Introduction
Ischemia/reperfusion (I/R) injury is a common
clinical pathological phenomenon that may occur in a wide range of
clinical settings, including body trauma with hemorrhagic shock,
cardiac coronary ischemia, cardiac cardiopulmonary bypass surgery,
partial hepatectomy and liver and kidney transplantation, and it
may significantly influence disease prognosis (1). Previous studies have revealed that
mitochondrial permeability transition pore (MPTP) opening serves as
a key nodal event mediating cell necrosis during early reperfusion
and inducing further mitochondrial-related apoptosis (2,3).
MPTP opening is regulated by multiple intracellular signal
transduction pathways (4-6). It has previously been reported that
cyclo-oxygenase-2 (COX-2) inhibitor pretreatment decreases MPTP
opening and liver damage in a rat I/R model (7). However, inhibiting COX-2 signaling
may aggravate ischemic injury in myocardial and renal I/R (8,9),
which may be associated with the ability of COX-2 to regulate
numerous prostaglandins and receptors downstream.
The COX-2 signaling pathway is activated during I/R
to catalyze the production of prostaglandin E2 (PGE2) from
arachidonic acid (AA). As a member of the eicosanoid family, PGE2
is an endogenous lipid mediator (10) that binds to four specific G
protein-coupled receptors on the cell membrane [prostaglandin E
receptor subtype 1 (EP1), EP2, EP3 and EP4] (11). Of these receptors, EP4 is the most
widely distributed among tissues and is involved in various
pathophysiological processes (12-17). After activation, EP4 is coupled
primarily to intracellular stimulated Gs-proteins and upregulates
intracellular cyclic adenosine monophosphate (cAMP) levels
(18). Changes in cAMP
concentrations further influence intracellular cAMP-dependent
protein kinases and their downstream signals, such as cAMP-protein
kinase A (PKA), phosphatidylinositol 3-kinase (PI3K) and
extracellular signal-regulated protein kinase (ERK) 1/2, to produce
corresponding biological effects (11,13). Recently, Kuzumoto et al
(12) reported that EP4
activation protects the mouse liver from ischemic injury by
mediating inflammation during early reperfusion. Nishizawa et
al (19) demonstrated that
the PGE2/EP4 pathway is enhanced during hepatic I/R in mice and is
closely associated with liver injury and repair. Additionally, the
PGE2/EP4 axis has been revealed to serve as a homeostatic mechanism
that regulates endoplasmic reticulum stress and autophagy in liver
transplant recipients (20).
However, the full mechanism underlying EP4 signaling in hepatic I/R
modulation, particularly the role of EP4 in mitochondrial function,
is yet to be elucidated. In our preliminary study (supplementary
data), it was revealed that the mRNA expression of EP4 is
significantly upregulated in a rat liver I/R model and
downregulated by COX-2 inhibition preconditioning, after 2 h of
reperfusion (Fig. S1). Further
studies on human hepatic specimens indicated that EP4 expression
was significantly higher in transplant allografts that underwent
~20 min of warm ischemia, 6 h of cold ischemia and 60 min of
reperfusion compared with non-ischemic liver specimens (Fig. S2). Considering that EP4 is a PGE2
receptor and a regulatory product downstream of COX-2, it was
hypothesized that EP4 influences COX-2-associated MPTP modulation
during I/R. Therefore, the present study was designed to further
investigate the role and mechanism underlying the action of EP4 in
MPTP modulation and hepatic I/R.
Materials and methods
Animals
A total of 132 Male Sprague-Dawley rats (6 weeks
old) that weighed 200-220 g were purchased from Sino-British
SIPPR/BK Lab Animal Ltd. (Shanghai, China). The animals were housed
in standard cages and maintained under standard conditions at a
constant room temperature of 20-25°C, a humidity of 40-70% and a 12
h/12 h light/dark cycle, with unrestricted access to food and
water. All experiments were performed in accordance with the
National Institutes of Health Guide for the Care and Use of
Laboratory Animals and were approved by the Changzheng Hospital
Ethics Committee [approval number, CZEC (2015)-01].
Hepatic I/R injury model
A rat model was constructed using 70% partial
hepatic ischemia for 60 min as described previously (21-24). Briefly, rats were fasted for 12 h
before surgery and anesthetized by intraperitoneal (i.p.) injection
of 40 mg/kg pentobarbital. After midline laparotomy, the
interlobular ligaments were dissected. The left hepatoduodenal
ligament containing the hepatic artery, portal vein and bile duct
leading to the left and median lobe was clamped in the liver hilum
using a microvascular clamp for 60 min. Reperfusion was initiated
by clamp removal. Sham-operated rats underwent the same surgical
procedures but without vascular occlusion. The animals were
sacrificed by an intraperitoneal injection of sodium pentobarbital
(100 mg/kg) at 2 or 6 h after reperfusion. Death of the rats was
verified by a combination of criteria, including lack of pulse,
breathing, corneal reflex, response to a firm toe pinch and graying
of the mucous membranes. Liver and serum samples were collected for
further analysis.
Experimental protocol
To increase EP4 activity, a dose of 0.1, 0.5, 1 or
10 mg/kg of an EP4 agonist (CAY10598 [CAY]; Cayman Chemical
Company) was administered subcutaneously to animals 0, 0.5 and 2.5
h prior to the onset of liver reperfusion. To increase MPTP
susceptibility, a single dose of carboxyatractyloside (CATR; 5
mg/kg, Sigma-Aldrich; Merck KGaA) was dissolved in 0.9% saline and
then administered intraperitoneally to animals 30 min prior to the
60 min ischemic insult. Additionally, to inhibit ERK1/2 activity,
rats received an intraperitoneal injection of an ERK1/2 inhibitor
[PD98059 (PD), Cell Signaling Technology, Inc.], 5 mg/kg dissolved
in dimethyl sulfoxide (DMSO) (Cell Signaling Technology, Inc.), 30
min prior to the onset of liver ischemia. The sham and I/R animals
received the same volume of the respective vehicles.
Histological assays
Liver samples were fixed in 4%
formaldehyde/phosphate-buffered saline overnight at 4°C. The
samples were dehydrated and embedded in paraffin. Hepatocellular
necrosis was calculated in hematoxylin and eosin (H&E)-stained
tissue using a semi-quantitative scale, as previously described
(21). Briefly, for each
H&E-stained tissue sample, a total of 30 randomly selected
high-power visual fields (magnification, ×400) were analyzed in a
blinded fashion to determine the percentage of necrotic cells. In
the current study, only grade 3 severe injury with hepatic cord
disintegration was categorized as necrosis (21).
To evaluate apoptosis, terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling (TUNEL) staining was
also performed using a commercial kit from Roche Diagnostics,
according to the manufacturer's instructions. Briefly,
paraffin-embedded tissue sections were dewaxed by heating at 60°C
followed by washing in xylene, rehydration using a graded series of
ethanol (100, 95, 90, 80 and 70%) and double-distilled water, and
then pretreatment with proteinase-k. Endogenous peroxidase activity
was blocked by immersing in 3% H2O2 in
methanol for 10 min at room temperature. TUNEL reaction mixture and
Converter-POD were then added, and incubated in a humidified
atmosphere for 60 and 30 min at 37°C in the dark, respectively.
Each slice was stained using DAB for 10 min at room temperature,
and liver cell apoptosis was observed under light microscopy. In
each section, areas without significant necrosis in 10 different
visual fields (magnification, ×400) were observed and analyzed for
TUNEL-positive cells. The TUNEL index was calculated by counting
the total nuclei and cells with brown nuclei and using the
following formula: [(number of stained cells)/(number of stained
cells + number of unstained cells)] ×100. Six sections of each
tissue sample were analyzed for each group.
For the immunohistochemical analysis of EP4, liver
tissue sections (5-µm thick) were then immunostained using
the EnVision method (DakoCytomation; Agilent Technologies, Inc.)
according to the manufacturer's instructions. Briefly, tissue
sections were pre-treated for 10 min with peroxidase-blocking
reagent (Dako; Agilent Technologies, Inc.) to suppress endogenous
peroxidase and pseudoperoxidase activity at room temperature,
washed again in phosphate-buffered saline(PBS) and incubated for 1
h at 37°C in a humid chamber with the anti-EP4 polyclonal antibody
(1:100; cat. no. SC-20677; Santa Cruz Biotechnology, Inc.). The
slides were rinsed twice and then incubated for 60 min with goat
anti-mouse immunoglobulins conjugated to a peroxidase-labeled
polymer (1:500; cat. no. P0447; DAKO EnVision™ + System; Dako;
Agilent Technologies, Inc.) at room temperature. After washing,
cells were treated with 3,3′-diaminobenzidine (Sigma-Aldrich; Merck
KGaA) as a chromogen substrate for 10 min at room temperature.
Slides were then washed again in PBS and counterstained with
hematoxylin for 2 min at room temperature. Sections stained with
H&E were used for comparison and compared using a light
microscope with a magnification of ×400.
Western blot analysis
The levels of EP4 (Santa Cruz Biotechnology, Inc.),
ERK1/2, phosphorylated (p-) ERK1/2, janus kinase 2 (JAK2), p-JAK2,
signal transducer and activator of transcription 3 (STAT3),
p-STAT3, glycogen synthase kinase-3β (GSK3β) and p-GSK3β (Ser9;
Cell Signaling Technology, Inc.) were evaluated in rat liver
lysates using western blot analysis, as previously reported
(25). Briefly, liver tissues
were homogenized in RIPA lysis buffer (Wuhan Servicebio Technology
Co., Ltd.), followed by centrifugation at 12,000 × g for 15 min at
4°C. The bicinchoninic acid method was used for quantitative
analysis the protein samples. A total of 40 µg protein was
applied to each lane and separated via 10% SDS-PAGE before being
transferred onto PVDF membranes. A total of 5% bovine serum albumin
(Sigma-Aldrich; Merck KGaA) and TBS with 0.1% Tween-20 (PBST) was
used to block the unspecific binding of antibodies for 2 h at 4°C.
Then, blots were incubated with the following primary antibodies:
EP4 (1:1,000; cat. no. SC-20677; Santa Cruz Biotechnology, Inc.),
ERK1/2 (1:1,000; cat. no. 4695; Cell Signaling Technology, Inc.),
p-ERK1/2 (1:2,000; cat. no. 4370; Cell Signaling Technology, Inc.),
JAK2 (1:1,000; cat. no. 3230; Cell Signaling Technology, Inc.),
p-JAK2 (1:1,000; cat. no. 3776; Cell Signaling Technology, Inc.),
STAT3 (1:1,000; cat. no. 30835; Cell Signaling Technology, Inc.),
p-STAT3 (1:2,000; cat. no. 9145; Cell Signaling Technology, Inc.),
GSK3β (1:1,000; cat. no. 5676; Cell Signaling Technology, Inc.),
p-GSK3β (1:1,000; cat. no. 9322; Cell Signaling Technology, Inc.)
and GAPDH (1:1,000; cat. no. 5174; Cell Signaling Technology,
Inc.), overnight at 4°C. Blots were then incubated with a
goat-anti-rabbit IgG-HRP (1:2,000; cat. no. 7074; Cell Signaling
Technology, Inc.) for 1.5 h at room temperature and washed three
times with TBST. An enhanced chemiluminescence (ECL) western
blotting detection reagent (cat. no. 34076; Thermo Fisher
Scientific, Inc.) was used for visualization. A digital gel image
analysis system (model GIS-1000; Tanon) were used to evaluate the
specific signals and the corresponding band intensities.
Plasma biochemical assays
Serum alanine aminotransferase (ALT) and aspartate
aminotransferase (AST) levels were determined after 2 and 6 h of
reperfusion using an AutoAnalyzer (H-7600, Hitachi Ltd.).
Evaluation of tissue reactive oxygen
species (ROS) levels
Liver tissue were collected after 2 and 6 h of
reperfusion. Tissue ROS levels were measured using a ROS
enzyme-linked immunosorbent assay (ELISA) kit (Cell Signaling
Technology, Inc.) according to the manufacturer's instructions. The
results are expressed as relative fluorescence units (U/ml).
Mitochondrial isolation and calcium
retention capacity (CRC) assessment
Mitochondria were isolated in liver tissue at 2 and
6 h of reperfusion using differential centrifugation at 4°C as
previously described (26).
Briefly, the fresh liver tissues (1 g) were homogenized with 8 ml
of isolation buffer A (ISA) containing 220 mmol/l d-mannitol, 70
mmol/l sucrose, 10 mmol/l Tris-HCl, 1 mmol/l EGTA and 0.4% bovine
serum albumin (pH 7.4). The homogenates were centrifuged at 850 × g
for 10 min to collect supernatants, followed by centrifugation at
10,000 × g for an additional 10 min. The mitochondrial pellet was
resuspended in a final washing isolation buffer B containing 220
mmol/l d-mannitol, 70 mmol/l sucrose and 10 mmol/l Tris-HCl (pH
7.4). Protein concentration was determined via the biuret method
and calibrated with bovine serum albumin (BSA). A CRC assay was
used to evaluate MPTP susceptibility, which was adapted from a
previously described method (27). CRC is defined as the amount of
Ca2+ required to trigger mass Ca2+ release
from isolated liver mitochondria. The extramitochondrial
Ca2+ concentration was recorded using a fluorescence
microplate reader controlled by SoftMax Pro 4.8 software (Molecular
Devices, LLC) in the presence of the Calcium Green-5N molecular
probe (1 µmol/l, Invitrogen; Thermo Fisher Scientific,
Inc.), with excitation and emission wavelengths of 500 and 530 nm,
respectively (7). The
fluorescence scan interval was set at 12 sec.
Electron microscopy
Electron microscopy was used to examine the
ultrastructure of the hepatocytes in liver tissue samples after 6 h
of reperfusion. Briefly, the liver samples were fixed in a 1% osmic
acid fixative solution (Wuhan Servicebio Technology Co., Ltd.) at
4°C for 3 h and then washed with phosphate-buffered saline (PBS)
three times at 4°C for 30 min. Dehydration was carried out in
graded ethanol solutions (50, 70 and 90%), in a 90% solution
(ethanol:acetone; 1:1; v/v) and finally in a 90% acetone solution
at 4°C for 20 min. Then, the samples were washed three times with
acetone at room temperature for 30 min. The tissue samples were
embedded in a solution [acetone: Optimal cutting temperature
compound (Wuhan Servicebio Technology Co., Ltd.); 2:1; v/v] for 4 h
and then stored overnight at room temperature. Subsequently, the
tissues were embedded in 100% embedding medium for 3 h at 37°C.
Next, the tissues were cured in a drying oven at 37°C overnight, at
45°C for 12 h, and at 60°C for 24 h. Finally, the tissues were cut
to 50-nm thick sections using an ultramicrotome and stained with 2%
uranyl acetate and lead citrate (Wuhan Servicebio Technology Co.,
Ltd.) with pH 12 at room temperature for 10 min. The ultrathin
sections were then examined and imaged using a Hitachi H-7650
transmission electron microscope (TEM; Hitachi, Ltd.), with
magnification ×3,500.
Tissue cAMP level assay
Liver tissue were collected at 6 h of reperfusion. A
cAMP complete ELISA kit (cat. no. ab133051; Abcam) was used to
determine the cAMP levels in liver tissue samples, according to the
manufacturer's instructions.
Statistical analysis
SPSS 11.0 (SPSS, Inc.) was used for the statistical
analysis. The data are expressed as the mean ± standard deviation
(SD). Statistical analyses were performed using unpaired two-tailed
Student's t-test or one-way ANOVA followed by Tukey's post-hoc
test, with P<0.05 considered to indicate statistical
significance.
Results
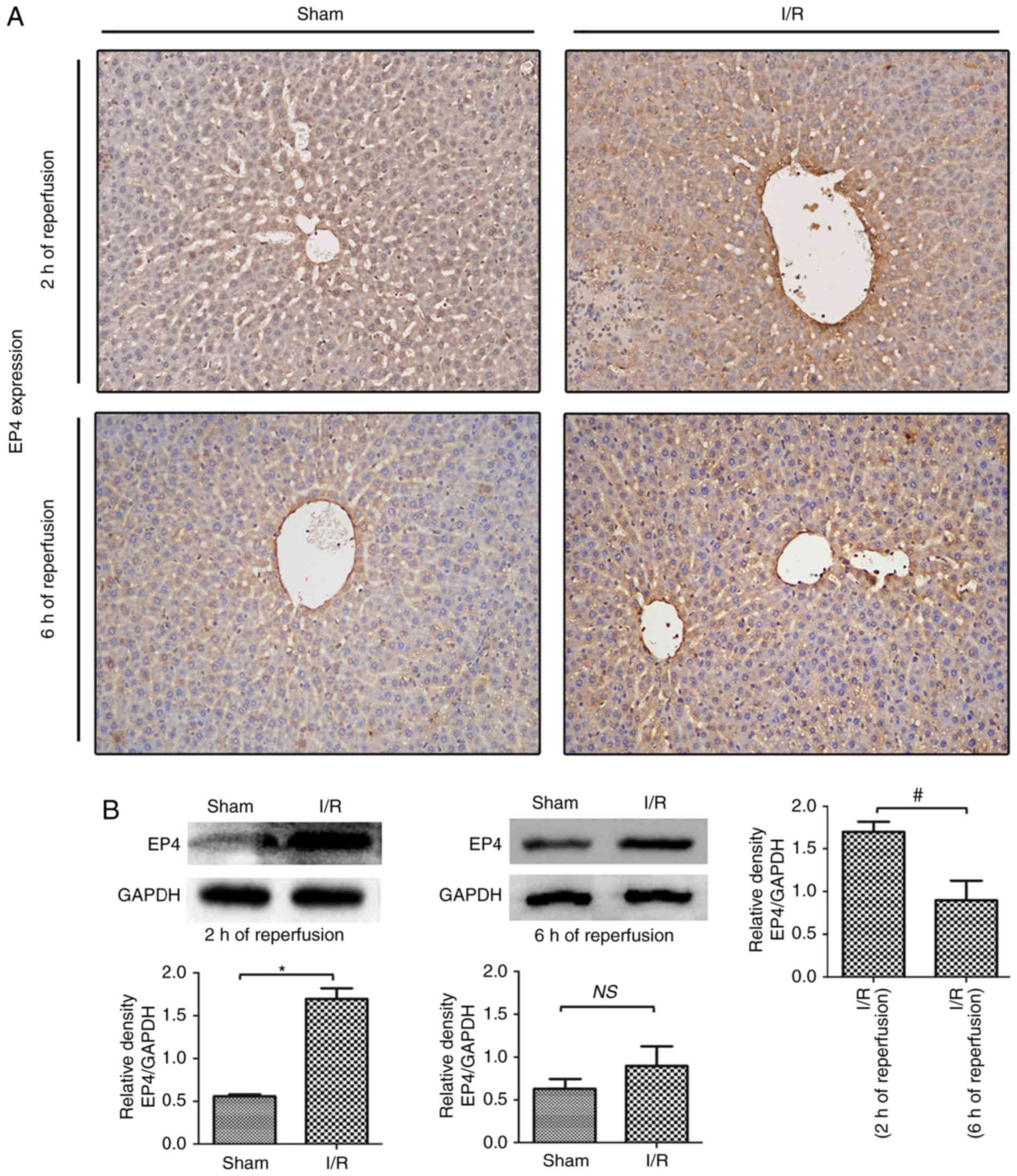
EP4 protein expression during hepatic I/R
injury
In the present study, EP4 protein expression during
rat hepatic I/R injury was examines using immunochemistry and
western blot analyses. As indicated in Fig. 1, EP4 expression was downregulated
in hepatocytes compared with the sham liver samples. After 2 h of
reperfusion following 60 min of ischemia, the EP4 protein was
prominently expressed in the membranes as well as in the cytoplasm
of hepatocytes, indicating that I/R induced EP4 protein expression
during early reperfusion. The expression of EP4 was lower in
I/R-injured livers after 6 h compared with after 2 h of reperfusion
(Fig. 1B), indicating that
decreased EP4 expression may be associated with liver injury during
reperfusion. Thus, in the following experiments, the effects of
activated EP4 signaling on hepatic I/R injury were examined by
subcutaneously injecting rats with the EP4-specific agonist CAY
prior to reperfusion.
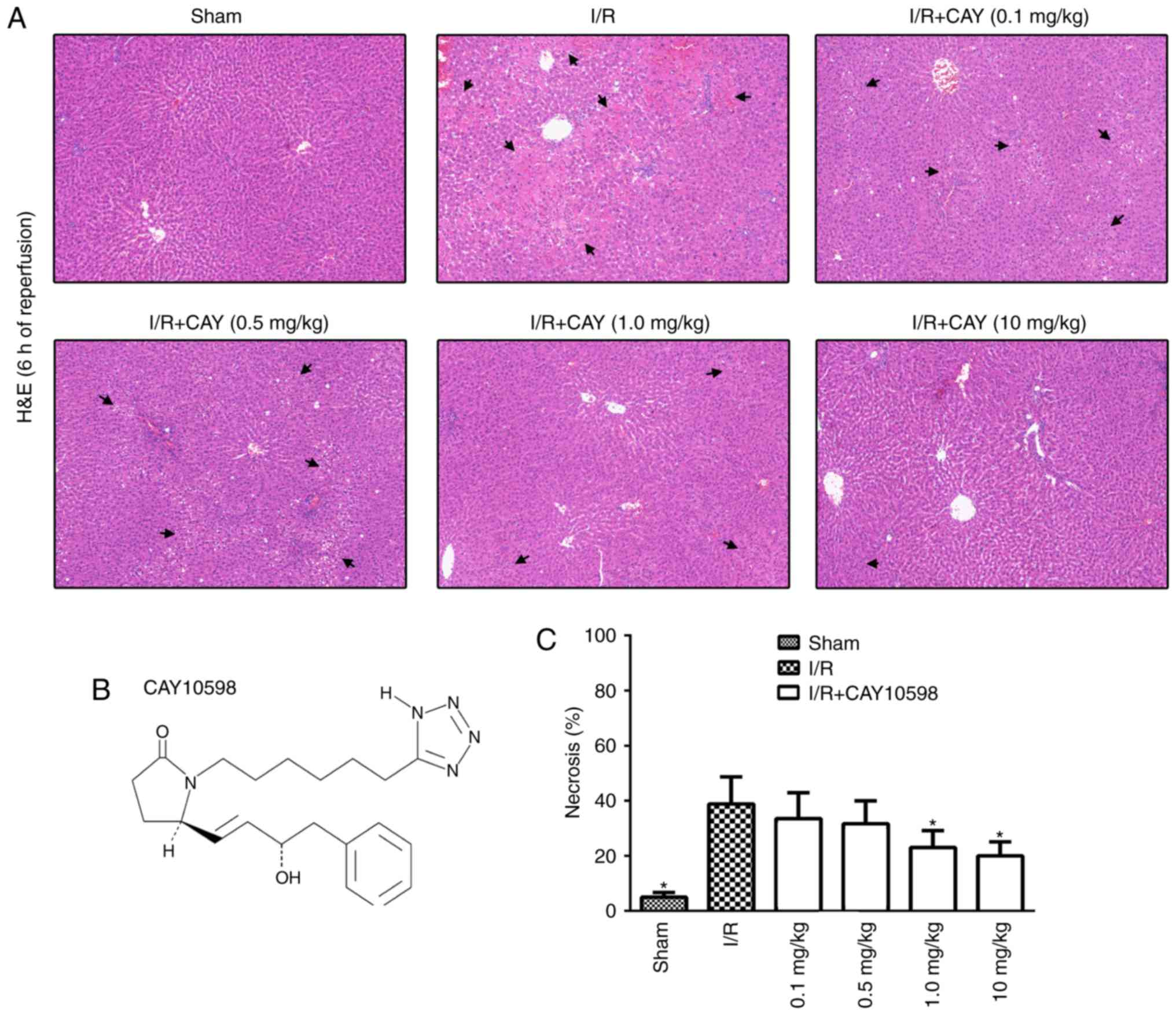
EP4 agonism protects against hepatic I/R
injury in a dose-dependent manner
To examine the dose-response relationship between
CAY and hepatic I/R, rats received CAY three times at a dose of
0.1, 0.5, 1 or 10 mg/kg. Liver sections obtained from the ischemic
lobe after 6 h of reperfusion were subjected to histopathological
analysis (Fig. 2A). A
semiquantitative scale was used to further determine the level of
hepatocellular necrosis. The necrosis rates after 6 h of
reperfusion following 60 min of ischemia for the 0.1, 0.5, 1 and 10
mg/kg treatments were 33.5±9.4, 31.7±8.3, 22.5±6.0 and 20.0±5.1,
respectively (Fig. 2C). The
current data indicate that this EP4 agonist inhibits hepatic I/R
injury in a dose-dependent manner. Therefore, the minimal effective
dose of CAY (1 mg/kg) was used to further investigate the
protective mechanisms of the EP4 agonist against hepatic I/R.
EP4 agonism protects the liver during
reperfusion by inhibiting MPTP opening
An MPTP opener, CATR (28), was used to determine the effect of
the EP4 agonist on MPTP modulation during I/R. As indicated in
Fig. 3A, I/R insult significantly
induced hepatic injury by increasing serum ALT and AST levels in
animals over 2 and 6 h of reperfusion, and treatment with 1 mg/kg
CAY markedly decreased the serum ALT and AST levels in I/R animals.
Next, hepatocyte necrosis and apoptosis were evaluated in each
group after 6 h of reperfusion using H&E and TUNEL staining. As
indicated in Fig. 3B and C, the
hepatocyte necrosis rates and TUNEL index values were significantly
lower in the I/R + CAY group compared with the I/R group. Notably,
the protective effect produced by CAY was partially reversed when
the MPTP opener CATR was administered 30 min prior to I/R insult.
The current results indicate that EP4 activation with CAY
significantly protects the liver, and that MPTP opening may serves
an important role in this effect.
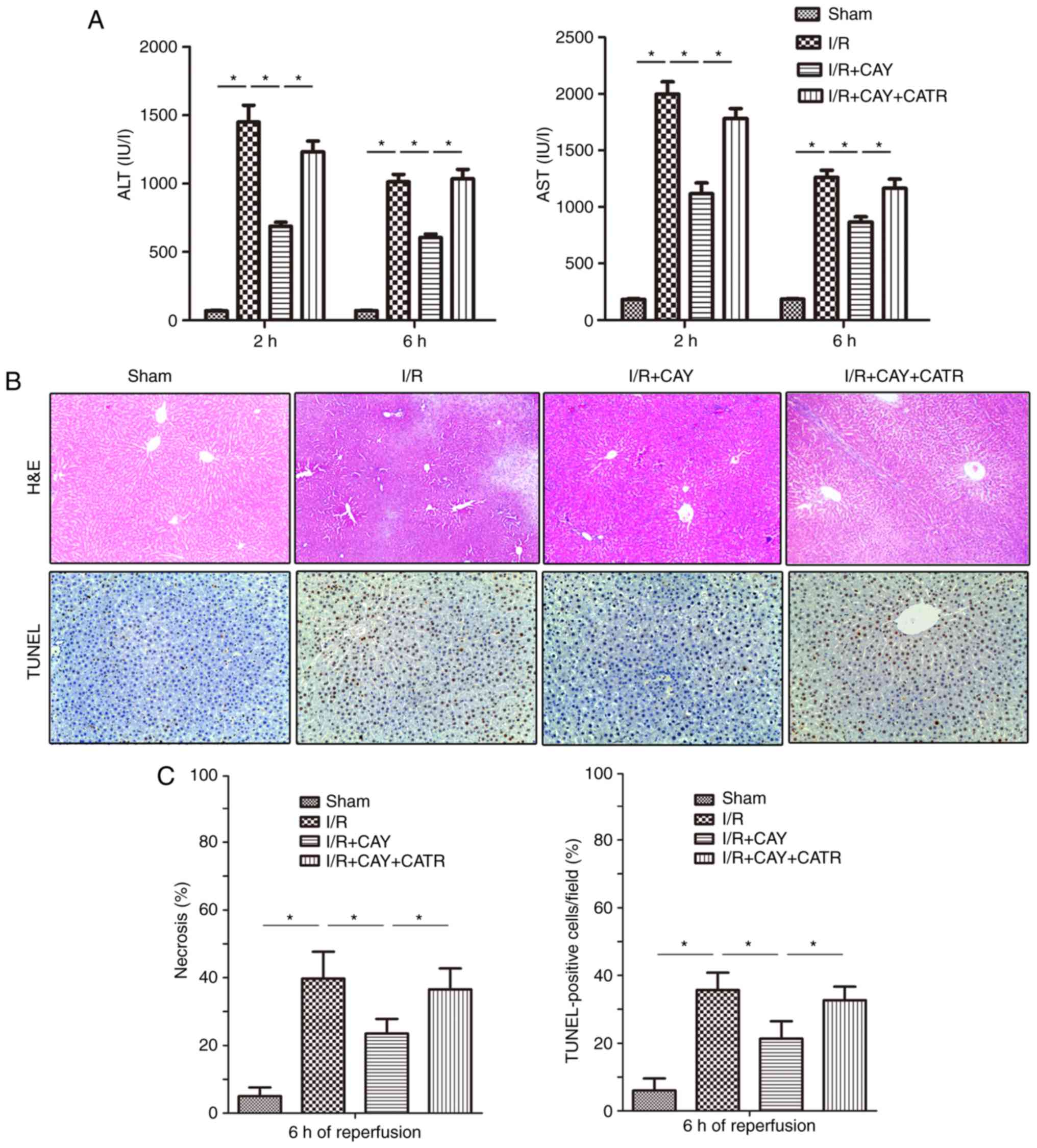 | Figure 3EP4 agonism significantly reduces
liver injury during I/R. CAY (1 mg/kg) was used to further
investigate the protective effect of an EP4 agonist on hepatic I/R.
(A) Serum ALT and AST levels in animals after 2 and 6 h of
reperfusion. (B) H&E staining and TUNEL staining of liver
tissue collected after 6 h of reperfusion; 100× magnification. In
the sham group, the morphology of the liver was regular, and intact
hepatocytes were observed. In contrast, large numbers of necrotic
hepatocytes and disordered hepatic sinusoidal morphology were
visible in the I/R groups. Only local hepatocyte necrosis was found
in the I/R+CAY treatment group. (C) Percentages of necrotic cells
measured in H&E-stained sections and percentages of apoptotic
cells measured in TUNEL-stained sections. n=6.
*P<0.05. I/R, ischemia/reperfusion; EP4,
prostaglandin E receptor subtype 4; H&E, hematoxylin and eosin;
TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick-end
labeling; CAY, CAY10598; ALT, alanine aminotransferase; AST,
aspartate aminotransferase; CATR, carboxyatractyloside. |
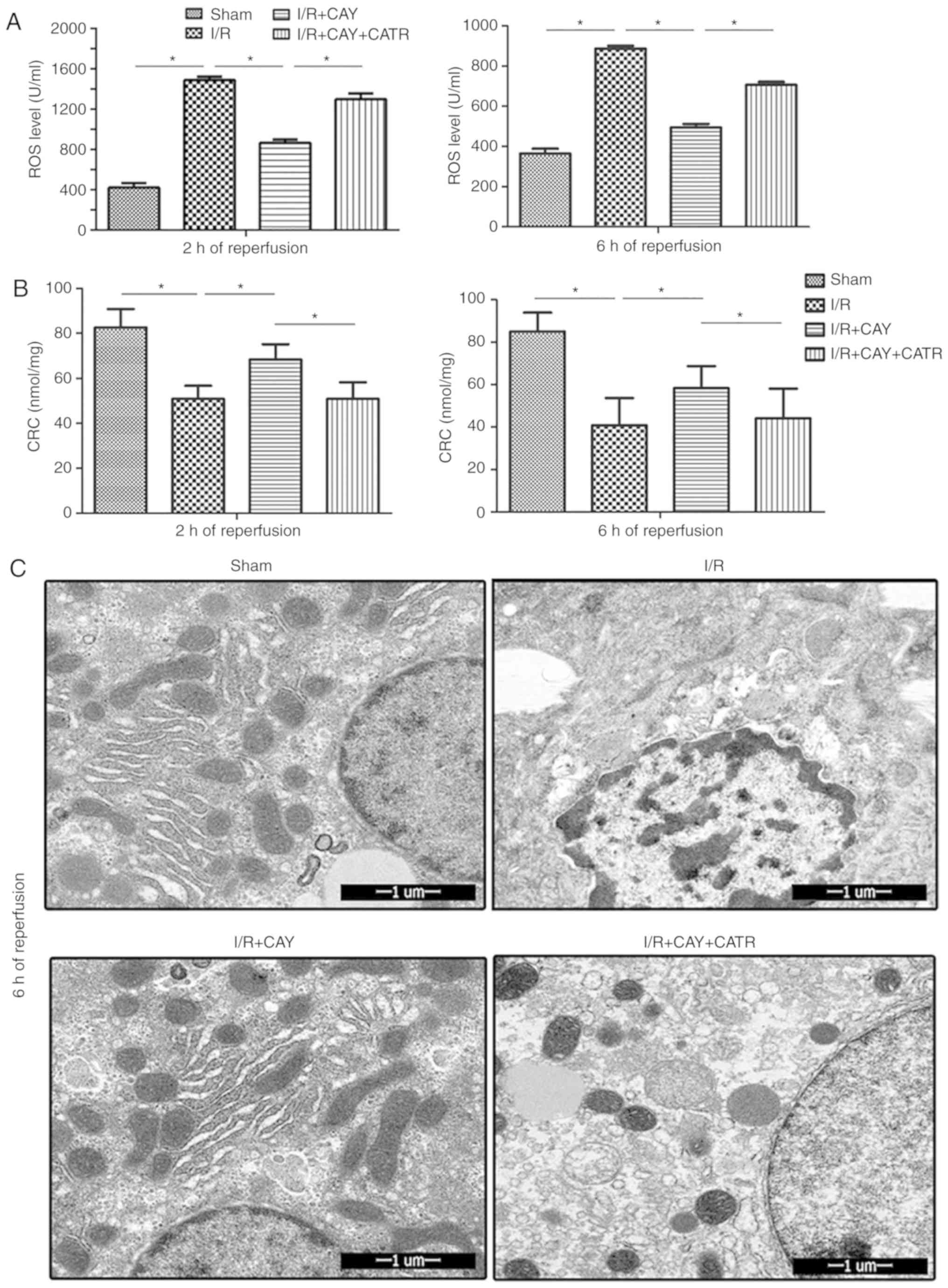
To further clarify the effect of EP4 signaling on
MPTP modulation during I/R, ROS levels were examined in hepatic
tissues and isolated hepatocyte mitochondria, in order to determine
the CRC, which is an index used to evaluate MPTP susceptibility,
after 2 and 6 h of reperfusion. As shown in Fig. 4, compared with the I/R group, the
I/R + CAY group exhibited significantly reduced liver ROS levels
(Fig. 4A) and increased
mitochondrial CRC values (Fig.
4B), indicating that MPTP opening is inhibited by EP4
activation. However, when the MPTP opener CATR was administered
prior to ischemia, the protective effects induced by the EP4
agonist were significantly reversed; the ROS levels in liver
tissues were significantly increased (Fig. 4A) and the CRC levels significantly
reduced (Fig. 4B) in the animals
treated with both CATR and CAY compared with those treated with CAY
alone, over 2 and 6 h of reperfusion. To further confirm this
result, hepatocyte ultrastructure was observed after 6 h of
reperfusion using transmission electron microscopy. As revealed in
Fig. 4C, hepatocyte nuclei
remained intact in both the sham and I/R + CAY groups, with most of
the mitochondrial morphology remaining intact. However, nuclear
chromatin condensation was found in the I/R group, and most of the
mitochondria were swollen or destroyed in both the I/R and I/R +
CAY + CATR groups. Given that MPTP opening serves a key role in
I/R-induced cell death, the current data further confirm that the
protective effect of EP4 activation on hepatic I/R is achieved by
inhibition of MPTP opening.
EP4 regulates MPTP opening via the
ERK1/2-GSK3β pathway
To further investigate the signaling cascades of
EP4-assicated MPTP modulation during I/R, the changes in cAMP
levels and ERK1/2 signaling were measured in hepatocytes. As
expected, EP4 activation significantly increased cAMP
concentrations and subsequent ERK1/2 signaling activation by
enhancing p-ERK1/2 levels in the liver tissue after 6 h of
reperfusion (Fig. 5A and B).
ERK1/2 is an intra-cellular signaling molecule that influences
various functions (including cell metabolism and development) and
interacts with a variety of intracellular signaling cascades.
Within these cascades, GSK3β and STAT3 have been confirmed to be
key molecules regulating MPTP opening (2,6,29).
Thus, changes in expression of GSK3β and JAK-STAT3 signaling
proteins were evaluated in hepatocytes after 6 h of reperfusion. As
indicated in Fig. 5C, the I/R
group exhibited significantly downregulated p-GSK3β expression,
compared with the sham group. However, compared with I/R alone, the
EP4 agonist significantly increased p-GSK3β expression in the liver
tissue, suggesting that EP4 activation may increase GSK3β
phosphorylation during reperfusion. In addition, as revealed in
Fig. 5D and E, I/R stimulated
increases in JAK2 phosphorylation and subsequent STAT3
phosphorylation, compared with sham treatment. However, compared
with I/R alone, the EP4 agonist failed to further stimulate the
phosphorylation of JAK2 and STAT3. The current results indicate
that EP4 activation-associated MPTP inhibition, and subsequent
hepatoprotection, may be associated with increased cAMP
concentration and subsequent ERK1/2-GSK3β signaling activation.
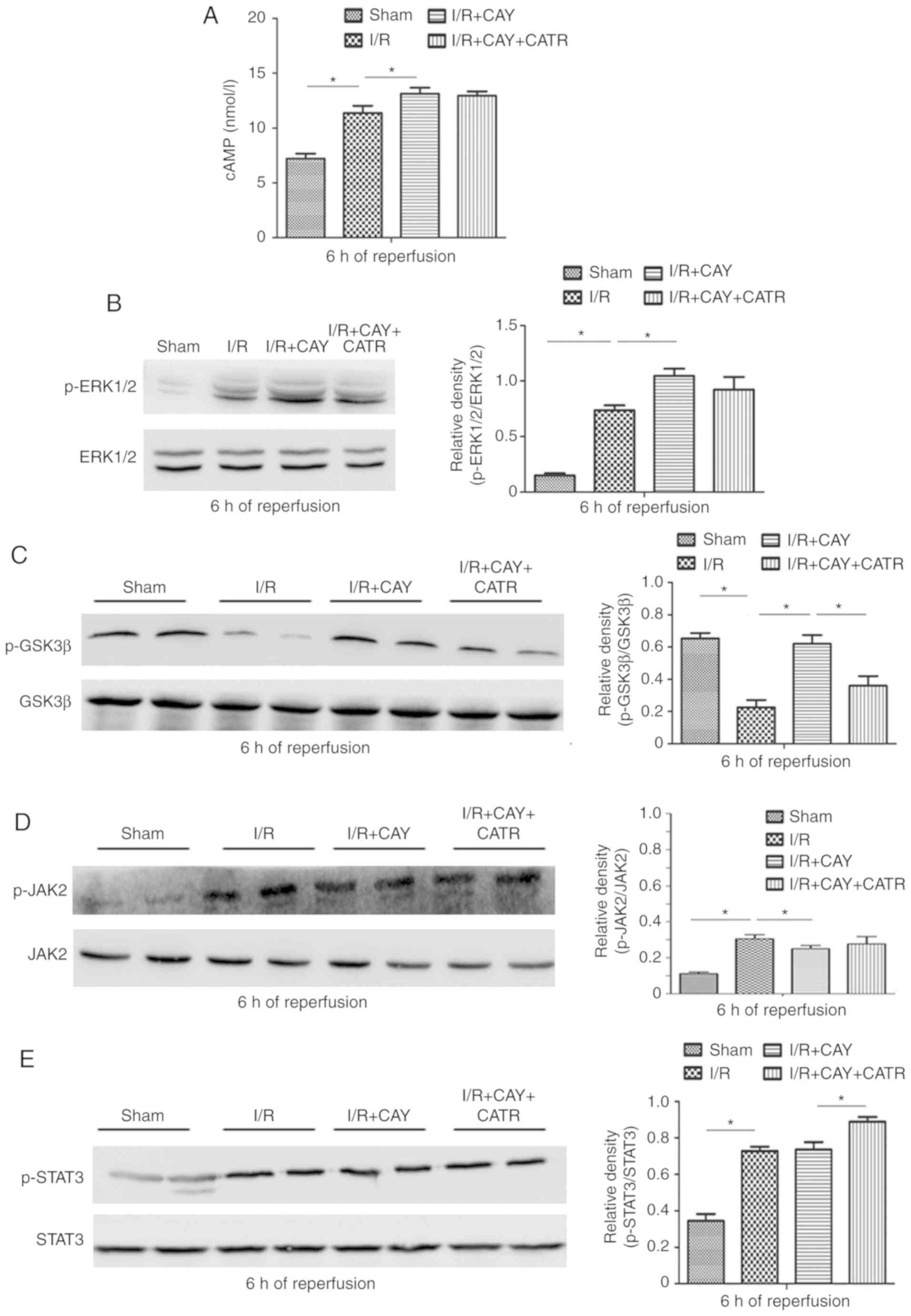 | Figure 5Effects of an EP4 agonist on
cAMP-ERK1/2-GSK3β and JAK2-STAT3 signaling after 6 h of
reperfusion. (A) cAMP levels in liver tissues. Western blot
analysis was performed to determine the protein expression of (B)
p-ERK1/2 and ERK1/2, (C) p-GSK3β and GSK3β, (D) p-JAK2 and JAK2 and
(E) p-STAT3 and STAT3 in liver tissues. n=6. *P<0.05.
I/R, ischemia/reperfusion; EP4, prostaglandin E receptor subtype 4;
CAY, CAY10598; CATR, carboxy-atractyloside; p-, phosphorylated;
JAK, janus kinase; STAT, signal transducer and activator of
transcription; GSK3β, glycogen synthase kinase 3β. |
Finally, to further confirm the role of ERK1/2-GSK3β
signaling in EP4-associated MPTP modulation, PD, an ERK1/2
inhibitor, was used to block the liver protection mediated by CAY.
As depicted in Fig. 6, the
necrotic hepatocyte percentages and ROS levels were significantly
higher in the I/R + CAY + PD group than in the I/R + CAY group
(Fig. 6A and D), while the CRC
level was significantly lower (Fig.
6C). Notably, p-GSK3β expression was inhibited in the animals
pretreated with PD after 6 h of reperfusion (Fig. 6B). Thus, the intraperitoneal
injection of the ERK1/2 inhibitor prior to ischemia significantly
reversed the protective effect of CAY during hepatic I/R, further
confirming that EP4 regulates the signaling cascade of MPTPs, via
the ERK1/2-GSK3β pathway.
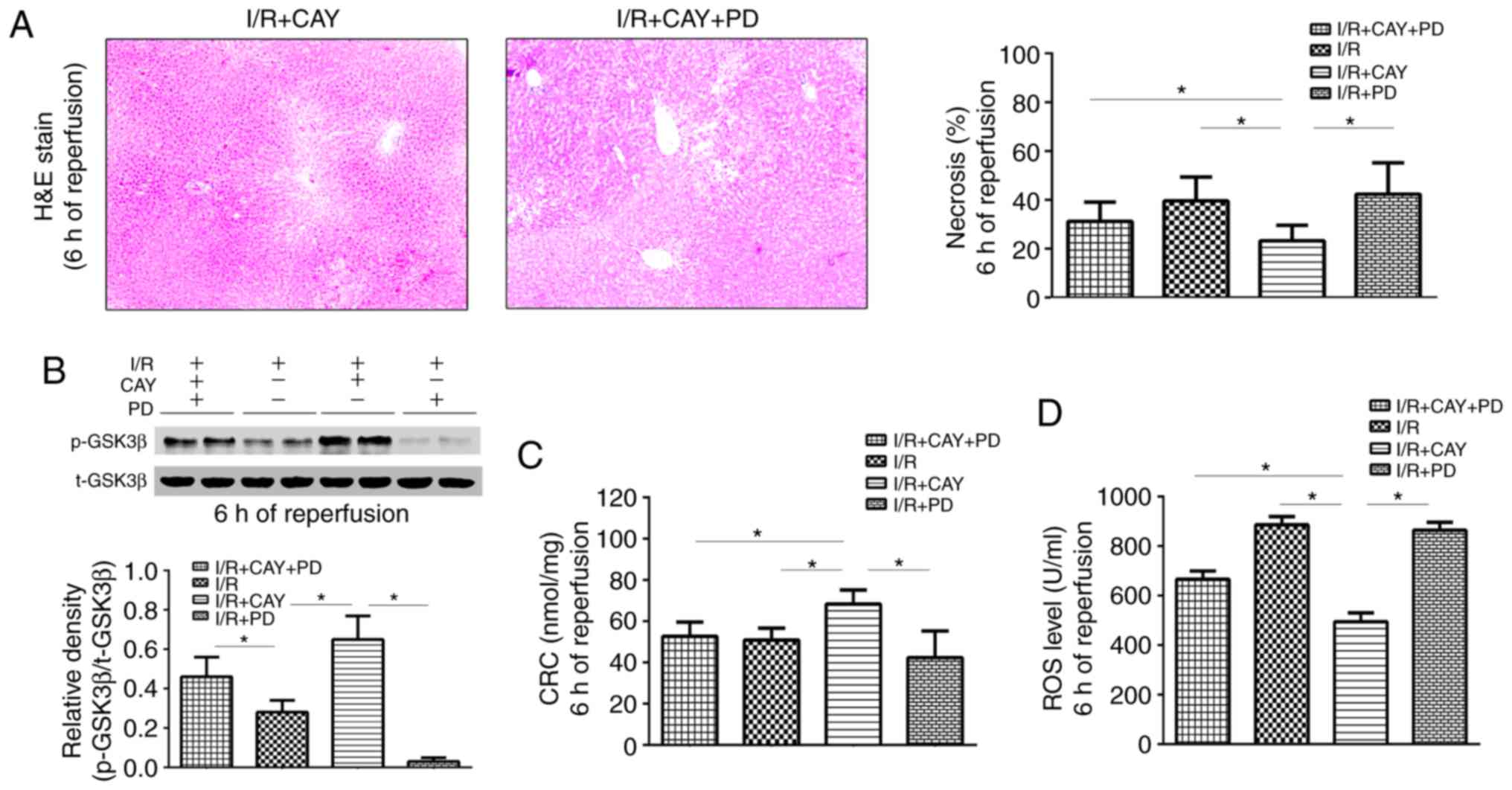 | Figure 6ERK1/2 inhibition decreases EP4
agonist-mediated hepatoprotection after 6 h of reperfusion. (A)
H&E staining of liver tissues (magnification, ×100) and
percentages of necrotic cells measured in the H&E-stained
sections. (B) Western blot analysis of p-GSK3β and GSK3β protein
expression in liver tissues. (C) CRC levels in liver mitochondria.
(D) ROS levels in liver tissues. n=6. *P<0.05. I/R,
ischemia/reperfusion; EP4, prostaglandin E receptor subtype 4;
H&E, hematoxylin and eosin; CAY, CAY10598; PD, PD98059; CATR,
carboxyatractyloside; ROS, reactive oxygen species; CRC, calcium
retention capacity; GSK3β, glycogen synthase kinase 3β. |
Taken together, the present results indicate that
EP4 activation protects the liver against I/R by activating
ERK1/2-GSK3β signaling and further inhibiting MPTP opening.
Discussion
The mechanism of I/R is complex and involves
extensive ROS production, inflammatory responses mediated by innate
immune cells (including endothelial cells, macrophages and
neutrophils) and apoptosis (1,3,7,9,12).
The factors that contribute to the initiation of this pathological
process are a disrupted supply of oxygen to cells and a lack of ATP
production (1). Mitochondria are
organelles that regulate oxidative phosphorylation-mediated ATP
production, and I/R injury is closely associated with the
regulation of mitochondrial function (2,30).
It has previously been revealed that COX-2 inhibition reduces
hepatic I/R injury via modulation of MPTP opening (7). The present study further
investigated the regulation of mitochondrial function and the
signaling cascade initiated by EP4 signaling, downstream of
COX-2.
EP4 is a Gαs (Gs) protein-coupled receptor that
serves an important role in various pathophysiological processes by
increasing the levels of the secondary messenger cAMP (12-16). There is evidence that endogenous
EP4 signaling is involved in I/R injury, as studies on I/R in the
liver and brain have revealed that an EP4 agonist can reduce organ
damage (12,15). Consistent with a report by
Kuzumoto et al (12), our
preliminary study also revealed that EP4 expression was
significantly upregulated in liver samples from rats and humans,
following I/R. Notably, the expression level of EP4 was lower in
I/R-injured livers after 6 h of reperfusion than after 2 h of
reperfusion, indicating the possibility that decreased EP4
expression may be associated with liver injury during reperfusion.
Indeed, alterations to intracellular signaling may be dynamic
during reperfusion. A similar trend has also been reported in GSK3β
signaling. Increased phosphorylation of GSK3β on Ser9 has been
demonstrated to reduce I/R damage in numerous organs, especially in
the liver and heart (2,4,6,29,31). However, during early reperfusion,
increased p-GSK3β expression was reported in myocardial I/R tissue,
when physical and pharmacological increases in p-GSK3β expression
conferred a protective effect during I/R (32,33).
Mitochondria are organelles that rely on cellular
oxygen uptake and are the core utilizers of cellular oxygen. PGE2
regulates the oxygen uptake of hepatocytes by interfering with the
intracellular cAMP-dependent protein kinase pathway (34), it is also possible that EP4
signaling regulates mitochondrial function during hepatic I/R.
Thus, the effect of EP4 on hepatic I/R and the mechanism underlying
the EP4 signaling-associate modulation of MPTPs were investigated
in the present study.
In an animal model of 70% partial liver I/R, CAY (an
EP4 agonist) was used to observe the effect of EP4 activation on
MPTP opening and liver damage. CAY is a very potent EP4 agonist and
is highly selective of EP4 over EP1-3 or other prostanoid receptors
(35). Studies have indicated
that CAY effectively activates endogenous EP4 signaling by
increasing cAMP production in various animal and human tissues
(36-40). In the present study, CAY protected
the liver from I/R in a dose-dependent manner. Subcutaneous
injection of CAY at a dose of 1 mg/kg significantly reduced serum
ALT and AST levels, and necrotic and apoptotic cell percentages,
during reperfusion. Furthermore, the EP4 agonist significantly
reduced liver ROS levels, increased mitochondrial CRC values and
protected mitochondrial morphology. CATR is an MPTP opener and has
been demonstrated to protect MPTP opening by stabilizing ADP/ATP
translocase on the cytosolic side of the mitochondrial inner
membrane (28). In the current
study, CATR also partially reversed the protective effects of the
EP4 agonist on the liver and mitochondria, further confirming the
key role of EP4 signaling in MPTP modulation during hepatic
I/R.
MPTPs are functional channels located on the inner
mitochondrial membrane that are usually closed. However, under
specific circumstances, such as hypoxia or I/R, a large number of
free radicals promote MPTP opening, further causing mitochondrial
damage and proapoptotic factor release (2). Numerous studies have confirmed that
the mitochondrial dysfunction caused by MPTP opening serves an
important role in cell necrosis and apoptosis during I/R (2,7,30,41). In the early reperfusion stage, I/R
damage can be directly ameliorated if MPTP opening is effectively
inhibited (4,30).
MPTP susceptibility is affected by many factors.
GSK3β and STAT3 have been demonstrated to be important regulators
of MPTP opening, as increased phosphorylation of GSK3β and STAT3
inhibits MPTP opening (2,6,29,42,43). GSK3β, a highly conserved
serine/threonine protein kinase, is ubiquitously expressed and
active in quiescent cells; however, both the Wnt and PI3K/protein
kinase B (Akt) signaling pathways downregulate its activity via
inhibitory phosphorylation of serine residues (5,42).
Several previous studies have confirmed that protection against I/R
injury is enhanced via Akt-GSK3β signaling activation (32,44,45). A classic protective strategy,
ischemia preconditioning (IPC), is considered to trigger multiple
protective signaling pathways during I/R, including the PKA,
PI3K/Akt, PKC and ERK1/2 pathways, of which GSK3β is a crucial
downstream regulator (23,46,47).
It has previously been revealed that enhanced phosphorylation of
GSK3β on Ser9 in rats with liver I/R inhibits MPTP opening and
reduces liver damage (4,31). Varela et al (48) also reported that a GSK3β inhibitor
(indirubin-3′-oxime) prevents hepatic I/R damage by inhibiting MPTP
opening. Phosphorylated GSK3β inhibits MPTP opening via multiple
mechanisms, including preservation of hexokinase II in the MPTP
complex, prevention of the interaction of cyclophilin-D with
adenine nucleotide translocase, inhibition of p53 activation and
attenuation of ATP hydrolysis during ischemia (42). In the present study, it was
revealed that an EP4 agonist also significantly increased p-GSK3β
expression in liver tissue during reperfusion, indicating that EP4
modulates MPTP opening via GSK3β. GSK3β is a convergence point for
multiple cell signaling cascades (49), such as the classic reperfusion
injury salvage kinase (RISK) pathway and the survivor activating
factor enhancement (SAFE) pathway (4-6),
justifying the choice of this signal transducer as a target for
potentiating pro-survival cascades and maximizing organ protection
(50,51). The RISK pathway consists of
Akt-GSK3β and ERK1/2 signaling events, whereas the SAFE pathway
involves the activation of tumor necrosis factor (TNF)-α and the
JAK-STAT axis (52). Thus,
pharmacological EP4 activation may directly or indirectly affect
one or more of these signals.
EP4 is described as a Gs protein-coupled receptor
that induces intracellular cAMP production, resulting in
PI3K-ERK1/2 signaling activation (14). The ERK1/2 signaling cascade was
previously revealed to influence EP4-mediated cardiac hypertrophy
(16). Wang et al
(53) found that remote ischemic
preconditioning (RIPC) protects the rat's liver from I/R injury by
inducing the heme oxygenase-1/ERK1/2-dependent autophagy. In
addition, Yang et al (54)
revealed that triiodo-thyronine preconditioning protects against
liver I/R injury in mice by regulating autophagy through the
MEK/ERK axis. Similar to the present findings, increased p-ERK1/2
expression was also found in I/R livers both in vivo and
in vitro in these two experiments, indicating that ERK1/2
signaling may mediate I/R-induced liver damage (53,54). Furthermore, pharmacological or
physical activation of ERK1/2 signaling via further enhancement of
p-ERK1/2 levels has been shown to exert protective effects during
I/R (46,53-55). In the current study, CAY treatment
significantly increased cAMP concentrations and subsequent ERK1/2
signaling activation by enhancing p-ERK1/2 levels in liver tissue
after 6 h of reperfusion, whereas the ERK1/2 signaling inhibitor PD
significantly inhibited the EP4-mediated GSK3β phosphorylation and
hepatoprotective effects, further confirming that activation of
ERK1/2 signaling may alleviate I/R-induced liver damage and mediate
the protective effect of CAY.
Previous studies have confirmed the role of the
JAK-STAT pathway in myocardial injury (29,43). For example, Gross et al
(51) found that the
JAK/STAT/GSK3β pathway is essential for opioid-induced
cardioprotection. Boengler et al (6) reported that STAT3 may exert
cardioprotective effects by stimulating respiration and inhibiting
MPTP opening. In the current study, the use of an EP4 agonist did
not significantly increase the expression of p-JAK2 and p-STAT3
during reperfusion in the context of liver I/R, which does not
support the conclusion that EP4 regulates MPTPs and liver function
via JAK2-STAT3 signaling.
In conclusion, the current data reveal a novel
function of active EP4; the induction of ERK1/2-GSK3β signaling and
subsequent effects of MPTP inhibition and hepatoprotection.
However, considering the diversity and functional complexity of
prostaglandin receptors and the complicated process of I/R-induced
hepatocyte death, the present study only considered the relevant
major mechanistic pathways. Nevertheless, these findings may shed
new light on the clinical use of EP4 agonists for liver protection
in surgical settings.
Supplementary Data
Abbreviations:
|
EP
|
prostaglandin E receptor
|
|
PGE2
|
prostaglandin E2
|
|
COX-2
|
cyclooxygenase-2
|
|
AA
|
arachidonic acid
|
|
MPTP
|
mitochondrial permeability transition
pore
|
|
I/R
|
ischemia/reperfusion
|
|
CATR
|
carboxyatractyloside
|
|
CAY
|
CAY10598
|
|
ALT
|
alanine aminotransferase
|
|
AST
|
aspartate aminotransferase
|
|
PKA
|
protein kinase A
|
|
Akt
|
protein kinase B
|
|
GSK3β
|
glycogen synthase kinase 3β
|
|
ATP
|
adenosine triphosphate
|
|
PI3K
|
phosphoinositide 3′-OH kinase
|
|
ERK1/2
|
extracellular signal-regulated protein
kinase 1/2
|
|
RISK
|
reperfusion injury salvage kinase
|
|
SAFE
|
survivor activating factor
enhancement
|
|
JAK
|
janus kinase
|
|
STAT
|
signal transducer and activator of
transcription
|
|
ROS
|
reactive oxygen species
|
|
H&E
|
hematoxylin and eosin
|
|
TUNEL
|
terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling
|
|
CRC
|
calcium retention capacity
|
|
cAMP
|
cyclic adenosine monophosphate
|
|
TNF-α
|
tumor necrosis factor-α
|
Acknowledgements
Not applicable.
Funding
The present study was partially supported by grants
from the National Natural Science Foundation of China (grant nos.
81670564, 81300344, 81671304 and 81873945). The funders did not
play any role in the study design, data collection and analysis,
decisions regarding data release or manuscript preparation.
Availability of data and materials
The datasets used during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
LLC, HTX and QLW wrote the manuscript, designed the
experiment, and contributed equally to this work. YQZ, WC, DYZ, FL
and YHL helped conduct the experiment and data analyses; and HLF,
YHL and HBY conducted the experiments and assisted with paper
writing. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experiments were approved by the Changzheng
Hospital Ethics Committee [approval number, CZEC (2015)-01] and
were performed in compliance with the Guide for the Care and Use of
Laboratory Animals published by the US National Institutes of
Health (NIH publication no. 85-23, revised 1996).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Howard TK, Klintmalm GB, Cofer JB, Husberg
BS, Goldstein RM and Gonwa TA: The influence of preservation injury
on rejection in the hepatic transplant recipient. Transplantation.
49:103–107. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Halestrap AP: What is the mitochondrial
permeability transition pore? J Mol Cell Cardiol. 46:821–831. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhu J, Rebecchi MJ, Glass PS, Brink PR and
Liu L: Cardioprotection of the aged rat heart by GSK-3beta
inhibitor is attenuated: Age-related changes in mitochondrial
permeability transition pore modulation. Am J Physiol Heart Circ
Physiol. 300:H922–H930. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fu H, Xu H, Chen H, Li Y, Li W, Zhu Q,
Zhang Q, Yuan H, Liu F, Wang Q, et al: Inhibition of glycogen
synthase kinase 3 ameliorates liver ischemia/reperfusion injury via
an energy-dependent mitochondrial mechanism. J Hepatol. 61:816–824.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gomez L, Paillard M, Thibault H, Derumeaux
G and Ovize M: Inhibition of GSK3beta by postconditioning is
required to prevent opening of the mitochondrial permeability
transition pore during reperfusion. Circulation. 117:2761–2768.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Boengler K, Hilfiker-Kleiner D, Heusch G
and Schulz R: Inhibition of permeability transition pore opening by
mitochondrial STAT3 and its role in myocardial
ischemia/reperfusion. Basic Res Cardiol. 105:771–785. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fu H, Chen H, Wang C, Xu H, Liu F, Guo M,
Wang Q and Shi X: Flurbiprofen, a cyclooxygenase inhibitor,
protects mice from hepatic ischemia/reperfusion injury by
inhibiting GSK-3β signaling and mitochondrial permeability
transition. Mol Med. 18:1128–1135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kwak HJ, Park KM, Choi HE, Lim HJ, Park JH
and Park HY: The cardioprotective effects of zileuton, a
5-lipoxygenase inhibitor, are mediated by COX-2 via activation of
PKC delta. Cell Signal. 22:80–87. 2010. View Article : Google Scholar
|
|
9
|
Hwang HS, Yang KJ, Park KC, Choi HS, Kim
SH, Hong SY, Jeon BH, Chang YK, Park CW, Kim SY, et al:
Pretreatment with paricalcitol attenuates inflammation in
ischemia-reperfusion injury via the up-regulation of
cyclooxygenase-2 and prostaglandin E2. Nephrol Dial Transplant.
28:1156–1166. 2013. View Article : Google Scholar
|
|
10
|
Ho ATV, Palla AR, Blake MR, Yucel ND, Wang
YX, Magnusson KEG, Holbrook CA, Kraft PE, Delp SL and Blau HM:
Prostaglandin E2 is essential for efficacious skeletal muscle
stem-cell function, augmenting regeneration and strength. Proc Natl
Acad Sci USA. 114:6675–6684. 2017.PubMed/NCBI
|
|
11
|
Narumiya S and FitzGerald GA: Genetic and
pharmacological analysis of prostanoid receptor function. J Clin
Invest. 108:25–30. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kuzumoto Y, Sho M, Ikeda N, Hamada K,
Mizuno T, Akashi S, Tsurui Y, Kashizuka H, Nomi T, Kubo A, et al:
Significance and therapeutic potential of prostaglandin E2 receptor
in hepatic ischemia/reperfusion injury in mice. Hepatology.
42:608–617. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nataraj C, Thomas DW, Tilley SL, Nguyen
MT, Mannon R, Koller BH and Coffman TM: Receptors for prostaglandin
E(2) that regulate cellular immune responses in the mouse. J Clin
Invest. 108:1229–1235. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Konya V, Marsche G, Schuligoi R and
Heinemann A: E-type prostanoid receptor 4 (EP4) in disease and
therapy. Pharmacol Ther. 138:485–502. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liang X, Lin L, Woodling NS, Wang Q,
Anacker C, Pan T, Merchant M and Andreasson K: Signaling via the
prostaglandin E(2) receptor EP4 exerts neuronal and vascular
protection in a mouse model of cerebral ischemia. J Clin Invest.
121:4362–4371. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pang L, Cai Y, Tang EH, Irwin MG, Ma H and
Xia Z: Prostaglandin E receptor subtype 4 signaling in the heart:
Role in ischemia/reperfusion injury and cardiac hypertrophy. J
Diabetes Res. 2016:13243472016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mo C, Zhao R, Vallejo J, Igwe O, Bonewald
L, Wetmore L and Brotto M: Prostaglandin E2 promotes proliferation
of skeletal muscle myoblasts via EP4 receptor activation. Cell
Cycle. 14:1507–1516. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Naribayashi-Inomoto Y, Ding M, Nakata H,
Narumiya S, Sugimoto Y, Honda A, Ichikawa A, Chiba T and Kinoshita
Y: Copresence of prostaglandin EP2 and EP3 receptors on gastric
enterochromaffin-like cell carcinoid in African rodents.
Gastroenterology. 109:341–347. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nishizawa N, Ito Y, Eshima K, Ohkubo H,
Kojo K, Inoue T, Raouf J, Jakobsson PJ, Uematsu S, Akira S, et al:
Inhibition of microsomal prostaglandin E synthase-1 facilitates
liver repair after hepatic injury in mice. J Hepatol. 69:110–120.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakamura K, Kageyama S, Ito T, Hirao H,
Kadono K, Aziz A, Dery KJ, Everly MJ, Taura K, Uemoto S, et al:
Antibiotic pretreatment alleviates liver transplant damage in mice
and humans. J Clin Invest. 129:3420–3434. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Selzner N, Selzner M, Jochum W and Clavien
PA: Ischemic preconditioning protects the steatotic mouse liver
against reperfusion injury: An ATP dependent mechanism. J Hepatol.
39:55–61. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu A, Dirsch O, Fang H, Dong W, Jin H,
Huang H, Sun J and Dahmen U: HMGB1 translocation and expression is
caused by warm ischemia reperfusion injury, but not by partial
hepatectomy in rats. Exp Mol Pathol. 91:502–508. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yamada F, Saito T, Abe T, Tsuchiya T, Sato
Y, Kenjo A, Kimura T and Gotoh M: Ischemic preconditioning enhances
regenerative capacity of hepatocytes in long-term ischemically
damaged rat livers. J Gastroenterol Hepatol. 22:1971–1977. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Limani P, Linecker M, Oberkofler CE,
Barmettler G, Kaech A, Graf R, Humar B and Clavien PA: Remote
ischemic preconditioning: A novel strategy in rescuing older livers
from ischemia-reperfusion injury in a rodent model. Ann Surg.
264:797–803. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yamamoto M, Morita T, Ishikawa M and
Sakamoto A: Specific microRNAs are involved in the renoprotective
effects of sevoflurane preconditioning and ischemic preconditioning
against ischemia reperfusion injury in rats. Int J Mol Med.
45:1141–1149. 2020.PubMed/NCBI
|
|
26
|
Parks RJ, Murphy E and Liu JC:
Mitochondrial permeability transition pore and calcium handling.
Methods Mol Biol. 1782:187–196. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ichas F, Jouaville LS, Sidash SS, Mazat JP
and Holmuhamedov EL: Mitochondrial calcium spiking: A transduction
mechanism based on calcium-induced permeability transition involved
in cell calcium signalling. FEBS Lett. 348:211–215. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Novgorodov SA, Gudz TI, Kushnareva YE,
Zorov DB and Kudrjashov YB: Effect of ADP/ATP antiporter
conformational state on the suppression of the nonspecific
permeability of the inner mitochondrial membrane by cyclosporine A.
FEBS Lett. 277:123–126. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Barry SP, Townsend PA, Latchman DS and
Stephanou A: Role of the JAK-STAT pathway in myocardial injury.
Trends Mol Med. 13:82–89. 2007. View Article : Google Scholar
|
|
30
|
Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu
Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, et al:
Glycogen synthase kinase-3beta mediates convergence of protection
signaling to inhibit the mitochondrial permeability transition
pore. J Clin Invest. 113:1535–1549. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cai L, Li Y, Zhang Q, Sun H, Yan X, Hua T,
Zhu Q, Xu H and Fu H: Salidroside protects rat liver against
ischemia/reperfusion injury by regulating the GSK-3β/Nrf2-dependent
antioxidant response and mitochondrial permeability transition. Eur
J Pharmacol. 806:32–42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Song JQ, Teng X, Cai Y, Tang CS and Qi YF:
Activation of Akt/GSK-3beta signaling pathway is involved in
intermedin(1-53) protection against myocardial apoptosis induced by
ischemia/reperfusion. Apoptosis. 14:1061–1069. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nishino Y, Webb IG, Davidson SM, Ahmed AI,
Clark JE, Jacquet S, Shah AM, Miura T, Yellon DM, Avkiran M and
Marber MS: Glycogen synthase kinase-3 inactivation is not required
for ischemic preconditioning or postconditioning in the mouse. Circ
Res. 103:307–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qu W, Graves LM and Thurman RG: PGE(2)
stimulates O(2) uptake in hepatic parenchymal cells: Involvement of
the cAMP-dependent protein kinase. Am J Physiol. 277:G1048–G1054.
1999.PubMed/NCBI
|
|
35
|
Billot X, Chateauneuf A, Chauret N, Denis
D, Greig G, Mathieu MC, Metters KM, Slipetz DM and Young RN:
Discovery of a potent and selective agonist of the prostaglandin
EP4 receptor. Bioorg Med Chem Lett. 13:1129–1132. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Luo R, Kakizoe Y, Wang F, Fan X, Hu S,
Yang T, Wang W and Li C: Deficiency of mPGES-1 exacerbates renal
fibrosis and inflammation in mice with unilateral ureteral
obstruction. Am J Physiol Renal Physiol. 312:F121–F133. 2017.
View Article : Google Scholar
|
|
37
|
Carboneau BA, Allan JA, Townsend SE,
Kimple ME, Breyer RM and Gannon M: Opposing effects of
prostaglandin E2 receptors EP3 and EP4 on mouse and human β-cell
survival and proliferation. Mol Metab. 6:548–559. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Quan Y, Jiang J and Dingledine R: EP2
receptor signaling path-ways regulate classical activation of
microglia. J Biol Chem. 288:9293–9302. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang F, Lu X, Peng K, Du Y, Zhou SF, Zhang
A and Yang T: Prostaglandin E-prostanoid4 receptor mediates
angiotensin II-induced (pro)renin receptor expression in the rat
renal medulla. Hypertension. 64:369–377. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Machado-Carvalho L, Torres R,
Perez-Gonzalez M, Alobid I, Mullol J, Pujols L, Roca-Ferrer J and
Picado C: Altered expression and signalling of EP2 receptor in
nasal polyps of AERD patients: Role in inflammation and
remodelling. Rhinology. 54:254–265. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tanaka T, Saotome M, Katoh H, Satoh T,
Hasan P, Ohtani H, Satoh H, Hayashi H and Maekawa Y: Glycogen
synthase kinase-3β opens mitochondrial permeability transition pore
through mitochondrial hexokinase II dissociation. J Physiol Sci.
68:865–871. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Miura T and Tanno M: Mitochondria and
GSK-3beta in cardio-protection against ischemia/reperfusion injury.
Cardiovasc Drugs Ther. 24:255–263. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bolli R, Dawn B and Xuan YT: Role of the
JAK-STAT pathway in protection against myocardial
ischemia/reperfusion injury. Trends Cardiovasc Med. 13:72–79. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gracia-Sancho J, Casillas-Ramirez A and
Peralta C: Molecular pathways in protecting the liver from
ischaemia/reperfusion injury: A 2015 update. Clin Sci (Lond).
129:345–362. 2015. View Article : Google Scholar
|
|
45
|
Izuishi K, Tsung A, Hossain MA, Fujiwara
M, Wakabayashi H, Masaki T, Billiar TR and Maeta H: Ischemic
preconditioning of the murine liver protects through the Akt kinase
pathway. Hepatology. 44:573–580. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cao CM, Zhang Y, Weisleder N, Ferrante C,
Wang X, Lv F, Zhang Y, Song R, Hwang M, Jin L, et al: MG53
constitutes a primary determinant of cardiac ischemic
preconditioning. Circulation. 121:2565–2574. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Centurion SA, Centurion LM, Souza ME,
Gomes MC, Sankarankutty AK, Mente ED and Castro e Silva O: Effects
of ischemic liver preconditioning on hepatic ischemia/reperfusion
injury in the rat. Transplant Proc. 39:361–364. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Varela AT, Simoes AM, Teodoro JS, Duarte
FV, Gomes AP, Palmeira CM and Rolo AP: Indirubin-3′-oxime prevents
hepatic I/R damage by inhibiting GSK-3beta and mitochondrial
perme-ability transition. Mitochondrion. 10:456–463. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Beurel E, Grieco SF and Jope RS: Glycogen
synthase kinase-3 (GSK3): Regulation, actions, and diseases.
Pharmacol Ther. 148:114–131. 2015. View Article : Google Scholar
|
|
50
|
Gross ER, Hsu AK and Gross GJ: GSK3beta
inhibition and K(ATP) channel opening mediate acute opioid-induced
cardio-protection at reperfusion. Basic Res Cardiol. 102:341–349.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gross ER, Hsu AK and Gross GJ: The
JAK/STAT pathway is essential for opioid-induced cardioprotection:
JAK2 as a mediator of STAT3, Akt, and GSK-3 beta. Am J Physiol
Heart Circ Physiol. 291:H827–H834. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lecour S: Activation of the protective
survivor activating factor enhancement (SAFE) pathway against
reperfusion injury: Does it go beyond the RISK pathway? J Mol Cell
Cardiol. 47:32–40. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang Y, Shen J, Xiong X, Xu Y, Zhang H,
Huang C, Tian Y, Jiao C, Wang X and Li X: Remote ischemic
preconditioning protects against liver ischemia-reperfusion injury
via heme oxygenase-1-induced autophagy. PLoS One. 9:e988342014.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang J, Wang Y, Sui M, Liu F, Fu Z and
Wang QX: Tri-iodothyronine preconditioning protects against liver
ischemia reperfusion injury through the regulation of autophagy by
the MEK/ERK/mTORC1 axis. Biochem Biophys Res Commun. 467:704–710.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yu J, Wang L, Akinyi M, Li Y, Duan Z, Zhu
Y and Fan G: Danshensu protects isolated heart against ischemia
reperfusion injury through activation of Akt/ERK1/2/Nrf2 signaling.
Int J Clin Exp Med. 8:14793–14804. 2015.PubMed/NCBI
|