Introduction
Acute lung injury (ALI) is a severe clinical
condition with a high mortality rate among critically ill patients
(1,2). Although there have been significant
advancements in the therapeutic strategies of ALI, the morbidity
and mortality rates remains high (3,4).
In recent years, it has become widely accepted that inflammation is
an important pathological characteristic of ALI, and contributes to
the initiation and development of ALI (5). Therefore, inhibition of the
inflammatory response may be key to the improvement of ALI patient
outcomes.
It is well known that toll-like receptors (TLRs)
play an important role in the inflammatory process triggered by
lipopolysaccharide (LPS) (6,7).
After TLR4 recognizes LPS, a series of cascades, including myeloid
differentiation factor 88 (MyD88), are initiated, followed by
activation of the nuclear factor (NF)-κB pathway and the secretion
of pro-inflammatory cytokines, including tumor necrosis factor
(TNF)-α, interleukin (IL)-1β and IL-6, ultimately resulting in ALI
(8). Once the TLR4 pathway is
inhibited, the inflammatory response can be alleviated, thereby
attenuating ALI. Extensive studies have demonstrated, using an LPS
induced in vivo ALI model, that inhibition of the TLR4
pathway is beneficial in ALI (9,10).
For example, Zhang et al reported that inhibition of the
TLR4/NF-κB signaling pathway improved the oxidative stress and
inflammatory response in the lung tissues of ALI rats (11). Therefore, suppression of the
activation of the TLR4/NF-κB pathway may alleviate
inflammation-induced ALI.
MicroRNAs (miRNAs) are a family of short non-coding
RNAs (with a mean size of 22 nucleotides), which suppress target
gene expression through either translation repression or RNA
degradation (12). Accumulating
evidence has demonstrated that miRNAs potentially contribute to the
development of ALI via regulation of target genes (13-15). For example, Yang et al
observed that miR-140-5p inhibited LPS-induced inflammatory
response in ALI via blocking the TLR4 pathway (16). Ling et al demonstrated that
miR-494 inhibition improved lung injury through suppressing the
inflammatory response in ALI rats (17). miR-17, a member of the miR-17-92
cluster, has been found to play an important role in ameliorating
inflammatory response, particularly pulmonary inflammation
(18,19). More importantly, a recent study
has identified decreased expression of miR-17 in ALI mice, and
miR-17 negatively regulates lung FOXA1 expression, which plays an
important role in ALI by promoting the apoptosis of alveolar type
II epithelial cells in vitro and in vivo (20). However, the function of miR-17 in
inflammatory response in ALI has yet to be fully elucidated.
In the present study, an in vivo mice model
of ALI and an in vitro LPS-induced RAW264.7 cell injury
model were established to investigate the role and underlying
mechanism of action of miR-17 in the regulation of inflammation in
ALI. The aim was to determine whether miR-17 may hold promise as a
novel treatment target for the prevention and treatment of ALI.
Materials and methods
Ethics statement
The protocol of the present study was approved by
the Ethics Committee of the Affiliated Hospital of Inner Mongolia
University for Nationalities (permit no. 2018-0139). The mice were
treated humanely, and all measures were undertaken to minimize
animal suffering. The mice were monitored every 12 h over a period
of 1 week for health and behavior. A humane endpoint was used in
our experiments according to previous report (21). The specific signs used to
determine the endpoint included: i) Loss of >25% body weight
compared with the starting weight; ii) decreased food or water
intake; iii) decreased mobility/activity, lethargy, rough hair
coat. Sacrifice was performed by intraperitoneal injection of
sodium pentobarbital (50 mg/kg) followed by cervical dislocation,
and death was confirmed when no spontaneous breathing for 2-3 min
and no blinking reflex were observed (22). No animals died before meeting
these endpoints. All mice (n=60) were euthanized as mentioned
above.
Animals
A total of 60 male BALB/c mice (6-8 weeks old,
weighing 18-22 g) were obtained from the Shanghai SLAC Laboratory
Animal Co. Ltd. BALB/c mice were housed under standard conditions
(12-h light-dark cycle, 25-27°C, ~40% humidity) with free access to
food and water throughout the duration of the experiments. A total
of 20 mice were randomly divided into four groups (n=5/group) as
follows: i) Control, ii) LPS, iii) LPS + agomir-17 and iv) LPS +
agomir-negative control (NC) groups. LPS group mice were injected
through the tail vein with 2 mg/kg LPS. The control group received
the same volume of normal saline. Mice in the agomir-17 or agomir
NC groups were injected intravenously with agomir-17 or agomir NC
(8 mg/kg), respectively (20), 24
h prior to LPS induction. All mice were anesthetized with
pentobarbital sodium (50 mg/kg, intraperitoneal injection), and
euthanized by cervical dislocation at 24 h after LPS treatment
(except those included in the survival experiment), then the lung
tissues and bronchoalveolar lavage fluid (BALF) were collected for
subsequent analysis.
For the survival rate analysis, 40 mice were divided
into four groups (n=10/group) as mentioned above. The survival rate
from 0 to 7 days was observed and calculated using the Kaplan-Meier
method. Agomir-17/NC were designed and synthesized by RiboBio. The
sequences were as follows: Agomir-17: 5′-CAA AGU GCU UAC AGU GCA
GGU AG-3′; and agomir NC: 5′-GUC CUG AGA AGG CUA GCA UAG AU-3′.
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
Total RNA was isolated from lung tissues or cells
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
RT of miR-17 was performed using the miScript II RT kit
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 30 min.
miR-17 expression was measured using the Exiqon SYBR Green Master
Mix (Exiqon; Qiagen, Inc.) on a Light Cycler instrument (Bio-Rad
Laboratories, Inc.). The thermocycling conditions were as follows:
A hot start step at 95°C for 10 min, followed by 40 cycles at 95°C
for 15 sec and 60°C for 1 min. The primers for RT-qPCR analysis
were as follows: miR-17 forward, 5′-AGGCCCAAAGTGCTGTTCGT-3′ and
reverse, 5′-GTGCAGGGTCCGAGGT-3′; U6 forward, 5′-TGC GGG TGC TCG CTT
CGC AGC-3′ and reverse, 5′-CCA GTG CAG GGT CCG AGG T-3′. The miRNA
relative expressions were analyzed using the 2−ΔΔCq
method (23).
Lung histology
After a 24-h LPS challenge, the mice were sacrificed
and the lung tissues were harvested and fixed in 10% formalin for
24 h at 4°C, embedded in paraffin, and cut into 4-µm
sections. The sections were stained using hematoxylin for 5 min and
rinsed with distilled water, followed by color separation with
alcohol hydrochloric acid at room temperature. Subsequently, the
samples were stained with eosin for 2 min, dehydrated, cleared,
dried and mounted at room temperature. Histological changes were
observed and photographed under a light microscope (E-800M, Nikon
Corporation) at a magnification of ×200 and 400.
Evaluation of lung permeability
The Evans Blue (EB) dye extravasation method was
used to assess pulmonary permeability as previously described
(24). Briefly, EB dye (20 mg/kg,
Sigma-Aldrich; Merck KGaA) at a concentration of 0.5% (5 mg/ml) in
normal saline was injected into the mice of each group through the
tail vein. After 2 h, the mice were sacrificed and then the dye was
extracted by incubation in formamide for 24 h at 60°C. The light
absorbance at 620 nm was measured, and the dye concentration in
lung homogenate was calculated against a standard curve and was
expressed as µg of Evans blue dye per g of lung tissue.
Lung wet/dry (W/D) ratio
The W/D ratio was used to assess pulmonary edema.
After a 24-h LPS challenge, the right lung of the mice was
harvested and immediately weighted, then dried in an incubator at
80°C for 60 h.
Measurement of IL-6, IL-1β and TNF-α
The supernatant from the BALF was collected by
centrifugation at 12,000 x g for 10 min at 4°C. For cultured cells,
the supernatant was carefully collected by centrifugation 12,000 ×
g for 10 min at 4°C. The concentrations of IL-6, IL-1β and TNF-α
were analyzed by using IL-6 (cat. no. p1330), IL-1β (cat. no.
p1305) and TNF-α (cat. no. pt518) ELISA kits, respectively,
according to the instructions of the manufacturer (Beyotime
Institute of Biotechnology).
Measurement of myeloperoxidase (MPO)
activity
Lung tissue homogenate was subjected to MPO assay
using a commercial kit (cat. no. ab105136, Abcam) and the
absorbance at 460 nm was detected using a microplate reader
(Bio-Tek Instruments, Inc.).
Cell culture and transfection
RAW264.7 cells were obtained from Sciencell Research
Laboratories and maintained in DMEM supplemented with 10% FBS
(Sigma-Aldrich; Merck KGaA), 1% penicillin and streptomycin
(Sigma-Aldrich; Merck KGaA) at 37°C and 5% CO2 in an
incubator.
After RAW264.7 cells in 6-well plates had grown to
~80% confluence, 20 nM miR-17 mimics, 20 nM miR-17 inhibitor, 2
µg pcDNA-TLR4 or 30 nM si-TLR4 were transfected into the
cells at 37°C for 24 h using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). miR-17 inhibitor,
miR-17 mimics and the corresponding control vectors were purchased
from RiboBio Co., Ltd. The TLR4 overexpressing vector pcDNA-TLR4
and empty vector pcDNA were constructed by Qiagen, Inc. In
addition, TLR4 siRNA (si-TLR4) and corresponding negative control
siRNA (si-Scramble) were purchased from RiboBio Co., Ltd. The
sequences were as follows: miR-17-5p mimic: 5′-CAA AGU GCU UAC AGU
GCA GGU AG-3′; mimics NC: 5′-GUC CUG AGA AGG CUA GCA UAG AU-3′;
miR-17-5p inhibitor: 5′-CUA CCU GCA CUG UAA GCA CUU UG-3′,
inhibitor NC 5′-CUA UGC U AGC CUU CUC AGG ACU U-3′.
Cell proliferation
The effect of LPS (2 µg/ml) on RAW264.7 cells
was measured by using the MTT assay. At the end of the
transfection, 20 µl MTT solution (Sigma-Aldrich; Merck KGaA)
was added into each well (2x105/well), and RAW264.7
cells were cultured for another 2 h. Then, the absorbance at 450 nm
was detected by a microplate reader (Bio-Tek Instruments,
Inc.).
Luciferase assay
miRNA target prediction tools, including PicTar
version 2007 (https://pictar.mdc-berlin.de/) and TargetScan Release
7.0 (http://targetscan.org/) were used to
search for the putative targets of miR-17. The dual luciferase
reporter assay was performed as described previously (25). RAW264.7 cells were treated with
miR-17 mimics, miR-17 inhibitor the luciferase reporter plasmids
(wt-TLR4-UTR-pGL3 or mt-TLR4-UTR-pGL3), and pRL-TK-Renilla control
plasmid (Promega Corporation) using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.). At 48 h post-transfection,
lucif-erase activity was detected using the Dual Luciferase
Reporter kit (Beyotime Institute of Biotechnology).
Western blot analysis
Protein was extracted from the mouse lung tissues
using the Nuclear and Cytoplasmic Protein Extraction kit (Beyotime
Institute of Biotechnology). The protein concentration was measured
using a protein analysis kit (Bio-Rad Laboratories, Inc.).
Extracted protein samples (40 µg) were separated by 12%
SDS-PAGE (w/v) and transferred onto a PVDF membrane (EMD
Millipore). The membrane was blocked with 5% skimmed milk for 2 h
at room temperature, followed by incubation with primary antibodies
against TLR4 (cat. no. 14358, 1:2,000 dilution), nuclear p-p65
(Ser-468, cat. no. 3039, 1:1,000 dilution), p-IκB-α (Ser-32, cat.
no. 2859, 1:1,000 dilution), IκB-α (cat. no. 4814, 1:1,000
dilution), Histone H3 (cat. no. 9728, 1:1,000 dilution) and β-actin
(cat. no. 3700, 1:1,000) (all from Cell Signaling Technology, Inc.)
at 4°C overnight, followed by HRP-conjugated goat anti-rabbit IgG
(1:10,000; cat. no. 205718; Abcam). β-actin and Histone H3 served
as internal controls. The protein bands were developed using an ECL
kit (GE Healthcare) and blot bands were quantified with ImageJ
version 1.46 (Rawak Software, Inc.).
Statistical analysis
GraphPad Prism 5.0 (GraphPad Software, Inc.) was
used to perform the statistical analyses. When only two groups were
compared, Student's t-test was used. One-way analysis of variance
followed by Tukey's post hoc test was applied to compare
differences between multiple groups. Survival analysis was
performed using the Kaplan-Meier method. All data are presented as
the mean ± standard deviation. P<0.05 was considered to indicate
statistically significant differences.
Results
miR-17 is downregulated in lung tissues
of ALI mice
First, an LPS-induced ALI mouse model was
established, which is widely used to simulate the pathological
conditions of severe lung injury in vivo (26). Following LPS treatment, the
pathological changes of lung tissues, lung W/D ratio (an indicator
of lung edema), pulmonary capillary permeability, MPO activity and
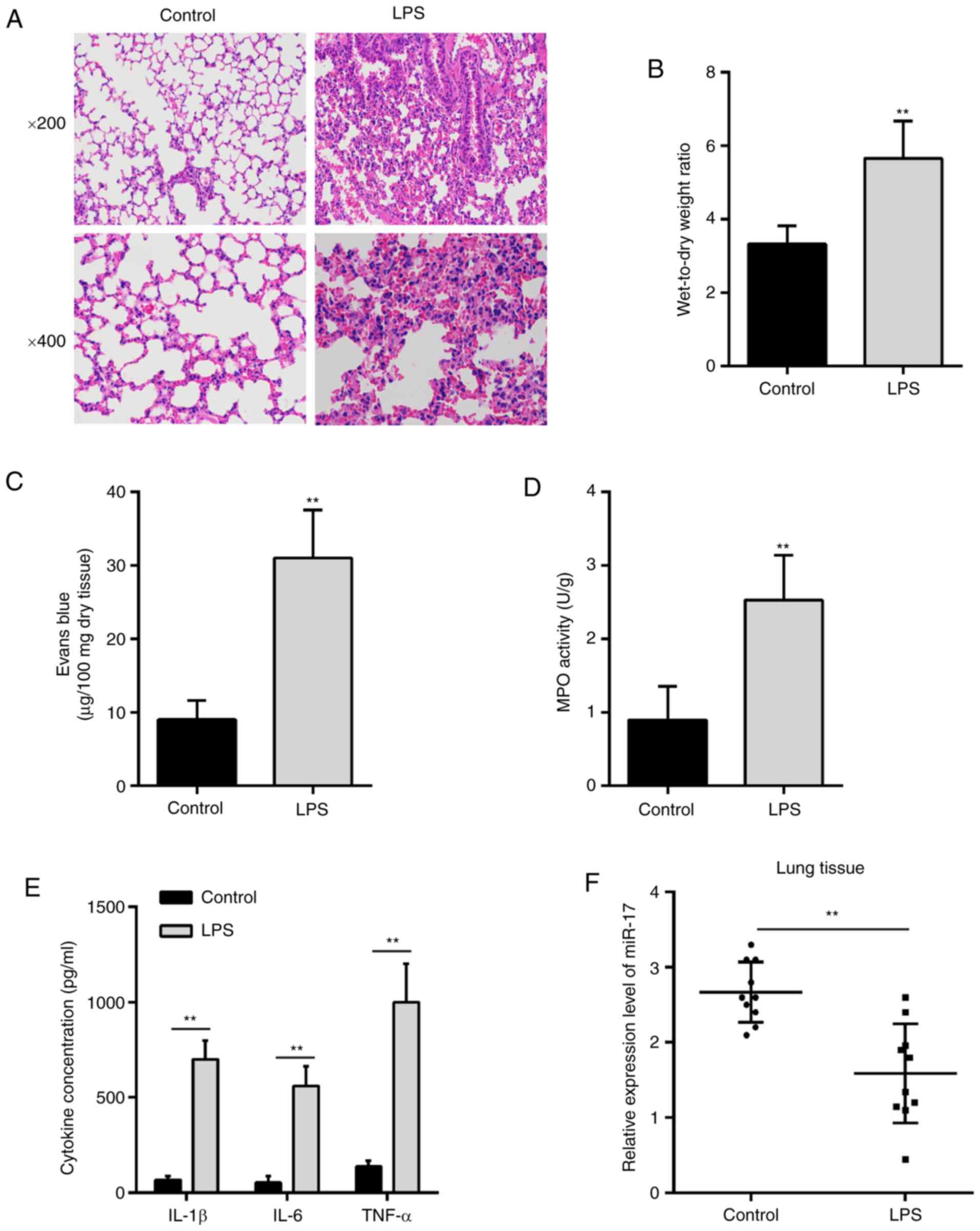
the inflammatory response were evaluated. As shown in Fig. 1A, the H&E staining results
revealed that LPS treatment caused obvious pathological changes,
including inflammatory cell infiltration and widespread increased
alveolar wall thickness, compared with the control group
(n=5/group). The W/D ratio of mice in the LPS group (n=5/group) was
markedly higher compared with that in the control group (Fig. 1B). The pulmonary capillary
permeability was measured by the EB dye method (n=5/group) and the
results demonstrated that LPS treatment led to a significant
increase in EB dye extravasation compared with the control group
(Fig. 1C). The MPO activity,
determined by a commercial assay (n=5/group), was markedly
upregulated following LPS stimulation compared with that in the
control group (Fig. 1D).
Furthermore, the expression of IL-1β, IL-6 and TNF-α was measured
by ELISA (n=5/group). As shown in Fig. 1E, the levels of these cytokines
were obviously increased in the LPS group compared with those in
the control group (Fig. 1E). All
these data indicated that the LPS-induced ALI mouse model was
successfully constructed.
To investigate the potential involvement of miR-17
in ALI, the expression level of miR-17 was measured in lung tissues
from ALI mice (n=10/group). It was observed that miR-17 was
markedly downregulated in the lung tissues from ALI mice (Fig. 1F), suggesting that miR-17 may play
an important role in the pathogenesis of ALI.
Overexpression of miR-17 improves
LPS-induced ALI in vivo
To further examine the therapeutic effect of miR-17
in LPS-induced ALI, agomiR-17 was intravenously administered to the
mice, followed by LPS treatment. The miRNA transfection efficiency
was first evaluated using RT-qPCR. The results demonstrated that
the miR-17 expression in lung tissues from ALI mice was increased
following agomiR-17 injection (n=5/group; Fig. 2A). Subsequently, the pathological
changes, pulmonary edema, pulmonary capillary permeability and MPO
activity were determined with the use of H&E staining, EB
extravasation, W/D ratio and MPO assays, respectively (n=5/group).
It was observed that LPS treatment was associated with a higher
extent of lung injury compared with the control group, while
agomir-17 alleviated the severity and distribution of lung lesions
caused by LPS (Fig. 2B). As shown
in Fig. 2C and D, agomir-17
treatment markedly reduced W/D ratio and EB extravasation in ALI
mice, suggesting that miR-17 effectively improved the pulmonary
edema and pulmonary capillary permeability. It was also observed
that the increased MPO activity induced by LPS was obviously
inhibited by agomir-17 (Fig. 2E).
In addition, the Kaplan-Meier method revealed that 50% of mice in
the LPS group died within 7 days, whereas the 7-day survival rate
after agomiR-17 injection was markedly higher compared with the LPS
group (n=10/group; Fig. 2F). More
importantly, the protein levels of cytokines, including IL-1β, IL-6
and TNF-α, induced by LPS were significantly decreased following
agomir-17 injection (n=5/group; Fig.
2G). Collectively, these data indicate that miR-17 upregulation
may improve LPS-induced ALI.
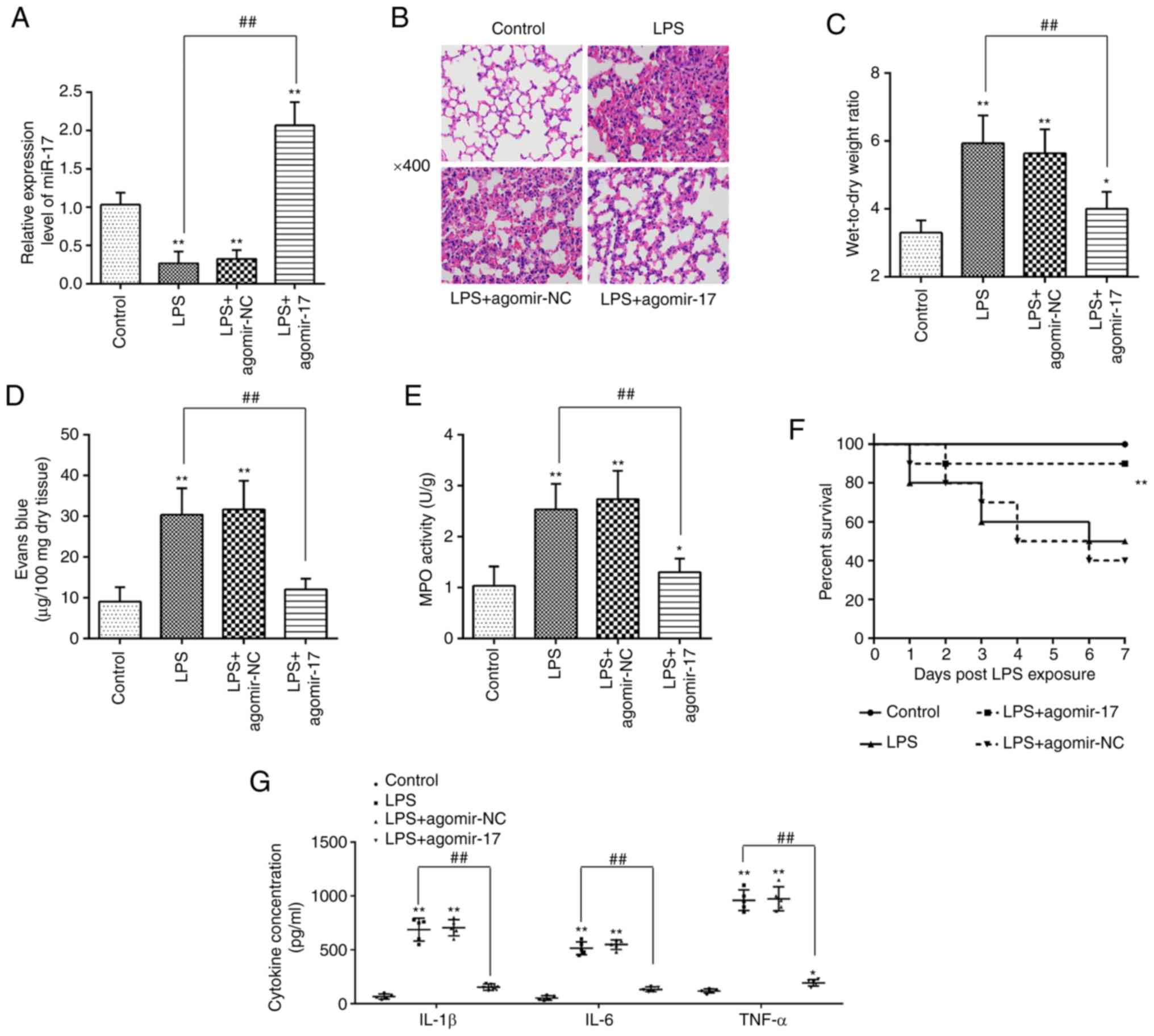 | Figure 2Overexpression of miR-17 improved
LPS-induced ALI in vivo. The mice were injected
intravenously with agomir-17 or agomiR-NC 24 h prior to LPS
treatment. The mice were sacrificed after LPS administration for 24
h, and then the lung tissues and BALF were collected for analysis
(n=5 mice per group). (A) The miR-17 expression level was measured
by RT-qPCR analysis. (B) Histopathological analysis of lung tissue
was performed following with hematoxylin and eosin staining.
Original magnification, x400. (C) Pulmonary edema was evaluated
using the lung W/D ratio. (D) Lung permeability was assessed using
the Evans Blue dye extravasation method. (E) Infiltration of
neutrophils into the lung tissues was assessed by MPO activity. (F)
Kaplan-Meier survival curves of mice in all treatment groups (n=10
mice per group). (G) The levels of the cytokines IL-1β, IL-6 and
TNF-α were detected by ELISA. Data are expressed as mean ± standard
deviation of three independent experiments. *P<0.05,
**P<0.01 vs. control group. ##P<0.01
vs. LPS group. LPS, lipopolysaccharide; ALI, acute lung injury;
MPO, myeloperoxidase; RT-qPCR, reverse transcription-quantitative
PCR; W/D ratio, wet/dry ratio; BALF, bronchoalveolar lavage fluid;
IL, interleukin; TNF, tumor necrosis factor. |
TLR4 is a direct target of miR-17
To investigate the potential molecular mechanism of
miR-17-regulated inflammatory events in ALI, TargetScan and PicTar
analyses were conducted to predict the target genes of miR-17. As
shown in Fig. 3A and B, TLR4 was
identified as a potential target gene of miR-17, with the target
site located in the 3′-UTR of TLR4 mRNA. It is well known that
TLR-4 is a common receptor of LPS, and its downstream signaling
effector, NF-κB, is closely associated with inflammatory response
in ALI (14,27-30). Thus, TLR4 was selected for the
subsequent investigation. The potential targeting interaction
between miR-17 and TLR-4 was verified by dual luciferase reporter
assays. The results demonstrated that the miR-17 mimics
significantly inhibited the luciferase activity of the TLR4-3′UTR
wt, and that miR-17 inhibitor transfection resulted in a
significant increase in luciferase activity; however, no changes
were observed in the cells following co-transfection of TLR4
3′-UTR-mut with either miR-17 mimics or inhibitor (Fig. 3C). miR-17 was then introduced into
RAW264.1 cells and the expression of TLR4 was analyzed by western
blotting. It was observed that miR-17 overexpression suppressed the
protein expression of TLR4, while miR-17 inhibition promoted TLR4
expression in RAW264.1 macrophages (Fig. 3D). Of note, the protein expression
of TLR4 was also found to be decreased in lung tissues of ALI mice
after agomiR-17 injection (n=5/group; Fig. 3E). These results indicate that
TLR4 may be a functional target of miR-17 against lung inflammatory
response in ALI mice.
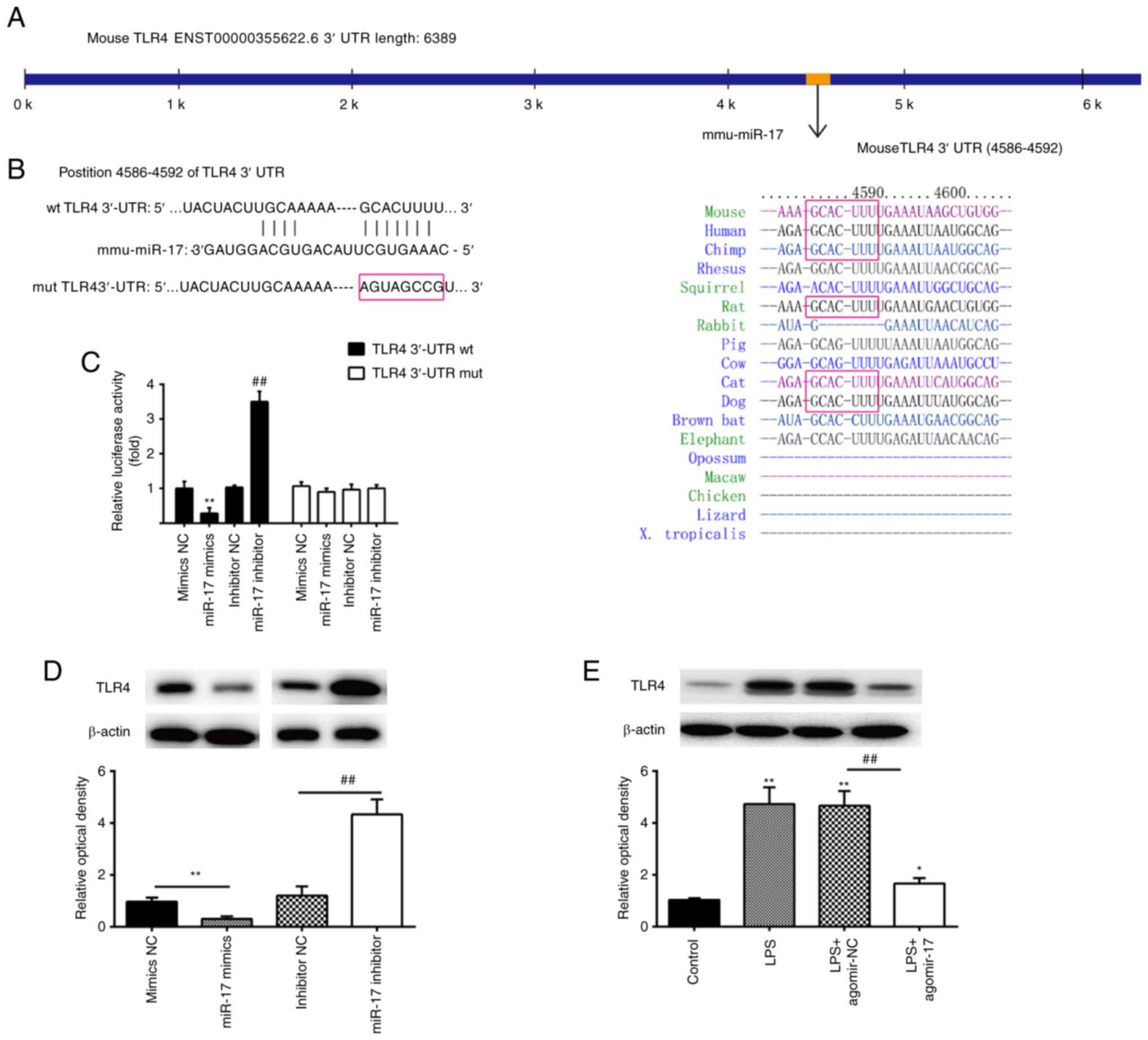 | Figure 3TLR4 is a direct target of miR-17. (A
and B) Putative binding site of miR-17 and TLR4. (C) Luciferase
activity was detected by a dual luciferase assay in RAW264.7 cells
co-transfected with firefly luciferase constructs containing the
TLR4 wt or mut 3′-UTRs and miR-17 mimics, mimics NC, miR-17
inhibitor or inhibitor NC, as indicated (n=3). (D) The expression
of the TLR4 protein was detected by western blotting following
transfection with miR-17 mimics and miR-17 inhibitor. Data are
expressed as mean ± standard deviation of three independent
experiments, **P<0.01 vs. mimics NC,
##P<0.01 vs. inhibitor NC. (E) The expression of the
TLR4 protein was measured by western blotting in lung tissues from
agomir-17- and agomir-NC-injected ALI mice. β-actin was used as the
internal control. Data are expressed as mean ± standard deviation
of three independent experiments (n=5 mice per group).
*P<0.05, **P<0.01 vs. mimics NC,
##P<0.01 vs. inhibitor NC. ALI, acute lung injury;
TLR, toll-like receptor; wt, wild-type; mut, mutant; UTR,
untranslated region. |
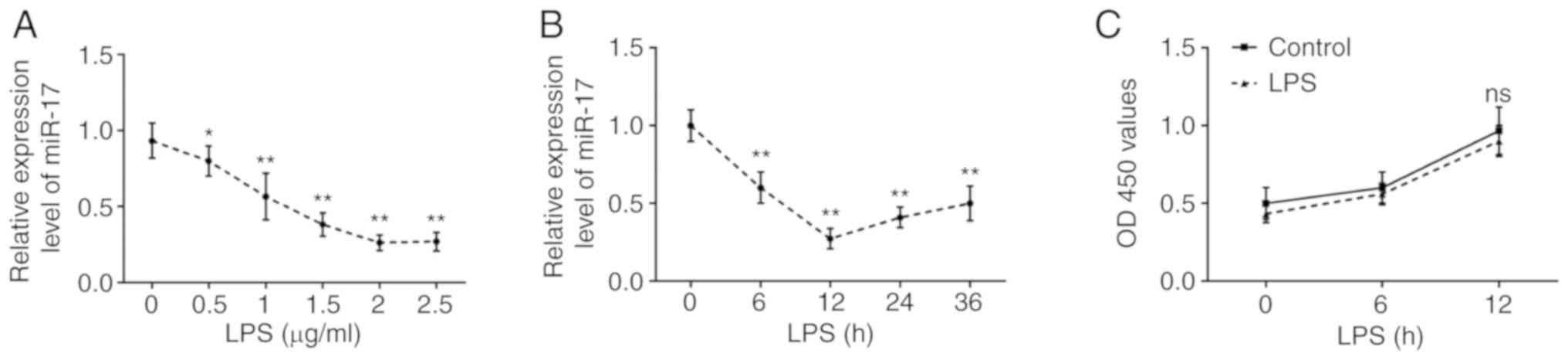
miR-17 is downregulated in LPS-treated
RAW264.1 macrophages
In the physiopathology of ALI, macrophages play an
important role in the regulation of inflammation (31,32). To further confirm whether miR-17
is involved in LPS-mediated immune response, RAW264.1 macrophages
were treated with LPS at different concentrations (0-2.5
µg/ml) for different times (0-36 h), as previously described
(33). After LPS treatment of
RAW264.1 cells, the expression level of miR-17 decreased in a
dose-dependent manner (Fig. 4A).
Furthermore, it was also observed that miR-17 expression
time-dependently decreased, while the expression of miR-17 reached
a nadir at 12 h after LPS treatment (Fig. 4B). Additionally, the MTT assay was
used to assess the effect of LPS (2 µg/ml) on the viability
of RAW264.7 cells, and the results demonstrated that cell viability
was not affected by LPS at any of the different time points
(Fig. 4C). Based on the data
mentioned above, treatment with 2 µg/ml LPS for 12 h was
selected as the optimal conditions for subsequent experiments.
Overexpression of miR-17 improves the
inflammatory response by targeting TLR4 in LPS-treated RAW264.7
cells
As mentioned above, TLR4 was found to be a direct
target of miR-17 in RAW264.7 cells; therefore, it was further
investigated whether miR-17 regulates the inflammatory response in
ALI through targeting TLR4 by co-transfecting miR-17 mimics and
pcDNA-TLR4 into RAW264.7 cells, followed by treatment with LPS.
Consistently with the results of the inflammatory cytokine
generation in vivo, LPS treatment resulted in significant
increases in the levels of IL-1β, IL-6 and TNF-α, whereas these
effects caused by LPS were attenuated by overexpression of miR-17.
Interestingly, TLR4 overexpression reversed the inhibitory effects
of miR-17 overexpression on the expression of these cytokines in
this cell model (Fig. 5A-C). In
addition, RAW264.7 cells were transfected with si-TLR4 to determine
the role of TLR4 in the regulation of inflammatory response. As
shown in Fig. 5D-F, the
production of these cytokines induced by LPS was markedly reduced
by TLR4 knockdown, which was similar to the effect of miR-17 mimics
on LPS-treated RAW264.7 cells. These findings demonstrated that
miR-17 improves the inflammatory response by targeting TLR4 in an
ALI cell model.
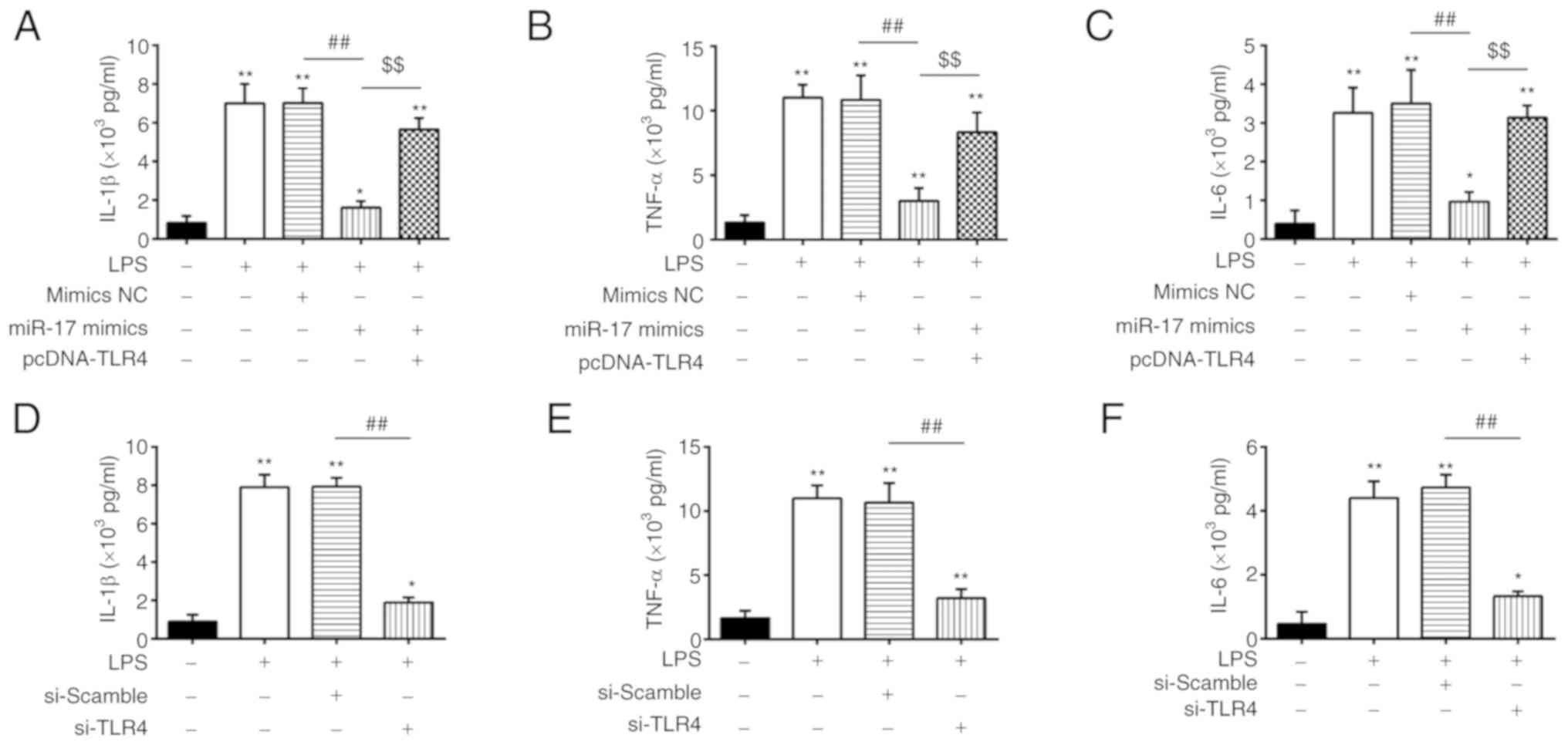 | Figure 5miR-17 regulates the inflammatory
response in LPS-treated RAW264.7 cells through targeting TLR4.
(A-C) IL-1β, IL-6 and TNF-α levels were measured using ELISA in
RAW264.7 cells co-transfected with pcDNA-TLR4 and miR-17 mimics, in
addition to LPS treatment. (D-F) IL-1β, IL-6 and TNF-α levels were
measured using ELISA in RAW264.7 cells transfected with si-TLR4, in
addition to LPS treatment. Data are expressed as mean ± standard
deviation of three independent experiments. *P<0.05,
**P<0.01 vs. control group. ##P<0.01
vs. LPS + mimics NC group or LPS + si-scramble,
$$P<0.01 vs. LPS + miR-17 mimics group. LPS,
lipopolysaccharide; TLR, toll-like receptor; IL, interleukin; TNF,
tumor necrosis factor. |
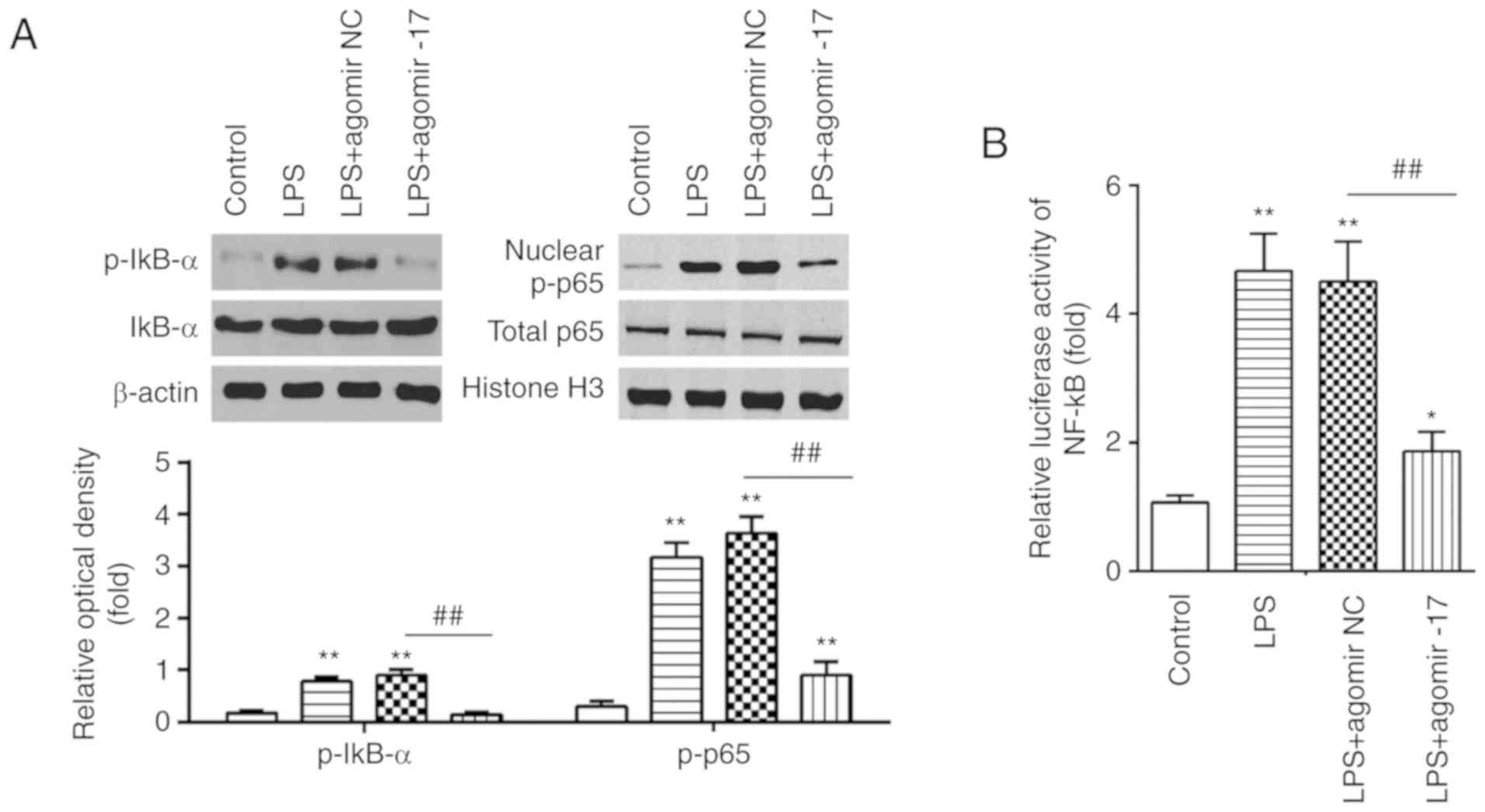
Overexpression of miR-17 blocks the
activation of the NF-κB signaling pathway
According to previous research, TLR4 is a common
receptor of LPS, and its downstream signaling effector, the NF-κB
pathway, plays a crucial role in the inflammatory response in ALI
(34,35). Therefore, to further investigate
the possible mechanisms underlying the role of miR-17 in the
inflammatory response, we focused on the phosphorylation/activation
of the NF-κB signaling pathway in ALI mice using western blot assay
(n=5/group). The data demonstrated that LPS treatment significantly
upregulated the levels of p-IκBα and nuclear p-p65 compared with
the control group. However, compared with the LPS group, the levels
of these proteins were markedly downregulated in the LPS +
agomiR-17 group (Fig. 6A). In
addition, it was also observed that LPS led to a significant
increase in NF-κB activity, whereas agomiR-17 markedly reduced the
increased NF-κB activity caused by LPS (Fig. 6B). These results demonstrated that
miR-17 exerts its potent inhibitory effects against LPS-induced
lung injury via the NF-κB signaling pathway.
Discussion
In the present study, miR-17 was significantly
downregulated in both LPS-induced ALI mice and LPS-treated RAW264.7
cells. Further analysis revealed that miR-17 improved LPS-induced
lung injury through inhibition of the inflammatory response through
blocking the activation of the TLR4/NF-κB pathway. These results
suggest that miR-17 may be of value as a potential therapeutic
approach to ALI.
Accumulating evidence has demonstrated that miRNAs
participate in the regulation of the inflammatory response in
several types of diseases (36,37). A number of miRNAs, such as miR-9,
miR-147 and miR-132, have been reported to exert an inhibitory
effect on inflammatory response (38-40). Rao et al demonstrated that
miR-17 attenuated staphylococcal enterotoxin B-induced lung injury
via suppressing the pulmonary inflammatory response (41). Wang and Zhang found that miR-17
overexpression mediated the protective effects of resveratrol
against LPS-induced cell injury through reducing the production of
pro-inflammatory cytokines in the human keratinocyte cell line
HaCaT (18). However, the
mechanism underlying the association between miR-17 and LPS-induced
ALI remains elusive. Therefore, it is necessary to further
investigate whether miR-17 affects the inflammatory response in
ALI. The present study demonstrated that the expression of miR-17
was low in the lung tissues of ALI mice, whereas agomiR-17
injection reduced tissue damage and lung edema, and inhibited the
LPS-induced inflammatory response in an LPS-induced ALI mouse
model. Furthermore, the results of the present study demonstrated
that agomiR-17 markedly improved the survival rate of mice with
LPS-induced ALI. Using an in vitro RAW264.7 macrophage
model, downregulation of miR-17 in RAW264.7 cells was observed
after LPS stimulation. Consistently with the results in
vivo, this demonstrated that miR-17 can attenuate LPS-induced
inflammatory response. Collectively, these data suggest that
upregulation of miR-17 may improve LPS-induced ALI by inhibition of
the inflammatory response.
TLR4 is an essential LPS signaling receptor, which
is known to play an important role in inflammatory response in
animal or cell models of ALI (42,43). For example, Hu et al
reported that the inhibition of the TLR4 signaling pathway
suppressed the production of inflammatory cytokines, thereby
improving ALI in rats (28). He
et al demonstrated that the LPS/TLR4 axis can activate the
NF-κB signaling pathway, resulting in inflammatory cell
infiltration (44). A previous
study also reported that miR-27a alleviated LPS-induced ALI in mice
via inhibiting inflammation through modulating the TLR4/MyD88/NF-κB
pathway (45). Interestingly, the
expression levels of TLR4 were previously reported to be regulated
by miR-17 in an LPS-induced sepsis mouse model (46); however, it is ambiguous whether
miR-17 serves as a protective factor in LPS-induced ALI through
regulation of TLR4. In the present study, TLR4 was identified as a
target of miR-17. Furthermore, TLR4 overexpression abrogated the
inhibitory effects of miR-17 on inflammatory response in
LPS-induced RAW 264.7 cells. By contrast, silencing TLR4 by siTLR4
had a similar effect to that of miR-17 on LPS-treated RAW 264.7
cells. Collectively, these findings indicate that the miR-17/TLR4
axis may serve as a novel therapeutic target for LPS-induced
ALI.
Activation of canonical NF-κB plays important roles
in LPS-induced ALI (47,48). In ALI, NF-κB is constitutively
activated and is involved in promoting the inflammatory response,
which suggests that inhibiting the activity of NF-κB may constitute
a promising therapeutic approach to ALI (49). According to previous research,
TLR4 is a recognized inducer of the NF-κB signaling pathway, and it
has been demonstrated that inhibition of TLR-4/NF-κB signaling may
improve LPS-induced ALI in a mouse model (50,51). Therefore, the mechanism through
which miR-17 affects the TLR-4/NF-κB signaling pathway requires
further investigation. In the present study, it was observed that
the overexpression of miR-17 significantly suppressed the
LPS-induced activation of NF-κB in vivo. These results
indicate that miR-17 may alleviate the LPS-induced inflammatory
response by inhibiting the TLR4/NF-κB signaling pathway.
In conclusion, the present study demonstrated that
miR-17 can alleviate LPS-induced ALI in mice through inhibiting the
inflammatory response via the TLR4/NF-κB signaling pathway.
Therefore, the miR-17/TLR4 axis may be a candidate target for the
treatment of patients with ALI.
Funding
No funding was received.
Availability of data and materials
All data generated and/or analyzed during the
present study are included in this published article.
Authors' contributions
SF conceptualized the study design, obtained the
experimental materials, performed the experiments, analyzed the
data and wrote the paper. The author has read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
All individuals provided informed consent for the
use of human specimens for clinical research. The present study was
approved by the Affiliated Hospital of Inner Mongolia University
for the Nationalities Ethics Committees.
Patient consent for publication
Not applicable.
Competing interests
The author declares that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Herridge MS, Tansey CM, Matté A, Tomlinson
G, Diaz-Granados N, Cooper A, Guest CB, Mazer CD, Mehta S, Stewart
TE, et al: Functional disability 5 years after acute respiratory
distress syndrome. N Engl J Med. 364:1293–1304. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mendez JL and Hubmayr RD: New insights
into the pathology of acute respiratory failure. Curr Opin Crit
Care. 11:29–36. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang J, Cao J, Feng J, Wu Q and Chen BY:
A study of noninvasive positive-pressure mechanical ventilation in
the treatment of acute lung injury with a complex critical care
ventilator. J Int Med Res. 42:788–798. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang Z, Chen L and Ni H: The
effectiveness of Corticosteroids on mortality in patients with
acute respiratory distress syndrome or acute lung injury: A
secondary analysis. Sci Rep. 5:176542015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Deng X, Jin K, Li Y, Gu W, Liu M and Zhou
L: Platelet-derived growth factor and transforming growth factor β1
regulate ARDS-associated lung fibrosis through distinct signaling
pathways. Cell Physiol Biochem. 36:937–946. 2015. View Article : Google Scholar
|
|
6
|
Wang X, Zhang L, Duan W, Liu B, Gong P,
Ding Y and Wu X: Anti-inflammatory effects of triptolide by
inhibiting the NF-κB signalling pathway in LPS-induced acute lung
injury in a murine model. Mol Med Rep. 10:447–452. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hoshino K, Takeuchi O, Kawai T, Sanjo H,
Ogawa T, Takeda Y, Takeda K and Akira S: Cutting edge: Toll-like
receptor 4 (TLR4)-deficient mice are hyporesponsive to
lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J
Immunol. 162:3749–3752. 1999.PubMed/NCBI
|
|
8
|
Chang X, He H, Zhu L, Gao J, Wei T, Ma Z
and Yan T: Protective effect of apigenin on Freund's complete
adjuvant-induced arthritis in rats via inhibiting P2X7/NF-κB
pathway. Chem Biol Interact. 236:41–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wei D and Huang Z: Anti-inflammatory
effects of triptolide in LPS-induced acute lung injury in mice.
Inflammation. 37:1307–1316. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang Q, Yi M, Guo Q, Wang C, Wang H, Meng
S, Liu C, Fu Y, Ji H and Chen T: Protective effects of polydatin on
lipopolysac-charide-induced acute lung injury through
TLR4-MyD88-NF-κB pathway. Int Immunopharmacol. 29:370–376. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang ZM, Wang YC, Chen L and Li Z:
Protective effects of the suppressed NF-κB/TLR4 signaling pathway
on oxidative stress of lung tissue in rat with acute lung injury.
Kaohsiung J Med Sci. 35:265–276. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tao Z, Yuan Y and Liao Q: Alleviation of
lipopolysac-charides-induced acute Lung injury by MiR-454. Cell
Physiol Biochem. 38:65–74. 2016. View Article : Google Scholar
|
|
14
|
Yang Y, Yang F, Yu X, Wang B, Yang Y and
Zhou X, Cheng R, Xia S and Zhou X: miR-16 inhibits NLRP3
inflammasome activation by directly targeting TLR4 in acute lung
injury. Biomed Pharmacother. 112:1086642019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou T, Garcia JG and Zhang W: Integrating
microRNAs into a system biology approach to acute lung injury.
Transl Res. 157:180–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang Y, Liu D, Xi Y and Li J, Liu B and Li
J: Upregulation of miRNA-140-5p inhibits inflammatory cytokines in
acute lung injury through the MyD88/NF-κB signaling pathway by
targeting TLR4. Exp Ther Med. 16:3913–3920. 2018.PubMed/NCBI
|
|
17
|
Ling Y, Li ZZ, Zhang JF, Zheng XW, Lei ZQ,
Chen RY and Feng JH: MicroRNA-494 inhibition alleviates acute lung
injury through Nrf2 signaling pathway via NQO1 in sepsis-associated
acute respiratory distress syndrome. Life Sci. 210:1–8. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang X and Zhang Y: Resveratrol alleviates
LPS-induced injury in human keratinocyte cell line HaCaT by
up-regulation of miR-17. Biochem Biophys Res Commun. 501:106–112.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oglesby IK, Vencken SF, Agrawal R, Gaughan
K, Molloy K, Higgins G, McNally P, McElvaney NG, Mall MA and Greene
CM: miR-17 overexpression in cystic fibrosis airway epithelial
cells decreases interleukin-8 production. Eur Respir J.
46:1350–1360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu Z, Zhang C, Cheng L, Hu M, Tao H and
Song L: The microRNA miR-17 regulates lung FoxA1 expression during
lipopolysaccharide-induced acute lung injury. Biochem Biophys Res
Commun. 445:48–53. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Toyama M, Kudo D, Aoyagi T, Miyasaka T,
Ishii K, Kanno E, Kaku M, Kushimoto S and Kawakami K: Attenuated
accumulation of regulatory T cells and reduced production of
interleukin 10 lead to the exacerbation of tissue injury in a mouse
model of acute respiratory distress syndrome. Microbiol Immunol.
62:111–123. 2018. View Article : Google Scholar
|
|
22
|
Wen N, Guo B, Zheng H, Xu L, Liang H, Wang
Q, Wang D, Chen X, Zhang S, Li Y and Zhang L: Bromodomain inhibitor
jq1 induces cell cycle arrest and apoptosis of glioma stem cells
through the VEGF/PI3K/AKT signaling pathway. Int J Oncol.
55:879–895. 2019.PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Duan Y, Learoyd J, Meliton AY, Leff AR and
Zhu X: Inhibition of Pyk2 blocks lung inflammation and injury in a
mouse model of acute lung injury. Respir Res. 13:42012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yao Y, Sun F and Lei M: miR-25 inhibits
sepsis-induced cardiomyocyte apoptosis by targetting PTEN. Biosci
Rep. 38:BSR201715112018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cai ZG, Zhang SM, Zhang Y, Zhou YY, Wu HB
and Xu XP: MicroRNAs are dynamically regulated and play an
important role in LPS-induced lung injury. Can J Physiol Pharmacol.
90:37–43. 2012. View
Article : Google Scholar
|
|
27
|
Dong Z and Yuan Y: Accelerated
inflammation and oxidative stress induced by LPS in acute lung
injury: Iotanhibition by ST1926. Int J Mol Med. 41:3405–3421.
2018.PubMed/NCBI
|
|
28
|
Hu X, Liu S, Zhu J and Ni H: Dachengqi
decoction alleviates acute lung injury and inhibits inflammatory
cytokines production through TLR4/NF-κB signaling pathway in vivo
and in vitro. J Cell Biochem. 120:8956–8964. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu JX, Li X, Yan FG, Pan QJ, Yang C, Wu
MY, Li G and Liu HF: Protective effect of forsythoside B against
lipo-polysaccharide-induced acute lung injury by attenuating the
TLR4/NF-κB pathway. Int Immunopharmacol. 66:336–346. 2019.
View Article : Google Scholar
|
|
30
|
Deng G, He H, Chen Z, OuYang L, Xiao X, Ge
J, Xiang B, Jiang S and Cheng S: Lianqinjiedu decoction attenuates
LPS-induced inflammation and acute lung injury in rats via
TLR4/NF-κB pathway. Biomed Pharmacother. 96:148–152. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zeng Z, Gong H, Li Y, Jie K, Ding C, Shao
Q, Liu F, Zhan Y, Nie C, Zhu W and Qian K: Upregulation of miR-146a
contributes to the suppression of inflammatory responses in
LPS-induced acute lung injury. Exp Lung Res. 39:275–282. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu WD, Xu J, Zhang M, Zhu TM, Zhang YH
and Sun K: MicroRNA-21 inhibits lipopolysaccharide-induced acute
lung injury by targeting nuclear factor-κB. Exp Ther Med.
16:4616–4622. 2018.PubMed/NCBI
|
|
33
|
Jiang K, Guo S, Zhang T, Yang Y, Zhao G,
Shaukat A, Wu H and Deng G: Downregulation of TLR4 by miR-181a
provides negative feedback regulation to lipopolysaccharide-induced
inflammation. Front Pharmacol. 9:1422018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu YL, Yu G, Ding ZY, Li SJ and Fang QZ:
Overexpression of miR-145-5p alleviated LPS-induced acute lung
injury. J Biol Regul Homeost Agents. 33:1063–1072. 2019.PubMed/NCBI
|
|
35
|
Wang B, Wang J, Lu D, Qi N and Liu Q: The
defensive action of LYRM03 on LPS-induced acute lung injury by
NF-κB/TLR4/NLRP3 signals. J Invest Surg. 1–13. 2019.
|
|
36
|
Li W, Qiu X, Jiang H, Han Y, Wei D and Liu
J: Downregulation of miR-181a protects mice from LPS-induced acute
lung injury by targeting Bcl-2. Biomed Pharmacother. 84:1375–1382.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wei C, Li L, Kim IK, Sun P and Gupta S:
NF-κB mediated miR-21 regulation in cardiomyocytes apoptosis under
oxidative stress. Free Radic Res. 48:282–291. 2014. View Article : Google Scholar
|
|
38
|
Zhou Z and You Z: Mesenchymal stem cells
alleviate LPS-induced acute lung injury in mice by
MiR-142a-5p-controlled pulmonary endothelial cell autophagy. Cell
Physiol Biochem. 38:258–266. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee HM, Kim TS and Jo EK: MiR-146 and
miR-125 in the regulation of innate immunity and inflammation. BMB
Rep. 49:311–318. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bao MH, Li JM, Luo HQ, Tang L, Lv QL, Li
GY and Zhou HH: NF-κB-regulated miR-99a modulates endothelial cell
inflammation. Mediators Inflamm. 2016:53081702016. View Article : Google Scholar
|
|
41
|
Rao R, Nagarkatti PS and Nagarkatti M:
Δ(9) Tetrahydrocannabinol attenuates Staphylococcal enterotoxin
B-induced inflammatory lung injury and prevents mortality in mice
by modulation of miR-17-92 cluster and induction of T-regulatory
cells. Br J Pharmacol. 172:1792–1806. 2015. View Article : Google Scholar :
|
|
42
|
Lu YC, Yeh WC and Ohashi PS: LPS/TLR4
signal transduction pathway. Cytokine. 42:145–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Meng L, Li L, Lu S, Li K, Su Z, Wang Y,
Fan X, Li X and Zhao G: The protective effect of dexmedetomidine on
LPS-induced acute lung injury through the HMGB1-mediated TLR4/NF-κB
and PI3K/Akt/mTOR pathways. Mol Immunol. 94:7–17. 2018. View Article : Google Scholar
|
|
44
|
He X, Qian Y, Li Z, Fan EK, Li Y, Wu L,
Billiar TR, Wilson MA, Shi X and Fan J: TLR4-upregulated IL-1β and
IL-1RI promote alveolar macrophage pyroptosis and lung inflammation
through an autocrine mechanism. Sci Rep. 6:316632016. View Article : Google Scholar
|
|
45
|
Ju M, Liu B, He H, Gu Z, Liu Y, Su Y, Zhu
D, Cang J and Luo Z: MicroRNA-27a alleviates LPS-induced acute lung
injury in mice via inhibiting in fl ammation and apoptosis through
modulating TLR4/MyD88/NF-κB pathway. Cell cycle. 17:2001–2018.
2018. View Article : Google Scholar :
|
|
46
|
Ji ZR, Xue WL and Zhang L: Schisandrin B
attenuates inflammation in LPS-induced sepsis through
miR-17-5pdownregulating TLR4. Inflammation. 42:731–739. 2019.
View Article : Google Scholar
|
|
47
|
Bollenbach M, Salvat E, Daubeuf F, Wagner
P, Yalcin I, Humo M, Letellier B, Becker LJ, Bihel F, Bourguignon
JJ, et al: Phenylpyridine-2-ylguanidines and rigid mimetics as
novel inhibitors of TNFα overproduction: Beneficial action in
models of neuropathic pain and of acute lung inflammation. Eur J
Med Chem. 147:163–182. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lorne E, Dupont H and Abraham E: Toll-like
receptors 2 and 4: Initiators of non-septic inflammation in
critical care medicine? Intensive Care Med. 36:1826–1835. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Narukawa M: Physiological responses to
taste signals of functional food components. Biosci Biotechnol
Biochem. 82:200–206. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang J, Fan SM and Zhang J:
Epigallocatechin-3-gallate ameliorates lipopolysaccharide-induced
acute lung injury by suppression of TLR4/NF-kappaB signaling
activation. Braz J Med Biol Res. 52:e80922019. View Article : Google Scholar
|
|
51
|
Wang M, Niu J, Ou L, Deng B, Wang Y and Li
S: Zerumbone protects against carbon tetrachloride (CCl4)-induced
acute liver injury in mice via inhibiting oxidative stress and the
inflammatory response: Involving the TLR4/NF-κB/COX-2 pathway.
Molecules. 24:E19642019. View Article : Google Scholar
|