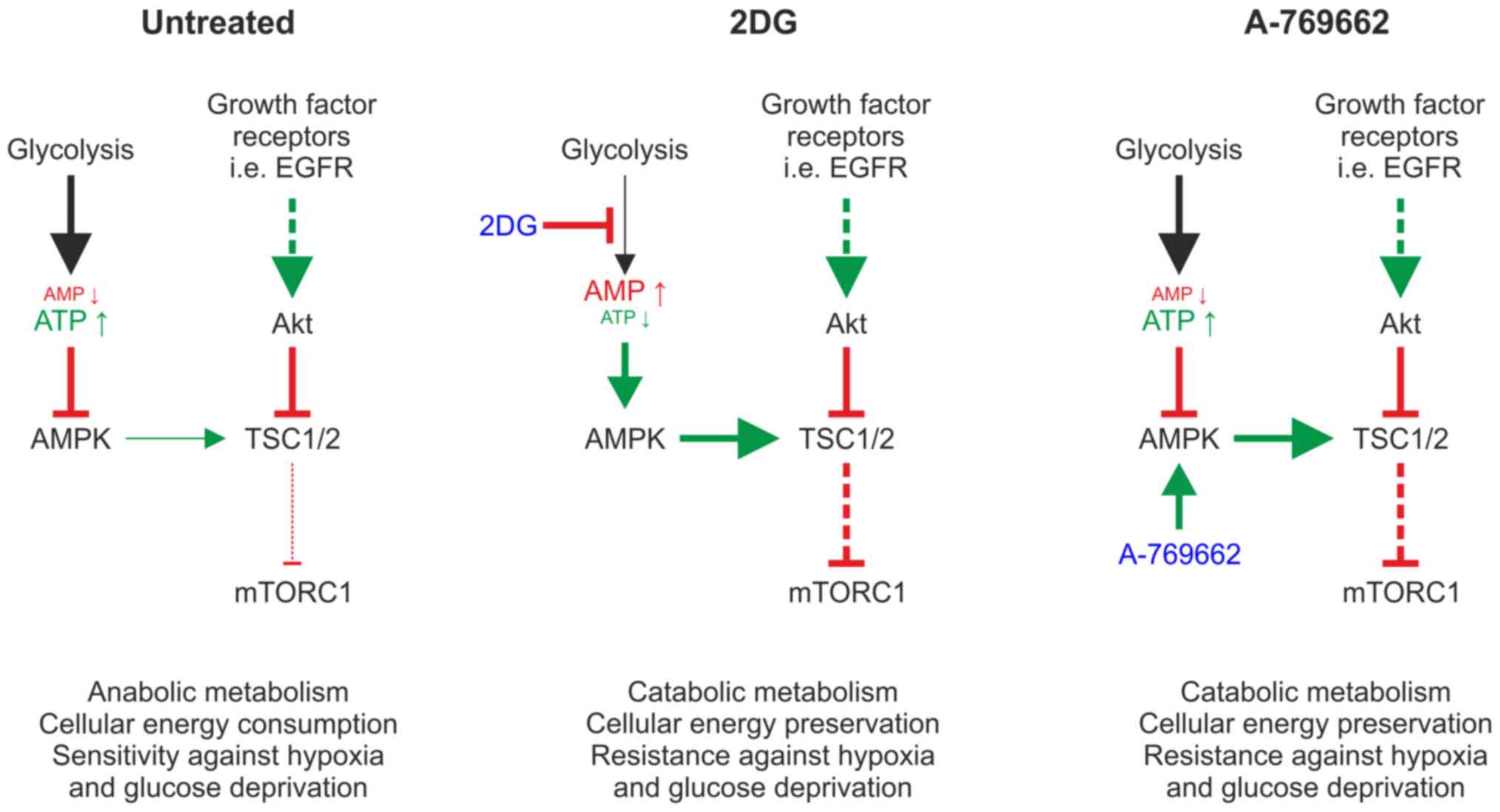
Introduction
Adenosine monophosphate (AMP)-activated protein
kinase (AMPK) is a major cellular energy sensor, which is activated
by an intracellular increase in the AMP/adenosine triphosphate
(ATP) ratio. AMPK activation induces the initiation of adaptive
cellular programs, decreasing ATP consumption by inhibition of
anabolic pathways and increasing ATP production. Subsequently, AMPK
indirectly inhibits the mammalian target of rapamycin (mTOR)
complex 1 (mTORC1) (1), which
integrates signals from growth factor receptors and nutrient
sensors, such as AMPK to adjust cell growth and metabolism. mTORC1
is activated by growth factor signals conveyed by receptor tyrosine
kinases, such as epidermal growth factor receptor (EGFR) (2). Conversely, mTORC1 receives
inhibitory signals when glucose, amino acids, ATP or oxygen are
depleted under starvation conditions. mTORC1 adjusts cellular
energy consumption to the available energy supply via the
regulation of highly energy-intensive anabolic cellular processes,
such as cap-dependent translation and cellular proliferation
(3). A direct AMPK target protein
also highly relevant for cellular energy consumption is the enzyme
acetyl-CoA carboxylase (ACC), which is deactivated via
phosphorylation (1). The
inhibition of ACC activity, as a result of AMPK activation, results
in a decrease in fatty acid synthesis (4). In summary, AMPK activation inhibits
energy-consuming anabolic pathways and activates ATP-generating
catabolic pathways through various targets, including the
inhibition of mTOR and ACC activation, thereby stabilizing cellular
energy homeostasis.
AMPK activity can be stimulated pharmacologically by
direct activators, as well as indirectly by an increase in the
concentration of AMP. A-769662 is a direct activator of AMPK
(5). In glioblastoma (GB) cells,
A-769662 has been shown to decrease glucose consumption and lactate
production (6), and to protect GB
cells from hypoxia-induced cell death, as well as from glucose
deprivation (Lorenz et al, unpublished data). However,
results obtained from neoplastic cells cannot easily be transferred
into non-neoplastic astrocytic cells, as GB cells need to adapt to
specific metabolic challenges in the tumor microenvironment and
commonly rely on aerobic glycolysis for energy homeostasis
(7). This phenotype has not been
described for non-tumor glial cells.
The competitive glycolysis inhibitor,
2-deoxy-d-glucose (2DG), causes a decrease in ATP levels inducing
phosphorylation of AMPK and hence its indirect activation. 2DG
differs from glucose in having a hydrogen instead of a 2’hydroxyl
residue. Due to this structural similarity, 2DG is phosphorylated
by hexokinase and then competitively binds to the glycolytic
enzymes, hexokinase and phosphohexose isomerase. As a consequence,
both aerobic and anaerobic glycolysis are blocked by 2DG (8,9).
Glutamine is metabolized as an alternative to
glucose by conversion to glutamate and α-ketoglutarate (αKG). Under
aerobic conditions, αKG is used in the Krebs cycle to produce ATP.
The metabolization of glutamine to replenish ATP levels is known as
‘glutaminolysis’ (10).
Furthermore, αKG produced from glutamine is used to replace
biosynthetic intermediates from the Krebs cycle needed for the
production of macromolecules, such as fatty acids and nucleic
acids. The replacement of these biosynthetic intermediates
(anaplerosis) permits its continued function and takes place both
under aerobic and anaerobic conditions (11,12).
Neuroprotection by definition seeks to preserve
neuronal structure and function. However, non-neuronal cells, such
as astrocytes play an important neuroprotective role in the
pathophysiology leading to the cell death of neurons during
ischemia (13). The overall goal
of the present study was to evaluate the effects of AMPK activation
on energy preservation and cell protection in astrocytes. This
approach may indirectly be applied for neuroprotective purposes.
Such an intervention could potentially be employed under several
conditions: i) In the case of a cerebral ischemia, the periphery of
the ischemic lesion (penumbra) is typically still supplied with
some blood flow by collateral arteries. However, this blood flow is
not sufficient to maintain neurologic function or even preserve the
tissue affected from ischemic destruction longer than a short time
interval of a few hours (14,15); ii) furthermore, in patients where
a vascular intervention in the carotid arteries or the aortic arch
is planned, pre-conditioning the tissue at risk for a temporal
interruption of the blood circulation through AMPK activation or
mTOR inhibition may be feasible. Similarly, hypothermic perfusion
is used as a feasible and effective neuroprotective approach during
these interventions (16,17).
The role of astrocytes in ischemia and in
neuroprotection has recently received increasing attention
(13), as it has become
increasingly evident that astrocytes play central roles in the
energy homeostasis of the brain, controlling cerebral blood flow
(18,19), the transport of extracellular
water (20), as well as the
uptake, synthesis and secretion of neurotransmitters (21). Previous studies have focused on
neuronal protection, while the role of astrocytes has been
neglected, at least to the best of our knowledge. For example, Xie
et al demonstrated that AMPK activation with A-769662 and
silibinin protected murine cortical neurons and SH-SY5Y
neuroblastoma cells from oxygen-glucose deprivation and
re-oxygenation (22). The effect
of AMPK activation by A-769662 was thus investigated in astrocytic
cells under starvation conditions. As an established model for
astrocytic cells, the SV40-immortalized astrocytic cell line, SVG,
was used (23,24). In the present study, the effects
of direct AMPK activation via A-769662 and glycolysis inhibition
via 2DG were investigated in SVG cells in an established paradigm
of hypoxia-induced cell death.
It was hypothesized that a preventive induction of
energy deprivation-activated signaling pathways via AMPK will
protect astrocytes from permanent hypoxia and glucose
deprivation.
Materials and methods
Cells and cell culture
SV40-immortalized astrocytic SVG cells were
purchased from the American Type Culture Collection (ATCC ; cat.
no. CRL-8621). SVG cells were maintained in a cell culture
incubator (Binder) at 37°C under a 5% CO2 atmosphere.
SVG cells were cultivated in Dulbecco modified Eagle’s minimal
essential medium (DMEM; Life Technologies; Thermo Fisher
Scientific, Inc.) containing 10% fetal calf serum (FCS; Biochrom
KG) supplemented with 100 IU/ml penicillin and 100 μg/ml
streptomycin (Life Technologies; Thermo Fisher Scientific, Inc.).
For the experimental conditions, DMEM glucose-free medium (Life
Technologies; Thermo Fisher Scientific, Inc.) without FCS was
supplemented with glucose to obtain the appropriate concentration.
2DG (Sigma-Aldrich; Merck KGaA) was used at a concentration of 10
mM and A-769662 (R&D Systems, Inc.) at a concentration of 100
μM. As the medium containing 2DG and A-769662 was left on the
cells, these were treated until the read-out of the respective
experiments (for details please see figure legends). Staurosporin
(Sigma-Aldrich; Merck KGaA) was used as a positive control for
poly(ADP-ribose) polymerase (PARP) and caspase-3 cleavage and the
induction of apoptotic cell death at a concentration of 1 μg/ml.
The duration of treatment with staurosporin was 12 h.
SVG cells were seeded and allowed to attach in DMEM
containing 10% FCS overnight under normoxic conditions (21%
oxygen). Thereafter, the medium was, prior to incubating the cells
under hypoxic conditions, replaced with serum-free DMEM containing
the respective glucose concentrations supplemented with 2DG,
A-769662 or the vehicle, dimethyl sulfoxide (DMSO). Hypoxia of 0.1%
oxygen was induced by incubation in BD GasPak pouches (BD
Biosciences) (25).
Cell viability assays
Cell viability was analyzed by propidium iodide (PI)
staining and lactate dehydrogenase (LDH) release assay. For PI
staining, 120,000 cells were seeded in a 24-well plate, and for LDH
assay, 20,000 SVG cells per well were seeded in a 96-well plate.
Cells were examined under a light microscope (Wilovert A, Helmut
Hund GmbH) following approximately 24 h (t1), 48 h (t2) or 96 h
(t3) in a cell culture incubator (Binder) at 37°C under a 5%
CO2 atmosphere. The exact time points of measurements
were fine-tuned to the respective extent of cell death. LDH
activity was determined in the supernatant and after cell lysis.
The ratio of LDH activity in the supernatant and after cell lysis
was calculated as a surrogate marker of cell death as published
earlier (26). LDH release assays
were only performed at the time points of 24 and 48 h of
incubation, as the results of the LDH measurements at the time
point of 96 h with the glucose concentration of 25 mM were
unreliable due to high background enzyme activity. PI uptake was
analyzed by flow cytometry using a BD Canto II flow cytometer and
BD FACS Diva software version 6.1.3 (BD Biosciences), as previously
described (26).
Western blot analysis
For western blot analysis, 3.4×106 SVG
cells were seeded in 10-cm plastic dishes, allowed to attach
overnight and then treated for 1 h with the vehicle, DMSO, 2DG,
A-769662, or staurosporin as indicated in the respective figure
legends. After washing with ice-cold PBS, the cells were scraped
off the dish with a cell scraper and lysed with lysis buffer
comprising 50 mM Tris-HCl, 130 mM NaCl, 5 mM EDTA, 0.5% Nonidet
P-40 and supplemented with 1% Halt® Protease and
Phosphatase Inhibitor (Thermo Fisher Scientific, Inc.). Protein was
quantified with the Bradford protein assay and aliquoted so that
each lane contained 10 μg for the PARP/caspase-3 western blot or 20
μg for all other western blots. The lysates were diluted in Laemmli
buffer and subjected to sodium dodecyl sulfate 12% polyacrylamide
gel electrophoresis. After blotting with 5% skim milk in TBST for 1
h at room temperature, the blotting membranes (nitrocellulose
blotting membrane 0.45 μm, cat. no. 10600016; GE Healthcare Life
Sciences) were incubated for 12 h at 7°C with antibodies diluted as
indicated: Rabbit anti-PARP (cat. no. 9542), rabbit anti-cleaved
caspase-3 Asp175 (cat. no. 9664), rabbit anti-p-ACC Ser79 (cat. no.
3661), rabbit anti-phospho-AMPKα Thr172 (cat. no. 2531), rabbit
Pathscan Multiplex Western Cocktail I (cat. no. 5301; containing
antibodies against p-p90RSK, p-Akt, p-p42/44, MAPK, p-RPS6 and
Rab11) (all diluted 1:1,000; all from Cell Signaling Technology) or
goat anti-actin (cat. no. sc-1616, diluted 1:2,000; Santa Cruz
Biotechnology, Inc.). Secondary goat anti-rabbit antibody and
donkey anti-goat antibodies were used respectively (diluted
1:5,000, cat. no. sc-2004 and cat. no. sc-2020; Santa Cruz
Biotechnology, Inc.). The secondary antibodies were incubated for 1
h at room temperature (21°C). For detection, a chemiluminescence
solution composed of 1 ml solution A (50 mg luminol in 200 ml 0.1 M
Tris-HCl, pH 8.6), 100 μl solution B (11 mg p-hydroxy-coumaric acid
in 10 ml DMSO) and 0.3 μl H2O2 (30%) was
added. The quantification of the western blot bands was performed
by measuring the pixel density of the scanned films using ImageJ
1.51f software (NIH). All western blot analyses were performed at
least 3 times, and a representative blot was selected for
presentation and quantification.
Measurement of glucose, lactate and ATP
concentrations
Glucose and lactate concentrations were measured in
cell-free supernatants with a biochemistry analyzer Hitachy 917
(Roche). For the analysis of oxygen consumption, cultured cells
were overlaid with paraffin oil and oxygen concentrations were
measured every 15 min utilizing a fluorescence-based assay with a
sensor at the bottom of each well (OxoDish, PreSens), as previously
described (27).
For ATP assays, plates (96-well plates with 20,000
SVG cells per well) were placed on ice immediately following
incubation at 37°C under normoxic or hypoxic (0.1% O2)
conditions for 6 h. The SVG cells were pelleted by centrifugation
and lysed with ATP releasing agent (Sigma-Aldrich; Merck KGaA). The
ATP concentration was then measured with a luciferase-based CLS II
kit (Boehringer), as previously described (25).
Statistical analysis
Data are presented as the mean values in the
figures, ± the standard deviation, which were calculated with Excel
version 2010 (Microsoft, Inc.). Glucose and lactate concentrations
were measured from pooled supernatant of 5 wells. For western blot
analysis quantifications, the intensities of an exemplary blot were
normalized to actin expression. For all other experiments,
statistical analyses were performed with GraphPad Prism 7 (GraphPad
Software, Inc.). Values of P≥0.05, P<0.05, P<0.01 and
P<0.001 were regarded as not significant (n.s.), significant,
highly significant and extremely significant, respectively. For
oxygen consumption analyses, a non-linear regression analysis
followed by ANOVA and the Tukey’s multiple comparison test was
used. For all other statistical calculations, ANOVA and Dunnett’s
multiple comparison test was used.
Results
Inhibition of glycolysis and activation
of AMPK protects SVG cells from hypoxia-induced cell death in the
absence of glutamine
Glucose and oxygen concentrations are two decisive
determinants in cerebral ischemic lesions. Therefore, the present
study first employed an established paradigm (26–28) of severe hypoxia with 0.1% oxygen,
varied glucose concentrations and complete glutamine deprivation.
Cell death was measured by flow cytometry [fluorescence-activated
cell sorting (FACS)] as a percentage of PI-positive cells (for
exemplary PI FACS plots please see Fig. S1). A partial glucose restriction
with a glucose concentration of 2 mM, a roughly normoglycemic
concentration of 5 mM, as well as a highly supraphysiological
glucose concentration of 25 mM were tested. Both glycolysis
inhibition with 2DG and direct AMPK activation with A-769662
protected the SVG cells from hypoxia-induced cell death (Fig. 1A, C and E). This effect was most
pronounced at glucose concentrations of 2 and 5 mM, although it was
still detectable at the supraphysiological high glucose
concentration of 25 mM. The earlier time points were selected for
the experiments with lower glucose concentrations to adjust for the
earlier induction of cell death under these lower glucose
concentrations. 2DG had a toxic effect at a glucose concentration
of 5 mM, but not at 2 or 25 mM (Fig.
1B, D and F). A-769662 protected the SVG cells from glutamine
deprivation at glucose concentrations of 5 and 25 mM under normoxic
conditions (Fig. 1B, D and
F).
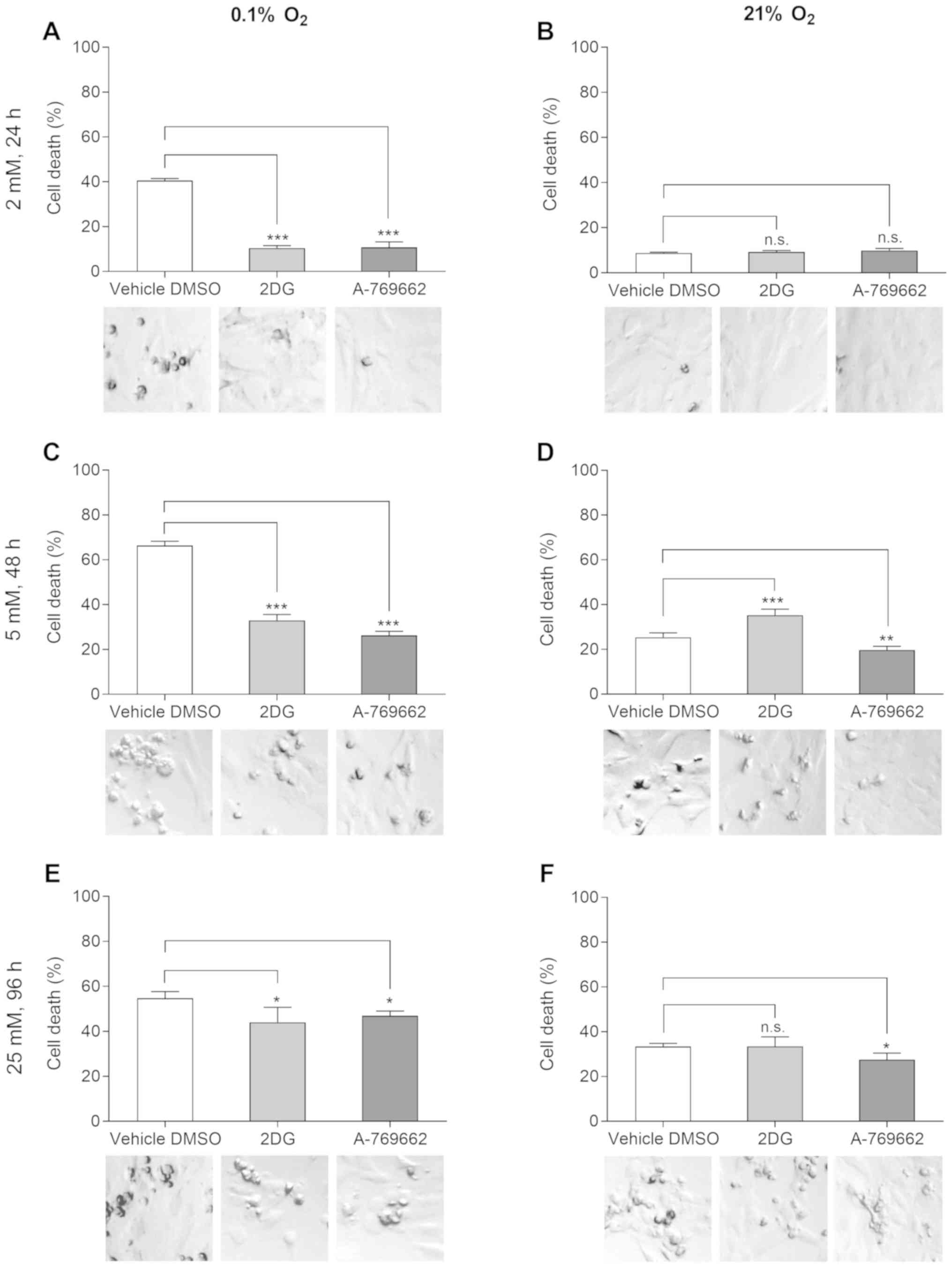 | Figure 1Inhibition of glycolysis and
activation of AMPK protects SVG cells from hypoxia-induced cell
death in the absence of glutamine. Effects of 2DG and A-769662 on
cell viability under hypoxic (0.1% O2) and normoxic (21%
O2) conditions were measured with PI-FACS. SVG cells
were exposed to serum-free DMEM with 2, 5 or 25 mM glucose, vehicle
DMSO, 2DG or A-769662 and hypoxia or normoxia. PI-positive cells
are presented as percentage of the total cell count. Exposure to 2
mM glucose and (A) hypoxia or (B) normoxia, 24 h treatment.
Exposure to 5 mM glucose and (C) hypoxia or (D) normoxia, 48 h
treatment. Exposure to 25 mM glucose and (E) hypoxia or (F)
normoxia, 96 h treatment [n=4; n.s., not significant (P≥0.05);
*P<0.05, **P<0.01,
***P<0.001, ANOVA with Dunnett’s multiple comparisons
test]. Light microscopic images (original magnification, ×40; scale
bar, 25 μm) of the SVG cells are presented in the photo inlays.
2DG, 2-deoxy-d-glucose. |
The protective effects of 2DG and A-769662 under
hypoxic conditions were also confirmed by light microscopic
imaging. Cells treated with the vehicle, DMSO, detached more
rapidly and commonly displayed condensation and pyknosis under
hypoxic conditions, as compared to the cells treated with 2DG and
A-769662 (Fig. 1, photo inlays).
Western blot analysis detected an increased caspase-3 and PARP
cleavage (cCaspase 3 and cPARP) in the SVG cells treated with the
vehicle (DMSO) as compared to the cells treated with 2DG or
A-769662, indicating an apoptotic cell death mechanism (Fig. S2).
Inhibition of glycolysis and activation
of AMPK protects SVG cells from hypoxia-induced cell death in the
presence of glutamine
To examine the possibility that glutamine may be
metabolized to replenish ATP levels and to thus protect the cells
from glucose deprivation, the experiments described above were
repeated in the presence of glutamine. Comparable to the results
obtained with glutamine withdrawal, 2DG and A-769662 protected the
SVG cells from hypoxia (Fig. S3A and
C) at glucose concentrations of 2 and 5 mM, and glutamine
concentrations of 4 mM. Under the supraphysiologically high glucose
concentration of 25 mM and a glutamine concentration of 4 mM, 2DG
did not protect the SVG cells from hypoxia; however, A-769662 still
had a protective effect (Fig.
S3E). Under normoxic conditions and glucose concentrations of 5
mM, 2DG and A-769662 exerted a toxic effect, while at a glucose
concentration of 25 mM, only A-769662 was toxic (Fig. S3B, D and F). In summary, glutamine
was unable to compensate for the withdrawal of glucose under these
conditions.
Effect of 2DG and A-769662 on cell
viability under hypoxic conditions
The results obtained by PI staining were confirmed
by LDH release assays. The protective effect of 2DG and A-769662
was verified under hypoxic conditions supplemented with 2 and 5 mM
glucose without the addition of glutamine, as evidenced by a
reduced cellular LDH release (Fig.
S4).
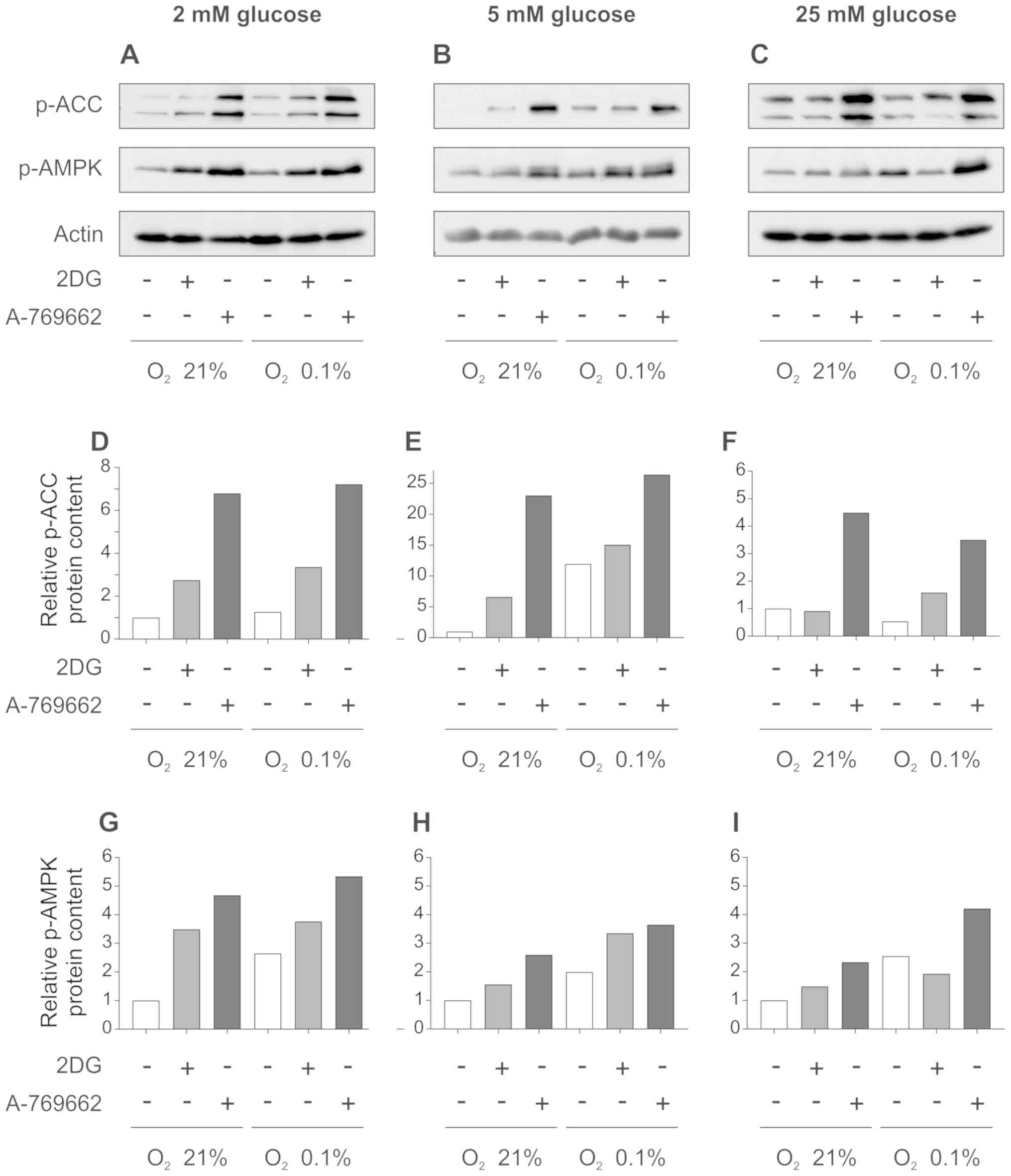
Effect of 2DG and A-769662 on AMPK and
the target enzyme ACC
To examine the effect of A-769662 and 2DG on
metabolic pathways critical for fatty acid biosynthesis in
astrocytes, western blot analysis was performed for p-ACC (Ser79),
as a readout of its enzymatic activity. Fatty acid biosynthesis is
highly energy-intensive and, alongside protein biosynthesis,
consumes a large portion of the ATP produced by the cell. Treatment
of the SVG cells with 2DG resulted in ACC phosphorylation and
therefore, in deactivation under normoxic and hypoxic conditions
with glucose concentrations of 2 and 5 mM (Fig. 2A and B, and quantification of
intensities in D and E). At 25 mM glucose, 2DG increased ACC
phosphorylation only under hypoxic, but not under normoxic
conditions (Fig. 2C, and
quantification of intensities in F). 2DG increased the
phosphorylation and therefore, the activation of AMPK both under
normoxic and hypoxic conditions at glucose concentrations of 2 and
5 mM (Fig. 2A and B, and
quantification of intensities in G and H). In medium supplemented
with 25 mM glucose, there was a slight increase in AMPK
phosphorylation under normoxic, but not under hypoxic conditions
(Fig. 2C, and quantification of
intensities in I).
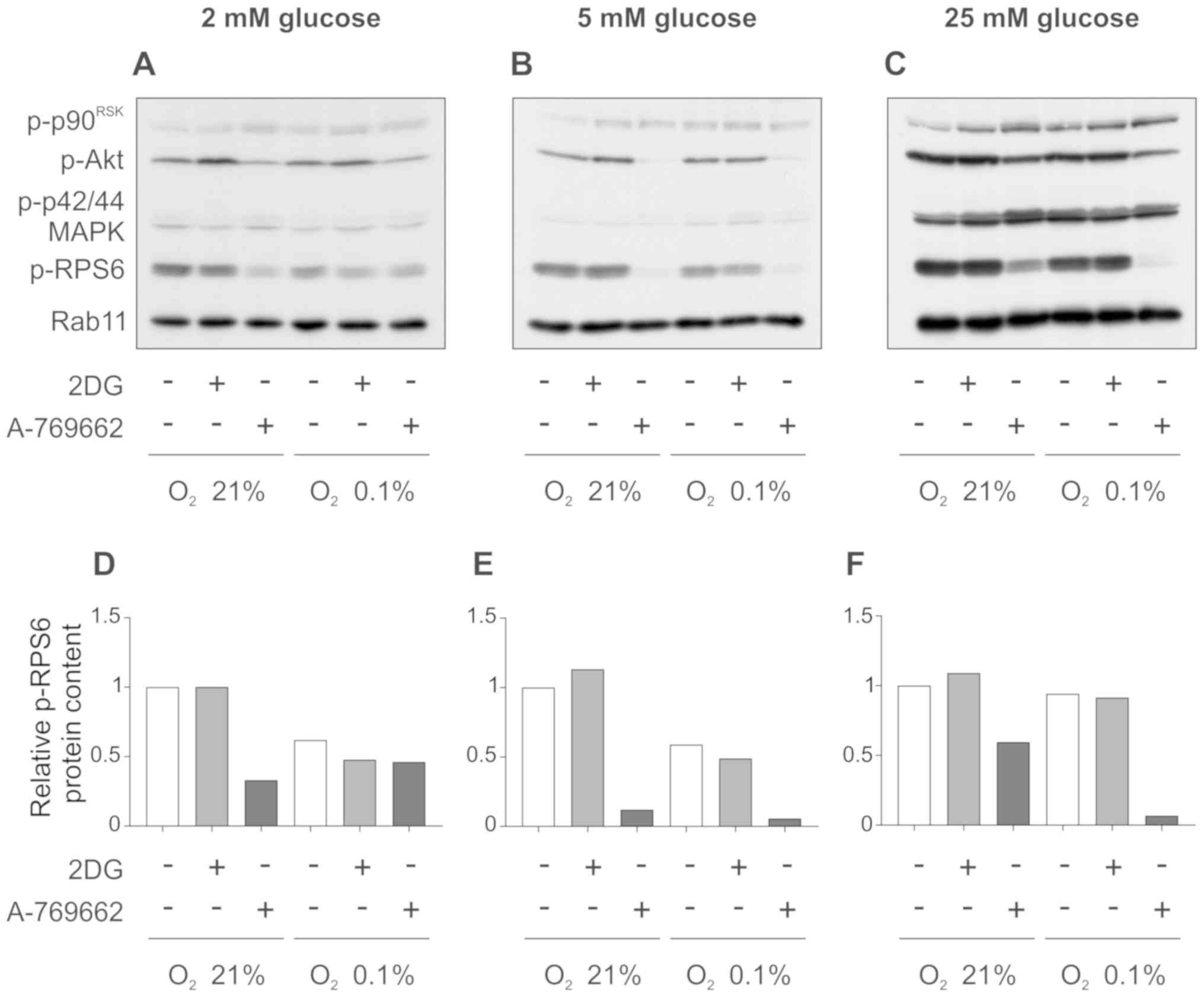
Effect of 2DG and A-769662 on Akt and
ribosomal protein S6 (RPS6, mTORC1 downstream target)
To examine the effect of 2DG and A-769662 on mTORC1
signaling, western blot analysis for key components of this pathway
was performed. Treatment of the SVG cells with 2DG did not alter
the phosphorylation of serine/threonine kinase Akt/protein kinase B
(Akt) and the mTORC1 downstream target, ribosomal protein S6
(p-RPS6), under normoxic and hypoxic conditions at glucose
concentrations of 2, 5 and 25 mM (Fig. 3A–C). The exposure of the SVG cells
to A-769662 decreased the phosphorylation of RPS6 under normoxic
and hypoxic conditions at glucose concentrations of 5 and 25 mM
(Fig. 3B and C, and
quantification of intensities in E and F). At the low glucose
concentration of 2 mM, RPS6 phosphorylation was decreased by
A-769662 only in normoxic, but not hypoxic conditions (Fig. 3A, and quantification of
intensities in D). Overall, these results indicate an inactivation
of mTORC1 in response to 2DG and A-769662 through AMPK activation.
Similarly, the phosphorylation of Akt was reduced by A-769662
treatment under normoxic and hypoxic conditions at glucose
concentrations of 2, 5 and 25 mM (Fig. 3A–C). As expected, no relevant
changes in the levels of pospho-p90RSK (p-p90RSK) and p-p42/44
MAPK, which are not regulated by mTORC1, were observed.
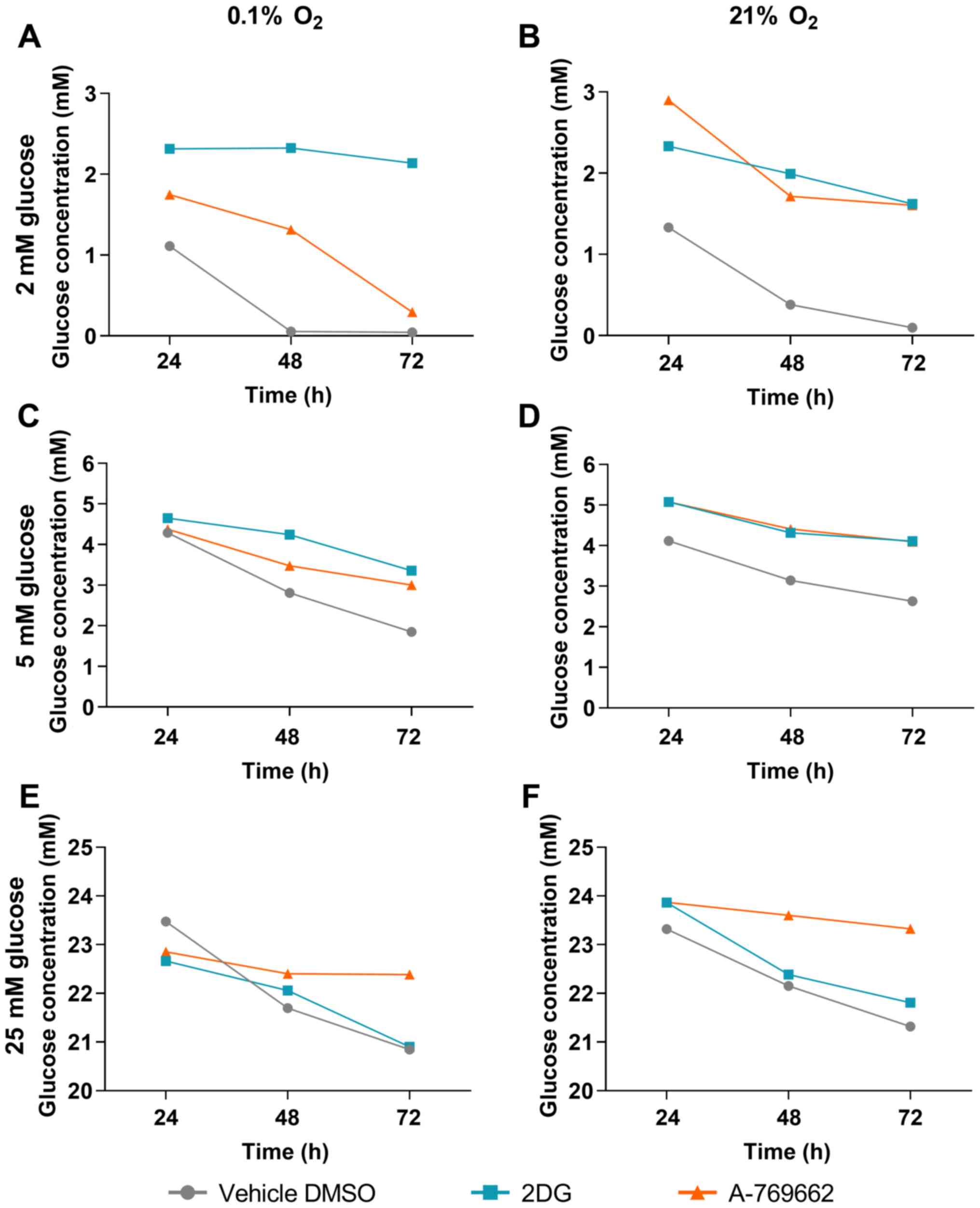
Glucose concentration of SVG cells
treated with 2DG and A-769662
The extracellular glucose concentration in the cell
culture medium was measured in the absence of glutamine to examine
the effects of 2DG and A-769662 on glucose consumption in
astrocytes under hypoxic conditions. Glucose concentrations in the
cell culture medium were measured after 24, 48 and 72 h. The
results following treatment with 2DG and A-769662 were compared to
treatment with the vehicle, DMSO, at the respective time points. A
higher glucose concentration in the medium following treatment with
2DG or A-769662 as compared to treatment with the vehicle, DMSO, at
the same time point is equivalent to a decrease in glucose
consumption by 2DG or A-769662 treatment, respectively. Treatment
of the SVG cells with 2DG led to reduced glucose consumption under
normoxic and hypoxic conditions at glucose concentrations of 2 and
5 mM (Fig. 4A–D). Under the
supraphysiologically high glucose concentration of 25 mM, no effect
of 2DG treatment on glucose consumption was observed (Fig. 4E and F). Treatment with A-769662
led to reduced glucose consumption under normoxic and hypoxic
conditions under all glucose concentrations examined (2, 5 and 25
mM; Fig. 4).
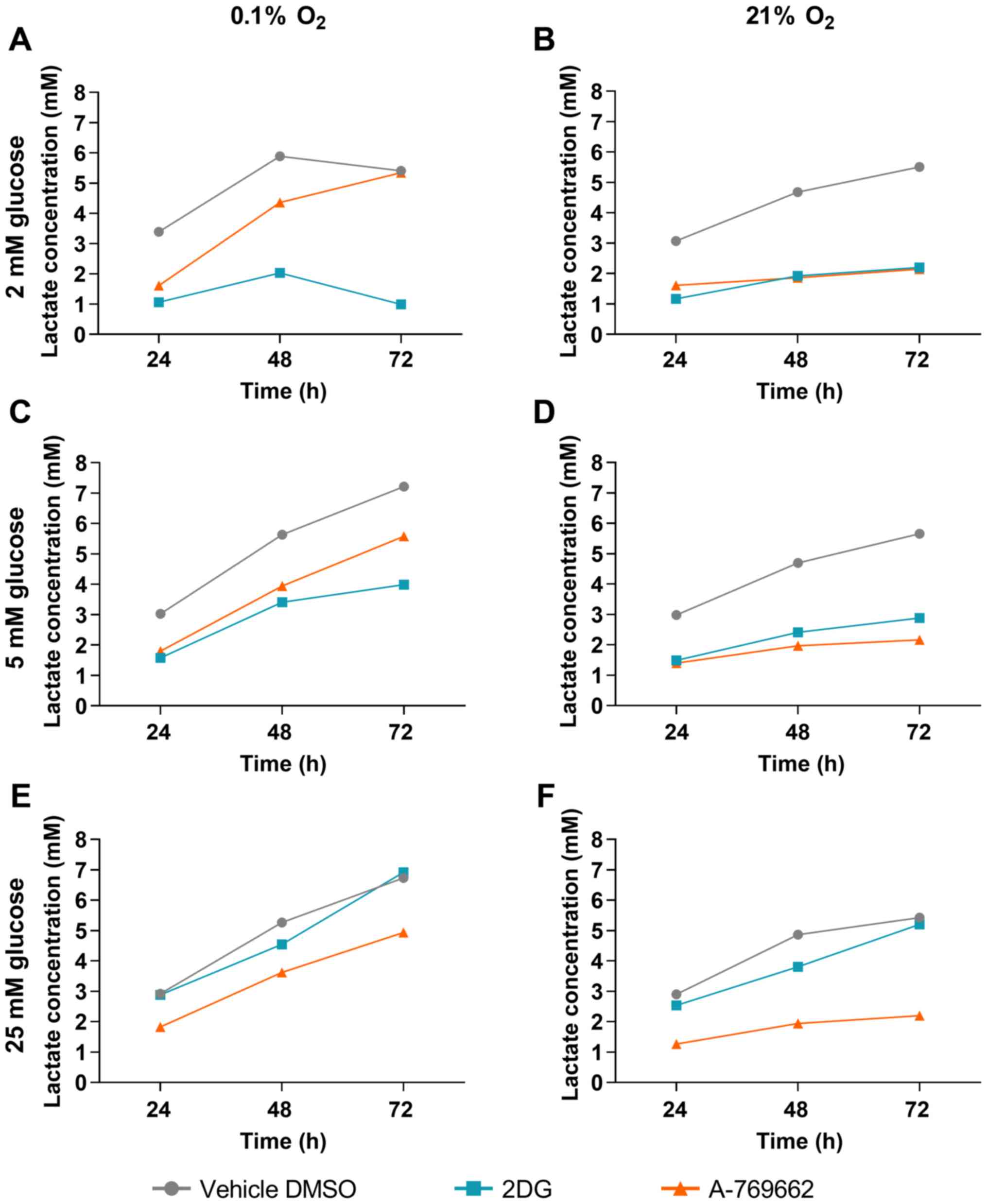
Lactate concentration in SVG cells
treated with 2DG and A-769662
To investigate the effects of 2DG and A-769662 on
glycolysis under hypoxic and normoxic conditions, the extracellular
lactate concentrations in the absence of glutamine were determined.
Lactate concentrations in the cell culture medium were measured
after 24, 48 and 72 h. The results following treatment with 2DG and
A-769662 were compared to those of treatment with the vehicle,
DMSO, at the respective time points. A lower lactate concentration
in the medium following treatment with 2DG or A-769662 as compared
to treatment with the vehicle, DMSO, at the same time point is
equivalent to a decrease in lactate production by 2DG or A-769662
treatment, respectively. Treatment of the SVG cells with 2DG
reduced lactate production at 2 and 5 mM under both hypoxic and
normoxic conditions (Fig. 5). At
the supraphysiologically high glucose concentration of 25 mM, no
effect of treatment with 2DG on lactate production was observed
under hypoxic and normoxic conditions. Lactate production was
reduced by A-769662 treatment both under normoxic and hypoxic
conditions at glucose concentrations of 2, 5 and 25 mM.
Activation of AMPK via A-769662 increases
O2 consumption and the ATP concentration
The cellular oxygen consumption was measured to
determine the proportion of aerobic metabolism. The results
following treatment with 2DG and A-769662 were compared to
treatment with the vehicle, DMSO, at the respective time points. A
lower O2 concentration in the medium following treatment
with 2DG or A-769662 as compared to treatment with the vehicle,
DMSO, at the same time point is equivalent to an increase in
O2 consumption by 2DG or A-769662 treatment,
respectively. The activation of AMPK via A-769662 increased the
cellular oxygen consumption of SVG cells at 2, 5 and 25 mM glucose
(Fig. 6A, C and D). The
inhibition of glycolysis with 2DG did not increase oxygen
consumption significantly. The increased oxygen consumption in
conjunction with the decreased glucose consumption and the
decreased lactate production of SVG cells treated with A-769662
indicate a switch to more efficient glucose metabolism through
oxidative phosphorylation. Glycolysis inhibition with 2DG under
normoxic conditions did not alter the ATP concentration (Fig. 6B). However, under hypoxic
conditions, treatment with 2DG reduced the ATP concentration.
Direct AMPK activation with A-769662 increased the ATP
concentration under both hypoxic and normoxic conditions.
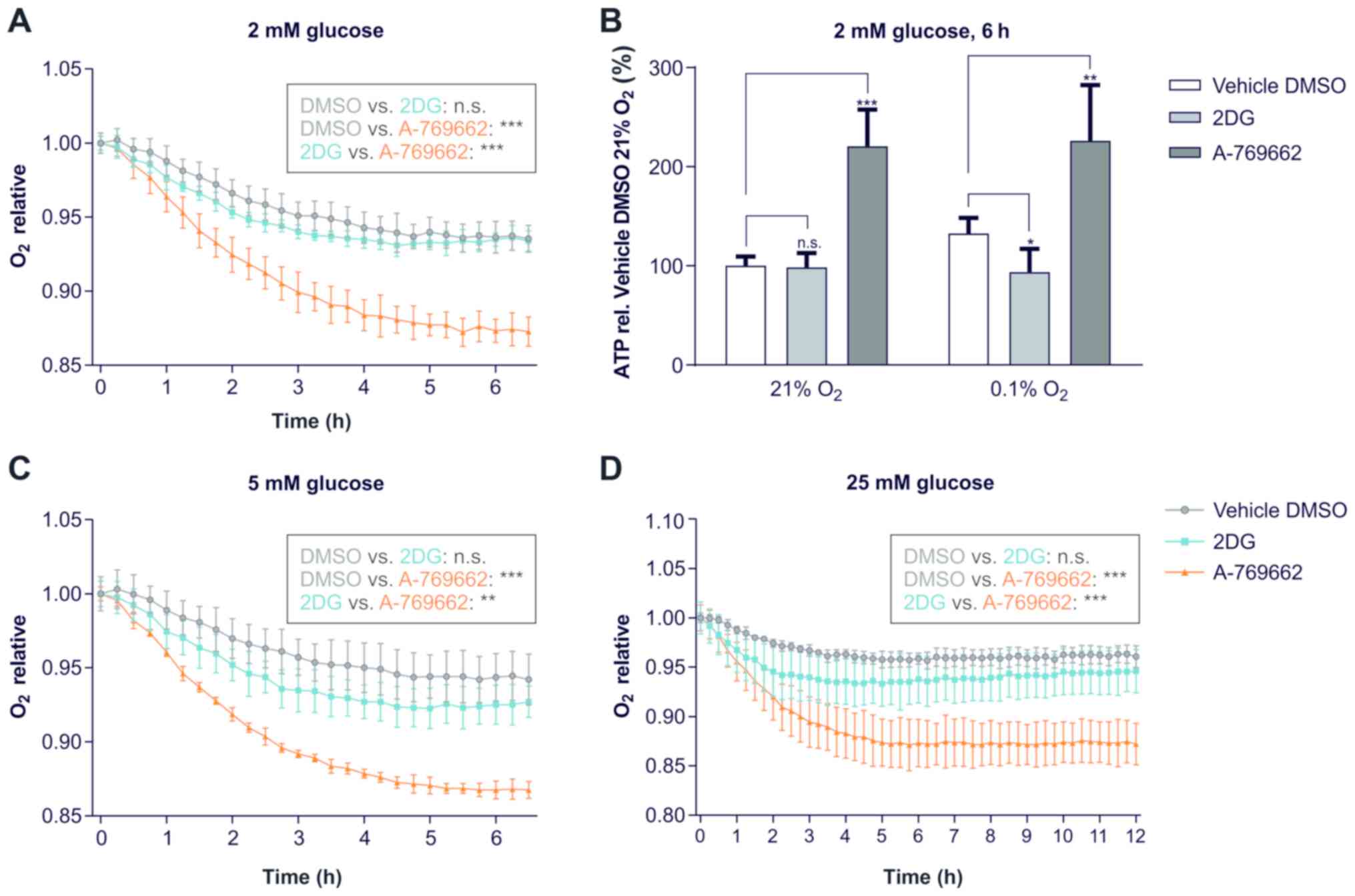 | Figure 6Activation of AMPK via A-769662
increases O2-consumption and ATP concentrations. (A, C
and D) Effect of 2DG and A-769662 on oxygen consumption. SVG cells
were exposed to serum-free DMEM with 2, 5 or 25 mM glucose, 0 mM
glutamine, vehicle DMSO, 2DG or A-769662 and overlayed with sterile
paraffin oil [n=3; n.s., not significant (P≥0.05);
**P<0.01, ***P<0.001, non-linear
regression analysis followed by ANOVA with Tukey’s multiple
comparisons test]. Oxygen was measured with a fluorescence-based
assay as the proportion of the initial oxygen concentration under
normoxic conditions. (A) Exposure to 2 mM glucose. (C) Exposure to
5 mM glucose. (D) Exposure to 25 mM glucose. (B) ATP concentrations
of SVG cells treated with 2DG or A-769662 normalized to the vehicle
DMSO under normoxic conditions. SVG cells were exposed to
serum-free DMEM with 2 mM glucose, 0 mM glutamine and the vehicle
DMSO, 2DG or A-769662 and incubated for 6 h. ATP concentrations
were measured and normalized to the vehicle DMSO. [n=5; n.s., not
significant (P≥0.05); *P<0.05,
**P<0.01, ***P<0.001, ANOVA with
Dunnett’s multiple comparisons test). 2DG, 2-deoxy-d-glucose. |
Discussion
In the present study, it was hypothesized that a
preventive induction of energy deprivation-activated signaling
pathways via AMPK may protect astrocytes from hypoxia and glucose
deprivation. The results revealed that both direct and indirect
AMPK activation resulted in a potent protective effect. An overview
of the effects of 2DG and A-769662 on SVG cells under oxygen and
partial glucose deprivation conditions is illustrated in Fig. 7.
Direct AMPK activation with A-769662, as well as
glycolysis inhibition with 2DG protected the SVG cells from
hypoxia-induced cell death. Even though this effect was still
detectable at high glucose concentrations, it was most potent with
low glucose concentrations. This accentuated protective effect of
A-769662 at low glucose concentrations indicated a synergistic
effect with the intrinsic activation of AMPK induced by the
relative lack of glucose.
2DG and A-769662 protected the SVG cells from
hypoxia both in the presence and in absence of glutamine. Under
hypoxic conditions, ATP cannot be generated from the metabolization
of glutamine in the process of glutaminolysis (10). However, under hypoxic conditions,
glutamine can still be metabolized through anaplerosis (29). As the addition of glutamine had no
effect on the extent of cell death, this demonstrates that
anaplerosis did not protect the astrocytes in our paradigm from
hypoxia and partial glucose deprivation.
In cortical rat astrocytes, it has been previously
demonstrated that AMPK activation induced by metformin exerts a
protective effect against oxygen and glucose deprivation (30), and conversely that AMPK inhibition
by compound C increases cell death through a reduced rate of
autophagy (31). Furthermore,
AMPK activation with AICAR has been shown to protect rodent
astrocytes via peroxisome proliferator-activated receptor gamma
co-activator 1α (PGC-1α) activation (32). The present study confirmed the
protective effect of AMPK activation with the more specific AMPK
activator, A-769662 (5,33,34), in a paradigm of permanent oxygen
and glucose deprivation.
Western blot analysis revealed that treatment with
A-769662 and 2DG activated AMPK, which resulted in an increased
phosphorylation of ACC. The effect of A-769662 was more potent than
that of 2DG. This difference was expected due to the direct
mechanism of action of A-769662, as compared to the indirect
mechanism of 2DG. The inhibition of ACC is known to reduce cellular
energy consumption, decreasing the highly energy intensive anabolic
process of lipid biosynthesis (35,36). Furthermore, AMPK activation with
A-769662 led to an inhibition of Akt and the mTORC1 target protein,
RPS6. The inhibition of mTORC1 and its target protein RPS6 have
previously been demonstrated to reduce the energy consuming
anabolic processes of translation and cellular proliferation
(25,26,28).
In contrast to A-769662, 2DG had no marked effect on
the phosphorylation of AMPK, ACC, Akt and RPS6 in SVG cells exposed
to a supraphysiologically high glucose concentration of 25 mM. This
discrepancy may be explained by the mechanism of glycolysis
inhibition, as 2DG displaces glucose, competitively binds to
phosphoglucoisomerase and thus inhibits glycolysis. Due to this
competitive mechanism, 2DG does not inhibit glycolysis at high
glucose to 2DG ratios (8).
In the present study, direct and indirect AMPK
activation led to reduced glucose consumption and lactate
production both under normoxic and hypoxic conditions. Furthermore,
an increase in oxygen consumption in cells exposed to A-769662 was
observed. This increase in oxygen consumption, the decrease in
glucose consumption and the decrease in lactate production point to
a metabolic switch towards an augmented oxidative phosphorylation.
It has already been known that the A-769662 treatment of astrocytes
increases extracellular ATP levels (37). The present study demonstrated that
direct AMPK activation with A-769662 increased the intracellular
ATP concentrations as well, potentially due to a reduced rate of
ATP consumption in anabolic processes. In contrast to glycolysis
inhibition with 2DG, direct AMPK activation with A-769662 is not
expected to restrict ATP production. By contrast, glycolysis
inhibition with 2DG reduced cellular ATP concentrations under
hypoxic conditions when SVG cells fully depend on anaerobic
glycolysis for ATP production. Nevertheless, even though ATP levels
were reduced by 2DG treatment under hypoxic conditions, the
protective effect was still present. It was thus speculated that
the negative effect of indirect AMPK activation by 2DG on ATP
consumption was sufficiently potent to balance the lower ATP levels
due to relatively higher glucose levels for a longer period of time
and to thus delay the subsequently resulting disequilibrium of ATP
production and consumption finally leading to cell death.
Under the paradigm of sublethal oxygen/glucose
deprivation with subsequent reoxygenation, a reactive induction of
astrocytic proliferation after reoxygenation is observed (38,39). The experimental conditions of this
study differed in that the glucose/oxygen deprivation applied was
permanent and no reoxygenation was performed to specifically
measure the amount of cell death occurring under hypoxia and
glucose deprivation. This paradigm of hypoxia- and glucose
deprivation-induced cell death was intentionally selected in order
to focus on the extent of cell death occurring under these static
adverse conditions. This is indeed a simplification of the complex
processes occurring during ischemic brain infarction. However, the
penumbra of an ischemic lesion is characterized by a reduced
perfusion as observed in perfusion-weighted magnetic resonance
imaging. Even though this residual perfusion is not sufficient to
preserve the tissue affected from ischemic cell death, it may be
sufficient to allow for pharmacologic interventions (40,41). Therefore, the preventive induction
of energy deprivation-activated signaling pathways via AMPK
activation is a promising research approach with which to prolong
the time window during ischemic stroke.
The limitations of the present study include the use
of the immortalized astrocytic cell line, SVG. While of human
origin, it should be noted that SVG is a SV40-transformed cell line
and therefore may exhibit metabolic differences as compared to
normal human astrocytes.
In conclusion, the findings of the present study
demonstrate that direct, as well as indirect AMPK activation
protects astrocytic cells from hypoxic cell death. AMPK activation
may thus prove to be an indirect neuroprotective strategy for the
management of cerebral ischemia via the preservation of function
and integrity of astrocytes.
Supplementary Information
Acknowledgements
Not applicable.
Funding
The present study was supported by the Dr.
Senckenberg Institute of Neurooncology which is supported by the
Senckenberg foundation.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article or are available from the
corresponding author on reasonable request.
Authors’ contributions
LB, MWR, JPS and MCB were involved in the
conceptualization of the study. LB, MIS, ALL, MJ, IB, ACMS, ICM,
SMH, AD, NIL, MV, MWR, JPS and MCB were involved in the study
methodology. LB was involved in the investigative aspect of the
study, and in the writing and preparation of the original draft of
the manuscript. LB, MIS, ALL, MJ, IB, ACMS, ICM, SMH, AD, NIL, MV,
MWR, JPS and MCB were involved in the writing, reviewing and
editing of the manuscript. MWR, JPS and MCB were involved in study
supervision. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
2DG
|
2-deoxy-d-glucose
|
|
αKG
|
α-ketoglutarate
|
|
ACC
|
acetyl-CoA carboxylase
|
|
Akt
|
serine/threonine kinase Akt/protein
kinase B
|
|
AMP
|
adenosine monophosphate
|
|
AMPK
|
5’adenosine monophosphate-activated
protein kinase
|
|
ATP
|
adenosine triphosphate
|
|
DMEM
|
Dulbecco modified Eagle’s minimal
essential medium
|
|
DMSO
|
dimethyl sulfoxide
|
|
EGFR
|
epidermal growth factor receptor
|
|
FACS
|
fluorescence-activated cell
sorting
|
|
FCS
|
fetal calf serum
|
|
GB
|
glioblastoma
|
|
LDH
|
lactate dehydrogenase
|
|
mTOR
|
mammalian target of rapamycin
|
|
mTORC1
|
mTOR complex 1
|
|
n.s.
|
not significant
|
|
p-p42/44 MAPK
|
phospho-p42/44 mitogen-activated
protein kinase
|
|
p-p90RSK
|
phospho-p90RSK (MAPK-activated protein
kinase-1)
|
|
PI
|
propidium iodide
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
Rab11
|
Ras-related protein Rab-11
|
|
RPS6
|
ribosomal protein S6
|
References
|
1
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mitsudomi T and Yatabe Y: Epidermal growth
factor receptor in relation to tumor development: EGFR gene and
cancer. FEBS J. 277:301–308. 2010. View Article : Google Scholar
|
|
3
|
Wullschleger S, Loewith R and Hall MN: TOR
signaling in growth and metabolism. Cell. 124:471–484. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hardie DG and Pan DA: Regulation of fatty
acid synthesis and oxidation by the AMP-activated protein kinase.
Biochem Soc Trans. 30:1064–1070. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Goransson O, McBride A, Hawley SA, Ross
FA, Shpiro N, Foretz M, Viollet B, Hardie DG and Sakamoto K:
Mechanism of action of A-769662, a valuable tool for activation of
AMP-activated protein kinase. J Biol Chem. 282:32549–32560. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hartel I, Ronellenfitsch M, Wanka C,
Wolking S, Steinbach JP and Rieger J: Activation of AMP-activated
kinase modulates sensitivity of glioma cells against epidermal
growth factor receptor inhibition. Int J Oncol. 49:173–180. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Strickland M and Stoll EA: Metabolic
reprogramming in glioma. Front Cell Dev Biol. 5:432017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Woodward GE and Hudson MT: The effect of
2-desoxy-D-glucose on glycolysis and respiration of tumor and
normal tissues. Cancer Res. 14:599–605. 1954.PubMed/NCBI
|
|
9
|
Wick AN, Drury DR, Nakada HI and Wolfe JB:
Localization of the primary metabolic block produced by
2-deoxyglucose. J Biol Chem. 224:963–969. 1957.PubMed/NCBI
|
|
10
|
Yang C, Ko B, Hensley CT, Jiang L, Wasti
AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M, et al:
Glutamine oxidation maintains the TCA cycle and cell survival
during impaired mitochondrial pyruvate transport. Mol Cell.
56:414–424. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sellers K, Fox MP, Bousamra M II, Slone
SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R,
et al: Pyruvate carboxylase is critical for non-small-cell lung
cancer proliferation. J Clin Invest. 125:687–698. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Csibi A, Fendt SM, Li C, Poulogiannis G,
Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T,
et al: The mTORC1 pathway stimulates glutamine metabolism and cell
proliferation by repressing SIRT4. Cell. 153:840–854. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Becerra-Calixto A and Cardona-Gomez GP:
The role of astrocytes in neuroprotection after brain stroke:
Potential in cell therapy. Front Mol Neurosci. 10:882017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Heiss WD: The concept of the penumbra: Can
it be translated to stroke management? Int J Stroke. 5:290–295.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Candelario-Jalil E: Injury and repair
mechanisms in ischemic stroke: Considerations for the development
of novel neurotherapeutics. Curr Opin Investig Drugs. 10:644–654.
2009.PubMed/NCBI
|
|
16
|
Khaladj N, Peterss S, Oetjen P, von
Wasielewski R, Hauschild G, Karck M, Haverich A and Hagl C:
Hypothermic circulatory arrest with moderate, deep or profound
hypothermic selective antegrade cerebral perfusion: Which
temperature provides best brain protection? Eur J Cardiothorac
Surg. 30:492–498. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pacini D, Leone A, Di Marco L, Marsilli D,
Sobaih F, Turci S, Masieri V and Di Bartolomeo R: Antegrade
selective cerebral perfusion in thoracic aorta surgery: Safety of
moderate hypothermia. Eur J Cardiothorac Surg. 31:618–622. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Abbott NJ, Ronnback L and Hansson E:
Astrocyte-endothelial interactions at the blood-brain barrier. Nat
Rev Neurosci. 7:41–53. 2006. View Article : Google Scholar
|
|
19
|
Newman EA: Glial cell regulation of
neuronal activity and blood flow in the retina by release of
gliotransmitters. Philos Trans R Soc Lond B Biol Sci. 370:2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kitchen P, Day RE, Taylor LH, Salman MM,
Bill RM, Conner MT and Conner AC: Identification and molecular
mechanisms of the rapid tonicity-induced relocalization of the
aquaporin 4 channel. J Biol Chem. 290:16873–16881. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kaczor P, Rakus D and Mozrzymas JW:
Neuron-astrocyte interaction enhance GABAergic synaptic
transmission in a manner dependent on key metabolic enzymes. Front
Cell Neurosci. 9:1202015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xie Z, Ding SQ and Shen YF: Silibinin
activates AMP-activated protein kinase to protect neuronal cells
from oxygen and glucose deprivation-re-oxygenation. Biochem Biophys
Res Commun. 454:313–319. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Major EO, Miller AE, Mourrain P, Traub RG,
de Widt E and Sever J: Establishment of a line of human fetal glial
cells that supports JC virus multiplication. Proc Natl Acad Sci
USA. 82:1257–1261. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bachis A, Major EO and Mocchetti I:
Brain-derived neurotrophic factor inhibits human immunodeficiency
virus-1/gp120-mediated cerebellar granule cell death by preventing
gp120 internalization. J Neurosci. 23:5715–5722. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Steinbach JP, Wolburg H, Klumpp A, Probst
H and Weller M: Hypoxia-induced cell death in human malignant
glioma cells: Energy deprivation promotes decoupling of
mitochondrial cytochrome c release from caspase processing and
necrotic cell death. Cell Death Differ. 10:823–832. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Steinbach JP, Klumpp A, Wolburg H and
Weller M: Inhibition of epidermal growth factor receptor signaling
protects human malignant glioma cells from hypoxia-induced cell
death. Cancer Res. 64:1575–1578. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Thiepold AL, Lorenz NI, Foltyn M, Engel
AL, Divé I, Urban H, Heller S, Bruns I, Hofmann U, Dröse S, et al:
Mammalian target of rapamycin complex 1 activation sensitizes human
glioma cells to hypoxia-induced cell death. Brain. 140:2623–2638.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ronellenfitsch MW, Brucker DP, Burger MC,
Wolking S, Tritschler F, Rieger J, Wick W, Weller M and Steinbach
JP: Antagonism of the mammalian target of rapamycin selectively
mediates metabolic effects of epidermal growth factor receptor
inhibition and protects human malignant glioma cells from
hypoxia-induced cell death. Brain. 132:1509–1522. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wise DR, Ward PS, Shay JE, Cross JR,
Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC and
Thompson CB: Hypoxia promotes isocitrate dehydrogenase-dependent
carboxylation of α-ketoglutarate to citrate to support cell growth
and viability. Proc Natl Acad Sci USA. 108:19611–19616. 2011.
View Article : Google Scholar
|
|
30
|
Gabryel B, Kost A, Kasprowska D, Liber S,
Machnik G, Wiaderkiewicz R and Łabuzek K: AMP-activated protein
kinase is involved in induction of protective autophagy in
astrocytes exposed to oxygen-glucose deprivation. Cell Biol Int.
38:1086–1097. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gabryel B and Liber S: Metformin limits
apoptosis in primary rat cortical astrocytes subjected to oxygen
and glucose deprivation. Folia Neuropathol. 56:328–336. 2018.
View Article : Google Scholar
|
|
32
|
Guo X, Jiang Q, Tuccitto A, Chan D,
Alqawlaq S, Won GJ and Sivak JM: The AMPK-PGC-1α signaling axis
regulates the astrocyte glutathione system to protect against
oxidative and metabolic injury. Neurobiol Dis. 113:59–69. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Viollet B, Guigas B, Leclerc J, Hébrard S,
Lantier L, Mounier R, Andreelli F and Foretz M: AMP-activated
protein kinase in the regulation of hepatic energy metabolism: From
physiology to therapeutic perspectives. Acta Physiol (Oxf).
196:81–98. 2009. View Article : Google Scholar
|
|
34
|
Dasgupta B and Seibel W: Compound
c/dorsomorphin: Its use and misuse as an AMPK inhibitor. Methods
Mol Biol. 1732:195–202. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shin SY, Kim TH, Wu H, Choi YH and Kim SG:
SIRT1 activation by methylene blue, a repurposed drug, leads to
AMPK-mediated inhibition of steatosis and steatohepatitis. Eur J
Pharmacol. 727:115–124. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wu L, Zhang L, Li B, Jiang H, Duan Y, Xie
Z, Shuai L and Li J and Li J: AMP-activated protein kinase (AMPK)
regulates energy metabolism through modulating thermogenesis in
adipose tissue. Front Physiol. 9:1222018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vlachaki Walker JM, Robb JL, Cruz AM,
Malhi A, Weightman Potter PG, Ashford MLJ, McCrimmon RJ, Ellacott
KLJ and Beall C: AMP-activated protein kinase (AMPK) activator
A-769662 increases intracellular calcium and ATP release from
astrocytes in an AMPK-independent manner. Diabetes Obes Metab.
19:997–1005. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhao X, Zhou KS, Li ZH, Nan W, Wang J, Xia
YY and Zhang HH: Knockdown of Ski decreased the reactive astrocytes
proliferation in vitro induced by oxygen-glucose
deprivation/reoxygenation. J Cell Biochem. 119:4548–4558. 2018.
View Article : Google Scholar
|
|
39
|
He M, Shi X, Yang M, Yang T, Li T and Chen
J: Mesenchymal stem cells-derived IL-6 activates AMPK/mTOR
signaling to inhibit the proliferation of reactive astrocytes
induced by hypoxic-ischemic brain damage. Exp Neurol. 311:15–32.
2019. View Article : Google Scholar
|
|
40
|
Lin L, Bivard A and Parsons MW: Perfusion
patterns of ischemic stroke on computed tomography perfusion. J
Stroke. 15:164–173. 2013. View Article : Google Scholar
|
|
41
|
Lee DH, Kang DW, Ahn JS, Choi CG, Kim SJ
and Suh DC: Imaging of the ischemic penumbra in acute stroke.
Korean J Radiol. 6:64–74. 2005. View Article : Google Scholar : PubMed/NCBI
|