Introduction
Cells and the extracellular matrix (ECM) in the
airways are subjected to various mechanical stimuli, such as the
continuous cyclic stretch of breathing and the contraction of the
airway smooth muscle, due to the dynamic nature of lung function
(1). Pathologically high levels
of stretch exerted on lung tissues play a key role in many
pathological situations. This process can induce the expression of
various inflammatory mediators, including interleukin (IL)-6, tumor
necrosis factor (TNF)-α, IL-13 and matrix metalloproteinase (MMP-9)
(2,3), and an increase in goblet cell number
and mucin5AC (MUC5AC) protein secretion (4,5).
Furthermore, excessive stretching is a major cause of airway
remodeling (2,6-8).
Over the past few years, extensive research has been conducted to
investigate the role of abnormal mechanical stress in airway
remodeling in asthma; however, very little attention has been
devoted to stress-induced airway remodeling in chronic obstructive
pulmonary disease (COPD), which is characterized by chronic
coughing, sputum production and recurrent episodes of wheezing that
result in the pathological upregulation of airway pressure.
Airway remodeling is a critical feature of COPD,
characterized by the aberrant repair of the epithelium and the
accumulation of fibroblasts, which contribute to ECM deposition,
leading to irreversible airway obstruction and progressive high
pressure in the airways (9,10).
Thus, airway remodeling and increased airway mechanical stress
function in a vicious cycle that may contribute to the ongoing
decline in lung function and quality of life and, ultimately, the
poor prognosis of patients with COPD. Thus, further elucidation of
the mechanisms of mechanical stress-induced airway remodeling in
COPD is important for the treatment of this disease.
Recently, epithelial-mesenchymal transition (EMT)
has been identified as a new source of fibroblasts that can
contribute to the remodeling of the airways (11). EMT is a biological process that
allows a polarized epithelial cell with cell-cell contacts that is
attached to the basal membrane to acquire the characteristics of
mesenchymal cells through multiple biochemical alterations
(12,13). Significant EMT has been
demonstrated in the airway epithelial cells of patients with COPD
(14-16), and EMT has been suggested as an
important factor for airway remodeling in COPD (17,18). Previous studies have demonstrated
that primary airway epithelial cells subjected to mechanical
stretch acquire EMT phenotypes (19-21). However, the specific molecular
mechanisms of EMT in response to mechanical stress remain poorly
understood.
Transient receptor potential canonical 1 (TRPC1), a
member of the vertebrate mechanosensitive cation (MscCa) channels,
plays a critical role in converting the sensed mechanical stimuli
into biological signals by transuding stretch into Ca2+
flux across the cell membrane (22,23). TRPC1 has been reported to be
abundantly expressed in airway epithelial cells and its expression
is markedly increased in patients with COPD (18). Furthermore, the recent study by Xu
et al indicated that the increase in TRPC1 expression was
closely related to the occurrence of EMT in patients with COPD
where they exposed human bronchial epithelial (HBE; 16HBE cells) to
5% cigarette smoking extract (CSE) (18). Therefore, it was hypothesized that
TRPC1 plays a vital role in the process of mechanical
stress-induced airway remodeling in COPD via the occurrence of EMT.
To test this hypothesis, in vivo, the present study examined
the expression of TRPC1 in patients with COPD. In vitro,
16HBE cells we exposed to mechanical stretch for up to 48 h to
mimic the effects of high airway pressure, and TRPC1 expression was
then measured by RT-qPCR and western blot analysis. The function of
TRPC1 was assessed by Ca2+ imaging and siRNA
transfection, and the occurrence of EMT was identified by
immunofluorescence, western blot analysis and RT-qPCR. It was that
TRPC1 expression was upregulated in patients with COPD and in 16HBE
cells subjected to mechanical stretch. The TRPC1-mediated increase
in intracellular Ca2+ played a key role in the
occurrence of EMT in human lung epithelial cells in response to
mechanical stretch. Thus, this molecule may serve as a novel
therapeutic target for progressive airway remodeling in COPD.
Materials and methods
Reagents
16HBE cells were purchased from the American Type
Culture Collection (ATCC). Lipofectamine RNAiMAX reagent was
obtained from Invitrogen; Thermo Fisher Scientific, Inc. Rabbit
polyclonal anti-TRPC1 antibody (ab75322) was purchased from Abcam.
Mouse monoclonal anti-cytokeratin 8 (sc-58736) and E-cadherin
(sc-8426) antibodies were purchased from Santa Cruz Biotechnology,
Inc. Rabbit anti-α-smooth muscle actin (α-SMA) monoclonal
antibodies (ab32575) were purchased from Abcam. Rabbit monoclonal
anti-GAPDH antibody (AF1186) was from Beyotime Institute of
Biotechnology. Cy3-labeled goat anti-mouse antibodies (A0521) and
FITC-labeled goat anti-rabbit antibodies (A0562) were purchased
from Beyotime Institute of Biotechnology. TRPC1 and NC siRNAs were
purchased from Ribo Biotechnology Co, Ltd. SYBR Premix EX Taq was
purchased from Takara Biotechnology. The bicinchoninic acid (BCA)
assay kit and Fluro-3 AM were purchased from Beyotime Institute of
Biotechnology, and TRIzol reagent was purchased from Invitrogen;
Thermo Fisher Scientific, Inc. The PCR primers for TRPC1, GAPDH,
cytokeratin 8, E-cadherin and α-SMA were from Sangon Biotech. The
Revert Aid First Strand cDNA synthesis kits were purchased from
Fermentas; Thermo Fisher Scientific, Inc. 3,3′-Diaminobenzidine
tetrahydrochloride (DAB) was from Sigma-Aldrich; Merck KGaA;
1,2-bis(2-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid
(BAPTA-AM) was purchased from Santa Cruz Biotechnology, Inc. ECL
reagent was from Beyotime Institute of Biotechnology.
Tissue samples
Normal human lung tissues were obtained from
surgical specimens that were resected in the Department of Thoracic
Surgery, the 2nd Clinical Hospital of Chongqing Medical University
from 2014 to 2015. The test population consisted of 20 patients who
were pathologically diagnosed with stage I non-small cell lung
cancer (NSCLC) according to the 8th edition of the TNM
classification for lung cancer. All patients were informed of the
research content of the project and signed an agreement to
participate in the present study. Ethics committee approval was
obtained for the Second Clinical Hospital of Chongqing Medical
University. None of the patients had a history of any disease other
than COPD and lung cancer. All the normal lung tissues that were
obtained from the organization were separated from the tumor by at
least 5 cm and were examined by a pathologist to confirm that they
were non-cancerous. The subjects were divided into the COPD group
(8 males, 5 females) with a mean age of 58.7 years (SD, 3.9 years)
and the control group (4 males, 3 females) with a mean age of 60.4
years (SD, 5.4 years). The diagnosis of COPD was based on the
Global Initiative for Chronic Obstructive Lung Disease (GOLD)
criteria (24) namely a post
bronchodilator FEV1/FVC <0.7 [forced expiratory volume in the
first second (FEV1); forced vital capacity (FVC)] with a history of
long-term cigarette smoking or significant biomass exposure.
According to the GOLD criteria, the patients with COPD were defined
as stage I COPD (FEV1 >80% of the predicted value, n=8) and
stage II COPD (50< FEV1 >80%, n=5). None of the patients with
COPD were classified as GOLD stage III or GOLD stage IV. The
control group exhibited no underlying chronic inflammatory airway
disease. Patients with COPD were more likely to be smokers (COPD
vs. control, 10/13 vs. 2/7) and had more pack-years of smoking
(COPD vs. control, 37.11±6.53 vs. 9.81±1.21) than control. The
resected tissues were then immediately frozen in liquid nitrogen.
Human bronchi were analyzed using immunohistochemistry, and each
bronchial mucosa was evaluated by western blot analysis and
RT-qPCR.
Immunohistochemical localization of TRPC1
in human lung tissues
Human lung tissues were immunohistochemically
stained to assess the protein expression of TRPC1. The tissues were
fixed with formalin, embedded in paraffin, and cut into
5-μm-thick serial sections. Endogenous peroxide activity was
quenched using 3% H2O2, and non-specific
binding was minimized by incubating the tissues in 1% normal goat
serum. The sections were then incubated at room temperature with
rabbit anti-TRPC1 polyclonal antibody at a final dilution of 1:200
for 1 h. This step was followed by incubation with goat anti-rabbit
biotinylated secondary antibodies (1:200 dilution) for 30 min at
37°C and incubation with a solution of streptavidin-peroxidase
complexes at a dilution of 1:100 for 45 min. Finally, color
reactions were developed using the chromogen DAB. Five fields were
randomly selected for the slices observed under a light microscope
(Olympus Corp.). The dark brown cells were regarded as positive
cells. Image-Pro Plus 6.0 software (MediaCybernetics) was employed
to detect the staining intensity for further analysis.
Cell culture and grouping
The 16HBE cells were cultured in RPMI-1640 medium
containing 10% fetal bovine serum and incubated at 37°C in a
humidified water-jacketed incubator containing 95% air and 5%
CO2 during the subsequent experiments. Cultured cells
were divided into a control group and a group exposed to mechanical
stretch. Cells in the control group were cultured on similar plates
in the same incubator without any additional intervention, whereas
cells in the mechanical stretch group were subjected to sinusoidal
stretching for 48 h at a frequency of 60 cycles per min at a 15%
elongation using a FX-4000 Flexcell tension system (Flexcell
International Corp.) (please see the mechanical stretch system
section below for more details). Inhibition experiments were
performed by transfection of the 16HBE cells with TRPC1 siRNA/NC
siRNA or by the addition of the intracellular calcium chelator,
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
(BAPTA-AM).
Transient transfection with TRPC1
siRNA
The 16HBE cells were plated into 6-well plates at
200,000 cells per well in medium containing 10% fetal bovine serum
and incubated overnight. The cells were then cultured in serum-free
medium before being divided into 3 groups as follows: The control
group, the TRPC1 siRNA group and the NC siRNA group. A total of 100
pmol TRPC1 siRNA or NC siRNA were transfected into the 16HBE cells
using Lipofectamine RNAiMAX reagent (10:1) according to the
manufacturer's instructions. The detailed sequence of TRPC1 siRNA
and NC siRNA are shown as follows: TRPC1 siRNA sense, 5′-GGA UGU
GCG GGA GGU GAA Gtt-3′ and antisense, 5′-CUU CAC CUC CCG CAC AUC
Ctt-3′; NC siRNA sense, 5′-UUC UCC GAA CGU GUC ACG UTT-3′ and
antisense, 5′-ACG UGA CAC GUU CGG AGA ATT-3′. At 6 h following
transfection, the cells were fed with medium containing 10% fetal
bovine serum and incubated at 37°C for a further 48 h. RT-qPCR and
western blot analysis were then performed to determine the
efficiency of TRPC1 knockdown.
Mechanical stretch system
In the in vitro experiments, a FX-4000
Flexcell tension system (Flexcell International Corp.) was used to
apply sinusoidal strain to the 16HBE cells as previously described
(2,8) to imitate the pathological conditions
that are associated with airway mechanical stress. Briefly, the
16HBEcells were first separately seeded at 4×105
cells/well in Flexcell biaxial 6-well plates coated with type I
collagen (Flexcell International Corp.) in medium containing 10%
fetal bovine serum for 24 h before it was replaced with serum-free
medium. The cells were then divided into a control group and a
group exposed to mechanical stretch. Cells in the control group
were cultured on similar plates in the same incubator without any
additional intervention, including mechanical loading, whereas
cells in the mechanical stretch group were subjected to sinusoidal
stretching for 48 h at a frequency of 60 cycles per min at a 15%
elongation.
Ca2+ imaging was used to measure the
activation of TRPC1 in 16HBE cells after the cells were exposed to
sinusoidal stretching for 0.5, 1, 2, 4, 8, 12, 24 and 48 h. After
the cells had been stretched for 48 h at a 15% elongation, western
blot analysis, immunofluorescence and RT-qPCR were performed to
determine the protein and mRNA expression profiles of EMT-related
genes. Inhibition experiments were performed with 16HBE cells
transfected with TRPC1 siRNA/NC siRNA.
Ca2+ imaging
The levels of intracellular Ca2+ were
measured using the membrane- and cell-permeable
Ca2+-indicator, Fluo-3-AM. Following mechanical
stretching, 16HBE cells were seeded into confocal dishes at a
density of 1,000,000 cells per dish. After the cells were washed 3
times with PBS, they were incubated with PBS containing 3 mmol/l of
Fluo-3-AM for 45 min at 37°C. The cells were then washed 3 times
with modified Krebs-Ringer HEPES buffer and incubated at 37°C for a
further 30 min prior to analysis. Images were collected and
analyzed at multiple time points with a laser scanning confocal
microscope system (TCSSP2, Leica Microsystems GmbH). The
fluorescence intensity was analyzed with the quantification tools
available in the confocal microscope software.
RT-qPCR
The mRNA levels of TRPC1, cytokeratin 8, E-cadherin
and α-SMA were measured by RT-qPCR. TRIzol reagent was used to
extract total RNA from the cultured 16HBE cells according to the
manufacturer's instructions. A total of 5 μg total RNA was
reverse transcribed into complementary DNA (cDNA) with a
first-strand cDNA synthesis kit. The specific primer sequences for
RT-qPCR were as follows: TRPC1 sense, 5′-TCG TGG TTG TGA TTG TGC
TT-3′ and anti-sense, 5′-TGG TGA GGG AAT GAT GTT GA-3′; GAPDH
sense, 5′-AGA AGG CTG GGG CTC ATT TG-3′ and antisense, 5′-AGG GGC
CAT CCA CAG TCT TC-3′; cytokeratin 8 sense, 5′-GAG GCA TCA CCG CAG
TTA C-3′ and antisense, 5′-TTG CTT CGA GCC GTC TTC T-3′; E-cadherin
sense, 5′-GCC AAA GAC AGA GCG GAA CTA T-3′ and antisense, 5′-ATG
TGT TCA GCT CAG CCA GC-3′; and α-SMA sense, 5′-CCG ACC GAA TGC AGA
AGG A-3′ and antisense, 5′-ACA GAG TAT TTG CGC TCC GAA-3′. The PCR
reaction was performed for 35 cycles under the following
conditions: Pre-denaturation at 94°C for 2 min; denaturation at
94°C for 30 sec, annealing at 60°C (TRPC1 and α-SMA), 65°C
(cytokeratin 8), or 58°C (E-cadherin) for 30 sec and extension at
72°C for 10 min. The comparative Cq method (2−ΔΔCq)
(25) was used to determine the
relative mRNA quantification, and GAPDH was used as the endogenous
control gene.
Western blot analysis
First, the 16HBE cells were homogenized on ice in
RIPA lysis buffer containing protease inhibitors. After the mixture
was centrifuged at 11,000 rpm for 10 min at 4°C, the supernatant
was used for western blot analysis. The total protein was assessed
using a BCA protein assay. Equivalent amounts of protein (30
μg) were obtained from each sample and fractionated using
10% SDS-PAGE. The blots were then transferred onto polyvinylidene
fluoride membranes. The membranes were blocked in skim milk for 1 h
at room temperature and then incubated overnight at 4°C with rabbit
anti-TRPC1 polyclonal antibodies (1:1,000), mouse anti-cytokeratin
8 monoclonal antibodies (1:1,000), mouse anti-E-cadherin monoclonal
antibodies (1:1,000), rabbit anti-α-SMA monoclonal antibodies
(1:1,000) and rabbit anti-GAPDH monoclonal antibodies (1:1,000).
The membranes were then incubated with the corresponding
horseradish peroxidase-conjugated secondary antibodies (1:2,000)
for 1 h at 37°C. Finally, the membranes were incubated with western
blot ECL reagent in the dark for 5 min. Quantitative results were
acquired by measuring the optical density of the labeled bands
using Quantity One software (Bio-Rad Laboratories, Inc.). The
values were normalized to the intensity level of GAPDH.
Immunofluorescence
First, the 16HBE cells were seeded into 24-well
plates at a density of 100,000 cells per well and cultured in
medium containing 10% fetal bovine serum overnight. The cells were
then washed 3 times with PBS, fixed with 4% paraformaldehyde for 20
min and permeabilized using 0.3% Triton X-100 for 5 min. After the
cells were blocked with goat serum for 1 h at room temperature,
they were incubated with rabbit anti-TRPC1 polyclonal antibodies
(1:200), mouse anti-cytokeratin 8 monoclonal antibodies (1:100),
mouse anti-E-cadherin monoclonal antibodies (1:100), or rabbit
anti-α-SMA monoclonal antibodies (1:500) at 4°C overnight. Finally,
all the culture dishes were incubated with Cy3-labeled goat
anti-mouse antibodies (1:500) and FITC-labeled goat anti-rabbit
antibodies (1:500) for 1 h at 37°C. The cells were then stained
with DAPI (10 mg/ml) for 15 min. Fluorescent labeling was analyzed
using a fluorescence upright microscope (BX51, Olympus Corp.).
Fluorescence intensities were evaluated using Image-Pro Plus
software (Media Cybernetics). The results are presented as the fold
control of fluorescence intensity.
Statistical analysis
All data are expressed as the means ± SD. Data
analyses were performed using SPSS 22.0 software (IBM Corp.).
Statistical analysis was performed using a Student's t-test for
comparisons between 2 groups and one-way analysis of variance
(ANOVA) followed by Least Significant Difference (LSD) for
comparisons involving >2 groups. The level of statistical
significance was established at P<0.05.
Results
TRPC1 protein and mRNA levels in
bronchial epithelial cells are increased in patients with COPD
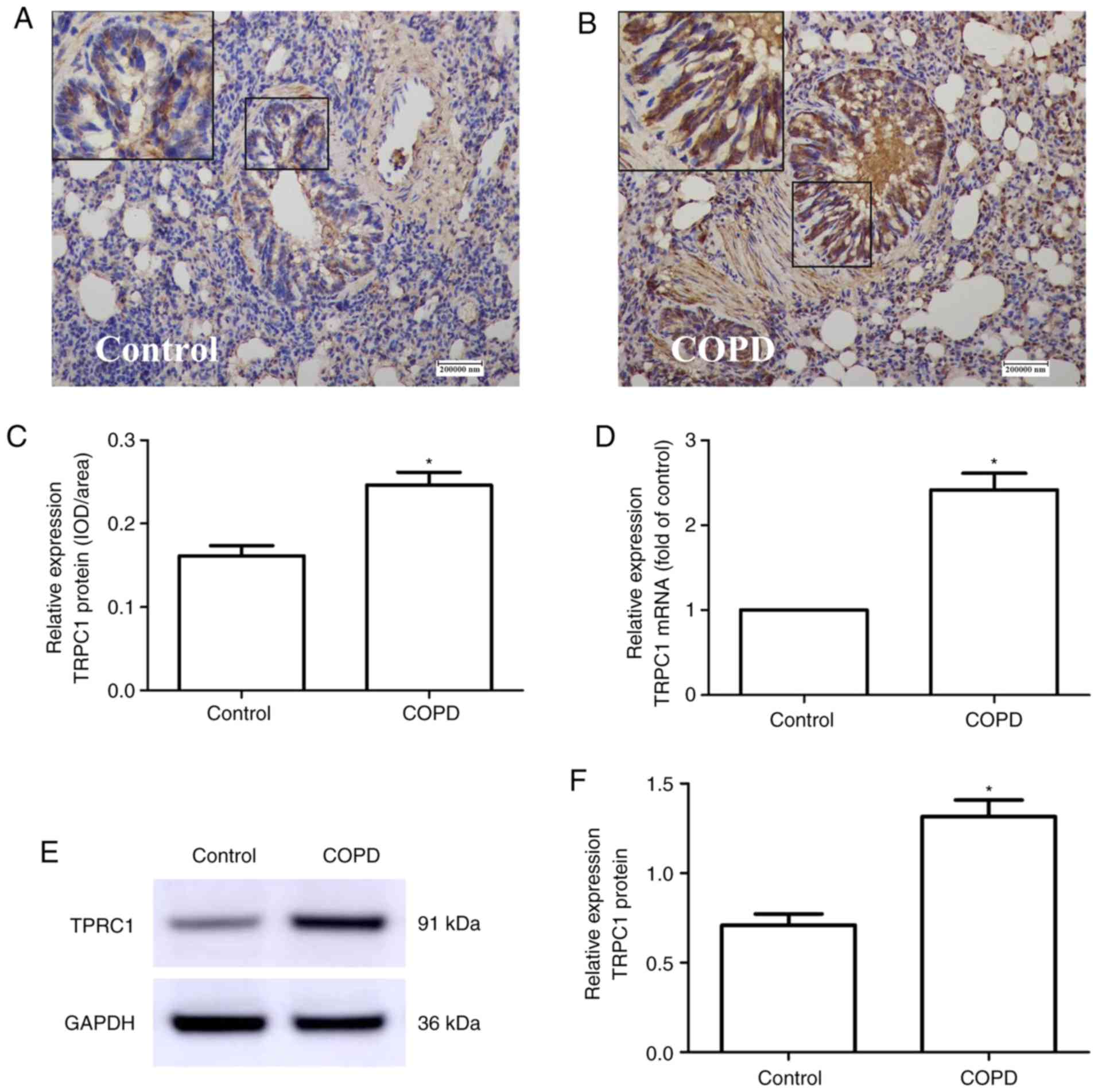
The results of immunohistochemistry revealed that
the majority of TRPC1 immunoreactivity was localized at the basal
surface in epithelial cells (Fig. 1A
and B). The value of the integrated optical density/area of the
TRPC1 protein in the bronchial epithelium was higher in patients
with COPD (0.246±0.027) than in patients in the control group who
had no underlying chronic inflammatory airway disease (0.161±0.021,
P=0.012; Fig. 1C). The results of
RT-qPCR and western blot analysis revealed that the TRPC1 mRNA
(COPD vs. control, 2.42±0.702 vs. 1±0.00, P<0.001, Fig. 1D) and protein (COPD vs. control,
1.32±0.337 vs. 0.710±0.164, P=0.002, Fig. 1E and F) levels in the lung tissues
of patients with COPD were significantly higher than those of the
control group.
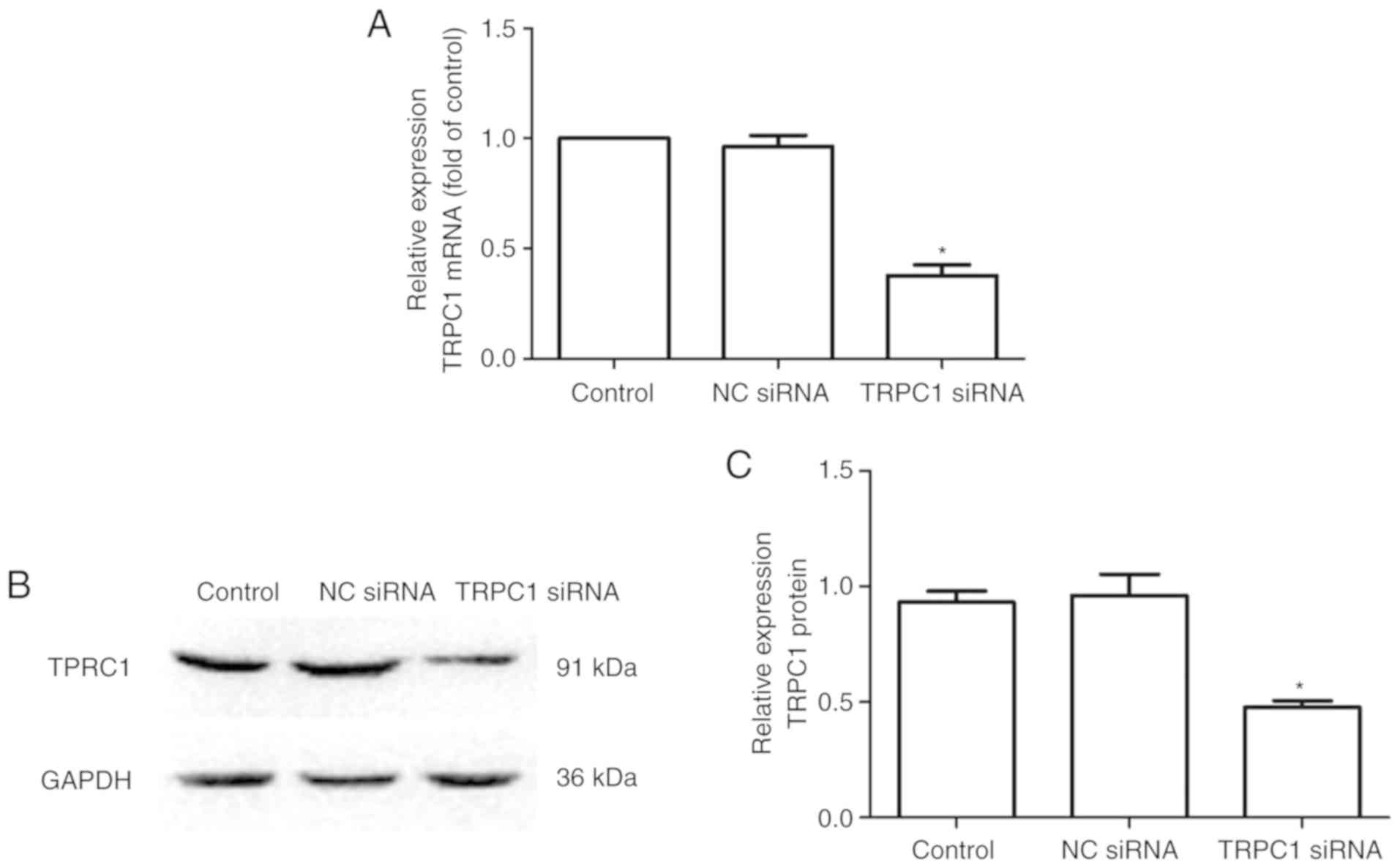
Transfection with TRPC1 siRNA suppresses
TRPC1 protein expression in 16HBE cells
The knockdown efficiency was determined by RT-qPCR
and western blot analysis. A significantly lower level of TRPC1
mRNA (control vs. TRPC1 siRNA, 1±0.00 vs. 0.378±0.086, P<0.001,
Fig. 2A) and protein (control vs.
TRPC1 siRNA, 0.930±0.085 vs. 0.476±0.051, P=0.0014, Fig. 2B and C) was observed in the 16HBE
cells transiently transfected with TRPC1 siRNA than in the control
cells. However, there was no significant difference in the TRPC1
mRNA and protein levels between the NC siRNA transfection group and
the control group (P>0.6726, Fig.
2).
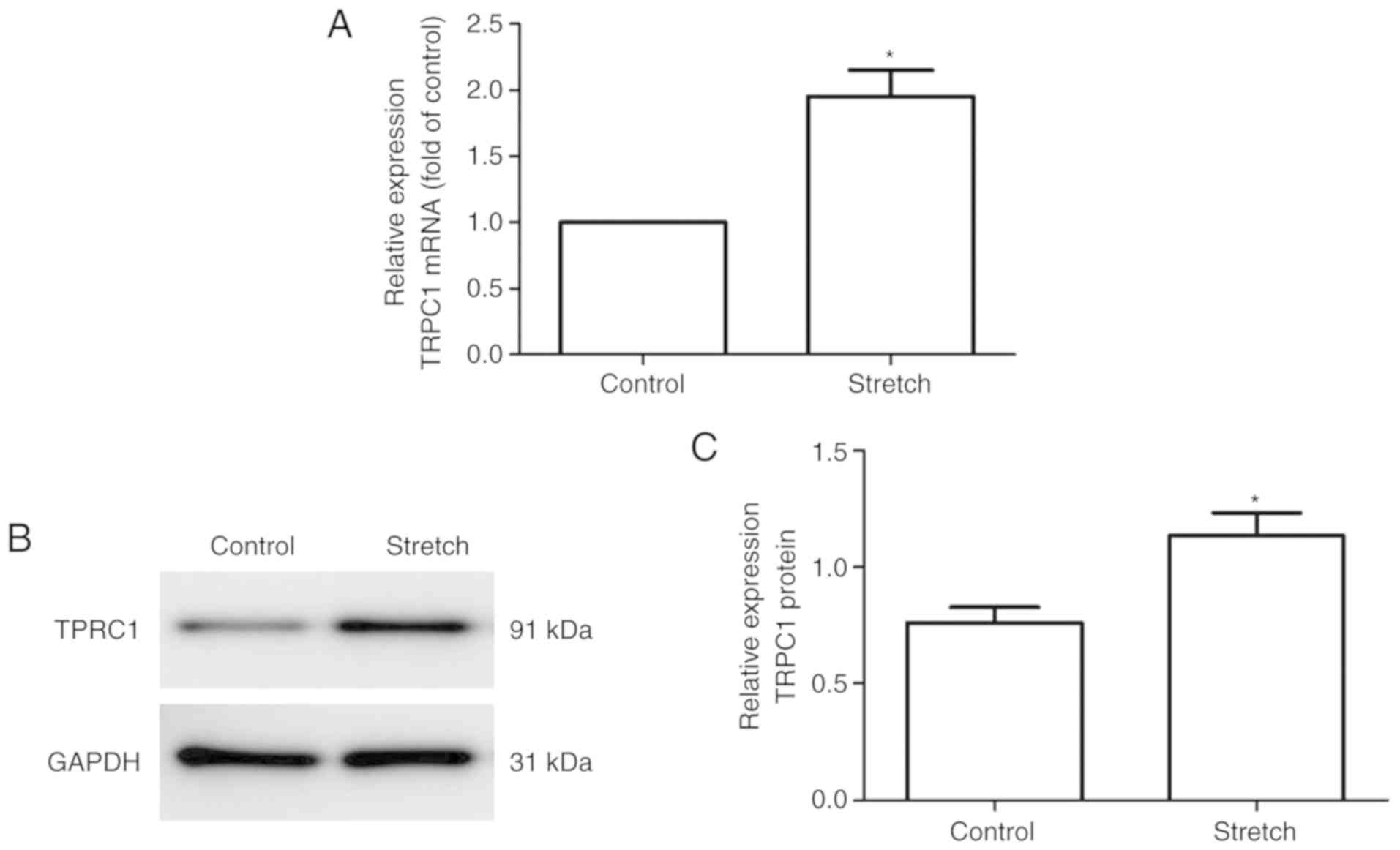
Mechanical stretch upregulates the mRNA
and protein expression of TRPC1 levels in 16HBE cells
RT-qPCR and western blot analysis were used to
quantify changes in the mRNA and protein levels of TTRPC1 following
exposure of the 16HBE cells to cyclic stretch for 48 h. As shown in
Fig. 3, significant increases in
the mRNA (control vs. stretch, 1±0.00 vs. 1.95±0.343, P=0.004,
Fig. 3A) and protein levels
(control vs. stretch, 0.762±0.111 vs. 1.13±0.173, P=0.035, Fig. 3B and C) of TRPC1 were observed in
the 16HBE cells following exposure to cyclic stretch.
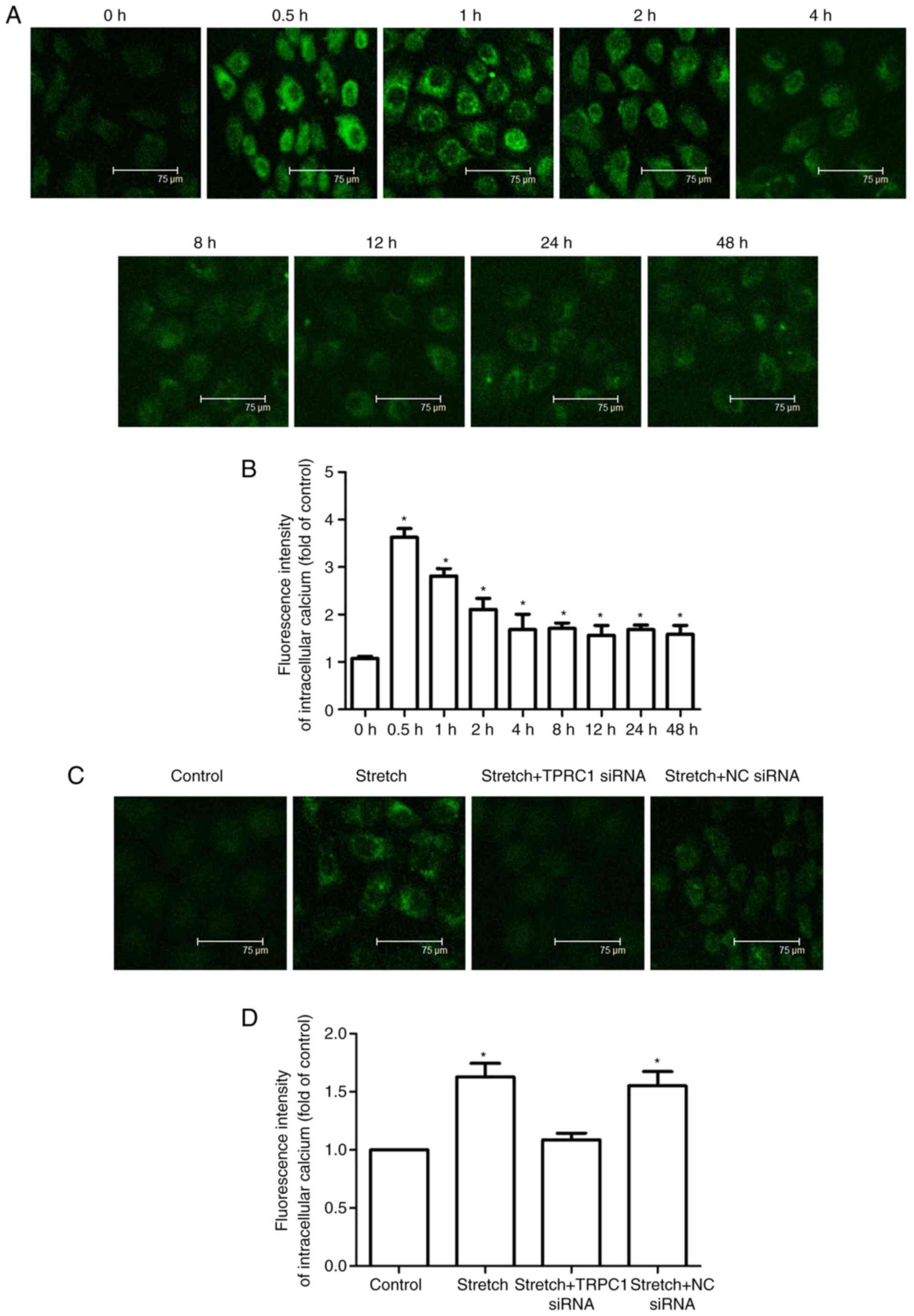
Mechanical stretch activated the TRPC1
channels in16HBE cells
Ca2+ imaging was used to assess TRPC1
function in the 16HBE cells stimulated with cyclic stretch. As
shown in Fig. 4A and B, cyclic
stretch produced a robust increase in the intracellular
Ca2+ of the 16HBE cells. The intracellular
Ca2+ levels reached a maximum at approximately 0.5 h
after the cells were exposed to stretch and then declined slowly to
a plateau level that was still higher than the baseline level at
approximately 4 h following stretch initiation. As shown in
Fig. 4C and D, the
stretch-induced increase in intracellular Ca2+ levels at
48 h after stretching was substantially attenuated in the 16HBE
cells that were transfected with TRPC1 siRNA (stretch vs. stretch +
TRPC1 siRNA, 1.63±0.205 vs. 1.08±0.096, P=0.03), but not in the
16HBE cells transfected with NC siRNA (stretch vs. stretch + NC
siRNA, 1.63±0.205 vs. 1.56±0.211, P=0.572). These results indicated
that TRPC1 plays a vital role in mechanical stress-induced increase
in intracellular Ca2+ in 16HBE cells.
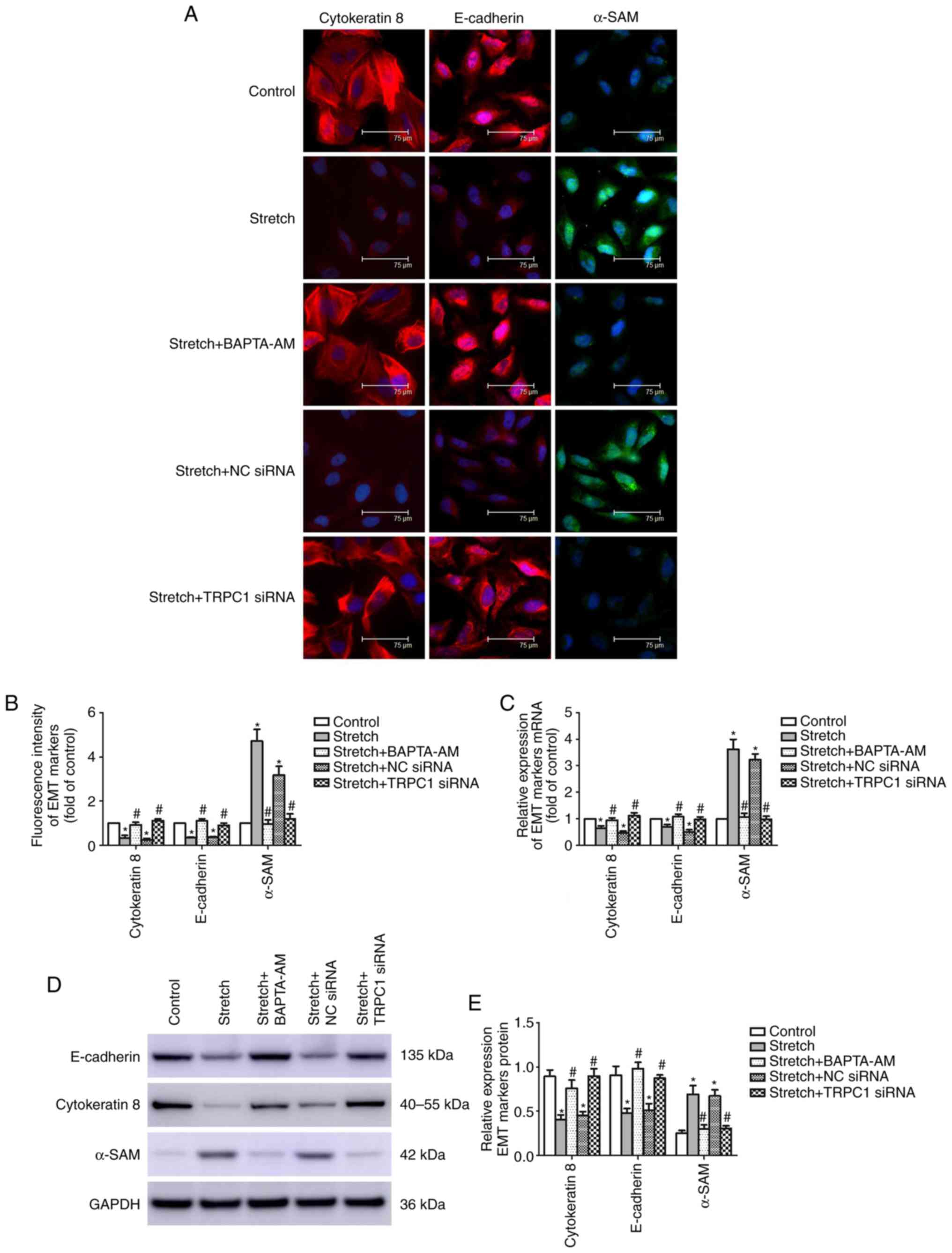
Mechanical stretch alters the expression
of EMT-related markers in 16HBE cells
After the 16HBE cells were exposed to cyclic
stretch, changes in the protein expression of lung epithelial cell
markers (cytokeratin 8 and E-cadherin) and mesenchymal marker
(α-SMA) were observed using immunofluorescence staining, RT-qPCR
and western blot analysis. The results of immunofluorescence
staining revealed that the protein levels of cytokeratin 8 (control
vs. stretch, 1±0.00 vs. 0.335±0.111, P<0.001) and E-cadherin
(control vs. stretch, 1±0.00 vs. 0.345±0.033, P<0.001) were
significantly decreased after the cells were stretched to 15%
elongation for 48 h at a frequency of 60 cycles per min, and the
protein expression of α-SMA (control vs. stretch, 1±0.00 vs.
4.71±0.530, P<0.001) was increased (Fig. 5A and B). Moreover, the altered
fluorescence intensities of cytokeratin 8, E-cadherin and α-SMA
induced by mechanical stretch were significantly attenuated by
transfection with TRPC1 siRNA (stretch vs. stretch + TRPC1 siRNA,
cytokeratin 8, 0.335±0.111 vs. 1.11±0.081, E-cadherin, 0.345±0.033
vs. 0.909±0.008, α-SMA, 4.71±0. 53 vs. 1.19±0.235, P<0.001).
RT-qPCR and western blot analysis were used to quantify the changes
in the mRNA and protein levels of cytokeratin 8, E-cadherin and
α-SMA in the 16HBE cells following mechanical stretch. As shown in
Fig. 5C-E, mechanical stretch
substantially enhanced both the mRNA and protein levels of α-SMA
and attenuated the mRNA and protein levels of cytokeratin 8 and
E-cadherin (P≤0.001). Additionally, these changes were
significantly attenuated by transfection with TRPC1 siRNA
(P≤0.001). However, transfection with NC siRNA did not alter the
immunofluorescence intensity, mRNA or protein levels of cytokeratin
8, E-cadherin or α-SMA (P≥0.069).
The Ca2+ imaging results revealed that
mechanical stretch induced an increase in intracellular
Ca2+ levels. To further explore the role of
intracellular Ca2+ in the expression of EMT markers
under the condition of mechanical stretch, an inhibitory experiment
was also performed by the addition of the intracellular
Ca2+ chelator, BAPTA-AM. As shown in Fig. 5, changes in immunofluorescence
intensity, and in the mRNA and protein levels of cytokeratin 8,
E-cadherin and α-SMA induced by mechanical stretch were markedly
attenuated by BAPTA-AM (P<0.001).
Discussion
In the present study, it was observed that the
expression of TRPC1 was increased in airway epithelial cells in
patients with COPD. In vitro, a Flexcell FX-4000 Tension
System was used to stretch the 16HBE cells at a 15% elongation to
imitate the pathological increased airway mechanical pressure in
COPD. The decreased expression of cytokeratin 8 and E-cadherin and
the simultaneous increase in the expression of α-SMA supported the
occurrence of EMT. Furthermore, the mechanisms underlying these
changes was mediated, at least in part, by a TRPC1-mediated
intracellular Ca2+ increase since transfection with
TRPC1 siRNA or treatment with intracellular Ca2+
chelator BAPTA-AM abolished these alterations in EMT marker
expression induced by mechanical stress. These results may thus
provide a novel mechanistic insight into the treatment or
alleviation of airway remodeling in COPD.
Cells and the ECM in the lung exist in a
mechanically dynamic environment. Mechanical stretch is essential
for the regulation of respiratory physiology and pathophysiology.
Pathologically increased mechanical stretch exerted on lung tissues
is one of the most common characteristics of chronic inflammatory
airway diseases, such as COPD and asthma due to
bronchoconstriction, mucus hypersecretion and airway remodeling. In
turn, increased mechanical stretch has been reported to increase
goblet cell number and MUC5AC protein expression (5), upregulate the expression levels of
IL-13, transforming growth factor (TGF-β1) and MMP-9 in bronchial
epithelial cells (2), finally
further aggravating mucus hypersecretion as well as airway
remodeling in these conditions. Thus, a better understanding of the
mechanisms through which how lung cells respond to mechanical
stretch is of key importance for identifying targets for the
treatment and prevention of chronic inflammatory airway
diseases.
Mechanotransduction is a fundamental process of the
conversion of mechanical stimuli into biochemical or/and electrical
signals (26,27). This phenomenon is widely studied
in several areas of medical science, including vascular biology
(6,28,29) and skeletal biology (7,30).
It has been proposed that a number of TRP isoforms exhibit
mechanosensitivity (TRPA1; TRPV1, 2, 4; TRPC1, 5, 6; TRPM3, 7;
TRPP2) (31,32). Among these, TRPC1 was the first
cloned TRP channel (33) and
exhibited high mechanosensitivity in vertebrates (22). It is widely present in the heart,
arteries, skeletal muscle and was reported to be plentifully
expressed in airway epithelial cells and show a significant
increase in patients with COPD (18). Previous studies have demonstrated
that mechanical stretch can induce an influx in Ca2+
levels and can upregulate the expression of airway
remodeling-associated factors, IL-13, MMP-9 and TGF-β1 in the 16HBE
cells via the activation of TRPC1, indicating its critical role in
mechanical stretch induced airway remodeling (2,8).
Currently, the EMT mechanism for human airway
remodeling in COPD has attracted the attention of various
researchers. Emerging evidence suggests that primary human lung
epithelial cells subjected to mechanical stretch develop EMT
phenotypes (19,20,34); Xu et al recently reported
that a high expression of TRPC1 in the airway epithelia of patients
with COPD was accompanied by an increased level of vimentin and a
simultaneously decreased level of E-cadherin, as well as
morphological changes from a typical epithelial cobblestone
appearance with close connection to a more loosely connected
elongated fusiform appearance, demonstrating its role in occurrence
of EMT in COPD (18). Given the
mechanosensitive properties of TRPC1 and the fact that TRPC1
overexpression in COPD promoted EMT process, the present study
investigated the role of TRPC1 in the occurrence of COPD-associated
EMT in response to mechanical stress. The results demonstrated that
TRPC1-mediated intracellular Ca2+ increase plays an
important role in this process.
As a well-established mechanosensitive,
Ca2+-permeable TRP channels (35), the activation of TRPC1 depends on
both phosphatidylinositol-4,5-bisphosphate(PIP2) and
protein kinase C (PKC) (36). The
intracellular Ca2+ increase following TRPC1 activation
can activate phospholipase C(PLC), which subsequently hydrolyze
PIP2, generating inositoltrisphosphate (IP3) and
diacylglycerol (DAG). DAG can activate PKC, and IP3 may bind to its
calcium channel-coupled receptor on the ER membrane and induce
Ca2+ release from endoplasmic reticulum (37). Thus, it was hypothesized that the
intracellular Ca2+ increase observed in the present
study may result from both an influx of extracellular
Ca2+ and Ca2+ release from the ER, and the
reduced level of PIP2 and the simultaneous increase of
PKC may explain the plateau of the intracellular calcium
concentration when exposed to chronic mechanical stress. However,
further studies are required to address these issues.
Studies on in vitro alveolar epithelial cell
cultures using mechanical stretch suggest that a 5 to 12%
elongation is physiological, while mechanical stretch at 37 to 50%
elongation is associated with pathophysiological conditions
produced by mechanical ventilation (34,38,39). Therefore, in the present study, a
15% elongation for 48 h was used as a stimulus for 16HBE cells.
This magnitude of stimulation and timing likely reflects the
pathologic conditions of chronic airway diseases such as COPD.
A higher TRPC1 expression was observed level in the
lung tissues of patients with COPD compared with those of the
control group, which was consistent with the findings of a previous
study (18). It has previously
been reported that 5% CSE for 48 h significantly increased TRPC1
protein expression in 16HBE cells (18), and nicotine treatment obviously
upregulated TRPC1 expression in cultured rat distal pulmonary
arterial smooth muscle (PASMCs) (40), indicating a role of cigarette
smoke in upregulation of TRPC1 expression. However, Jiang et
al found no significant difference in TRPC1 expression between
patients with NSCLC who were smokers and non-smokers (41). Thus, it is currently not clear
whether the upregulation of TRPC1in COPD is due to cigarette smoke
or other factors associated with COPD.
In conclusion, the present study demonstrated that
the TRPC1-mediated increase in intracellular Ca2+ levels
play a key role in mechanical stress-induced occurrence of EMT.
There were some limitations to the present study. First, the
possible involved signaling pathway downstream TRPC1-mediated
intracellular Ca2+ in EMT occurrence induced by
mechanical stretch was not investigated. Second, in the present
study, 16HBE cells subjected to a Flexcell FX-4000 Tension System
used to mimic the effects of high airway pressure. This model may
not fully reflect the in vivo condition, where other cell
types are present as well. In addition, primary normal human
bronchial epithelial (NHBE) cells may be more suitable since they
were established without any genetic background changes (42). Therefore, further studies are
required to address these questions thoroughly, which will aid in
the elucidation of the mechanisms of mechanical stretch-induced
airway remodeling in COPD and may provide a novel therapeutic
target for patients with COPD.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant no. 81270102), the National
Natural Science Foundation of Chongqing (grant no.
cstc2019jcyj-msxmX0849) and the Joint Fund of science and health
Medicine of Chongqing, China (grant no. 2019QNXM004).
Availability of data and materials
The analyzed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ML and MZ designed the research; JW wrote the
manuscript and analyzed data, YH, NL and GY performed the research
and analyzed data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Ethics committee approval was obtained for the
Second Clinical Hospital of Chongqing Medical University [reference
no. 2014(65)], and all subjects provided written informed
consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition
|
|
TRPC1
|
transient receptor potential canonical
1
|
|
COPD
|
chronic obstructive pulmonary
disease
|
|
ECM
|
extracellular matrix
|
|
MUC5AC
|
mucin5AC
|
|
MscCa
|
mechanosensitive cation
|
|
Ca2+
|
calcium
|
|
HBE
|
human bronchial epithelial
|
|
PIP2
|
phosphatidylinositol-4,5-bisphosphate
|
|
PKC
|
protein kinase C
|
|
BAPTA-AM
|
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
|
|
GOLD
|
Global Initiative for Chronic
Obstructive Lung Disease
|
|
FEV1
|
forced expiratory volume in the first
second
|
|
FVC
|
forced vital capacity
|
References
|
1
|
Garcia CS, Prota LF, Morales MM, Romero
PV, Zin WA and Rocco PR: Understanding the mechanisms of lung
mechanical stress. Braz J Med Biol Res. 39:697–706. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yu Q and Li M: Effects of transient
receptor potential canonical 1 (TRPC1) on the mechanical
stretch-induced expression of airway remodeling-associated factors
in human bronchial epithelioid cells. J Biomech. 51:89–96. 2017.
View Article : Google Scholar
|
|
3
|
Birukova AA, Tian Y, Meliton A, Leff A, Wu
T and Birukov KG: Stimulation of Rho signaling by pathologic
mechanical stretch is a 'second hit' to Rho-independent lung injury
induced by IL-6. Am J Physiol Lung Cell Mol Physiol. 302:L965–L975.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park JA and Tschumperlin DJ: Chronic
intermittent mechanical stress increases MUC5AC protein expression.
Am J Respir Cell Mol Biol. 41:459–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li N, Li Q, Zhou XD, Kolosov VP and
Perelman JM: Chronic mechanical stress induces mucin 5AC expression
in human bronchial epithelial cells through ERK dependent pathways.
Mol Biol Rep. 39:1019–1028. 2012. View Article : Google Scholar
|
|
6
|
Tschumperlin DJ and Drazen JM: Mechanical
stimuli to airway remodeling. Am J Respir Crit Care Med.
164:S90–S94. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Suki B, Sato S, Parameswaran H, Szabari
MV, Takahashi A and Bartolák-Suki E: Emphysema and mechanical
stress-induced lung remodeling. Physiology (Bethesda). 28:404–413.
2013.
|
|
8
|
Li N, He Y, Yang G, Yu Q and Li M: Role of
TRPC1 channels in pressure-mediated activation of airway
remodeling. Respir Res. 20:912019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ito JT, Lourenço JD, Righetti RF, Tibério
IFLC, Prado CM and Lopes FDTQS: Extracellular matrix component
remodeling in respiratory diseases: What has been found in clinical
and experimental studies? Cells. 8:pii: E342. 2019. View Article : Google Scholar
|
|
10
|
Jones RL, Noble PB, Elliot JG and James
AL: Airway remodelling in COPD: It's not asthma! Respirology.
21:1347–1356. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pain M, Bermudez O, Lacoste P, Royer PJ,
Botturi K, Tissot A, Brouard S, Eickelberg O and Magnan A: Tissue
remodelling in chronic bronchial diseases: From the epithelial to
mesenchymal phenotype. Eur Respir Rev. 23:118–130. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sohal SS, Reid D, Soltani A, Ward C,
Weston S, Muller HK, Wood-Baker R and Walters EH: Reticular
basement membrane fragmentation and potential epithelial
mesenchymal transition is exaggerated in the airways of smokers
with chronic obstructive pulmonary disease. Respirology.
15:930–938. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sohal SS, Reid D, Soltani A, Ward C,
Weston S, Muller HK, Wood-Baker R and Walters EH: Evaluation of
epithelial mesenchymal transition in patients with chronic
obstructive pulmonary disease. Respir Res. 12:1302011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gohy ST, Hupin C, Fregimilicka C, Detry
BR, Bouzin C, Gaide Chevronay H, Lecocq M, Weynand B, Ladjemi MZ,
Pierreux CE, et al: Imprinting of the COPD airway epithelium for
dedifferentiation and mesenchymal transition. Eur Respir J.
45:1258–1272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Milara J, Peiró T, Serrano A and Cortijo
J: Epithelial to mesenchymal transition is increased in patients
with COPD and induced by cigarette smoke. Thorax. 68:410–420. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu F, Liu XC, Li L, Ma CN and Zhang YJ:
Effects of TRPC1 on epithelial mesenchymal transition in human
airway in chronic obstructive pulmonary disease. Medicine
(Baltimore). 96:e81662017. View Article : Google Scholar
|
|
19
|
Heise RL, Stober V, Cheluvaraju C,
Hollingsworth JW and Garantziotis S: Mechanical stretch induces
epithelial-mesenchymal transition in alveolar epithelia via
hyaluronan activation of innate immunity. J Biol Chem.
286:17435–17444. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mao P, Li J, Huang Y, Wu S, Pang X, He W,
Liu X, Slutsky AS, Zhang H and Li Y: MicroRNA-19b mediates lung
epithelial-mesenchymal transition via
phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase in response
to mechanical stretch. Am J Respir Cell Mol Biol. 56:11–19. 2017.
View Article : Google Scholar
|
|
21
|
Yang Y, Hu L, Xia H, Chen L, Cui S, Wang
Y, Zhou T, Xiong W, Song L, Li S, et al: Resolvin D1 attenuates
mechanical stretch-induced pulmonary fibrosis via
epithelial-mesenchymal transition. Am J Physiol Lung Cell Mol
Physiol. 316:L1013–L1024. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maroto R, Raso A, Wood TG, Kurosky A,
Martinac B and Hamill OP: TRPC1 forms the stretch-activated cation
channel in vertebrate cells. Nat Cell Biol. 7:179–185. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Banner KH, Igney F and Poll C: TRP
channels: Emerging targets for respiratory disease. Pharmacol Ther.
130:371–384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Global Initiative for Chronic Obstructive
Lung Disease: Global strategy for the diagnosis, management, and
prevention of chronic obstructive pulmonary disease 2019 report.
https://goldcopd.org/gold-reports/.
Accessed December 2, 2018.
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Gillespie PG and Walker RG: Molecular
basis of mechanosensory transduction. Nature. 413:194–202. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Marshall KL and Lumpkin EA: The molecular
basis of mechanosensory transduction. Adv Exp Med Biol.
739:142–155. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Humphrey JD, Schwartz MA, Tellides G and
Milewicz DM: Role of mechanotransduction in vascular biology: Focus
on thoracic aortic aneurysms and dissections. Circ Res.
116:1448–1461. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yin J and Kuebler WM: Mechanotransduction
by TRP channels: General concepts and specific role in the
vasculature. Cell Biochem Biophys. 56:1–18. 2010. View Article : Google Scholar
|
|
30
|
Burkholder TJ: Mechanotransduction in
skeletal muscle. Front Biosci. 12:174–191. 2007. View Article : Google Scholar
|
|
31
|
Inoue R, Jian Z and Kawarabayashi Y:
Mechanosensitive TRP channels in cardiovascular pathophysiology.
Pharmacol Ther. 123:371–385. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Plant TD: TRPs in mechanosensing and
volume regulation. Handb Exp Pharmacol. 223:743–766. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nesin V and Tsiokas L: TRPC1. Handb Exp
Pharmacol. 222:15–51. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cabrera-Benítez NE, Parotto M, Post M, Han
B, Spieth PM, Cheng WE, Valladares F, Villar J, Liu M, Sato M, et
al: Mechanical stress induces lung fibrosis by
epithelial-mesenchymal transition. Crit Care Med. 40:510–517. 2012.
View Article : Google Scholar
|
|
35
|
Rychkov G and Barritt GJ: TRPC1
Ca(2+)-permeable channels in animal cells. Handb Exp Pharmacol.
23–52. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Albert AP: Gating mechanisms of canonical
transient receptor potential channel proteins: Role of
phosphoinositols and diacylglycerol. Adv Exp Med Biol. 704:391–411.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bosanac I, Michikawa T, Mikoshiba K and
Ikura M: Structural insights into the regulatory mechanism of IP3
receptor. Biochim Biophys Acta. 1742:89–102. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Letsiou E, Sammani S, Zhang W, Zhou T,
Quijada H, Moreno-Vinasco L, Dudek SM and Garcia JG: Pathologic
mechanical stress and endotoxin exposure increases lung endothelial
microparticle shedding. Am J Respir Cell Mol Biol. 52:193–204.
2015. View Article : Google Scholar :
|
|
39
|
Suryadevara V, Fu P, Ebenezer DL,
Berdyshev E, Bronova IA, Huang LS, Harijith A and Natarajan V:
Sphingolipids in ventilator induced lung injury: Role of
sphingosine-1-phosphate lyase. Int J Mol Sci. 19:pii: E114. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang J, Chen Y, Lin C, Jia J, Tian L, Yang
K, Zhao L, Lai N, Jiang Q, Sun Y, et al: Effects of chronic
exposure to cigarette smoke on canonical transient receptor
potential expression in rat pulmonary arterial smooth muscle. Am J
Physiol Cell Physiol. 306:C364–C373. 2014. View Article : Google Scholar :
|
|
41
|
Jiang HN, Zeng B, Zhang Y, Daskoulidou N,
Fan H, Qu JM and Xu SZ: Involvement of TRPC channels in lung cancer
cell differentiation and the correlation analysis in human
non-small cell lung cancer. PLoS One. 8:e676372013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Feng W, Guo J, Huang H, Xia B, Liu H, Li
J, Lin S, Li T, Liu J and Li H: Human normal bronchial epithelial
cells: A novel in vitro cell model for toxicity evaluation. PLoS
One. 10:e01235202015. View Article : Google Scholar : PubMed/NCBI
|