Introduction
Cardiovascular diseases, particularly coronary heart
disease (CHD), have a high mortality rate worldwide (1). Atherosclerosis (AS) is the primary
pathological basis of CHD. Endothelial function is considered to
serve as an excellent benchmark of underlying vascular health, as
it represents an orchestrated response to the numerous known and
unknown processes that contribute to the development, progression
and clinical presentation of AS (2). Oxidized low-density lipoprotein
(ox-LDL) can induce endothelial cell apoptosis (3), which promotes the occurrence and
development of AS. In this context, B-cell lymphoma extra-large
(Bcl-xL) plays a pivotal role in mediating cellular apoptosis by
maintaining the mitochondrial membrane potential and preventing the
release of pro-apoptotic enzymes, such as caspases, from the
mitochondria (4). Caspase-3 acts
as a regulatory hub to control the onset of cellular apoptosis and
plays a central role in activating further apoptotic enzymes.
Therefore, preventing cellular apoptosis of the endothelial lining
has become an attractive strategy for AS treatment, and drugs that
effectively inhibit endothelial cell apoptosis are considered to
have important clinical significance in future anti-AS
therapies.
MicroRNAs (miRNAs or miRs) are single-stranded,
short non-coding RNAs with an average length of 22-24 nucleotides
that regulate gene expression by complementary base-pairing with
their target mRNAs. Accumulating evidence indicates that miRNAs
play an active role in the development of AS, participating in all
steps, from initial plaque formation to plaque instability and
rupture, and regulate endothelial and immune function (5). In particular, miRNA-133a has been
proposed to play a central role in the development of CHD and AS
plaque stability (6,7). Several factors, including
hyperglycemia, hyperlipidemia and hyperhomocysteinemia, increase
the expression of miR-133a in endothelial cells, thereby promoting
vascular endothelial dysfunction. Importantly, the inhibition of
miR-133a targets the regulation of GTP cyclo-hydrolase-1 and
protects vascular endothelial function to ultimately prevent AS
(8). Despite its role in
endothelial cell function, the role of miR-133a in the regulation
of apoptosis remains unclear.
Salidroside (SAL; 2-4-hydroxyphenyl
ethyl-β-D-glucopyranoside) is the main active component of the
herb, Rhodiola, which has attracted substantial attention in
recent years owing to its significant anti-AS effect (9,10),
particu-larly with respect to its effects on preserving endothelial
cell function. SAL can protect the endothelium against oxidative
stress, inflammation and endoplasmic reticulum stress; inhibit
endothelial apoptosis; and delay endothelial senescence,
consequently preventing the occurrence and development of AS
(11). A recent study
demonstrated that SAL promoted the expression of the anti-apoptotic
protein, Bcl-xL, thereby inhibiting the induction of apoptosis
mediated by H2O2 oxidative stress (12). Of note, miR-133a specifically
regulates the expression of the Bcl-xL gene (13-15); however, it remains unclear as to
whether SAL affects the miR-133a expression levels to, in turn,
regulate the levels of anti-apoptotic proteins. Therefore, the aim
of the present study was to explore the regulation of endothelial
cell apoptosis by SAL through the miR-133a pathway and to provide
reference data for further studies on the mechanisms of AS that
could lead to the development of novel drug targets.
Materials and methods
Cells and cell culture
Cultured human coronary artery endothelial cells
(HCAECs; ScienCell Research Laboratories, Inc.) were seeded in
fibronectin-coated plates (BD Biosciences), and cultured in
endothelial cell medium (ScienCell Research Laboratories, Inc.)
supplemented with 5% fetal bovine serum and endothelial cell growth
supplement (ECGS) The cells were maintained at 37°C in a humidified
atmosphere with 5% CO2. Cells from passages 4-7 were
used in the experiments.
Cell treatment
HCAECs were seeded in 96-well plates and exposed to
various concentrations of ox-LDL (5, 15, 25, 50 and 75
µg/ml; Beijing Solarbio Science & Technology Co., Ltd.)
and SAL (0.5, 1, 5, 10, 50, 100, 500, 1,000 and 5,000 µM;
Nanjing Zelang Biotechnology Co., Ltd.) for 12 h or 24 h. HCAECs
were also seeded in 6-well plates and exposed to 25, 50 and 75
µg/ml ox-LDL for 12 and 24 h. For each experiment, the cells
were divided into 3 experimental groups: The control (untreated)
group, ox-LDL-treated group and ox-LDL + SAL-treated group.
Detection of cell viability
Cells were seeded in 96-well plates for 12 and 24 h,
and 20 µl/well of 3-[4,5-dimethylthi-azol-2-yl]-2,5-diphenyl
tetrazolium bromide (MTT; Beyotime Institute of Biotechnology,
Inc.) were added to each well. The plates were incubated in a cell
incubator at 37°C for 4 h. Following incubation, the blue-purple
crystals were dissolved in 150 µl of dimethyl sulfoxide The
optical density (OD) of the solution was read at 490 nm, and cell
viability was calculated according to the following formula: (OD
value of treatment group-OD value of blank group)/(OD value of
control group-OD value of blank group) ×100%.
Lactate dehydrogenase (LDH) release
assay
To evaluate the cytotoxic effects of SAL on HCAECs,
the level of extracellular LDH was measured using the LDH release
test kit (Beyotime Institute of Biotechnology, Inc.) according to
the manufacturer's recommendations. In brief, the cells were
transferred to a 96-well plate and treated with various
concentrations of SAL (5, 10, 50, 100, 500 and 10,000 µM)
for 12 and 24 h. The supernatants were harvested and centrifuged
for 5 min at 400 × g in a porous plate centrifuge (centrifugation
was carried out at room temperature). Subsequently, 120 µl
of the supernatant were mixed with 60 µl of LDH detection
solution in a new 96-well plate, incubated for 30 min at room
temperature, and the absorbance at 490 nm was read on an
enzyme-linked immunosorbent assay plate reader (Molecular Devices,
LLC).
miRNA screening
Bioinformatics screening was performed using miRBase
(http://www.mirbase.org) to confirm the miRNAs
targeting the Bcl-xL gene. miRNA expression levels were detected by
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) after the HCAECs were exposed to ox-LDL for 24 h, as
described below.
Cell transfection
The cells were sub-cultured in a 6-well plate and
grown to 60-70% confluency. The cells were then transfected with
the miR-133a inhibitor (diluted in Opti-MEM) or miR-133a mimics,
and their respective negative controls (NC; 10 µM stock)
(GenePharma) using Lipofectamine® RNAiMAX Reagent
(Thermo Fisher Scientific, Inc.). Pre-mixed transfection reagents
and RNA were mixed at a 1:1 ratio and incubated at room temperature
for 5 min in a 6-well plate. At 48 h following transfection, the
cells were exposed to ox-LDL (75 µg/ml), and treated with
SAL (100 µM), or ox-LDL (75 µg/ml) + SAL (100
µM) for 24 h.
Detection of cell apoptosis
To determine cell apoptotic rates, the Annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) double
staining kit (BD Biosciences) was used according to the
manufacturer's instructions. In brief, the cells were detached from
the culture flasks with 0.05% trypsin and washed twice with chilled
phosphate-buffered saline (PBS). The cells were then resuspended in
100 µl 1X binding buffer and stained with 5 µl
Annexin V-FITC, which stains apoptotic cells (early apoptotic Q2
and late apoptotic Q3 quadrant), and PI for 15 min at room
temperature in the dark. The samples were analyzed using a flow
cytometer (BD FACSCalibur, BD Biosciences) and the data were
analyzed using FlowJo 7.6 software.
RT-qPCR
Total RNA was extracted from the cells using
phenol:chloroform. In brief, the cells were washed twice with
chilled PBS, and RNAiso plus reagent (Takara Bio USA, Inc.) was
used to lyse the cells for 10 min on ice. Following incubation,
chloroform and isopropanol were added to the lysates, and the
precipitate was washed with 75% ethanol. The final pellet was
resuspended in DEPC water, and the concentration was measured using
an ultramicro spectrophotometer to confirm that the A260/A280 ratio
was between 1.8 and 2.0.
The expression levels of both miR-133a and Bcl-xL
were measured using reverse transcription kit (638313 and RR047A,
Takara Bio USA, Inc.) according to the manufacturer's
recommendations and detected by quantitative PCR (miRNA and mRNA)
with TB green staining. The relative expression was determined
using the 2−ΔΔCq method (16), and the expression of U6 and GAPDH
was used as internal control. The primers for miR-133a and Bcl-xL
detection were synthesized by Shanghai Sangon Biotech. The primer
sequences used for PCR were as follows: Bcl-xL forward, 5′-GCA TAT
CAG AGC TTT GAA CGG-3′ and reverse, 5′-GAA GGA GAA AAA GGC CAC AAT
G-3′; GAPDH forward, 5′-CAT GAG AAG TAT GAC AAC AGC CT-3′ and
reverse, 5′-AGT CCT TCC ACG ATA CCA AAG T-3′; miR-133a forward,
5′-GGC CTT TGG TCC CCT TCA A-3′; miR-133b forward, 5′-GTT TGG TCC
CCT CAA CCA GCT A-3′; let-7c-5p forward, 5′-GGG TGA GGT AGT AGG TTG
T-3′; let-7g-5p forward, 5′-CCG GCT GAG GTA GTA GTT TGT ACA GTT-3;
miR-491 forward, 5′-CTA GTG GGG AAC CCT TCC ATG AG-3′; miR-326
forward, 5′-ACC TCT GGG CCC TTC CTC-3′; and miR-608 forward, 5′-AGG
GGT GGT GTT GGG ACA GCT CCG T-3′. The post-primer and internal
reference gene, U6, for miRNA were included in the Takara kit (cat.
no. 638313); however, the company did not provide the sequences for
confidentiality reasons. For Bcl-xL, according to the instructions
of the kit, the RNA extracted from samples was incubated at 42°C
for 5 min, 37°C for 15 min, and 85°C for 5 sec for reverse
transcription. PCR amplification was carried out using a two-step
method; the first step involved pre-denaturation for 30 sec at
95°C, and the second step involved 40 cycles of amplification. The
reaction conditions were 95°C for 5 sec followed by 60°C for 34
sec. For miRNA reverse transcription, the extracted RNA was
incubated at 37°C for 1 h, followed by 85°C for 5 min. PCR
amplification was carried out using a two-step method; the first
step involved pre-denaturation for 10 sec at 95°C, and the second
step involved 40 cycles of amplification. The reaction conditions
were 95°C for 5 sec followed by 60°C for 20 sec.
Western blot analysis
The cells were collected in chilled RIPA lysis
buffer (Beyotime Institute of Biotechnology, Inc.) containing
protease inhibitor (PMSF and cocktail) to extract total proteins,
and the protein concentration was detected using a BCA kit
(Beyotime Institute of Biotechnology. Inc). Proteins (20 or 30 mg)
were separated by 12 or 15% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis, and then transferred to polyvinylidene
fluoride membranes. The membranes were then incubated with the
following antibodies overnight at 4°C with rocking: Rabbit
anti-Bcl-xL (1:2,000, cat. no. 2764T, Cell Signaling Technology,
Inc.), rabbit anti-caspase-3 (1:500, cat. no. 9662, Cell Signaling
Technology, Inc.) and mouse anti-β-actin (1:2,000, cat. no.
60008-1-Ig, ProteinTech Group, Inc.). After washing, the membranes
were incubated with the corresponding secondary antibodies (diluted
1:5,000, cat. no. 115-155-075 and 111-545-144, Jackson Laboratory)
at room temperature for 1.5 h. A chemiluminescence gel imaging
analysis system was used to develop the image, and quantification
was conducted using ImageJ software (https://imagej.nih.gov/ij/docs/index.html).
Statistical analysis
Each experiment was repeated at least 3 times. All
data are presented as the means ± standard deviation. All
statistical analyses were conducted using SPSS 25.0 software (SPSS,
Inc.), and data plotting was conducted in GraphPad Prism version 6.
Differences between 2 groups were compared by unpaired t-tests, and
differences among multiple groups were compared by one-way analysis
of variance followed by Tukey's test as appropriate. Differences
with P<0.05 were considered statistically significant.
Results
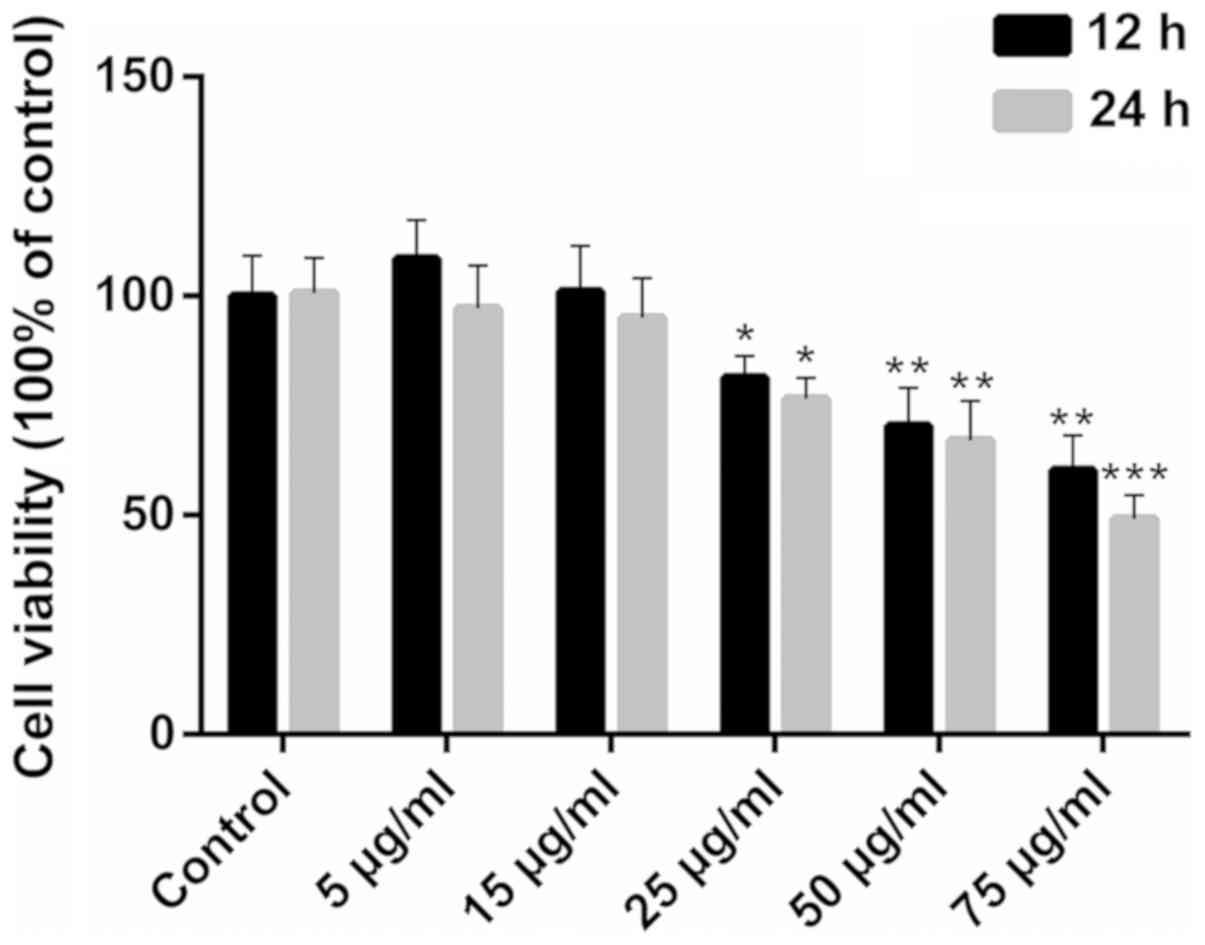
Ox-LDL inhibits the viability of
HCAECs
Exposure to ox-LDL impaired HCAEC viability only at
concentrations >25 µg/ml (Fig. 1; P<0.05). When exposure to 75
µg/ml ox-LDL was prolonged for 24 h, cell viability was
inhibited more significantly (P<0.001). Therefore, 25, 50 and 75
µg/ml were used as the screening concentrations for the
model of ox-LDL-induced apoptosis.
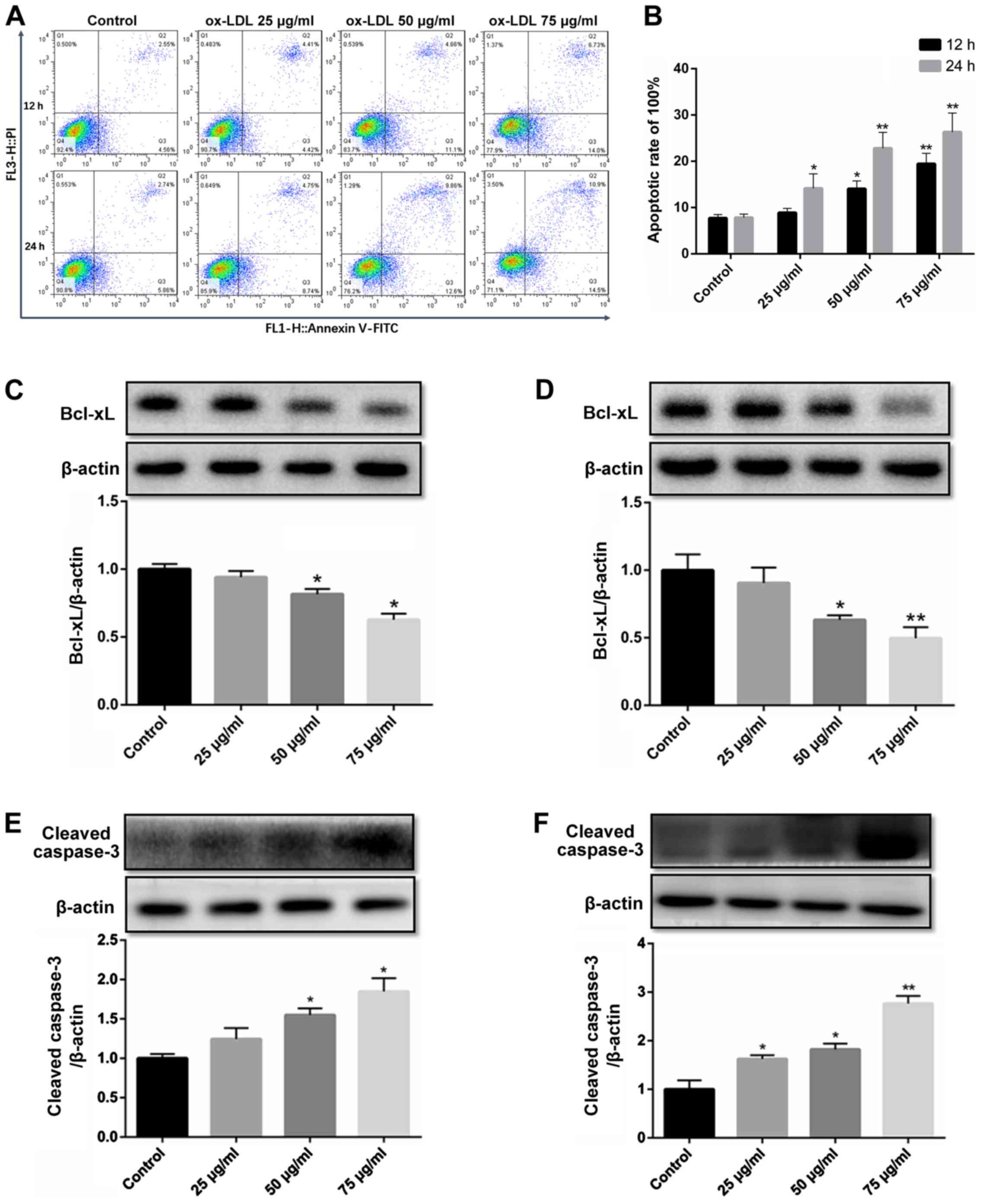
ox-LDL downregulates Bcl-xL expression
and activates caspase-3 to promote the apoptosis of HCAECs
ox-LDL-induced endothelial cell apoptosis is a
widely used model in the study of the mechanisms of AS (17,18). In the present study, flow
cytometry revealed that compared to the untreated control group,
apoptosis began to occur in the group exposed to 50 µg/ml
ox-LDL for 12 h, while the apoptotic rate increased in the group
exposed to 25 µg/ml for 24 h. In addition, the apoptotic
level of the HCAECs was significantly increased at an concentration
of 75 µg/ml (Fig. 2A and
B).
The expression level of the anti-apoptotic protein,
Bcl-xL, decreased gradually with the increasing ox-LDL
concentration (25-75 µg/ml) and treatment duration (12 and
24 h) (Fig. 2C and D).
The activation of caspase-3 by cleavage is the final
step in the induction of apoptosis. To confirm the induction of
apoptosis, HCAECs were exposed to ox-LDL for 12 and 24 h, and the
level of cleaved caspase-3 was determined. Similarly, the level of
cleaved caspase-3 increased at the concentration of 50 µg/ml
in the group exposed to ox-LDL for 12 h, and the activation of
caspase-3 in the group exposed ox-LDL (25 µg/ml) for 24 h,
while the concentration of 75 µg/ml ox-LDL significantly
increased the level of cleaved caspase-3 (Fig. 2E and F).
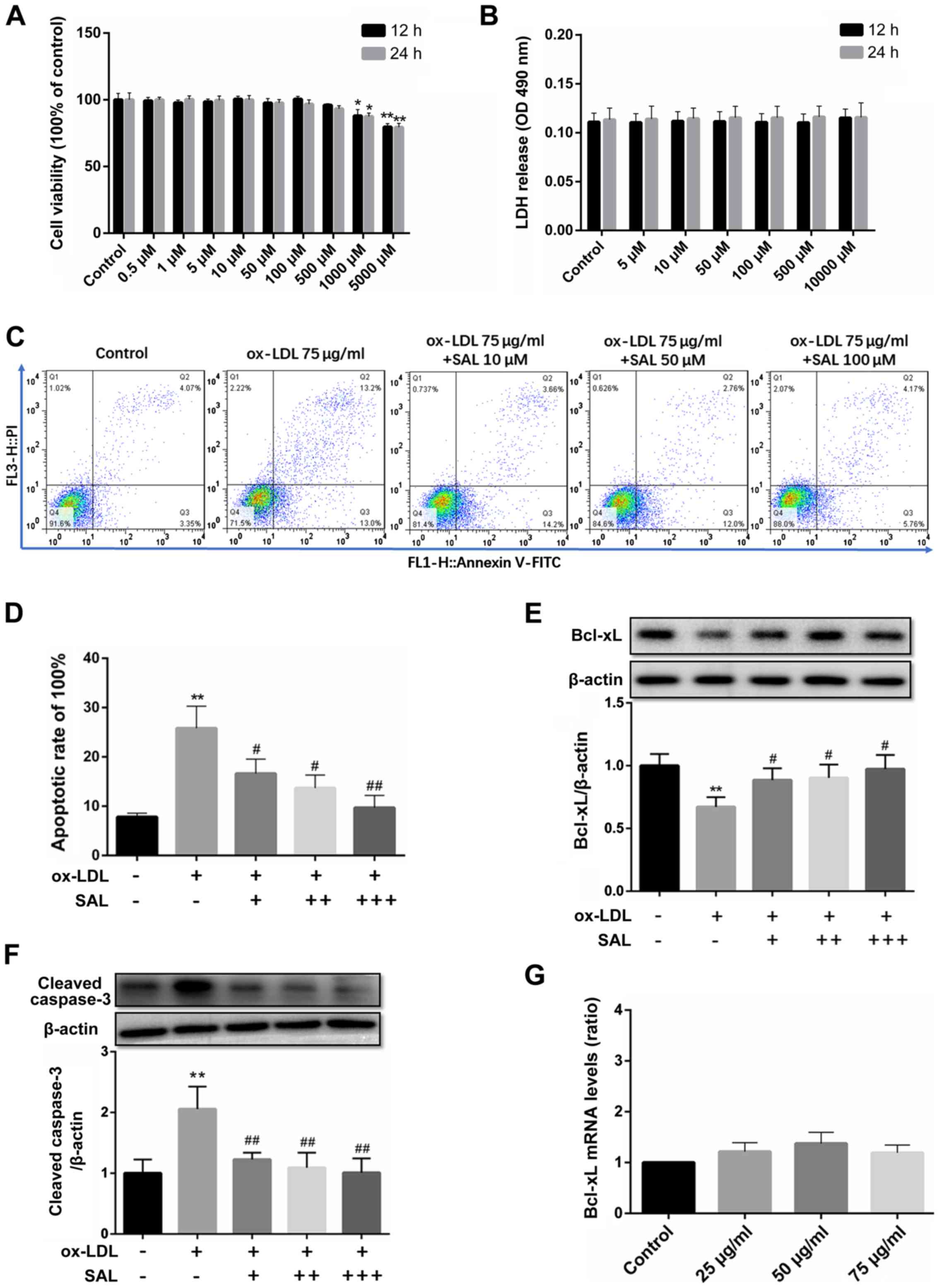
SAL protects the HCAECs from
ox-LDL-induced apoptosis
SAL inhibited cell viability only at concentrations
>1,000 µM (Fig. 3A;
P<0.05). Therefore, the low, medium and high concentrations of
SAL at 10, 50 and 100 µM, respectively were selected for use
in further experiments.
In addition to the MTT assay, LDH release assay was
used to evaluate the effects of ox-LDL and SAL on cell toxicity. An
increase in the SAL concentration (5-10,000 µM) did not
affect the amount of LDH released (Fig. 3B). Taken together, these results
indicated that SAL did not have any potential toxicity at 10, 50
and 100 µM.
SAL prevented ox-LDL-mediated apoptosis in a
concentration-dependent manner, with the concentrations >10
µM exerting the most potent anti-apoptotic effect (Fig. 3C and D). Following treatment with
SAL, the expression of Bcl-xL was upregulated in a
concentration-dependent manner (Fig.
3E). Importantly, SAL also reversed the increase in the cleaved
caspase-3 levels in a concentration-dependent manner (Fig. 3F).
However, no significant increase or decrease in the
mRNA level of Bcl-xL under ox-LDL induction was observed (Fig. 3G). Since bioinformatics analyses
predicted that miRNAs may regulate approximately one-third of all
human genes (19,20), in the present study, the related
literature for the targeted regulation of Bcl-xL by miRNAs was
screened as the starting point of the research.
Abnormal increase in miR-133a expression
in apoptotic HCAECs is reversed by SAL
A literature search confirmed a total of 7 miRNAs
[miR-133a (13), miR-133b
(13), let-7c (21), let-7g (21), miR-491 (22), miR-326 (23) and miR-608 (24)] that target Bcl-xL transcripts.
Among these, the expression level of miR-133a was abnormally
increased in ox-LDL-exposed cells (Fig. 4A). However, co-treatment with SAL
impaired miR-133a overexpression under the induction of apoptosis
(Fig. 4B). In addition, by
searching the literature, it was found that Liu et al, Ji
et al and Ma et al (13-15) used luciferase assay and target
gene mutation to confirm that Bxl-xL was the target gene of
miR-133a.
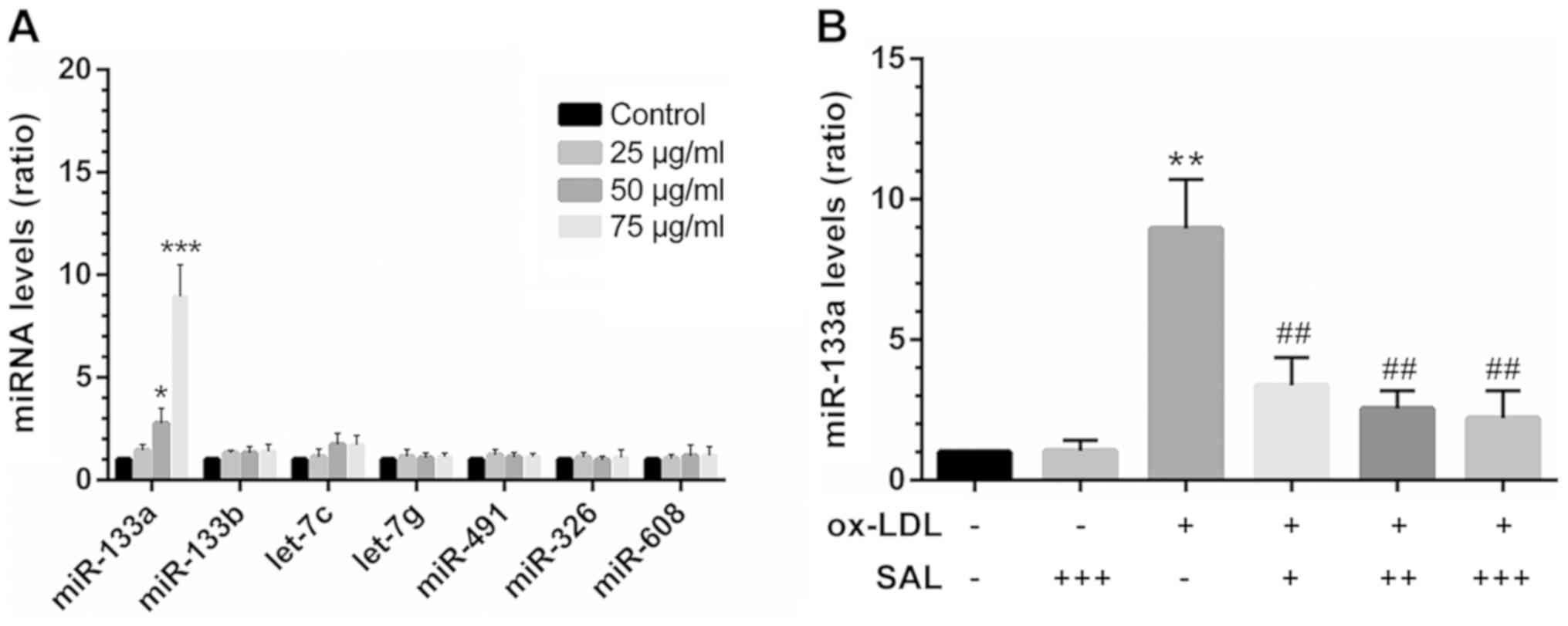 | Figure 4Abnormal increase in the miR-133a
levels in the model of HCAEC apoptosis induced by ox-LDL is
reversed by SAL. (A) At 24 h after HCAEC apop-tosis was induced by
various concentrations of ox-LDL, miRNAs targeting Bcl-xL were
screened by RT-qPCR. (B) SAL reversed the increase in miR-133a
levels in HCAECs exposed to ox-LDL (24 h). *P<0.05,
**P<0.01, ***P<0.001 vs. control;
##P<0.01 treatment with ox-LDL alone vs. treatment
with ox-LDL+SAL, -, without any drug treatment; + 10 µM, ++
50 µM, +++ 100 µM SAL + 75 µg/ml ox-LDL. SAL,
salidroside; ox-LDL, oxidized low-density lipoprotein; HCAECs,
human coronary artery endothelial cells. |
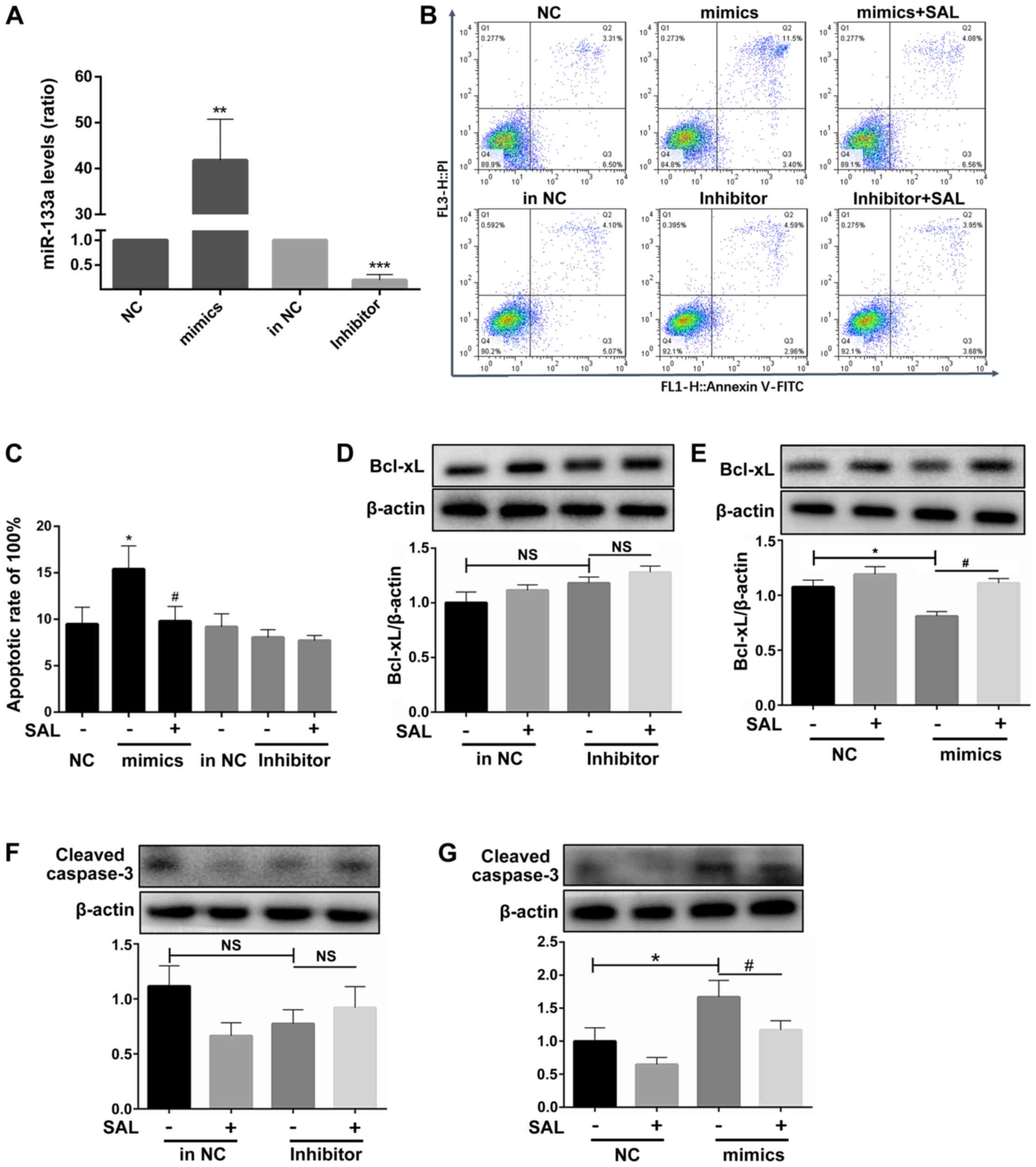
miR-133a participates in the
ox-LDL-induced apoptosis of HCAECs
The expression level of miR-133a was confirmed to be
increased following transfection with miR-133a mimics and decreased
following transfection with miR-133a inhibitor to obtain miR-133a
overexpression and silenced cell models, respectively (Fig. 5A).
Following the overexpression of miR-133a in
ox-LDL-exposed cells, the cell apoptotic rates significantly
increased compared to those of the untreated group, whereas no
significant difference was observed following the knock-down of
miR-133a (Fig. 5B and C). This
suggested that endogenous miR-133a may not be directly involved in
the process of apoptosis.
After miR-133a overexpression, Bcl-xL transcript
levels significantly decreased (Fig.
5D), whereas there was no significant change in Bcl-xL
transcript levels after knockdown of miR-133a (Fig. 5E). Treatment with SAL also
reversed the pro-apoptotic effect (Fig. 5D). Following the addition of SAL,
the mimics group exhibited an anti-apoptotic effect, whereas the
miR-133a inhibitor did not affect apoptosis.
To further confirm that miR-133a is involved in the
process of endothelial cell apoptosis, the protein expression of
cleaved caspase-3 was further examined. No significant difference
in the expression of cleaved caspase-3 was observed after the
silencing of miR-133a (Fig. 5F);
however, the activation of caspase-3 was significantly increased by
the overexpression of miR-133a, and SAL decreased the activation of
caspase-3 (Fig. 5G).
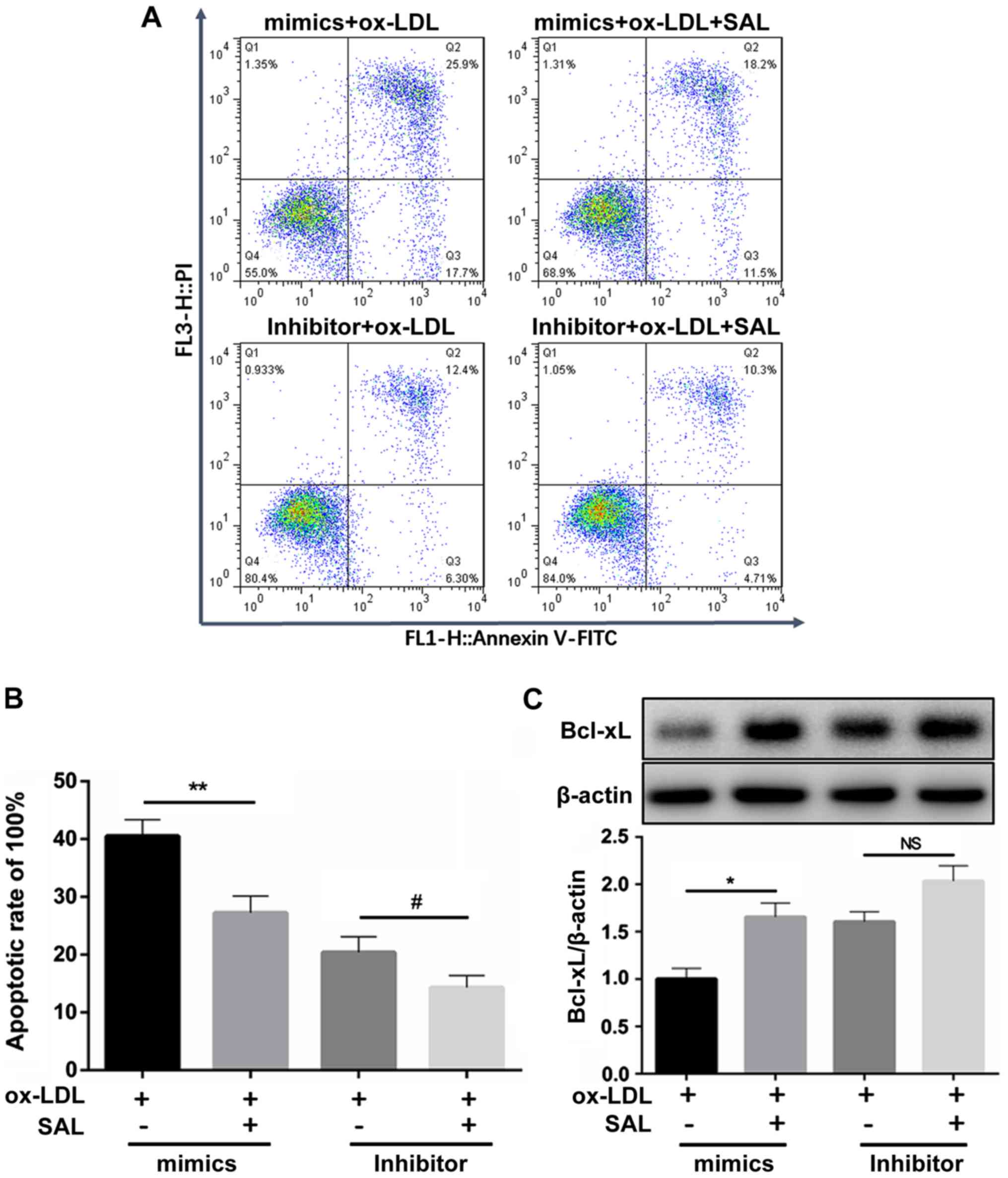
SAL reverses miR-133-induced apoptosis,
and this effect is suppressed following the knockdown of
miR-133a
The ox-LDL-induced apoptosis of HCAECs with miR-133a
overexpression was reversed by treatment with SAL. However, this
anti-apoptotic effect of SAL was attenuated following the knockdown
of miR-133a (Fig. 6A and B). This
finding suggested that SAL can inhibit endothelial cell apoptosis
induced by ox-LDL by downregulating miR-133a.
Following the knockdown of miR-133a, the decrease in
the Bcl-xL expression level induced by ox-LDL was not be reversed
by SAL, whereas the anti-apoptotic effect of SAL was partly
restored following the overexpression of miR-133a (Fig. 6C). Taken together, these results
suggest that SAL can directly act on miR-133a to regulate the
expression of Bcl-xL.
Discussion
The endothelium acts as a physiological barrier
between the blood and blood vessels, and plays a central role in
the development of AS. Endothelial cell apoptosis can increase the
proliferation and migration of smooth muscle cells, promote blood
coagulation and participate in the development of AS (25). The instability and rupture of
plaques are closely related to endothelial cell apoptosis (26). The present study aimed to explore
a novel approach and therapeutic target of salidroside against
endothelial cell apoptosis.
In general terms, apoptosis is initiated by a
signaling transduction cascade mediated by members of the Bcl-2
protein family. Of these, the apoptosis-initiating protein, Bid,
induces the release of cytochrome c by binding and isolating
the anti-apoptotic proteins Bcl-2 and Bcl-xL in the mitochondrial
outer membrane (27). The
increased cytochrome c efflux can then drive multimolecular
complexes to activate caspase-3 and caspase-7 to initiate apoptotic
cascades (28). Bcl-xL, encoded
by the BCL2L1 gene, is a major member of the Bcl-2 protein
family. Bcl-xL is superior to Bcl-2 and Mcl-1 in preventing DNA
damage-induced apoptosis (29).
The overexpression of Bcl-xL protects endothelial cells from
apoptosis mediated by DNA damage agents and pro-inflammatory
factors (30). Therefore, Bcl-xL
is more valuable to the study of apoptosis. The present study
demonstrated that endothelial cells exposed to ox-LDL exhibit a
decreased Bcl-xL level, caspase-3 activation and an increased
apoptotic rate, which are related to the concentration of ox-LDL
and the duration of exposure. This result is consistent with the
findings of Xu et al and Qin et al (18,31).
Several miRNAs have been demonstrated to play a
critical role in the pathogenesis of AS. For example, miRNA-181b
has been shown to inhibit inflammatory injury in endothelial cells
by regulating the NF-κB signal transduction pathway (32), whereas the effective blockage of
the miR-92a pathway can prevent endothelial dysfunction and
inflammation, thereby suppressing the development of AS (33). Furthermore, miR-126-5p can inhibit
the expression of DIK1 to promote tissue repair and prevent the
development of AS (34). In
addition, the miR-26a-targeted regulation of TRPC6 has been found
to prevent endothelial cell apoptosis (35). The present study demonstrated that
miR-133a regulated the expression of Bcl-xL and promoted
endothelial cell apoptosis, providing a new understanding of a new
pathway of endothelial cell apoptosis and a target for
anti-apoptotic therapy.
Several reports have highlighted the effects of
drugs on miRNA activity. For example, rapamycin has been reported
to enhance the expression of miRNA-155 to promote autophagy and
delay the formation of AS plaques (36). Similarly, Wujisan, a traditional
Chinese medicine, has been found to regulate ICAM-1, VCAM-1 and
E-selectin expression through miR10a and miR126-3p, thereby
inhibiting the development of AS (37). However, to the best of our
knowledge, no study available to date has examined whether or not
SAL can regulate the activity of miRNAs in the context of AS. The
present study found that SAL upregulated the protein expression of
Bcl-xL, whereas it not affect the mRNA level of Bcl-xL. Therefore,
it was hypothesized that there may be a non-coding RNA regulating
the process of Bcl-xL translation. Bioinformatics analysis was used
to screen the miRNAs confirmed to target Bcl-xL and regulate its
expression. Among these, it was found that only the miR-133a levels
were abnormally increased following ox-LDL-induced apoptosis.
The miR-133 family consists of 2 members, miR-133a
and miR-133b, which are located on different chromosomes (38). Previous studies have found that
miR-133a is a muscle-specific miRNA that plays an essential role in
the normal development and function of the skeletal muscle and
myocardium (38,39). In addition, epithelial cell
miR-133a expression has been reported to be upregulated by
neurotensin, which mediates colitis (40). The present study found that
miR-133a can regulate the transcript levels of the anti-apoptotic
protein Bcl-xL, thereby highlighting its role in the process of
endothelial cell apoptosis. Specifically, it was found that
following the overexpression of miR-133a, the Bcl-xL protein levels
significantly decreased, and the apoptotic rate increased in the
HCAECs. Importantly, no significant differences in the expression
of Bcl-xL and apoptotic rates were observed following the silencing
of miR-133a compared to those of the control group. Thus, Bcl-xL
may not be regulated by endogenous miR-133a in the normal
physiological state of endothelial cells, but rather only in a
pathologic condition.
Rhodiola is recognized to be effective in the
treatment of CHD, and SAL is its main active ingredient. Animal
experiments have confirmed that SAL has a definite anti-AS effect
(41,42). Further mechanistic studies have
demonstrated that SAL exerts antioxidant effects (9,43),
inhibits endoplasmic reticulum stress (44), exerts anti-inflammatory effects
(45), inhibits foam cell
formation (46) and exerts
anti-proliferative effects on vascular smooth muscle (47). Accumulating studies have also
indicated the role of SAL in endothelial cell apoptosis. SAL can
upregulate the expression of the anti-apoptotic protein Bcl-2;
downregulate the expression of the pro-apoptotic protein Bax;
increase the content of ATP (45); inhibit the activation of
caspase-3, caspase-9 and PARP (48); and activate AMPK to promote the
PI3K/AKT signaling pathway, which in turn affects the expression of
mammalian target of rapamycin (mTOR) (49). However, it is not clear whether
SAL can regulate miR-133a and then affect the expression of the
anti-apoptotic protein Bcl-xL to protect the endothelium. The
present study found that SAL impaired the ox-LDL-induced
upregulated expression of miR-133a in HCAECs, thereby affecting the
level of Bcl-xL. Significantly, the effect of SAL was decreased
following the silencing of miR-133a, whereas the overexpression of
miR-133a restored the effects of SAL treatment. Taken together, the
results demonstrated that SAL inhibited the decrease in the
expression of Bcl-xL via miR-133a, thereby preventing
ox-LDL-induced endothelial cell apoptosis. Therefore, SAL preserves
vascular function and confers protection against AS. However, the
mechanisms through which SAL regulates miR-133a warrant further
investigation, and animal experiments are required to confirm these
effects in vivo.
In conclusion, the findings of the present study
indicate that SAL inhibits the ox-LDL-induced upregulation of
miR-133a expression, while promoting the expression of Bcl-xL,
thereby preventing endothelial cell apoptosis. Therefore, it is
suggested that SAL can preserve vascular function and prevent the
development of AS.
Acknowledgments
Not applicable.
Abbreviations:
|
CHD
|
coronary heart disease
|
|
HCAECs
|
human coronary artery endothelial
cells
|
|
NC
|
negative control
|
|
OD
|
optical density
|
|
PBS
|
phosphate-buffered saline
|
|
PI
|
propidium iodide
|
Funding
The present study was supported by the Foundation of
Key Scientific Research Projects of Higher Education Institutions
in Henan Province (grant nos. 18A320005 and 19A360032).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
YoZ, FL and GZ conceived and designed the study. YoZ
and FL performed the experiments with the assistance of ZY, ZC, YC,
and YiZ. ZY and YoZ analyzed the data. YoZ wrote the manuscript.
All authors discussed the manuscript and all authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Benjamin EJ, Virani SS, Callaway CW,
Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling
FN, Deo R, et al: Heart disease and stroke statistics-2018 update:
A report from the American heart association. Circulation. 137. pp.
e67–e492. 2018, View Article : Google Scholar
|
|
2
|
Vita JA and Keaney JF Jr: Endothelial
function: A barometer for cardiovascular risk? Circulation.
106:640–642. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Trpkovic A, Resanovic I, Stanimirovic J,
Radak D, Mousa SA, Cenic-Milosevic D, Jevremovic D and Isenovic ER:
Oxidized low-density lipoprotein as a biomarker of cardiovascular
diseases. Crit Rev Clin Lab Sci. 52:70–85. 2015. View Article : Google Scholar
|
|
4
|
Zhou F, Yang Y and Xing D: Bcl-2 and
Bcl-xL play important roles in the crosstalk between autophagy and
apoptosis. FEBS J. 278:403–413. 2011. View Article : Google Scholar
|
|
5
|
Menghini R, Stohr R and Federici M:
MicroRNAs in vascular aging and atherosclerosis. Ageing Res Rev.
17:68–78. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fichtlscherer S, De Rosa S, Fox H,
Schwietz T, Fischer A, Liebetrau C, Weber M, Hamm CW, Röxe T,
Müller-Ardogan M, et al: Circulating microRNAs in patients with
coronary artery disease. Circ Res. 107:677–684. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cipollone F, Felicioni L, Sarzani R,
Ucchino S, Spigonardo F, Mandolini C, Malatesta S, Bucci M,
Mammarella C, Santovito D, et al: A unique microRNA signature
associated with plaque instability in humans. Stroke. 42:2556–2563.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li P, Yin YL, Guo T, Sun XY, Ma H, Zhu ML,
Zhao FR, Xu P, Chen Y, Wan GR, et al: Inhibition of aberrant
MicroRNA-133a expression in endothelial cells by statin prevents
endothe-lial dysfunction by targeting GTP cyclohydrolase 1 in vivo.
Circulation. 134:1752–1765. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xing SS, Yang XY, Zheng T, Li WJ, Wu D,
Chi JY, Bian F, Bai XL, Wu GJ, Zhang YZ, et al: Salidroside
improves endothelial function and alleviates atherosclerosis by
activating a mitochondria-related AMPK/PI3K/Akt/eNOS pathway.
Vascul Pharmacol. 72:141–152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Panossian A, Hamm R, Wikman G and Efferth
T: Mechanism of action of Rhodiola, salidroside, tyrosol and
triandrin in isolated neuroglial cells: An interactive pathway
analysis of the down-stream effects using RNA microarray data.
Phytomedicine. 21:1325–1348. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang YJ, Zhao GA, Lin F, et al: Research
progress on anti-atherosclerotic mechanism of salidroside. Chin J
Arteriosclerosis. 6:547–552. 2019.In Chinese.
|
|
12
|
Tang Y, Vater C, Jacobi A, Liebers C, Zou
X and Stiehler M: Salidroside exerts angiogenic and cytoprotective
effects on human bone marrow-derived endothelial progenitor cells
via Akt/mTOR/p70S6K and MAPK signalling pathways. Br J Pharmacol.
171:2440–2456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu Y, Zhang X, Zhang Y, Hu Z, Yang D,
Wang C, Guo M and Cai Q: Identification of miRNomes in human
stomach and gastric carcinoma reveals miR-133b/a-3p as therapeutic
target for gastric cancer. Cancer Lett. 369:58–66. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ji F, Zhang H, Wang Y, Li M, Xu W, Kang Y,
Wang Z, Wang Z, Cheng P, Tong D, et al: MicroRNA-133a,
downregulated in osteosarcoma, suppresses proliferation and
promotes apoptosis by targeting Bcl-xL and Mcl-1. Bone. 56:220–226.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma J, Wang T, Guo R, Yang X, Yin J, Yu J,
Xiang Q, Pan X, Zu X, Peng C, et al: MicroRNA133a and microRNA326
co-contribute to hepatocellular carcinoma 5-fluorouracil and
cisplatin sensitivity by directly targeting B-cell lymphoma-extra
large. Mol Med Rep. 12:6235–6240. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
17
|
Qin B, Shu Y, Long L, Li H, Men X, Feng L,
Yang H and Lu Z: MicroRNA-142-3p induces atherosclerosis-associated
endothelial cell apoptosis by directly targeting rictor. Cell
Physiol Biochem. 47:1589–1603. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu K, Liu P and Zhao Y: Upregulation of
microRNA-876 induces endothelial cell apoptosis by suppressing
Bcl-Xl in development of atherosclerosis. Cell Physiol Biochem.
42:1540–1549. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Friedman RC, Farh KK, Burge CB and Bartel
DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009. View Article : Google Scholar :
|
|
20
|
Eulalio A, Huntzinger E and Izaurralde E:
Getting to the root of miRNA-mediated gene silencing. Cell.
132:9–14. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shimizu S, Takehara T, Hikita H, Kodama T,
Miyagi T, Hosui A, Tatsumi T, Ishida H, Noda T, Nagano H, et al:
The let-7 family of microRNAs inhibits Bcl-xL expression and
potentiates sorafenib-induced apoptosis in human hepatocellular
carcinoma. J Hepatol. 52:698–704. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo R, Wang Y, Shi WY, Liu B, Hou SQ and
Liu L: MicroRNA miR-491-5p targeting both TP53 and Bcl-XL induces
cell apoptosis in SW1990 pancreatic cancer cells through
mitochondria mediated pathway. Molecules. 17:14733–14747. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu S, Huang H, Deng G, Xie Z, Ye Y, Guo R,
Cai X, Hong J, Qian D, Zhou X, et al: miR-326 targets antiapoptotic
Bcl-xL and mediates apoptosis in human platelets. PLoS One.
10:e01227842015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Y, Schiff D, Park D and Abounader R:
MicroRNA-608 and microRNA-34a regulate chordoma malignancy by
targeting EGFR, Bcl-xL and MET. PLoS One. 9:e915462014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Choy JC, Granville DJ, Hunt DW and McManus
BM: Endothelial cell apoptosis: Biochemical characteristics and
potential implications for atherosclerosis. J Mol Cell Cardiol.
33:1673–1690. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kavurma MM, Bhindi R, Lowe HC, Chesterman
C and Khachigian LM: Vessel wall apoptosis and atherosclerotic
plaque instability. J Thromb Haemost. 3:465–472. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shamas-Din A, Kale J, Leber B and Andrews
DW: Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb
Perspect Biol. 5:a0087142013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Galluzzi L, Kepp O, Trojel-Hansen C and
Kroemer G: Mitochondrial control of cellular life, stress, and
death. Circ Res. 111:1198–1207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen HC, Kanai M, Inoue-Yamauchi A, Tu HC,
Huang Y, Ren D, Kim H, Takeda S, Reyna DE, Chan PM, et al: An
interconnected hierarchical model of cell death regulation by the
BCL-2 family. Nat Cell Biol. 17:1270–1281. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fuchsluger TA, Jurkunas U, Kazlauskas A
and Dana R: Anti-apoptotic gene therapy prolongs survival of
corneal endothelial cells during storage. Gene Ther. 18:778–787.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qin B, Xiao B, Liang D, Li Y, Jiang T and
Yang H: MicroRNA let-7c inhibits Bcl-xl expression and regulates
ox-LDL-induced endothelial apoptosis. BMB Rep. 45:464–469. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun X, He S, Wara AKM, Icli B, Shvartz E,
Tesmenitsky Y, Belkin N, Li D, Blackwell TS, Sukhova GK, et al:
Systemic delivery of microRNA-181b inhibits nuclear factor-κB
activation, vascular inflammation, and atherosclerosis in
apolipoprotein E-deficient mice. Circ Res. 114:32–40. 2014.
View Article : Google Scholar
|
|
33
|
Loyer X, Potteaux S, Vion AC, Guérin CL,
Boulkroun S, Rautou PE, Ramkhelawon B, Esposito B, Dalloz M, Paul
JL, et al: Inhibition of microRNA-92a prevents endothelial
dysfunction and atherosclerosis in mice. Circ Res. 114:434–443.
2014. View Article : Google Scholar
|
|
34
|
Schober A, Nazari-Jahantigh M, Wei Y,
Bidzhekov K, Gremse F, Grommes J, Megens RT, Heyll K, Noels H,
Hristov M, et al: MicroRNA-126-5p promotes endothelial
proliferation and limits atherosclerosis by suppressing Dlk1. Nat
Med. 20:368–376. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang Y, Qin W, Zhang L, Wu X, Du N, Hu Y,
Li X, Shen N, Xiao D, Zhang H, et al: MicroRNA-26a prevents
endothelial cell apoptosis by directly targeting TRPC6 in the
setting of atherosclerosis. Sci Rep. 5:94012015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ma J, Yang S, Ma A, Pan X, Wang H, Li N,
Liu S and Wu M: Expression of miRNA-155 in carotid atherosclerotic
plaques of apolipoprotein E knockout (ApoE−/−) mice and
the interventional effect of rapamycin. Int Immunopharmacol.
46:70–74. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Han BH, Seo CS, Yoon JJ, Kim HY, Ahn YM,
Eun SY, Hong MH, Lee JG, Shin HK, Lee HS, et al: The inhibitory
effect of ojeoksan on early and advanced atherosclerosis.
Nutrients. 10:12562018. View Article : Google Scholar :
|
|
38
|
Yu H, Lu Y, Li Z and Wang Q: microRNA-133:
Expression, function and therapeutic potential in muscle diseases
and cancer. Curr Drug Targets. 15:817–828. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen JF, Mandel EM, Thomson JM, Wu Q,
Callis TE, Hammond SM, Conlon FL and Wang DZ: The role of
microRNA-1 and microRNA-133 in skeletal muscle proliferation and
differentiation. Nat Genet. 38:228–233. 2006. View Article : Google Scholar
|
|
40
|
Law IK and Pothoulakis C: MicroRNA-133α
regulates neurotensin-associated colonic inflammation in colonic
epithelial cells and experimental colitis. RNA Dis. 2:e4722015.
|
|
41
|
Zhang BC, Li WM, Guo R and Xu YW:
Salidroside decreases atherosclerotic plaque formation in
low-density lipoprotein receptor-deficient mice. Evid Based
Complement Alternat Med. 2012:6075082012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang LP: Effect of salidroside on
atherosclerosis induced by intermittent hypobaric hypoxia in
ApoE-/-mice. Chin J Arteriosclerosis. 7:675–679. 2014.In
Chinese.
|
|
43
|
Zhu Y, Zhang YJ, Liu WW, Shi AW and Gu N:
Salidroside suppresses HUVECs cell injury induced by oxidative
stress through activating the Nrf2 signaling pathway. Molecules.
21:10332016. View Article : Google Scholar
|
|
44
|
Zhu L, Jia F, Wei J, Yu Y, Yu T, Wang Y,
Sun J and Luo G: Salidroside protects against homocysteine-induced
injury in human umbilical vein endothelial cells via the regulation
of endoplasmic reticulum stress. Cardiovasc Ther. 35:33–39. 2017.
View Article : Google Scholar
|
|
45
|
Liu L, Zhang S and Zhang MQ: Protective
effect and mechanism of salidroside on endothelial cell surface
injury induced by high glucose. Chin Herb Med. 40:949–952. 2017.In
Chinese.
|
|
46
|
Ni J, Li Y, Li W and Guo R: Salidroside
protects against foam cell formation and apoptosis, possibly via
the MAPK and AKT signaling pathways. Lipids Health Dis. 16:1982017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhuang X, Maimaitijiang A, Li Y, Shi H and
Jiang X: Salidroside inhibits high-glucose induced proliferation of
vascular smooth muscle cells via inhibiting mitochondrial fission
and oxidative stress. Exp Ther Med. 14:515–524. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhao X, Jin L, Shen N, Xu B, Zhang W, Zhu
H and Luo Z: Salidroside inhibits endogenous hydrogen peroxide
induced cytotoxicity of endothelial cells. Biol Pharm Bull.
36:1773–1778. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xu MC, Shi HM, Wang H and Gao XF:
Salidroside protects against hydrogen peroxide-induced injury in
HUVECs via the regulation of REDD1 and mTOR activation. Mol Med
Rep. 8:147–153. 2013. View Article : Google Scholar : PubMed/NCBI
|