Introduction
Glioblastoma multiforme (GBM), is the most lethal
human cancer with very poor prognosis even following surgery and
chemotherapy. The majority of the primary GBM arises de novo
and develops progressively without recognisable symptoms or
precursor lesions. Despite advancement in GBM detection, radiation,
chemotherapy and surgery, the outcome of GBM remains poor, with an
overall survival of only 14 months (1-3).
A higher degree of infiltration is one of the
hallmarks of GBM. It rarely metastasises outside the brain but
actively migrates through two types of extracellular spaces in the
brain: The perivascular space around all blood vessels, and spaces
between the neurons and glial cells (2). In order to invade through these
spaces, GBM cells have to undergo several biological changes,
including gaining mobility, the ability to degrade the
extracellular matrix (ECM) and the ability to acquire stem cell
phenotype (4). Invasion involves
a complicated mechanism comprising cross-talk between canonical
pathways in cancer (2). For
example, activating epidermal growth factor receptor (EGFR)
mutations may result in increased expression and phosphorylation of
cell adhesion molecules, thereby increasing GBM invasiveness
(5). In addition, tumor protein
p53 (TP53) mutations accelerate GBM invasion by facilitating
the recycling of integrin, a class of cell surface receptor that
forms a network with ECM during cell migration (6). In addition, aberrant AKT signalling
increases metalloproteases activity, which subsequently increases
the tumour cell proteolytic capability to invade the border of the
normal brain tissue (7).
Research and development for improving GBM survival
by targeting kinases involved in specific cancer pathways are
active areas of investigation. For example, erlotinib targeting the
EGFR pathway and bevacizumab targeting the VEGFR and PI3K pathways
have both been used to treat GBM. However, clinical trials
(clinical trial nos. NCT01110876 and NABTT 0502) showed conflicting
results. Erlotinib has been successful in treating lung and breast
cancer but not GBM (8). Its poor
efficacy in treating GBM is due to the EGFR mutation sites,
which occur in the extracellular domain in GBM, whereas in lung
cancer, the mutations are at the kinase domain (9). Bevacizumab has received an
accelerated approval by the United States of America Food and Drug
Administration in 2009 due to its success in eradicating recurrent
GBM. However, bevacizumab treatment is not beneficial for newly
diagnosed patients with GBM (10). Another example of treatment
failure is PI3K inhibitors, which primarily target the PI3K class 1
subunit. Despite binding to the PI3K subunits, GBM cells continue
to proliferate due to activation of the alter-native
RAS/MAPK/mitogen-activated protein kinase kinase (MEK) pathway
(11). The combination of PI3K
and MEK pathway inhibitors has been demonstrated to improve
treatment efficacy in GBM (12).
However, inhibition of the PI3K will cause downstream independent
activation of the AKT pathways or restoration of AKT function
involving molecules such as upregulation of receptor tyrosine
kinase (RTK) and mTORC2 (13),
highlighting the insufficiency of inhibiting PI3K signalling
pathways as a singular treatment strategy, and the need to identify
an alternative target. Advanced biotechnology platforms as well as
integrative analysis tools allow the identification of novel kinome
pathways for GBM therapy. The results may provide an implicative
understanding to target GBM in a highly strategic manner, thereby
improving patient survival. The present study aimed to identify
novel kinase targets via RNA interference (RNAi) screening of
upregulated kinases identified from meta-analysis, and to validate
the functional role of 'hit' target genes, namely Tousled Like
Kinase 1 (TLK1), in GBM cells harbouring different types of PTEN
and TP53 status by investigating specifically its involvement in
GBM cell viability and survival regulations.
Materials and methods
In silico analysis via Oncomine
Meta-analysis was performed to identify kinases that
are involved in GBM using 5 micro-array datasets from Oncomine
Research Edition (14). Data were
obtained from Bredel Brain (15),
Liang Brain (16), Shai Brain
(17), Lee Brain (18) and Sun Brain (19) datasets. All significantly
upregulated kinase genes were selected based on their median rank
and P<0.05 (99% confidence interval). All identified kinases
were then compared with those in the Human Kinome Database
(www.kinase.com) (20). High-throughput RNAi knockdown of
the selected kinases was performed to validate these targets.
Cell culture
The human GBM LN18 (ATCC® CRL-2610™) and
U87MG (ATCC® HTB-14™) cell lines of an unknown origin
were obtained from American Type Culture Collection. The cells were
maintained in monolayers in Dulbecco's modified Eagle's medium
(DMEM; Sigma-Aldrich; Merk KGaA) containing 10% foetal bovine serum
(Lonza Group Ltd.). The cell lines were routinely maintained at
37°C in humidified 5% CO2. The cells were harvested by
removing the medium, washing with 5 ml 1X Dulbecco's PBS (Gibco;
Thermo Fisher Scientific, Inc.), and trypsinized using 1X TrypLE
(Gibco; Thermo Fisher Scientific, Inc.). Normal human astrocytes
(NHA; Lonza Group Ltd.) were maintained in monolayers using
astrocyte basal medium mixed with AGM™ BulletKit (Lonza Group
Ltd.). The NHA were also routinely maintained at 37°C in humidified
5% CO2. The NHA were harvested and washed using
ReagentPack™ (cat. no. CC-5034; Lonza Group Ltd.) containing
trypsin/ethylenediaminetetraacetic acid (EDTA), trypsin
neutralizing solution, and HEPES-buffered saline solution.
RNAi screening
RNAi screening was performed on a ON-TARGETplus
SMARTpool Custom reverse transfection format library containing 113
overexpressed kinases genes of interest (GE Healthcare Dharmacon,
Inc.) with 6.25 pmol lyophilized small interfering RNA (siRNA) per
well in 96-well plates. RNAi screening was performed on 2 different
GBM cell lines harbouring 2 different mutations: LN18 cells
harbouring TP53 mutation and wild-type PTEN, and
U87MG cells harbouring wild-type TP53 and mutant
PTEN. Both cell lines were wet reverse-transfected with 0.15
µl lipophilic base transfection reagent, DharmaFect1 (GE
Healthcare Dharmacon, Inc.) according to the manufacturer's
protocol, yielding a final concentration of 50 nM siRNA pool per
target gene per well. The cells were then incubated at 37°C in
humidified 5% CO2 for 48 h to allow efficient RNA
knockdown. Then, the cell culture medium was change and cell
viability was measured at 96 h by adding 1:10 resazurin-based
solution, PrestoBlue® (Invitrogen; Thermo Fisher
Scientific, Inc.), with 10 µl dropped into each well. The
mixture was incubated for 1 h at 37°C. Relative fluorescence unit
(RFU) were measured using a Varioskan Flash multimode plate reader
(Thermo Fisher Scientific, Inc.) at 560 and 590 nm. All experiments
were performed in triplicate. Non-targeting siRNA (si-NTG; GE
Healthcare Dharmacon, Inc.) was used as the negative control; PLK1
siRNA (si-PLK1; GE Healthcare Dharmacon, Inc.) was included as the
positive control.
Data analysis for RNAi screening
Raw RFU data were log-transformed and the k-Median
Absolute Deviation (kMAD) was used for hits identification analysis
as it is resistant to outliers in the samples. The kMAD identifies
weak hits in RNAi data efficiently compared with the B-score,
Z-score, SSMD or mean + k (standard deviations) whilst also
capturing strong hits and controlling false positive hits (21-23). As this was a selective RNAi
screening strategy, a cut-off of median ± 'kMAD' was selected for
kinase genes to be considered a hit for each screen. Unpaired
t-test against the NTG control was also performed to ensure that
the target serves a significant role in cancer cell
vulnerabilities.
RNA extraction and reverse
transcription-quantitative poly- merase chain reaction
(RT-qPCR)
Transfected cells were harvested at 48 h, and RNA
extraction was performed using the RNeasy Plus Mini kit (Qiagen
GmbH). cDNA was synthesized using 100 ng of RNA, and generation of
cDNA was performed using an iScript™ cDNA Synthesis kit containing
iScript reverse transcriptase (Bio-Rad Laboratories, Inc.). The
qPCR was performed using iTaq Universal SYBR Green master mix
(Bio-Rad Laboratories, Inc.) on a Rotor-Gene 6000 qPCR platform
(Corbett Life Science; Qiagen, Inc.). The forward and reverse
TLK1 primers used were 5′-CAGTGGAAGTTTGGAGGGGCCG-3′ and
5′-CCGGATGGCGGCGTGTGAT-3′, respectively. β-actin (ACTB) was
used as the housekeeping gene, and the forward and reverse primer
sequences were 5′-CATGTACGTTGCTATCCAGGC-3′ and
5′-CTCCTTAATGTCACGCACGAT-3′, respectively. The relative gene
expression was determined using the comparative threshold cycle
(2−∆∆Cq) method (24).
The thermocycling conditions for iTaq polymerase activation and
denaturation were 95°C for 30 sec, followed by 40 cycles of
denaturation at 95°C (5 sec) and annealing/extension at 60°C (30
sec), and a final extension at 72°C for 60 sec. Melt curve analysis
was performed at 65-95°C with 0.5°C increments (2-5 sec/step). For
microarray validation, qPCR was performed on the selected genes
namely; TLK1, Rac family small GTPase 2 (RAC2), Rho
associated coiled-coil containing protein kinase 2, paxillin,
collagen type IV α chain, actin related protein 2/3 complex subunit
2, thrombospondin 2 and FYN proto-oncogene, Src family tyrosine
kinase.
Protein extraction and western blot
analysis
For protein extraction, cells were lysed with
radioimmunoprecipitation assay buffer (Thermo Fisher Scientific,
Inc.) supplemented with protease cocktail inhibitor (Roche
Diagnostics), 25 µM sodium fluoride (New England Biolabs,
Inc.) and 25 µM sodium orthovanadate (New England Biolabs,
Inc.), followed by sonication for 1 min and finally centrifugation
at 15,000 × g for 30 min at 4°C. The supernatant containing the
total protein lysates was collected and the protein concentrations
were measured using a Bradford assay (Bio-Rad Laboratories,
Inc.).
A total 30 µg proteins were resolved by
SDS-PAGE using Mini-PROTEAN precast gels (4-20%) and 1X
Tris-glycine buffer (Bio-Rad Laboratories, Inc.), and transferred
to polyvinylidene difluoride membranes (Bio-Rad Laboratories,
Inc.). Subsequently, the membranes were probed with primary mouse
anti-human TLK1 antibody (1:200; cat. no. sc-100345, Santa Cruz
Biotechnology, Inc.). Then, the membranes were washed 4 times with
1X TBS-0.05% Tween-20 (TBST) buffer (Thermo Fisher Scientific,
Inc.) and incubated with goat anti-mouse antibody conjugated to
horseradish peroxidase (HRP; 1:2,000; cat. no. sc-2005, Santa Cruz
Biotechnology) using casein blocker (Bio-Rad Laboratories, Inc.)
for 12 h at 4°C. Following incubation, the membranes were washed 4
times for 5 min with 1X TBS Tween-20 buffer (Thermo Fisher
Scientific, Inc.), on an orbital shaker. The loading control was
mouse anti-human β-actin (cat. no. sc-69879; Santa Cruz
Biotechnology, Inc.); the secondary antibody was goat anti-mouse
IgG-HRP (cat. no. sc-2005, Santa Cruz Biotechnology, Inc.). Signals
were measured using Pierce™ ECLPlus substrate (Thermo Fisher
Scientific, Inc.) and viewed under a chemiluminescence imager
(Bio-Rad Laboratories). Analysis of the software for densitometric
analysis was performed using Image Lab™ version 4.1 (Bio-Rad
Laboratories, Inc.).
Pooled TLK1 siRNA (si-TLK1) transient
transfection
Resuspension of lyophilized ON-TARGETplus pool
si-TLK1 and pool si-NTG (20 nmol, cat. no. D-001810-10; GE
Healthcare Dharmacon, Inc.) was performed using 1X siRNA buffer to
yield 100 µM stock. Prior to reverse-transfection, 5
µM siRNA solution in 1X siRNA buffer was prepared. For
experiments requiring 96-well plates, the transfection reagents
were prepared in separate tubes. In tube 1, 10 µl siRNA in
serum-free medium was added to 0.5 µl 5 µM siRNA in
9.5 µl serum-free medium. In tube 2, 10 µl of diluted
DF1 transfection reagent (GE Healthcare Dharmacon, Inc.) was
prepared in serum-free medium (0.1 µl DF1 with 9.85
µl serum-free medium). Both tubes were gently mixed and
incubated for 5 min at room temperature. Subsequently, the contents
of tube 1 and 2 were mixed for 20 min at room temperature. GBM
cells (5,000 cells/well) seeded in 96-well plates and a suspension
was made with 80 µl antibiotic-free complete DMEM per well.
The cell suspensions in the antibiotic-free medium were mixed with
the previously prepared siRNA, serum-free medium, and transfection
reagent in each plate. The cells were incubated for 48 h (RNA) and
72 h (protein) at 37°C in 5% CO2. The ON-TARGETplus
si-TLK1 sequences used were 5′-GAGUAUGCAAGAUCGAUUA-3′,
5′-GAAGCUCGGUCUAUUGUAA-3′, 5′-GCAAUGACUUGGAUUUCUA-3′ and
5′-GUUCAAAGAUCACCCAACA-3′.
ORF clones and short hairpin RNA (shRNA)
transduction
Precision LentiORF lentiviral particle individual
clones (cat. no. V13121301) and LentiORF red fluorescent protein
(RFP) control (cat. no. OHS5832; Open Biosystems Inc.; Thermo
Fisher Scientific, Inc.) were used to overexpress TLK1 in
U87MG and LN18 cells following the manufacturer's protocol. The
GIPZ shRNA individual clones (cat. nos. V3LHS_637461, V3LHS_637455
and V3LHS_335655) and GIPZ NTG shRNA control clones were purchased
in viral particle format (Open Biosystems Inc.; Thermo Fisher
Scientific, Inc.) to knockdown TLK1 in the U87MG and LN18
cells. The day prior to transduction, 5×104 U87MG or
LN18 cells at 40-50% confluence were seeded onto 24-well tissue
culture plates with their respective media. The LentiORF control
viral stock was diluted in a round-bottom 96-well plate using
serum-free medium in a series of 5-fold dilutions to reach a final
dilution of 390,625-fold. Following dilution, the virus stock was
pre-incubated for 5 min at room temperature.
Next, 24-well plates were labelled, using one row
per replicate. The culture medium in the 24-well plates was
removed, and then 225 µl serum-free medium was added to each
well. The cells were transduced by adding 25 µl diluted
control LentiORF lentivirus from the original 96-well plate to 1
well on the 24-well destination plate containing the cells. The
transduced cultures were incubated at 37°C for 4 to 6 h.
Subsequently, 1 ml normal concentration DMEM was added and the
cells were incubated for 72 h at 37°C. The green fluorescent
protein (GFP)-expressing cells or cell colonies were counted to
determine the functional titre and relative transduction
efficiency. Similar methods were used to perform shRNA transduction
in both the LN18 and U87MG cell lines. qPCR was then performed as
described above to determine successful knockdown and
overexpression of the gene of interest. Methods for gene expression
by qPCR have been mentioned previously.
Cell viability assay
U87MG and LN18 cells were transfected with 25 nM
si-TLK1 in 6-well plates. A total of 10 µl
PrestoBlue® cell viability reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) was added to each well plate. After 1 h of
incubation at 37°C readings were taken using a microplate reader,
SkanIt RE, for Varioskan Flash 2.4 (Thermo Fisher Scientific, Inc.)
at 560/590 nm excitement/emission. Readings were taken at 24, 48,
72 and 96 h post-transfection. Experiments were performed in
triplicate.
Human HT-12 v4 expression BeadChip
microarray
Total RNA from U87MG cells was isolated using a
RNeasy Plus Mini kit (Qiagen GmbH) after 48 h of si-TLK1
transfection. A total of 150 ng purified RNA was amplified using an
Illumina Total Prep RNA amplification kit (Life Technologies;
Thermo Fisher Scientific, Inc.). Biotin-labelled cRNA was directly
hybridized on an Illumina Human HT-12 v4 Expression BeadChip arrays
kit (Illumina, Inc.) according to the manufacturer's protocol.
Microarray bead chips were scanned using an iScan array scanner
(Illumina, Inc.) and the intensity of the data were processed using
Genome Studio version 2008.1 (Illumina, Inc.). Analysis was
performed using GeneSpring GX 12.6 software (Agilent Technologies,
Inc.). Microarray data analysis was performed whereby fluorescent
intensities were log-transformed, quantile-normalized, and
pre-filtered to remove low-quality data. Principal component
analysis was used to assess the data quality control. A moderated
unpaired t-test was used to determine the underlying pathways
involved following TLK1 knock-down in U87MG cells. P-value
computation was conducted using asymptotic theory and
Benjamini-Hochberg multiple testing corrections were applied. A
significant gene list was obtained where P<0.05 and fold change
>1.1 were chosen for pathway analysis. Clustering heat maps of
differentially expressed genes were constructed using a hierarchy
based on Euclidean similarity measure and complete linkage. Gene
Set Enrichment Analysis was performed using filtered probe signals
with q-value <0.3. Pathway analysis was performed using
WebGestalt (25) and Pathway
Studio 8.0 software (Ariadne Genomics; Elsevier). Protein-protein
interaction network analysis was performed to identify possible
TLK1 interactors using NetworkAnalyst 3.0 (26).
Single-stranded DNA (ssDNA) apoptosis
assay
The cells were seeded in 96-well plates
(3×103 cells/well) and transfected with si-TLK1.
After 48 h incubation, the apoptotic activity assay was performed
using the ssDNA apoptosis ELISA kit (cat. no. APT225; Chemicon
International) according to the manufacturer's protocol.
Experiments were performed in triplicate.
Annexin V fluorescence assay
Cells were transfected with si-TLK1 for 48 h in
6-well plates. After replacing the media, 2.5 mM temozolomide (TMZ;
cat. no. T2577-25MG; Sigma-Aldrich; Merck KGaA) was added to each
well. After 48 h of incubation with TMZ at 37°C, cells were
harvested. The cells were then washed with PBS and centrifuged at
200 × g for 5 min. The cell pellet was suspended with 100 µl
of Annexin V-FLUOS (cat. no. 11988549001; Roche Applied Science)
labelling solution. Annexin V-FLUOS (20 µl) was initially
diluted in 1 ml incubation buffer with 20 µl propidium
iodide solution. The suspension was incubated for 10-15 min at
15-25°C. Analysis was performed using a Tali® Image
Cytometer (Thermo Fisher Scientific, Inc.). Experiments were
performed in triplicate.
Cell cycle assay
The cell cycle assay was performed using
1×106 LN18 or U87MG cells transfected with
si-TLK1 or si-NTG. The cells were harvested using a standard
protocol as described in the Cycletest™ PLUS DNA Reagent kit
protocol (BD Biosciences). Flow cytometric analysis was performed
using BD FACSAria™ (Becton, Dickinson and Company). Data were
analysed using ModFit LT software version 2.0 (Verity Software
House, Inc.). Experiments were performed in triplicate.
Colony formation assay
Cells were initially transfected with siRNA or shRNA
or pLOC LentiORF-expression vector for 48 h in T45 flasks prior to
the assessment of cell colony formation in the monolayers. At 96 h,
cells were trypsinized and washed. Subsequently, 100 cells were
counted and seeded on 6-well plates. Colonies were allowed to grow
for 14 days. Later, the DMEM was removed and the cells were washed
gently with PBS prior to fixation at 25°C with 50% methanol for 10
min and then staining for 1 h at room temperature with 0.5% crystal
violet that had been diluted in 50% methanol. The stain was removed
and the cells were washed with distilled water. Colonies was
observed using light microscopy (magnification, ×40); Nikon
Corporation. Colonies containing >50 cells were counted to as a
single colony. Images of the colonies were captured using ChemiDoc
Imager (Bio-Rad Laboratories, Inc.) and automatically counted using
openCFU software version 4.0 (27). Experiments were performed in
triplicate.
Total/phosphorylated TP53,
Erk/AKT/ribosomal protein S6 kinase β-1 (p70s6k) activation
assay
Total/phosphorylated TP53 and Erk/AKT/p70s6k
activation assays were performed using InstantOne™ ELISA kits (cat.
nos. 85-86123 and 85-86018, respectively; eBioscience, Inc.).
Initially, GBM cell lines were transfected with si-TLK1 as
described above and incubated for 72 h. Subsequently,
0.4×106 cells were resuspended in 500 µl DMEM and
incubated for 2 h at 37°C. After that, cells were harvested and
lysed with agitation (300 RPM) at room temperature for 10 min.
Samples, and negative and positive controls supplied by the
manufacturer were added to the microplate assay wells.
Total/phosphorylated TP53 and Erk/AKT/p70s6k antibodies were added
to each well. The microplates were sealed and incubated for 60 min
at room temperature on a plate shaker at 300 RPM. Wells were washed
three times with wash buffer and detection reagent was added to
each well. After 30 min, the reaction was stopped by adding stop
solution. The absorbance at 450 nm was detected using a VarioSkan
Flash multimode plate reader (Thermo Fisher Scientific, Inc.).
Xenograft mouse model
A total of 42 immunodeficient nude mice (female
BALB/c-nu; 6 weeks old; 16-18 g) obtained from BioLASCO
Taiwan Co., Ltd. were randomly divided into 7 groups and maintained
in pathogen-free environments. All animal experiments performed in
the present study complied with the international guidelines for
the care and treatment of laboratory animals adapted from WHO
CHRONICLE, 1985 (28). The animal
experiments were approved by the UKM Animal Ethics Committee under
the ethics registration number
UMBI/2014/ROSLAN/24-SEPT./607-SEPT.-201 4-DEC.-2014. Injection of
TLK1-overexpressing and TLK1-knockdown U87MG cells was performed
using equal numbers of log-phase U87MG-pGIPZ-NTG,
U87MG-pGIPZ-shTLK1-455, U87MG-shTLK1-461, U87MG-pLOC-RFP control or
U87MG-pLORF-TLK1-301 cells (3×107). The cells were
harvested, washed in PBS and suspended in 200 µl PBS. The
cell suspension was injected subcutaneously into the right
subdorsal flank tissue of the nude mice to establish subcutaneous
xenograft models. The mice were monitored daily 47-day observation
period and the sizes of the transplanted tumours were measured by
slide calliper every 7 days. A growth curve for the transplanted
tumours was plotted after calculating the tumour volume.
The experiment was terminated at day 47 following
the completion of observation. The mice were sacrificed and all
tumours that had formed subcutaneously were removed and weighed
using a Pioneer weighing machine (OHAUS Europe GmbH). Tumour
tissues and the adjacent tissues were snap-frozen and stored in
liquid nitrogen for future study.
Statistical analysis
For the in vitro functional experiments, all
data are expressed as the mean ± standard deviation, and data
analysis was performed using appropriate statistical analysis
tests, such as Kruskal-Wallis with Dunn's post hoc test, or two-way
analysis of variance (ANOVA) with Bonferroni's multiple correction
post hoc test. For the xenograft in vivo monitoring
experiments, comparisons among all groups were performed using a
mixed ANOVA with Bonferroni multiple comparisons test. Tumour
weight measurement was performed using Kruskal-Wallis analysis
followed by Dunn's multiple comparisons test. For comparisons
between two groups, a Mann Whitney U test was utilised. All
statistical analyses were performed using GraphPad Prism version
8.0 (GraphPad Software, Inc.). P<0.05 was considered to indicate
a statistically significant difference.
Results
RNAi screening in LN18 and U87MG cell
lines
In total, 113 upregulated kinase genes were screened
and the -kMAD score of each cell line was calculated and sorted
using scatter plots (Fig. S1).
The scatter plot visualization of the -kMAD clearly demonstrated
that Polo-like kinase 1 (PLK1) consistently exhibited the lowest
-kMAD value, which was expected as the si-PLK1 was the positive
control for each experimental plate. RNAi screening of the LN18
cells identified 20 potential hits involved in cell cycle and
checkpoint control regulation, namely aurora kinase A, cyclin
dependent kinase inhibitor 1A, cyclin dependent kinase 1
(CDC2), NIMA related kinase (NEK)2, NEK8 and
TLK1. However, in the U87MG cells, 22 potential hits were
identified to serve critical roles in various cellular pathways.
Functional clustering analysis using the Database for Annotation,
Visualization and Integrated Discovery (DAVID) revealed that the
kinases were involved in the cell cycle pathway and focal adhesion
pathway. Only CDC2, serine/threonine kinase 33, TLK1,
CDC like kinase 2, and cyclin dependent kinase inhibitor 3 were
associated with cell cycle and mitosis regulation.
To identify the relevant kinase target for further
functional investigations, the 'hit lists' identified from the
earlier statistical analysis was overlapped using a Venn diagram
(Fig. 1A and B). A total of 4
kinases, CDC2, TLK1, Fms related tyrosine kinase 3
(FLT3) and α kinase 1 (ALPKI) were overlapped in both
cell lines. Further validation was performed on these genes on a
small-scale basis. Fig. 1C
demonstrated that only TLK1 knockdown significantly
decreased the number of viable cells to between 10 and 40%
(P<0.05). Therefore, TLK1 was selected as the potential
investigative target in the present study, and its functional role
in GBM has also not been established. Fig. S2 demonstrates the efficiency of
CDC2, TLK1, FLT3 and ALPK1 mRNA
knockdown with their respective siRNA.
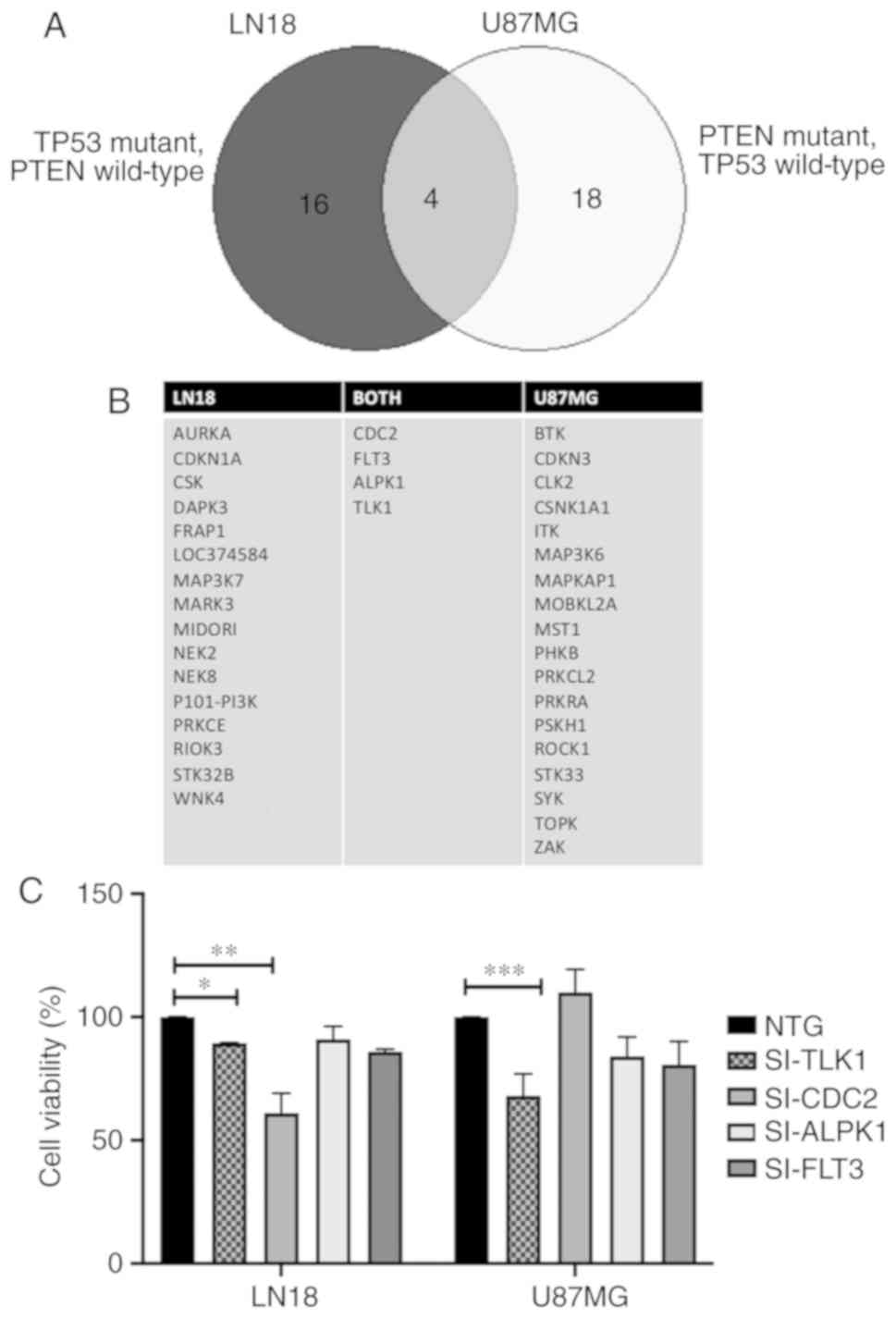 | Figure 1Potential hit genes from RNAi screen
targeting the GBM kinome in LN18 and U87MG cells. (A) Venn diagram
of the 16 and 18 genes identified by the k-Median Absolute
Deviation statistical analysis in LN18 cells and U87MG cells,
respectively. A total of 4 genes were consistently present in both
cell lines: CDC2, TLK1, FLT3 and ALPK1.
(B) The complete list of genes identified from RNAi screen. (C)
Validation of the 4 genes identified from RNAi screen using Presto
Blue assay for cell viability analysis. Readings were performed at
96 h post-transfection. Analysis were performed using
Kruskal-Wallis non-parametric test with Dunn's post hoc test.
Values are presented as mean ± standard deviation of 3 biological
replicates. *P<0.05, **P<0.01 and
***P<0.005. RNAi, RNA interference; GBM, glioblastoma
multiforme; TP53, tumor protein 53; CDC2, cyclin dependent
kinase 1; TLK1, tousled like kinase 1; FLT3, Fms
related tyrosine kinase 3; ALPK1, α kinase 1; si, small
interference RNA; NTG, non-targeting RNA. |
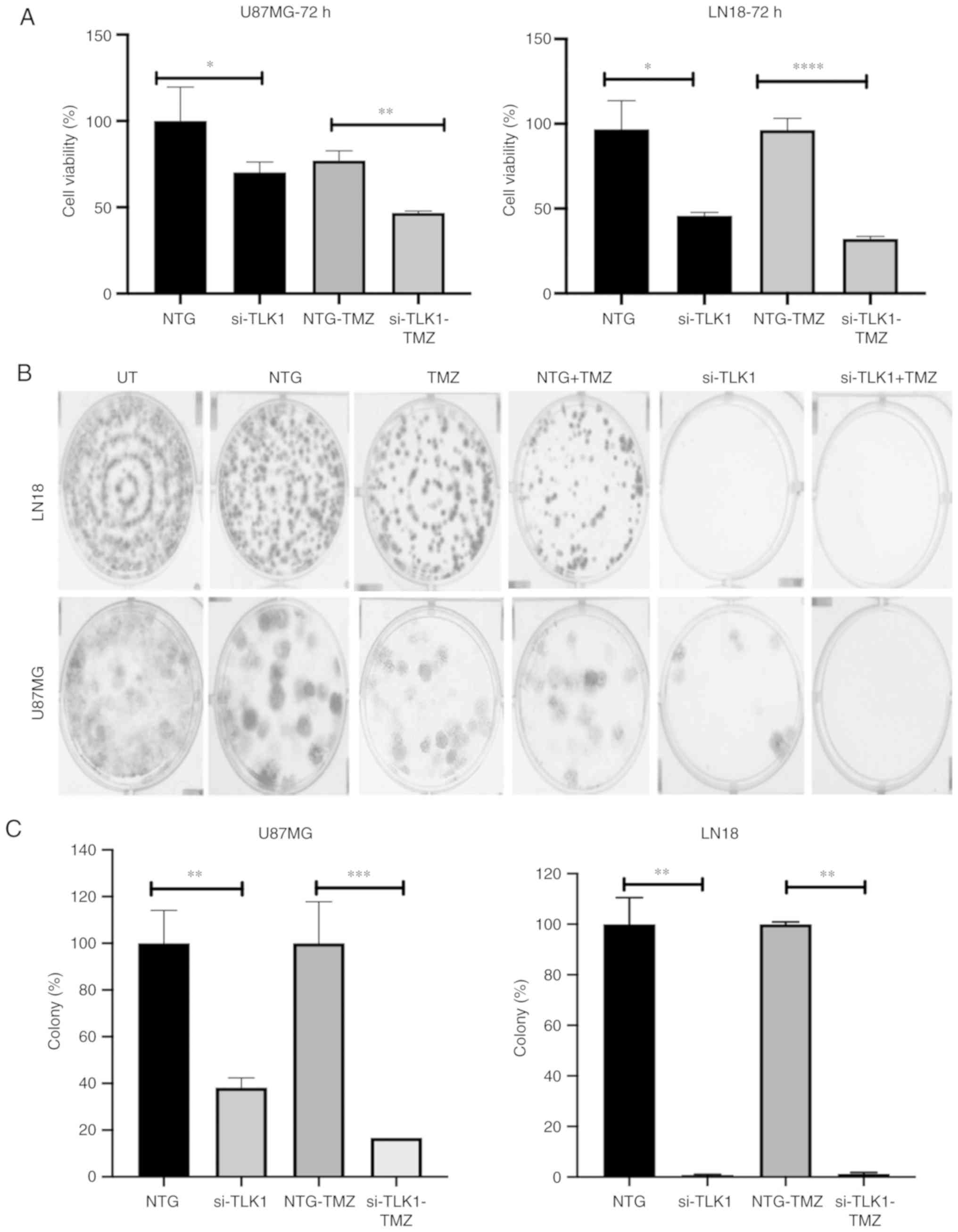
Role of TLK1 in modulation of survival
and apoptosis path- ways
Transient transfection of specific si-TLK1 on
U87MG and LN18 cells was performed to analyse the effect of
TLK1 on GBM cell proliferation and apoptosis. TLK1
mRNA levels were decreased by >80% and TLK1 protein expression
decreased significantly in the si-TLK1-transfected GBM cells
(Fig. 2A). There were
significantly fewer viable si-TLK1 cells compared with the
si-NTG cells at 72 h post-transfection (Fig. 2B); however, the significant
decrease in cell viability observed only began at 96 h after
si-TLK1 transfection (Fig.
S3).
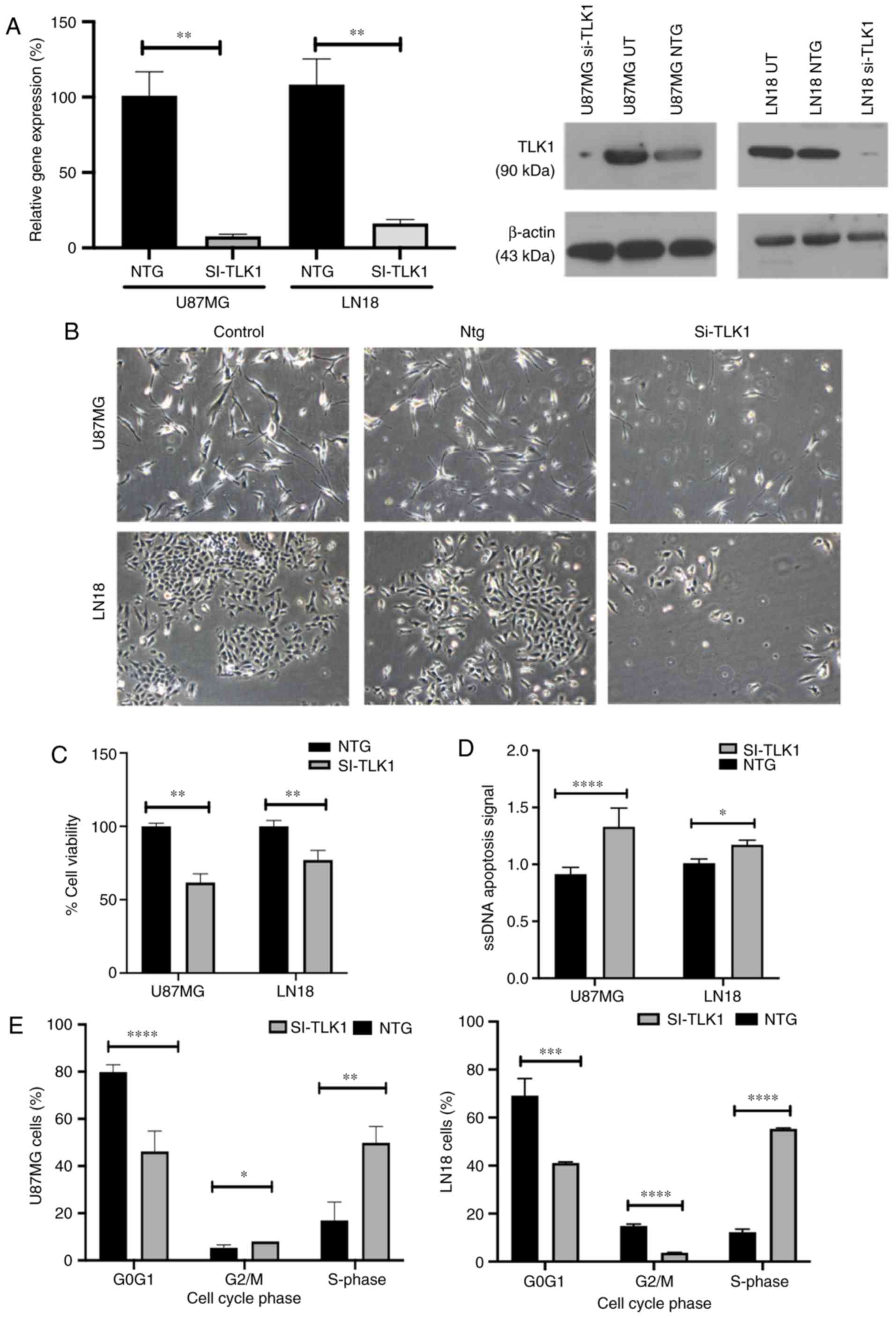 | Figure 2TLK1 knockdown in GBM cells
decreases cell viability. (A) Reverse transcription-quantitative
polymerase chain reaction and western blot analysis were performed
to determine transfection efficiency. TLK1 knockdown in both
cell lines was effective, with >80% transfection efficiency and
decreased TLK1 protein levels at 72 h post-transfection. (B)
TLK1 knockdown caused a decrease in the number of viable
cells under an inverted light microscope. (C) Significant decreases
in viable U87MG and LN18 cells by 40 and 20%, respectively, were
observed following TLK1 knockdown at 72 h. (D) Increased
apoptotic ssDNA signaling in U87MG cells compared with LN18 cells.
(E) Cell cycle analysis indicating a marked increase in S-phase and
a decrease in G0/G1 using two-way analysis of variance with
Bonferroni's multiple correction post hoc test. All experiments
were performed in triplicate and the results were compared with the
si-NTG control. Values are presented as mean ± standard deviation
of 3 biological replicates. *P<0.05,
**P<0.01, ***P<0.005 and
****P<0.001. TLK1, tousled like kinase 1; GBM,
glioblastoma multiforme; ssDNA, single-stranded DNA; si-, small
interfering RNA: NTG, non-targeting RNA. |
Apoptosis signals were increased in the 2 GBM cell
lines (Fig. 2C and D), mediated
by caspase-3 and caspase-7 activation. Qualitative fluorescence
imaging using confocal microscopy indicated increased DEVD complex
formation based on the presence of fluorescence green dots
(Fig. S4). TLK-1 inhibition also
resulted in cell cycle arrest, as demonstrated by a marked increase
in S-phase cells and a decrease in G0/G1 cells (P<0.05) in both
cell lines. Notably, there were significant decreases in the
numbers of LN18 cells (P<0.05) at the G2/M phase, which was the
opposite to the pattern observed in the U87MG cells
(Fig. 2E). A marked
increase in the number of S-phase cells and a decrease in the
number of cells in G0/G1 phase, as indicated by the flow cytometry
analysis data (Fig. 2E) may
suggest that the cells underwent extensive replication, or that
there was an S-phase arrest. By incorporating brdU analysis, it was
confirmed that cell replication and proliferation had halted. It
was demonstrated that the brdU signal was significantly decreased
following TLK1 knockdown in U87MG, as indicated in Fig. S5A.
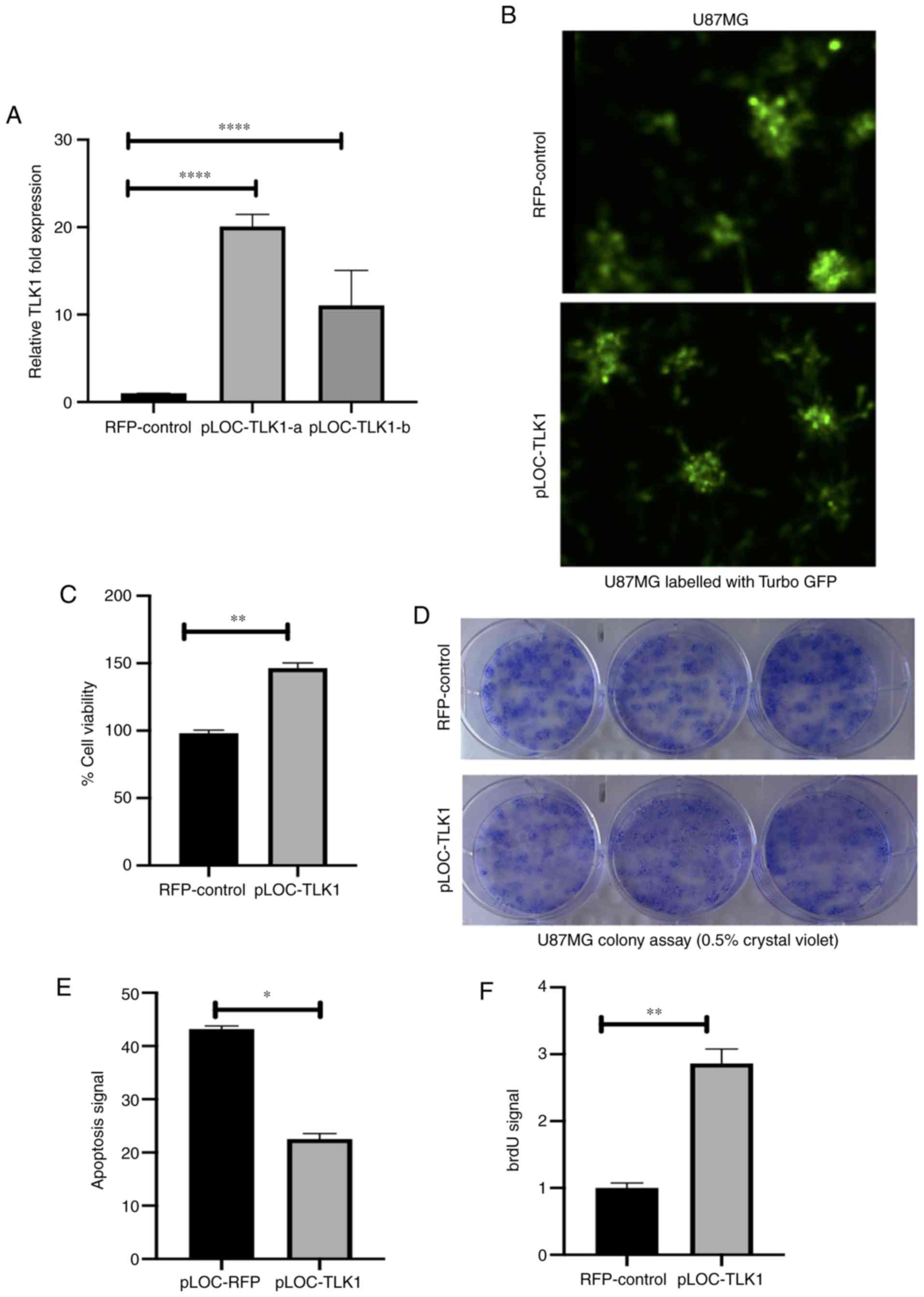
To determine the effects of TLK1 overexpression on
GBM cell proliferation, U87MG cells were transduced with pLOC-ORF
clones using lentiviral delivery. Fig. 3A demonstrates the up-to-nearly
20-fold increase in TLK1 expression in U87MG cells observed
compared to empty RFP vector control cells. Transduction efficiency
of the TLK1 ORF clones was confirmed by the presence of GFP signals
were obtained via fluorescence imaging (Fig. 3B). TLK1 overexpression
significantly increased cell viability (Fig. 3C). In Fig. 3D, clonogenicity potential was
identified to be increased in the pLOC-TLK1 group. Staining of
these colonies was performed using 0.5% crystal violet (w/v) in 25%
methanol. In addition, the overexpression of TLK1 decreased the
levels of apoptosis, as demonstrated by a decrease in Annexin V
signal when incubated with 2.5 mM TMZ (Fig. 3E). BrdU signal levels were
increased, suggesting increased DNA synthesis and increased cell
proliferation (Fig. 3F); this
observation was also supported by increases in percentages of cells
in the S-phase, as demonstrated in Fig. S5B.
Chemosensitisation effects of TLK1
inhibition
TLK1 knockdown increased sensitivity of GBM
cells to sublethal dose of TMZ at 72 h (Fig. 4A). A significant sensitisation
effect was observed (P<0.05), as demonstrated by a decrease in
the colony growth of both cell lines after 14 days incubation
following TMZ and si-TLK1 treatment in U87MG cells (Fig. 4B and C). This was due to the
decrease in the cellular proliferation through the sublethal dose
of TMZ and apoptosis regulation during TLK1 knockdown.
Microarray gene expression analysis
identifies downstream pathways of TLK1
It is important to characterize the genome-wide
effect of TLK1 inhibition and to identify cancer cell
phenotypes targeted by TLK1 modulation. Microarray analysis
identified 527 genes with at least 1.5-fold change (P<0.05),
with 300 upregulated genes and 227 downregulated genes, and 2,632
genes with 1.1-fold change (P<0.05). The cell cycle-associated
pathways, including cell cycle, DNA replication and G1-S cell cycle
control, were significantly sensitive to TLK1 knockdown
(Tables I and SI). Fig.
5A demonstrates the network gene interaction between
differentially expressed genes identified from the TLK1 knockdown
microarray experiment with its potential protein-protein
interactome, obtained from network analysis using NetworkAnalyst
3.0. Additional information on nodes and edges are presented in
Table SII. Fig. 5B describes the TLK1 hypothetical
pathway involved in GBM. We hypothesized that TLK1 may interact
with CDC-42 and RAC2 to activate TLK1 downstream signalling for GBM
cells to survive.
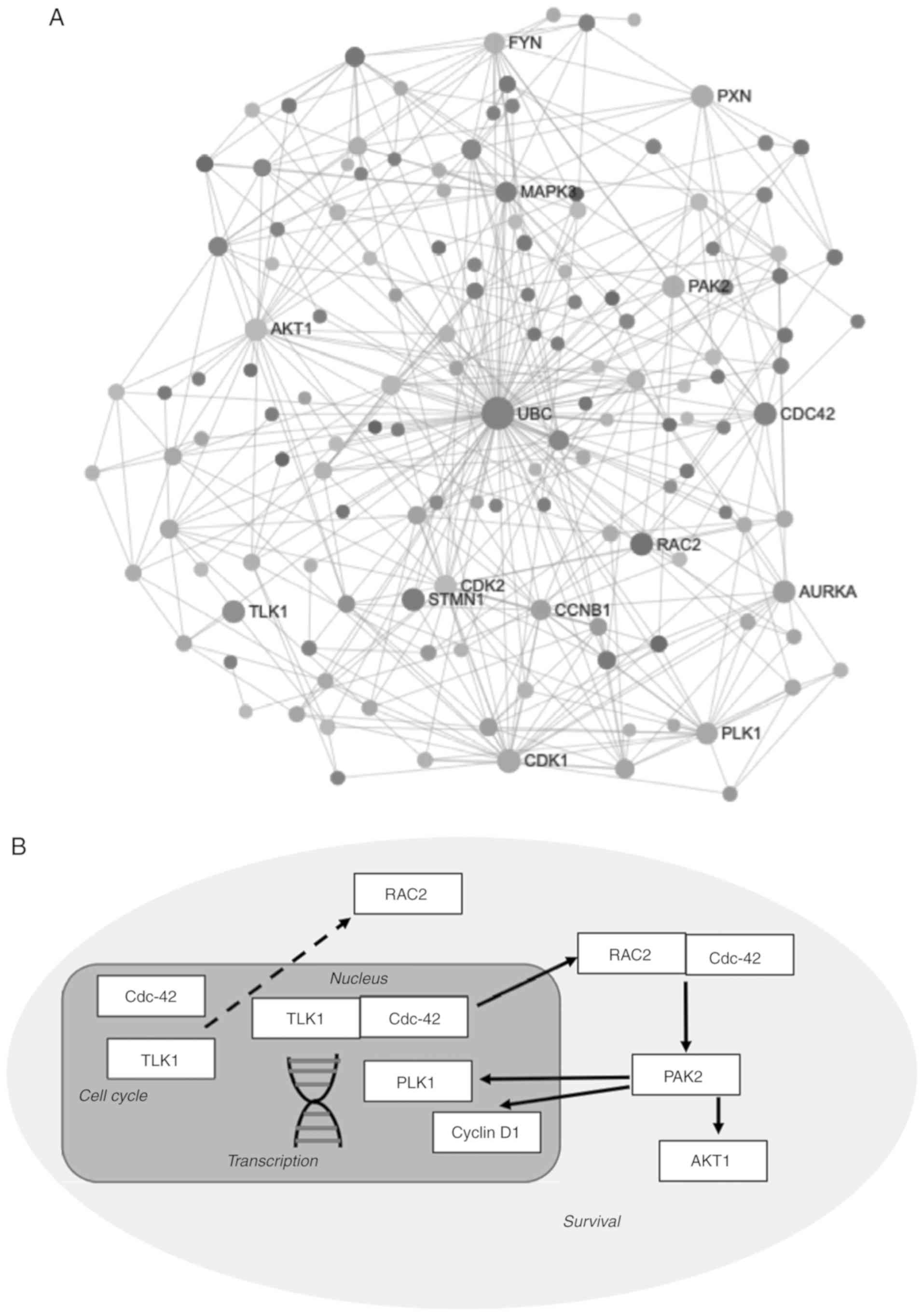 | Figure 5Analysis of TLK1 interactome and
hypothetical pathway. (A) TLK1 network identified from network
analysis identifies 1 subnetwork consists of 626 nodes and 1,831
edges. Nodes were then filtered based on enriched KEGG pathways
P<0.01. (B) Hypothetical model of the TLK1 RAC2 CDC42 PAK2
regulation pathways in GBM. CDC42 is localized within the nucleus
together with TLK1. CDC42, being a small Ras GTP binding protein,
may bind to TLK1 and be a substrate for TLK1. Activated CDC42 in
the nucleus binds or interacts with activated TLK1 protein kinase
to emit signals to the other CDC42 binding protein that is
localized in the cytoplasm, to induce binding with activated RAC2.
In other cases, activated CDC42 may bind to activate TLK1 and later
be released into the cytoplasm to emit signal while binding with
RAC2. In a homeostatic environment, CDC42 and RAC2 will bind to p21
protein, which becomes activated PAK2 and emits signals for other
downstream molecules. PAK2 signals AKT1 for the regulation of
survival pathways. PAK2 also regulates cell cycle activity with
PLK1 and cyclin D1 for initiation of transcriptional machinery.
TLK1, tousled like kinase 1; CDC42, cell division cycle 42; RAC2,
Rac family small GTPase 2; PAK2, p21 (RAC1) activated kinase 2;
AKT1, AKT serine/threonine kinase 1. |
 | Table IFunctional pathway analysis
identified statistically significant pathways affected by
TLK1 knockdown in U87MG cells. |
Table I
Functional pathway analysis
identified statistically significant pathways affected by
TLK1 knockdown in U87MG cells.
| Pathway name | Gene | Statistics |
|---|
| Cell cycle | 39 |
2.70×10−31 |
| DNA
replication | 27 |
7.26×10−31 |
| G1 to S cell cycle
control | 26 |
1.78×10−20 |
| DNA damage
response | 17 |
5.30×10−11 |
| TGF β signaling
pathway | 19 |
1.53×10−7 |
| TSH signaling
pathway | 10 | 0.0001 |
| Wnt signaling
pathway | 8 | 0.0003 |
| Focal adhesion | 16 | 0.0003 |
| Parkin-ubiquitin
proteosomal system pathway | 9 | 0.0006 |
| TOR signaling | 6 | 0.0013 |
| Wnt signaling
pathway and pluripotency | 10 | 0.0013 |
| Interleukin-11
signaling pathway | 7 | 0.0013 |
| Integrin-mediated
cell adhesion | 10 | 0.0013 |
| Signaling pathways
in glioblastoma | 9 | 0.0016 |
| Sphingolipid
metabolism | 5 | 0.0016 |
| Mismatch
repair | 4 | 0.0016 |
| TNF-α signaling
pathway | 10 | 0.0019 |
| Apoptosis | 9 | 0.0026 |
| Regulation of actin
cytoskeleton | 12 | 0.0035 |
| MAPK signaling
pathway | 12 | 0.0051 |
Role of TLK1 in TP53, AKT and MAPK
pathway signalling
Further validation was performed by gene knockdown
in GBM cells with shRNA. Data showing that shRNAs were able to
decrease the expression level of TLK1 in the tissue targeted are
presented in Fig. S6. TP53, AKT
and MAPK have key genes in GBM pathogenesis (29). To validate the downstream
functional activation of TLK1, the protein markers TP53, AKT1/2/3,
p70s6k and extracellular signal-regulated kinase (ERK)1/2/3 were
examined using ELISA-based assays. In the TLK1-depleted U87MG
cells, the ratio of phosphorylated TP53 to total TP53 protein was
significantly increased in the sh-TLK1 GBM cells. However, in the
TLK1-overexpressing U87MG cells, the phosphorylated to total TP53
protein signal ratio was decreased significantly. The p70s6k
protein signal was significantly decreased in the sh-TLK1 U87MG
cells but not in the LN18 cells, suggesting that the inhibition of
p70s6k occurred in a TP53-dependant manner. Notably, the protein
signals were increased in both TLK1 knockdown cell lines (Fig. S7).
TLK1 knockdown in normal human astrocytes
functional analysis
The most important aspect of determining a specific
cancer target is its ability to target the specific cancer cells
but not their neighbouring normal cells. Knockdown of TLK1 did not
cause any significant changes in NHA viability, morphology and
apoptosis signals (Fig. S8).
These results suggest that TLK1 specifically targets cancer cell
lines but not normal astrocytes.
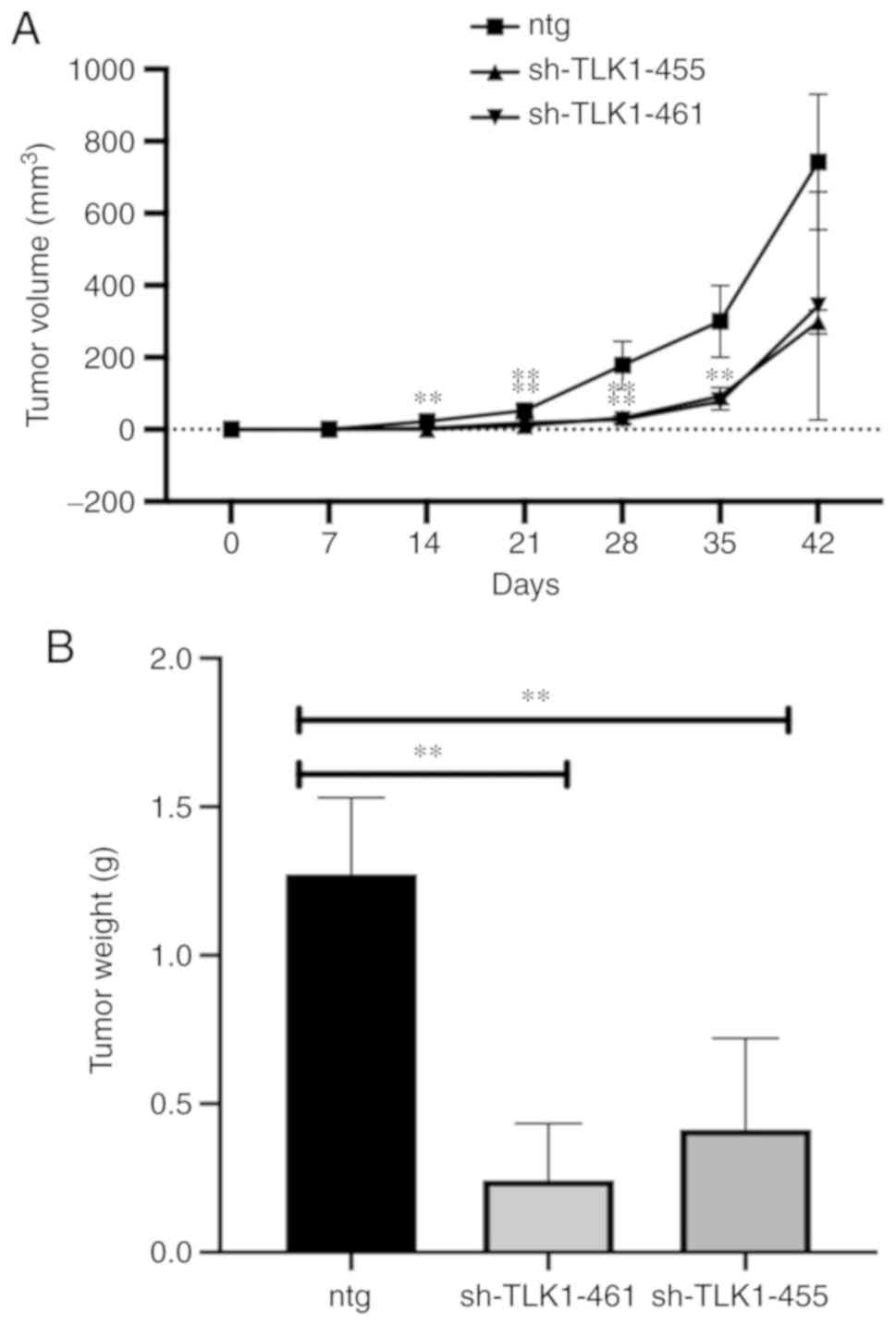
TLK1 knockdown decreases GBM tumour
growth in mouse xenografts
Tumour growth was significantly decreased in mice
transplanted subcutaneously with U87MG-sh-TLK1-455 or sh-TLK1-461
cells, as observed on days 21, 28 and 35 (P<0.05). However, on
day 42, the results were not significant (Fig. 6A). On day 47, the mice were
sacrificed, and the tumour weights were recorded. The mean tumour
weights of the U87MG-sh-455 and U87MG-sh-461 groups were
significantly decreased compared to the U87MG-sh-NTG cells
(P<0.05; Fig. 6B).
Discussion
Identification of novel targets by
integration of in silico analysis and functional genomics
approach
Previous studies have identified several signalling
pathways involved in GBM including the p53, retinoblastoma protein,
PI3K-PTEN-AKT-mTOR and RAS-ERK pathways (30). In the present study, 2 different
GBM cell lines harbouring different mutations were used; LN18 with
a TP53 mutation and wild type PTEN and U87MG cells with PTEN
mutation and wild-type TP53, which were constructed to mimic common
mutations affecting primary GBM. The high-throughput RNAi
loss-of-function screen on these 2 different GBM cell lines
identified four common targets: CDC2, FLT3,
ALPK1 and TLK1. These genes are overexpressed in
gliomas, particularly GBM (14).
CDC2 or CDK1 is a key player in cell cycle
regulation and highly conserved protein that functions as a
serine/threonine kinase. Overexpression of CDC2 promotes
oncogenesis and progression of human gliomas and CDC2 knockdown
decreases cell proliferation by causing an increase in the number
of G2M cell cycle arrest (31).
ALPK1 comes from the family atypical kinases. The α
kinase family has been demonstrated to be mutated in various types
of cancer (32). ALPK1 has been
implicated in epithelial cell polarity and exocytic vesicular
transport towards the apical plasma membrane (33). ALPK1 is located on Golgi-derived
vesicles, where it phosphorylates myosin IA, an apical vesicle
transport motor protein that regulates the delivery of vesicles to
the plasma membrane (33).
FLT3, a class 3 RTK, serves an important role in the
regulation of normal haematopoiesis. It was identified in
relatively high abundance in astrocytic tumours but without
detection of any activating FLT3 mutations involved (34). Activating mutations in FLT3,
particularly involving internal tandem duplications in the juxta
membrane domain, are detected in ~30% of adult and 15% of childhood
AMLs (35) suggesting that the
types of FLT3 abnormalities in GBM differ from leukaemia. Bleeker
et al (36) identified
that FLT3 was one of the frequent genes that contained somatic
mutations in GBM. Overexpression of FLT3 ligand (FLT3LG) in GBM
cells may provide a positive advantage in activating the
anti-tumour immune response (37). King et al (38) indicated that combination therapy
with adenoviral FLT3LG and adenoviral thymidine kinase therapy had
successfully eradicated multi-focal brain tumour disease in a
syngeneic, intracranial GBM model (38).
In the present study, less commonly reported
kinase-associated genes were also identified as positive hits as
well-established kinases such as EGFR, platelet derived
growth factor receptor and mitogen-activated protein kinase 3 were
intentionally excluded from the RNAi screening during the candidate
target selection, as the major aim was to identify novel kinase
targets in GBM. Revalidation of these 4 candidate targets revealed
that knockdown of TLK1 consistently resulted in decreased cell
viability. In addition, the role of TLK1 in GBM is not fully
understood. TLK1 is a serine threonine kinase (STK) homologous to
the Tousled gene in Arabidopsis thaliana, which is required
for normal flower and leaf development (39,40). There are 2 types of TLK: TLK1 and
TLK2, which are highly conserved in mammals (41). TLK1 activity is cell
cycle-dependent and regulates chromatin dynamics, including DNA
replication, DNA repair, transcription and chromosome segregation
(42,43). TLK1 is a potential novel
therapeutic target in GBM, as this gene may control GBM cell growth
and survival. TLK is absent in yeast, suggesting that TLK homologs
are present only in higher forms of eukaryotes (41). A massive screening of 125 STKs
identified frequently altered STKs in different tumour types.
However, due to the lack of glioblastoma samples, the study was
unable to identify the significance of TLK1 in glioblastoma
(44).
TLK1-regulated mechanisms of GBM cell
survival, proliferation and apoptosis
Knockdown of TLK1 in GBM cells significantly
decreased cell viability, clonogenicity potential and DNA
synthesis. In addition, TLK-1 inhibition also induced downstream
activation of caspase-3 and caspase-7 intrinsic apoptosis pathway.
Similar results were also reported by Carrera et al
(41), whereby TLK1
activity was decreased in a loss-of-function mutation induced in
Drosophila, causing nuclear division arrest at interphase
accompanied by apoptosis. Alternatively, it may activate a
different novel form of programmed cell death that is independent
of caspase activation (45).
Notably, NHA cells were not affected by TLK1 knockdown,
suggesting that the effects of TLK1 inhibition maybe
specific to TLK1-overexpressing cancer cells and the growth
and survival of the adjacent normal cells will not be disrupted.
The results of the present study were in concordance with those
reported by Segura-Bayona et al, in which a decreased colony
formation in TLK1-knockdown cells, particularly in the mouse
embryonic fibroblasts cell lines, was observed, suggesting that
TLK1 is required to maintain genomic instability and integrity as
well as cellular viability (46).
A limitation of the present study was the inability to check for
TLK1 protein expression due to time and funding limitations,
although the efficiency of TLK1 knockdown at the mRNA level in NHA
was examined. Also, a limitation of the present study was that cell
viability was not examined at different time points (24, 48 or 96
h), which would have been necessary to gain insight in the
proliferative capability of GBM cells.
TLK1 serves an important role in regulating DNA
replication, mitosis and cytokinesis (47,48). TLK1 knockdown inhibits DNA
replication and this was supported by the results of the present
study. However, the mechanisms of inhibition in U87MG and LN18
cells differ slightly. TLK1 knockdown in U87MG cells
decreased the number of G0/G1 cells, slightly increased the G2/M
cells, and increased S-phase arrest, whilst LN18 cells at G0/G1 and
G2/M were decreased but the S-phase arrest increased significantly
(49). The mechanism of S-phase
arrest and decrease in G0/G1 cells concurs with the microarray
results of the present study, which involved downregulation of
proliferative genes, namely CCND1, and the upregulation of the
tumor suppressor gene transforming growth factor β1. G2/M was
observed to be slightly increased in U87MG cells with wild-type
TP53 expression, suggesting that these cells undergo normal DNA
repair (50). However, with the
downregulation of the cell division cycle (CDC)25A and CDC25B
genes, it may suggest that these cells cannot undergo further
mitosis, but alternatively undergo apoptosis. An increase in the
number of G2/M cells may provide evidence of G2/M phase cell cycle
arrest, supported by molecular data (50). These results were observed in a
fibroblast cell line (BSF), in which TLK1 knockdown depleted
spindle formation and chromosome segregation (48), but not in fibroblasts with
TLK2 knockdown (47),
thereby disrupting cellular mitotic activity. Wang et al
(51) suggested that TP53
status affected the patients' response towards DNA topoisomerase
inhibitor CPT-11. They also demonstrated that in a primary culture
of glioblastoma cells with wild-type TP53 expression, CDC2
phosphorylation was disrupted in a TP53-dependent manner.
It was also demonstrated that TLK1
overexpression in GBM cells increased proliferation and resistance
to apoptosis. This is in line with previous data, where TLK1
overexpression in breast and prostate cancer resulted in resistance
to radio-therapy (52). By
contrast, Zhang et al (53) observed that TLK1 overexpression in
Drosophila eyes induced cell death and pigmentation loss,
suggesting that TLK1 may serve a different role in eye
development.
Regulation of downstream TLK1 survival
pathways
Different key kinases were implicated in the U87MG
and LN18 cells. In wild-type TP53 and mutant PTEN
U87MG cells, TLK1 knockdown significantly downregulated and
deactivated p70s6k (Thr389). The 70-kDa ribosomal P70S6 kinase is a
downstream target of the PI3K/mTOR pathway that regulates cell
growth and G1 cell cycle progression by inducing the cellular
translational machinery (54).
P70s6k is commonly upregulated in breast cancer and GBM (55,56). Harada et al (57) demonstrated that the function of
p70s6k was not limited to just protein synthesis and growth
maintenance. The IGF-1 cytokine signals p70s6k to control cell
survival by inhibiting the proapoptotic BAD through
phosphorylation. Therefore, p70s6k deficiency activates the
proapoptotic BAD, and vice versa (56).
The levels of phosphorylated TP53 was increased,
confirming that the mechanism of apoptosis occurs via a
TP53-dependent pathway. Simultaneously, CDC42 (Rho GTPase)
activation following TLK1 knockdown indicated activation of
the apoptosis signalling pathway. Volarević and Thomas (58) suggested that activation of the
TP53-dependent apoptosis activation does not only occur by
BAX induction, but also via upregulation of the activation
of the membrane-bound CDC42 in cancer cells with wild-type
TP53 (58). By contrast,
Tu and Cerione (59) suggested
that CDC42 is actually a substrate for caspase-3 and caspase-7,
which is associated with Fas-induced apoptosis via the NF-κB
pathway. Decreased CDC42 activity in TLK1-overexpressing cells
increased cell growth, was in concordant with the results reported
by Warner et al (60).
Chemo sensitization of GBM cells by TLK1
knockdown
Activation of the O-6-methylguanine-DNA
methyltransferase (MGMT) repair enzyme, which repairs TMZ-generated
O-6-methylguanine-DNA adducts, causes TMZ resistance in GBM
cells (61). The presence of MGMT
inherently activates base excision repair and mismatch repair
(62). A synthetic lethality
screen identified genes such as MRP1 and WEE1 that
increase chemo sensitization towards TMZ (63,64). In the present study, TLK1
was identified to be an important target for sensitizing GBM cells
to TMZ. TLK1 is also involved in DNA damage and DNA repair,
whereby TLK1 kinase activity decreases in response to genotoxic
stress, such as ionizing radiation or hydroxyurea treatment
(65,66).
Takayama et al (67) demonstrated that TLK1
inhibition sensitized cholangiocarcinoma cells to the
platinum-based cytotoxic compound cisplatin (67). Ronald et al (52) indicated that TLK1 knockdown
significantly sensitized DU45 prostate cancer cells to radiation.
In the present study, knockdown of TLK1 sensitized GBM cells
to TMZ, in particular the U87MG cells harbouring PTEN
mutations but with wild-type TP53 expression. TMZ induces
DNA damage by forming DNA adducts and promotes G2 and S-phase
arrest (68). In glioma cells,
wild-type TP53 expression has the capability of sensitizing
the GBM cells to anti-cancer drugs, and this supports the results
of the present study demonstrating that knockdown of TLK1
sensitized U87MG cells to TMZ, which is consistent with other
studies (69-71). In contrast, GBM cells with mutant
TP53 are resistant to TMZ (72). Simultaneous inhibition of
TLK1 may cause global and enhanced cell cycle arrest at both
G2-M and S phases, synergizing the effects of TMZ on GBM cell
growth inhibition. However, not all chemotherapeutic agents are
suitable for TLK1 sensitization (67).
TLK1 inhibition decreases GBM growth in a
subcutaneous xenograft mouse model
The functional role of TLK1 in regulating GBM
growth in female BALB/c-nu mice was investigated. The
subcutaneous xenograft growth rate of TLK1-knockdown GBM cells was
significantly decreased. Ronald et al (73) performed a PC-3 xenograft model
study in SCID/bg mice to examine the in vivo effects of a
TLK1 inhibitor, the anti-psychotic thioridazine. Notably, oral
administration of thioridazine significantly decreased the rate of
tumour growth. Therefore, future work may involve combining
thioridazine and TMZ in GBM models to examine its effectiveness and
efficacy.
In conclusion, our in vitro functional
analyses suggest TLK1 serves important roles in GBM cell survival,
cell cycle, proliferation and apoptosis. The in vivo animal
model also demonstrated the role of TLK1 in tumour growth.
Therefore, TLK1 may therefore serve as a promising potential target
in GBM therapy.
Supplementary Data
Acknowledgments
The authors would like to thank Professor Rodney J
Scott from University of Newcastle, Australia for his critical
comments in this project.
Funding
The present study was funded by a Higher
Institution Centre of Excellence (HICoE) research grant (grant no.
JJ-015-2011), Ministry of Higher Education, Malaysia.
Availability of data and materials
All data generated or analysed during this study
are included in this published article.
Authors' contributions
KI conceived and designed the experiments,
performed the experiments, analysed the data, contributed
reagents/materials/analysis tools, wrote the manuscript and
prepared figures and tables. NAAM performed the microarray
experiments and reviewed drafts of the manuscript. RH conceived and
designed the experiments, and reviewed drafts of the manuscript. RJ
was the principal investigator, and was involved in data analysis
and reviewing of manuscript drafts. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
The animal experiments were approved by the UKM
Animal Ethics Committee (approval no. UMBI/2014/ROSLAN/24-
SEPT./607-SEPT.-2014-DEC.-2014).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Johnson DR and O'Neill BP: Glioblastoma
survival in the United States before and during the temozolomide
era. J Neurooncol. 107:359–364. 2012. View Article : Google Scholar
|
|
2
|
Paw I, Carpenter RC, Watabe K, Debinski W
and Lo HW: Mechanisms regulating glioma invasion. Cancer Lett.
362:1–7. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Petropoulos C, Guichet P-O, Masliantsev K,
Wager M and Karayan-Tapon L: Functional invadopodia formed in
glioblastoma stem cells are important regulators of tumor
angiogenesis. Oncotarget. 9:20640–20657. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Micallef J, Taccone M, Mukherjee J, Croul
S, Busby J, Moran MF and Guha A: Epidermal growth factor receptor
variant III-induced glioma invasion is mediated through
myristoylated alanine-rich protein kinase C substrate
overexpression. Cancer Res. 69:7548–7556. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Muller PAJ and Vousden KH: p53 mutations
in cancer. Nat Cell Biol. 15:2–8. 2013. View Article : Google Scholar
|
|
7
|
Kubiatowski T, Jang T, Lachyankar MB,
Salmonsen R, Nabi RR, Quesenberry PJ, Litofsky NS, Ross AH and
Recht LD: Association of increased phosphatidylinositol 3-kinase
signaling with increased invasiveness and gelatinase activity in
malignant gliomas. J Neurosurg. 95:480–488. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brandes AA, Franceschi E, Tosoni A, Hegi
ME and Stupp R: Epidermal growth factor receptor inhibitors in
neuro-oncology: Hopes and disappointments. Clin Cancer Res.
14:957–960. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vivanco I, Robins HI, Rohle D, Campos C,
Grommes C, Nghiemphu PL, Kubek S, Oldrini B, Chheda MG, Yannuzzi N,
et al: Differential sensitivity of glioma-versus lung
cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer
Discov. 2:458–471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mrugala MM and Chamberlain MC: Mechanisms
of disease: Temozolomide and glioblastoma-look to the future. Nat
Clin Pract Oncol. 5:476–486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Foukas LC, Berenjeno IM, Gray A, Khwaja A
and Vanhaesebroeck B: Activity of any class IA PI3K isoform can
sustain cell proliferation and survival. Proc Natl Acad Sci USA.
107:11381–11386. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yuan TL and Cantley LC: PI3K pathway
alterations in cancer: Variations on a theme. Oncogene.
27:5497–5510. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Faes S and Dormond O: PI3K and AKT:
Unfaithful partners in cancer. Int J Mol Sci. 16:21138–21152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rhodes DR, Yu J, Shanker K, Deshpande N,
Varambally R, Ghosh D, Barrette T, Pandey A and Chinnaiyan AM:
ONCOMINE: A cancer microarray database and integrated data-mining
platform. Neoplasia. 6:1–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bredel M, Bredel C, Juric D, Harsh GR,
Vogel H, Recht LD and Sikic BI: High-resolution genome-wide mapping
of genetic alterations in human glial brain tumors. Cancer Res.
65:4088–4096. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liang Y, Diehn M, Watson N, Bollen AW,
Aldape KD, Nicholas MK, Lamborn KR, Berger MS, Botstein D, Brown PO
and Israel MA: Gene expression profiling reveals molecularly and
clinically distinct subtypes of glioblastoma multiforme. Proc Natl
Acad Sci USA. 102:5814–5819. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shai R, Shi T, Kremen TJ, Horvath S, Liau
LM, Cloughesy TF, Mischel PS and Nelson S: Gene expression
profiling identifies molecular subtypes of gliomas. Oncogene.
22:4918–4923. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee J, Kotliarova S, Kotliarov Y, Li A, Su
Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al:
Tumor stem cells derived from glioblastomas cultured in bFGF and
EGF more closely mirror the phenotype and genotype of primary
tumors than do serum-cultured cell lines. Cancer Cell. 9:391–403.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun L, Hui AM, Su Q, Vortmeyer A,
Kotliarov Y, Pastorino S, Passaniti A, Menon J, Walling J, Bailey
R, et al: Neuronal and glioma-derived stem cell factor induces
angiogenesis within the brain. Cancer Cell. 9:287–300. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Manning G, Whyte DB, Martinez R, Hunter T
and Sudarsanam S: The protein kinase complement of the human
genome. Science. 298:1912–1934. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Birmingham A, Selfors LM, Forster T,
Wrobel D, Kennedy CJ, Shanks E, Santoyo-Lopez J, Dunican DJ, Long
A, Kelleher D, et al: Statistical methods for analysis of
high-throughput RNA inter-ference screens. Nat Methods. 6:569–575.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chung N, Zhang XD, Kreamer A, Locco L,
Kuan PF, Bartz S, Linsley PS, Ferrer M and Strulovici B: Median
absolute deviation to improve hit selection for genome-scale RNAi
screens. Biomol Screen. 13:149–158. 2008. View Article : Google Scholar
|
|
23
|
Zhang XD, Yang XC, Chung N, Gates A, Stec
E, Kunapuli P, Holder DJ, Ferrer M and Espeseth AS: Robust
statistical methods for hit selection in RNA interference
high-throughput screening experiments. Pharmacogenomics. 7:299–309.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Wang J, Duncan D, Shi Z and Zhang B:
WEB-based GEne SeT analysis toolkit (WebGestalt): Update 2013.
Nucleic Acids Res. 41:W77–W83. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou G, Soufan O, Ewald J, Hancock REW,
Basu N and Xia J: NetworkAnalyst 3.0: A visual analytics platform
for comprehensive gene expression profiling and meta-analysis.
Nucleic Acids Res. 47:234–241. 2019. View Article : Google Scholar
|
|
27
|
Geissmann Q: OpenCFU, a new free and
open-source software to count cell colonies and other circular
objects. PLoS One. 8:e540722013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Howard-Jones N: A CIOMS ethical code for
animal experimentation. WHO Chron. 39:51–56. 1985.PubMed/NCBI
|
|
29
|
Crespo I, Vital AL, Gonzalez-Tablas M,
Patino Mdel C, Otero A, Lopes MC, de Oliveira C, Domingues P, Orfao
A, Tabernero MD, et al: Molecular and genomic alterations in
glioblastoma multiforme. Am J Pathol. 185:1820–1833. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cancer Genome Atlas Research Network:
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen H, Huang Q, Dong J, Zhai DZ, Wang AD
and Lan Q: Overexpression of CDC2/CyclinB1 in gliomas, and CDC2
depletion inhibits proliferation of human glioma cells in vitro and
in vivo. BMC Cancer. 8:292008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Greenman C, Stephens P, Smith R, Dalgliesh
GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C,
et al: Patterns of somatic mutation in human cancer genomes.
Nature. 446:153–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Middelbeek J, Clark K, Venselaar H, Huynen
MA and van Leeuwen F: The alpha-kinase family : An exceptional
branch on the protein kinase tree. Cell Mol Life Sci. 67:875–890.
2010. View Article : Google Scholar
|
|
34
|
Eßbach C, Andrae N, Pachow D, Warnke JP,
Wilisch-Neumann A, Kirches E and Mawrin C: Abundance of Flt3 and
its ligand in astrocytic tumors. Onco Targets Ther. 6:555–561.
2013.
|
|
35
|
Kottaridis PD, Gale RE, Frew ME, Harrison
G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett
AK, et al: The presence of a FLT3 internal tandem duplication in
patients with acute myeloid leukemia (AML) adds important
prognostic information to cytogenetic risk group and response to
the first cycle of chemotherapy: Analysis of 854 patients from the
United Kingdom Medical Research Council AML 10 and 12 trials.
Blood. 98:1752–1759. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bleeker FE, Lamba S, Zanon C, Molenaar RJ,
Hulsebos TJ, Troost D, van Tilborg AA, Vandertop WP, Leenstra S,
van Noorden CJ and Bardelli A: Mutational profiling of kinases in
glioblastoma. BMC Cancer. 14:7182014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ali S, King G, Curtin J, Candolfi M, Xiong
W, Liu C, Puntel M, Cheng Q, Prieto J, Ribas A, et al: Combined
immunostimulation and conditional cytotoxic gene therapy provide
long-term survival in a large glioma model. Cancer Res.
65:7194–7204. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
King GD, Muhammad AK, Curtin JF, Barcia C,
Puntel M, Liu C, Honig SB, Candolfi M, Mondkar S, Lowenstein PR and
Castro MG: Flt3L and TK gene therapy eradicate multifocal glioma in
a syngeneic glioblastoma model. Neuro Oncol. 10:19–31. 2008.
View Article : Google Scholar
|
|
39
|
Roe JL, Rivin CJ, Sessions RA, Feldmann KA
and Zambryski PC: The Tousled gene in A. thaliana encodes a protein
kinase homolog that is required for leaf and flower development.
Cell. 75:939–950. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Roe JL, Nemhauser JL and Zambryski PC:
TOUSLED participates in apical tissue formation during gynoecium
development in Arabidopsis. Plant Cell. 9:335–353. 1997.PubMed/NCBI
|
|
41
|
Carrera P, Moshkin YM, Gronke S, Sillje
HHW, Nigg EA, Jackle H and Karch F: Tousled-like kinase functions
with the chromatin assembly pathway regulating nuclear divisions.
Genes Dev. 17:2578–2590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Silljé HH, Takahashi K, Tanaka K, Van
Houwe G and Nigg EA: Mammalian homologues of the plant Tousled gene
code for cell-cycle-regulated kinases with maximal activities
linked to ongoing DNA replication. EMBO J. 18:5691–5702. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
De Benedetti A: The tousled-like kinases
as guardians of genome integrity. ISRN Mol Biol. 2012:6275962012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Capra M, Nuciforo PG, Confalonieri S,
Quarto M, Bianchi M, Nebuloni M, Boldorini R, Pallotti F, Viale G,
Gishizky ML, et al: Frequent alterations in the expression of
serine/threonine kinases in human cancers. Cancer Res.
66:8147–8154. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xiang W, Zhang D, Montell DJ and Brill J:
Tousled-like kinase regulates cytokine-mediated communication
between cooperating cell types during collective border cell
migration. Mol Biol Cell. 27:12–19. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Segura-Bayona S, Knobel PA, González-Burón
H, Youssef SA, Peña-Blanco A, Coyaud É, López-Rovira T, Rein K,
Palenzuela L, Colombelli J, et al: Differential requirements for
Tousled-like kinases 1 and 2 in mammalian development. Cell Death
Differ. 24:1872–1885. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li Z, Gourguechon S and Wang CC:
Tousled-like kinase in a microbial eukaryote regulates spindle
assembly and S-phase progression by interacting with Aurora kinase
and chromatin assembly factors. J Cell Sci. 120:3883–3894. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li Z, Umeyama T and Wang CC: The
chromosomal passenger complex and a mitotic kinesin interact with
the tousled-like kinase in trypanosomes to regulate mitosis and
cytokines. PLoS One. 3:e38142008. View Article : Google Scholar
|
|
49
|
Zhao T, Sun Q, del Rincon SV, Lovato A,
Marques M and Witcher M: Gallotannin imposes S phase arrest in
breast cancer cells and suppresses the growth of triple-negative
tumors in vivo. PLoS One. 9:e928532014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
DiPaola RS: To arrest or not to
G2-M cell-cycle arrest. Clin Cancer Res. 8:3311–3314.
2002.PubMed/NCBI
|
|
51
|
Wang Y, Zhu S, Cloughesy TF, Liau LM and
Mischel PS: p53 disruption profoundly alters the response of human
glioblastoma cells to DNA topoisomerase I inhibition. Oncogene.
23:1283–1290. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ronald S, Sunavala-Dossabhoy G, Adams L,
Williams B and De Benedetti A: The expression of tousled kinases in
CaP cell lines and its relation to radiation response and DSB
repair. Prostate. 71:1367–1373. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhang Y, Cai R, Zhou R, Li Y and Liu L:
Tousled-like kinase mediated a new type of cell death pathway in
Drosophila. Cell Death Differ. 23:146–157. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Minami Y, Endo S, Okumura S, Shibukawa K,
Sasaki T and Ohsaki Y: Activating the prostaglandin
I2-IP signaling suppresses metastasis in lung cancer.
Cancer Res. 72:2012.
|
|
55
|
Heinonen H, Nieminen A, Saarela M,
Kallioniemi A, Klefström J, Hautaniemi S and Monni O: Deciphering
downstream gene targets of PI3K/mTOR/p70S6K pathway in breast
cancer. BMC Genomics. 9:3482008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Pelloski CE, Lin E, Zhang L, Yung WK,
Colman H, Liu JL, Woo SY, Heimberger AB, Suki D, Prados M, et al:
Prognostic associations of activated mitogen-activated protein
kinase and Akt pathways in glioblastoma. Clin Cancer Res.
12:3935–3942. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Harada H, Andersen JS, Mann M, Terada N
and Korsmeyer SJ: p70S6 kinase signals cell survival as well as
growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad
Sci USA. 98:9666–9670. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Volarević S and Thomas G: Role of S6
phosphorylation and S6 kinase in cell growth. Prog Nucleic Acid Res
Mol Biol. 65:101–127. 2001. View Article : Google Scholar
|
|
59
|
Tu S and Cerione RA: Cdc42 is a substrate
for caspases and influences Fas-induced apoptosis. J Biol Chem.
276:19656–19663. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Warner SJ, Yashiro H and Longmore GD: The
Cdc42/Par6/aPKC polarity complex regulates apoptosis-induced
compensatory proliferation in epithelia. Curr Biol. 20:677–686.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhang X, Zhang W, Cao WD, Cheng G and
Zhang YQ: Glioblastoma multiforme : Molecular characterization and
current treatment strategy (Review). Exp Ther Med. 3:9–14. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhang J, Stevens MFG and Bradshaw TD:
Temozolomide: Mechanisms of action, repair and resistance. Curr Mol
Pharmacol. 5:102–114. 2012. View Article : Google Scholar
|
|
63
|
Pokorny JL, Calligaris D, Gupta SK,
Iyekegbe DO Jr, Mueller D, Bakken KK, Carlson BL, Schroeder MA,
Evans DL, Lou Z, et al: The efficacy of the Wee1 inhibitor MK-1775
combined with temozolomide is limited by heterogeneous distribution
across the blood-brain barrier in glioblastoma. Clin Cancer Res.
21:1916–1924. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tivnan A, Zakaria Z, O'Leary C, Kögel D,
Pokorny JL, Sarkaria JN and Prehn JH: Inhibition of multidrug
resistance protein 1 (MRP1) improves chemotherapy drug response in
primary and recurrent glioblastoma multiforme. Front Neurosci.
9:2182015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Groth A, Lukas J, Nigg EA, Silljé HH,
Wernstedt C, Bartek J and Hansen K: Human Tousled like kinases are
targeted by an ATM- and Chk1-dependent DNA damage checkpoint. EMBO
J. 22:1676–1687. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Krause DR, Jonnalagadda JC, Gatei MH,
Sillje HH, Zhou BB, Nigg EA and Khanna K: Suppression of
Tousled-like kinase activity after DNA damage or replication block
requires ATM, NBS1 and Chk1. Oncogene. 22:5927–5937. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Takayama Y, Kokuryo T, Yokoyama Y, Ito S,
Nagino M, Hamaguchi M and Senga T: Silencing of Tousled-like kinase
1 sensitizes cholangiocarcinoma cells to cisplatin-induced
apoptosis. Cancer Lett. 296:27–34. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Cui B, Johnson SP, Bullock N, Ali-Osman F,
Bigner DD and Friedman HS: Decoupling of DNA damage response
signaling from DNA damages underlies temozolomide resistance in
glioblastoma cells. J Biomed Res. 24:424–435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hermisson M, Klumpp A, Wick W, Wischhusen
J, Nagel G, Roos W, Kaina B and Weller M: O6-methylguanine DNA
methyltransferase and p53 status predict temozolomide sensitivity
in human malignant glioma cells. J Neurochem. 96:766–776. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Martin S, Janouskova H and Dontenwill M:
Integrins and p53 pathways in glioblastoma resistance to
temozolomide. Front Oncol. 2:1572012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Roos WP, Batista LF, Naumann SC, Wick W,
Weller M, Menck CFM and Kaina B: Apoptosis in malignant glioma
cells triggered by the temozolomide-induced DNA lesion
O6-methylguanine. Oncogene. 26:186–197. 2007. View Article : Google Scholar
|
|
72
|
Blough MD, Beauchamp DC, Westgate MR,
Kelly JJ and Cairncross JG: Effect of aberrant p53 function on
temozolomide sensitivity of glioma cell lines and brain tumor
initiating cells from glioblastoma. J Neurooncol. 102:1–7. 2011.
View Article : Google Scholar
|
|
73
|
Ronald S, Awate S, Rath A, Carroll J,
Galiano F, Dwyer D, Kleiner-Hancock H, Mathis JM, Vigod S and De
Benedetti A: Phenothiazine inhibitors of TLKs affect double-strand
break repair and DNA damage response recovery and potentiate tumor
killing with radiomimetic therapy. Genes Cancer. 4:39–53. 2013.
View Article : Google Scholar : PubMed/NCBI
|