Introduction
Chronic myeloid leukemia (CML) is a
myeloproliferative disease that is characterized by an increase in
immature myeloid cells in the bone marrow and peripheral blood
(1). CML is associated with a
reciprocal translocation t(9;22) (q34;q11), which results in the
generation of a BCR/ABL fusion gene (2). The BCR-ABL fusion gene encodes a
tyrosine kinase oncoprotein that induces cell proliferation and
leukemogenesis by activating the RAS, PI3K/AKT, ERK, MYC and
JAK/STAT signaling pathways (3).
The tyrosine kinase inhibitor (TKI) imatinib (IM) has been
demonstrated to inhibit the kinase activity of BCR-ABL and has been
used as a first-line treatment strategy for CML (4). However, ~35% of CML patients exhibit
disease progression, relapse and/or intolerance to IM (5). The majority of primary and secondary
resistance mechanisms include the amplification of BCR-ABL and
point mutations within the ABL kinase domain (6-8).
Second and third generation TKIs, such as dasatinib and ponatinib,
are able to overcome IM resistance in some patients with T315I
mutations (9-11). However, these TKIs have severe
side effects, such as cardiovascular thrombotic events, and have
therefore been removed from the market. There is an urgent need to
develop novel therapeutic strategies with high efficacy and low
toxicity for the treatment of patients with CML.
Interferon α (IFNα) has been the treatment choice
for certain patients with CML (12). Therefore, TKIs, IFNα or a
combination strategy may emerge as a treatment option for CML
(13). Interferon-induced protein
with tetratricopeptide repeats 2 (IFIT2), also known as ISG54, is a
member of the IFN-stimulated genes. IFIT2 was originally identified
as a direct response target to type I IFN, and it has been reported
to serve a crucial role in host antiviral defense in the innate
immune response (14). IFIT2 is
considered a tumor suppressor in several tumor types, such as oral
squamous cell carcinoma, breast cancer and leukemia, as it has been
identified to inhibit cancer cell growth and migration, and promote
cell apoptosis. In addition, IFIT2 has been demonstrated to be
associated with clinical parameters and therapeutic outcomes
(15,16). Our previous study (16) revealed that IFIT2 overexpression
could enhance curcumin-induced apoptosis in the CML cell line K562.
However, whether IFIT2 expression inhibits CML progression remains
to be determined.
Using RNA sequencing arrays, the present observed
that specific interferon signaling pathway-associated genes,
including IFIT2, had reduced expression levels in CML patients
compared with healthy controls. Therefore, the current study
investigated the role of IFIT2 in CML.
Materials and methods
CML and normal bone marrow cells
Bone marrow cells that were isolated from 26
patients with primary CML were obtained from the department of
Hematology, First Affiliated Hospital of Nanchang University
(Nanchang, China) between January 2016 and December 2017. All
patients were diagnosed with CML during the chronic phase using the
Morphology, Immunology, Cytogenetics and Molecular (MICM) criteria
of the World Health organization classification of myeloid
neoplasms (17). Patients were
identified as harboring BCR-ABL transcripts using karyotype
analysis, fluorescence in situ hybridization and/or reverse
transcription (RT)-PCR. All patients included in the study were
administered IM (400 mg p.o. q.d.) as a front-line treatment, and
the copies of BCR/ABL were monitored by PCR every 3 months.
Additionally, 16 bone marrow samples as controls were collected
from patients with normal bone marrow, diagnosed by the MICM
criteria, at the department of Hematology, First Affiliated
Hospital of Nanchang University between January 2016 and December
2017. No significant differences were identified between the sex
and age of the two groups (Table
I). The present study was approved by the Medical Ethics
Committee of the First Affiliated Hospital of Nanchang University
(Nanchang, China). All the patients provided written informed
consent.
 | Table IAge and sex distribution of controls
and patients with CML. |
Table I
Age and sex distribution of controls
and patients with CML.
| Control (n=16) | CML (n=26) | P-value |
|---|
| Sex | | | 0.9998 |
| Male | 7 | 11 | |
| Female | 9 | 15 | |
| Age, years | 19-70 | 24-71 | 0.3861 |
Library construction and RNA
sequencing
Total RNA from the bone marrow of patients was
extracted using TRIzol reagent (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. RNA integrity was
determined using the Agilent Bioanalyzer 2100 (Agilent
Technologies, Inc.). RNA-sequencing library construction was
performed using the standard TruSeq RNA sample preparation v2
protocol (Illumina, Inc.). Libraries were then sequenced using the
Illumina HiSeq2500 platform at Shanghai Biotechnology Corporation
(Shanghai, China). Sequencing and real-time data analyses were
performed using the StringTie software (version 1.3.0) provided by
Illumina.
Cell culture and reagents
The human CML cell line K562 was cultured in
RPMI-1640 media (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (Hyclone; ge Healthcare
Life Sciences). The 293T cell line was cultured in DMEM (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine
serum. Cells were cultured in a 37°C incubator with 5%
Co2. Doxycycline (DOX) and puromycin were purchased from
Sigma-Aldrich; Merck KgaA and Invitrogen; Thermo Fisher Scientific,
Inc., respectively. RT reagent (cat. no. KR103), Super-Real qPCR
PreMix (SYBR-Green) reagent kit (cat. no. FP205) and PCR 2xMix
reagent (cat. no. KT201) were purchased from Tiangen Biotech Co.,
Ltd. pLVX-tetonepuro lentiviral expression vector and the
lentiviral packaging plasmids were purchased from Clontech
Laboratories, Inc. Antibodies against p27 (cat. no. sc-53906;
1:500) and IFIT2 (cat. no. sc-390724; 1:500), goat anti-mouse
IgG-HRP (cat. no. sc-2005; 1:10,000) and goat anti-rabbit IgG-HRP
(cat. no. SC-2004; 1:10,000) were purchased from Santa Cruz
Biotechnology. Anti-cullin-1 (cat. no. 71-8700; 1:1,000) was
purchased from Invitrogen; Thermo Fisher Scientific, Inc. Anti-CSN3
(cat. no. gTX33109; 1:5,000) and anti-CSN5 (cat. no. GTX70203;
1:1,000) were purchased from GeneTex, Inc. Antibodies against mTOR
(cat. no. 2972; 1:1,000), phosphorylated (p)-mTOR (cat. no. 2971;
1:1,000), AKT (cat. no. 9272; 1:1,000) and p-AKT (cat. no. 9271;
1:1,000) were purchased from Cell Signaling Technology, Inc.
Antibodies against hemagglutinin (HA) (cat. no. Ae008; 1:5,000),
ABL proto-oncogene 1, non-receptor tyrosine kinase (ABL1; cat. no.
A0282; 1:500), c-Jun (cat. no. A0246; 1:500), p-c-Jun-ser63 (cat.
no. AP0048; 1:500) and β-actin (cat. no. AC038; 1:50,000) were
purchased from Abclonal Technology, Inc.
Plasmid construction, transfection and
lentiviral transduction
The IFIT2 open reading frame (ORF) was cloned
in-frame to the HA tag at the N-terminal to generate the
pEGFP-C1-IFIT2 plasmid. The vector pEGFP-C1 was from Clontech
Laboratories, Inc. The IFIT2 ORF was amplified using PCR with PCR
2xMix reagent and the following primers: Sense, 5′-AGT CCA ATT GCC
ACC ATG GTG TAC CCA TAC GAC GTC CCA GAC TAC GCT AGT GAG AAC AAT
AAC-3′ and antisense, 5′-TAT GGA TCC TCA GCA GTA GC C TA-3′. The
HA-IFIT2 complementary DNA (cDNA) was then inserted into the
lentiviral vector pLVX-tetone-puro, and then transfected into 293T
cells together with the packing plasmids pSPAX2 and pMD2G at a
ratio of 5:3:2 using EndoFectin™ (GeneCopoeia, Inc.) based on the
manufacturer's protocol. A total of 48 h post-transfection, virus
was harvested and used to infect K562 cells. Stable clones
expressing HA-IFIT2 (IFIT2-K562) and negative control (K562-TETONE)
were then selected using puromycin. Stable cell clones transduced
with the empty vector were used as the controls. The expression of
HA-IFIT2 was measured after treating with 2 µg/ml
Doxycycline (cat. no. 24390-14; MedChemExpress).
Cell cycle analysis
Cell cycle analysis was performed as described
previously (18). Briefly,
1×106 cells stably expressing IFIT2 or control cells
were harvested and then stained using the COULTER DNA Prep Reagents
kit (Beckman Coulter, Inc.), according to the manufacturer's
protocol. Stained cells were analyzed using a flow cytometer and
cell cycle analysis was performed using MODFIT LT 4.1 software
(Verity Software House).
Immunofluorescence staining
Stable cells were transferred onto slides using
cytospin and fixed with 4% paraformaldehyde for 15 min at room
temperature. Following permeabilization with 0.5% Triton X-100 in
PBS for 5 min at room temperature, cells were incubated with
primary antibody anti-IFIT2 (1:200) for 2 h at 37°C. Slides were
then washed three times with PBST and incubated with
FITC-conjugated anti-mouse IgG (1:100; cat. no. AB_2769475;
Abclonal Technology, Inc.) for 1 h at room temperature. Nuclear
staining was performed using 4,6-diamidino-2-phenylindole for 5 min
at room temperature. Slides were then washed with PBST and
fluorescent images were observed using a fluorescence microscope
(Olympus Corporation).
RT-quantitative PCR (qPCR)
Total RNA was extracted using TRIzol (cat. no.
15596026; Invitrogen; Thermo Fisher Scientific, Inc.) and reverse
transcribed to cDNA at 42°C for 1 h. Gene expression levels for
IFIT2 were measured by qPCR with Super-Real qPCR PreMix reagent at
the following conditions: 95°C for 5 min, and 95°C for 5 sec and
60°C for 30 sec for 40 cycles. Normalization was performed using
β-actin expression levels. Primers used for qPCR were as follows:
IFIT2 forward, 5′-CTG CAA CCA TGA GTG AGA AC-3′ and reverse,
5′-CAG-GTG ACC AGA CTT CTG AT-3′; and β-actin forward, 5′-CAT GTA
CGT TGC TAT CCA GGC-3′ and reverse, 5′-CTC CTT AAT GTC ACG CAC
GAT-3′. Relative expression levels were calculated using the
comparative 2−ΔΔCq method (19).
Western blot analysis
Total proteins were extracted using lysis buffer [1%
Triton X-100, 50 mM Tris (pH 8.0), 150 mM NaCl, 1 mM PMSF, 1 mM
Na3VO4 and protease inhibitor cocktail). Protein concentrations
were quantified using the Bio-Rad Protein assay kit II (cat. no.
5000002; Bio-Rad Laboratories, Inc.). Total protein (20
µg/lane) was separated using 10% SDS-PAGE gel, and then
electro-transferred to a nitrocellulose membrane. Membranes were
blocked for 1 h at room temperature in Tris-buffered saline-0.05%
Tween-20 (TBST) containing 5% non-fat dry milk. Subsequently, the
membranes were incubated with primary antibodies overnight at 4°C
The membranes were washed three times with TBS for 10 min, and then
incubated with peroxidase-conjugated secondary antibody at room
temperature for 1 h. Following three additional 10 min washes with
TBST, target proteins were detected using enhanced
chemiluminescence detection reagent (EMD Millipore) and imaged
using the Bio-Rad ChemiDoc XRS+ chemiluminescence imaging system
(Bio-Rad laboratories, Inc.).
Statistical analysis
Student's t-test was used to compare the expression
of IFIT2 in patients and control groups. One-way ANOVA was used to
analyze the expression of IFIT2 in the bone marrow of the patients
following treatment with TKI for different durations. Two-way ANOVA
was used to analyze the differences in proliferation and cell cycle
of the cells following stable expression of IFIT2 and treatment
with DOX. Following ANOVA, Bonferroni's post hoc test was used to
determine significant differences. All data are presented as mean ±
standard deviation. P<0.05 was considered to indicate a
statistically significant difference. Statistical analyses were
performed using GraphPad Prism 6 (GraphPad Software, Inc.).
Results
IFIT2 expression levels are downregulated
in CML patients and upregulated following TKI administration
To determine the mechanism of leukemogenesis in CML,
the present study performed RNA-sequencing analysis of bone marrows
obtained from three primary CML patients and health donors. Gene
expression analysis demonstrated that a number of IFN signal
pathway-related genes were differentially expressed in CML compared
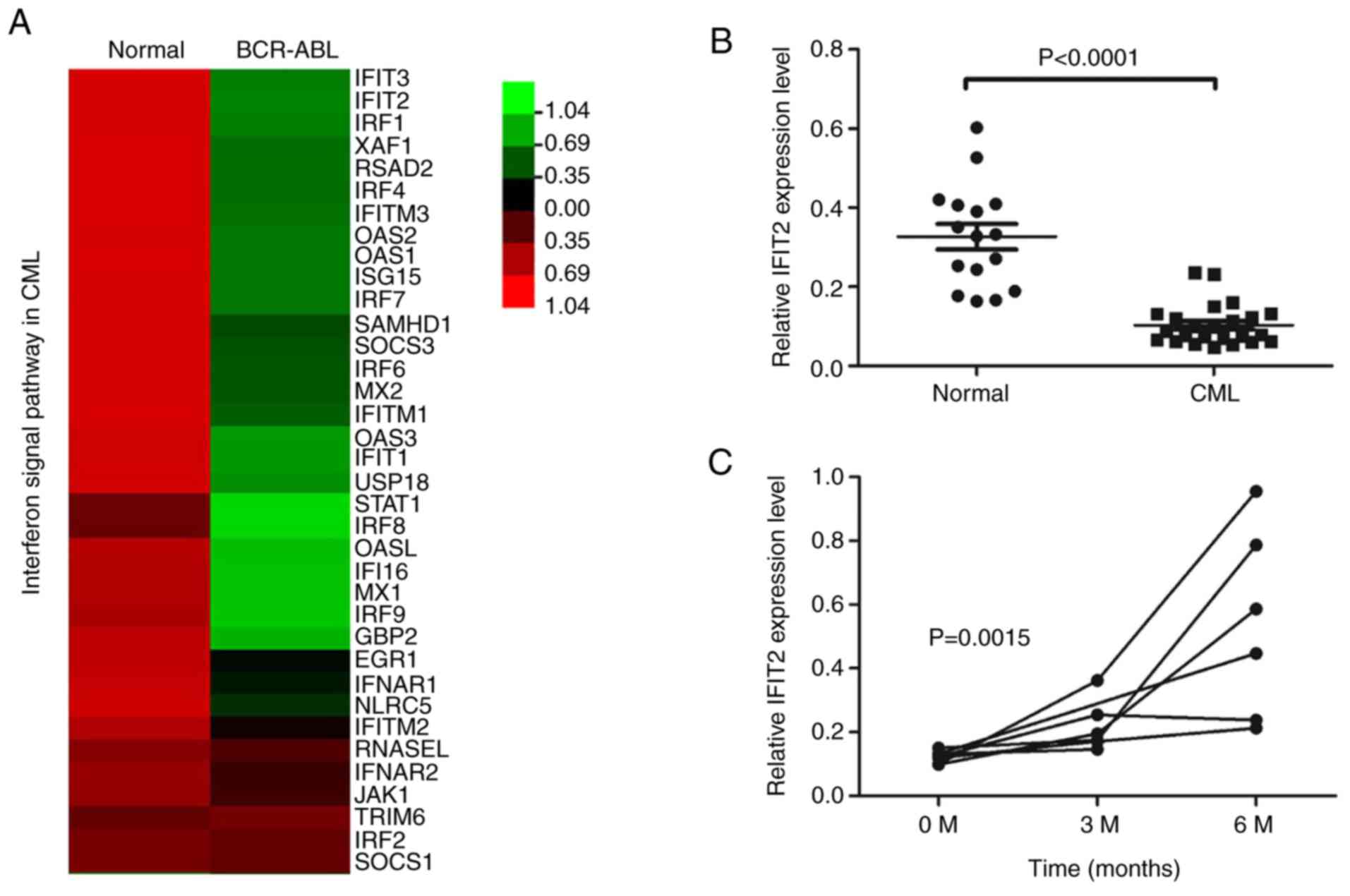
healthy controls. This included several well-known IFN-induced
genes, such as IFIT3, IFIT2, IRF1, IFITM3, OAS2 and OAS1 (Fig. 1A). Among them, IFIT2 mRNA levels
were markedly downregulated in CML patients. To validate these
results, IFIT2 mRNA expression levels were measured in bone marrow
cells from 26 patients with primary CML and 16 healthy controls by
RT-qPCR. As presented in Fig. 1B,
IFIT2 expression levels were significantly lower in CML patients
compared with controls. In addition, IFIT2 expression levels were
measured in CML patients treated with TKI. In 6 patients with CML
who had been treated with TKI for 6 months, IFIT2 expression levels
were higher compared with newly diagnosed patients (Fig. 1C). These results indicated that
IFIT2 levels were decreased in CML patients and upregulated in
patients treated with TKIs.
Stable K562 cell lines expressing
IFIT2
To functionally investigate the role of IFIT2 in
CML, the present study established a stable IFIT2-K562 cell line
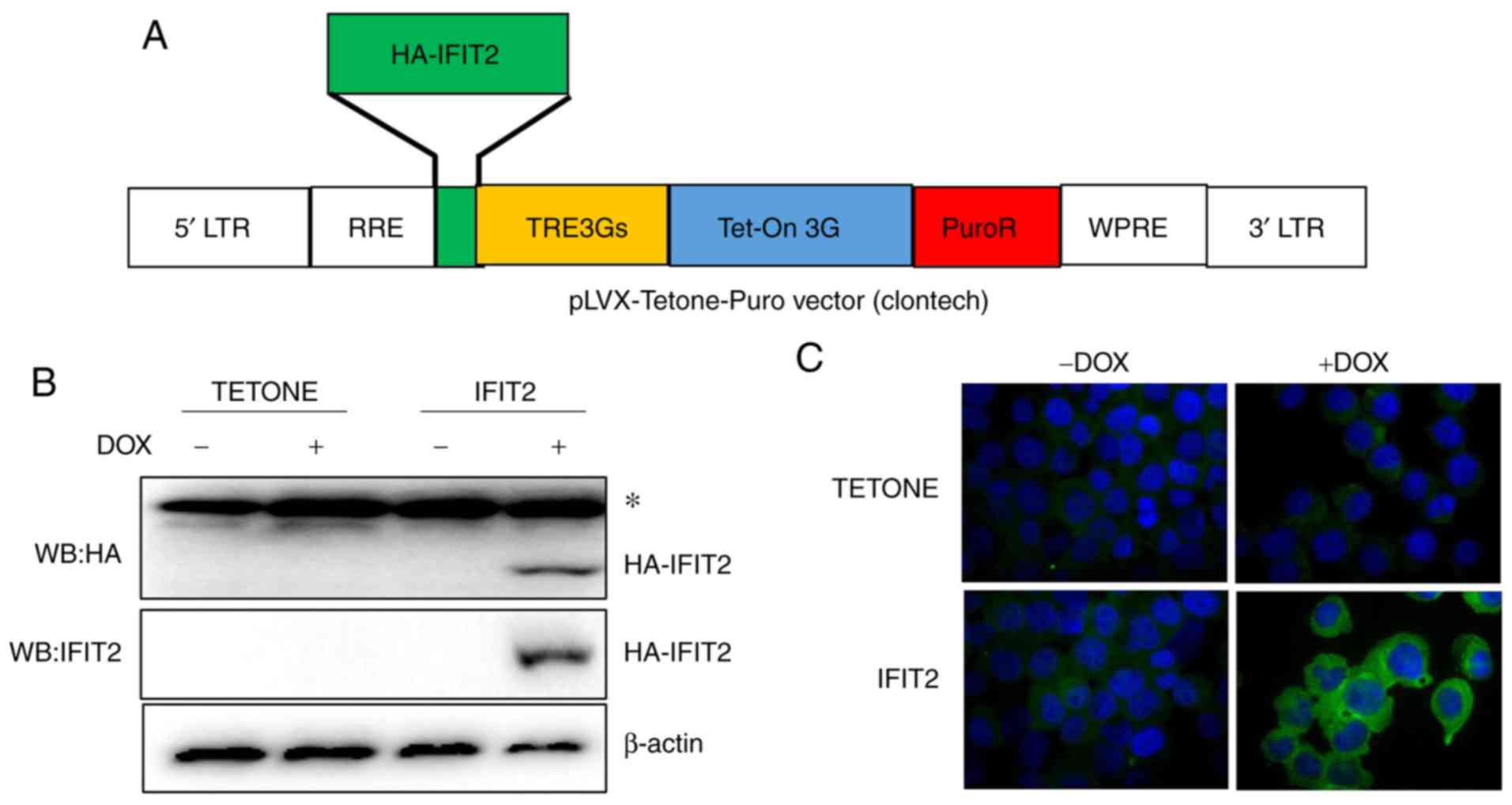
that overexpressed IFIT2 following DOX exposure. HA-IFIT2 ORF was
cloned into pLVX-Tetone-Puro vector with the Tet-On system, which
induced gene expression in the presence of DOX (Fig. 2A). Recombinant lentiviruses were
produced using 293T cells that were subsequently used to infect
K562 cells. Following puromycin selection, western blot and
immunofluorescence assays were performed to confirm IFIT2
expression in the selected stable cell lines. As presented in
Fig. 2B, the HA-IFIT2 protein was
detected using either IFIT2 or HA antibodies in IFIT2-K562 cell
lines after 24 h of DOX exposure (2 µg/ml). IFIT2 was not
detected in the vector control cell lines K562-TETONE (Fig. 2B). Immunofluorescence staining
using IFIT2 antibody also demonstrated increased IFIT2 protein
expression in the cytoplasm of IFIT2-K562 cells following DOX
exposure (Fig. 2C).
IFIT2 inhibits cell proliferation by
inducing G1 phase arrest in K562 cells
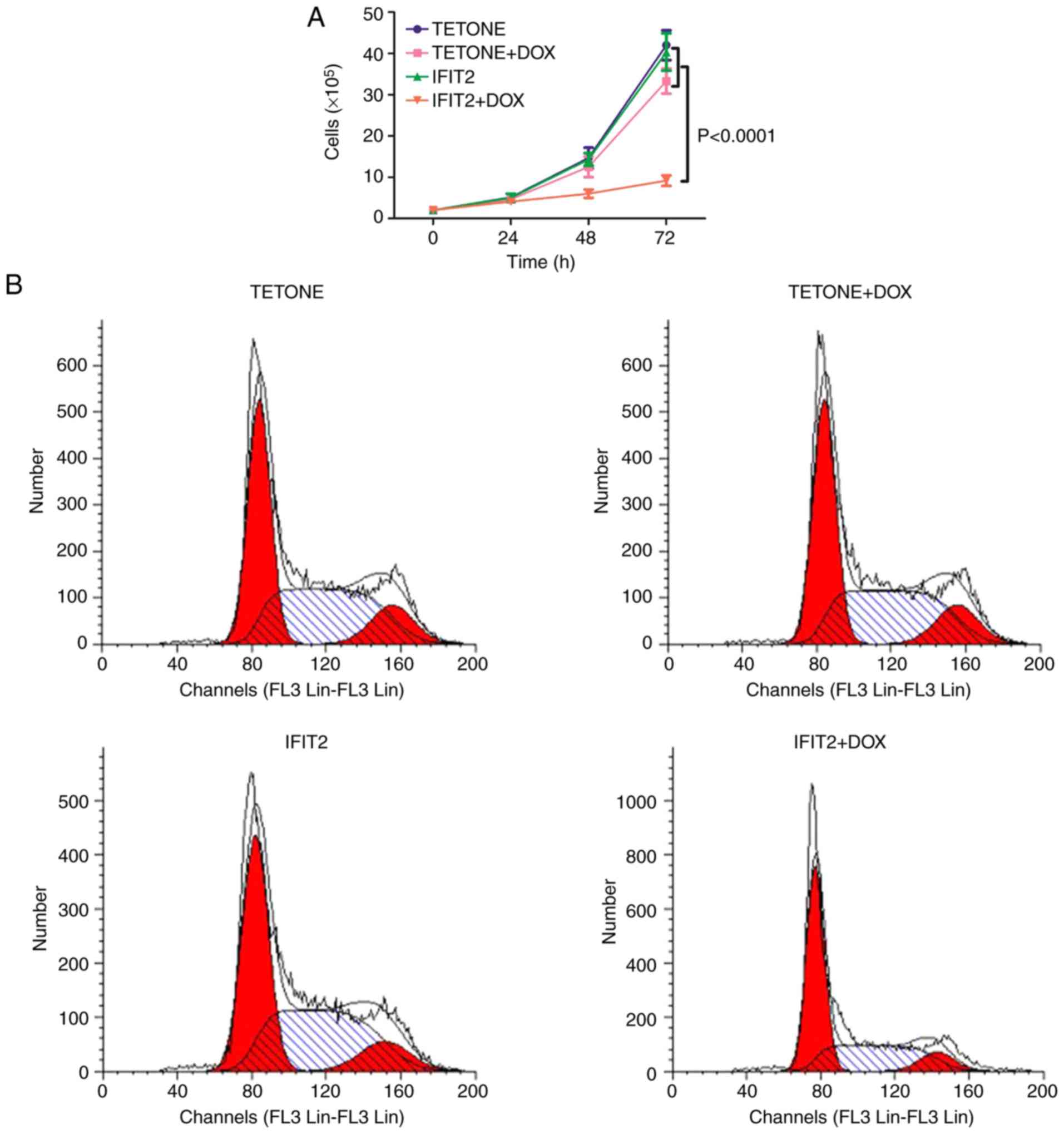
The effect of IFIT2 overexpression on the
proliferation of K562 cells was then measured. The proliferation
rates of the K562-TETONE and K562-IFIT2 cell lines were similar
(Fig. 3A). Following exposure to
2 µg/ml DOX, cell proliferation in K562-TETONE cells was
only slightly reduced due to DOX cytotoxicity; however,
proliferation of K562-IFIT2 cells was significantly reduced at 48
and 72 h compared with K562-TETONE cells treated with DOX
(P<0.0001). Cell cycle analysis demonstrated that IFIT2
overexpression induced growth arrest at the G1 phase in K562 cells.
The two cell lines had a similar cell cycle phase distribution in
the absence of DOX (Fig. 3B and
C). However, treatment of K562-IFIT2 cells with DOX for 48 h
resulted in an obvious accumulation of the cells at G1 transition
phase (p<0.01). Moreover, a significant increase of K562-IFIT2
cells was present in G1 phase following treatment with DOX for 72 h
(P<0.001; Fig. 3D). These
results demonstrated that IFIT2 inhibits cell proliferation and
arrests cells at the G1 phase in CML cells.
IFIT2 reduces BCR-ABL expression and
inhibits phosphorylation of AKT/mTOR
The present study then assessed the effect of IFIT2
on the expression of BCR/ABL fusion protein and its downstream
targets in K562 cells. As presented in Fig. 4, the two cell lines had similar
BCR-ABL expression levels in the absence of DOX treatment. However,
BCR-ABL protein expression was markedly decreased in IFIT2-K562
cells following DOX exposure, while ABL1 expression levels were
unchanged for both stable cell lines. It has been demonstrated that
several signal transduction pathways are activated by BCR/ABL, such
as the JAK/STAT/AKT, PI3K/AKT/mTOR and ERK/MAPK/JNK pathways
(3). These are critical pathways
required for CML cell proliferation. To determine whether IFIT2
affects any of these signaling pathways, western blot analysis was
performed. IFIT2 overexpression markedly reduced the protein levels
of c-Jun and p-c-Jun. However, it had no effect on AKT or mTOR
protein levels, but reduced the phosphorylation levels of p-AKT and
p-mTOR. These results demonstrated that inhibition of cell
proliferation induced by IFIT2 in CML cells was associated with
reduced expression of BCR/ABL and inhibition of the
BCR-ABL/AKT/mTOR signaling pathway.
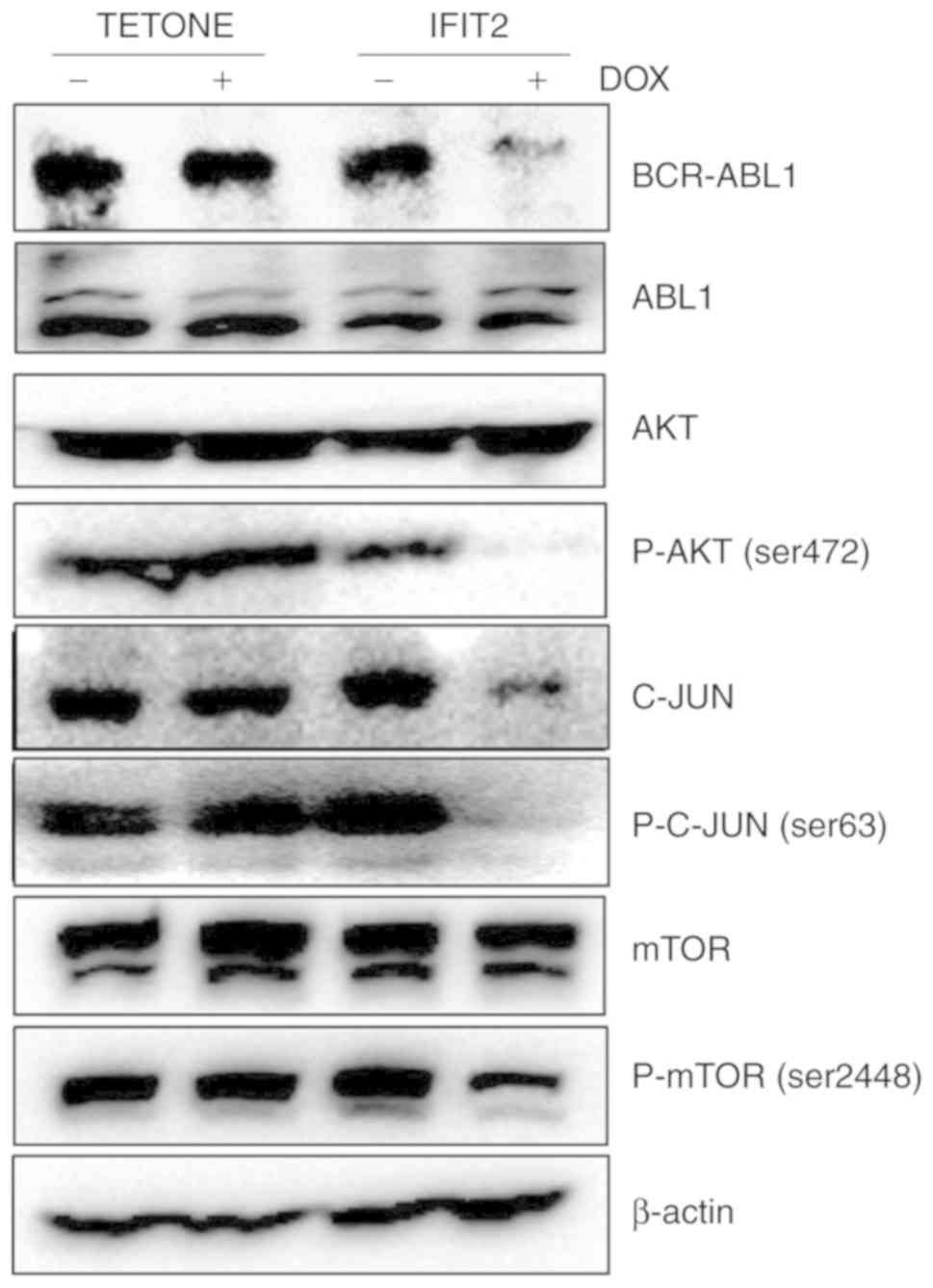 | Figure 4IFIT2 overexpression reduces BCR-ABL
expression and inhibits phosphorylation of mTOR and c-Jun.
K562-TETONE and K562-IFIT2 cells were treated with or without DOX
(2 µg/ml) for 72 h, and then the expression levels of ABL1,
BCR-ABL1, AKT, p-AKT (ser472), c-Jun, p-c-Jun (ser63), mTOR and
p-mTOR were analyzed by western blotting. β-actin expression levels
were used as the loading controls. IFIT2, interferon-induced
protein with tetratricopeptde repeats 2; DOX, doxycycline; p-,
phosphorylated; ABL, ABL proto-oncogene 1, non-receptor tyrosine
kinase. |
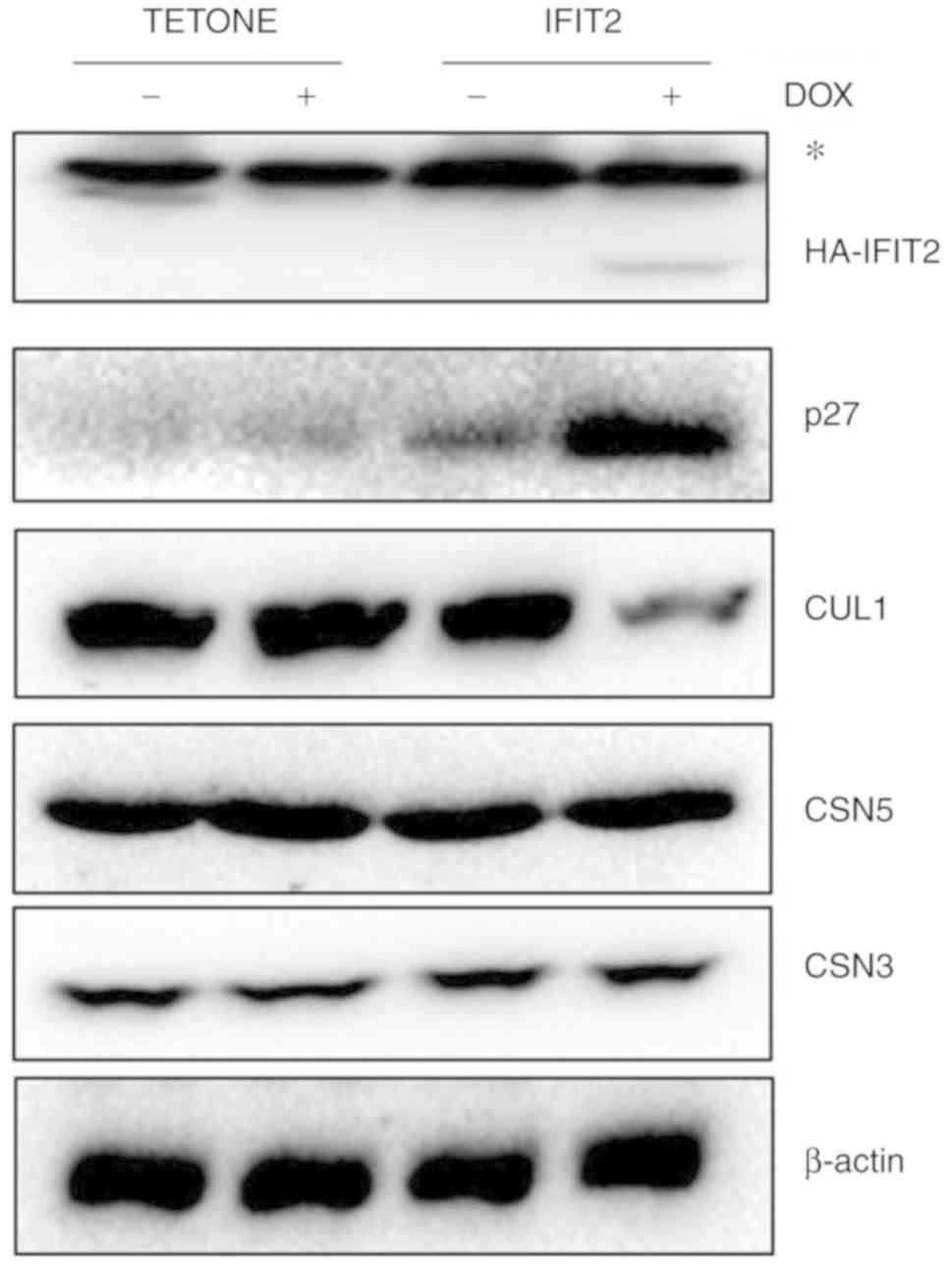
IFIT2 increases p27kip1
expression by inhibiting CRL1-E3 ligase
BCR-ABL inhibits p27kip1 via the MAPK and
PI3K signaling pathways and accelerates abnormal cell proliferation
leading to leukemogenesis (20).
The present study hypothesized that IFIT2 could upregulate
p27kip1 protein expression by inhibiting BCR-ABL. Using
western blot analysis, it was observed that p27kip1
protein levels were markedly increased in K562-IFIT2 cells
following DOX exposure (2 µg/ml). Cell-cycle inhibitor p27
is a known substrate of cullin1-RING ligase (CRL1). CRL1 belongs to
a large family of ubiquitin E3 ligases that regulate protein
degradation via the ubiquitin proteasome system (21). As presented in Fig. 5, CUL1 levels were markedly
decreased in K562-IFIT2 cells following DOX exposure compared with
control cells. However, CSN3 and CSN5 levels were similar between
the two cell lines, in the presence or absence of DOX. These
results demonstrated that IFIT2 promotes the accumulation of
p27kip1 by inhibiting BCR-ABL tyrosine kinase activity
and degrading cullin1-mediated E3 ligases to suppress CML cell
proliferation.
Discussion
In humans, IFITs consist of four members, IFIT1,
IFIT2, IFIT3 and IFIT5. In addition to antiviral activity, IFIT2
has been reported to inhibit tumor cell growth, migration and
metastasis in a variety of tumor types (22-24). Our previous study demonstrated
that curcumin induces cell apoptosis via an IFIT2-dependent
signaling pathway in U937 leukemia cells but not in K562 cells. In
addition, our previous study demonstrated that upregulation of
IFIT2 by exogenous methods or treatment with IFNγ in K562 cells
increases apoptosis and enhances the anticancer effects of curcumin
(25). As the extraction of bone
marrow is an invasive method, it was difficult to collect bone
marrow specimens from healthy volunteers as controls. In the
present study, the expression of IFIT2 in the bone marrow of CML
patients was compared with that in normal controls that were
diagnosed with iron deficiency anemia and immune thrombocytopenia,
and whose bone marrow samples were normal according to the MICM
criteria. It was demonstrated that IFIT2 levels are decreased in
the bone marrow of CML patients, while patients treated with TKI
therapy exhibited increased levels. This suggests that IFIT2 may
play an important role in leukemogenesis and could be a potential
therapeutic target for CML.
The present study established a CML stable cell
line, which was a K562 cell line that stably expressed IFIT2. Using
this stable cell line, it was demonstrated that IFIT2
overexpression inhibited cell proliferation and arrested cells at
the G1 phase. In addition, it was demonstrated that IFIT2 decreased
BCR-ABL expression and inhibited the phosphorylation of AKT, mTOR
and c-JUN. Previous studies have demonstrated that constitutive
tyrosine kinase activity of BCR-ABL contributes significantly to
leukemogenesis in CML. BCR-ABL promotes the survival, proliferation
and adhesion of leukemic cells by regulating downstream pathways.
Multiple intracellular signal transduction pathways, including the
JAK/STAT, ERK/MAPK and PI3K/AKT pathways, have been implicated in
this process. Huang et al (26) demonstrated that EPS8 regulates the
proliferation, apoptosis and chemo-sensitivity of BCR-ABL positive
cells via the BCR-ABL/PI3K/AKT/mTOR pathway. This suggests that
IFIT2 may suppress the proliferation of CML cells by regulating the
BCR-ABL/AKT/mTOR pathway.
The present study also demonstrated that IFIT2
overexpression induced an accumulation of p27kip1
protein and inhibited CRL1-E3 ligase. p27 kip1 is a
potent inhibitor of cyclin-dependent kinases that drives G1 to S
phase transition (27). The
degradation of p27kip1 protein is mainly regulated
through an ubiquitination proteasome dependent pathway associated
with cullin 1-mediated E3 ligases (21). The current results demonstrated
the involvement of cullin 1-mediated E3 ligases in the upregulation
of p27kip1. A previous study has demonstrated that
p27kip1 deficiency increases the HSC-containing
Lin−Sca-1+c-Kit+ cell population
and accelerates leukemogenesis in CML mouse models (28). Therefore, increased
p27kip1 levels may inhibit K562 proliferation. Previous
studies have reported that inhibition of the nuclear import of p27
kip1 by protein kinase B/Akt-mediated phosphorylation is
associated with poor prognosis in numerous breast cancer patients
(29,30). Tomoda et al (20) demonstrated that BCR-ABL tyrosine
kinase facilitates the downregulation of p27 kip1 by
modulating complex formation of Jab1/CSN through the MAPK and PI3K
signaling pathways. The present study found that overexpression of
IFIT2 in K562 cells decreased c-Jun, p-c-Jun and p-AKT levels.
c-Jun, a critical component of the transcription factor AP-1, plays
an important role in cell cycle progression, differentiation and
cell transformation, and is a downstream target of the PI3K/AKT
signaling pathway (31). AP-1
blockade has been shown to arrest the cell cycle by inducing the
expression of p27 in cancer cells (31,32). This suggests that the
PI3K/AKT/c-Jun signaling pathway may be involved in the increased
expression of p27.
In conclusion, the present study demonstrated that
IFIT2 inhibits CML cell proliferation and arrests the cell cycle at
the G1 phase. The underlying mechanism is via regulation of p27 and
inhibition of the BCR-ABL/AKT/mTOR signaling pathway. Therapeutic
targeting of the signaling pathways that modulate IFIT2 expression
may improve clinical outcomes for patients with CML.
Funding
This work was supported by the Natural Science
Foundation of China (grant. nos. 81760539 and 81760381), Natural
Science Foundation of Jiangxi Province (grant. no. 20151BAB205020)
and Science and Technology Plan Project of Jiangxi Provincial
Health planning Commission (grant. no. 20171045).
Availability of data and materials
The datasets generated and/or analyzed during this
study are included in this published article.
Authors' contributions
AL contributed to the study design. ZZ, NL, SL, MJ
and JW preformed the in vitro and in vivo
experiments. YZ, LW and CX performed the data analysis. ZZ, CX and
AL wrote the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Medical Ethics
Committee of the First Affiliated Hospital of Nanchang University
(Nanchang, China). All patients provided written informed
consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Faderl S, Kantarjian HM and Talpaz M:
Chronic myelogenous leukemia: update on biology and treatment.
Oncology (Williston Park). 13:169–180; discussion 181, 184.
1999.
|
|
2
|
Ben-Neriah Y, Daley GQ, Mes-Masson AM,
Witte ON and Baltimore D: The chronic myelogenous leukemia-specific
p210 protein is the product of the bcr/abl hybrid gene. Science.
233:212–214. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ren R: Mechanisms of BCR-ABL in the
pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer.
5:172–183. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Druker BJ, Guilhot F, O'Brien SG, Gathmann
I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM,
Stone RM, et al: Five-year follow-up of patients receiving imatinib
for chronic myeloid leukemia. N Engl J Med. 355:2408–2417. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Santos FP, Kantarjian H, Quintas-Cardama A
and Cortes J: Evolution of therapies for chronic myelogenous
leukemia. Cancer J. 17:465–476. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zabriskie MS, Eide CA, Tantravahi SK,
Vellore NA, Estrada J, Nicolini FE, Khoury HJ, Larson RA, Konopleva
M, Cortes JE, et al: BCR-ABL1 compound mutations combining key
kinase domain positions confer clinical resistance to ponatinib in
Ph chromosome-positive leukemia. Cancer Cell. 26:428–442. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shah NP, Nicoll JM, Nagar B, Gorre ME,
Paquette RL, Kuriyan J and Sawyers CL: Multiple BCR-ABL kinase
domain mutations confer polyclonal resistance to the tyrosine
kinase inhibitor imatinib (STI571) in chronic phase and blast
crisis chronic myeloid leukemia. Cancer Cell. 2:117–125. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Apperley JF: Part I: Mechanisms of
resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol.
8:1018–1029. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Talpaz M, Shah NP, Kantarjian H, Donato N,
Nicoll J, Paquette R, Cortes J, O'Brien S, Nicaise C, Bleickardt E,
et al: Dasatinib in imatinib-resistant Philadelphia
chromosome-positive leukemias. N Engl J Med. 354:2531–2541. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
O'Hare T, Shakespeare WC, Zhu X, Eide CA,
Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, et al:
AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia,
potently inhibits the T315I mutant and overcomes mutation-based
resistance. Cancer Cell. 16:401–412. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Poch Martell M, Sibai H, Deotare U and
Lipton JH: Ponatinib in the therapy of chronic myeloid leukemia.
Expert Rev Hematol. 9:923–932. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Talpaz M, Kantarjian H, Kurzrock R,
Trujillo JM and Gutterman JU: Interferon-alpha produces sustained
cytogenetic responses in chronic myelogenous leukemia. Philadelphia
chromosome-positive patients. Ann Intern Med. 114:532–538. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
El Eit R, Itani AR, Nassar F, Rasbieh N,
Jabbour M, Santina A, Zaatari G, Mahon FX, Bazarbachi A and Nasr R:
Antitumor efficacy of arsenic/interferon in preclinical models of
chronic myeloid leukemia resistant to tyrosine kinase inhibitors.
Cancer. 125:2818–2828. 2019.PubMed/NCBI
|
|
14
|
Zhou X, Michal JJ, Zhang L, Ding B, Lunney
JK, Liu B and Jiang Z: Interferon induced IFIT family genes in host
antiviral defense. Int J Biol Sci. 9:200–208. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stawowczyk M, Van Scoy S, Kumar KP and
Reich NC: The interferon stimulated gene 54 promotes apoptosis. J
Biol Chem. 286:7257–7266. 2011. View Article : Google Scholar :
|
|
16
|
Chen L, Liu S, Xu F, Kong Y, Wan L, Zhang
Y and Zhang Z: Inhibition of proteasome activity induces
aggregation of IFIT2 in the centrosome and enhances IFIT2-induced
cell apoptosis. Int J Biol Sci. 13:383–390. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vardiman JW, Harris NL and Brunning RD:
The world health organization (WHo) classification of the myeloid
neoplasms. Blood. 100:2292–2302. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu S, Wan J, Kong Y, Zhang Y, Wan L and
Zhang Z: Inhibition of CRL-NEDD8 pathway as a new approach to
enhance ATRA-induced differentiation of acute promyelocytic
leukemia cells. Int J Med Sci. 15:674–681. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
20
|
Tomoda K, Kato JY, Tatsumi E, Takahashi T,
Matsuo Y and Yoneda-Kato N: The Jab1/COP9 signalosome subcomplex is
a downstream mediator of Bcr-Abl kinase activity and facilitates
cell-cycle progression. Blood. 105:775–783. 2005. View Article : Google Scholar
|
|
21
|
Morimoto M, Nishida T, Honda R and Yasuda
H: Modification of cullin-1 by ubiquitin-like protein Nedd8
enhances the activity of SCF(skp2) toward p27(kip1). Biochem
Biophys Res Commun. 270:1093–1096. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen L, Zhai W, Zheng X, Xie Q, Zhou Q,
Tao M, Zhu Y, WU C and Jiang J: Decreased IFIT2 expression promotes
gastric cancer progression and predicts poor prognosis of the
patients. Cell Physiol Biochem. 45:15–25. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shen H, Zhan M, Zhang Y, Huang S, Xu S,
Huang X, He M, Yao Y, Man M and Wang J: PLZF inhibits proliferation
and metastasis of gallbladder cancer by regulating IFIT2. Cell
Death Dis. 9:712018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ohsugi T, Yamaguchi K, Zhu C, Ikenoue T
and Furukawa Y: Decreased expression of interferon-induced protein
2 (IFIT2) by Wnt/β-catenin signaling confers anti-apoptotic
properties to colorectal cancer cells. Oncotarget. 8:100176–100186.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Y, Kong Y, Liu S, Zeng L, Wan L and
Zhang Z: Curcumin induces apoptosis in human leukemic cell lines
through an IFIT2-dependent pathway. Cancer Biol Ther. 18:43–50.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang R, Liu H, Chen Y, He Y, Kang Q, Tu
S, He Y, Zhou X, Wang L, yang J, et al: EPS8 regulates
proliferation, apoptosis and chemosensitivity in BCR-ABL positive
cells via the BCR-ABL/PI3K/AKT/mTOR pathway. Oncol Rep. 39:119–128.
2018.
|
|
27
|
Toyoshima H and Hunter T: p27, a novel
inhibitor of g1 cyclin-Cdk protein kinase activity, is related to
p21. Cell. 78:67–74. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang H, Peng C, Hu Y, Li H, Sheng Z, Chen
Y, Sullivan C, Cerny J, Hutchinson L, Higgins A, et al: The Blk
pathway functions as a tumor suppressor in chronic myeloid leukemia
stem cells. Nat Genet. 44:861–871. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakao T, Geddis AE, Fox NE and Kaushansky
K: PI3K/Akt/ FOXO3a pathway contributes to thrombopoietin-induced
proliferation of primary megakaryocytes in vitro and in vivo via
modulation of p27(Kip1). Cell Cycle. 7:257–266. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shin I, Yakes FM, Rojo F, Shin NY, Bakin
AV, Baselga J and Arteaga CL: PKB/Akt mediates cell-cycle
progression by phosphorylation of p27(Kip1) at threonine 157 and
modulation of its cellular localization. Nat Med. 8:1145–1152.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wisdom R, Johnson RS and Moore C: c-Jun
regulates cell cycle progression and apoptosis by distinct
mechanisms. EMBO J. 18:188–197. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Khattar E and Kumar V: Mitogenic
regulation of p27(Kip1) gene is mediated by AP-1 transcription
factors. J Biol Chem. 285:4554–4561. 2010. View Article : Google Scholar
|