Introduction
Treatment of low birthweight neonates with
supplemental oxygen is crucial to their survival, but it is often
accompanied by bronchopulmonary dysplasia (BPD), a chronic lung
disease predominantly observed in premature infants requiring
oxygen and/or ventilation therapy (1). In 2010, the United States National
Institute of Child Health and Human Development reported an
estimated prevalence of BPD of ≤68% among premature infants with
birthweight of 401-1,500 g and a gestational age of 22-28 weeks
(2). Children with BPD experience
not only pulmonary dysfunction, but also cardiovascular and nervous
system complications that can persist to adulthood (3). Current understanding suggests that
the underlying mechanism of BPD in neonates may be related to
oxidative stress induced by the shift from the hypoxic environment
of the mother to the relatively high-oxygen environment of normal
air and/or supplemental oxygen (1,4).
The lungs of premature infants have not yet developed a robust
protection mechanism and are more vulnerable to damage compared
with lungs of infants at full term (4). In addition, oxygen treatment or
mechanical ventilation can potentially cause additional lung damage
and may interrupt the development of the alveolar and pulmonary
vasculature, eventually leading to BPD (4). Therefore, there is an urgent
requirement to understand the pathogenesis of BPD in premature
infants.
BPD is thought to be caused mainly by intrauterine
inflammation, acute lung injury caused by oxidative stress and
abnormal differentiation of pulmonary progenitor and stem cells,
such as AT-II cells (5,6). However, there are currently no safe
and effective treatments for BPD. For example, treatments such as
antibiotics for pregnant women or anti-oxidants for neonates have
shown no clinical efficacy (7).
Glucocorticoids can suppress inflammation and reduce the incidence
of BPD, but can also increase the risk of mortality and the
incidence of cerebral palsy (1).
Autophagy is a highly conserved cellular recycling
process that participates in the degradation of proteins,
organelles and pathogens (8). The
rate of autophagy is influenced by numerous physiological and
pathological conditions, including inflammation, cellular stress,
apoptosis, transdifferentiation and aging. However, abnormal
autophagy activity can also perturbate the balance between cell
survival and apoptosis (8). Our
previous study showed that AT-II cells from the lungs of neonatal
rats with BPD had elevated levels of aggregated autophagosomes and
inhibition of the autophagic flux, which contributes to the
development of pulmonary tissue dysplasia (9). It was also found that
autophagy-inducing drugs may improve lung development in rats with
BPD. However, pharmacological modulators of autophagy are not yet
in clinical use, and managing their administration to neonates
would be more challenging than in adults. Nevertheless,
understanding the mechanisms via which BPD inhibits the autophagic
flux in pulmonary cells could provide novel approaches for the
development of interventions that prevent and treat BPD.
Blockade of the autophagic flux is mainly caused by
a failure of autophagosomes and lysosomes to fuse and abnormal
degradation of autophagosomes (10). Syntaxin 17 (STX17) is a soluble
N-ethylmaleimide-sensitive factor attachment protein receptor
(SNARE) protein that promotes the maturation of autophagosomes and
binds to another SNARE protein, Vesicle-associated membrane protein
8 (VAMP8), which is essential for the fusion of autophagosomes and
lysosomes (10). Failure of this
event leads to the accumulation of lysosomes and autophagosomes.
Consistent with this, cells deficient in STX17 have been reported
to exhibit premature aggregation of autophagosomes (11,12). Based on these previous findings,
the aim of the present study was to determine whether STX17 may be
abnormally regulated during BPD, leading to the hyperoxia-induced
defects in autophagy in the lungs.
Materials and methods
Animal model of hyperoxia-induced
BPD
Sprague-Dawley rats (40 female rats; 8 male rates;
weight, 200-240 g; age, 45-65 days) were provided by the
Experimental Animal Center of Shengjing Hospital of China Medical
University. All newborn rats used for experiments were born to dams
at 21-23 days of gestation. Dams were fed a normal diet ad
libitum. and housed at a temperature of 22±2°C and a relative
humidity of 60-70%, and exposed to light for 12 h a day. Exposure
to hyperoxia was performed as previously described (13). Newborn rats and their mothers were
placed in oxygen boxes with either a continuous input of oxygen
[fraction of inspired oxygen (FiO2), 0.8] for the model
group or normal air (FiO2, 0.21) for the control group.
Rats were grouped using random number generation program of SPSS
v22.0 software (SPSS, Inc.), with ten rats in each group. The
oxygen concentration was monitored continuously and CO2
was absorbed with soda lime to maintain a concentration of
<0.5%. The maternal rats in the model and control groups were
exchanged once every 24 h to avoid differences in feeding ability.
Every day, the boxes were opened for 30 min, the padding was
replaced and fresh drinking water and food were provided. On
postnatal days 1, 3, 7, 14 and 21, ten rats from each group were
selected, anesthetized with pentobarbital sodium (50 mg/kg;
intra-peritoneal) and sacrificed. The chest was opened and the
lungs were immediately resected. Sections of the left lung were
fixed in 4% paraformaldehyde >24 h and 2.5% glutaraldehyde >2
h at room temperature for hematoxylin and eosin staining (24 h
later; observe under an optical microscope; hematoxylin for 5 min
at room temperature; eosin for 10 sec at room temperature) and
immunofluorescence staining, respectively. The right lung was
frozen at -80°C before analysis by western blotting and reverse
transcription-quantitative PCR (RT-qPCR).
No female rats or newborn rats died in this
experiment. However, the BPD newborn rats were not as healthy as
the control group newborn rats in terms of weight and breathing
status (data not shown). This study has passed and been approved by
the Ethical Review of Scientific Research Projects from Shengjing
Hospital of China Medical University (approval no. 2019PS321K).
Transmission electron microscope
(TEM)
Lung tissues were cut into 1-mm3 tissue
blocks, fixed in 2.5% glutaral for >2 h at room temperature,
rinsed with PBS and then fixed in 1% osmic acid for 2-3 h at room
temperature. Tissues were dehydrated with gradient alcohols
(50-90%, 4°C), embedded with acetone embedding medium (TEDIA
Company) for 3-4 h at room temperature, sectioned (thickness, 50-60
nm) and double-stained with 3% uranyl acetate-lead citrate for 30
min at room temperature. Then, sections were observed at an
accelerating voltage of 80-100 kV under JEOL JEM-1200EX TEM
(magnification, ×20,000; JEOL Ltd.).
Dissociation, purification and culture of
primary AT-II cells
The preparation and culture of AT-II cells were
performed as described previously (13). On postnatal day 3 and 7, the
newborn rats were sacrificed, soaked in 75% alcohol for 2 sec and
the lungs were excised aseptically. Tissue was washed in PBS, cut
into 1-mm3 blocks, washed again three times with PBS,
mixed with 4 ml trypsin and incubated in at 37°C water bath for 30
min with shaking. DMEM (Gibco; Thermo Fisher Scientific, Inc.)
containing 10% FBS (Gibco; Thermo Fisher Scientific, Inc.) was
added at 4 ml per sample and the cells were gently homogenized into
a single-cell suspension, filtered with a sterile 200-mesh sieve
and centrifuged for 5 min at 200 × g at room temperature. The
supernatant was discarded and the cells were resuspended in 5 ml
type I collagenase, shaken in a water bath at 37°C for 40 min and
centrifuged for 5 min at 200 × g. The supernatant was discarded and
the cells were resuspended in DMEM containing 10% FBS, 10,000 U/ml
penicillin and 25 µg streptomycin. The cell suspension was
adjusted to a density of 5.0×105/ml, placed in a
25-cm3 culture flask and incubated at 37°C in a 5%
CO2 atmosphere to adhere.
Hyperoxia exposure and treatment of AT-II
cells
Primary AT-II cells prepared as described in the
aforementioned section were incubated at 37°C at FiO2
0.8 for 0, 6, 12, 24 or 48 h and then harvested for analysis.
Rapamycin (RAPA; in DMSO and PBS), LiCl (in PBS), 3-methyladenine
(3-MA; in PBS) and/or chloroquine (CQ; in PBS) were added at final
concentrations of 5 µM, 5 mM, 5 µM and 5 µM at
37°C for 24 h, respectively. Cells were collected for analysis at
the indicated time point of 24 h. All of the aforementioned drugs
were purchased from Sigma-Aldrich (Merck KGaA). Preparation of the
main reagents is presented in Table
I.
 | Table IPreparation of the main reagents. |
Table I
Preparation of the main reagents.
| Reagent name | Preparation
method |
|---|
| RAPA | In total, 50 mg
RAPA was weighed, dissolved in 1 ml DMSO solution and prepared into
55 mM stock solution with PBS, which was filtered and sterilized
for use. The final drug concentration was 5 µM. |
| LiCl | In total, 2.129 g
LiCl was weighed, dissolved in 50 ml PBS solution and prepared into
1 M stock solution, which was filtered and sterilized for use. The
final drug concentration was 5 mM. |
| 3-MA | In total, 0.0075 g
3-MA was weighed, dissolved in 50 ml PBS solution and prepared into
1 mM stock solution, which was filtered and sterilized for use. The
final drug concentration was 5 µM. |
| CQ | In total, 0.1289 g
CQ was weighed, dissolved in 50 ml PBS solution and prepared into 5
mM stock solution, which was filtered and sterilized for use. The
final drug concentration was 5 µM. |
Immunofluorescence
Lung tissues were fixed in 4% paraformaldehyde at
room temperature for 24 h, dehydrated with gradient alcohol,
vitrified with xylene, embedded in paraffin and sectioned
(thickness, 5 µm). Paraffin-embedded lung tissue sections
were deparaffinized, hydrated, soaked in formaldehyde at room
temperature for 30 min, washed three times with PBS, retrieved by
microwave with citric acid, washed again with PBS three times and
incubated with 10% goat serum (MXB Biotechnologies, Inc.) at 37°C
for 30 min to block antibodies. Then, sections were incubated with
primary antibodies (rabbit polyclonal STX17 antibody; cat. no.
17815-1-AP; 1:100; ProteinTech Group, Inc.) overnight at 4°C. The
negative control was incubated with PBS. The following day,
sections were washed with PBS, incubated with secondary antibody
(cat. no. KIT-9720; 1:5,000; MXB Biotechnologies, Inc.) at 37°C for
4 h, washed again with PBS three times and stained with DAPI at
23°C for 5 min. Sections were washed with PBS and observed under a
fluorescence confocal microscope (magnifications, ×200 and ×400;
MTC-600; Bio-Rad Laboratories, Inc.). Results were evaluated as
follows: Red fluorescence indicated STX17 positivity.
Western blotting
Lung tissues were cut into pieces, incubated in
lysis buffer at 4°C for 1 h and centrifuged at 4°C at 15,000 × g
for 10 min. Protein concentration determination using bicinchoninic
acid assay. The supernatant was collected, diluted in Laemmli
buffer (Sigma-Aldrich; Merck KGaA) and boiled for 3 min. Proteins
(10-15 µl/lane)were separated on 12% SDS-PAGE for 2 h at 80
V at room temperature and then transferred to PVDF membranes for 90
min at 80 V at 4°C. The membranes were blocked with 5% albumin
bovine serum at room temperature for 1 h (Beijing Solarbio Science
& Technology Co., Ltd.) and then incubated overnight at 4°C
with the following primary antibodies: Rabbit monoclonal
Microtubule-associated protein 1A/1B-light chain 3B (LC3B; cat. no.
3868; 1:100; Cell Signaling Technology, Inc.), rabbit monoclonal
cleaved caspase3 (cat. no. 9664; 1:1,000; Cell Signaling
Technology, Inc.), rabbit monoclonal p62 antibody (cat. no. 5114;
1:1,000; Cell Signaling Technology, Inc.) and mouse monoclonal
lysosomal-associated membrane protein 1 (Lamp1; cat. no. sc17768;
1:1,000; Santa Cruz Biotechnology, Inc.). As a negative control,
membranes were incubated with Tris-HCl buffer plus Tween-20 (0.1%)
overnight at 4°C. The following day, the membranes were incubated
with secondary antibodies (horseradish peroxidase conjugate;
anti-mouse IgG; cat. no. 7076; 1:2,000, Cell Signaling Technology,
Inc.; Anti-rabbit IgG; cat. no. 7074; 1:2,000; Cell Signaling
Technology, Inc.) at 37°C for 2 h, and proteins were then detected
using ECL substrate (Thermo Fisher Scientific, Inc.). β-actin (cat.
no. 4970; 1:2,000; Cell Signaling Technology, Inc.) was probed as a
loading control. Band densities were calculated using Image-pro
Plus software6.0 (Media Cybernetics, Inc.).
RT-qPCR
RNA was extracted from frozen lung tissues using the
TRIzol®-acetone (Takara Bio, Inc.) method and then
reverse transcribed (37°C for 15 min and 85°C for 5 sec) using a
Takara reverse-transcription kit (cat. no. RR047A; Takara Bio,
Inc.) according to the manufacturer's instructions. qPCR was
performed using a Takara SYBR-Green kit (cat. no. RR420A; Takara
Bio, Inc.) in 20-µl reaction volume with primers designed
and synthesized by Takara Bio, Inc. (Table II). The thermocycling conditions
were as follows: Initial denaturation at 95°C for 10 sec, followed
by 40 cycles at 55°C for 5 sec and 60°C for 34 sec. The results
were automatically analyzed by the PCR analyzer 1.02 (Applied
Biosystems; Thermo Fisher Scientific, Inc.) The relative amount of
transcripts was calculated using the 2−ΔΔCq formula,
normalized to GAPDH or actin transcript as an internal control
(14).
| Table IIPrimer sequences. |
Table II
Primer sequences.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| LC3B |
AGAGCGATACAAGGGTGAGAA |
CACTTCAGAGATGGGTGTGG |
| STX17 |
CTTGTCATCGTCCGCATTCTG |
TCAAGCCTGCGTAGCTTCACC |
| p62 |
CTGTGGTGGGAACTCGCTAT |
AAGGGGTTGGGAAAGATGAG |
| Lamp1 |
TCCAGGCTTTCAGGGTAGAA |
ATGAGGACGATGAGGACCAG |
| β-actin |
CGTGCGTGACATTAAAGAG |
TTGCCGATAGTGATGACCT |
MTT cell cytotoxicity assay
Primary AT-II cells were cultured for 24 h until
70-80% confluent, collected and seeded in 96-well plates at
4×103 cells/well (n=5 per condition). Cells were
cultured in an incubator under hyperoxic (FiO2, 0.8) or
normal conditions (FiO2, 0.21) at 37°C for 24 h in the
presence or absence of autophagy inducers or inhibitors (Table I). At the end of the incubation,
20 µl (5 mg/ml) MTT was added per well and the cells were
incubated at 37°C for 4 h. The supernatant was removed and 200
µl DMSO was added per well to dissolve the purple formazan
crystals. The absorbance at 490 nm was measured using a microplate
reader and the data were subsequently analyzed.
Statistical analysis
SPSS v22.0 software (SPSS, Inc.) was used to analyze
the data, which are presented as the mean ± standard deviation. The
homogeneity of variance of two samples was analyzed with the F
test, and multiple comparisons were performed using one-way ANOVA
with Bonferroni's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Morphological and ultrastructural changes
in the lungs of rats with BPD
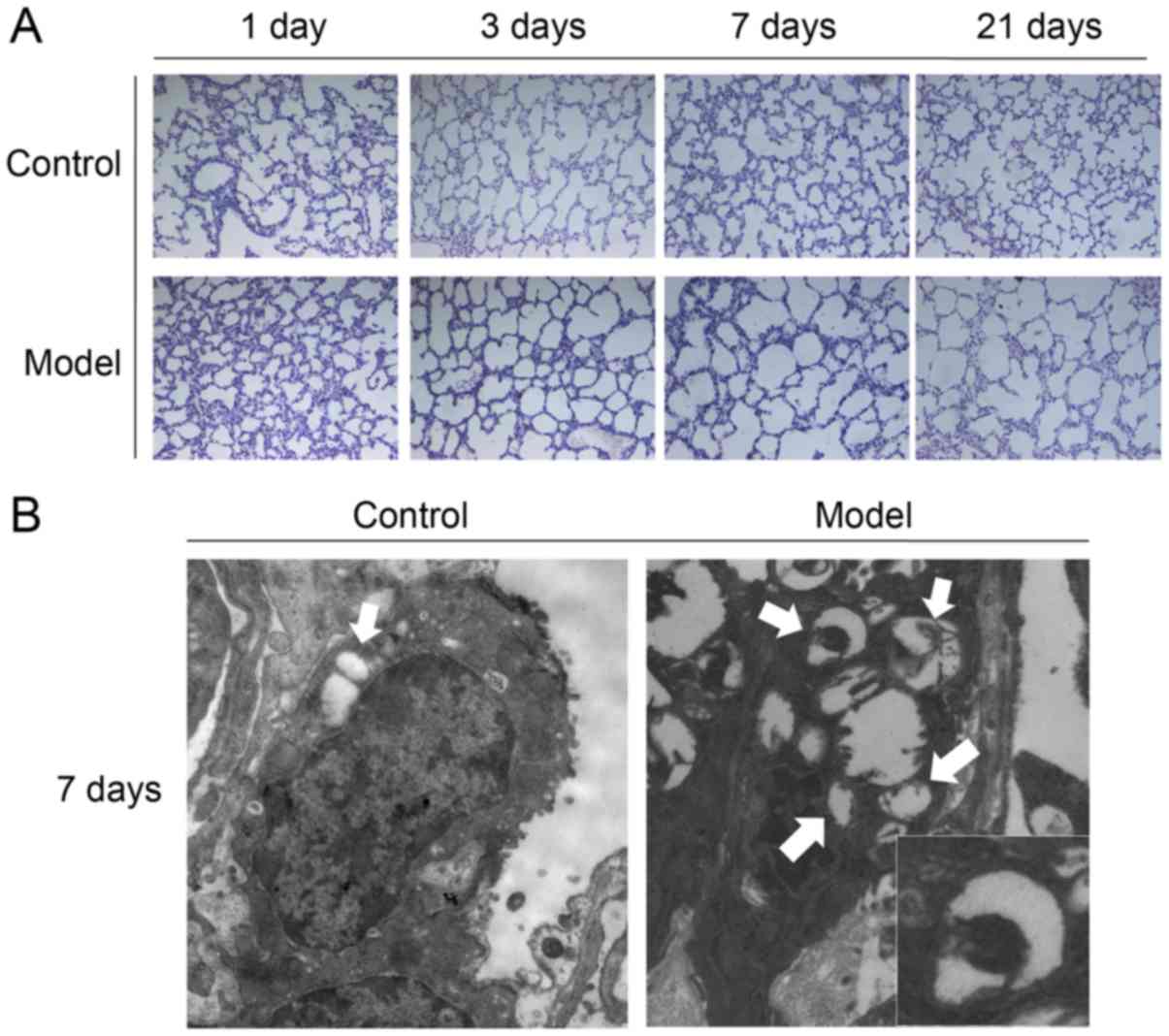
Light microscopy of lung sections from control rats
on postnatal day 1 identified pulmonary alveoli with a large volume
and uniform size, as well as moderate alveolar septa (Fig. 1A). Moreover, the ridge-like
structures gradually increased, alveoli and capillaries were more
abundant and the alveolar septa thinned as the age increased.
However, the lungs of BPD rats exhibited notably widened and
edematous alveolar septa on day 3, and by day 7 there were fewer
but larger alveoli, fewer ridge-like structures and thicker
alveolar septa compared with control rats; these trends continued
to day 14 (Fig. 1A).
Autophagosomes were observed in pulmonary cells of
both control and BPD neonatal rats by TEM. Compared with the
control group, the number of autophagosomes in the BPD group
gradually increased, peaking at day 7, at which time point
autophagosome aggregates were visible (Fig. 1B).
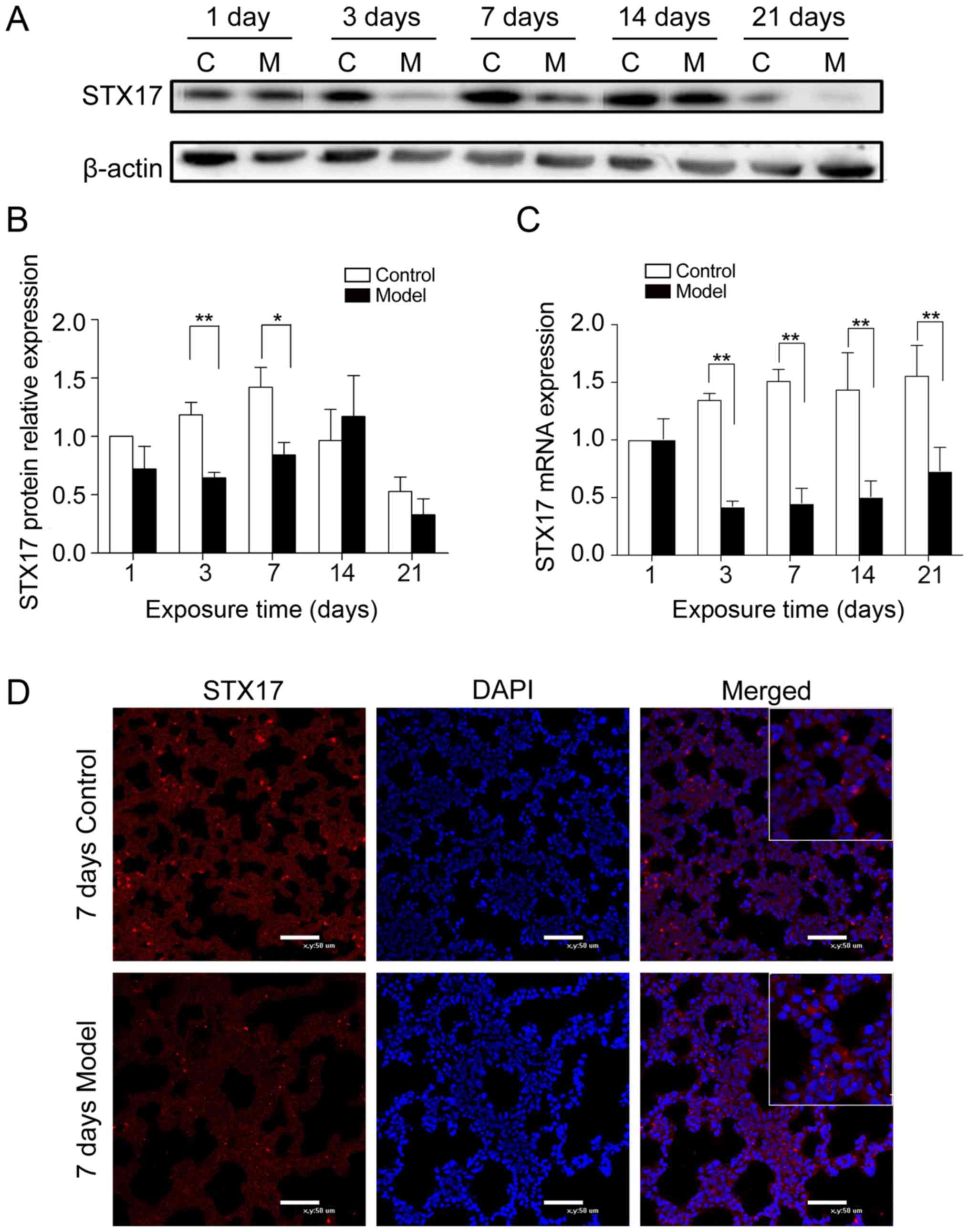
Changes in STX17 expression in lung
tissues of rats with BPD
Immunofluorescence staining and western blot
analysis of STX17 protein expression demonstrated that it was
expressed in nearly all pulmonary cells and was mainly cytoplasmic.
On postnatal days 3 and 7, STX17 expression was significantly lower
in the lungs of the BPD rats compared with the control rats, but
afterwards the expression increased and reached a peak at day 14
(Fig. 2A, B and D). Consistent
with the protein analysis, it was found that STX17 mRNA expression
was significantly lower in the lung tissues of BPD rats compared
with control rats on postnatal day 3. However, unlike STX17 protein
expression, the mRNA expression of STX17 remained low (Fig. 2C).
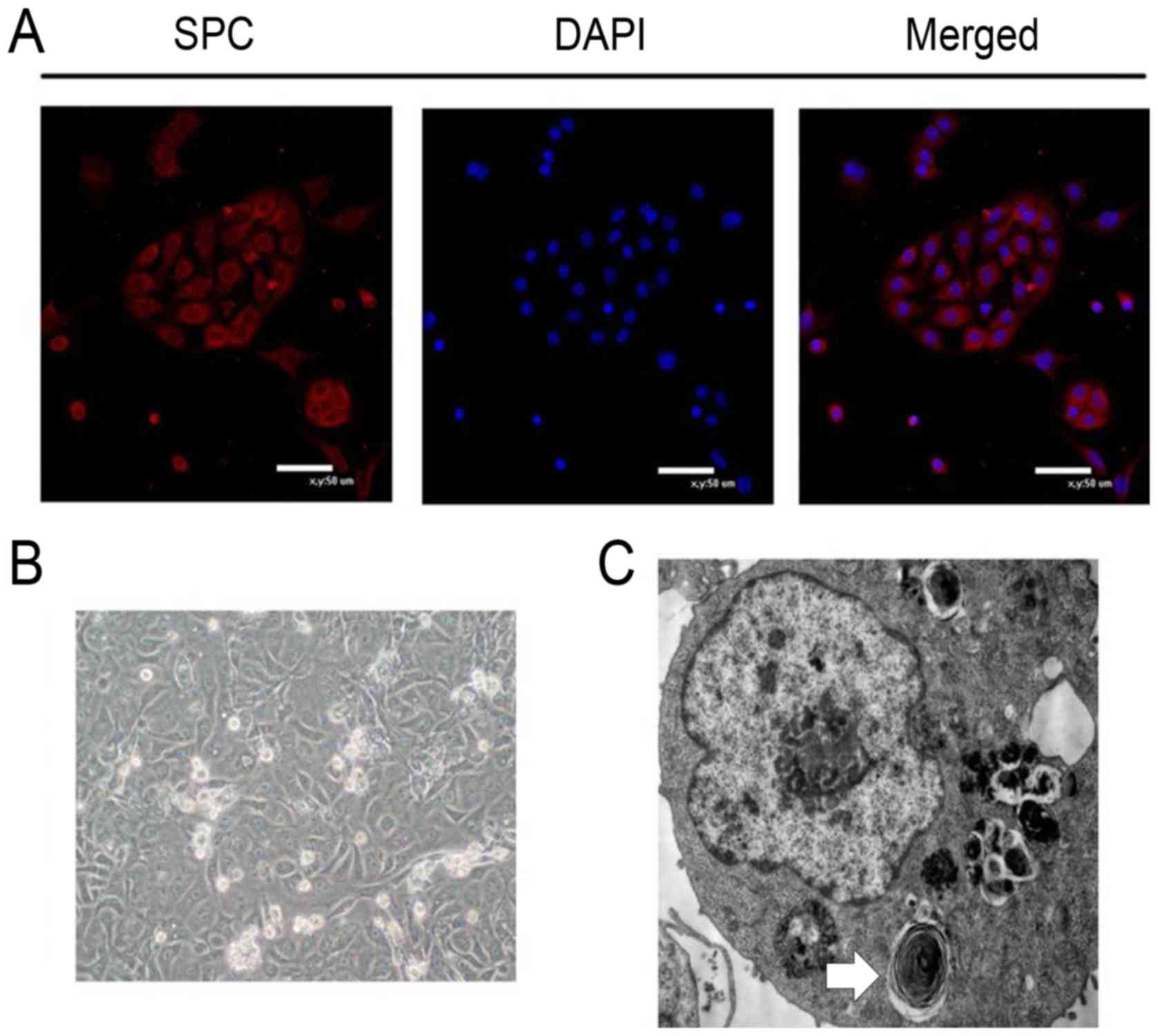
Hyperoxia-induced changes in STX17 and
autophagy protein expression in primary AT-II cells in vitro
To examine the effects of hyperoxia on AT-II cells
directly, these cells were isolated from the lungs of control or
BPD rats on postnatal day 1. Using light microscopy, the purified
cells were elliptical in shape and had formed islets with paving
stone-like changes (Fig. 3B).
Using TEM, lamellar body structures were visible in the cells.
These findings were consistent with the morphological
characteristics of AT-II cells (Fig.
3C). The identity of the cells was assessed by
immunofluorescence staining for pro-surfactant, an AT-II
cell-specific marker, which indicated that ≥90% of the cells
(calculate by Image J) exhibited positive cytoplasmic staining
(Fig. 3A).
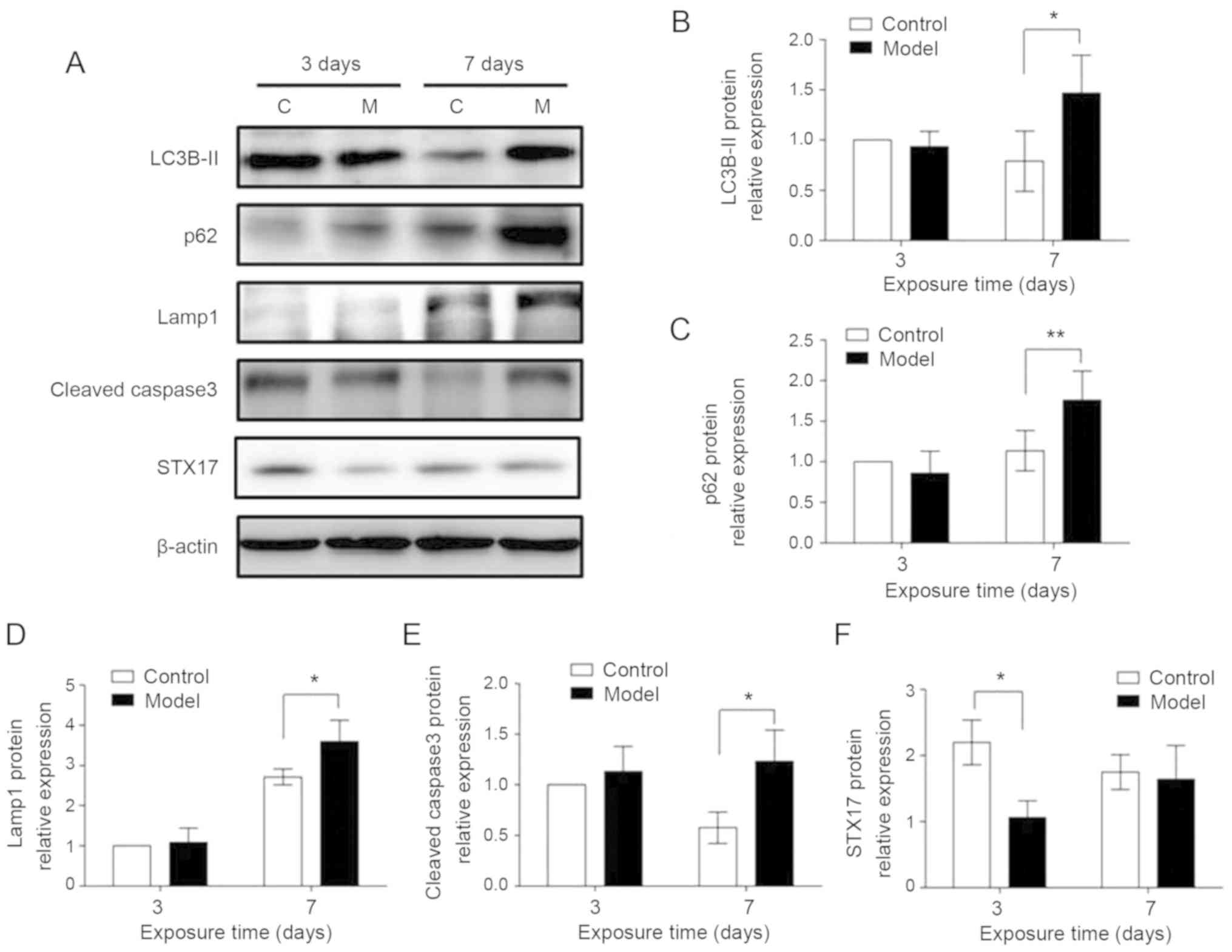
Western blot analysis was performed to examine STX17
protein expression. Lower STX17 protein expression was found in
AT-II cells isolated from BPD rats compared with control rats on
postnatal day 3. In contrast, the expression levels of autophagy
proteins LC3B-II, p62 and Lamp1, and the apoptotic protein cleaved
caspase3, were all significantly higher in AT-II cells from BPD
rats compared with control rats on postnatal day 7, indicating that
the BPD-induced change in STX17 protein expression preceded the
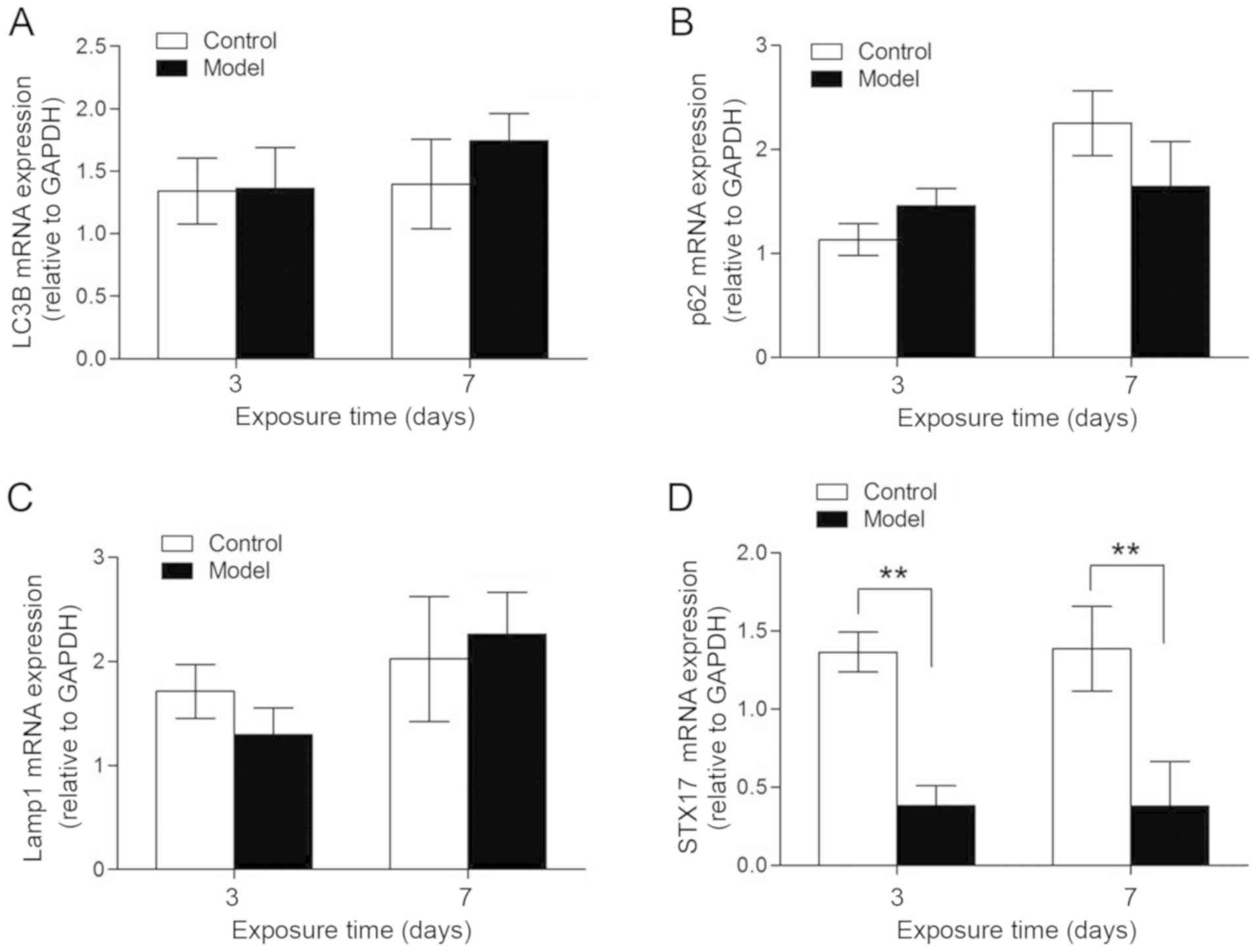
effects on autophagy- and apoptosis-related proteins (Fig. 4). Furthermore, while the same
reduction was identified for STX17 mRNA expression in AT-II cells
from BPD rats compared with control rats on postnatal days 3 and 7,
there were no significant BPD-associated differences in the mRNA
expression levels of LC3B, p62 or Lamp1 (Fig. 5).
Next, the present study examined the effects of
in vitro exposure to hyperoxia on primary AT-II cells
isolated from BPD rats. The results indicated an early decrease in
STX17 expression (6 h), followed by an increase in
autophagy-related protein expression, in hyperoxic cells (12 h)
compared with normoxic cells. In addition, STX17 expression was
decreased by hyperoxia, reaching the lowest point at 6 h, while
LC3B-II and p62 protein expression levels were increased by
hyperoxia, peaked after 12 h exposure and then gradually decreased
(Fig. 6A).
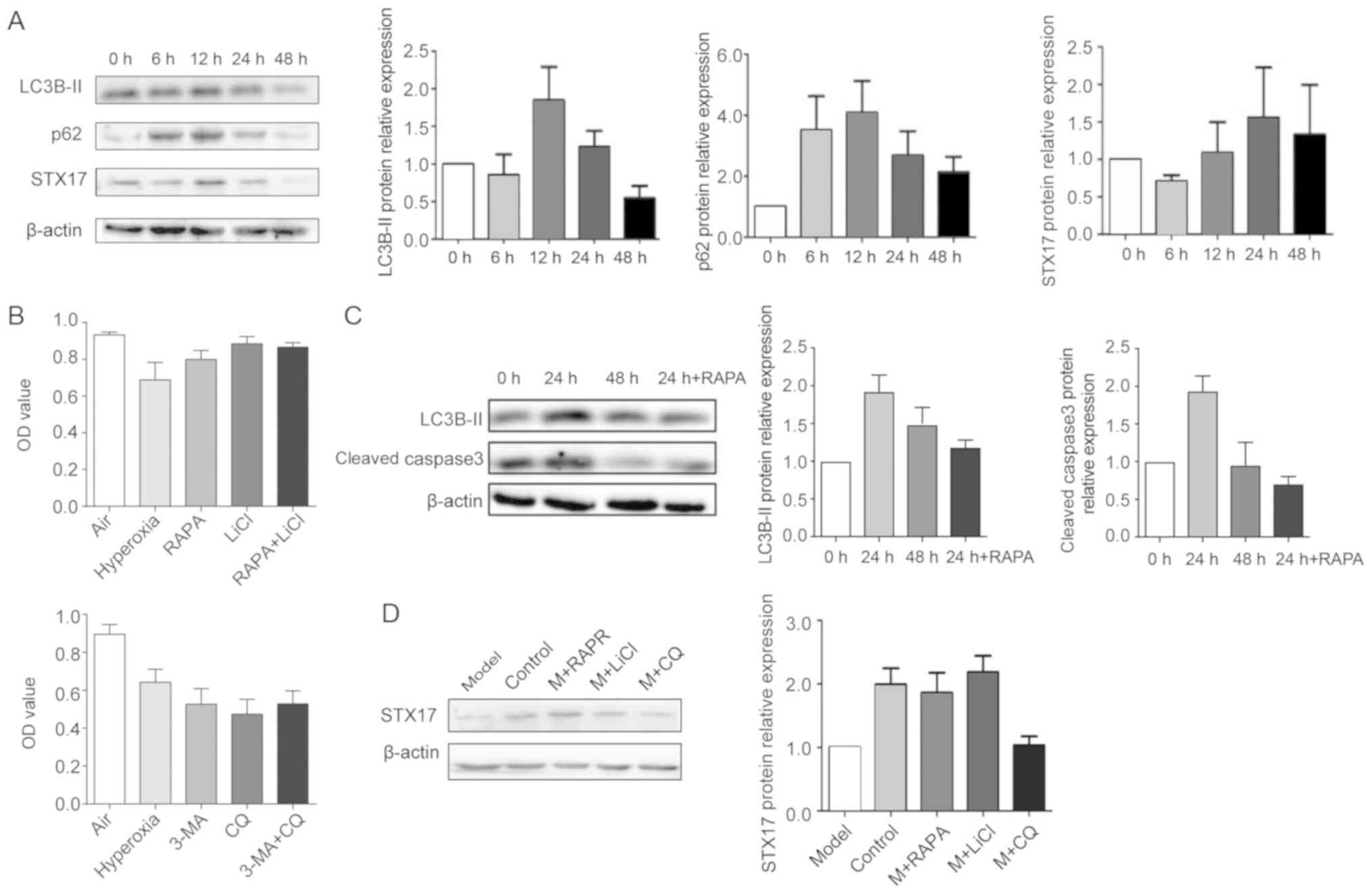 | Figure 6Expression of STX17 and autophagy-
and apoptosis-related proteins in primary AT-II cells exposed to
hyperoxia. (A) Western blot analysis of LC3B-II, p62 and STX17 in
AT-II cells exposed to hyperoxia for the indicated times. (B) MTT
proliferation assay of primary AT-II cells incubated with RAPA,
LiCl, 3-MA and/or CQ. (C) Western blot analysis of LC3B-II and
cleaved caspase3 in AT-II cells. (D) Western blot analysis of STX17
expression in AT-II cells incubated in the presence or absence of
RAPA, LiCl or CQ. RAPA, rapamycin; 3-MA, 3-methyladenine; CQ,
chloroquine; M, model; AT-II, alveolar type II; STX17, syntaxin 17;
LC3B, Microtubule-associated protein 1A/1B-light chain 3B; Lamp1,
Lysosomal-associated membrane protein 1; OD, optical density. |
Autophagy inhibitors reverse the effects
of hyperoxia on primary AT-II cells in vitro
Whether modulation of autophagy affected AT-II cell
survival under hyperoxia was also determined using AT-II cells
exposed to hyperoxia in the presence or absence of the autophagy
promoters RAPA (5 µM) and LiCl (5 mM) or the autophagy
inhibitors 3-MA (5 µM) and CQ (5 µM). Exposure to
RAPA promoted cell survival in hyperoxia condition (Fig. 6B) and decreased the expression
levels of LC3B-II and cleaved caspase3 (Fig. 6C) compared with control cells. In
addition, while STX17 protein expression was significantly
decreased in cells after 6 h of hyperoxia, this was reversed by
incubation with RAPA or LiCl, but not with CQ (Fig. 6D). Thus, autophagy inhibitors
restored the autophagic flux and promoted cell survival in AT-II
cells exposed to hyperoxia.
Discussion
The development of BPD is influenced by multiple
factors, including premature birth, oxidative stress, inflammation,
stem cell damage, abnormal cell differentiation and
trans-differentiation (2).
However, the underlying molecular pathogenesis of BPD is not fully
understood, and there are currently no safe and effective
preventive or therapeutic clinical interventions (15). Thus, the development and analysis
of animal models of BPD are important for investigating the
pathogenesis of BPD.
Prolonged exposure of neonatal rats to high oxygen
concentrations can influence alveolar and pulmonary vascular
development and trigger pathological changes similar to those
observed in human BPD (16,17). The present study established a
hyperoxia exposure model in neonatal rats that simulated the
characteristic changes in BPD alveoli, and this was used to
investigate the underlying mechanism of BPD (9).
Our previous studies reported that BPD in neonatal
rats caused a block in the autophagic flux, causing increased
pulmonary apoptosis and abnormal alveolar development (9,18).
Autophagy is crucial to embryonic development (19), and blockade of the pathway can
result in aggregation of toxic abnormal proteins, which triggers an
inflammatory response and eventually leads to cell apoptosis
(20,21). Abnormalities in the autophagic
flux are speculated to be involved in the development of several
pathologies, including cancer, neurode-generative diseases,
cardiovascular diseases and autoimmunity, as well as in aging
(22). In respiratory system
diseases, inhibition of the autophagic flux has been suggested to
be associated with chronic inflammation and possibly the
development of chronic obstructive pulmonary disease (23). Aberrant autophagy may also
interfere with the epithelial-mesenchymal transition (EMT). When
autophagic flux is blocked, epithelial cells can cause chronic
inflammation due to clearance failure of a large amount of
misfolded proteins, which is also one of the causes of EMT in
epithelial cells (24), and
reduced autophagic flux is known to be directly related to the
development of pulmonary fibrosis (25). In our previous studies, it was
shown that BPD caused a block in autophagic flux in pulmonary
tissues, which was characterized by aggregation of autophagosomes
and lysosomes, along with increased expression of the autophagosome
marker LC3B-II, the autophagy substrate p62 and the lysosome marker
Lamp1 (9,18). These results are consistent with
the present findings, as well as those reported by Zhang et
al (26) and Sureshbabu et
al (27), in which pulmonary
epithelial cells exhibited autophagosome aggregation and increased
LC3B-II expression after exposure to hyperoxia. It has also been
shown that treatment with an autophagy inducer rescues the
autophagic flux in pulmonary tissues under hyperoxia and improves
lung development (9). However,
the specific mechanism via which autophagic flux is blocked in BPD
remains unknown.
Autophagy occurs via a series of steps, including
the formation of autophagosomes, encapsulation of cellular cargo,
binding and fusion of autophagosomes and lysosomes and the
degradation of the lysosomal contents (11). Abnormalities occurring at any
stage can influence the pathway function. Previous studies have
reported that STX17 binds with two other SNARE proteins,
Synaptosomal-associated protein 29 (SNAP29) and VAMP8, to enable
the recognition and fusion of autophagosomes and lysosomes
(28,29). Thus, when STX17 expression or
function is reduced, autophagosome-lysosome fusion is disrupted,
resulting in aggregation of lysosomes and autophagosomes and
inhibition of the autophagic flux (12). Furthermore, the SNAP29-STX17-VAMP8
complex is a key target for dysregulation of the autophagic flux
occurring in numerous diseases. O-linked β-N-acetylglucosamine
glycosylation of SNAP29 has been revealed to block autophagy and
aggravate myocardial damage in type I diabetes by interfering with
the SNAP29-STX17-VAMP8 complex (30). Another study reported that the
toxicity of Coxsackie virus B3 may be related to reduced STX17
expression and blockade of the autophagic flux in HeLa cells
(31). This study also revealed
that overexpression of STX17 in HeLa cells restored autophagy and
reduced apoptosis and the toxicity of Coxsackie virus (31). Certain bacteria, such as
Legionella, can also block autophagy and increase apoptosis
via the degradation of STX17 (32). In the present study, it was
demonstrated that STX17 mRNA and protein expression levels were
decreased at an early time point (postnatal day 3) in both the lung
tissues and AT-II cells of neonatal rats with hyperoxia-induced
BPD.
As alveolar stem cells, AT-II cells play an
important role in alveolarization and recovery from alveolar
damage, and AT-II cell dysfunction or defects may eventually result
in alveolar dysplasia (33). In
the present study, using an established model of hyperoxia-exposed
AT-II cells, it was found that hyperoxia caused a decrease in STX17
expression followed by a significant increase in LC3B-II, p62 and
cleaved caspase3 expression, which is consistent with the results
of the experiments with BPD pulmonary tissues. These findings
suggested that the decrease in STX17 may be related to the
subsequent blockade of autophagic flux following hyperoxia
exposure. Furthermore, treatment of hyperoxia-exposed AT-II cells
with the autophagy inducers RAPA and LiCl reduced the expression of
LC3B-II and p62, partially restored the autophagic flux and may
reduce apoptosis. In addition, the autophagy inducers restored
STX17 expression. Therefore, it was speculated that hyperoxia may
interfere with the autophagic flux of AT-II cells via the reduction
in STX17 expression. Thus, restoring STX17 expression with
pharmacological agents may be a potential therapeutic approach to
alleviate BPD-induced defects in autophagy in AT-II cells.
In conclusion, the present results indicated that
expression of STX17 was decreased in both pulmonary tissues and
AT-II cells in response to hyperoxia during the crucial early
neonatal stage of lung development. Moreover, the reduction in
STX17 may be related to a subsequent block in autophagy. RAPA and
LiCl rescued the reduced STX17 expression and improved the
autophagic flux; however, it is not fully understood whether the
effect on autophagy was mediated via the change in STX17 expression
itself or via another route. Therefore, further investigation is
required to clarify the precise molecular mechanisms involved and
to fully understand the pathogenesis of BPD.
Funding
This study was funded by the Natural Science
Foundation of China (grant nos. 81901520 and 81571479), Basic
research projects of Key Laboratory of Liaoning Provincial
Department of Education (grant no. LZ2015070) and China
Postdoctoral Science Foundation (grant no. 2019M661164).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JF and XX participated in the design of the study.
XZ conceived of the study, and participated in its design. DiZ
participated in its design and coordination and helped to draft the
manuscript. SG participated in the immunoassays and performed the
statistical analysis. DaZ participated in RT-qPCR and western
blotting, and draft the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
This study has passed and been approved by the
Ethical Review of Scientific Research Projects from Shengjing
Hospital of China Medical University (approval no. 2019PS321K).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank the TEM technical
support provided by Professor Fuhui Zhang from Department of
Cytobiology, China Medical University, and the technical support in
western blotting and RT-qPCR provided by Professor Zhihong Zong
from Department of Biochemistry, China Medical University. The
authors would also like to thank Dr Anne M. O'Rourke, from Liwen
Bianji, Edanz Group China, for editing the English text of a draft
of this manuscript.
References
|
1
|
Thébaud B, Goss KN, Laughon M, Whitsett
JA, Abman SH, Steinhorn RH, Aschner JL, Davis PG, McGrath-Morrow
SA, Soll RF and Jobe AH: Bronchopulmonary dysplasia. Nat Rev Dis
Primers. 5:782019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stoll BJ, Hansen NI, Bell EF, Shankaran S,
Laptook AR, Walsh MC, Hale EC, Newman NS, Schibler K, Carlo WA, et
al: Neonatal outcomes of extremely preterm infants from the NICHD
Neonatal research network. Pediatrics. 126:443–456. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jobe AH: The new bronchopulmonary
dysplasia. Curr Opin Pediatr. 23:167–172. 2011. View Article : Google Scholar
|
|
4
|
Kinsella JP, Greenough A and Abman SH:
Bronchopulmonary dysplasia. Lancet. 367:1421–1431. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rock JR and Hogan BL: Epithelial
progenitor cells in lung development, maintenance, repair, and
disease. Annu Rev Cell Dev Biol. 27:493–512. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rawlins EL: The building blocks of
mammalian lung development. Dev Dyn. 240:463–476. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Willis KA, Siefker DT, Aziz MM, White CT,
Mussarat N, Gomes CK, Bajwa A, Pierre JF, Cormier SA and Talati AJ:
Perinatal maternal antibiotic exposure augments lung injury in
offspring in experimental bronchopulmonary dysplasia. Am J Physiol
Lung Cell Mol Physiol. 318:L407–L418. 2020. View Article : Google Scholar
|
|
8
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang D, Wu L, Du Y, Zhu Y, Pan B, Xue X
and Fu J: Autophagy inducers restore impaired autophagy, reduce
apoptosis, and attenuate blunted alveolarization in
hyperoxia-exposed newborn rats. Pediatr Pulmonol. 53:1053–1066.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yim WW and Mizushima N: Lysosome biology
in autophagy. Cell Discov. 6:62020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Itakura E and Mizushima N: Syntaxin 17:
The autophagosomal SNARE. Autophagy. 9:917–919. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hubert V, Peschel A, Langer B, Gröger M,
Rees A and Kain R: Lamp-2 is required for incorporating syntaxin-17
into autophagosomes and for their fusion with lysosomes. Biol Open.
5:1516–1529. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu Y, Fu J, Yang H, Pan Y, Yao L and Xue
X: Hyperoxia-induced methylation decreases RUNX3 in a newborn rat
model of bronchopulmonary dysplasia. Respir Res. 16:752015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
15
|
Jain D and Bancalari E: Bronchopulmonary
dysplasia: Clinical perspective. Birth Defects Res A Clin Mol
Teratol. 100:134–144. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Manji JS, O'Kelly CJ, Leung WI and Olson
DM: Timing of hyperoxic exposure during alveolarization influences
damage mediated by leukotrienes. Am J Physiol Lung Cell Mol
Physiol. 281:L799–L806. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen CM, Wang LF, Chou HC, Lang YD and Lai
YP: Up-regulation of connective tissue growth factor in
hyperoxia-induced lung fibrosis. Pediatr Res. 62:128–133. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao X, Shi Y, Zhang D, Tong X, Sun Y, Xue
X and Fu J: Autophagy inducer activates Nrf2-ARE pathway to
attenuate aberrant alveolarization in neonatal rats with
bronchopulmonary dysplasia. Life Sci. 252:1176622020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tsukamoto S, Kuma A, Murakami M, Kishi C,
Yamamoto A and Mizushima N: Autophagy is essential for
preimplantation development of mouse embryos. Science. 321:117–120.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Q, Kang R, Zeh HJ III, Lotze MT and
Tang D: DAMPs and autophagy: Cellular adaptation to injury and
unscheduled cell death. Autophagy. 9:451–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Boya P, Gonzalez-Polo RA, Casares N,
Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D,
Souquere S, Yoshimori T, et al: Inhibition of macroautophagy
triggers apoptosis. Mol Cell Biol. 25:1025–1040. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu J and Debnath J: The evolving,
multifaceted roles of autophagy in cancer. Adv Cancer Res.
130:1–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vij N, Chandramani-Shivalingappa P, Van
Westphal C, Hole R and Bodas M: Cigarette smoke-induced autophagy
impairment accelerates lung aging, COPD-emphysema exacerbations and
pathogenesis. Am J Physiol Cell Physiol. 314:C73–C87. 2018.
View Article : Google Scholar :
|
|
24
|
Lu WH, Wang G, Li Y, Li S, Song XY, Wang
XY, Chuai M, Lee KK, Cao L and Yang X: Autophagy functions on EMT
in gastrulation of avian embryo. Cell Cycle. 13:2752–2764. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meng Y, Pan M, Zheng B, Chen Y, Li W, Yang
Q, Zheng Z, Sun N, Zhang Y and Li X: Autophagy attenuates
angiotensin ii-induced pulmonary fibrosis by inhibiting redox
imbalance-mediated NOD-like receptor family pyrin domain containing
3 inflamma-some activation. Antioxid Redox Signal. 30:520–541.
2018. View Article : Google Scholar
|
|
26
|
Zhang L, Zhao S, Yuan LJ, Wu HM, Jiang H,
Zhao SM, Luo G and Xue XD: Autophagy regulates hyperoxia-induced
intracellular accumulation of surfactant protein C in alveolar type
II cells. Mol Cell Biochem. 408:181–189. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sureshbabu A, Syed M, Das P, Janér C,
Pryhuber G, Rahman A, Andersson S, Homer RJ and Bhandari V:
Inhibition of regulatory-associated protein of mechanistic target
of rapamycin prevents hyperoxia-induced lung injury by enhancing
autophagy and reducing apoptosis in neonatal mice. Am J Respir Cell
Mol Biol. 55:722–735. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Viret C and Faure M: Regulation of
syntaxin 17 during autophagosome maturation. Trends Cell Biol.
29:1–3. 2019. View Article : Google Scholar
|
|
29
|
Shen Q, Shi Y, Liu J, Su H, Huang J, Zhang
Y, Peng C, Zhou T, Sun Q, Wan W and Liu W: Acetylation of STX17
(syntaxin 17) controls autophagosome maturation. Autophagy. 1–13.
2020.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Supplemental therapeutic oxygen for
prethreshold retinopathy of prematurity (Stop-Rop), a randomized,
controlled trial. I: Primary outcomes. Pediatrics 1. 05:295–310.
2000.
|
|
31
|
Tian L, Yang Y, Li C, Chen J, Li Z, Li X,
Li S, Wu F, Hu Z and Yang Z: The cytotoxicity of coxsackievirus B3
is associated with a blockage of autophagic flux mediated by
reduced syntaxin 17 expression. Cell Death Dis. 9:2422018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Arasaki K and Tagaya M: Legionella blocks
autophagy by cleaving STX17 (syntaxin 17). Autophagy. 13:2008–2009.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Del Riccio V, van Tuyl M and Post M:
Apoptosis in lung development and neonatal lung injury. Pediatr
Res. 55:183–189. 2004. View Article : Google Scholar
|