Introduction
Microglia are considered to be the largest
population of residual immune cells, which play a major role in
maintaining brain homeostasis. Microglia not only protect neurons
by phagocytosing pathogens and harmful particles in the tissue, but
also, they exhibit toxic effects upon neurons by secreting
proinflammatory cytokines (1).
Age-associated microglial activation contributes to increasing
sensitivity of dopaminergic neurons to neurotoxins (2). Microglia play essential roles in the
numerous types of neuropathogenesis, including Alzheimer's disease
and Parkinson's disease (PD) among other aging-related
neurodegenerative diseases (3-5).
Previous studies (6-8) have demonstrated that microglia
activated by lipopolysaccharide, reduces dopaminergic (DA) neuron
death caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, also
known as a neurotoxin in newborn rats. However, DA neuron death
increases in aged rats under the same conditions, suggesting a
functional switch of microglia from neuroprotection to
neurotoxicity. Microglial senescence may potentially explain this
functional switch. Thus, microglial senescence is likely to be
responsible for pathological progression and is considered as a
pivotal step involved in age-associated neuro-degenerative diseases
(9).
A variety of internal and external stimuli, such as
oxidative stress (10), injury
(11), radiation (12), tumor protein activation (13), and chemical mutagens (14) can induce cellular senescence.
Cellular senescence is basically defined as termination of, or a
decrease in cell division and expression of senescence-related
proteins, including elevated endogenous β-galactosidase activity
(15) and p21 (16). The most common pathological
senescence is called oncogene-induced senescence (OIS). The
initiation of OIS is an endogenous protective mechanism in the
body, by which tumor growth and progression are restrained.
Threatened by malignant transformation, cells cease their abnormal
proliferation and initiate the senescence progression, due to
activation oncogenes mutations or inactivation of tumor suppressor
genes (13,17,18). Phorbol 12-myristate 13-acetate
(PMA), capable of effectively activating protein kinase C as well
as being a potential carcinogen, has a significant role in
promoting cellular differentiation, mitogenesis, survival,
apoptosis and transformation (19) through the activation of oncogenes
(20,21).
Therefore, the present study hypothesized that: i)
Senescent microglia accumulate and release inflammatory cytokines,
which drastically alter the cerebral microenvironment; and ii)
microglial senescence can be over-activated by other pathological
stimuli and cause damage to DA neurons. A previous study on adult
rats found that microglial senescence can be induced by repeatedly
intra-nigrostriatal injection of PMA (20). In this study, in vitro
experiments were performed with microglial cultures to assess the
effects of malignant transformation on the occurrence of microglia
senescence and to explore the potential mechanism(s) underlying OIS
signaling. The current study provides a basis to explore the
possible mechanisms of PD pathogenesis further and to develop
interventions through the regulation of dynamic
cell-microenvironmental interactions.
Materials and methods
Microglia and PC12 Culture
Primary microglia were obtained from Wuhan Pricells
Biotechnology and Medicine Co., Ltd. All the procedures regarding
animal experiments were carried out in compliance of the guidelines
for the Care and Use of Laboratory Animals, which were approved and
supervised by the Ethics Committee of China Medical University. As
with previous studies, microglia cultures were isolated from brain
tissues of Sprague-Dawley rat pups (Wuhan Pricells Biotechnology
and Medicine Co., Ltd.) (22-24). In brief, after sacrificing the
rats, the whole brains were dissected out in pre-cold minimum
essential medium supplemented with L-glutamine (MEM; Invitrogen;
Thermo Fisher Scientific, Inc.). The meninges were peeled off,
followed by rinsing the remaining tissue. Afterwards, the tissues
were in turn strained, digested with dispase (1.5 U/ml; Gibco;
Thermo Fisher Scientific, Inc.), and pelleted by centrifugation at
300 x g for 10 min at room temperature. Following re-suspension in
MEM, cells were seeded in medium containing MEM combined with 10%
heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich; Merck
KGaA) and 0.05 mg/ml of gentamycin (Invitrogen; Thermo Fisher
Scientific, Inc.) in an incubator with 5% CO2 at 37°C
for 48 h. Afterwards, the medium was refreshed to remove cellular
debris as well as non-adherent cells. In order to harvest microglia
after 5-6 days culture, the flasks were shaken for 3-4 h on an
orbital shaker at 70 x g (37°C, 5% CO2), followed by
collecting the supernatant that contained microglia and
centrifugation at 300 x g for 10 min at room temperature. The
pellet was re-suspended in freshly prepared medium containing MEM,
2% heat-inactivated FBS (GE Healthcare; Hyclone™; cat. no.
SH30088.03), 0.002 mg/ml of penicillin and 0.002 mg/ml of
streptomycin. After seeding, it normally took 2-3 days for
microglia to settle at 37°C in an incubator containing 5%
CO2. PC12 cell lines obtained from Chinese Academy of
Science, were employed as a substitute for neurons. They were
cultured at 37°C in a humidified incubator containing 5%
CO2, utilizing RPMI 1640 medium (Thermo Fisher
Scientific, Inc.; Gibco™; cat. no. A1049101) supplemented with 5%
FBS and 10% horse serum (Thermo Fisher Scientific, Inc.; Gibco™;
cat. no. 026050088). All the in vitro experiments were
performed in compliance with guidelines approved and supervised by
the Ethics Committee of China Medical University.
Cell proliferation test (MTT)
Microglia were seeded onto a 96-well plate at the
density of 1×104 cell per well and cultured in MEM media
with 0.4 % FBS for 24 h prior to the addition of PMA at various
concentrations and different time periods (24, 48, 72, and 96 h).
Cells of different groups were stained with MTT (0.5 mg/ml) for 4 h
at 37°C. The optical density at 570 nm was determined, and counted
with a Countstar Automated Cell Counter, which uses trypan blue
staining for less than three minutes at room temperature to exclude
live and dead cells. The proliferation rate (X) was determined
using the following formula: X=cell counts of treatment group/ cell
counts of control group.
Experimental procedure and groups
Either PMA stimulation (100 ng/ml in saline, which
was diluted from stock solution in DMSO) or saline were added to
the microglia culture for 24, 48 or 72 h, respectively. They were
subdivided into four groups as follows: i) Microglia stimulated
with normal saline for 24, 48 and 72 h as controls; ii) microglia
stimulated by 24-h PMA administration; iii) microglia stimulated by
48-h PMA administration, and iv) microglia stimulated by 72-h PMA
administration.
Microglia-PC12 co-culture
Microglia were seeded onto permeable transwell
chamber (BD Falcon; Becton, Dickinson and Company) at the density
of 1×104 cell per well while PC12 cells onto 24-well
culture plates at 5×104 cell per well. They were both
allowed to adhere for 48 h. Cell media had been refreshed 24 h
prior to treatment. Microglia were treated with PMA stimulation
(100 ng/ml) with 72-h administration. Subsequently, activated
microglia underwent aging-related alteration(s). The transwell
chamber with over-activated microglia can be easily transferred
into the 24-well plate that had already been cultured with PC12
cells for co-culture for 24 h, thereby allowing material and/or
energy exchange between PC12 cells and microglia activated by
aging-related alteration(s). Finally, the transwell chamber and
microglia were simultaneously removed from the system of the
co-culture experiment, and the remaining PC12 cells were then
tested alone on the 24-well plate under ×400 magnification using a
light microscope.
Cell cycle test (flow cytometry)
When reaching 70-80% confluence, cells were treated
with 0.25% trypsin (Hyclone; GE Healthcare) after rinses, followed
by fixation with ice-cold 70% cold ethanol overnight. Samples were
then re-suspended in 0.5 ml of PBS and then underwent treatment
with RNase (Sigma-Aldrich; Merck KGaA) to remove RNA. Afterwards,
they were ready for staining with propidium iodide (Sigma-Aldrich;
Merck KGaA) for 1 h at 4°C in the darkness. Using a Becton
Dickinson FACScan system, their DNA was assessed by
fluorescence-activated cell sorting. For analysis, first the pulse
width-pulse area map was used to set the gate, a single cell
population was selected, then this gating was applied to the
scatter plot and significant cell debris was excluded. CellFIT
software (a Windows-based 2D version of CellFIT, known as Zazu was
downloaded from webpage of Dr. Shane Hutson in Vanderbilt
University, https://my.vanderbilt.edu/shanehutson/
software/cellfit-cellular-force-inference-toolkit/) was used to
calculate the percentage of cells in each phase. The G0/G1-phase
cell ratio was calculated as
G0/G1/(G0/G1+S+G2/M).
The S-phase cell ratio was calculated as
S/(G0/G1+S+G2/M).
Senescence-associated β-galactosidase
(SA-β-Gal) staining
After PBS rinses, microglia were fixed with 4%
paraformal-dehyde (PFA) for 15 min at room temperature, followed by
removal of the remaining PFA solution. The incubated cells were
kept in darkness overnight at 37°C in a sealed and wet container.
Those cells were covered by fresh staining buffer containing 1
mg/ml of X-gal, 40 mM of citric acid/sodium phosphate (Ph 6.0), 5
mM of potassium ferricyanide, 150 mM of NaCl and 2 mM of
MgCl2, which was substituted with normal saline
immediately before capturing using a light microscope under ×400
magnification. Total and SA-β-Gal positive cells were counted,
based on at least three distinct fields that were randomly
selected, each of which contained 100 cells in total under each
condition.
Immunofluorescence staining
After PBS rinses, microglia were fixed with 4% PFA
for 20 min at room temperature, followed by PBS rinses again and
permeabilization with 0.2% Triton X-100 for 10 min. After blocking
with 10% goat serum (Thermo Fisher Scientific, Inc.; Gibco™; cat.
no. 16210064) in PBS at room temperature for 30 min, cells were
then incubated overnight at 4°C with primary antibodies against p53
(1:400; Cell Signaling Technology, Inc., cat. no. 48818) and p21
(1:400; Abcam, cat. no. ab218311), and incubated at room
temperature for 2 h with fluorescent secondary antibodies (Thermo
Fisher Scientific, Inc.; Invitrogen™; cat. no. A-11030 for goat
anti-mouse IgG conjugated with Alexa Fluor 546, and A-11035 for
goat anti-rabbit IgG conjugated with Alexa Fluor 546) in the
darkness. Cells were then stained with DAPI for 3 min at room
temperature. After rinses with PBS, the cells were analyzed under a
microscope using an inverted mode or a confocal laser scanning mode
(LX71; Olympus Corporation).
Western blot analysis
Radioimmune precipitation assay buffer containing 1%
Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 100 mg/ml of
phenylmethylsulfonyl fluoride, 50 kallikrein inactivating units/ml
aprotinin and 1 mM of sodium orthovanadate in PBS, was freshly
prepared. Using the bicinchoninic acid assay Protein Assay
(Beyotime Institute of Biotechnology), the protein concentration of
each sample was evaluated. Accordingly, 100 mg of protein were
loaded upon a 15% SDS-polyacrylamide gel and then they were
transferred to polyvinylidene difluoride membranes (EMD Millipore).
Immediately following blocking at room temperature for 60 min or
4°C overnight, using western blocking buffer (Beyotime Institute of
Biotechnology; western blocking buffer; cat. no. P0023B-100 ml),
the membrane was then incubated with primary antibodies against p53
(1:1,000; Cell Signaling Technology, Inc.; cat. no. 48818) and p21
(1:1,000; Abcam; cat. no. ab218311) in a 5% skimmed milk-TBST
solution containing 10 mM of Tris-Cl (pH 7.5), 150 mM of NaCl and
0.05% Tween 20 at 4°C overnight. After rinses, all blots were then
incubated by corresponding secondary antibody conjugated to
horseradish peroxidase (1:2,500; Santa Cruz Biotechnology, Inc.;
cat. no. sc-2357 for mouse anti-rabbit IgG conjugated with HRP, and
sc-516102 for anti-mouse IgG Kappa binding protein secondaries) for
1 h at room temperature. Proteins were visualized via
chemiluminescent methods (Beyotime Institute of Biotechnology,
Chemiluminescent EMSA kit; cat. no. GS009) based on the
manufacturer's protocol. Quantitative analyses for protein levels
were carried out by gray value analysis in Gel-Pro-Analyzer (Media
Cybernetics, Inc.; Gel-Pro Analyzer Version 6.3 image analysis
software).
Measurement of intracellular ROS
formation
After rinses and re-suspension in FBS-free RPMI,
microglia were incubated with 10 µM of
dichlorodihydrofluorescein-diacetate (DCFH-DA; Beyotime Institute
of Biotechnology) at 37°C for 20 min, followed by rinsing again
with FBS-free RPMI. The fluorescence intensity of DCF was evaluated
in a microplate-reader under 485 nm excitation wavelength and 535
nm emission wavelength. Regarding cell observation using an
inverted fluorescence microscope under ×200 magnification, they
were stained with 10 µM of DCFH-DA for 20 min at 37°C after rinses
with FBS-free RPMI and then they were captured using confocal
microscopy under ×200 magnification.
Cytokine secretion test (ELISA)
Microglia were stimulated with PMA at the indicated
time (0, 24, 48, 72 or 96 h following administration) and culture
media removed. FBS-free RPMI was used for microglia culture for 24
h and then microglial supernatants were collected for ELISA assay.
According to the standard protocols provided by manufacturers,
ELISA kits were adopted to detect the TNF-α (Wuhan Boster
Biological Technology, Ltd.; PicoKine™; cat. no. EK0525), IL-1β
(Wuhan Boster Biological Technology, Ltd.; PicoKine™; cat. no.
EK0392), serial dilutions of the protein standards and supernatant
samples were added in a 96-well ELISA plate pre-coated with TNF-α
and IL-1β antibody, respectively. Samples then underwent incubation
with shaking at 4°C overnight. Subsequently following rinses in
each well, the primary antibodies were applied for further
incubation at 37°C for another 2 h, followed by incubating with
HRP-conjugated secondary antibody for 1 h. Afterwards,
3,3′-diaminobenzidine solution was added at room temperature and
the mixture was kept in the darkness for ~15-30 min. Afterwards,
the reaction was terminated by stopping solution and the densities
of mixture were measured at 450 nm absorbance. Triplicates were
used for each sample.
Detection of apoptosis of PC12 (flow
cytometry)
Apoptotic and necrotic cells were quantified by
Annexin V binding and PI uptake. An apoptosis kit (Neobioscience
Technology Co., Ltd.; cat. no. FAK015.50) was adopted for apoptosis
detection. Briefly, PC12 cells were centrifuged at 300 × g for 5
min at 4°C and collected in a 1.5 ml Eppendorf-tube and then washed
twice with cold PBS for 2 times, after being resuspended in 195 µl
binding buffer, 5 µl FITC conjugated with Annexin V were applied to
each sample and incubated for 3 min in the darkness, 5 µl PI were
added 10 min before flow cytometric detection, another 300 µl
binding buffer was then supplemented till a total volume of 500 µl
mixture. Triplicates were used for each sample and at least 10,000
cell events were recorded from each sample. Cells were analyzed
using an automated flow cytometer (BD Biosciences; FACScan™) to
determine the percentages of apoptotic cells with either Annexin
V+/PI+ or Annexin
V-/PI+ labeling.
Statistical analysis
Using SPSS 13.0 software (SPSS, Inc.), all data
obtained were presented as mean ± standard error. Statistical
differences between groups (n=10) were determined using one-way or
two-way analysis of variance in addition to Bonferroni's test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
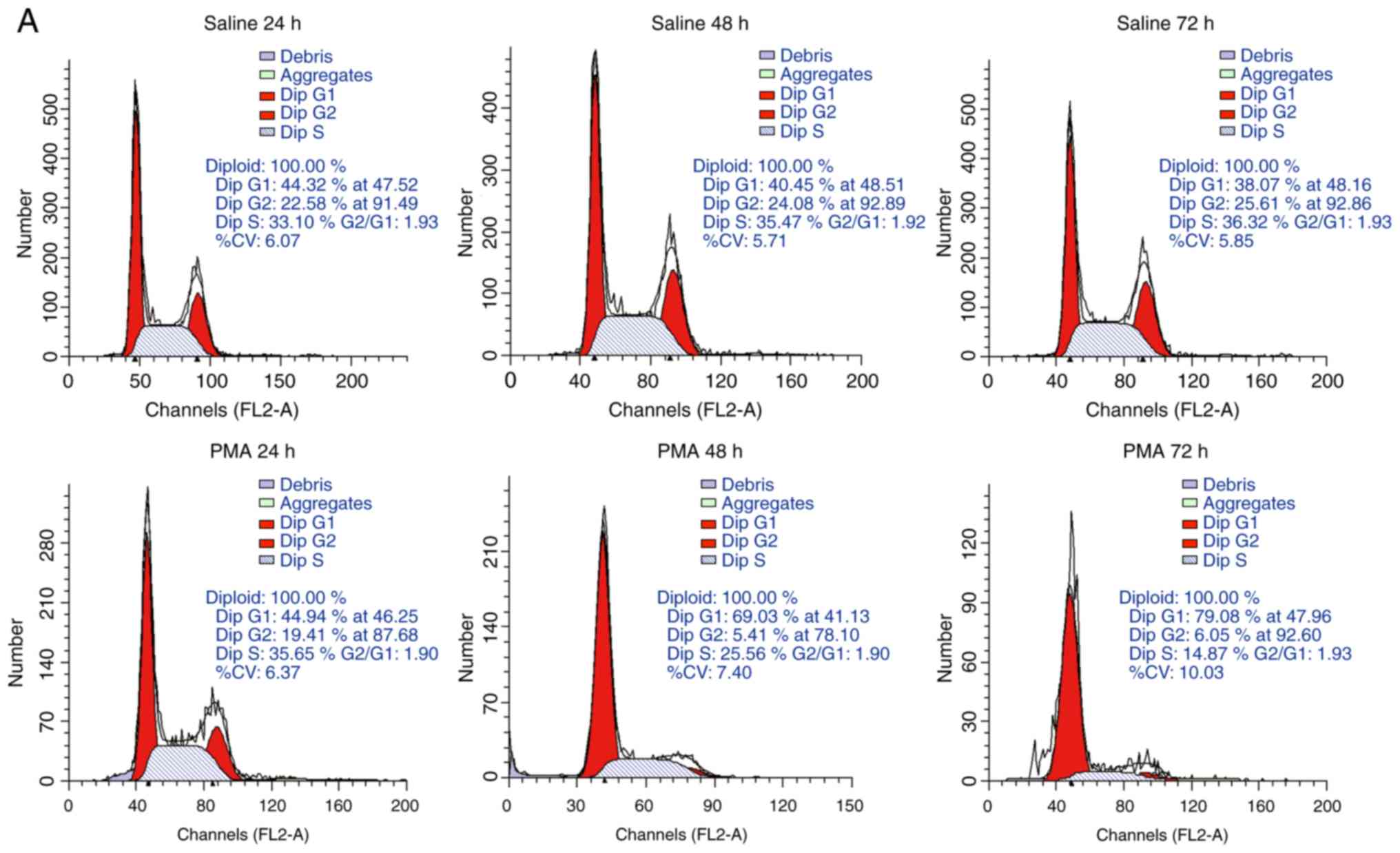
PMA-induced alterations in microglial
cell cycle
A decrease in the cell's proliferative ability is
another biomarker of senescence and changes in the cell cycle
reflect the proliferative potential of cells. Results from flow
cytometry analysis demonstrated that, compared with the control
group, the majority of cells from PMA-treated group were found to
be arrested at the G0/G1 phase, along with a reduced number of
cells at the S phase (Fig. 1A and
1B). Additionally, there was also
a rise in the proportion of cells at the G0/G1 phase, when they
received a longer duration of PMA stimulation, whereas the
percentage of S phase cells decreased over time (Fig. 1A and 1B). Moreover, statistical significance
also indicated an obvious decrease in cell number in PMA-treated
groups with higher concentration over time, compared with the
corresponding saline-treated groups (P<0.05), indicating that
PMA treatment induced a gradual stagnation of cell growth and
caused a significant decrease in cell proliferation (Fig. 1C). Even though the proliferation
of cell number in the control groups increased steadily over time,
cells treated with PMA (100 and 200 ng/ml) showed a significant
inhibition compared with the corresponding control group at 48, 72
and 96 h. In particular, 72 h of administration with 100 ng/ml PMA
was the most effective time point in terms of inhibition of
proliferation, given that conditions of 96 h group showed increased
cellular death (Fig. 1C).
Therefore, the longest treatment time was set as 72 h in the
following experiments.
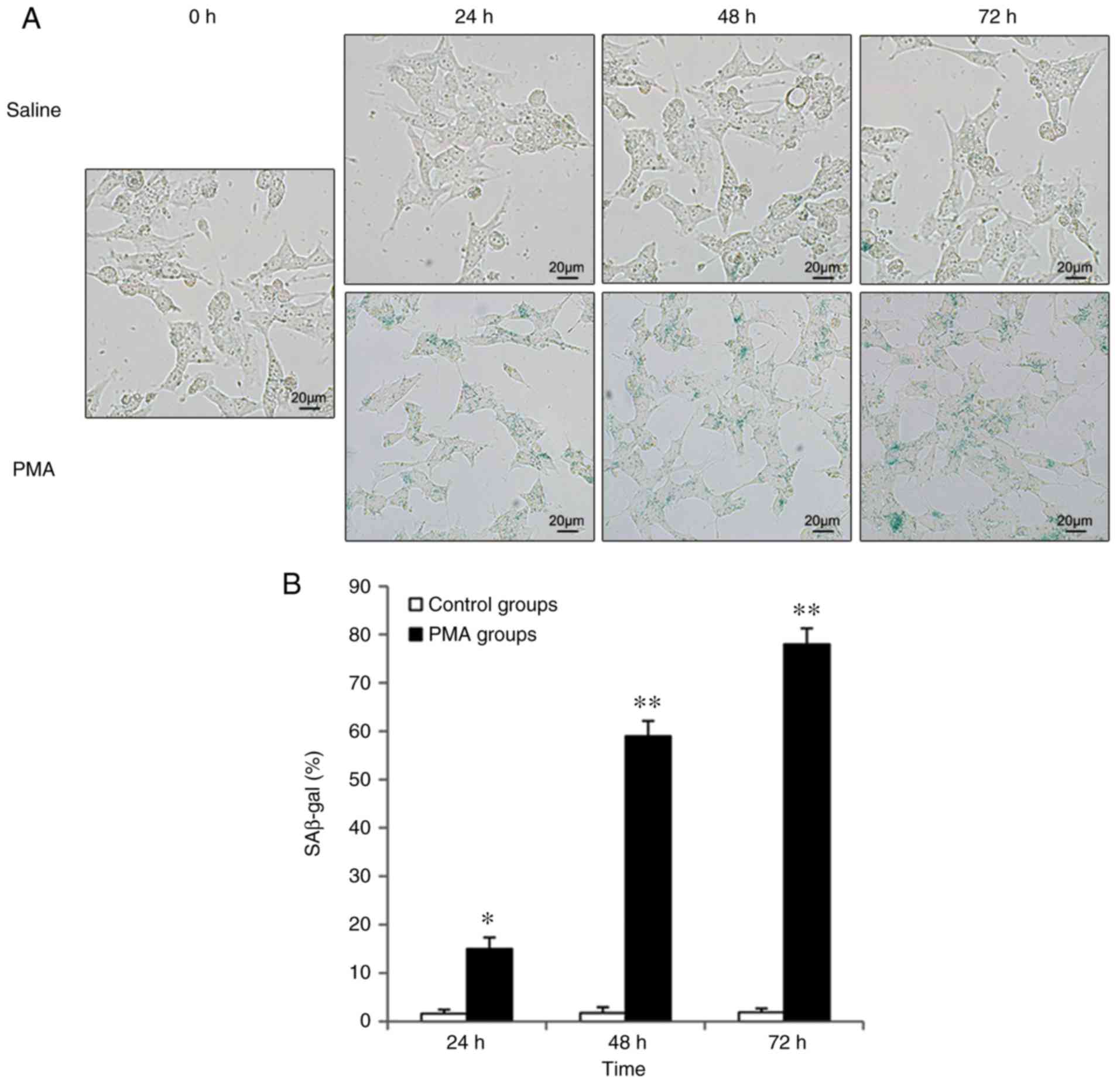
Expression of SA-β-Gal in response to PMA
induction
β-Gal is a biomarker that was used to detect
cellular senescence. Under conditions of Ph 6.0, a blue signal
indicates that the cells have become senescent. Increased blue
signals were detected in intracellular particles during cellular
senescence (Fig. 2). By
comparison, quiescent cells, immortalized cells and tumor cells did
not show any positive signal induced by β-Gal activity. The present
study found that β-Gal-stained cells were nearly undetectable in
each control group, but levels of positively stained cells
increased in microglia stimulated with phorbol 12-myristate
13-acetate (PMA) over time (Fig.
2). Additionally, the most robust staining was observed in
cells incubated with PMA for 72 h (Fig. 2).
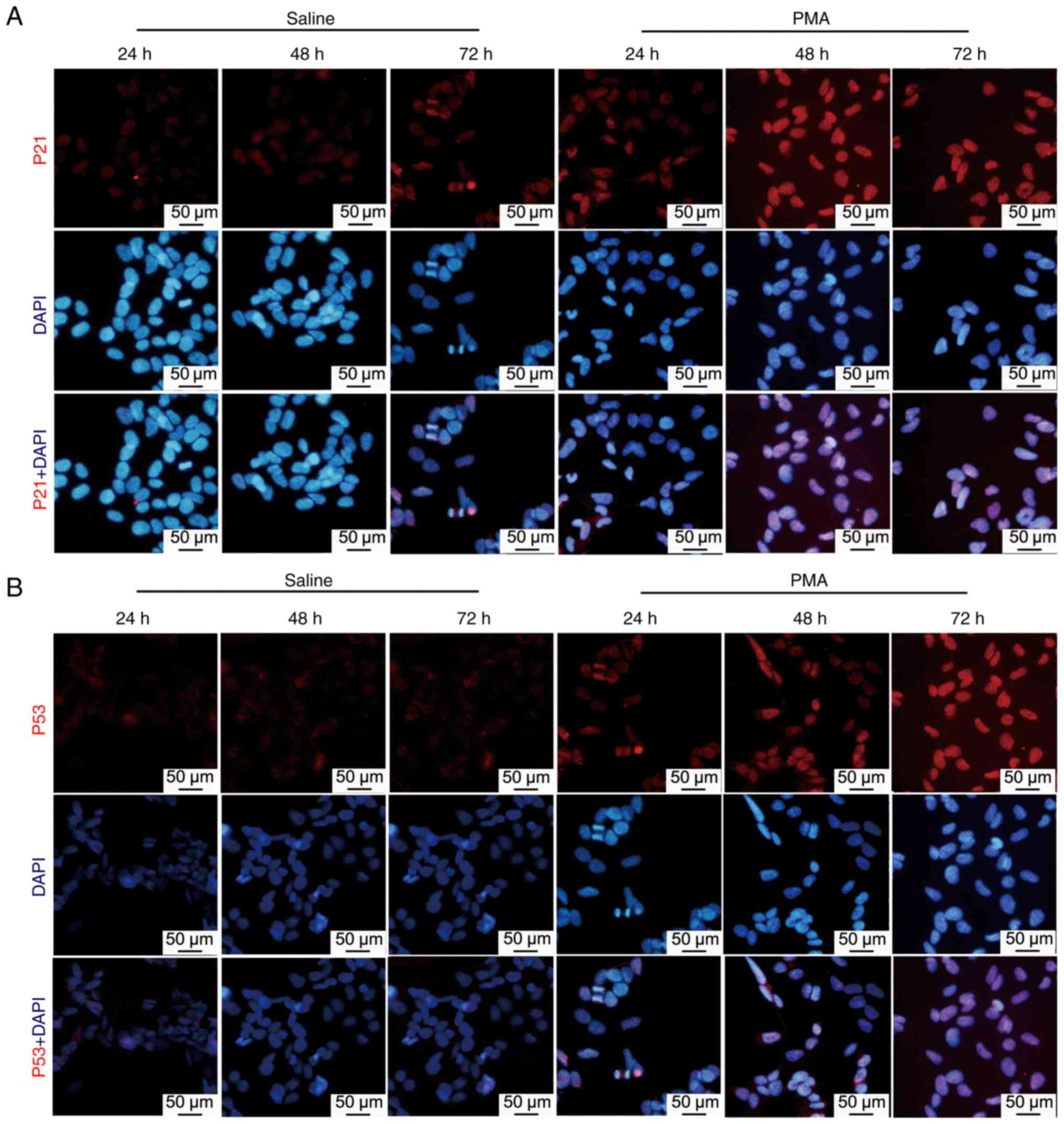
Expression of P53 and P21 protein in
response to PMA induction
Utilizing immunofluorescence and western blotting,
protein levels of p53 and p21 were examined to detect certain
cellular responses to PMA induction. The p21 and p53 proteins
(marked in red) were tested using immunofluorescence microscopy.
very low levels of p21 and p53 were observed in the control group
(Fig. 3). However, 24 h
microglial stimulation with PMA increased p21 fluorescence
intensity. The intensity of p21 fluorescence gradually increased
over time and reached its maximum intensity at 72 h. similar
changes were observed in the p53 protein level intensity (Fig. 3).
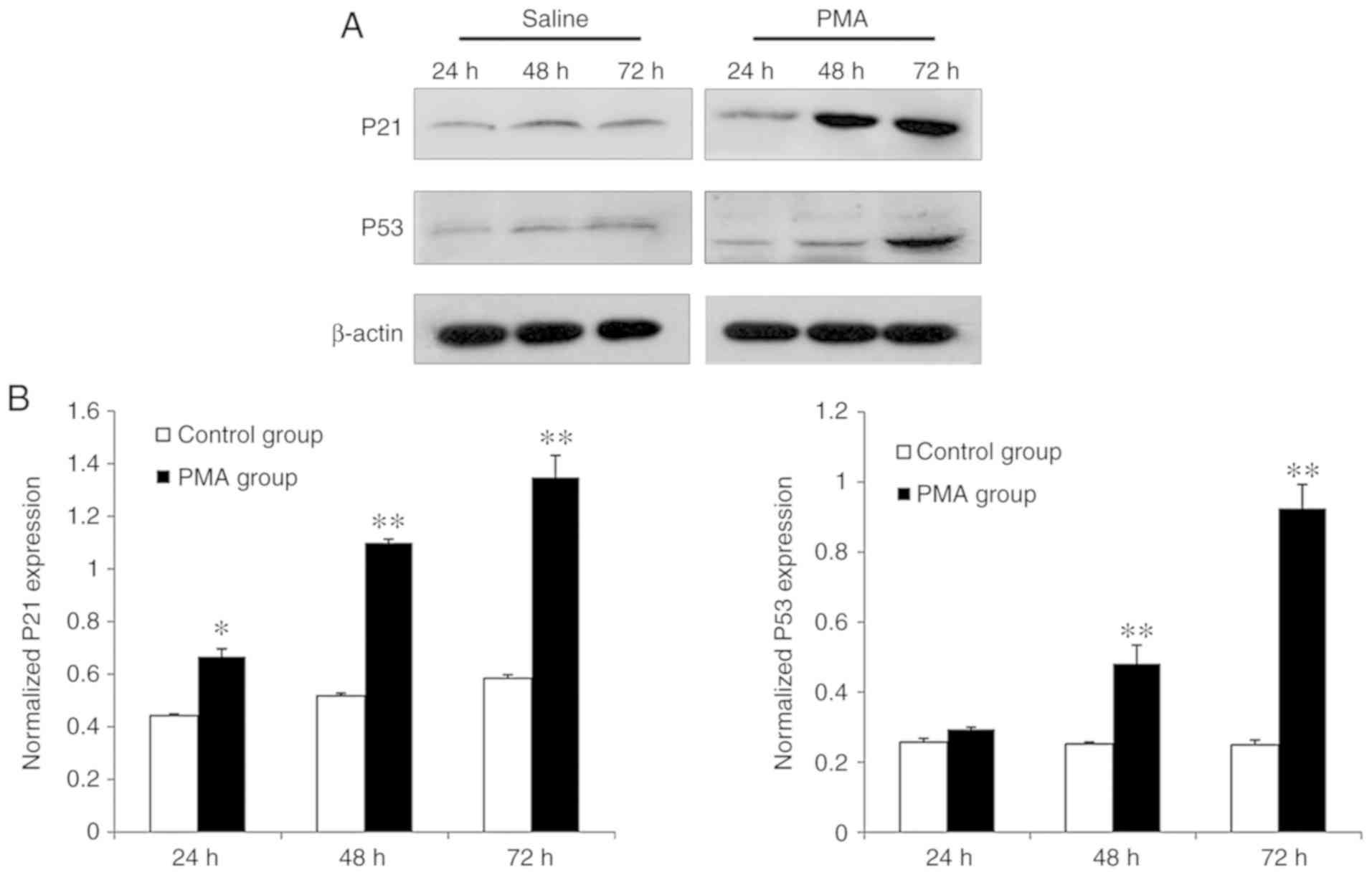
Through quantitative analyses for western blotting
conducted for assessing protein level alterations in p53 and p21
from each group, the results showed that only trace amounts of p53
and p21 protein were detected in cells of the control group
(Fig. 4). Compared with the
control group, PMA treatment upregulated p53 and p21 protein levels
in a time-dependent fashion. When microglia were tested after 24-h
PMA treatment, p21 protein expression gradually increased
(P<0.05) and reached its peak amount at 72 h (P<0.01).
Comparatively, the p53 protein level was upregulated in a
time-dependent manner and moreover, a significantly enhanced
expression level of p53 protein was first observed at 48 h of PMA
stimulation and peaked at 72 h (P<0.01), respectively
(Fig. 4).
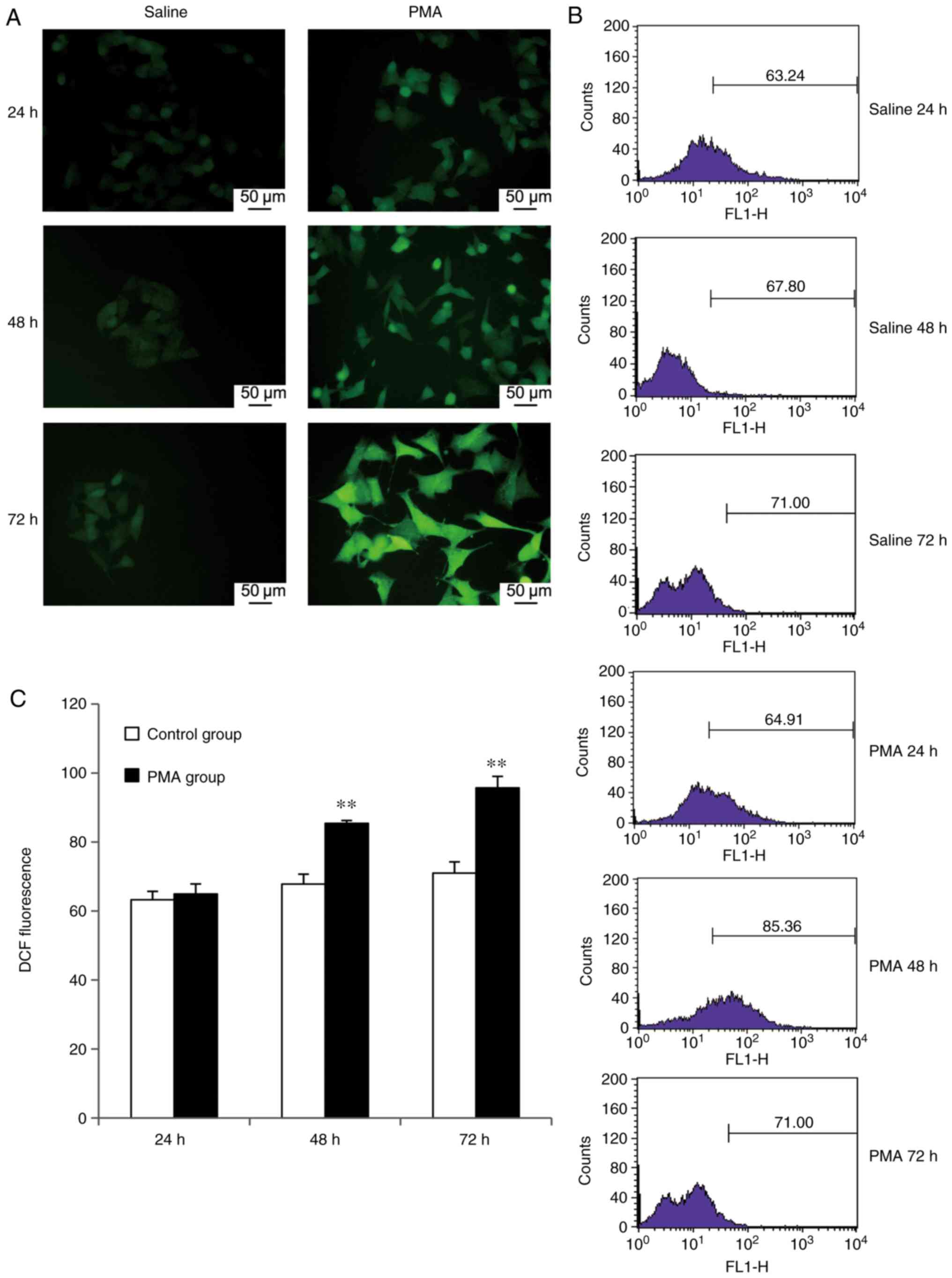
Expression of intracellular ROS in
response to PMA treatment
In this study, flow cytometry was used to measure
fluorescence intensities of intracellular DCF in microglia exposed
to PMA and the mean fluorescence intensity values (mean values)
were used to evaluate the level of ROS within microglia. Microglia
ROS levels in the PMA-treated group displayed an apparent increase,
compared with the control group and showed varying degrees of
increase after 24, 48 and 72 h (Fig.
5B). The increase in DCF fluorescence intensity occurred after
24 h PMA treatment and showed a statistically significant
difference regarding intensity values (P<0.01), compared with
the saline-treated 24-h control group. The increase in the DCF
fluorescence intensity was time-dependent and peaked after 72 h of
PMA treatment (Fig. 5B). A
similar pattern was observed using immunofluorescence microscopy.
The control group displayed weak DCF-mediated fluorescence, whereas
fluorescence intensity gradually increased after 24 h in a
time-dependent manner and the qualitative intensity was highest at
72 h (Fig. 5A).
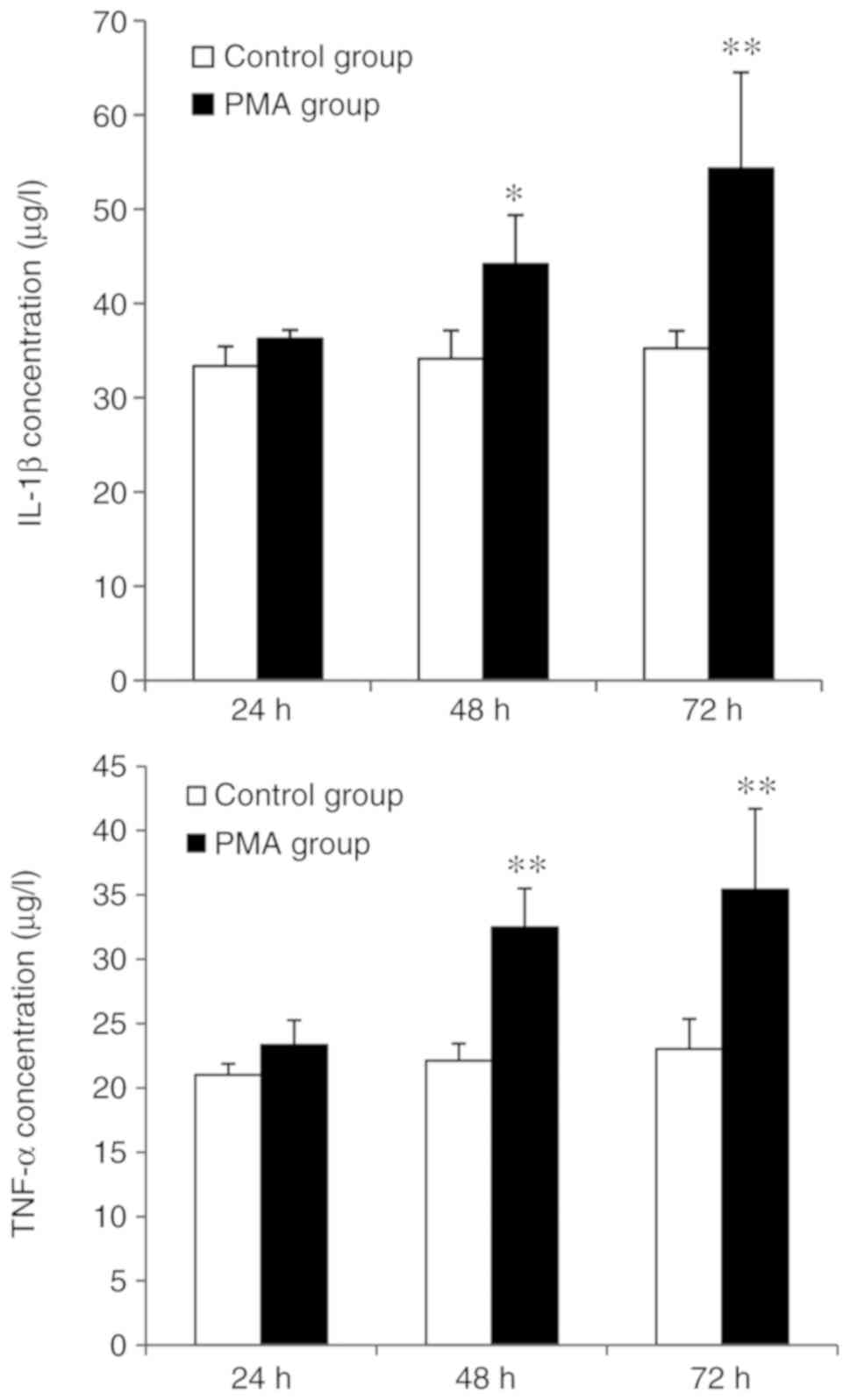
Detection of IL-1β and TNF-α in microglia
treated with PMA for various time points
ELISAs were used to detect the levels of IL-1β as
well as TNF-α, both of which were secreted by microglia from both
the control and PMA-treated groups (Fig. 6). There were also significant
differences, regarding levels of TNF-α from the control and
PMA-treated groups, both of which were exposed for 48 and 72 h,
respectively (P<0.05). Similarly, significant differences were
also noted in the amount of IL-1β secreted by microglia at 48 and
72 h following PMA stimulation, as compared with the control group
(P<0.01).
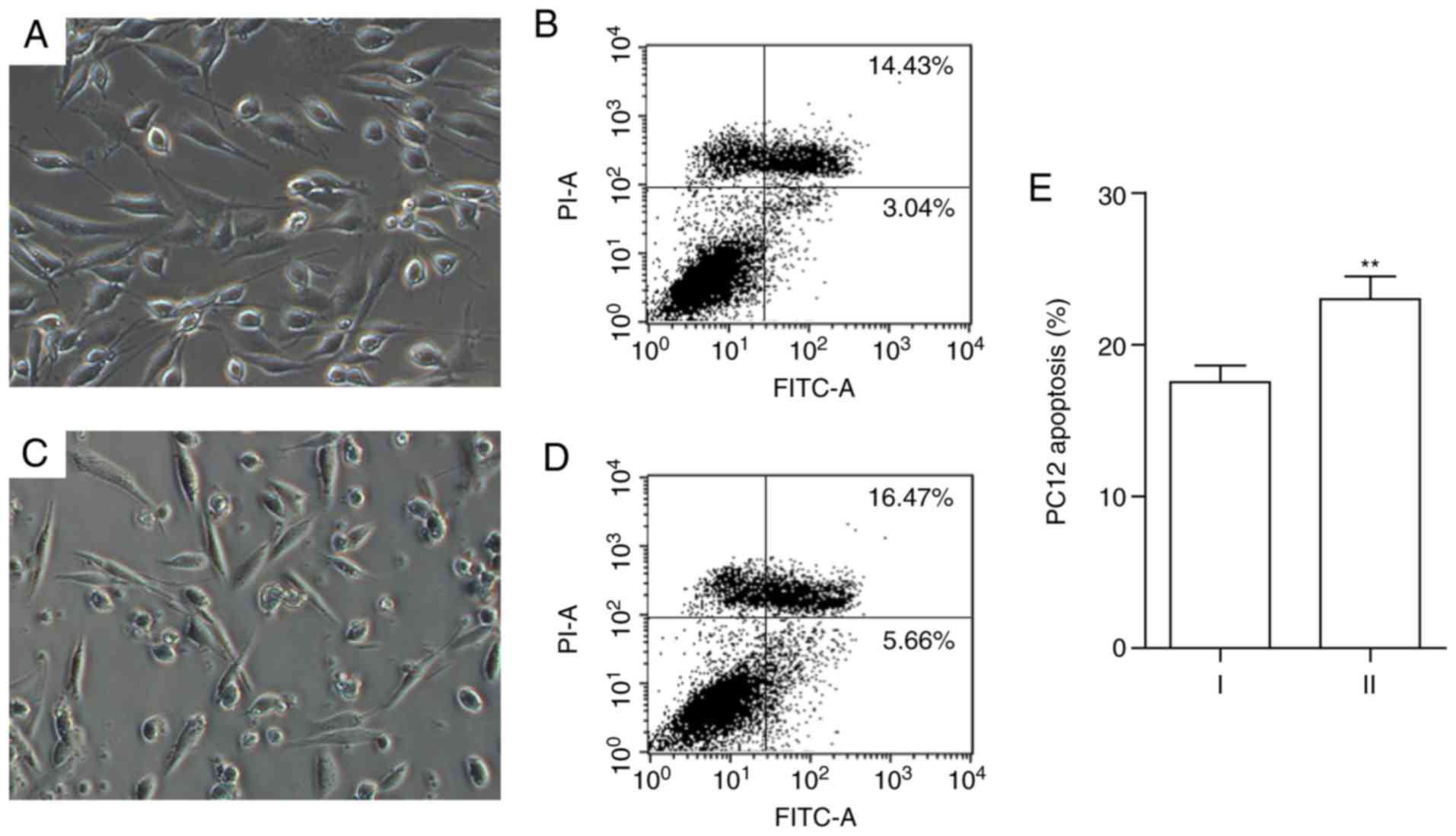
Abnormal morphology and apoptosis in PC12
cells following co-culturing with PMA-stimulated microglia
Under brightfield microscopy, PC12 cells in the
control group were assessed and found to exhibit spindle- or
triangle-shaped morphology and adherent growth (Fig. 7). In contrast, PC12 cells
co-cultured with senescent microglia showed irregular cell
morphology. Protruding structures gradually disappeared or were
shortened and the cells became smaller and rounded. Flow cytometry
analysis of the apoptotic rate demonstrated that, after being
co-cultured with senescent microglia, in PC12 cells the apoptosis
was apparently enhanced compared to the saline-treated 24-h control
group (P<0.05; Fig. 7).
Discussion
Under physiological conditions, senescence occurs
naturally and refers to a relatively stable state of dividing cells
that irreversibly lose their proliferative ability and experience
morphological changes with increasing age in human patients or
animal models (25). Based on
whether it is related to telomere shortening, senescence is divided
into physiological senescence and pathological senescence. The
former is also known as replicative senescence because it is
characterized by an increase in cell passages and the gradual loss
of telomeric sequences. Pathological senescence is due to the
stimulation of non-telomeric signals and is not related to telomere
shortening and decreased proliferative capacity. It occurs in
response to multiple stimuli under pathological conditions, such as
oxidative stress (10) injury
(11), radiation (12), tumor protein activation (13), and chemical mutagens (14), and is characterized by the
expression of senescence-related proteins, such as p53, p21, and
β-Gal and G0/G1 cell cycle arrest induced by the depletion of
ribonucleotide pools without detectable DNA damage under certain
conditions (26). This study
investigated the possible mechanisms by which carcinogens induce
OIS in primary cultures of rat microglia. After stimulating primary
microglia with the carcinogen PMA for 24, 48 and 72 h, it was found
that microglia displayed a typical pattern of cellular senescence,
including cell cycle-arrest phenotypes in the G0/G1 phase, slowed
cell proliferation, and reduced numbers of S-phase cells. In
addition, time-dependent increases in β-Gal activity and expression
levels of senescence-related proteins p53 and p21 concurrent with
PMA-induced inflammation were observed, both of which indicate
cellular senescence. During cellular senescence, functional
alterations also occurred and hence normal cellular function is
compromised. In the present study, it was found that the secretion
of inflammatory mediators, including IL-1β, TNF-α and ROS,
increased with longer PMA induction times and that the viability of
PC12 cells decreased after being co-cultured with microglia
stimulated by PMA, further confirming that the carcinogen PMA could
induce pathological senescence in microglia. Senescent microglia
not only show a significant increase in the release of inflammatory
mediators (9), but they also have
significantly different responses following injury, as compared to
that of cultured neurons (27).
OIS is the most common form of pathological
senescence. An endogenous protective mechanism used to prepare the
body fighting against cancer (13,17). When cells are threatened by
malignant transformation, OIS guides cells to terminate their
abnormal proliferation and to initiate senescence. Subsequently,
OIS-induced senescent cells secrete cytokines, which in turn
further promote cellular senescence, thus potentially forming a
positive feedback loop (28).
Several other studies also pointed out that OIS can also be induced
by oncogenes activation (29-32). For instance, moles are considered
as cancerous and/or pre-cancerous tissues and are often accompanied
by the occurrence of mutations in oncogenes. Oncogenic mutations
can induce cell senescence by OIS and thus can prevent benign nevi
from developing into a malignant melanoma (33). OIS is, therefore, an endogenous
protective mechanism by which the body prevents cancer
progression.
Senescent microglia are a cardinal cell type of the
brain's inflammatory microenvironment. During senescence,
over-activated microglia will produce and release a large number of
cytotoxic factors, such as TNF-α, IL-6, NO, and ROS, which have
toxic effects on neurons and lead to neuronal degeneration and cell
death (27,34). IL-1β and TNF-α are two important
proinflammatory cytokines released by microglia during the
inflammatory process in the CNS. Two previous studies found that
inflammatory cytokines are important mediators regulating cellular
senescence (35,36). The current study showed that
microglia cultured in vitro produced a large amount of IL-1β
and TNF-α when stimulated by the carcinogen PMA, suggesting that
PMA can induce microglial senescence and promote the release of
proinflammatory cytokines, including TNF-α and IL-1β, resulting in
CNS neuronal damage. Moreover, as shown by the in vitro
assays, oxidative stress can also result from PMA administration
(20,37). It has been reported previously
that ROS are critical factors during the senescence and that
subsequent oxidative damage, resulting from ROS accumulation in
senescent cells, can cause dysfunctional phenotype(s) (38). Long-term oxidative stress leads to
the accumulation of oxidative damage, which elicits cytotoxicity
against natural senescence and leads to senescence-related diseases
(39,40). Overall, the current study verified
these previous findings on the basis of generally consistent
results.
The mechanisms underlying senescence are complex and
not fully understood. Numerous studies have found a variety of
signaling pathways (p16INK4a/Rb, p53/p21 and oxidative
stress) involved in OIS signal transduction pathways (41-44). As a tumor suppressor, gene p53 is
an important regulator of cellular senescence and is triggered by
senescence-inducing stimuli. Activated p53 promotes the expression
of a variety of downstream target genes, including p21, thereby
inhibiting the activity of cyclin A/E and cyclin-dependent kinase
2, preventing Rb phosphorylation, and leading to the cessation of
cell proliferation (45,46). It has been reported that p53 is
only transiently increased in senescent cells. In contrast, p21
expression level is increased in senescent cells and even can
remain stable under different conditions (47), suggesting that p53 causes cell
senescence by activating stable levels of p21 (48,49). The present results also indicate
that in PMA-treated rat microglia, p21 protein levels gradually
increased during the senescence and were positively correlated with
p53 expression.
As a simplified in vitro model, PC12 cells
did provide solid as well as reproducible results within a
relatively short period of time, however, the authors admit that
primary neuron cultures would have been most ideal, which will
definitely be employed in future investigations. Moreover, a
significant reduction in cell proliferation during PMA-induced
microglia senescence was not found. In addition to individual
differences in experiment operational performance, culture time may
affect cell proliferation. In addition, although common indicators
of cellular senescence show sign of aging, they appear to initiate
at different time points and increase at different speeds,
indicating that they emerge during senescence-associated sequelae.
However, the initiating factor remains to be further explored. In
short, the current study believes that an in-depth investigation of
the mechanism underlying microglia senescence will lay the
foundation for future explorations of the role of microglia in PD
pathogenesis and lead to the translational application of PD and
cancer treatments.
Funding
This study was supported by grants from the China
National Nature Science Fund (grant no. 30973153), the Liaoning
Doctoral Starting Fund (grant no. 20071042), the Foundation of the
Liaoning Educational Committee (grant nos. L202013136 and
L2010560), and the Liaoning Foundation for outstanding young
scientist, which was awarded to Dr Ren Yan (grant no. LJQ2011081),
respectively.
Availability of data and materials
During the present study, all the data obtained as
well as analyzed are available, based on reasonable request to the
corresponding author.
Authors' contributions
YR and XGL designed the majority of the experiments,
DC performed investigation data analysis and wrote the manuscript;
LW and LSW provided experimental technique assistance; XHL and HMY
contributed to interpretation of the data and analyses. All of the
authors have read and approved the manuscript.
Ethics approval and consent to
participate
All animal experiments conducted were based on
applicable international/national/institutional guidelines for the
care and use of laboratory animals, which were approved and
supervised by the ethics committee of China Medical University. All
data published here are under the consent for publication. Written
informed consent was obtained from all individual participants
included in the study.
Patient consent for publication
All data published in the present study are covered
under the consent for publication. Written informed consent was
obtained from all individual participants included in the
study.
Competing interests
The authors declare they have no competing
interests.
Acknowledgments
Not applicable.
Abbreviations:
|
OIS
|
oncogene-induced senescence
|
|
PMA
|
phorbol 12-myristate 13-acetate
|
|
β-Gal
|
β-galactosidase
|
|
TNF-α
|
tumor necrosis factor-α
|
|
IL-1β
|
interleukin-1β
|
|
PD
|
Parkinson's disease
|
|
DA
|
dopaminergic
|
|
PFA
|
paraformaldehyde
|
|
DCF
|
dichlorodihydrofluorescein
|
|
ROS
|
reactive oxygen species
|
|
CNS
|
central nervous system
|
References
|
1
|
Lively S and Schlichter LC: Microglia
responses to pro-inflammatory stimuli (LPS, IFNγ+TNFα) and
reprogramming by resolving cytokines (IL-4, IL-10). Front Cell
Neurosci. 12:2152018. View Article : Google Scholar
|
|
2
|
Wang GQ, Li DD, Huang C, Lu DS, Zhang C,
Zhou SY, Liu J and Zhang F: Icariin reduces dopaminergic neuronal
loss and microglia-mediated inflammation in vivo and in vitro.
Front Mol Neurosci. 10:4412018. View Article : Google Scholar :
|
|
3
|
Heneka MT, Carson MJ, El Khoury J,
Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T,
Vitorica J and Ransohoff RM: et al Neuroinflammation in Alzheimer's
disease. Lancet Neurol. 14:388–405. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Peterson LJ and Flood PM: Oxidative stress
and microglial cells in Parkinson's disease. Mediators Inflamm.
2012:4012642012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Streit WJ and Xue QS: Human CNS immune
senescence and neurodegeneration. Curr Opin Immunol. 29:93–96.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu J, Wang MW, Gu P, Ma QY, Wang YY, Geng
Y, Yuan ZY, Cui DS, Zhang ZX and Ma L: et al Microglial activation
and age-related dopaminergic neurodegeneration in MPTP-treated
SAMP8 mice. Brain Res. 1345:213–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qin L, Wu X, Block ML, Liu Y, Breese GR,
Hong JS, Knapp DJ and Crews FT: Systemic LPS cause chronic
neuroinflammation and progressive neurodegeneration. Glia.
55:453–462. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sawada H, Hishida R, Hirata Y, Ono K,
Suzuki H, Muramatsu S, Nakano I, Nagatsu T and Sawada M: Activated
microglia affect the nigro-striatal dopamine neurons differently in
neonatal and aged mice treated with
1-methyl-4-phenyl-1,2,3,6-tetrahydropyr-idine. J Neurosci Res.
85:1752–1761. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luo XG, Ding JQ and Chen SD: Microglia in
the aging brain: Relevance to neurodegeneration. Mol Neurodegener.
5:122010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bu H, Wedel S, Cavinato M and Jansen-Durr
P: MicroRNA regulation of oxidative stress-induced cellular
senescence. Oxid Med Cell Longev. 2017:23986962017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Borlon C, Chretien A, Debacq-Chainiaux F
and Toussaint O: Transient increased extracellular release of H2O2
during establishment of UVB-induced premature senescence. Ann NY
Acad Sci. 1119:72–77. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Debacq-Chainiaux F, Leduc C, Verbeke A and
Toussaint O: UV, stress and aging. Dermatoendocrinol. 4:236–240.
2012. View Article : Google Scholar
|
|
13
|
Mooi WJ and Peeper DS: Oncogene-induced
cell senescence-halting on the road to cancer. N Engl J Med.
355:1037–1046. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kovatcheva M, Liu DD, Dickson MA, Klein
ME, O'Connor R, Wilder FO, Socci ND, Tap WD, Schwartz GK, Singer S,
et al: MDM2 turnover and expression of ATRX determine the choice
between quiescence and senescence in response to CDK4 inhibition.
Oncotarget. 6:8226–8243. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Itahana K, Campisi J and Dimri GP: Methods
to detect biomarkers of cellular senescence: The
senescence-associated beta-galactosidase assay. Methods Mol Biol.
371:21–31. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Macip S, Igarashi M, Fang L, Chen A, Pan
ZQ, Lee SW and Aaronson SA: Inhibition of p21-mediated ROS
accumulation can rescue p21-induced senescence. EMBO J.
21:2180–2188. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Serrano M: Cancer regression by
senescence. N Engl J Med. 356:1996–1997. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Serrano M, Lin AW, McCurrach ME, Beach D
and Lowe SW: Oncogenic ras provokes premature cell senescence
associated with accumulation of p53 and p16INK4a. Cell. 88:593–602.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li YH, Bi HC, Huang L, Jin J, Zhong GP,
Zhou XN and Huang M: Phorbol 12-myristate 13-acetate inhibits
P-glycoprotein-mediated efflux of digoxin in MDCKII-MDR1 and Caco-2
cell monolayer models. Acta Pharmacol Sin. 35:283–291. 2014.
View Article : Google Scholar
|
|
20
|
Liu L, Luo XG, Yu HM, Feng Y, Ren Y, Yin
YF, Shang H and He ZY: Repeated intra-nigrostriatal injection of
phorbol myristate acetate induces microglial senescence in adult
rats. Mol Med Rep. 12:7271–7278. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mowla S, Pinnock R, Leaner VD, Goding CR
and Prince S: PMA-induced up-regulation of TBX3 is mediated by AP-1
and contributes to breast cancer cell migration. Biochem J.
433:145–153. 2011. View Article : Google Scholar
|
|
22
|
Aschner M and Kimelberg HK: The use of
astrocytes in culture as model systems for evaluating
neurotoxic-induced-injury. Neurotoxicology. 12:505–517.
1991.PubMed/NCBI
|
|
23
|
Moreno JA, Sullivan KA, Carbone DL,
Hanneman WH and Tjalkens RB: Manganese potentiates nuclear
factor-kappaB-dependent expression of nitric oxide synthase 2 in
astrocytes by activating soluble guanylate cyclase and
extracellular responsive kinase signaling pathways. J Neurosci Res.
86:2028–2038. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Carbone DL, Popichak KA, Moreno JA, Safe S
and Tjalkens RB: Suppression of
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced nitric-oxide
synthase 2 expression in astrocytes by a novel diindolylmethane
analog protects striatal neurons against apoptosis. Mol Pharmacol.
75:35–43. 2009. View Article : Google Scholar :
|
|
25
|
Cichowski K and Hahn WC: Unexpected pieces
to the senescence puzzle. Cell. 133:958–961. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim KH and Sederstrom JM: Assaying cell
cycle status using flow cytometry. Curr Protoc Mol Biol.
111:28.6.1–28.6.11. 2015. View Article : Google Scholar
|
|
27
|
Polazzi E and Contestabile A: Reciprocal
interactions between microglia and neurons: From survival to
neuropathology. Rev Neurosci. 13:221–242. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lehmann AR and Carr AM: The
ataxia-telangiectasia gene: A link between checkpoint controls,
neurodegeneration and cancer. Trends Genet. 11:375–377. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chinta SJ, Lieu CA, Demaria M, Laberge RM,
Campisi J and Andersen JK: Environmental stress, ageing and glial
cell senescence: A novel mechanistic link to Parkinson's disease? J
Intern Med. 273:429–436. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Collado M, Gil J, Efeyan A, Guerra C,
Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM,
Barbacid M, et al: Tumour biology: Senescence in premalignant
tumours. Nature. 436:6422005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dhomen N, Reis-Filho JS, da Rocha Dias S,
Hayward R, Savage K, Delmas V, Larue L, Pritchard C and Marais R:
Oncogenic Braf induces melanocyte senescence and melanoma in mice.
Cancer Cell. 15:294–303. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sarkisian CJ, Keister BA, Stairs DB, Boxer
RB, Moody SE and Chodosh LA: Dose-dependent oncogene-induced
senescence in vivo and its evasion during mammary tumorigenesis.
Nat Cell Biol. 9:493–505. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Michaloglou C, Vredeveld LC, Soengas MS,
Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi
WJ and Peeper DS: BRAFE600-associated senescence-like cell cycle
arrest of human naevi. Nature. 436:720–724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
More SV, Kumar H, Kim IS, Song SY and Choi
DK: Cellular and molecular mediators of neuroinflammation in the
pathogenesis of Parkinson's disease. Mediators Inflamm.
2013:9523752013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Acosta JC, O'Loghlen A, Banito A, Guijarro
MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N,
et al: Chemokine signaling via the CXCR2 receptor reinforces
senescence. Cell. 133:1006–1018. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kuilman T, Michaloglou C, Vredeveld LC,
Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ and Peeper DS:
Oncogene-induced senescence relayed by an interleukin-dependent
inflammatory network. Cell. 133:1019–1031. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kumar A, Chen SH, Kadiiska MB, Hong JS,
Zielonka J, Kalyanaraman B and Mason RP: Inducible nitric oxide
synthase is key to peroxynitrite-mediated, LPS-induced protein
radical formation in murine microglial BV2 cells. Free Radic Biol
Med. 73:51–59. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Irani K: Oxidant signaling in vascular
cell growth, death, and survival: A review of the roles of reactive
oxygen species in smooth muscle and endothelial cell mitogenic and
apoptotic signaling. Circ Res. 87:179–183. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Harman D: The free radical theory of
aging. Antioxid Redox Signal. 5:557–561. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Park ES, Kim SR and Jin BK: Transient
receptor potential vanilloid subtype 1 contributes to mesencephalic
dopaminergic neuronal survival by inhibiting microglia-originated
oxidative stress. Brain Res Bull. 89:92–96. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chandeck C and Mooi WJ: Oncogene-induced
cellular senescence. Adv Anat Pathol. 17:42–48. 2010. View Article : Google Scholar
|
|
42
|
Coppe JP, Rodier F, Patil CK, Freund A,
Desprez PY and Campisi J: Tumor suppressor and aging biomarker
p16(INK4a) induces cellular senescence without the associated
inflammatory secretory phenotype. J Biol Chem. 286:36396–36403.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Moiseeva O, Bourdeau V, Roux A,
Deschenes-Simard X and Ferbeyre G: Mitochondrial dysfunction
contributes to oncogene-induced senescence. Mol Cell Biol.
29:4495–4507. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Reddy JP and Li Y: Oncogene-induced
senescence and its role in tumor suppression. J Mammary Gland Biol
Neoplasia. 16:247–256. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fitzgerald AL, Osman AA, Xie TX, Patel A,
Skinner H, Sandulache V and Myers JN: Reactive oxygen species and
p21Waf1/Cip1 are both essential for p53-mediated senescence of head
and neck cancer cells. Cell Death Dis. 6:e16782015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gatza C, Moore L, Dumble M and Donehower
LA: Tumor suppressor dosage regulates stem cell dynamics during
aging. Cell Cycle. 6:52–55. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
John R, Chand V, Chakraborty S, Jaiswal N
and Nag A: DNA damage induced activation of Cygb stabilizes p53 and
mediates G1 arrest. DNA Repair (Amst). 24:107–112. 2014. View Article : Google Scholar
|
|
48
|
Chandler H and Peters G: Stressing the
cell cycle in senescence and aging. Curr Opin Cell Biol.
25:765–771. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kim YY, Jee HJ, Um JH, Kim YM, Bae SS and
Yun J: Cooperation between p21 and Akt is required for
p53-dependent cellular senescence. Aging Cell. 16:1094–1103. 2017.
View Article : Google Scholar : PubMed/NCBI
|