26°C ambient
temperature with free access to food and water. The mice (2 male
and 2 female, 20-25 g) received a daily topical dose of 40 mg
commercially available IMQ cream (5%; Zhuhai United Laboratories
Co., Ltd.) on the shaved back, equivalent of a daily dose of 2.083
mg of the active compound. The control shaved skin nearby the
IMQ-treated area (distance, 1.5-2 cm) was treated similarly with a
control cream (Vaseline Lanette cream; Fagron). During the 7-day
experiment, the topical IMQ treatment did not lead pain, and the
health of all 4 mice was monitored daily. On day 7, all 4 mice were
sacrificed by rapid cervical vertebra dislocation, and the skin
specimens of mice were collected once the cervical tissue
separation was ensured.
Reverse transcription-quantitative PCR
(RT-qPCR) and mRNA sequencing
Total RNA from model mice, clinical tissues or cells
was isolated using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.), and first-strand cDNA for RT-qPCR was
synthesized using the PrimeScript™ RT reagent kit with gDNA Eraser
according to the manufacturer's protocol (Takara Bio, Inc.). qPCR
analysis was performed using the QuantStudio™ 7 Flex Real-Time PCR
system (Thermo Fisher Scientific, Inc.) using SYBR Premix Ex Taq
(Takara Bio, Inc.) with gene-specific primers listed in Table SII.
The following thermocycling conditions were used: Initial
denaturation at 95°C for 1 min; followed by 40 cycles of 95°C for
15 sec and 60°C for 30 sec. The disassociation stage was added to
check the amplicon specificity. For transcript detections in scales
tissues, UPF1 was quantified using the allele-specific
primers UPF1 insA for c.2935_2936insA mutant, or UPF1
del corresponding to the c.2030-2081del UPF1 mRNA (Table
SII). Specificity and efficiency of allele-specific PCR were tested
with corresponding UPF1 constructs, and further details are
described in Data S1. Each mRNA quantification value represented an
average of at least three measurements, and mRNA expression levels
were calculated using the 2−ΔΔCq method (27) after normalization to the
endogenous housekeeping gene 18S for human samples or
GAPDH for mouse samples.
For RT-PCR, RT was performed using 1 µg of
total RNA with the PrimeScript™ 1st Strand cDNA Synthesis kit
according to the manufacturer's protocol (Takara Bio, Inc.). PCR
amplification was performed with nested primers (Table SIII)
overlapping the coding sequence region of the UPF1 mRNA
(NCBI Reference Sequence, NM_002911.3). The following thermocycling
conditions were used: Initial denaturation at 96°C for 2 min;
followed by 35 cycles of 96°C for 10 sec, 55°C for 30 sec and 72°C
for 2 min. The sanger sequencing was performed at Tsingke
Biological Technology.
Protein detection
For immunohistochemical (IHC), skin tissue from the
model mice was fixed in 10% formalin for 24 h at room temperature
and embedded in IHC-grade paraffin. Formalin-fixed
paraffin-embedded sections were cut into 4-6-µm sections
with a microtome and deparaffinized two times in xylene, followed
by serial dilutions of ethanol. After heat-induced antigen
retrieval using the antigen unmasking solution (Vector
Laboratories, Inc.; Maravai LifeSciences) for 30 min at 95°C, the
internal peroxidase activity was quenched by incubation with 3%
hydroperoxide in methanol for 15 min at room temperature, then
sections were incubated in 3% bovine serum albumin (Beyotime
Institute of Biotechnology) for 1 h at room temperature. The
prepared sections were incubated overnight at 4°C with anti-UPF1
primary antibodies (cat. no. sc-390096; 1:100; Santa Cruz
Biotechnology, Inc.). The secondary antibody (cat. no. GAMPO; 1:1;
DAKO; Agilent Technologies, Inc.) was used according to the
manufacturer's instructions for 50 min at room temperature. The
intensity of IHC staining was measured using ImageJ software
(version 1.52a; Media Cybernetics, Inc.).
Protein extraction for western blotting was
performed using RIPA lysis buffer (Beyotime Institute of
Biotechnology) with phenylmethylsulfonyl fluoride serine protease
inhibitor (Beyotime Institute of Biotechnology). Protein extracts
were quantified using a BCA Protein Assay kit (Beyotime Institute
of Biotechnology) according to the manufacturer's instructions.
Total proteins (30 µg) were resolved by SDS-PAGE (8% gel)
and visualized using Odyssey® imaging system with IRDye
secondary antibodies (cat. no. 926-32210; 1:1,000; LI-COR
Biosciences) for 15 min at room temperature. The following primary
antibodies were used overnight at 4°C: Anti-UPF1 (cat. no.
sc-166092; 1:200; Santa Cruz Biotechnology, Inc.), anti-AREG (cat.
no. GTX100986; 1:200; GeneTex, Inc.), and anti-GAPDH (cat. no.
M20006L; 1:1,000; Abmart Pharmaceutical Technology Co., Ltd.).
Semi-quantification was performed using ImageJ software.
Cell culture and transfection
Primary cultured normal human keratinocytes (HEKα),
immortalized nontumorigenic human keratinocyte-derived cell line
(HaCaT) and 293T cell line were purchased from the Shanghai
Institute of Biochemistry and Cell Biology. HaCaT and 293T cells
were cultured in DMEM with 10% FBS (Biological Industries), and
HEKα cells were cultured in EpiLife Medium with 60 µM
calcium with Keratinocyte Growth Supplement (Gibco; Thermo Fisher
Scientific, Inc.). All cells were incubated at 37°C in a humidified
5% CO2 incubator.
For plasmid and small interfering (si)RNA
transfection, 4×104 cells/well in 6-well plates were
cultured overnight and then transfected using the GeneTran Reagent
(Biomiga, Inc.). For co-transfection targeting both UPF1 and
AREG, constructs were transfected at the same time; however,
when constructs targeting the same gene were co-transfected,
construct for knockdown was transfected first, then the
overexpression construct was transfected 24 h later. Total mRNA and
protein of cells were collected 48 h after transfection.
Constructs and oligonucleotides
A short hairpin RNA (shRNA) sequence specifically
targeting UPF1 (Table SIV) was cloned into Phblv-U6-puro
(provided by Professor Fan Handong, Hangzhou Normal University) by
BamHI and EcoRI digestion. The siRNA targeting
AREG (targeted sequence, CCACAAATACCTGGCTATA) and a
scrambled sequence were purchased from Guangzhou RiboBio Co., Ltd.
The UPF1 insA and UPF1 del plasmids were constructed
based on the pCMV-MYC-UPF1 vector (provided by Professor
Lynne E. Maquat, University of Rochester). Primers for
site-directed mutagenesis (Table SIV) were designed by QuickChange
Primer Design (Agilent Technologies, Inc.) and applied with the
KOD-Plus-PCR enzyme (Toyobo Life Science). Residual templates were
digested by DpnI (New England Biolabs, Inc.) at 37°C for 5
h. The AREG gene open reading frame (ORF) with or without
the 3′ untranslated region (3′UTR), which was referred as
AREG-3′UTR-pEGFP or AREG-ORF-pEGFP, respectively, was
cloned into the pEGFP-N1 vector (provided by Dr Wang Miao, Hangzhou
Normal University) using the HindIII and BamHI
restriction sites. The primer sequences used are listed in Table
SIV. The AREG 3′UTR sequence was amplified and cloned into
the dual-luciferase reporter construct pEZX-FR02 (GeneCopoeia,
Inc.) by double digestion with EcoRI and SpeI (the
primer sequences used are listed in Table SIV) to generate the
pEZX-AREG-3′UTR construct.
Half-life analysis
The 293T cells were transfected with or without
shUPF1 and the transcription in cells was inhibited by
incubation with 20 mg/ml
5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB; Enzo Life
Sciences, Inc.) dissolved in DMSO. Total RNA was extracted at 0, 3,
5, 7, and 10 h following the inhibition of transcription, and AREG
mRNA levels were measured by RT-qPCR, as described above. Quantity
of RNA at each time point was determined by comparison with
standard curves generated by amplification of the time-zero RNA
sample.
Dual-luciferase reporter assays
The pEZX-AREG-3′UTR transfected 293T cells
were co-transfected with or without shUPF1 using the
GeneTran™ III reagent (Biomiga, Inc.). At 80% confluence and 48 h
post transfection, cells were collected and the activities of both
firefly luciferase (FLuc) and Renilla luciferase (RLuc) were
measured according to the instructions of the dual luciferase
reporter assay system (GeneCopoeia, Inc.). The internal standard
for transfection efficiency was normalized to RLuc activity.
Cell proliferation and migration
assays
HaCaT and HEKα cells were plated at a density of
2×104 cells/well in 96-well plates and incubated
overnight. The proliferation index was measured using a Cell
Counting Kit-8 (Dojindo Molecular Technologies, Inc.) at 48 h
according to the manufacturer's instructions. For the cell wound
healing assay, cells in the logarithmic growth phase were seeded in
24 well plates at a density of 5×105 cells/ well. When
the cells reached 100% confluence, a straight line was drawn in the
cells in each well with a 10-µl pipette tip and the cells
were serum-starved. Representative images (magnification, ×20) of
wound closure were captured at 0, 6, 12, 24, and 48 h with an
inverted microscope, and the wound area was measured using ImageJ
software. The percentage of wound coverage was calculated by the
difference in area between indicated time point and 0 h, divided by
the area at 0 h.
Accession codes
The gene sequences used in the current study are
publicly available at the GenBank [UPF1 mRNA (NM_002911.3)
and AREG mRNA (NM_001657.4)]. Data on UPF1 mutations
were deposited in the GenBank under accession numbers MH183202
(UPF1 c.2935_2936 insA) and MK089816 (UPF1
c.2030_2081 del 50 nts).
Results
Abnormal expression of UPF1 in psoriasis
and psoriasis-like mouse model
First, a paired samples test for UPF1
expression was concluded using GEO datasets GDS5392, GDS4602,
GDS2518, GDS3539 and GDS4600. An analysis of 186 paired psoriasis
and corresponding normal tissues showed that the UPF1 mRNA
expression increased significantly in psoriasis (Fig. 1A). Increased UPF1 mRNA
levels were also observed in sporadic patients' skin scales
obtained in the current study, including four paired tissues (PA,
PB, PC and PD; Fig. 1B), six
independent scales and four independent normal tissues. The IMQ
induced psoriasis-like skin showed morphological characteristics
consistent with the symptoms of human psoriasis (28), such as skin thickening (Fig. S1 and Data S1) and hyperkeratosis
(Fig. 1D), and RT-qPCR showed
that the psoriasis-like skin expressed higher mRNA levels of
proinflammatory cytokines and chemokines associated with psoriasis
including AREG, interleukin (IL)-17A, IL-6,
tumor necrosis factor-α and C-X-C motif chemokine 2
(data not shown). All 4 paired tissues from IMQ-induced
psoriasis-like skin exhibited increased UPF1 mRNA levels
(Fig. 1C), while protein levels
were significantly decreased in 2/4 pairs, as detected by IHC
(Fig. 1D and E). Abnormal
UPF1 expression levels were also revealed by GEO database
analysis for three other skin diseases tissues, including the
Marfan syndrome, squamous cell carcinoma and melanoma tissues
(Fig. S2 and Data S1). As
UPF1 mRNA is a natural substrate of the NMD pathway, as well
as a rate limiting element for NMD (6), the abnormal expression of
UPF1 in those skin disease tissues was presumed to indicate
a disturbed NMD pathway in the keratinocyte cells. According to the
cell-type specific manner of NMD, multiple reported NMD substrates,
which have been reported to be sensitive to UPF1 depletion
in different cell lines (6-8),
were tested by RT-qPCR in shUPF1 treated keratinocyte cells
in a preliminary study. Three well-characterized endogenous NMD
substrates, NIK, TBL2 and NAT9 mRNAs, were verified
to increase in level in response to UPF1-depletion in the
studied 293T and keratinocyte cells (data not shown), thus, the
NIK, TBL2 and NAT9 mRNAs were selected to
presented the NMD endogenous substrates. In the psoriasis-like
mouse model, a disturbed NMD was hinted by the accumulation of NIK,
TBL2 and NAT9 mRNAs in some paired lesions (Fig. 1F).
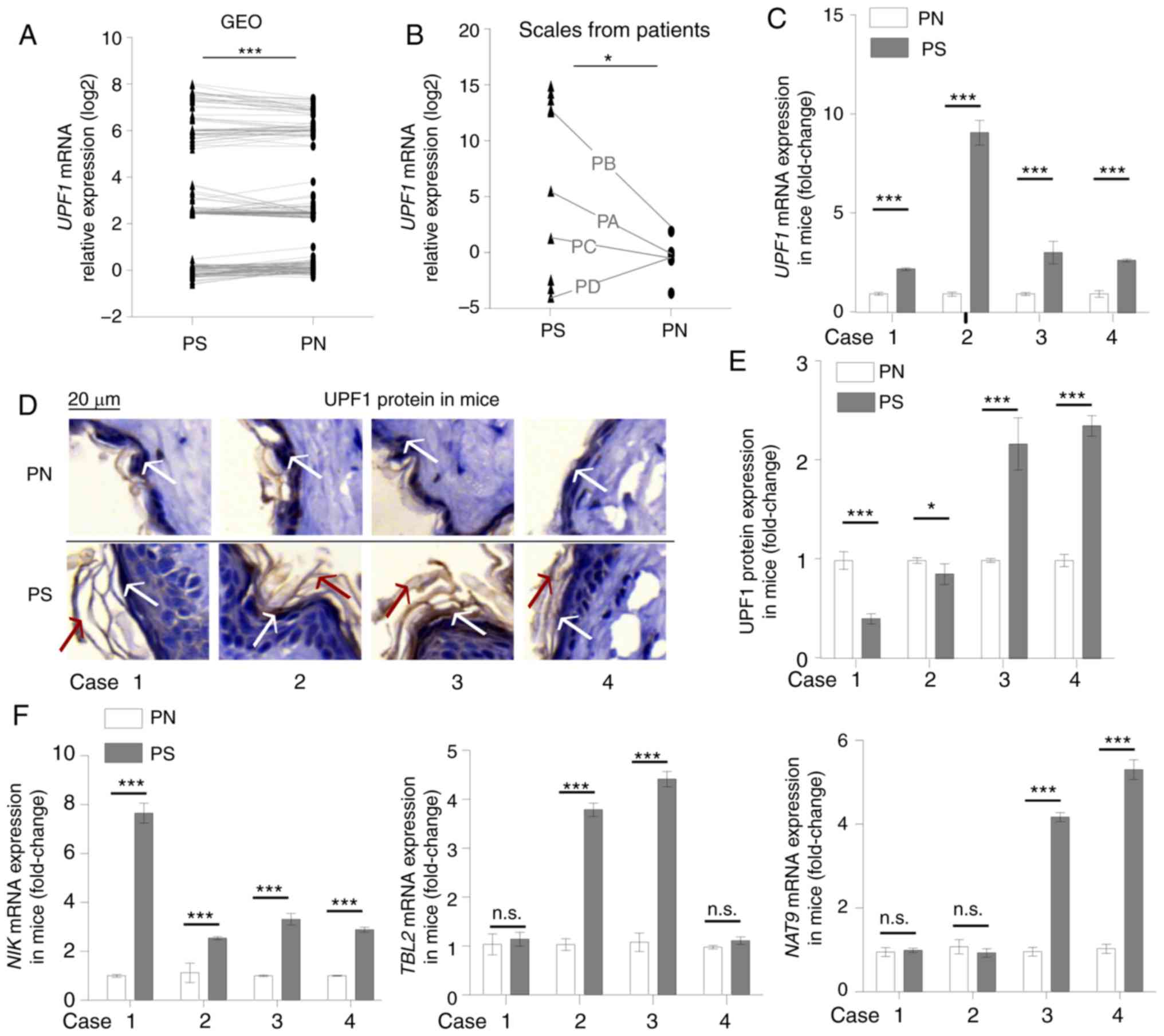 | Figure 1Abnormal expression of UPF1 in
psoriasis. (A) Paired sample t-test for 186 paired psoriasis
samples from GEO datasets (GDS5392, GDS4602, GDS2518, GDS3539 and
GDS4600). (B) Student's t-test for UPF1 mRNA levels in 10 psoriasis
scales and 8 healthy cornified epidermal layer samples. Among the
samples, four paired tissues were indicated with black lines. The
UPF1 expression increased in the psoriasis group; 3/4 paired
samples showed an upregulated expression level of UPF1. (C)
Mice received a daily topical IMQ cream to induce psoriasis-like
skin. The expression of UPF1 increased in all four
psoriasis-like tissues, as compared with the paired normal skin
tissue samples. (D) The expression of UPF1 protein distributed
throughout the cytoplasm in mouse skin was detected by IHC. The
white arrows indicate positive UPF1 protein staining, and the red
arrows indicate skin hyperkeratosis in IMQ-treated samples. (E) IHC
staining was suantified using ImageJ software. The UPF1 protein
decreased in 2/4 paired samples. (F) The mRNA expression of three
NMD substrates, NIK, TBL2 and NAT9, were
higher in psoriasis-like skin compared with normal tissue. Data are
presented as the mean ± SD from three independent experiments.
*P<0.05 and ***P<0.001. n.s., not
significant; UPF1, up-frameshift suppressor 1 homolog; IMQ,
imiquimod; IHC, immunohistochemistry; NMD, nonsense-mediated RNA
decay; NIK, NF-κ-B-inducing kinase; TBL2, transducing
β like 2; NAT9, N-Acetyltransferase 9; PS, psoriasis or
IMQ-treated psoriasis-like skin; PN, normal cornified epidermal
layer. |
Aberrant transcripts of UPF1 in psoriasis
scales
The skin scales are mostly composed of corneocytes
(29); thus, the tissues may have
been fragile for RNA sequencing. Five out of ten psoriasis scales
from sporadic psoriasis patients were demonstrated to be reliable
sources of RNA, as PCR fragments of UPF1 and AREG
mRNAs were amplified successfully in these tissues (Fig. S3 and Data S1). Among the 5
available scales for RNA sequencing, 2 aberrant UPF1 transcripts
were identified in patients A and B, and neither mutant was
detected in the corresponding healthy epidermal tissue. The
heterozygous mutation c.2935_2936insA (Fig. 2A) identified in patient A (a
36-year-old man with an affected buttock) generated an in-frame PTC
located 136 nts upstream of the exon 22-23 junction (Fig. 2B). The heterozygous mutation
c.2030-2081del (Fig. 2A) in
patient B (a 48-year-old woman with affected arms) was spliced by
the use of noncanonical splice donor/acceptor within exon 15
sharing a GGCC sequence, which generated a PTC 93 nts upstream of
the exon 16-17 junction (Fig.
2B). There was no detectable mutation in the corresponding DNA
or flanking introns, and, to the best of our knowledge, these two
mutations had not been reported prior to the present study. The
canonical ORF of UPF1 was disrupted in both aberrant
transcripts, and the mutants were predicted to produce truncated
proteins losing serine-glutamine cluster domain or the helicase
domain (Fig. 2B).
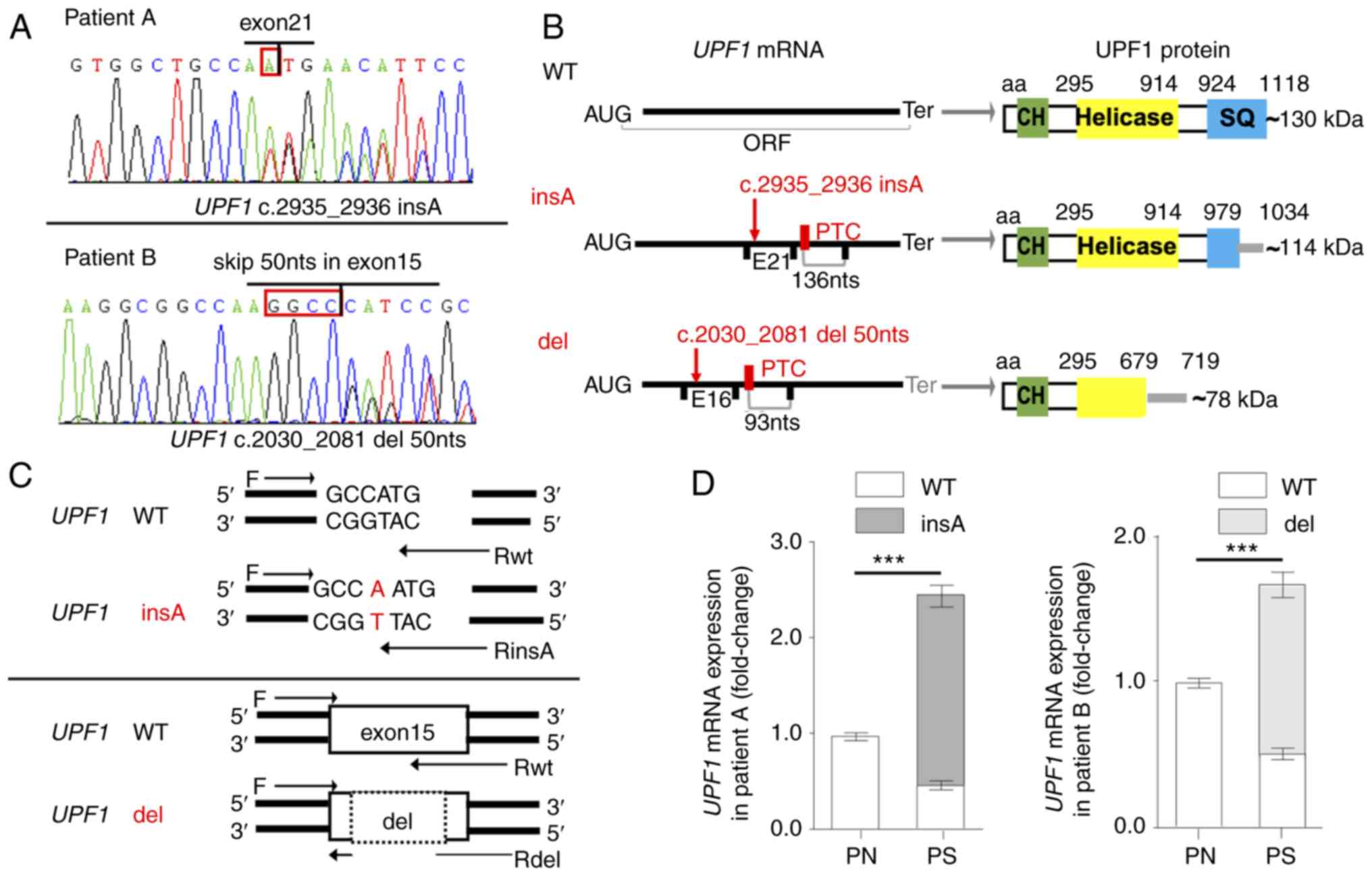 | Figure 2Somatic mutations of UPF1
transcripts in patients with psoriasis vulgaris. (A) Sequencing
traces corresponding to UPF1 insA (GenBank ID, MH183202) in patient
A and UPF1 del (GenBank ID, MK089816) in patient B. The red
frame indicates the insertion or shared sequence for splicing. The
sequencing primers used were F4/R4 and F3/R3 from Table SIII. (B)
Schematic representation of the aberrant mRNAs and proteins. The
mutations and PTCs are indicated in red, while the EJs and Ters are
presented in black in the mRNA schematic. The functional domains of
UPF1 are shown in the protein schematic, and the gray lines
indicate that the amino acid sequence was changed until the Ter.
(C) Primers specific to UPF1 mutants or UPF1 WT were
designed to detect the expression of UPF1 transcripts in the
2 patients. (D) The portions of UPF1 insA and UPF1
del were evaluated by reverse transcription-quantitative PCR.
Mutated UPF1 transcripts were only detectable in the
psoriasis scales. Data are presented as the mean ± SD from three
independent experiments. ***P<0.001. UPF1,
up-frameshift suppressor 1 homolog; PTC, premature
termination codon; EJ, exon-exon junction; Ter, termination codon;
WT, wild-type; PS, psoriasis scales; PN, normal cornified epidermal
layer; SQ, serine-glutamine; CH, calponin homology. |
To analyze the expression of UPF1 transcripts
in psoriasis scales, specific primers were designed, according to
the aberrant sequences (Fig. 2C).
The specificity and efficiency of RT-qPCR for UPF1 insA were
tested with allele-specific amplification. Transcript analysis
demonstrated that the aberrant mutants were only detectable in
psoriasis scales, whereas the wild-type (WT) transcripts were
expressed in both the scale lesions and healthy cornified epidermal
layer, showing a decrease in lesions (Fig. 2D).
Perturbation of NMD results from UPF1
insA and UPF1 del transcripts
The selective accumulation of the PTC-harboring
transcripts in lesions but not in the adjacent normal tissues
raised the possibility that the NMD pathway was perturbed in the
two scales. Consistent with this hypothesis, it was found that the
expression level of preselected NMD substrates, NIK,
TBL2 and NAT9, was elevated in the psoriasis scales
compared with normal control samples (Fig. 3A).
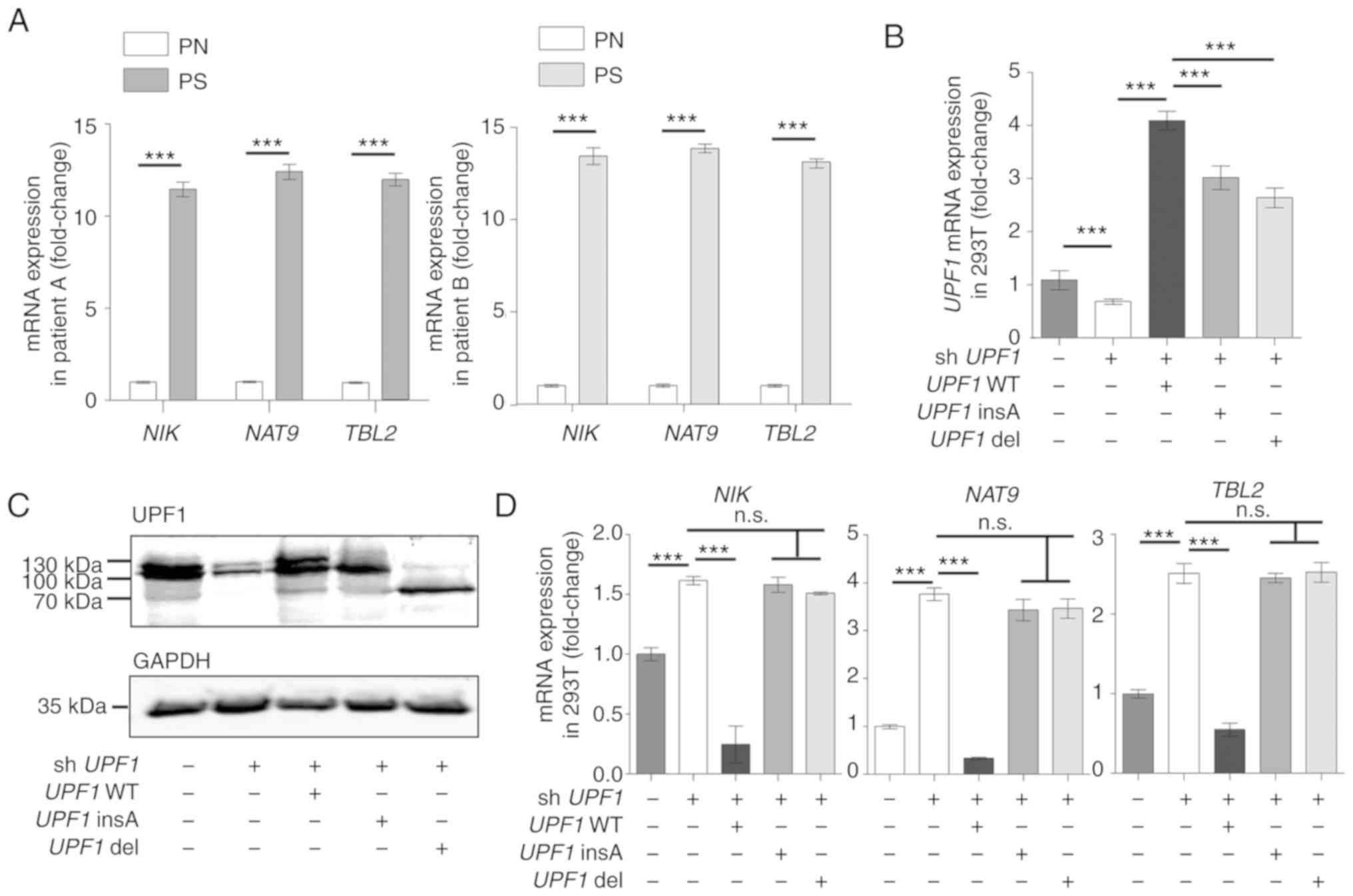 | Figure 3Aberrant UPF1 transcripts
disrupt the function of the NMD pathway. (A) The expression level
of NIK, TBL2 and NAT9 showed an increase in
psoriasis scales from patients A and B compared with PN samples.
(B) 293T cells were transfected with the shUPF1 construct,
and with the UPF1 WT, UPF1 insA or UPF1 del
vector 24 h later. UPF1 transcript expression was quantified
48 h after transfection. The expression levels of UPF1 insA
and UPF1 del were lower than that of UPF1 WT
following transfection with the respective constructs. (C) Western
blot analysis was used to measure the expression of UPF1 protein
isoforms, and GAPDH was used as a control. The UPF1 insA and UPF1
del generated minor protein bands, the predicted molecular weights
are indicated. (D) mRNA expression levels of NMD substrates
NIK, TBL2 and NAT9 were increased in
UPF1-depleted 293T cells, as detected by reverse
transcription-quantitative PCR. Data are presented as the mean ± SD
from three independent experiments. ***P<0.001. n.s.,
not significant. UPF1, up-frameshift suppressor 1 homolog;
NMD, nonsense-mediated RNA decay; NIK, NF-κ-B-inducing
kinase; TBL2, transducing β like 2; NAT9,
N-Acetyltransferase 9; WT, wild-type; PS, psoriasis scales; PN,
normal cornified epidermal layer. |
Successful overexpression and knockdown of
UPF1 was confirmed (Fig. S5A
and Data S1). The subsequent transfection of the UPF1
insA, UPF1 del or UPF1 WT constructs into
UPF1-depleted 293T cells revealed that the mutated
transcripts were expressed at a slightly lower steady-state level
than UPF1 WT at both the mRNA and protein levels (Fig. 3B and C; lanes 3-5). As predicted
above, the mutants generated minor protein bands that were
predicted to be ~114 kDa for UPF1 insA and 78 kDa for
UPF1 del (Fig. 3C; lanes
4-5), as compared with the 128 kDa full-length UPF1 protein
(Fig. 3C; lane 3). The truncated
UPF1 protein in psoriasis scales was not confirmed, as no protein
sample could be collected. Subsequently, the expression levels of
NMD substrates, which depended on mutated UPF1, were
examined. Unlike UPF1 WT, which could rescue NMD activity in
UPF1-depleted cells to decrease the level of NMD substrates,
UPF1 insA and UPF1 del did not impact the NIK,
TBL2 or NAT9 mRNA expression levels, indicating that
the two mutants lacked a detectable NMD function (Fig. 3D).
Identification of AREG mRNA as an NMD
substrate depend on the 3′UTR of AREG
After verifying the disruption of the NMD pathway by
UPF1 mutants, gene regulation under a deficient NMD pathway
was explored. As NMD targets diverse substrates and governs RNA
homeostasis in a cell-specific manner (6), it was hypothesized that NMD
deficiency would allow partial targets to be stabilized, leading to
a disordered gene expression in a keratinocyte-specific manner. As
the most abundant EGFR ligand gene in keratinocytes (30), AREG mRNA was identified as
a candidate for the NMD pathway, since it harbors an EJ in its
3′UTR, which could trigger moderate NMD (Fig. 4A) (1). The expression level of AREG
mRNA was increased in psoriasis tissues and UPF1-depleted
keratinocyte cells, while UPF1 overexpression conversely
decreased AREG mRNA levels in keratinocytes (Fig. 4B and C). Consistent with the
well-established NMD substrates noted above, the dysregulation of
AREG mRNA under UPF1 depletion could be rescued by
UPF1 WT, but not by UPF1 insA or UPF1 del
(Fig. 4D). This effect was also
observed at the protein level, as shown by western blotting
(Fig. 4E).
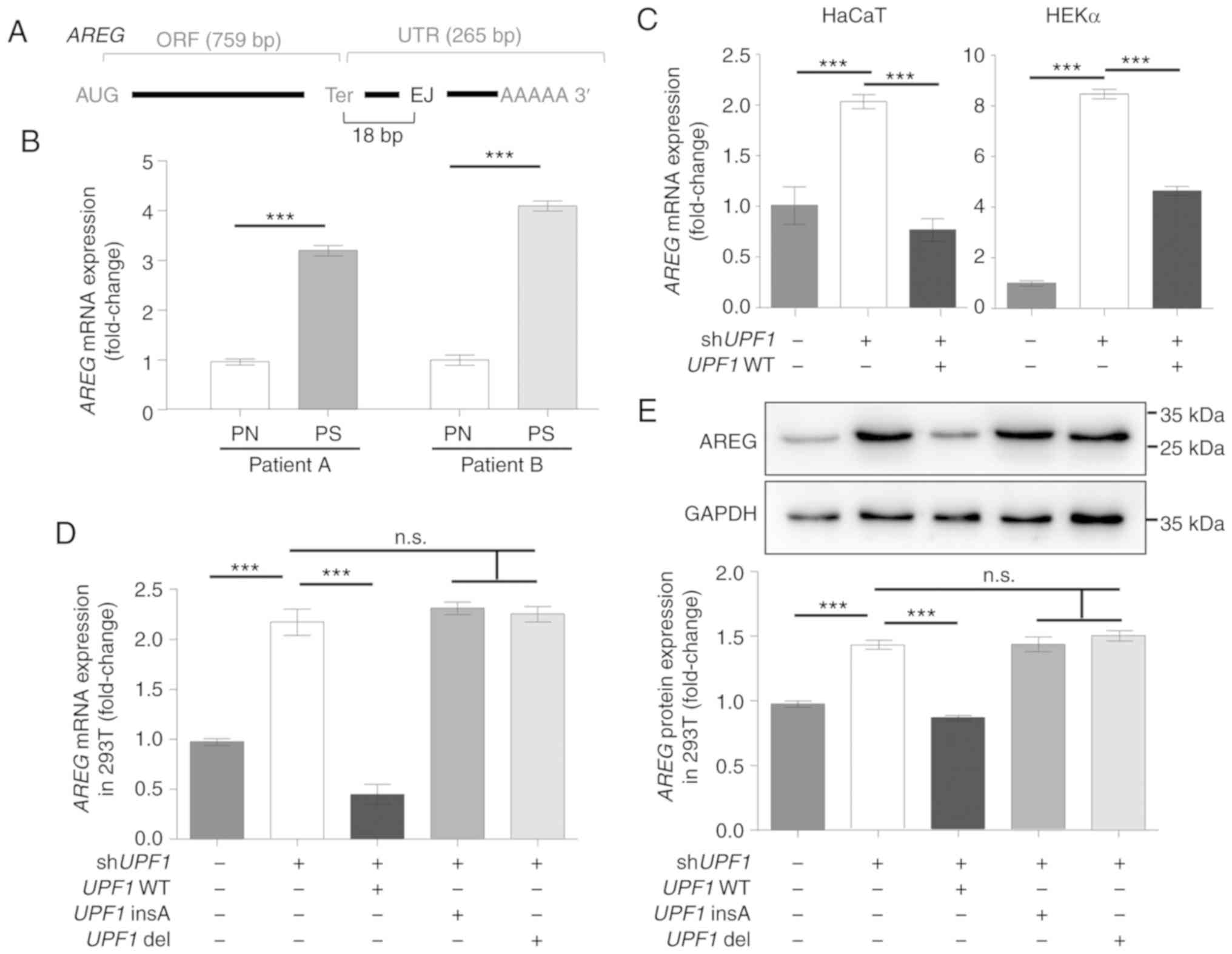 | Figure 4Expression of AREG is
regulated by the NMD pathway. (A) Schematic representation of
AREG mRNA. The EJ in the 3′UTR was presumed to be an NMD
pathway triggering feature. (B) AREG expression was
increased in the psoriasis scales from patients A and B compared
with their respective PN samples. (C) Keratinocytes were
transfected with shUPF1 or UPF1 WT constructs. The
AREG mRNA level was regulated by UPF1 expression in
HEKα and HaCaT cells, as quantified 48 h after transfection. (D)
293T cells were transfected with UPF1 expression vectors after
transfection with shUPF1, and AREG mRNA levels were
quantified 48 h after transfection. (E) AREG protein expression
levels were measured by western blotting, and GAPDH was used as a
control. AREG levels were only decreased by UPF1 WT. Data
are presented as the mean ± SD from three independent experiments.
***P<0.001. AREG, amphiregulin; NMD,
nonsense-mediated RNA decay; EJ, exon-exon junction; WT, wild-type;
UPF1, up-frameshift suppressor 1 homolog; n.s., not
significant; PS, psoriasis scales; PN, normal cornified epidermal
layer. |
Given that transcript destabilization is the
hallmark of direct NMD targets (31), RNA half-life analysis was
performed on DRB-treated 293T cells with an inhibited
transcriptional activity (32);
it was found that AREG mRNA was stabilized ~1.23-fold in
response to UPF1 depletion (Fig. 5A). To verify the NMD-triggering
effect of AREG mRNA, AREG vector with or without its
3′UTR sequence was constructed (Fig.
5B), and the overexpression and knockdown of AREG was
confirmed (Fig. S5B and Data
S1). After knocking down endogenous UPF1 and AREG
in 293T cells, no significant change was identified in the
expression of exogenous AREG following transfection with
AREG without the 3′UTR (Fig.
5C; lanes 5 and 6); however, the expression of AREG
following transfection with AREG with 3′UTR was increased
significantly (Fig. 5C; lanes 7
and 8), which indicated that the 3′UTR region was indispensable for
NMD targeting. The current study further examined whether the 3′UTR
of AREG was targeted by NMD via the dual-luciferase reporter
system pEZX-FR02 (Fig. 5D). The
results showed that the insertion of the AREG 3′UTR to
pEZX-FR02 increased the FLuc activity following UPF1
depletion, indicating that this sequence was adequate for the
triggering NMD (Fig. 5E).
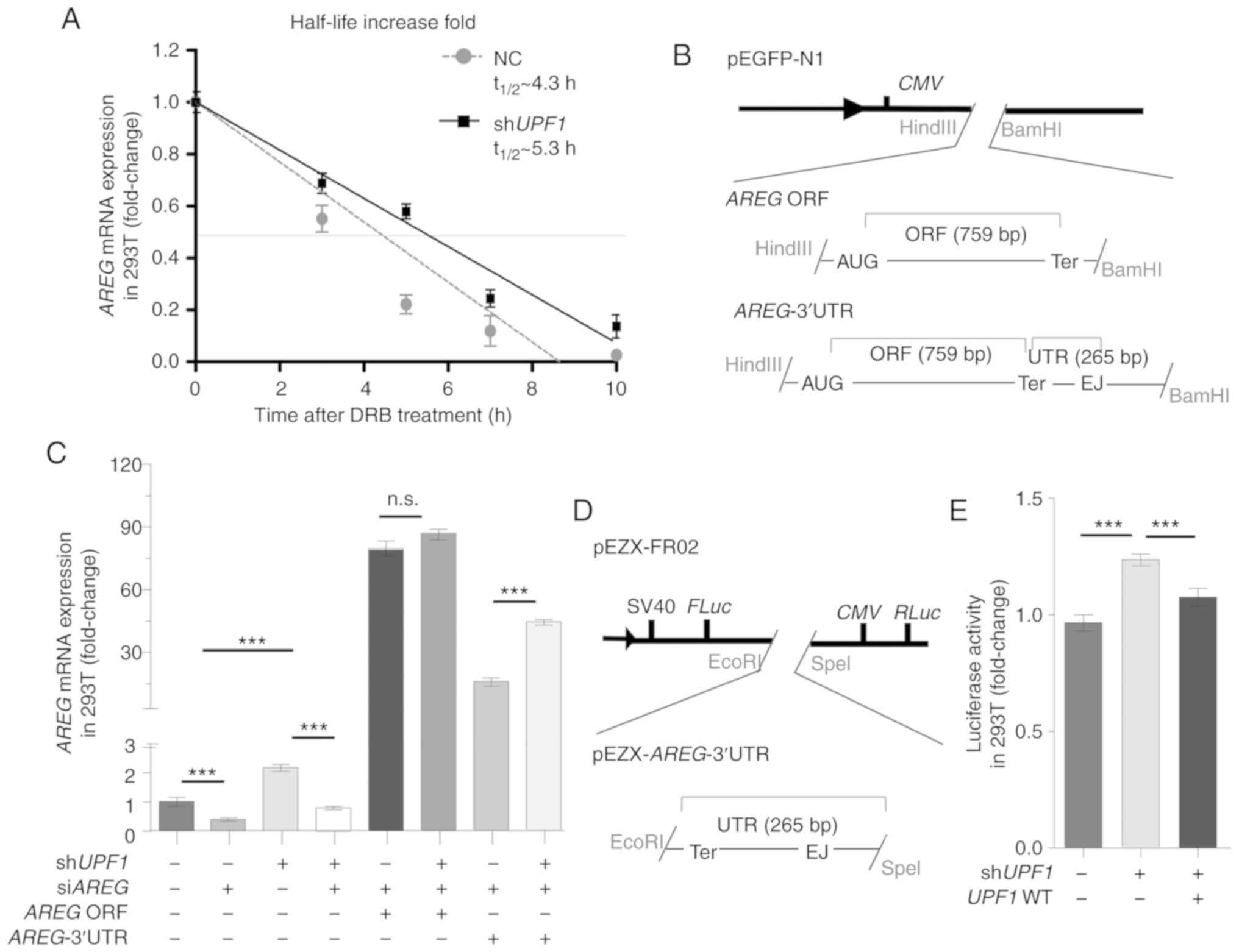 | Figure 5AREG triggers the NMD pathway
post-transcriptionally, depending on its 3′UTR. (A) 293T cells were
treated with DRB, and AREG mRNA levels were evaluated by
RT-qPCR at 0, 3, 5, 7 and 10 h. The t1/2 represents the
half-life of AREG mRNA. The AREG mRNA was stabilized
by UPF1 depletion, as detected by RT-qPCR. (B) The
structures represent the expression vector for AREG ORF and
AREG-3′UTR based on pEGFP-N1. (C) 293T cells were
transfected with shUPF1, and AREG expression vectors were
transfected 24 h later. AREG mRNA levels were quantified 48
h after transfection. The 3′UTR region was required for NMD
triggering by AREG. (D) Schematic representation of the
dual-luciferase reporter system pEZX-AREG-3′UTR. (E) 293T
cells co-transfected with pEZX-AREG-3′UTR plasmid together
with shUPF1 or UPF1 WT vector. Cells were collected
and the FLuc activity was measured 48 h after transfection. RLuc
activity was used as a control. The 3′UTR region was sufficient to
trigger the NMD pathway. Data are presented as the mean ± SD from
three independent experiments. ***P<0.001.
AREG, amphiregulin; NMD, nonsense-mediated RNA decay; DRB,
5,6-dichloro-1-β-D-ribofuranosylben zimidazole; UPF1,
up-frameshift suppressor 1 homolog; firefly luciferase, FLuc;
Renilla luciferase, RLuc; ns, not significant; NC, normal
cells; RT-qPCR, reverse transcription-quantitative PCR. |
NMD regulates keratinocyte homeostasis by
regulating AREG
UPF1 knockdown has been reported to result in
cell apoptosis (33,34), while AREG positively
regulates the proliferation of keratinocytes (11). Successful target gene expression
regulation by UPF1 and AREG vectors was confirmed in
keratinocyte cells (Fig. S6 and Data
S1), and the cell morphology and area of cells did not change
significantly (Fig. S7 and Data
S1). The viability of UPF1-depleted HaCaT and HEKα cells
was decreased, and the inhibition of AREG further decreased
cell viability in these cells (Fig.
6A). These results were confirmed by determining the mRNA
expression levels of proliferation marker protein Ki67, and BCL2
associated agonist of cell death (BAD) (Fig. 6B). Epidermal acanthosis,
hyperkeratosis and angiogenesis have been previously detected in
the skin of AREG transgenic animals (35), which indicates an ability of
AREG to affect cell differentiation. UPF1 depletion
markedly inhibited the expression of the differentiation gene
keratin-5 (K5), the terminal differentiation marker
filaggrin (FLG) and keratin-10 (K10). The expression
levels of the differentiation markers were reversed in HaCaT and
HEKα cells when AREG inhibition was coupled with UPF1
depletion (Fig. 6C).
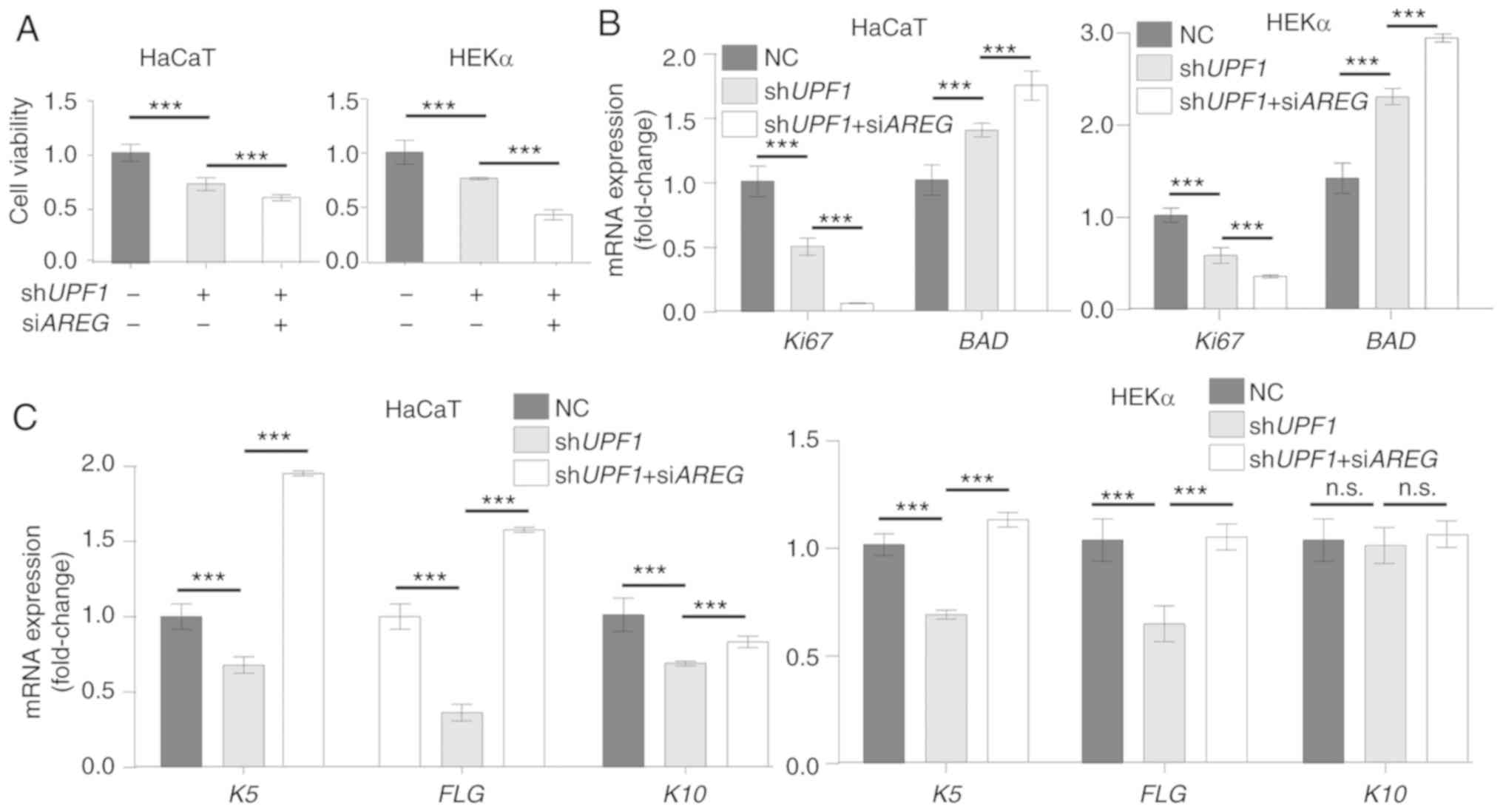 | Figure 6UPF1 depletion-induced
AREG disturbs keratinocyte homeostasis. (A) Cell counting
kit-8 assays were performed to measure cell viability 48 h after
UPF1 or AREG knockdown in HEKα and HaCaT cells.
UPF1 and AREG depletion both decreased cell viability
of keratinocytes. (B) The expression of Ki67 decreased in
keratinocytes with UPF1 or AREG knockdown, while the
expression of apoptosis-related gene BAD increased in both
groups. (C) mRNA expression level of K5, FLG and
K10 was qualified by reverse transcription-quantitative PCR
in keratinocytes. The expression levels of these differentiation
makers decreased with UPF1 depletion and were rescued by
supplementary AREG depletion. (D) Representative human
keratinocyte scratch wounds with cells treated with the
shUPF1 construct, or shUPF1 combined with
siAREG. The wound images at 0 and 24 h are presented, and
the epidermal sheet edge indicated by the yellow irregular curve
was identified by ImageJ software automatically. UPF1
depletion induced the migration of keratinocytes by stabilization
of AREG. (E) Quantitative analysis of the measured wound
coverage at 0, 6, 12, 24 and 48 h. ***P<0.001 vs. NC
group. (F) The motility markers SNAI1, COX-2 and
MMP1 and two chemokines associated with nuclear factor κB
activation, CCL20 and CXCL1, were identified at the
mRNA level in keratinocyte cells. Expression levels of the
migration makers and chemokines increased with UPF1
depletion and were rescued by supplementary AREG depletion.
The results are presented as the mean ± SD from three independent
experiments. ***P<0.001. UPF1, up-frameshift
suppressor 1 homolog; AREG, amphiregulin; BAD, BCL2
associated agonist of cell death; K5, keratin-5; FLG,
filaggrin; K10, keratin-10; COX-2, cyclooxygenase-2;
MMP1, matrix metalloproteinase 1; CCL20, chemokine
(C-C motif) ligand 20; CXCL1, human recombinant GRO-alpha;
NC, normal cells; Ki67, proliferation marker protein Ki67;
n.s., not significant. |
Keratinocytes in which terminal differentiation is
halted undergo changes in cell migration, and keratinocytes can
repair wounds partly by migration (36). A wound healing assay was therefore
performed to estimate the extent to which UPF1 depletion
promoted cutaneous wound healing. UPF1 depletion increased
the ability of both HaCaT and HEKα cells to migrate and
re-epithelialize as demonstrated by the changes in cell-free areas.
Following double knockdown of AREG and UPF1 in
keratinocytes, the cell re-epithelialization was impaired compared
with the shUPF1 group (Fig. 6D and
E). The perturbed NMD in the shUPF1 group induced the
expression of motility markers, cyclooxygenase-2 (COX-2),
matrix metalloproteinase 1 (MMP1) and snail family
transcriptional repressor 1 (SNAI1) compared with control
group, while double knockdown of AREG and UPF1 lost
this capacity (Fig. 6F).
Keratinocytes also produce proinflammatory mediators that react
against damage (37). In the
current study, an increased expression of chemokine (C-C motif)
ligand 20 (CCL20) and human recombinant GRO-α (CXCL1)
was observed following NMD disruption in HaCaT and HEKα cells. The
increased expression of these proinflammatory chemokines following
NMD disruption was restored by AREG knockdown in these cells
(Fig. 6F).
Discussion
To investigate the NMD pathway in skin diseases,
the core element UPF1 was examined in patients with
psoriasis and psoriasis-like mouse models. Psoriasis datasets from
the GEO, human psoriasis scales and IMQ-induced skin inflammation
samples all displayed increased UPF1 mRNA levels compared
with their respective controls. In addition, analysis for several
other skin diseases was conducted by analyzing UPF1
expression profiles acquired from the GEO database. An independent
sample test for Marfan syndrome (GDS2960) showed a significantly
increased UPF1 expression in lesions compared with normal
tissues, and multiple comparisons for squamous cell carcinoma
(GDS2200) and melanoma (GDS1375) showed a high UPF1
expression in squamous cell carcinoma compared with actinic
keratosis or normal tissues, and melanoma and benign nevi compared
with normal tissues (26-38). A negative feedback regulatory
network that directly acts on UPF1 in response to NMD
perturbation has been identified in a previous study which
demonstrated that UPF1 itself is a natural substrate of the
NMD pathway (6). Overexpression
of UPF1 indicated by the GEO datasets analysis herein hinted
at an abnormal NMD in several skin diseases besides psoriasis.
Furthermore, although increased UPF1 mRNA levels were
observed in IMQ-induced psoriasis-like skin, this result was not
confirmed at the protein levels in two mice. Expression levels of
well-characterized NMD substrates, NIK, TBL2 and
NAT9 mRNAs were also increased in mouse models of psoriasis,
suggesting the possibility of UPF1-related NMD perturbation
in psoriasis. The protein level of the NMD substrates was not
determined in the current study, as the NMD pathway regulates
substrates mainly by destabilizing the mRNAs, while protein level
may be affected by translation efficiency or protein degradation
(32).
By sanger sequencing in 5 scales, two somatic
UPF1 transcripts were identified in 2 patients with sporadic
psoriasis. The c.2935_2936insA and c.2030-2081del mutations in
UPF1 mRNAs both disrupted the conventional reading frame and
resulted in PTCs. The PTC-UPF1 transcripts should be
immediately degraded by NMD under well-balanced RNA surveillance,
while the abnormal mRNAs may escape degradation under exceptional
circumstances, and consequently be translated into truncated
proteins. Fully functional UPF1 is indispensable to the NMD pathway
(1), while UPF1 without
C-terminal domains has been assumed to fail in autophosphorylation
or RNA unwinding (38). The
truncated UPF1 protein disturbed NMD function, as demonstrated by
the upregulation of the NMD-sensitive mRNAs NIK, TBL2
and NAT9. The calponin homology domain retained in truncated
UPF1 protein has been shown to exert an inhibitory effect on
helicase activity, and the inhibition can be relieved by UPF2
binding (39). Although in the
current study RT-qPCR analysis showed that the expression of
UPF1 WT was retained in the psoriasis scales, it was
speculated that the truncated protein may competitively interact
with UPF2 and disturb the helicase activity of residual full-length
UPF1 protein. In UPF1-depleted 293T cells, the mutated
UPF1 exhibited slightly lower steady-state levels than
UPF1 WT, at both the mRNA and protein levels. One
explanation for this result is that the mutants were destabilized
by redundant endogenous UPF1. Of note, although the expression
level of UPF1 was found to be increased in psoriasis data
from the GEO database, IMQ-mice and psoriasis scale tissues, these
data were derived from chip detection or RT-qPCR and depended on
the specified nucleotide site binding, without considering the
overall sequences of the transcripts or proteins. In fact, the
increased expression of total UPF1 mRNA in psoriasis may be
due to the accumulation of aberrant transcripts, at least in these
2 psoriasis scales. It remains to be elucidated whether the
c.2935_2936insA and c.2030_2081del UPF1 could be
representative in psoriasis, and further data should be collected
to verity this hypothesis. In the present study, the abnormal
UPF1 transcripts, which were identified in two patients with
sporadic psoriasis, were employed to reduce UPF1 activity. These
finding indicated gene dysregulation related with NMD
deficiency.
It was also concluded that AREG is an
important substrate of the NMD pathway in keratinocytes. The EJ in
the 3′UTR of AREG mRNA, which is present at low levels among
cellular transcripts, could trigger the NMD pathway (1,3).
AREG has been reported to be the most abundant EGFR ligand in
keratinocytes (30) and
overexpressed in a wide spectrum of epithelial diseases, including
squamous cell carcinoma of the head and neck, colon cancer, lung
cancer and psoriasis (12-15).
A negative association was identified between the expression levels
of AREG and UPF1 in keratinocytes, and the
upregulation of AREG could be rescued by the recovery of
UPF1 WT, rather than the mutated UPF1. Based on the
premise that post-transcriptional regulation of the AREG
expression has little involvement in the NMD pathway, it was
empirically determined in a half-life assay under inhibited UPF1
transcription in vitro that AREG mRNA was directly
targeted by the NMD pathway. These results confirmed a previous
hypothesis that AREG expression was regulated by NMD
post-transcriptionally, rather than by impacting its transcription
level (32). In the current
study, only AREG transcripts with the 3′UTR sequence could
be regulated by NMD, and the results based on FLuc/RLuc
dual-luciferase reporter system indicated that the 3′UTR sequence
of AREG mRNA was sufficient to trigger the NMD pathway. It
was therefore concluded that AREG mRNA was a primary NMD
substrate, and that the mechanism through which AREG was
degraded by NMD was dependent on its 3′UTR.
According to the classic NMD mechanism, termination
codons situated >50-55 nts upstream of an EJ are typically
expected to trigger mRNA surveillance, depending on a multi-subunit
protein complex, the exon junction complex (EJC) (3). However, the distance between the
termination codon (Ter) and the EJ downstream in the AREG is
only 18 nts, which may prevent the mRNA from assembling a stable
EJC. According to a previous report, ~50% of EJCs were mapped to a
non-canonical position, where no binding sites were predicted to
exist (40), suggesting that the
AREG might be targeted by NMD by a 3′UTR-dependent but
EJC-independent pathway. The relative increase in the half-life of
AREG mRNA in UPF1-depleted cells was just 1.23-fold,
which was smaller than that of several documented substrates with a
similar NMD-inducing feature (Table SV) (8). As the NMD pathway targets natural
substrates in different magnitudes (8), it was speculated that the
recognition reaction between AREG and the NMD pathway may be
weaker compared with other endogenous substrates. AREG may
contribute to diverse biological mechanisms depending on its
dominance in keratinocytes, which is in line with the cell-specific
character of NMD.
The NMD pathway governs different subsets of
substrates and participates in diverse, essential homeostatic
mechanisms in different cells (6), and the perturbation of NMD
influences cellular homeostasis in a cell-specific manner. By
contrast, aberrant UPF1 may lead to reprogramming towards a
more malignant state in premalignant glandular pancreatic cells
(9), and may drive a
proinflammatory response in lung epithelial cells (7). As shown in HaCaT and HEKα
keratinocytes used in the current study, UPF1 depletion
decreased viability and differentiation, induced
re-epithelialization, with increased expression of motility markers
SNAI1, COX-2 and MMP1, and upregulated the
expression of the chemokines CCL20 and CXCL1.
NMD deficiency has been reported to result in cell
apoptosis though preferentially increasing the level of the growth
arrest and DNA damage inducible b in 293 cells (33). Double Homeobox 4 mRNA, an
NMD substrate, has also been shown to cause apoptosis in muscle
cells (34). The BAD protein has
been reported to be a proapoptotic factor inactivated by the AKT
signaling (41). In the present
study, the expression of BAD mRNA was upregulated in the
UPF1-depleted keratinocytes. As a major autocrine growth
factor for human keratinocytes, AREG plays an important, positive
role in keratinocyte proliferation (30). Unlike the previously reported role
of AREG in inducing hyperproliferation by itself (11), the disruption of NMD, along with
stabilization of AREG observed in the current study,
decreased the proliferation of keratinocytes, and following the
depletion of endogenous AREG, the viability of keratinocytes
further decreased, indicating an anti-apoptotic role of
AREG. NMD has been shown to extensively impact gene
expression either directly, by targeting degradation, or
indirectly, via downstream cascades (1). NMD deficiency in keratinocytes may
mediate a proapoptotic effect in advance and abrogate the
proliferation-promoting effect of AREG alone, as shown in a
previous study, in which oncogene-induced proliferation was
inhibited by cellular senescence (42).
AREG is considered to prevent keratinocyte
differentiation, and the suppression of AREG to irreversibly
upregulate genes involved in keratinocyte differentiation (11,43). The present data demonstrated that
the depletion of UPF1 in HaCaT and HEKα cells downregulated
the expression of differentiation markers by targeting AREG.
Skin diseases, such as psoriasis, can be triggered by physical
injury in susceptible patients (known as the Koebner phenomenon),
and it is critical to understand the elements participating in
processes occurring in cutaneous wounds (44,45). Additionally, it has been suggested
that endogenous AREG modulates tissue repair and regulates
the migration of several cell types, such as breast cancer cells
(46,47). The current findings confirmed that
NMD perturbation in keratinocytes promoted wound
re-epithelialization by adjusting endogenous AREG levels.
Previous reports have shown that the overexpression of exogenous
AREG in keratinocytes does not result in marked increases in
cell migration (48), and the
contrasting results in the present study highlight the unique
character of NMD deficiency in cellular balance. AREG has
also been reported to mediate inflammatory action in keratinocytes
(49,50). In the present study, NMD normally
suppressed inflammatory activation by targeting AREG mRNA;
when this suppression was alleviated due to UPF1 deficiency,
the stabilization of AREG mRNA led to a high expression of
the proinflammatory chemokines, which may contribute to triggering
immune cell infiltration and angiogenesis.
The present data supported a model in which NMD
normally regulated AREG abundance to maintain the
homeostasis in keratinocytes. NMD deficiency caused by UPF1
alterations could disrupt proliferation, inhibit cell
differentiation, promote wound healing, and activate inflammation
response, by regulating AREG post-transcriptionally in
keratinocytes, leading to indirect impacts on downstream gene
cascades.
To the best of our knowledge, this is the first
study revealing mutated UPF1 transcripts in psoriasis and
investigating the relationship between the AREG-NMD axis and
homeostasis of keratinocytes. These findings of aberrant
UPF1 expression in psoriasis and disruption of gene
regulation at the post-transcriptional level might be useful for
understanding the full development of keratinocyte morbidity.
Furthermore, these results suggested that therapies involving NMD
intervention may have the potential to ameliorate skin diseases or
even other diseases related to abnormal cell differentiation, wound
healing or inflammatory responses, through rescuing the
dysregulation of multiple NMD substrates, such as AREG,
either directly or via indirect regulation cascades. Mouse tissue
samples were suitable for analysis; however, the human scale
samples are fragile and were only selected due to the lack of
availability of biopsy samples. The role of the AREG-NMD
axis in treated psoriasis is still unknown, as all patients
recruited in the current study were not treated with any therapy
for at least 2 weeks before the sampling, and it was challenging to
follow the patients further without electronic medical records in
the hospital system. In order to determine whether the mutated
UPF1 could be representative in psoriasis, the quality and
quantity of samples should be improved in the future, and an animal
model with gene modification could be used for in vivo
verification. In addition, the role of the AREG-NMD axis in
skin diseases with abnormal UPF1 expression levels, such as
Marfan syndrome, non-melanoma skin cancer and malignant melanoma,
should be investigated further.
Supplementary Data
Funding
This research was funded by Natural Science
Foundation of Zhejiang Province, China (grant no. LQ18H110001) and
National Natural Science Foundation of China (grant no.
81802771).
Availability of data and materials
The datasets used or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC and QL were contributors in acquisition of data.
NS generated constructs for this study and contributed to revising
the manuscript. QZ provided resources and performed data analysis.
JR contributed to the construction of the mouse model and revised
the manuscript. LL contributed analytical tools and was involved in
the construction of the mouse model. LW contributed to designing
the research and writing the original draft. CL designed the
research, provided resources and was a major contributor in
interpretation of data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Ethics approval for sampling from patients with
psoriasis was obtained from the Ethics Committee of Hangzhou Normal
University, Hangzhou, China (approval no. 2017-010). Tissues were
scraped by blunt scalpels with patients' permission at the
Affiliated Hospital of Ningbo University, Ningbo, China. Ethics
approval for animal experimentation was obtained from the Ethics
Committee of Hangzhou Normal University, Hangzhou, China (approval
no. 2016008).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The pCMV-MYC-UPF1 was provided by Professor
Lynne E. Maquat (University of Rochester), the Phblv-U6-puro
construct was provided by Professor Fan Handong (Hangzhou Normal
University), and the pEGFP-N1 vector was provided by Dr Wang Miao
(Hangzhou Normal University).
References
|
1
|
Karousis ED and Mühlemann O:
Nonsense-mediated mRNA decay begins where translation ends. Cold
Spring Harb Perspect Biol. 11:a0328622019. View Article : Google Scholar
|
|
2
|
Huang L, Low A, Damle SS, Keenan MM, Kuntz
S, Murray SF, Monia BP and Guo S: Antisense suppression of the
nonsense mediated decay factor Upf3b as a potential treatment for
diseases caused by nonsense mutations. Genome Biol. 19:42018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jaffrey SR and Wilkinson MF:
Nonsense-mediated RNA decay in the brain: Emerging modulator of
neural development and disease. Nat Rev Neurosci. 19:715–728. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Celik A, He F and Jacobson A: NMD monitors
translational fidelity 24/7. Curr Genet. 63:1007–1010. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Popp MW and Maquat LE: Nonsense-mediated
mRNA decay and cancer. Curr Opin Genet Dev. 48:44–50. 2018.
View Article : Google Scholar :
|
|
6
|
Huang L, Lou CH, Chan W, Shum EY, Shao A,
Stone E, Karam R, Song HW and Wilkinson MF: RNA homeostasis
governed by cell type-specific and branched feedback loops acting
on NMD. Mol Cell. 43:950–961. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu J, Plank TD, Su F, Shi X, Liu C, Ji Y,
Li S, Huynh A, Shi C, Zhu B, et al: The nonsense-mediated RNA decay
pathway is disrupted in inflammatory myofibroblastic tumors. J Clin
Invest. 126:3058–3062. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mendell JT, Sharifi NA, Meyers JL,
Martinez-Murillo F and Dietz HC: Nonsense surveillance regulates
expression of diverse classes of mammalian transcripts and mutes
genomic noise. Nat Genet. 36:1073–1078. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu C, Karam R, Zhou Y, Su F, Ji Y, Li G,
Xu G, Lu L, Wang C, Song M, et al: The UPF1 RNA surveillance gene
is commonly mutated in pancreatic adenosquamous carcinoma. Nat Med.
20:596–598. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goetz AE and Wilkinson M: Erratum to:
Stress and the nonsense-mediated RNA decay pathway. Cell Mol Life
Sci. 74:40472017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stoll SW, Johnson JL, Li Y, Rittié L and
Elder JT: Amphiregulin carboxy-terminal domain is required for
autocrine keratinocyte growth. J Invest Dermatol. 130:2031–2040.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tinhofer I, Klinghammer K, Weichert W,
Knödler M, Stenzinger A, Gauler T, Budach V and Keilholz U:
Expression of amphiregulin and EGFRvIII affect outcome of patients
with squamous cell carcinoma of the head and neck receiving
cetuximab-docetaxel treatment. Clin Cancer Res. 17:5197–5204. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shao J, Lee SB, Guo H, Evers BM and Sheng
H: Prostaglandin E2 stimulates the growth of colon cancer cells via
induction of amphiregulin. Cancer Res. 63:5218–5223.
2003.PubMed/NCBI
|
|
14
|
Stabile LP, Rothstein ME, Keohavong P,
Lenzner D, Land SR, Gaither-Davis AL, Kim KJ, Kaminski N and
Siegfried JM: Targeting of both the c-Met and EGFR pathways results
in additive inhibition of lung tumorigenesis in transgenic mice.
Cancers (Basel). 2:2153–2170. 2010. View Article : Google Scholar
|
|
15
|
Cook PW, Pittelkow MR, Keeble WW,
Graves-Deal R, Coffey RJ and Shipley GD: Amphiregulin messenger RNA
is elevated in psoriatic epidermis and gastrointestinal carcinomas.
Cancer Res. 52:3224–3227. 1992.PubMed/NCBI
|
|
16
|
Cook PW, Piepkorn M, Clegg CH, Plowman GD,
DeMay JM, Brown JR and Pittelkow MR: Transgenic expression of the
human amphiregulin gene induces a psoriasis-like phenotype. J Clin
Invest. 9:2286–2294. 2004.
|
|
17
|
Chung E, Cook PW, Parkos CA, Park YK,
Pittelkow MR and Coffey RJ: Amphiregulin causes functional
downregulation of adherens junctions in psoriasis. J Invest
Dermatol. 124:1134–1140. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Swindell WR, Xing X, Stuart PE, Chen CS,
Aphale A, Nair RP, Voorhees JJ, Elder JT, Johnston A and Gudjonsson
JE: Heterogeneity of inflammatory and cytokine networks in chronic
plaque psoriasis. PLoS One. 7:e345942012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nair RP, Duffin KC, Helms C, Ding J,
Stuart PE, Goldgar D, Gudjonsson JE, Li Y, Tejasvi T, Feng BJ, et
al: Genome-wide scan reveals association of psoriasis with IL-23
and NF-kappaB pathways. Nat Genet. 41:199–204. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Reischl J, Schwenke S, Beekman JM,
Mrowietz U, Stürzebecher S and Heubach JF: Increased expression of
Wnt5a in psoriatic plaques. J Invest Dermatol. 127:163–169. 2007.
View Article : Google Scholar
|
|
21
|
Yao Y, Richman L, Morehouse C, de los
Reyes M, Higgs BW, Boutrin A, White B, Coyle A, Krueger J, Kiener
PA and Jallal B: Type I interferon: Potential therapeutic target
for psoriasis? PLoS One. 3:e27372008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Suárez-Fariñas M, Li K, Fuentes-Duculan J,
Hayden K, Brodmerkel C and Krueger JG: Expanding the psoriasis
disease profile: Interrogation of the skin and serum of patients
with moderate-to-severe psoriasis. J Invest Dermatol.
132:2552–2564. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qiu CC, Su QS, Zhu SY and Liu RC:
Identification of potential biomarkers and biological pathways in
juvenile dermatomyositis based on miRNA-mRNA network. Biomed Res
Int. 2019:78142872019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lv M, Deng J, Tang N, Zeng Y and Lu C:
Efficacy and safety of tripterygium wilfordii Hook F on psoriasis
vulgaris: A systematic review and meta-analysis of randomized
controlled trials. Evid Based Complement Alternat Med.
2018:26230852018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Medicine DC oPLAAoTC: Consensus on
diagnosis and treatment of Chinese integrative medicine for
psoriasis vulgaris. Chin J Dermatol Venerol Integ Tradit West Med.
8:3282009.
|
|
26
|
Oka T, Sugaya M, Takahashi N, Takahashi T,
Shibata S, Miyagaki T, Asano Y and Sato S: CXCL17 attenuates
imiquimod-induced psoriasis-like skin inflammation by recruiting
myeloid-derived suppressor cells and regulatory T cells. J Immunol.
198:3897–3908. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Ueyama A, Yamamoto M, Tsujii K, Furue Y,
Imura C, Shichijo M and Yasui K: Mechanism of pathogenesis of
imiquimod-induced skin inflammation in the mouse: A role for
interferon-alpha in dendritic cell activation by imiquimod. J
Dermatol. 41:135–143. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Amer M, Mostafa FF, Tosson Z and Nasr AN:
Corneocytes in scaly parakeratotic diseases. Int J Dermatol.
35:417–421. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stoll SW, Stuart PE, Lambert S,
Gandarillas A, Rittié L, Johnston A and Elder JT: Membrane-tethered
intracellular domain of amphiregulin promotes keratinocyte
proliferation. J Invest Dermatol. 136:444–452. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chan WK, Huang L, Gudikote JP, Chang YF,
Imam JS, MacLean JA and Wilkinson MF: An alternative branch of the
nonsense-mediated decay pathway. EMBO J. 26:1820–1830. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lou CH, Shao A, Shum EY, Espinoza JL,
Huang L, Karam R and Wilkinson MF: Posttranscriptional control of
the stem cell and neurogenic programs by the nonsense-mediated RNA
decay pathway. Cell Rep. 6:748–764. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nelson JO, Moore KA, Chapin A, Hollien J,
Metzstein MM. Degradation of Gadd45 mRNA by nonsense-mediated decay
is essential for viability. Elife. 5:e128762016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feng Q, Snider L, Jagannathan S, Tawil R,
van der Maarel SM, Tapscott SJ and Bradley RK: A feedback loop
between nonsense-mediated decay and the retrogene DUX4 in
facioscapu-lohumeral muscular dystrophy. Elife. 4:2015. View Article : Google Scholar
|
|
35
|
Li Y, Stoll SW, Sekhon S, Talsma C, Camhi
MI, Jones JL, Lambert S, Marley H, Rittié L, Grachtchouk M, et al:
Transgenic expression of human amphiregulin in mouse skin:
Inflammatory epidermal hyperplasia and enlarged sebaceous glands.
Exp Dermatol. 25:187–193. 2016. View Article : Google Scholar :
|
|
36
|
Patel GK, Wilson CH, Harding KG, Finlay AY
and Bowden PE: Numerous keratinocyte subtypes involved in wound
re-epitheli-alization. J Invest Dermatol. 126:497–502. 2006.
View Article : Google Scholar
|
|
37
|
Ruiz N, Wang B, Pentland A and Caparon M:
Streptolysin O and adherence synergistically modulate
proinflammatory responses of keratinocytes to group A streptococci.
Mol Microbiol. 27:337–346. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hurt JA, Robertson AD and Burge CB: Global
analyses of UPF1 binding and function reveal expanded scope of
nonsense-mediated mRNA decay. Genome Res. 23:1636–1650. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fiorini F, Boudvillain M and Le HH: Tight
intramolecular regulation of the human Upf1 helicase by its N- and
C-terminal domains. Nucleic Acids Res. 41:2404–2415. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schweingruber C, Rufener SC, Zünd D,
Yamashita A and Mühlemann O: Nonsense-mediated mRNA
decay-mechanisms of substrate mRNA recognition and degradation in
mammalian cells. Biochim Biophys Acta. 1829:612–623. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sun Y, Xiao S, Chen J, Wang M, Zheng Z,
Song S and Zhang L: Heat shock protein 90 mediates the apoptosis
and autophage in nicotinic-mycoepoxydiene-treated HeLa cells. Acta
Biochim Biophys Sin (Shanghai). 47:451–458. 2015. View Article : Google Scholar
|
|
42
|
Li X, Xu H, Xu C, Lin M, Song X, Yi F,
Feng Y, Coughlan KA, Cho WC, Kim SS and Cao L: The yin-yang of DNA
damage response: Roles in tumorigenesis and cellular senescence.
Int J Mol Sci. 14:2431–2448. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Stoll SW, Stuart PE, Swindell WR, Tsoi LC,
Li B, Gandarillas A, Lambert S, Johnston A, Nair RP and Elder JT:
The EGF receptor ligand amphiregulin controls cell division via
FoxM1. Oncogene. 35:2075–2086. 2016. View Article : Google Scholar :
|
|
44
|
Albanesi C, De Pità O and Girolomoni G:
Resident skin cells in psoriasis: A special look at the
pathogenetic functions of keratinocytes. Clin Dermatol. 25:581–588.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lai Y, Li D, Li C, Muehleisen B, Radek KA,
Park HJ, Jiang Z, Li Z, Lei H, Quan Y, et al: The antimicrobial
protein REG3A regulates keratinocyte proliferation and
differentiation after skin injury. Immunity. 37:74–84. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zaiss DMW, Gause WC, Osborne LC and Artis
D: Emerging functions of amphiregulin in orchestrating immunity,
inflammation, and tissue repair. Immunity. 42:216–226. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Schmucker H, Blanding WM, Mook JM, Wade
JF, Park JP, Kwist K, Shah H and Booth BW: Amphiregulin regulates
proliferation and migration of HER2-positive breast cancer cells.
Cell Oncol (Dordr). 41:159–168. 2018. View Article : Google Scholar
|
|
48
|
Stoll SW, Rittié L, Johnson JL and Elder
JT: Heparin-binding EGF-like growth factor promotes
epithelial-mesenchymal transition in human keratinocytes. J Invest
Dermatol. 132:2148–2157. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Farley SM, Purdy DE, Ryabinina OP,
Schneider P, Magun BE and Iordanov MS: Fas ligand-induced
proinflammatory transcriptional responses in reconstructed human
epidermis. Recruitment of the epidermal growth factor receptor and
activation of MAP kinases. J Biol Chem. 283:919–928. 2008.
View Article : Google Scholar
|
|
50
|
Kennedy-Crispin M, Billick E, Mitsui H,
Gulati N, Fujita H, Gilleaudeau P, Sullivan-Whalen M, Johnson-Huang
LM, Suárez-Fariñas M and Krueger JG: Human keratinocytes' response
to injury upregulates CCL20 and other genes linking innate and
adaptive immunity. J Invest Dermatol. 132:105–113. 2012. View Article : Google Scholar
|














