Introduction
Lung cancer is the leading cause of
cancer-associated deaths worldwide and has a low 5-year survival
rate after diagnosis (1,2). Non-small cell lung cancer (NSCLC)
accounts for ~85% of all lung cancer cases and lung adenocarcinoma
(LUAD) is the predominant histological subtype of NSCLC, which is
often exhibited by females and people who have never smoked
(3–5). The poor patient survival rate of
NSCLC is primarily due to the high frequency of late diagnosis
(6) and the prognosis of NSCLC
largely depends on the tumor stage. The lung tumor of patients with
NSCLC with pathological stage I disease (early stage) can be
completely removed through surgical resection and these patients,
therefore, have a 5-year survival rate of >70%. However,
mid-late-stage (stages II–IV) lung cancer is difficult and often
impossible to remove completely with surgery, and the 5-year
survival rate for patients with stage II–IV disease ranges from 40
to <10% (7,8). Thus, accurate staging is critical
for NSCLC treatment.
MicroRNAs (miRNAs or miRs) are a class of small (~22
nucleotides), often phylogenetically conserved noncoding RNAs that
are widely expressed and regulate the majority of biological
functions (9). Mammalian miRNA
binding sites are most commonly found in introns or the 3′
untranslated region of mRNAs (10). After miRNAs are cleaved and
activated by the Dicer complex, the activated miRNAs bind to a
complementary sequence in the 3′ untranslated region of the target
mRNAs, which results in decreased gene expression through
translational repression and mRNA destabilization and degradation
(11). Since miRNAs regulate gene
expression through incomplete base pairing, each miRNA has the
ability to regulate multiple genes. Unlike mRNAs, miRNAs are stable
and can be easily detected in archived formalin-fixed
paraffin-embedded specimens (12). Deregulation of miRNA expression
has been linked to the majority of cellular functions, especially
those involved in cancer initiation and progression (13), making miRNAs attractive biomarkers
for the detection, classification, and prognosis of multiple cancer
types (14–16).
Previous studies have attempted to identify miRNA
signatures as potential biomarkers for patients with lung cancer.
Patnaik et al (17)
reported a 6-miRNA-based classifier that could predict the
recurrence of localized stage I NSCLC based on the miRNA expression
profiles of 77 surgically treated pathologic stage I NSCLC cases.
Bishop et al (18)
reported that a miRNA-based method could be used to classify lung
squamous cell carcinoma and LUAD. Li et al (19) identified an 8-miRNA signature as a
potential biomarker for predicting survival in LUAD. Although
several miRNAs have been identified as predictors of clinical
diagnosis or outcome in lung cancer, due to the small patient
number and lack of external validation, the models predicted in
these studies (17–19) might not be reliable. Importantly,
whether miRNAs can be used as pathological staging markers remains
unclear. Therefore, a larger patient cohort and an external
independent validation cohort for investigation of LUAD
staging-specific classifiers are urgently required.
The Cancer Genome Atlas (TCGA) database provides a
collection of clinical data, DNA/RNA sequences and DNA methylation
profiles of ≥500 cases of 20 different tumor types, which is
publicly available (20), while
the Gene Expression Omnibus (GEO) is a public functional genomics
data repository supporting minimum information about a microarray
experiment-compliant data submissions (21). TCGA and GEO contain extensive
genomic data, including miRNA sequencing (miRNAseq) data and
related clinical information of LUAD cases. Yerukala Sathipati and
Ho reported an 18-miRNA signature associated with LUAD patient
survival based on the TCGA-LUAD dataset (22). This study used miRNAseq expression
profiles downloaded from TCGA and GEO to identify the differential
miRNA expression patterns in samples from patients with early and
mid-late pathological stage LUAD. Additionally, a 16-miRNA
signature that could distinguish early-stage LUAD from
mid-late-stage tumor, was constructed. Furthermore, Kyoto
Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO)
analyses were performed to understand the biological pathways
regulated by the prognostic miRNA signature.
Materials and methods
miRNA expression data collection and
preprocessing
The miRNA expression profiles and clinical
information from TCGA-LUAD dataset were downloaded from TCGA
database (https://portal.gdc.cancer.gov/), while raw data from
the GSE62182 (23) and GSE83527
(24) datasets were downloaded
from the GEO database (https://www.ncbi.nlm.nih.gov/geo). The expression
profiles of the 3 miRNA miRNAseq datasets used in the present study
were all generated by an Illumina HiSeq 2000 platform (Illumina,
Inc.). These full clinical datasets were assessed for eligibility
and nontumor samples (45 cases in TCGA-LUAD, 94 cases in GSE62182
and 41 cases in GSE83527), recurrent tumor samples (2 cases in
TCGA-LUAD) and samples lacking staging information (7 cases in
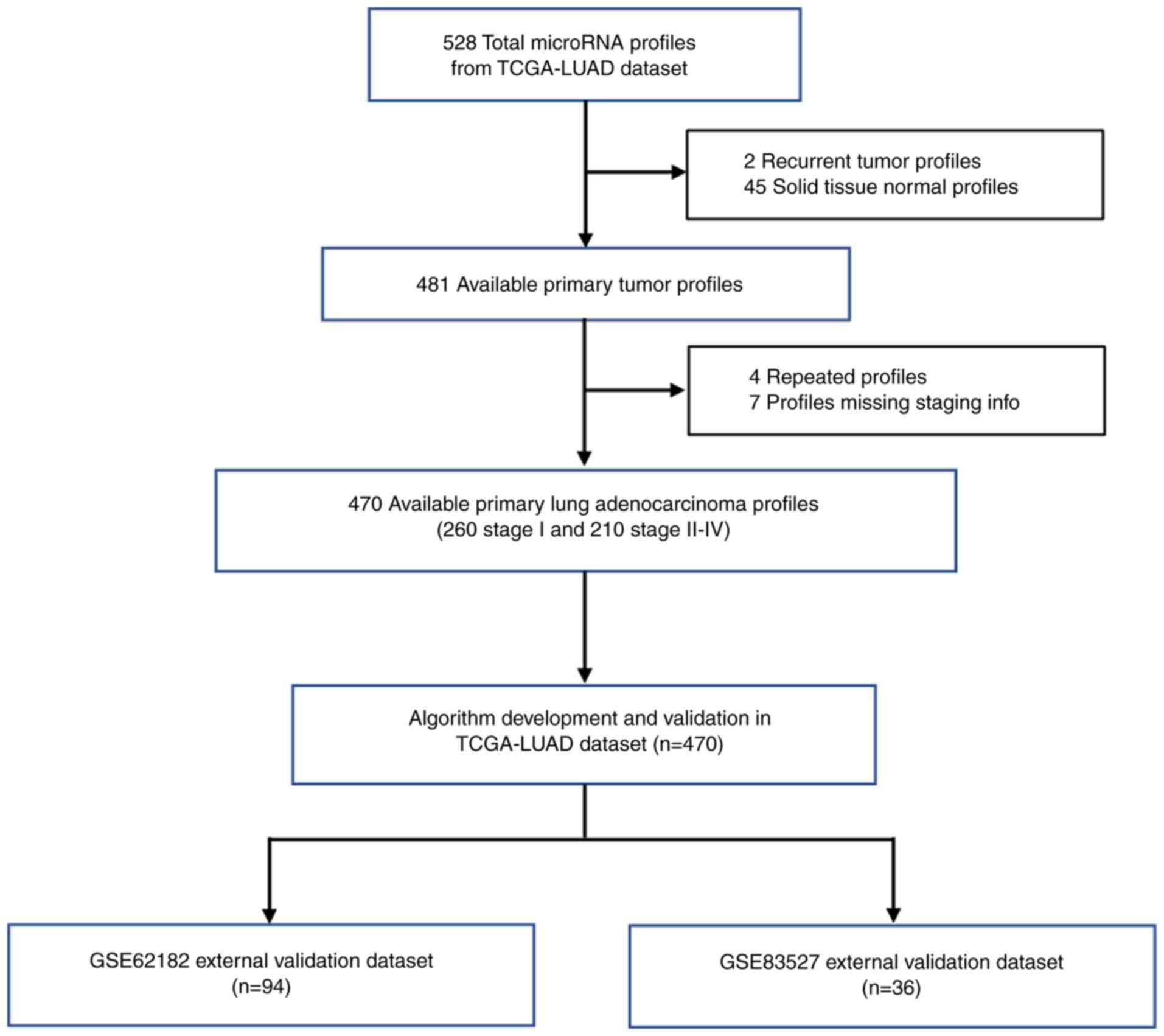
TCGA-LUAD) were removed. There were 470 available primary LUAD
samples with histopathological information in TCGA-LUAD dataset and
94 and 36 available primary LUAD samples in the GSE62182 and
GSE83527 datasets, respectively. Notably, 10 LUAD samples from the
GSE83527 dataset were originally classified using the
Tumor-Node-Metastasis (TNM) staging system and were therefore
reclassified following the 8th edition TNM stage classification
guide developed by the International Association for the Study of
Lung Cancer (25). All the
clinical information of the selected samples is summarized in
Table I.
 | Table IDemographic and histopathological
data of patients from TCGA and GEO databases. |
Table I
Demographic and histopathological
data of patients from TCGA and GEO databases.
| Variable | TCGA-LUAD (n=470)
Training and validation dataset | GSE62182 (n=94)
Test dataset 1 | GSE83527 (n=36)
Test dataset 2 |
|---|
| Sex, n (%) |
| Female | 255 (54.3) | 65 (69.1) | 15 (41.7) |
| Male | 215 (45.7) | 29 (30.9) | 21 (58.3) |
| Smoking, n (%) |
| Current or
former | 320 (68.1) | 67 (71.3) | 33 (91.7) |
| Never | 150 (31.9) | 27 (28.7) | 3 (8.3) |
| Stage, n (%) |
| I | 260 (55.3) | 58 (61.7) | 15 (41.6) |
| II | 109 (23.2) | 23 (24.5) | 15 (41.6) |
| III | 78 (16.6) | 10 (10.6) | 5 (13.9) |
| IV | 23 (4.9) | 3 (3.2) | 1 (2.8) |
The miRNA expression levels were reported as reads
per million miRNA mapped (RPM). Since a number of miRNAs were
differentially expressed by tumor subtype and differences in sample
procurement were observed, the RNA extraction quality, enzymatic
efficiency, and other sources might lead to systematic variability.
Consequently, the accuracy of the methods used for expression
analysis was critically dependent on the proper normalization of
the raw data. An ideal normalizer would be a single miRNA that is
stable and has invariant expression across all samples. In this
study, Homo sapiens (hsa)-miR-191 was used as the normalizer
because it was the most stable single miRNA and has been shown to
have the lowest expression variability in lung cancer tissues
(26). Finally, the
hsa-miR-191-normalized RPM values were used to represent the miRNA
expression levels in TCGA-LUAD, GSE62182 and GSE83527 datasets.
miRNA selection and model
construction
The goal of the miRNA feature selection is to remove
redundant features and to identify the most relevant features,
thereby improving the classification performance for early-stage
and mid-late-stage lung cancer. The expression heatmaps of the
selected miRNAs were generated using HemI v1.0, a toolkit for
illustrating heatmaps (http://hemi.biocuckoo.org). The present group
previously developed a feature selection scheme based on a DX score
and successfully evaluated its effectiveness in a classification
system (27,28). Briefly, the DX score is used to
measure the diversity between positive and negative classes for
each feature. The DX scores can be defined as follows:
DX=(m1−m0)2/d12
+ d02 + σ, where m1
(m0) and d1
(d0) are the mean value and standard deviation of
a feature in a positive (negative) sample, respectively. To avoid a
denominator equal to zero when both classes had constant features,
a small positive number, namely σ, was added. The larger the DX
score, the better the performance of differentiating between the
positive and negative samples by this feature.
Starting with the individual DX scores, the DX score
of each feature was ranked from high to low in order to form a
ranked feature set and its classification performance was evaluated
by 5-fold cross-validation (CV). This procedure yielded a curve of
CV accuracy with several top-ranked miRNAs. The optimized miRNAs
with the best accuracy were identified for later training and
testing.
The predictive model was established using the miRNA
feature selection scheme based on the DX score and a support vector
machine (SVM) classifier. The top-ranked miRNAs with the best
accuracy were identified as the optimized miRNAs that could capture
the subtle difference between early-stage and mid-late-stage lung
cancer. This feature selection scheme was confirmed by 5-fold CV
and area under the curve (AUC) analysis. In 5-fold CV, the dataset
was randomly divided into 5 subsets with ~the same size and each
subset had practically the same number of cases of the two types
(i.e., early-stage and mid-late-stage lung cancer). Then, 4 subsets
were used as the training data and the remaining subset was used to
validate the trained classifier. This process was repeated 5 times
and each subset was used as the validation data once. The accuracy
of the 5-fold CV was defined as the average classification accuracy
over the 5 rounds of validation.
The SVM algorithm (29) was selected for classification
since its superior performance is well established theoretically
and practically. In addition, SVM is a typical supervised machine
learning approach and is employed as a classifier in this
predictive model. For most classification and prediction systems,
SVM is superior to other machine learning methods, including the
neural network and decision tree classifiers (30).
More specifically, a well-established SVM tool,
LIBSVM (31), was selected as the
classifier. The radial basis function (RBF) was employed as the
Kernel function based on various trials. A grid search was also
implemented on the RBF parameter γ and the trade-off coefficient C.
To evaluate the accuracy and robustness of each classifier, a
receiver operating characteristic (ROC) curve for sensitivity and
specificity was calculated. Sensitivity was determined by
TP/(TP+FN), while specificity was computed by TN/(FP+TN), where TP,
FP, FN and TN refer to true positive, false positive, false
negative and true negative, respectively.
GO and KEGG pathway analyses
To investigate the significantly enriched functions
of the differentially expressed genes regulated by the miRNAs and
to better understand the significant pathways in which the
differentially expressed genes were involved, both GO and KEGG
analyses were performed by DIANA-miRPath v3.0 online software
(32). Predicted interactions
between target genes and biological pathways regulated by the
miRNAs were identified using DIANA-TarBase v7.0, enabling an
experimentally supported miRNA functional annotation (33). Two-sided Fisher’s exact test was
used to analyze the significance of the GO category and KEGG
pathway enrichment, and corrected P<0.05 was considered to
indicate a statistically significant difference.
Results
Study design
Although patients with stage I or stage II LUAD
could both be removed by surgery, their 5-year survival rate was
different. According to previous reports, stage I LUAD patients had
a 5-year survival rate of >70%, whereas the 5-year survival rate
of stage II patients was only ~40% (7,8).
Thus, the aim of the present study was to separate stage I from
stage II–IV LUAD, which was based on the 5-year survival rate above
or below 50%. In the present study, miRNA expression profiles and
tumor staging information were obtained from 3 public datasets that
had been sequenced using the same miRNAseq platform (TCGA-LUAD,
GSE62182 and GSE83527). Only LUAD tissues with pathological staging
information were used in this study. TCGA-LUAD dataset originally
contained 528 miRNA profiles of patients with LUAD, but 2 recurrent
tumor profiles, 45 solid tissue normal profiles, 4 repeated
profiles and 7 profiles without staging information were removed
according to the exclusion criteria. To minimize unwanted variation
between different datasets, the miRNA expression levels in each
profile were normalized to that of hsa-miR-191. The miRNA signature
associated with LUAD pathological grade was first trained and
validated through the normalized miRNA expression profiles of the
remaining 470 patients in TCGA-LUAD dataset. Since the miRNA
signature might be overfitted to the training dataset, it was
further evaluated in patients from the GSE62182 and GSE83527
independent datasets to test the robustness of the diagnostic
model. The study flow diagram is shown in Fig. 1.
miRNA selection, model training and
validation
Certain miRNA features were selected based on the
SVM method as described above. The top 50 LUAD staging-related
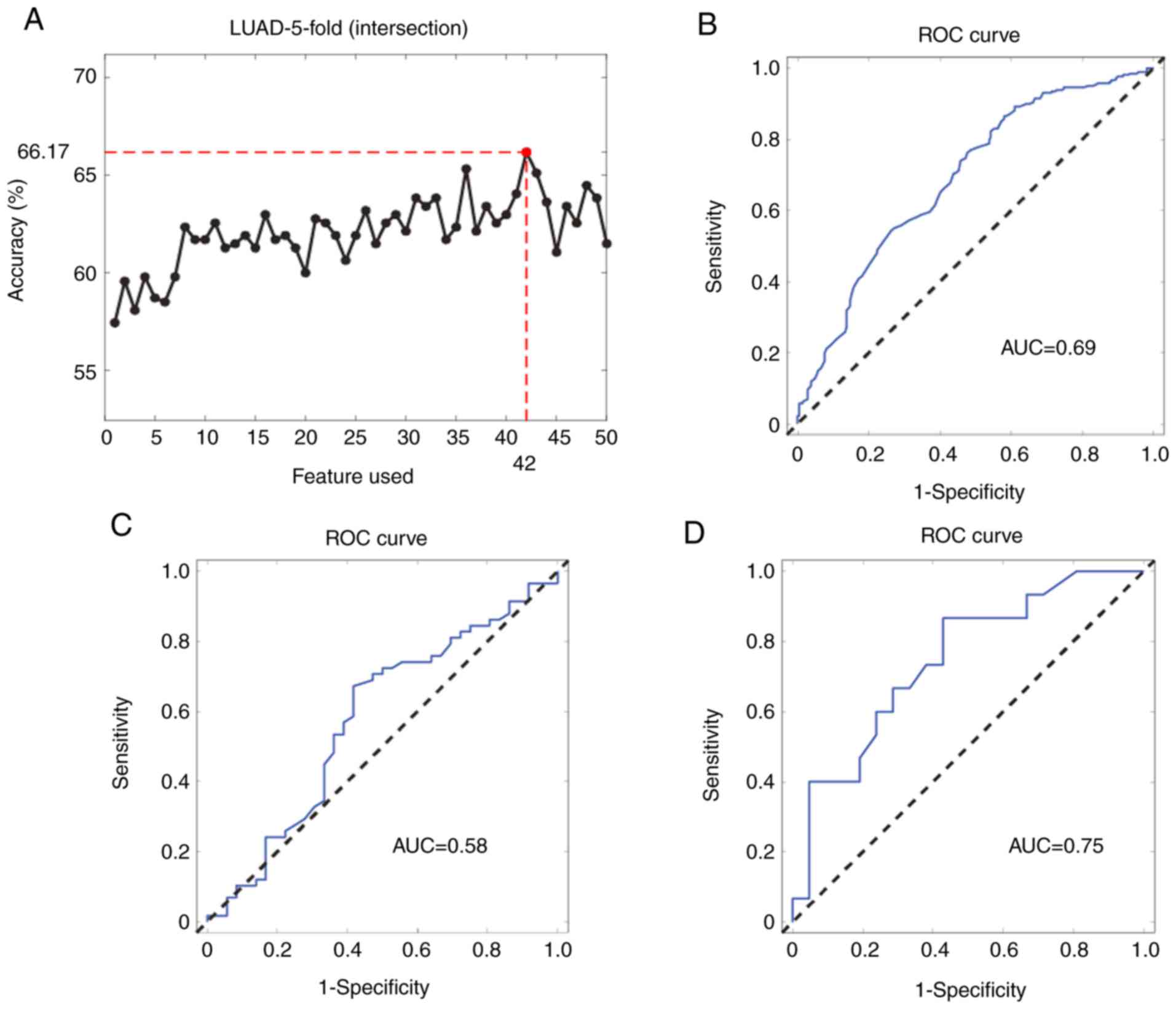
miRNAs were selected and it was observed that the combination of
the expression levels of the top 42 miRNAs produced the best model
for LUAD staging (Fig. 2A). The
AUC was used to evaluate the diagnostic ability of the miRNA
signature. The ROC curve for pathological diagnosis of LUAD was
plotted based on miRNA expression levels and the AUC curve for the
signature comprising the 42 miRNAs in the internal validation
dataset was 0.69 [95% confidence interval (CI): 0.64–0.73]
(Fig. 2B). The 42-miRNA model
showed a similar performance in the other two external validation
datasets [AUC, 0.75 (95% CI: 0.56–0.88) and 0.58 (95% CI:
0.45–0.69), respectively; Fig. 2C and
D].
However, the combination of 42 miRNAs was markedly
complex and could be difficult to use for clinical detection; thus,
the accuracy of the combination of other miRNAs to obtain a more
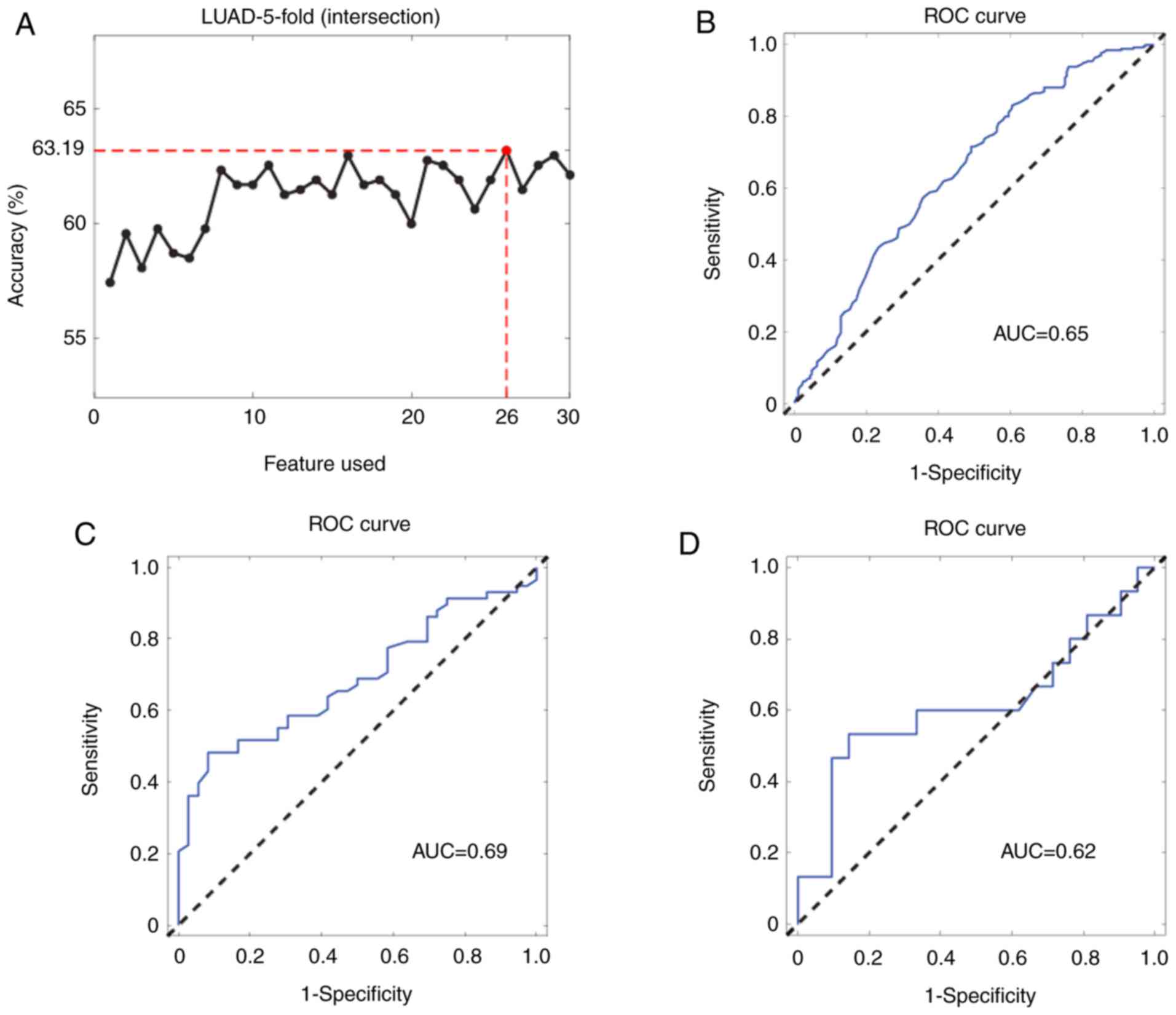
simplified miRNA signature was calculated. After permutation and
combination analyses, the number of miRNAs was first reduced to 26.
The results showed that the 26-miRNA signature had a similar
performance compared to that of all 42 miRNAs (Fig. 3A), with the AUCs in the internal
validation dataset and the two independent external validation
datasets calculated to be 0.65 (95% CI: 0.65–0.69), 0.69 (95% CI:
0.56–0.78) and 0.62 (95% CI: 0.40–0.82), respectively (Fig. 3B–D).
Then, the number of miRNAs was further reduced to
construct an easier and suitable model that could be a potential
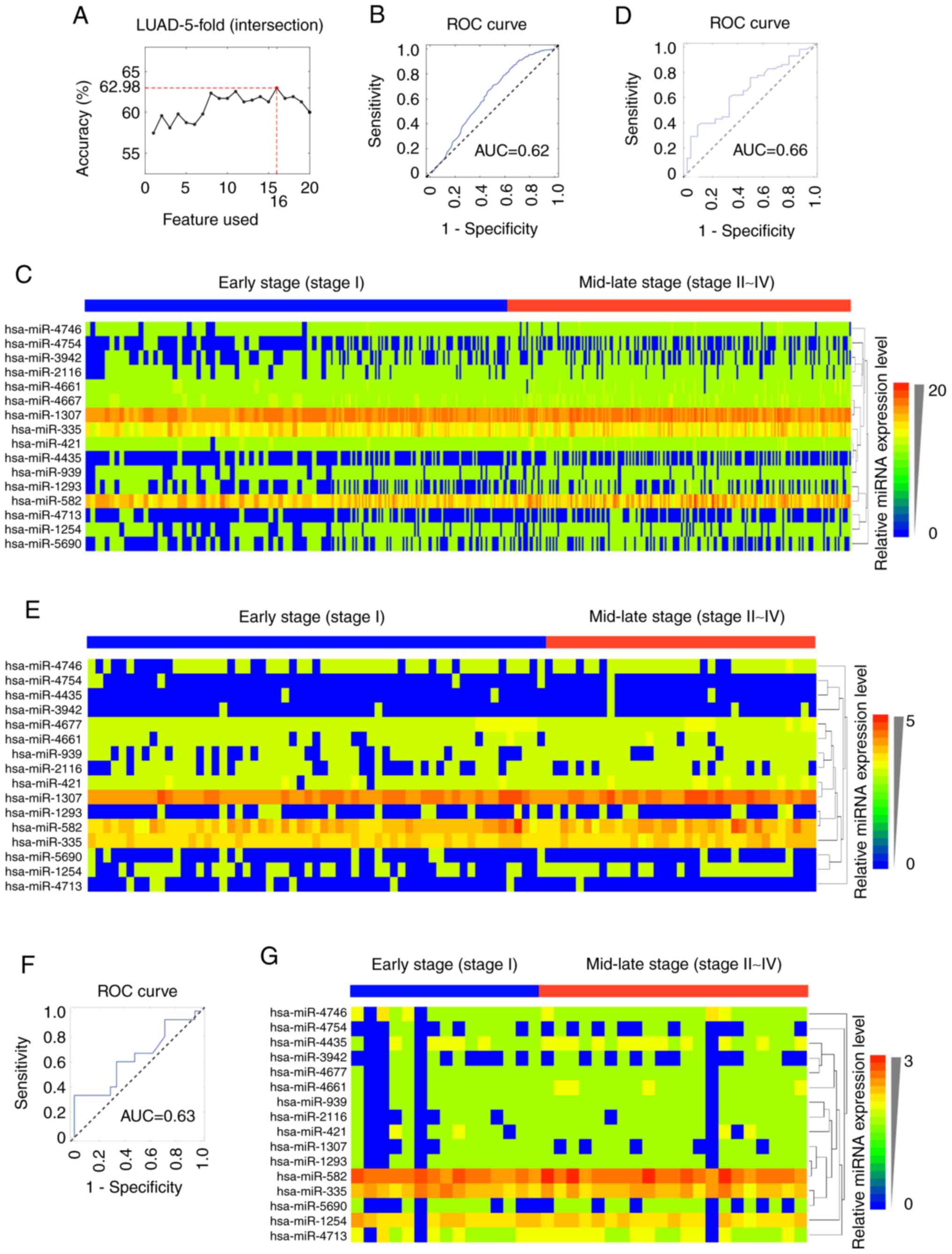
biomarker for the staging of LUAD. It was observed that the
16-miRNA signature (hsa-mir-2116, hsa-mir-4161, hsa-mir-3942,
hsa-mir-4435, hsa-mir-1307, hsa-mir-1254, hsa-mir-582,
hsa-mir-5690, hsa-mir-4713, hsa-mir-1293, hsa-mir-939, hsa-mir-421,
hsa-mir-335, hsa-mir-4677, hsa-mir-4754 and hsa-mir-4746; Table II) showed a similar ability to
classify LUAD pathological stages to that of the combinations of 42
or 26 miRNAs (Fig. 4A). The AUC
for the 16-miRNA signature was 0.62 (95% CI: 0.65–0.67) in the
internal validation dataset (Fig.
4B), 0.66 (95% CI: 0.54–0.76) in the GSE62182 external
validation dataset (Fig. 4D) and
0.63 (95% CI: 0.43–0.82) in the GSE83527 external validation
dataset (Fig. 4F). The expression
heatmaps of the 16 miRNAs in TCGA-LUAD, GSE62182 and GSE83527
datasets were generated using HemI v1.0 software (34) (Fig.
4C, E and G).
| Table IIList of the 16 miRNAs in the
signature. |
Table II
List of the 16 miRNAs in the
signature.
| miRNA ID | miRNA region | Mature miRNA |
|---|
| hsa-mir-2116 | MIMAT0011161 |
hsa-miR-2116-3p |
| hsa-mir-4661 | MIMAT0019729 |
hsa-miR-4661-5p |
| hsa-mir-3942 | MIMAT0018358 |
hsa-miR-3942-5p |
| hsa-mir-4435 | MIMAT0018951 | hsa-miR-4435 |
| hsa-mir-1307 | MIMAT0005951 |
hsa-miR-1307-3p |
| hsa-mir-1254 | MIMAT0005905 | hsa-miR-1254 |
| hsa-mir-582 | MIMAT0004797 | hsa-miR-582-3p |
| hsa-mir-5690 | MIMAT0022482 | hsa-miR-5690 |
| hsa-mir-4713 | MIMAT0019821 |
hsa-miR-4713-3p |
| hsa-mir-1293 | MIMAT0005883 | hsa-miR-1293 |
| hsa-mir-939 | MIMAT0004982 | hsa-miR-939-5p |
| hsa-mir-421 | MIMAT0003339 | hsa-miR-421 |
| hsa-mir-335 | MIMAT0000765 | hsa-miR-335-5p |
| hsa-mir-4677 | MIMAT0019760 |
hsa-miR-4677-5p |
| hsa-mir-4754 | MIMAT0019894 | hsa-miR-4754 |
| hsa-mir-4746 | MIMAT0019880 |
hsa-miR-4746-5p |
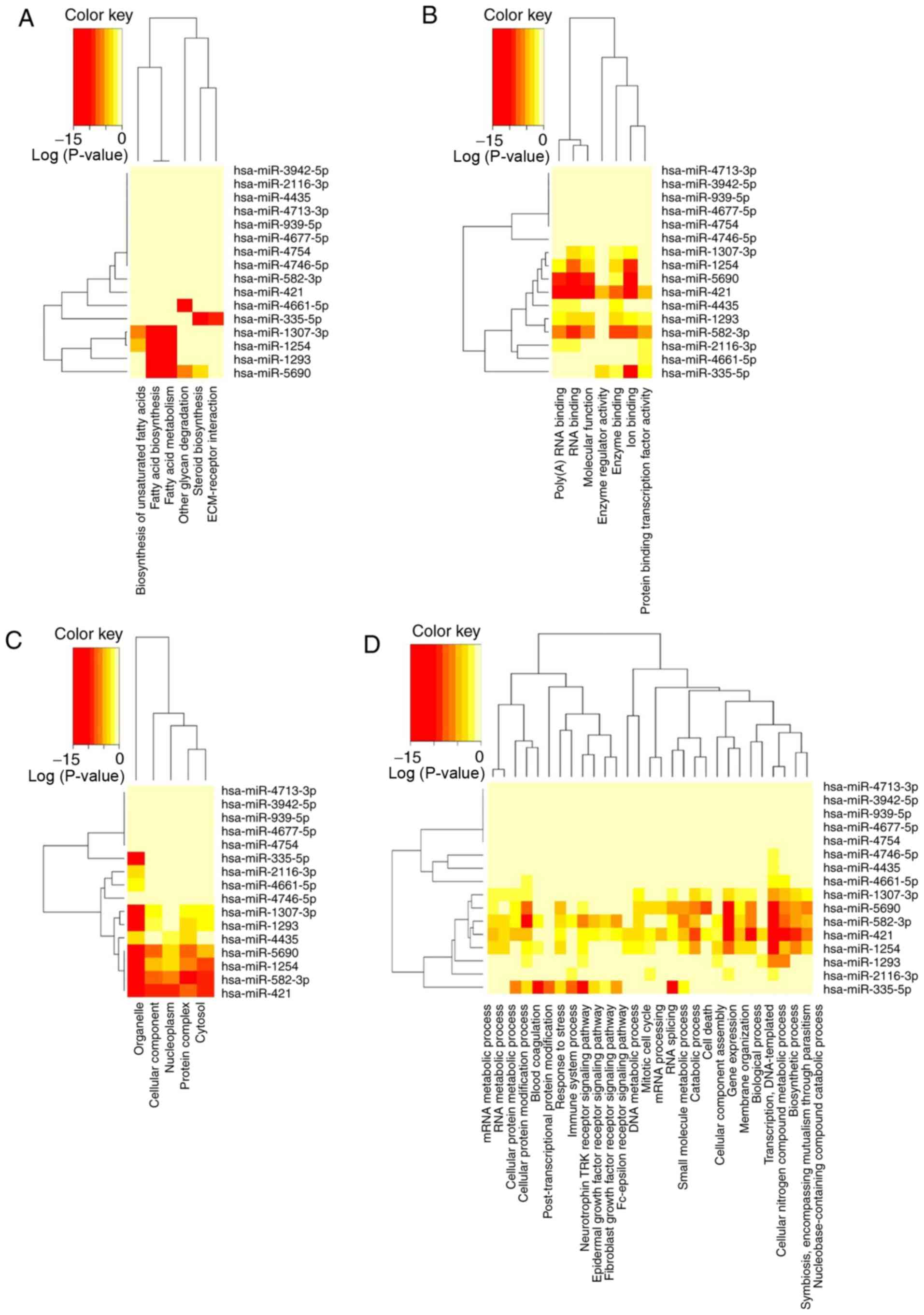
KEGG and GO analyses
Since the underlying molecular biology of different
stages of LUAD is still not very clear, the present study used KEGG
signaling pathway analysis and GO enrichment analysis to better
understand the potential biological function and mechanism of the
16-miRNA signature. By selecting P<0.05 as the cut-off criterium
in the KEGG pathway analysis, several comprehensive biological
pathways regulated by the 16-miRNA signature were revealed in
different stages of LUAD, including ‘fatty acid biosynthesis’
(hsa00061), ‘fatty acid metabolism’ (hsa01212), ‘other glycan
degradation’ (hsa00511), ‘steroid biosynthesis’ (hsa00100),
‘biosynthesis of unsaturated fatty acids’ (hsa01040) and
‘ECM-receptor interaction’ (hsa04512) (Table III), most of which are metabolic
pathways involving biosynthesis, metabolism, degradation of fatty
acids and other glycans. Therefore, these 16 miRNAs might be
involved in fatty acid metabolism, indicating that metabolic
pathways could play an important role in LUAD progression. The
significant categories according to GO results included ‘biological
process’ (Table IV), ‘cellular
component’ (Table V) and
‘molecular function’ (Table VI).
In these categories, various functions associated with RNA
processing, gene expression and multiple catabolic/metabolic
processes were identified. The clustered heatmap analysis of the
enriched KEGG pathways and GO categories was generated by
DIANA-miRPath v3.0 using the default settings (Fig. 5). The results indicated that the
16 miRNA classifiers might participate in the development of LUAD
through the regulation of a series of pathways, particularly
metabolism-related pathways.
| Table IIIEnriched biological pathways
identified in a KEGG pathway analysis. |
Table III
Enriched biological pathways
identified in a KEGG pathway analysis.
| No. | KEGG pathway | P-value | Genes | miRNAs |
|---|
| 1 | Fatty acid
biosynthesis (hsa00061) |
<1x10−325 | 1 | 4 |
| 2 | Fatty acid
metabolism (hsa01212) |
<1x10−325 | 6 | 4 |
| 3 | Other glycan
degradation (hsa00511) |
9.86x10−06 | 3 | 2 |
| 4 | Steroid
biosynthesis (hsa00100) |
6.13x10−05 | 13 | 2 |
| 5 | Biosynthesis of
unsaturated fatty acids (hsa01040) | 0.01643741 | 4 | 2 |
| 6 | ECM-receptor
interaction (hsa04512) | 0.04934272 | 26 | 1 |
| Table IVList of significant GO terms in the
biological process category. |
Table IV
List of significant GO terms in the
biological process category.
| No. | GO Term (Biological
Process) | P-value | Genes | miRNAs |
|---|
| 1 | Biological process
(GO:0008150) |
<1x10−325 | 1,353 | 5 |
| 2 | Symbiosis,
encompassing mutualism through parasitism (GO:0044403) |
<1x10−325 | 94 | 5 |
| 3 | Gene expression
(GO:0010467) |
<1x10−325 | 125 | 6 |
| 4 | Biosynthetic
process (GO:0009058) |
<1x10−325 | 450 | 7 |
| 5 | Cellular protein
modification process (GO:0006464) |
<1x10−325 | 562 | 8 |
| 6 | Cellular nitrogen
compound metabolic process (GO:0034641) |
<1x10−325 | 612 | 10 |
| 7 | Viral process
(GO:0016032) |
1.11x10−16 | 85 | 5 |
| 8 | Small molecule
metabolic process (GO:0044281) |
1.18x10−14 | 552 | 5 |
| 9 | Neurotrophin TRK
receptor signaling pathway (GO:0048011) |
3.90x10−13 | 99 | 6 |
| 10 |
Nucleobase-containing compound catabolic
process (GO:0034655) |
1.04x10−12 | 126 | 5 |
| 11 | Catabolic process
(GO:0009056) |
1.24x10−12 | 446 | 5 |
| 12 | Cellular protein
metabolic process (GO:0044267) |
2.49x10−08 | 131 | 5 |
| 13 | Response to stress
(GO:0006950) |
7.59x10−08 | 511 | 5 |
| 14 | Blood coagulation
(GO:0007596) |
1.66x10−06 | 115 | 3 |
| 15 | Cell death
(GO:0008219) |
1.79x10−06 | 81 | 3 |
| 16 | Mitotic cell cycle
(GO:0000278) |
1.87x10−06 | 60 | 5 |
| 17 | Membrane
organization (GO:0061024) |
1.99x10−06 | 93 | 5 |
| 18 | Fc-epsilon receptor
signaling pathway (GO:0038095) |
5.15x10−06 | 49 | 3 |
| 19 | mRNA metabolic
process (GO:0016071) | 6.09E-05 | 40 | 5 |
| 20 | Epidermal growth
factor receptor signaling pathway (GO:0007173) | 0.000209601 | 59 | 3 |
| 21 | Immune system
process (GO:0002376) | 0.000331715 | 319 | 3 |
| 22 | RNA metabolic
process (GO:0016070) | 0.000428269 | 38 | 4 |
| 23 | Cellular component
assembly (GO:0022607) | 0.000625565 | 124 | 4 |
| 24 | RNA splicing
(GO:0008380) | 0.002480079 | 35 | 2 |
| 25 | mRNA processing
(GO:0006397) | 0.002624066 | 61 | 4 |
| 26 | Activation of
signaling protein activity involved in unfolded protein response
(GO:0006987) | 0.01328594 | 22 | 1 |
| 27 | DNA metabolic
process (GO:0006259) | 0.02118011 | 54 | 2 |
| 28 | Post-translational
protein modification (GO:0043687) | 0.02692964 | 40 | 1 |
| 29 | Transcription,
DNA-templated (GO:0006351) | 0.02971434 | 131 | 2 |
| 30 | Fibroblast growth
factor receptor signaling pathway (GO:0008543) | 0.04102542 | 50 | 3 |
| Table VList of significant GO terms in the
cellular component category. |
Table V
List of significant GO terms in the
cellular component category.
| No. | GO Term (Cellular
Component) | P-value | Genes | miRNAs |
|---|
| 1 | Cellular component
(GO:0005575) |
<1x10−325 | 1,497 | 6 |
| 2 | Cytosol
(GO:0005829) |
<1x10−325 | 336 | 6 |
| 3 | Protein complex
(GO:0043234) |
<1x10−325 | 444 | 7 |
| 4 | Organelle
(GO:0043226) |
<1x10−325 | 2,361 | 10 |
| 5 | Nucleoplasm
(GO:0005654) |
1.99x10−13 | 158 | 5 |
| Table VIList of significant GO terms in the
molecular function category. |
Table VI
List of significant GO terms in the
molecular function category.
| No. | GO Term (Molecular
Function) | P-value | Genes | miRNAs |
|---|
| 1 | Molecular function
(GO:0003674) |
<1x10−325 | 1,491 | 6 |
| 2 | Ion binding
(GO:0043167) |
<1x10−325 | 1,414 | 7 |
| 3 | Poly(A) RNA binding
(GO:0044822) |
<1x10−325 | 242 | 7 |
| 4 | RNA binding
(GO:0003723) |
<1x10−325 | 297 | 8 |
| 5 | Enzyme binding
(GO:0019899) |
3.22x10−15 | 337 | 8 |
| 6 | Protein binding
transcription factor activity (GO:0000988) |
1.03x10−07 | 131 | 6 |
| 7 | Enzyme regulator
activity (GO:0030234) | 0.02094084 | 169 | 2 |
Discussion
Considering that abnormal miRNA expression affects
the molecular functions and biological processes of multiple
tumors, numerous attempts have been made to use miRNAs as
biomarkers for accurate prediction of lung cancer diagnosis and
prognosis (18,22,35). However, most previous studies have
focused on a small patient sample size. Moreover, most reported
lung cancer subtype models do not involve an external confirmation
dataset, therefore weakening the reliability of the results. In
other words, the miRNA signature might correctly classify tumor
status based on one particular dataset but might misclassify other
samples from another dataset that are likely to have a different
group of patients.
In this study, TCGA-LUAD, one of the largest miRNA
expression datasets of LUAD, was selected to establish the current
prediction model. Additionally, miRNA profiles from GEO62182 and
GEO83527 were used as independent external test datasets for
testing the robustness of the present algorithm. The current model
showed stable diagnostic capability, as the model achieved similar
accuracy in different datasets. However, the classification
performance was not very high, probably due to the difficulty of
the specific classification task. Unlike distinguishing squamous
cell carcinoma from adenocarcinoma, where the two histopathological
subtypes arise from different cells with distinct microenvironments
(36), there are often more
similarities than differences between the miRNA expression patterns
of early- and mid-late-stage LUAD. On the other hand, miRNA
expression might only provide limited information for tumor
staging. Multi-omics data, including mRNA expression and CpG
methylation, could lead to multi-omics integration and help to
discover more sensitive molecular features. In liver cancer patient
survival prediction, multi-omics data have shown better performance
than single-omics data for model building (37).
Among the aforementioned 16 miRNAs, the
overexpression of hsa-miR-939 was correlated with poor prognosis in
lung cancer (38), as well as
promoting epithelial to mesenchymal transition in epithelial
ovarian cancer (39). It has been
reported that hsa-miR421 promotes tumor progression in
hepatocellular carcinoma (40)
and osteosarcoma (41), and
hsa-miR-335 participates in the progression of gallbladder
carcinoma (42). On the other
hand, hsa-miR-582 functions as a tumor suppressor in colorectal
cancer (43) and hsa-miR-1254
inhibits cell migration and invasion in gastric cancer (44).
Based on the 16-miRNA signature, KEGG and GO
analyses were employed to predict the target genes and related
pathways, and the results showed that the diagnostic miRNAs
regulate metabolic processes such as glycan degradation, fatty acid
biosynthesis and metabolism, which is consistent with other reports
(45–47). Deregulated metabolism is
considered an important hallmark of cancer initiation, progression,
metastasis and immune evasion (45). miRNAs are involved in the
regulation of cell metabolism, which in turn regulates the
molecular mechanisms driving the Warburg effect in cancer cells,
including glucose uptake, glycolysis, lipid metabolism and amino
acid biogenesis (46). In
addition, changes in the tumor environment at different
pathological stages could alter the cell metabolism in NSCLC
(47). In conclusion, the present
results indicate that dysregulation of fatty acid and glycan
metabolism might be a critical change during the development of
LUAD.
There were several limitations in this study. First,
the current research only focused on LUAD, which means that the
16-miRNA pathological staging signature is not suitable for other
types of lung cancer, such as squamous cell carcinoma or small cell
lung cancer. Second, this study is based on TCGA and GEO public
datasets, which are retrospective; thus, the performance of the
16-miRNA signature needs to be validated in future clinical
studies. Third, the present diagnostic model only used miRNA
expression as single-omics data; thus, incorporating more
molecular-omics data such as mRNA expression, CpG methylation and
genomic information might help to improve the accuracy of the
model.
In conclusion, the present study identified a novel
16-miRNA signature based on a large sample size and multi-source
data, which is promising and effective at predicting the
pathological stages of patients with LUAD. In the authors’ future
studies, multi-omics information should be used to improve the
model.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data used in this study can be downloaded from
TCGA (https://portal.gdc.cancer.gov/) and
GEO (https://www.ncbi.nlm.nih.gov/geo)
databases.
Authors’ contributions
HY and XQ designed the study and wrote the
manuscript. ZY performed data analyses. LS was responsible for
interpretation of the results. XQ and LS provided the resources and
supervised the study. All authors have read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nanavaty P, Alvarez MS and Alberts WM:
Lung cancer screening: Advantages, controversies, and applications.
Cancer Control. 21:9–14. 2014. View Article : Google Scholar
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zappa C and Mousa SA: Non-small cell lung
cancer: Current treatment and future advances. Transl Lung Cancer
Res. 5:288–300. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yano T, Haro A, Shikada Y, Maruyama R and
Maehara Y: Non-small cell lung cancer in never smokers as a
representative ‘non-smoking-associated lung cancer’: Epidemiology
and clinical features. Int J Clin Oncol. 16:287–293. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hsu LH, Chu NM, Liu CC, Tsai SY, You DL,
Ko JS, Lu MC and Feng AC: Sex-associated differences in non-small
cell lung cancer in the new era: Is gender an independent
prognostic factor? Lung Cancer. 66:262–267. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goldstraw P, Chansky K, Crowley J,
Rami-Porta R, Asamura H, Eberhardt WE, Nicholson AG, Groome P,
Mitchell A and Bolejack V; International Association for the Study
of Lung Cancer Staging and Prognostic Factors Committee, Advisory
Boards, and Participating Institutions; International Association
for the Study of Lung Cancer Staging and Prognostic Factors
Committee Advisory Boards and Participating Institutions. The IASLC
lung cancer staging project: Proposals for revision of the TNM
stage groupings in the forthcoming (eighth) edition of the TNM
Classification for lung cancer. J Thorac Oncol. 11:39–51. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rami-Porta R, Crowley JJ and Goldstraw P:
The revised TNM staging system for lung cancer. Ann Thorac
Cardiovasc Surg. 15:4–9. 2009.PubMed/NCBI
|
|
9
|
Oliveto S, Mancino M, Manfrini N and Biffo
S: Role of microRNAs in translation regulation and cancer. World J
Biol Chem. 8:45–56. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shivdasani RA: MicroRNAs: Regulators of
gene expression and cell differentiation. Blood. 108:3646–3653.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Felekkis K, Touvana E, Stefanou CH and
Deltas C: MicroRNAs: A newly described class of encoded molecules
that play a role in health and disease. Hippokratia. 14:236–240.
2010.
|
|
12
|
Xi Y, Nakajima G, Gavin E, Morris CG, Kudo
K, Hayashi K and Ju J: Systematic analysis of microRNA expression
of RNA extracted from fresh frozen and formalin-fixed
paraffin-embedded samples. RNA. 13:1668–1674. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
MacFarlane LA and Murphy PR: MicroRNA:
Biogenesis, function and role in cancer. Curr Genomics. 11:537–561.
2010. View Article : Google Scholar
|
|
14
|
Lan H, Lu H, Wang X and Jin H: MicroRNAs
as potential biomarkers in cancer: Opportunities and challenges.
Biomed Res Int. 2015:125094. 2015. View Article : Google Scholar
|
|
15
|
Paranjape T, Slack FJ and Weidhaas JB:
MicroRNAs: Tools for cancer diagnostics. Gut. 58:1546–1554. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Grady WM and Tewari M: The next thing in
prognostic molecular markers: MicroRNA signatures of cancer. Gut.
59:706–708. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Patnaik SK, Kannisto E, Knudsen S and
Yendamuri S: Evaluation of microRNA expression profiles that may
predict recurrence of localized stage I non-small cell lung cancer
after surgical resection. Cancer Res. 70:36–45. 2010. View Article : Google Scholar
|
|
18
|
Bishop JA, Benjamin H, Cholakh H, Chajut
A, Clark DP and Westra WH: Accurate classification of non-small
cell lung carcinoma using a novel microRNA-based approach. Clin
Cancer Res. 16:610–619. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li X, Shi Y, Yin Z, Xue X and Zhou B: An
eight-miRNA signature as a potential biomarker for predicting
survival in lung adenocarcinoma. J Transl Med. 12:1592014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chandran UR, Medvedeva OP, Barmada MM,
Blood PD, Chakka A, Luthra S, Ferreira A, Wong KF, Lee AV, Zhang Z,
et al: TCGA expedition: A data acquisition and management system
for TCGA data. PLoS One. 11:e01653952016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41:D991–D995. 2013.
View Article : Google Scholar
|
|
22
|
Yerukala Sathipati S and Ho SY:
Identifying the miRNA signature associated with survival time in
patients with lung adenocarcinoma using miRNA expression profiles.
Sci Rep. 7:75072017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vucic EA, Thu KL, Pikor LA, Enfield KS,
Yee J, English JC, MacAulay CE, Lam S, Jurisica I and Lam WL:
Smoking status impacts microRNA mediated prognosis and lung
adenocarcinoma biology. BMC Cancer. 14:7782014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Becker-Santos DD, Thu KL, English JC,
Pikor LA, Martinez VD, Zhang M, Vucic EA, Luk MT, Carraro A,
Korbelik J, et al: Developmental transcription factor NFIB is a
putative target of oncofetal miRNAs and is associated with tumour
aggressiveness in lung adenocarcinoma. J Pathol. 240:161–172. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rami-Porta R, Bolejack V, Giroux DJ,
Chansky K, Crowley J, Asamura H and Goldstraw P; International
Association for the Study of Lung Cancer Staging and Prognostic
Factors Committee, Advisory Board Members and Participating
Institutions. The IASLC lung cancer staging project: The new
database to inform the eighth edition of the TNM classification of
lung cancer. J Thorac Oncol. 9:1618–1624. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Peltier HJ and Latham GJ: Normalization of
microRNA expression levels in quantitative RT-PCR assays:
Identification of suitable reference RNA targets in normal and
cancerous human solid tissues. RNA. 14:844–852. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qian X, Tan H, Zhang J, Zhuang X, Branch
L, Sanger C, Thompson A, Zhao W, Li KC, David L and Zhou X:
Objective classification system for sagittal craniosynostosis based
on suture segmentation. Med Phys. 42:5545–5558. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tan H, Bao J and Zhou X: A novel
missense-mutation-related feature extraction scheme for ‘driver’
mutation identification. Bioinformatics. 28:2948–2955. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cortes C and Vapnik V: Support-Vector
Networks. Mach Learn. 20:273–297. 1995. View Article : Google Scholar
|
|
30
|
You ZH, Yin Z, Han K, Huang DS and Zhou X:
A semi-supervised learning approach to predict synthetic genetic
interactions by combining functional and topological properties of
functional gene network. BMC Bioinformatics. 11:3432010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chang CC and Lin CJ: LIBSVM: A library for
support vector machines. ACM Trans Intell Syst Technol. 2011.
View Article : Google Scholar
|
|
32
|
Vlachos IS, Zagganas K, Paraskevopoulou
MD, Georgakilas G, Karagkouni D, Vergoulis T, Dalamagas T and
Hatzigeorgiou AG: DIANA-miRPath v3.0: Deciphering microRNA function
with experimental support. Nucleic Acids Res. 43:W460–W466. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vlachos IS, Paraskevopoulou MD, Karagkouni
D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos IL,
Maniou S, Karathanou K, Kalfakakou D, et al: DIANA-TarBase v7.0:
Indexing more than half a million experimentally supported
miRNA:mRNA interactions. Nucleic Acids Res. 43:D153–D159. 2015.
View Article : Google Scholar :
|
|
34
|
Deng W, Wang Y, Liu Z, Cheng H and Xue Y:
HemI: A toolkit for illustrating heatmaps. PLoS One. 9:e1119882014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Saito M, Schetter AJ, Mollerup S, Kohno T,
Skaug V, Bowman ED, Mathé EA, Takenoshita S, Yokota J, Haugen A and
Harris CC: The association of microRNA expression with prognosis
and progression in early-stage, non-small cell lung adenocarcinoma:
A retrospective analysis of three cohorts. Clin Cancer Res.
17:1875–1882. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Giangreco A, Groot KR and Janes SM: Lung
cancer and lung stem cells: Strange bedfellows? Am J Respir Crit
Care Med. 175:547–553. 2007. View Article : Google Scholar
|
|
37
|
Chaudhary K, Poirion OB, Lu L and Garmire
LX: Deep learning-based multi-omics integration robustly predicts
survival in liver cancer. Clin Cancer Res. 24:1248–1259. 2018.
View Article : Google Scholar
|
|
38
|
Han X, Du C, Chen Y, Zhong X, Wang F, Wang
J, Liu C, Li M, Chen S and Li B: Overexpression of miR-939-3p
predicts poor prognosis and promotes progression in lung cancer.
Cancer Biomark. 25:325–332. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tang M, Jiang L, Lin Y, Wu X, Wang K, He
Q, Wang X and Li W: Platelet microparticle-mediated transfer of
miR-939 to epithelial ovarian cancer cells promotes epithelial to
mesenchymal transition. Oncotarget. 8:97464–97475. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang W, Li Y, Li X, Liu B, Han S, Li X,
Zhang B, Li J and Sun S: Circular RNA circ-FOXP1 induced by SOX9
promotes hepatocellular carcinoma progression via sponging
miR-875-3p and miR-421. Biomed Pharmacother. 121:1095172020.
View Article : Google Scholar
|
|
41
|
Ren Z, He M, Shen T, Wang K, Meng Q, Chen
X, Zhou L, Han Y, Ji C, Liu S and Fu Q: MiR-421 promotes the
development of osteosarcoma by regulating MCPIP1 expression. Cancer
Biol Ther. 21:231–240. 2020. View Article : Google Scholar
|
|
42
|
Wang W, Chen LC, Qian JY and Zhang Q:
MiR-335 promotes cell proliferation by inhibiting MEF2D and
sensitizes cells to 5-Fu treatment in gallbladder carcinoma. Eur
Rev Med Pharmacol Sci. 23:9829–9839. 2019.PubMed/NCBI
|
|
43
|
Geng Y, Zheng X, Hu W, Wang Q, Xu Y, He W,
Wu C, Zhu D, Wu C and Jiang J: Hsa circ 0009361 acts as the sponge
of miR-582 to suppress colorectal cancer progression by regulating
APC2 expression. Clin Sci (Lond). 133:1197–1213. 2019. View Article : Google Scholar
|
|
44
|
Jiang M, Shi L, Yang C, Ge Y, Lin L, Fan
H, He Y, Zhang D, Miao Y and Yang L: MiR-1254 inhibits cell
proliferation, migration, and invasion by down-regulating Smurf1 in
gastric cancer. Cell Death Dis. 10:322019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lunt SY and Fendt SM: Metabolism-A
cornerstone of cancer initiation, progression, immune evasion and
treatment response. Curr Opin Syst Biol. 8:67–72. 2018. View Article : Google Scholar
|
|
46
|
Chen B, Li H, Zeng X, Yang P, Liu X, Zhao
X and Liang S: Roles of microRNA on cancer cell metabolism. J
Transl Med. 10:2282012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Davidson SM, Papagiannakopoulos T,
Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK,
O’Brien JP, Pierce KA, et al: Environment impacts the metabolic
dependencies of ras-driven non-small cell lung cancer. Cell Metab.
23:517–528. 2016. View Article : Google Scholar : PubMed/NCBI
|