1. Introduction
Nucleic acid amplification tests are core
technologies of clinical diagnosis. In pulmonary tuberculosis, such
testing is capable of identifying Mycobacterium species in
clinical respiratory samples more rapidly and accurately than
sputum specimen examinations and culture-based methods. This
advantage is key to appropriate treatment, prevention, and control
of transmission of tuberculosis. In HIV detection, the nucleic acid
amplification test is more sensitive and quantitative than other
methods based on HIV-1-specific antibody or viral antigens,
enabling the detection of HIV-1 at the initial stage of infection
and the monitoring of disease progression (1,2).
Various nucleic acid amplification technologies have
been devised, but the most widely used is PCR. In basic research,
most researchers use PCR primarily for amplification, possibly
because primer design is convenient and the enzymes are available
at a reasonable price (3). In
clinical diagnosis, on the other hand, isothermal nucleic acid
amplification methods such as nucleic acid sequence-based
amplification (NASBA) (4), strand
displacement amplification (SDA) (5), rolling circle amplification (RCA)
(6), helicase-dependent
isothermal DNA amplification (HAD) (7), and loop-mediated isothermal
amplification (LAMP) (8) are also
used. The advantage of isothermal amplifications over PCR is that
they do not require a complex device such as thermal cycler,
improving throughput in situations when large numbers of clinical
samples must be processed, as well as facilitating point-of-care
diagnosis (4-9).
The performance of a nucleic acid amplification test
depends largely on the performance of the enzymes involved.
Thermostable DNA polymerase, first identified in Thermus
aquaticus (Taq) in 1976 (10), has become widely used since the
discovery of PCR. Concerning performances of Taq polymerase,
it was initially reported that the activity decreased to 50% at
incubation at 95°C for 1.6 h; the rate of processing was 60-150
nucleotides/sec; and the error rate was 0.38−1.32×104
errors/base (11). Since then,
the performances of Taq polymerase were improved by genetic
engineering. For example, the mutation of Phe667 into Tyr increased
its efficiency of incorporation with ddNTP by 103-fold
(12), and fusion of the
helix-hairpin-helix motifs of DNA topoisom-erase V to Taq
polymerase increased the enzyme's stability and processivity
(13). The performances of DNA
polymerases from the hyperthermophilic archaeon Thermococcus
koda- karensis (KOD) or Pyrococcus furiosus(Pfu)
and that from thermophilic bacteria Thermus thermophilus
(Tth) have also been improved by genetic engineering. Today,
they are widely used in PCR along with Taq polymerase.
In addition to altering the enzymes, it is also
important to optimize the reaction conditions. In the amplification
techniques using multiple enzymes, such as RT-PCR and NASBA, this
process is more complicated because each enzyme has its own optimal
condition. Another concern is lowering the risk of contamination.
In this regard, it is preferable to perform one-tube reactions with
real-time monitoring (14).
The aim of the review is to outline recent advances
in nucleic acid amplification technologies. The foci of the study
are, reverse transcriptase as an example of an enzyme that has been
markedly improved by genetic engineering; recombination polymerase
amplification, an isothermal amplification which has attracted a
great deal of recent attention; and focus helicase, an enzyme which
increases specificity and decreases noise in the amplification.
Next-generation sequencing (NGS) was used to evaluate the fidelity
of cDNA synthesis and the statistical method to optimize the
reaction conditions.
2. Thermostabilization of reverse
transcriptase
Reverse transcriptase (RT) has RNA- and
DNA-dependent DNA polymerase and ribonuclease (RNase) H activities.
It is responsible for RNA viral genome replication. Moloney murine
leukemia virus (MMLV) RT and avian myeloblastosis virus (AMV) RT
are widely used in cDNA synthesis (15) (Table
I). MMLV RT is a 75-kDa monomer, and AMV RT is a heterodimer
consisting of an α subunit (63-kDa) and a β subunit (95-kDa)
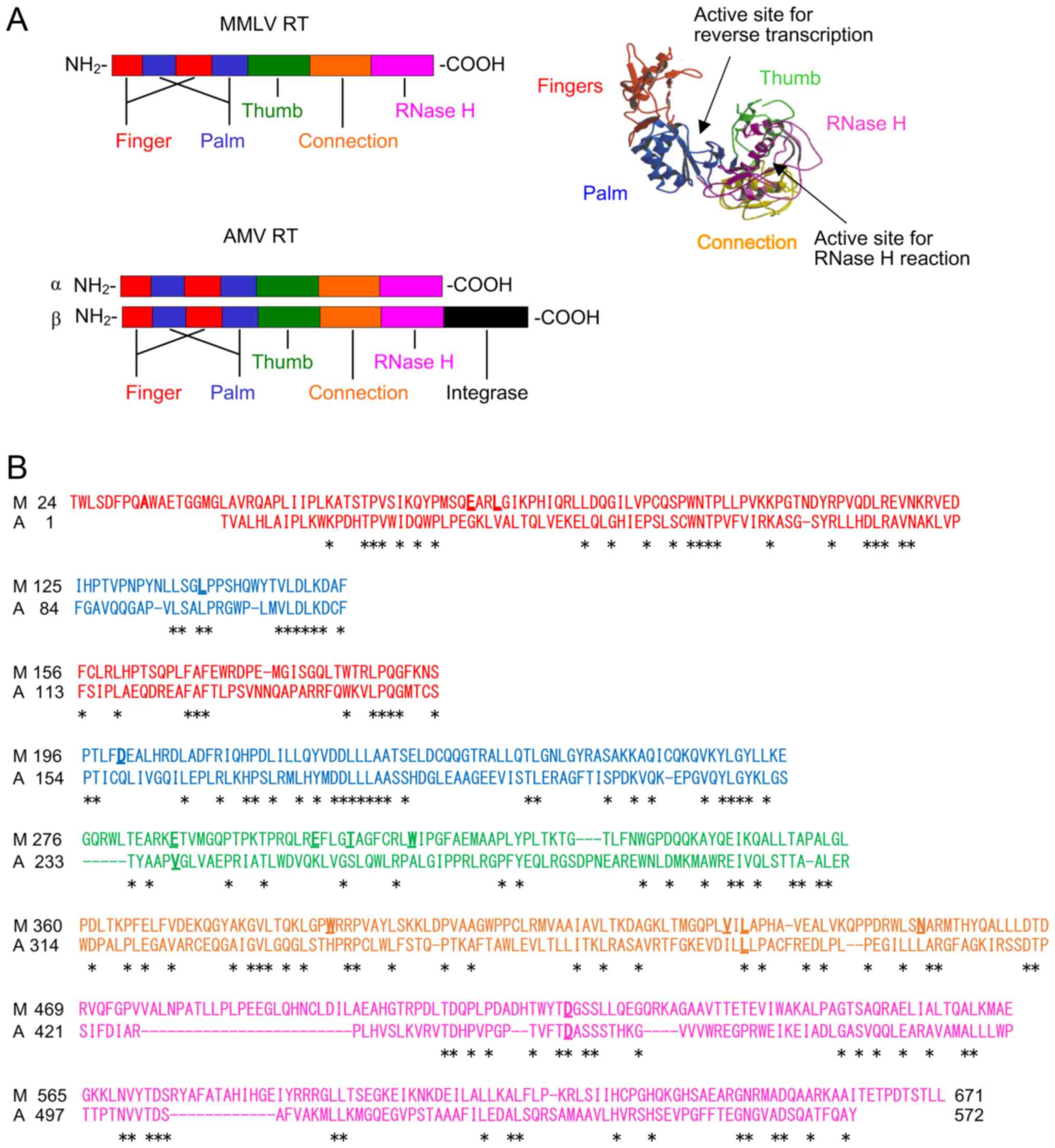
(16,17). The result of the homology search
performed using the search program DNA Data Bank of Japan (DDBJ;
https://www.ddbj.nig.ac.jp/index-e.html) CLUSTALW and
the crystal stuructures of MMLV RT is shown in Fig. 1. MMLV RT and the α subunit of AMV
RT comprise the fingers, palm, thumb, connection, and RNase H
domains. The β subunit of AMV RT includes these five domains along
with the C-terminal integrase domain. MMLV RT and AMV RT have two
active sites. The active site for the DNA polymerase reaction is in
the fingers/palm/thumb domain, and that for the RNase H reaction is
in the RNase H domain.
 | Table IEnzymes used for nucleic acid
amplification. |
Table I
Enzymes used for nucleic acid
amplification.
| Enzyme | Application | (Refs.) |
|---|
| Reverse
transcriptase (RT) | | |
| AMV RT | cDNA synthesis,
NASBA | (15,18,20,28,59) |
| MMLV RT | cDNA synthesis | (16-27,29,59,60) |
| DNA polymerase | | |
| Taq
polymerase | PCR | (10-13) |
| Tth
polymerase | PCR, cDNA
synthesis | (37,38) |
|
K4polL329Aa | PCR, cDNA
synthesis | (43,48) |
| RTXb | PCR, cDNA
synthesis | (47,48) |
| Bacillus
subtilis polymerase | RPA | (70) |
| DNA helicase | | |
|
Tk-EshA | PCR, cDNA
synthesis | (48,54,56) |
|
Tk-Upf1 | PCR | (56) |
| Single-strand
DNA-binding protein | | |
| T4 gp32 | RPA | (68,70) |
| Recombinase | | |
| T4 uvsY | RPA | (69,70) |
| T4 uvsX | RPA | (70) |
Thermostability of DNA polymerases is important for
their wide-range practical use. For cDNA synthesis, an elevated
reaction temperature is highly desirable because it reduces RNA
secondary structure and nonspecific binding of the primer. However,
RT is thermolabile. The initial activities of MMLV RT and AMV RT
are reduced by 50% at 44 and 47°C, respectively, during a 10-min
incubation (18). Thus, improving
the thermostability of RT has been an important subject. The
thermostabilities of MMLV RT (19-21) and AMV RT (20) were first improved by eliminating
the RNase H activity. The thermostability of MMLV RT was improved
by introducing the triple mutation E286R/E302K/L435R or
E286R/E302K/L435R/D524A in which the negatively charged (Glu286 and
Glu302) and hydrophobic (Leu435) residues that were thought to
interact with a template-primer were replaced with positively
charged residues, and the catalytic residue responsible for RNase H
activity Asp524 was replaced with Ala (22). The thermostability of MMLV RT was
also improved by the mutation of Val433 present on the molecular
surface to Arg (23). Finally, a
highly thermostable MMLV variant A32V/L72R/E286R/E302K/W388R/L435R
was generated by combining the triple mutation E286R/E302K/L435R
with the following mutations: The mutation of the internal residue,
Ala32 to Val in order to stabilize the hydrophobic core, the
mutation of the hydrophobic surface residue, Leu72 to Arg, and the
mutation of Trp388 which is close to the negatively charged
residues to Arg in order to introduce a salt bridge (24). In a random mutation assay followed
by a combination of stabilizing mutations,
E69K/E302R/W313F/L435G/N454K was generated using a filter assay
(25),
L139P/D200N/T330P/L603W/E607K was generated using emulsion PCR
(26), and D200C was obtained by
screening an amino acid scanning library (27). The amino acid residues mutated for
thermostabilization are widespread throughout the molecule
(Fig. 1B).
Recombinant MMLV RT is well expressed in the soluble
fractions in Escherichia coli, from which sufficient amounts
of active enzymes are purified. On the contrary, AMV RT has been
barely expressed in the soluble fractions of E. coli.
Instead, the active AMV RT α subunit was expressed in insect cells
(28), and its thermostability
was improved by introducing the triple mutation V238R/L388R/D450A,
corresponding to E286R/W388R/D524A in MMLV RT (29). Notably, recombinant AMV RT has
been successfully expressed in the soluble fractions in E.
coli since then, and is now commercially available.
cDNA synthesis, as with PCR, is a key technology
both in clinical diagnosis and basic research. However, cDNA
synthesis is less sensitive than PCR. To circumvent this problem, a
cDNA synthesis method using three enzymes, the thermostable MMLV RT
quadruple variant E286R/E302K/W388R/D524A (described above), the
genetically engineered family A DNA polymerase variant with RT
activity from the hyperthermophile Thermotoga petrophila K4
(K4polL329A) which will be described in the next section
and the DNA/RNA helicase from a hyperthermophilic archaeon
Thermococcus kodakarensis(Tk-EshA), was developed
(Table I). K4polL329A
and Tk-EshA will be described later. In amplification
techniques using multiple enzymes such as NASBA (1,30),
optimization is more complicated than when using a single enzyme as
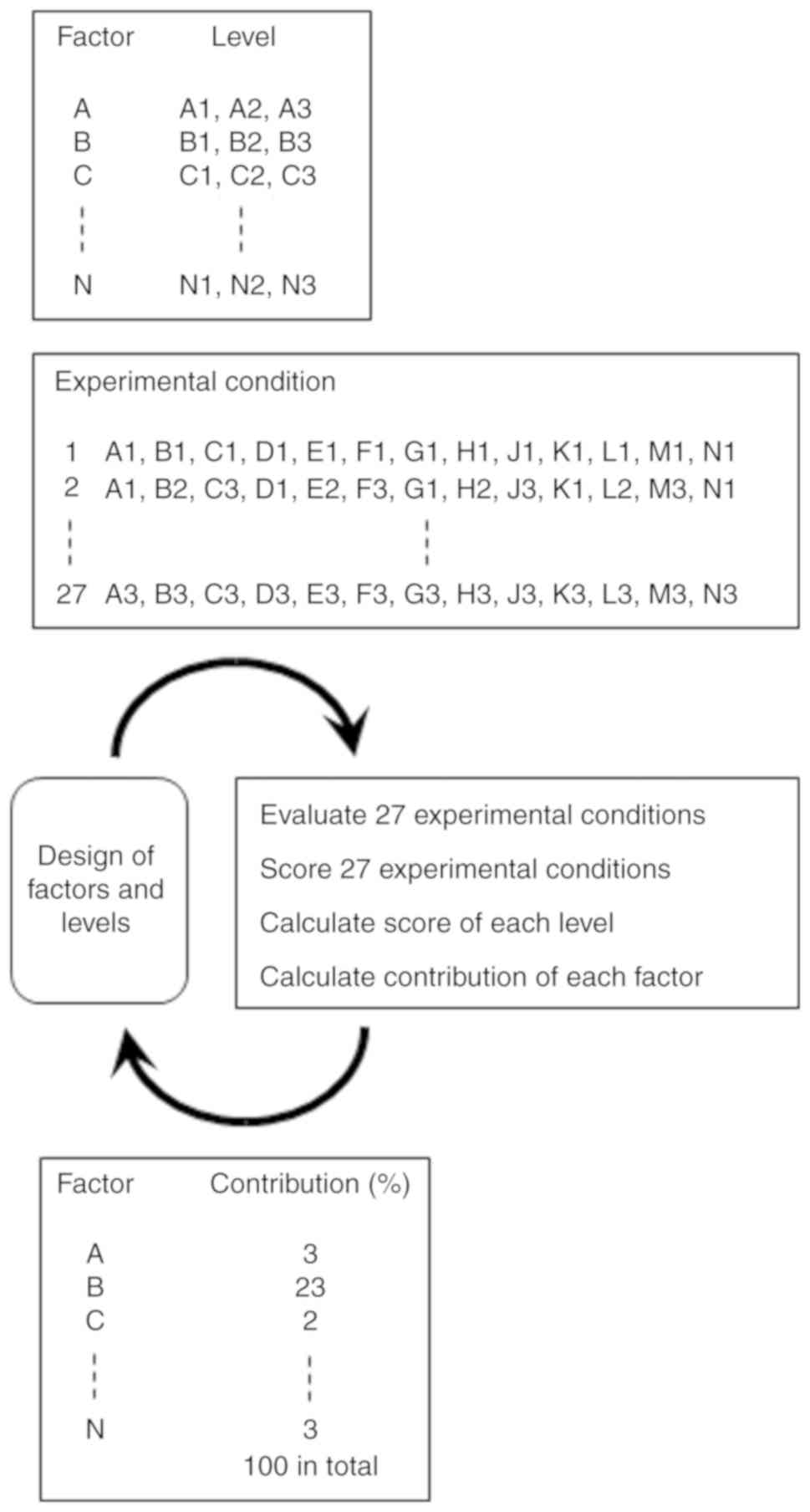
in the case of PCR. In this case, statistical methods such as
Taguchi's method have been successfully used for optimization
(31) (Fig. 2). The merit of statistical methods
is that many factors can be optimized at the same time with the
minimum number of experiments.
Stabilization of RT is desirable for cDNA synthesis.
Improvement in the thermostability of MMLV RT and AMV RT is an
important subject. Characterization of about 700 variants of phage
T4 lysozyme revealed that there can be various kinds of effective
stabilizing methods such as disulfide bridge, salt-bridge
interaction, metal binding, and hydrophobic stabilization (32). We consider that the
thermostabilities of MMLV RT and AMV RT may be further improved by
combining stabilizing mutations.
3. Creation of the reverse transcriptase
activity in thermostable DNA polymerase
The DNA-dependent DNA polymerase distinguishes
suitable substrates DNA and dNTPs from unsuitable RNA and rNTPs.
The exact mechanisms of this distinction are unknown, but two
mechanisms have been proposed. One mechanism is for rNTP/dNTP
distinction. In Klenow polymerase, the bulky 2′ hydroxyl group of
ribose interferes with the substrate-binding region of Klenow
polymerase: Glu710 sterically blocks the 2′ hydroxyl group of rNTP.
As a result, the enzyme accepts dNTP but excludes rNTP (33). A similar hindrance effect was
reported in archaeon Thermococcus litoralis family B DNA
polymerase: Tyr412 exludes rNTP by acting as a steric gate for the
2′ hydroxyl group of ribose (34). The other mechanism is for template
distinction. Archaeal family B DNA polymerase excludes
uracil-containing templates, and DNA synthesis is prematurely
arrested at the position where uracil is contained. By contrast,
bacterial DNA polymerase I ignores the absence of the 5' methyl
group in uracil, and accepts a uracil-containing template.
Therefore, the 2' hydroxyl group of ribose is considered a key
factor for the distinction of RNA/DNA for bacterial DNA-dependent
DNA polymerase (35). A similar
effect was reported in Klenow polymerase: Asn420 and Tyr423 in the
3′-5′ exonuclease domain play a role in RNA exlusion by interfering
with the 2′ hydroxyl group of the template molecule (36).
To generate thermostable RT using DNA polymerases
from thermophilic bacteria and archaea, several approaches have
been taken (37-42). Some bacterial DNA polymerases (Pol
I) show reverse transcriptase activity in the presence of
Mn2+. The Tth DNA polymerase from Thermus
thermophilus also shows the RT activity (37,38). It lacks a 3′-5′ exonuclease
domain, which contributes to fidelity in PCR. DNA polymerase I from
the hyperthermophilic bacterium, Thermotogasp, possesses a
3′-5′ exonuclease domain. A study on chimeric DNA polymerases from
Thermotoga sp and Thermus sp showed that chimeric DNA
polymerases with RT activity possessed attenuated 3′-5′ exonuclease
activity (42). Mutations were
introduced into another DNA polymerase from Thermotoga
petrophila K4 (K4PolI) to allow K4PolI to accept an RNA. Among
the variants constructed, T326A, L329A, Q384A, F388A, M408A, and
Y438A exhibited RT activity while their 3′-5′ exonuclease activites
were reduced. By contrast, K4PolN422A and K4PolF451A did
not exhibit RT activity but possessed full 3′-5′ exonuclease
activity (43). These results
suggest that there is a correlation between the gain of RT activity
and the loss of 3′-5′ exonuclease activity. On the other hand,
introduction of random mutations into Taqpolymerase showed
that mutations in domains other than the 3′-5′ exonuclease domain
generated the mutants with RT activity (39). Further structural studies are
needed to exlopre the mechnasim connecting RT and 3′-5′ exonuclease
activities.
Archaeal family B DNA polymerases, such as those
from Pyrococcus furiosus (44) or Thermococcus kodakarensis
(45), possess a higher fidelity
than thermophilic bacteria enzymes, such as those from T.
aquaticus and T. thermophilus. However, as mentioned
above, archaeal family B DNA polymerase excludes a template
containing uracil, which is different from bacterial DNA polymerase
(46). Family B DNA polymerase
recognizes DNA more precisely than bacterial DNA polymerase I.
Modified family B DNA polymerases with a Pol ζ fingers domain that
displayed RT activity were developed by the mutation experiment
into the 3′-5′ exonuclease domain of hybrid archaeal family B DNA
polymerases with a Pol ζ fingers domain (41). Recently, Ellefson et al
generated a 16-tuple variant of KOD DNA polymerase known as RTX
with RT activity from the hyperthermophilic archaeon,
Thermococcus kodakarensis, by a directed evolution method
(47). In this method, emulsion
PCR was carried out with primers containing various numbers of
ribonucleotides so that only DNA polymerase with RT activity
enabled self-replication (47).
These results indicate that family B DNA polymerases can be used as
a source to create reverse transcriptase.
DNA polymerases with RT activity enable one-step
RT-PCR without retroviral RT. The merit of one-step RT-PCR over
two-step RT-PCR is that multiple openings of reaction tubes and
reagent delivery are not necessary, leading to a decrease in DNA
contamination risk. Furthermore, artificially created reverse
transcriptase K4polL329A and RTX are applicable for high sensitive
RNA detection by one-step RT-PCR combining with the genetically
engineered MMLV-RT and thermostable DNA/RNA helicase (48). COVID-19 RNA was also detected from
clinical samples by using the system (data not shown). Details of
the helicase role are mentioned below.
4. Use of helicase to increase
specificity
DNA/RNA helicases exhibit nucleic acid binding, ATP
hydrolysis, translocation, and unwinding of nucleic acid duplex by
eliminating hydrogen bonds from the base-pairing between DNA/DNA,
DNA/RNA, and RNA/RNA hybrids from the 3′ or 5′ unpaired end
utilizing the energy generated upon ATP hydrolysis. Therefore,
helicases are expected to unwind the secondary structured template
and partially annealed primer/template duplexes in DNA and RNA
synthesis. DNA/RNA helicases are classified into several
superfamilies (SFs) according to their amino acid sequences
(49). The SF1 and SF2 helicases
are large and diverse groups, sharing catalytic cores with almost
identical folds and extensive structural similarities. UvrD, an SF1
DNA helicase that unwinds blunt-end substrates as well as nicked
circular DNA, was used in an isothermal DNA amplification at low
temperature, called helicase-dependent amplification (50-53). In this amplification, a mesophilic
DNA polymerase is applicable.
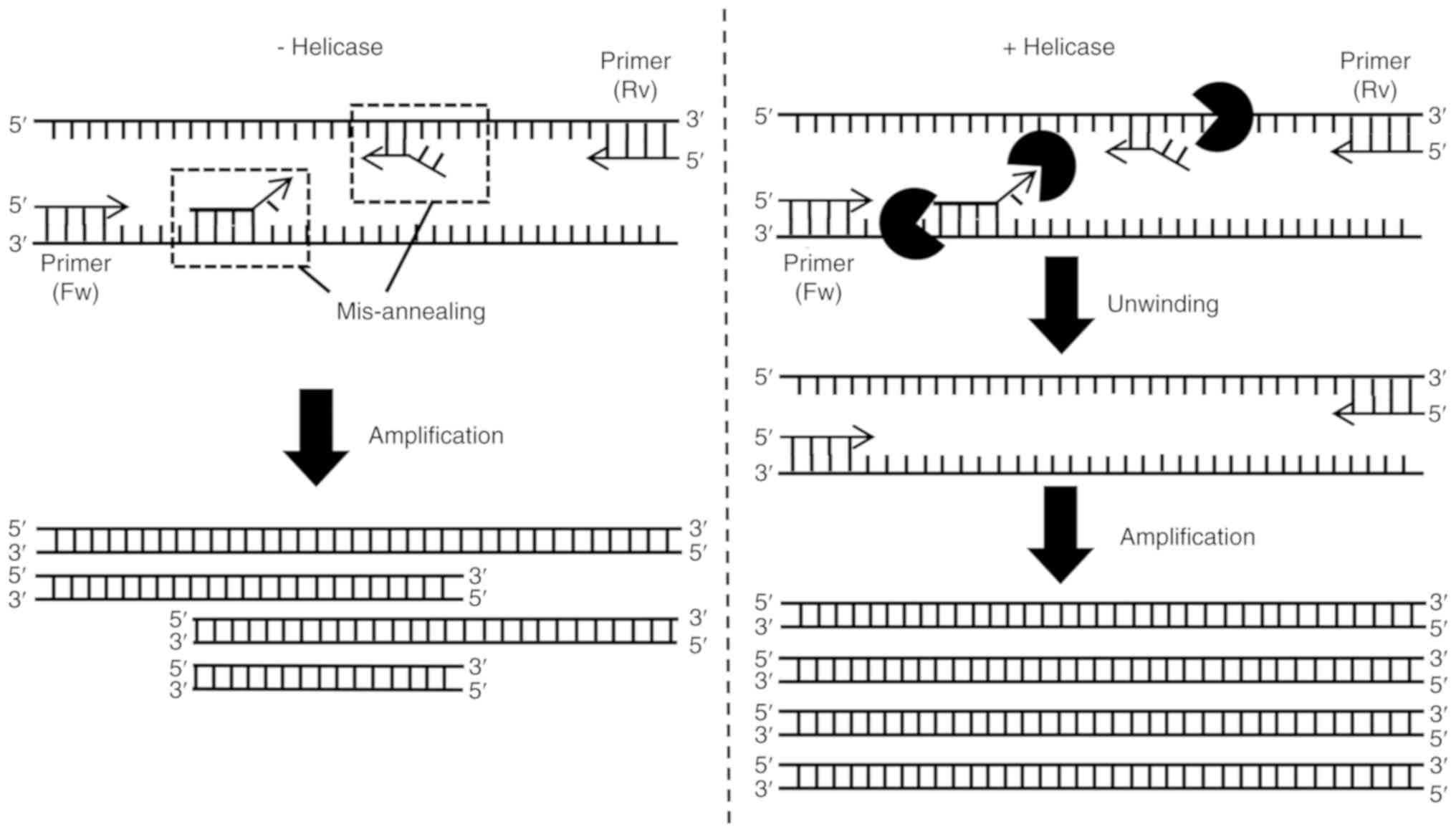
Unexpected DNAs sometimes get amplified due to
primer mis-annealing during PCR. In order to efficiently reduce
such mis-amplified products, an approach using helicase was devised
(54). Tk-EshA, a
euryarchaeota-specific SF2 helicase EshA from the hyperthermophilic
archaeon Thermococcus kodakarensis, was first used for this
purpose. In the presence of RNA, Tk-EshA exhibited maximal
ATPase activity at 80°C. Tk-EshA unwinds forked and 3′
overhung DNAs (54).
Tk-EshA also possesses euryarchaeal termination activity
(Eta), which disrupts the transcription elongation complex
(55). We hypothesized that
Tk-EshA unwinds the structured template and peels off
mis-annealed primers during PCR. To address this issue, PCR was
performed using various DNAs as a substrate. When 16S rDNA was
used, several mis-amplified products (noise DNAs) were detected in
the absence of Tk-EshA. However, they were eliminated in the
presence of Tk-EshA. These effects of Tk-EshA were
confirmed whether Taq DNA polymerase (a family A DNA
polymerase, PolI type) or KOD DNA polymerase (a family B DNA
polymerase, α type) was used. When toxA gene from
Pseudomonas aeruginosa DNA, which possesses high GC content
(69%), was used, mis-amplified bands were also eliminated by the
addition of Tk-EshA, suggesting that Tk-EshA was more
effective than increasing the annealing temperature to reduce
mis-amplified DNAs in the toxA amplification (54). The action of Tk-EshA is
shown in Fig. 3. Another type
(superfamily 1B) of helicase, Tk-Upf1 (TK0178) from T.
kodakarensis, was examined for the effects on conventional PCR
and digital PCR and compared with those of Tk-EshA. It is
important to eliminate nonspecific amplification for identification
of SNPs. Of four double-stranded DNA substrates, forked, 5′
overhung, 3′ overhung, and blunt-ended DNAs, the unwinding activity
of Tk-Upf1 was the highest towards 5′ overhung DNAs
(56). The concentration of
Tk-Upf1 required for noise DNA elimination was 10-fold lower
than that of Tk-EshA. The addition of Tk-Upf1 also
eliminated noise DNAs derived from the misannealed primer when a 5′
or 3′ overhung misannealed primer was included as a competitive
primer along with specific primers. In digital PCR, Tk-EshA
and Tk-Upf1 functioned as signal enhancers: Tk-EshA
or Tk-Upf1 increased the fluorescent intensities, improving
separation between the common and risk allele clusters. The amount
of Tk-Upf1 required to improve the performance of digital
PCR was smaller than that of Tk-EshA.
5. Fidelity evaluation with NGS
Fidelity indicates the performance in the
incorporation of correct nucleotides. Various methods have been
applied to analyze DNA polymerase fidelity such as misincorporation
(57), misextension (57), primer extension (58), and M13 lacZ mutation
(59) assays. In a
misincorporation assay, the reaction rates to incorporate correct
and incorrect nucleotides are compared, while in a misextension
assay, the reaction rates for extension from the mispaired end
(i.e., A:G) and from the paired end (i.e., A:T) are compared
(57). In these two assays, the
reactions are carried out under single-turnover conditions. In a
primer extension assay, the reaction in the absence of one dNTP is
compared with that in the presence of all four dNTPs (58). In the M13 lacZ mutation
assay, the error rates are calculated from the mutation frequency,
which is determined as the ratio of mutant plaques to all plaques
(59). The error rates of MMLV RT
and AMV RT determined by this assay were 3.3−5.9×10−4
errors/base and that of HIV-1 RT was 5.9×10−3
errors/base (59). The M13
lacZ mutation assay has been the only method used to
determine the error rate. However, it has some issues. Silent
mutation affects the calculation of error rates. Identification of
plaque color depends on the individual. In addition, the reaction
is DNA-dependent DNA synthesis, but not RNA-dependent DNA
synthesis, even for RT.
In NGS, hundreds of million sequences are obtained
in one NGS run. NGS has been widely used to identify rare
mutations, misincorporations, and base modifications introduced in
genomic DNA (60,61). One of the problems of NGS is that
a number of errors are introduced. To address this issue, a method
to identify ultra-rare mutations in the genomic DNA using NGS was
devised (62), which uses
adaptors containing two tags of 12 randomized bases for the
ligation of DNA fragments containing the sequences to be analyzed.
All sequence reads are grouped based on tag sequences and
orientations. By analyzing whether all sequence reads in the same
group had the same mutation or not, each mutation that was observed
via NGS indicated whether the error was already present in the
genome or was incorporated by PCR or NGS (62).
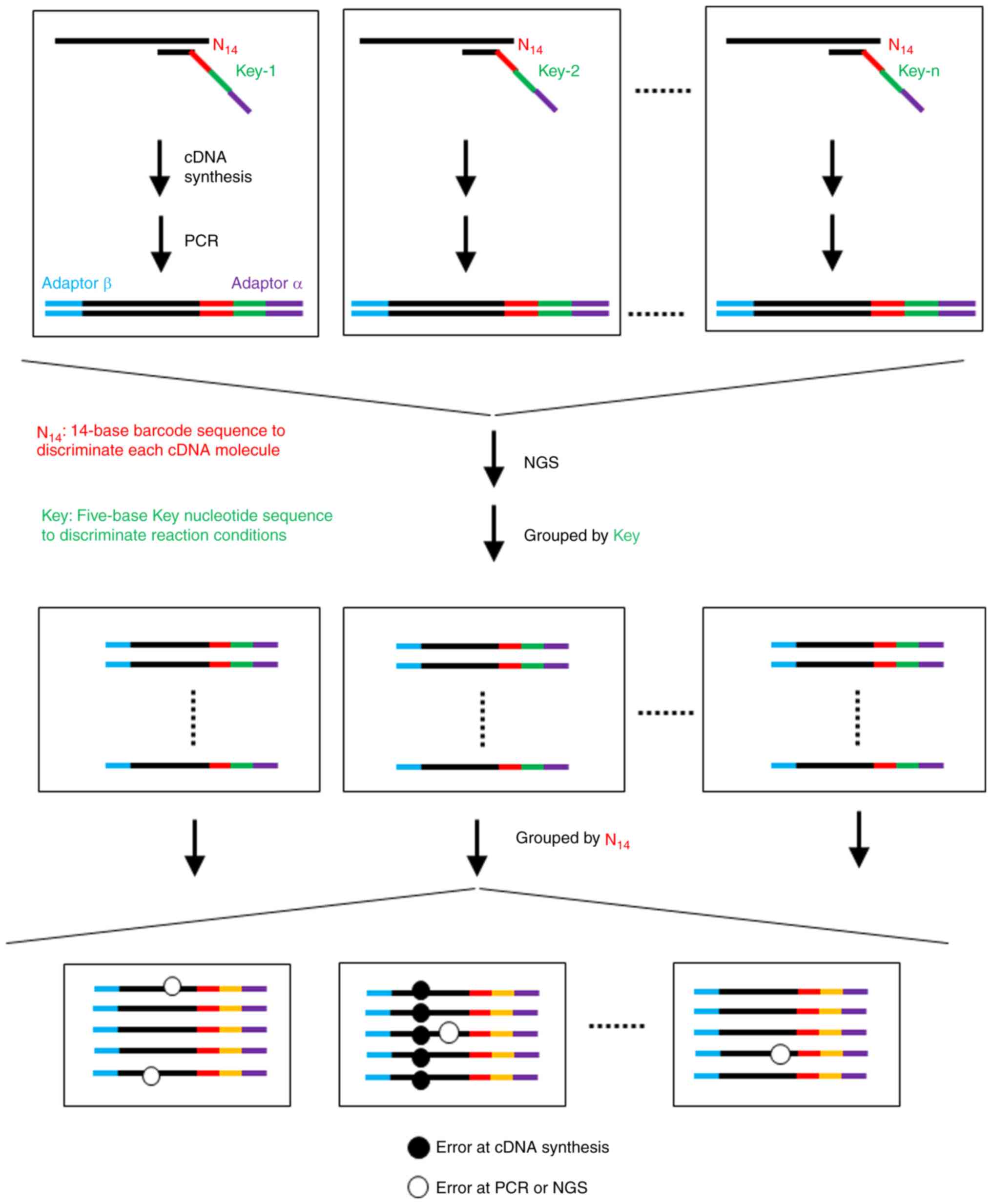
We used NGS to determine the error rate of cDNA
synthesis (63,64). As shown in Fig. 4, cDNA was synthesized from a
standard RNA with a primer possessing a tag of 14 randomized bases.
All sequence reads are grouped based on tag sequences. By analyzing
all sequence reads in the same group, each mutation revealed
whether the error was incorporated by cDNA synthesis or not. The
error rate obtained using this method of MMLV RT was
1.0×10−4 errors/base and that with HIV-1 RT was
2.6×10−4 errors/base (63), which was approximately 20% of
those reported using the M13 lacZ mutation assay (59). Notably, unlike the M13 lacZ
mutation assay, the NGS-based mutation assay reveals the mutation
species and the frequency at each nucleotide position (63). This method may be effective in the
assessment of the fidelity of various RTs with different reaction
conditions: We reported that high concentrations of dNTP,
MgCl2, and Mn(OCOCH3)2 decreased
the fidelity, and these effects were obvious in reactions using
HIV-1 RT (64).
Fidelity of cDNA synthesis is important in clinical
diagnosis and in life science research. The issue raised is how
fidelity of RT and DNA polymerase can be ameliorated. One strategy
is to optimize the concentrations of the enzyme, salts, and dNTP in
the reaction solution. Another strategy is based on the studies
conducted on HIV-1 RT (65-67). The fidelity of HIV-1 RT is lower
than that of MMLV RT and AMV RT. One of the consequences of low
fidelity of HIV-1 RT is the emergence of drug-resistant HIV-1 RT
variants, such as K65R, R78A, and V75I. Interestingly, the
mutations that confer drug resitance to these variants increase the
fidelity of HIV-1 RT (65-67).
This suggests that introduction of the corresponding mutations in
MMLV RT or AMV RT may increase the fidelity, although such evidence
has not yet been reported.
6. Use of recombinase and single-strand
binding protein for isothermal DNA amplification
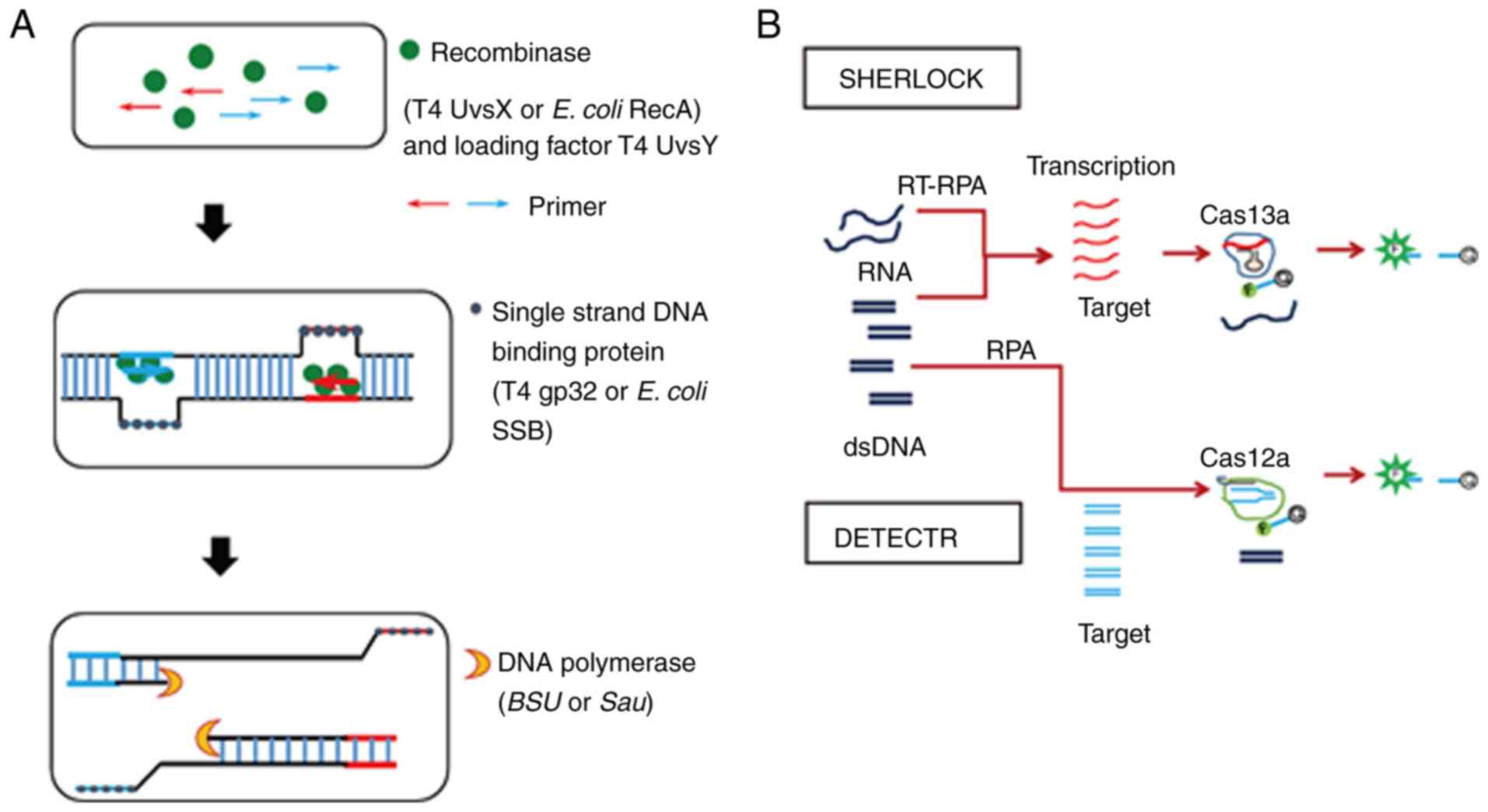
Recombinase polymerase amplification (RPA) is an
isothermal reaction that is conducted at a temperature between 37
and 42°C. RPA specifically amplifies a target DNA sequence with a
recombinase, a single-stranded DNA-binding protein (SSB), and a
strand-displacing polymerase (68). SSB binds to the primers and
prevents oligonucleotide primers from forming secondary structures.
Recombinase binds to the primers in the presence of ATP and with
the assistance of the loading factor, T4 UvsY, which was originally
identified as the T4 recombination mediator protein (69). The primers of the resulting
complex bind to the homologous sequences of the DNA template using
the ATP hydrolyzing activity of recombinase. In addition, SSB binds
to the dispatched strand, and strand-displacing polymerase extends
the primer. Thus, the synthesis of a new DNA strand occurs
(Fig. 5A).
In the first report of RPA in 2006 (70), T4 uvsX and T4 uvsY were used as
recombinase, T4 gp32 was used as SSB, and Bacillus subtilis
polymerase was used as strand-displacement DNA polymerase (Table I). Since then, RPA has been widely
used to detect various targets. At present, the RPA kit is
commercially available from TwistDX (Cambridge). One of the merits
of RPA over other isothermal nucleic acid amplification methods is
that the reaction occurs at the human body temperature (37°C). RPA
has the potential to eliminate the use of specialized equipment to
provide the required temperature. Thus, RPA may be the most ideal
nucleic acid amplification method for use in point-of-care
diagnosis. Indeed, a number of RPA targets reported to date are
pathogenic organisms including Mycobacterium tuberculosis
(71,72), Chlamydia trachomatis
(73), Streptococcus
pneumoniae (74), and
Leishmania donovani (75).
In accordance with this trend, various technologies
have been combined with RPA. For example, cutaneous leishmaniasis
was detected using an FTA card, a paper-based card commercialized
by GE Healthcare for the isolation and storage of nucleic acids,
and loop-mediated isothermal amplification (LAMP) (76,77). Lateral flow assay (78), enzyme-linked oligonucleotide assay
(79), and electrochemical method
(80) were used for end-point
detection of RPA amplicons, whereas solid phase amplification was
used for the real-time detection of RPA amplicons (81).
Clustered, regularly interspaced, short, palindromic
repeats (CRISPR)/CRISPR-associated (CAS) systems were originally
identified as an RNA-guided genetic silencing system in bacteria
and archaea (82). At present,
CRISPR/CAS9 is widely used in genome engineering. CRISPR-Cas13a and
CRISPR-Cas12a have been applied to RPA (Fig. 5B). Specific high sensitivity
enzymatic reporter unlocking (SHERLOCK) was established using
Cas13a, an RNA-guided RNase that cleaves its specific target as
well as the nearby non-targeted RNAs (collateral effect). The
collateral cleavage enables release of the quenched fluorescent
reporter (83). A multiplexed
detection system was also established using Cas13, Cas12a, and Csm6
(84). Use of SHERLOCK allowed
detection of Zika virus (sensitivity 2 aM) and that of a single
nucleotide polymorphism of a human gene (83,84). DNA endonuclease-targeted CRISPR
transreporter (DETECTR) was established using CAS12a, an RNA-guided
DNase. The DETECTR detected human papillomavirus (HPV) 16 and 18 at
attomolar levels (85). These
approaches may thus serve as valuable tools to increase the
sensitivity of RPA and provide a means for developing novel
point-of-care diagnosis with high sensitivity and rapidness.
7. Other considerable factors involved in
nucleic acid amplification
Various factors are known to be involved in
enzymatic reactions, and such factors include organic solvents.
Enzymes are generally inactivated by organic solvents, but use of
organic additives in enzymatic reactions can sometimes make
previously problematic processes feasible. Indeed, various organic
additives have been used to improve reaction efficiency and
specificity in PCR (86,87). Dimethyl sulfoxide (DMSO) and
formamide have been used to improve specificity for the reaction
with a G+C-rich DNA (88,89). In cDNA synthesis, DMSO and
formamide increased the reaction efficiency to some extent
(90).
Since nucleic acids are highly negatively charged,
they may be affected by positively charged small molecules such as
polyamines. It was initially reported that spermidine was not
beneficial in PCR (91). However,
subsequent reports showed that spermidine prevents PCR inhibition
problems encountered while analyzing clinical stool samples
(92,93). By optimizing the effects of these
polar molecules, the efficiency of nucleic acid amplification is
expected to further improve.
8. Conclusions and future perspectives
Despite being a widespread analytical method both in
fundamental research and clinical diagnosis, there are limitations
in nucleic acid amplification, which are represented by
false-positive and false-negative results. Many efforts are still
being devoted to improve the sensitivity, specificity, rapidness,
and accuracy of nucleic acid amplification. The catalytic mechanism
of nucleic acid-related enzymes has been extensively investigated
by means of X-ray crystallography, kinetic analysis, and
site-directed mutagenesis, leading to the generation of enzymes
exhibiting extremely high activity and stability. Such enzymes and
optimized reaction conditions offer many advantages that can be
expected to enhance the efficiency of nucleic acid amplification
tests, which may meet the increasing demand of point-of-care
diagnosis both in developed and developing countries.
Funding
This review was supported by SENTAN (K.Y., I.Y.,
S.F.) from Japan Science and Technology Agency, Grants-in-Aid for
Scientific Research (grant nos. 21580110, 18K19839 and 18K19839 for
K.Y.) from Japan Society for the Promotion of Science,
Emerging/re-emerging infectious disease project of Japan (K.Y.,
I.Y., S.F.) from Japan Agency for Medical Research and Development
(grant no. JP20fk0108143 for K.Y., I.Y., S.F.) and Grant Program
for Biomedical Engineering Research (K.Y., I.Y., S.F.) from
Nakatani Foundation, Japan.
Availability of data and materials
Not applicable.
Authors' contributions
KY, IY and SF contributed to conceiving and
designing the study, drafted and wrote the manuscript. All authors
have read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have not competing
interests.
Acknowledgements
We would like to thank Dr Kenji Kojima, Division of
Food Science and Biotechnology, Graduate School of Agriculture,
Kyoto University for insightful comments.
Abbreviations:
|
AMV
|
avian myeloblastosis virus
|
|
DETECTR
|
DNA endonuclease-targeted CRISPR
transreporter
|
|
HAD
|
helicase-dependent isothermal DNA
amplification
|
|
HIV
|
human immunodeficiency virus
|
|
HPV
|
human papillomavirus
|
|
LAMP
|
loop-mediated isothermal
amplification
|
|
MMLV
|
Moloney murine leukemia virus
|
|
NASBA
|
nucleic acid sequence-based
amplification
|
|
NGS
|
next-generation sequencing
|
|
RCA
|
rolling circle amplification
|
|
RPA
|
recombinase polymerase
amplification
|
|
RT
|
reverse transcriptase
|
|
SDA
|
strand displacement amplification
|
|
SHERLOCK
|
specific high sensitivity enzymatic
reporter unlocking
|
References
|
1
|
Seki M, Kim CK, Hayakawa S and Mitarai S:
Recent advances in tuberculosis diagnostics in resource-limited
settings. Eur J Clin Microbiol Infect Dis. 37:1405–1410. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhao J, Chang L and Wang L: Nucleic acid
testing and molecular characterization of HIV infections. Eur J
Clin Microbiol Infect Dis. 38:829–842. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mullis KB and Faloona FA: Specific
synthesis of DNA in vitro via a polymerase-catalyzed chain
reaction. Methods Enzymol. 155:335–350. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kievits T, van Gemen B, van Strijp D,
Schkkink P, Dircks M, Adriaanse H, Malek L, Sooknanan R and Lens P:
NASBA isothermal enzymatic in vitro nucleic acid amplification
optimized for the diagnosis of HIV-1 infection. J Virol Methods.
35:273–286. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Walker GT, Fraiser MS, Schram JL, Little
MC, Nadeau JG and Malinowski DP: Strand displacement
amplification-an isothermal, in vitro DNA amplification technique.
Nucleic Acids Res. 20:1691–1696. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lizardi PM, Huang X, Zhu Z, Bray-Ward P,
Thomas DC and Ward DC: Mutation detection and single-molecule
counting using isothermal rolling-circle amplification. Nat Genet.
19:225–232. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vincent M, Xu Y and Kong H:
Helicase-dependent isothermal DNA amplification. EMBO Rep.
5:795–800. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Notomi T, Okayama H, Masubuchi H, Yonekawa
T, Watanabe K, Amino N and Hase T: Loop-mediated isothermal
amplification of DNA. Nucleic Acids Res. 28:E632000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yan L, Zhou J, Zheng Y, Gamson AS, Roembke
BT, Nakayama S and Sintim HO: Isothermal amplified detection of DNA
and RNA. Mol Biosyst. 10:970–1003. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chien A, Edgar DB and Trela JM:
Deoxyribonucleic acid polymerase from the extreme thermophile.
Thermus aquaticus J Bacteriol. 127:1550–1557. 1976. View Article : Google Scholar
|
|
11
|
Pavlov AR, Pavlova NV, Kozyavkin SA and
Slesarev AI: Recent developments in the optimization of
thermostable DNA polymerases for efficient applications. Trends
Biotechnol. 22:253–260. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tabor S and Richardson CC: A single
residue in DNA polymerases of the Escherichia coli DNA polymerase I
family is critical for distinguishing between deoxy-and
dideoxyribonucleotides. Proc Natl Acad Sci USA. 92:6339–6343. 1995.
View Article : Google Scholar
|
|
13
|
Pavlov AR, Belova GI, Kozyavkin SA and
Slesarev AI: Helix-hairpin-helix motifs confer salt resistance and
processivity on chimeric DNA polymerases. Proc Natl Acad Sci USA.
99:13510–13515. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mallet F, Oriol G, Mary C, Verrier B and
Mandrand B: Continuous RT-PCR using AMV-RT and Taq DNA polymerase:
Characterization and comparison to uncoupled procedures.
Biotechniques. 18:678–687. 1995.PubMed/NCBI
|
|
15
|
Kimmel AR and Berger SL: Preparation of
cDNA and the generation of cDNA libraries: Overview. Methods
Enzymol. 152:307–316. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Georgiadis MM, Jessen SM, Ogata CM,
Telesnitsky A, Goff SP and Hendrickson WA: Mechanistic implications
from the structure of a catalytic fragment of Moloney murine
leukemia virus reverse transcriptase. Structure. 3:879–892. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Das D and Georgiadis MM: The crystal
structure of the monomeric reverse transcriptase from Moloney
murine leukemia virus. Structure. 12:819–829. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yasukawa K, Nemoto D and Inouye K:
Comparison of the thermal stabilities of reverse transcriptases
from avian myeloblastosis virus and Moloney murine leukaemia virus.
J Biochem. 143:261–268. 2008. View Article : Google Scholar
|
|
19
|
Kotewicz ML, Sampson CM, D'Alessio JM and
Gerard GF: Isolation of cloned Moloney murine leukemia virus
reverse transcriptase lacking ribonuclease H activity. Nucleic
Acids Res. 16:265–277. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gerard GF, Potter RJ, Smith MD, Rosenthal
K, Dhariwal G, Lee J and Chatterjee DK: The role of template-primer
in protection of reverse transcriptase from thermal inactivation.
Nucleic Acids Res. 30:3118–3129. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mizuno M, Yasukawa K and Inouye K: Insight
into the mechanism of the stabilization of Moloney murine leukaemia
virus reverse transcriptase by eliminating RNase H activity. Biosci
Biotechnol Biochem. 74:440–442. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yasukawa K, Mizuno M, Konishi A and Inouye
K: Increase in thermal stability of Moloney murine leukaemia virus
reverse transcriptase by site-directed mutagenesis. J Biotechnol.
150:299–306. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Konishi A, Ma X and Yasukawa K:
Stabilization of Moloney murine leukemia virus reverse
transcriptase by site-directed mutagenesis of the surface residue
Val433. Biosci Biotechnol Biochem. 78:147–150. 2014. View Article : Google Scholar
|
|
24
|
Baba M, Kakue R, Leucht C, Rasor P, Walch
H, Ladiges D, Bell C, Kojima K, Takita T and Yasukawa K: Further
increase in thermostability of Moloney murine leukemia virus
reverse transcriptase by mutational combination. Protein Eng Des
Sel. 30:551–557. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Arezi B and Hogrefe H: Novel mutations in
Moloney murine leukemia virus reverse transcriptase increase
thermostability through tighter binding to template-primer. Nucleic
Acids Res. 37:473–481. 2009. View Article : Google Scholar :
|
|
26
|
Baranauskas A, Paliksa S, Alzbutas G,
Vaitkevicius M, Lubiene J, Letukiene V, Burinskas S, Sasnauskas G
and Skirgaila R: Generation and characterization of new highly
thermostable and processive M-MuLV reverse transcriptase variants.
Protein Eng Des Sel. 25:657–668. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Katano Y, Li T, Baba M, Nakamura M, Ito M,
Kojima K, Takita T and Yasukawa K: Generation of thermostable
Moloney murine leukemia virus reverse transcriptase variants using
site saturation mutagenesis library and cell-free protein
expression system. Biosci Biotechnol Biochem. 81:2339–2345. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Konishi A, Nemoto D, Yasukawa K and Inouye
K: Comparison of the thermal stabilities of the αβ heterodimer and
the α subunit of avian myeloblastosis virus reverse transcriptase.
Biosci Biotechnol Biochem. 75:1618–1620. 2011. View Article : Google Scholar
|
|
29
|
Konishi A, Yasukawa K and Inouye K:
Improving the thermal stability of avian myeloblastosis virus
reverse transcriptase α-subunit by site-directed mutagenesis.
Biotechnol Lett. 34:1209–1215. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yasukawa K, Agata N and Inouye K:
Detection of cesA mRNA from Bacillus cereus by RNA-specific
amplification. Enzyme Microb Technol. 46:391–396. 2009. View Article : Google Scholar
|
|
31
|
Okano H, Katano Y, Baba M, Fujiwara A,
Hidese R, Fujiwara S, Yanagihara I, Hayashi T, Kojima K, Takita T
and Yasukawa K: Enhanced detection of RNA by MMLV reverse
transcriptase coupled with thermostable DNA polymerase and DNA/RNA
helicase. Enzyme. Microb Technol. 96:111–120. 2017. View Article : Google Scholar
|
|
32
|
Baase WA, Liu L, Tronrud DF and Matthews
BW: Lessons from the lysozyme of phage T4. Protein Sci. 19:631–641.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Astatke M, Ng K, Grindley ND and Joyce CM:
A single side-chain prevents Escherichia coli DNA polymerase I
(Klenow fragment) from incorporating ribonucleotides. Proc Natl
Acad Sci USA. 95:3402–3407. 1998. View Article : Google Scholar
|
|
34
|
Gardner AF and Jack WE: Determinants of
nucleotide sugar recognition in an archaeon DNA polymerase. Nucleic
Acids Res. 27:2545–2553. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lin TC, Wang CX, Joyce CM and Konigsberg
WH: 3′-5′ Exonucleolytic activity of DNA polymerases: Structural
features that allow kinetic discrimination between ribo- and
deoxyribo-nucleotide residues. Biochemistry. 40:8749–8755. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lam WC, Thompson EH, Potapova O, Sun XC,
Joyce CM and Millar DP: 3′-5′ Exonuclease of Klenow fragment: Role
of amino acid residues within the single-stranded DNA binding
region in exonucleolysis and duplex DNA melting. Biochemistry.
41:3943–3951. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shandilya H, Griffiths K, Flynn EK,
Astatke M, Shih PJ, Lee JE, Gerard GF, Gibbs MD and Bergquist PL:
Thermophilic bacterial DNA polymerases with reverse-transcriptase
activity. Extremophiles. 8:243–251. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Myers TW and Gelfand DH: Reverse
transcription and DNA amplification by a Thermus thermophilus DNA
polymerase. Biochemistry. 30:7661–7666. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ong JL, Loakes D, Jaroslawski S, Too K and
Holliger P: Directed evolution of DNA polymerase, RNA polymerase
and reverse transcriptase activity in a single polypeptide. J Mol
Biol. 361:537–550. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kranaster R, Drum M, Engel N, Weidmann M,
Hufert FT and Marx A: One-step RNA pathogen detection with reverse
tran-scriptase activity of a mutated thermostable Thermus aquaticus
DNA polymerase. Biotechnol J. 5:224–231. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jozwiakowski SK and Connolly BA: A
modified family-B archaeal DNA polymerase with reverse
transcriptase activity. Chembiochem. 12:35–37. 2011. View Article : Google Scholar
|
|
42
|
Schönbrunner NJ, Fiss EH, Budker O,
Stoffel S, Sigua CL, Gelfand DH and Myers TW: Chimeric thermostable
DNA polymerases with reverse transcriptase and attenuated 3′-5′
exonuclease activity. Biochemistry. 45:12786–12795. 2006.
View Article : Google Scholar
|
|
43
|
Sano S, Yamada Y, Shinkawa T, Kato S,
Okada T, Higashibata H and Fujiwara S: Mutations to create
thermostable reverse transcriptase with bacterial family A DNA
polymerase from Thermotoga petrophila K4. J Biosci Bioeng.
113:315–321. 2012. View Article : Google Scholar
|
|
44
|
Cline J, Braman JC and Hogrefe HH: PCR
fidelity of pfu DNA polymerase and other thermostable DNA
polymerases. Nucleic Acids Res. 24:3546–3551. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Takagi M, Nishioka M, Kakihara H,
Kitabayashi M, Inoue H, Kawakami B, Oka M and Imanaka T:
Characterization of DNA polymerase from Pyrococcus sp. strain KOD1
and its application to PCR. Appl Environ Microbiol. 63:4504–4510.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Firbank SJ, Wardle J, Heslop P, Lewis RJ
and Connolly BA: Uracil recognition in archaeal DNA polymerases
captured by X-ray crystallography. J Mol Biol. 381:529–539. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ellefson JW, Gollihar J, Shroff R, Shivram
H, Iyer VR and Ellington AD: Synthetic evolutionary origin of a
proofreading reverse transcriptase. Science. 352:1590–1593. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Okano H, Baba M, Kawato K, Hidese R,
Yanagihara I, Kojima K, Takita T, Fujiwara S and Yasukawa K: High
sensitive RNA detection by one-step RT-PCR using the genetically
engineered variant of DNA polymerase with reverse transcriptase
activity from hyperthermophilies. J Biosci Bioeng. 125:275–281.
2018. View Article : Google Scholar
|
|
49
|
Singleton MR, Dillingham MS and Wigley DB:
Structure and mechanism of helicases and nucleic acid translocases.
Annu Rev Biochem. 76:23–50. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
An L, Tang W, Ranalli TA, Kim HJ, Wytiaz J
and Kong H: Characterization of a thermostable UvrD helicase and
its participation in helicase-dependent amplification. J Biol Chem.
280:28952–28958. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jeong YJ, Park K and Kim DE: Isothermal
DNA amplification in vitro: The helicase-dependent amplification
system. Cell Mol Life Sci. 66:3325–3336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Artiushin S, Tong Y, Timoney J, Lemieux B,
Schlegel A and Kong H: Thermophilic helicase-dependent DNA
amplification using the IsoAmp™ SE experimental kit for rapid
detection of Streptococcus equi subspecies equi in clinical
samples. J Vet Diagn Invest. 23:909–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Runyon GT and Lohman TM: Escherichia coli
helicase II (UvrD) protein can completely unwind fully duplex
linear and nicked circular DNA. J Biol Chem. 264:17502–17512.
1989.PubMed/NCBI
|
|
54
|
Fujiwara A, Kawato K, Kato S, Yasukawa K,
Hides R and Fujiwara S: Application for a euryarchaeota-specific
helicase from Thermococcus kodakarensis and its application for
noise reduction in PCR. Appl Environ Microbiol. 82:3022–3031. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Walker JE, Luyties O and Santangelo TJ:
Factor-dependent archaeal transcription termination. Proc Natl Acad
Sci USA. 114:E6767–E6773. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hidese R, Kawato K, Nakura Y, Fujiwara A,
Yasukawa K, Yanagihara I and Fujiwara S: Thermostable DNA helicase
improves the sensitivity of digital PCR. Biochem Biophys Res
Commun. 495:2189–2194. 2018. View Article : Google Scholar
|
|
57
|
Gutiérrez-Rivas M, Ibáñez Á, Martínez MA,
Domingo E and Menéndez-Arias L: Mutational analysis of Phe160
within the 'palm' subdomain of human immunodeficiency virus type 1
reverse transcriptase. J Mol Biol. 290:615–625. 1999. View Article : Google Scholar
|
|
58
|
Kati WM, Johnson KA, Jerva LF and Anderson
KS: Mechanism and fidelity of HIV reverse transcriptase. J Biol
Chem. 267:25988–25997. 1992.PubMed/NCBI
|
|
59
|
Bebenek K and Kunkel TA: Analyzing
fidelity of DNA polymerase. Methods Enzymol. 262:217–232. 1995.
View Article : Google Scholar
|
|
60
|
Shendure J and Ji H: Next-generation DNA
sequencing. Nat Biotechnol. 26:1135–1145. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Iida K, Jin H and Zhu JK: Bioinformatics
analysis suggests base modifications of tRNAs and miRNAs in
Arabidopsis thaliana. BMC Genomics. 10:1552009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Schmitt MW, Kennedy SR, Salk JJ, Fox EJ,
Hiatt JB and Loeb LA: Detection of ultra-rare mutations by
next-generation sequencing. Proc Natl Acad Sci USA.
109:14508–14513. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Yasukawa K, Iida K, Okano H, Hidese R,
Baba M, Yanagihara I, Kojima K, Takita T and Fujiwara S:
Next-generation sequencing-based analysis of reverse transcriptase
fidelity. Biochem Biophys Res Commun. 492:147–153. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Okano H, Baba M, Hidese R, Iida K, Li T,
Kojima K, Takita T, Yanagihara I, Fujiwara S and Yasukawa K:
Accurate fidelity analysis of the reverse transcriptase by a
modified next-generation sequencing. Enzyme Microb Technol.
115:81–85. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Barrioluengo V, Álvarez M, Barbieri D and
Menéndez-Arias L: Thermostable HIV-1 group O reverse transcriptase
variants with the same fidelity as murine leukaemia virus reverse
transcritpase. Biochem J. 436:599–607. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Álvarez M, Barrioluengo V, Afonso-Lehmann
RN and Menéndez-Arias L: Altered error specificity of RNase
H-deficient HIV-1 reverse transcriptases during DNA-dependent DNA
synthesis. Nucleic Acid Res. 41:4601–4612. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Garforth SJ, Domaoal RA, Lwatula C, Landau
MJ, Meyer AJ, Anderson KS and Prasad VR: K65R and K65A
substitutions in HIV-1 reverse transcriptase enhance polymerase
fidelity by decreasing both dNTP misinsertion and mispaired primer
extension efficiencies. J Mol Biol. 401:33–44. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Li J, Macdonald J and von Stetten F:
Review: A comprehensive summary of a decade development of the
recombinase polymerase amplification. Analyst. 144:31–67. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bleuit JS, Xu H, Ma Y, Wang T, Liu J and
Morrical SW: Mediator proteins orchestrate enzyme-ssDNA assembly
during T4 recombination-dependent DNA replication and repair. Proc
Natl Acad Sci USA. 98:8298–8305. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Piepenburg O, Williams CH, Stemple DL and
Armes NA: DNA detection using recombination proteins. PLoS Biol.
4:e2042006. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Boyle DS, McNerney R, Teng Low H, Leader
BT, Pérez-Osorio AC, Meyer JC, O'Sullivan DM, Brooks DG, Piepenburg
O and Forrest MS: Rapid detection of Mycobacterium tuberculosis by
recombinase polymerase amplification. PLoS One. 9. pp. e1030912014,
View Article : Google Scholar
|
|
72
|
Shin Y, Perera AP, Tang WY, Fu DL, Liu Q,
Sheng JK, Gu Z, Lee TY, Barkham T and Kyoung Park M: A rapid
amplification/detection assay for analysis of Mycobacterium
tuberculosis using an isothermal and silicon bio-photonic sensor
complex. Biosens Bioelectron. 68:390–396. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Krõlov K, Frolova J, Tudoran O,
Suhorutsenko J, Lehto T, Sibul H, Mäger I, Laanpere M, Tulp I and
Langel Ü: Sensitive and rapid detection of Chlamydia trachomatis by
recombinase polymerase amplification directly from urine samples. J
Mol Diagn. 16:127–135. 2014. View Article : Google Scholar
|
|
74
|
Clancy E, Higgins O, Forrest MS, Boo TW,
Cormican M, Barry T, Piepenburg O and Smith TJ: Development of a
rapid recombinase polymerase amplification assay for the detection
of Streptococcus pneumoniae in whole blood. BMC Infect Dis.
15:4812015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Mondal D, Ghosh P, Khan MA, Hossain F,
Böhlken-Fascher S, Matlashewski G, Kroeger A, Olliaro P, Abd El and
Wahed A: Mobile suitcase laboratory for rapid detection of
Leishmania donovani using recombinase polymerase amplification
assay. Parasit Vectors. 9:2812016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Sriworarat C, Phumee A, Mungthin M,
Leelayoova S and Siriyasatien P: Development of loop-mediated
isothermal amplification (LAMP) for simple detection of Leishmania
infection. Parasit Vectors. 8:5912015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Nzelu CO, Cáceres AG, Guerrero-Quincho S,
Tineo-Villafuerte E, Rodriquez-Delfin L, Mimori T, Uezato H,
Katakura K, Gomez EA, Guevara AG, et al: A rapid molecular
diagnosis of cutaneous leishmaniasis by colorimetric malachite
green-loop-mediated isothermal amplification (LAMP) combined with
an FTA card as a direct sampling tool. Acta Trop. 153:116–119.
2016. View Article : Google Scholar
|
|
78
|
Jauset-Rubio M, Tomaso H, El-Shahawi MS,
Bashammakh AS, Al-Youbi AO and O'Sullivan CK: Duplex lateral flow
assay for the simultaneous detection of yersinia pestis and
francisella tularensis. Anal Chem. 90:12745–12751. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Toldrà A, Jauset-Rubio M, Andree KB,
Fernández-Tejedor M, Diogène J, Katakis I, O'Sullivan CK and Campàs
M: Detection and quantification of the toxic marine microalgae
karlodinium veneficum and karlodinium armiger using recombinase
polymerase amplification and enzyme-linked oligonucleotide assay.
Anal Chim Acta. 1039:140–148. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Al-Madhagi S, Joda H, Jauset-Rubio M,
Ortiz M, Katakis I and O' Sullivan CK: Isothermal amplification
using modified primers for rapid electrochemical analysis of
coeliac disease associated DQB1*02 HLA allele. Anal
Biochem. 556:16–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Sabaté del Río J, Steylaerts T, Henry OYF,
Bienstman P, Stakenborg T, Van Roy W and O′Sullivan CK: Real-time
and label-free ring-resonator monitoring of solid-phase recombinase
polymerase amplification. Biosens Bioelectron. 73:130–137. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wiedenheft B, Sternberg SH and Doudna JA:
RNA-guided genetic silencing systems in bacteria and archaea.
Nature. 482:331–338. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Gootenberg JS, Abudayyeh OO, Lee JW,
Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer
NM, Freije CA, et al: Nucleic acid detection with
CRISPR-Cas13a/C2c2. Science. 356:438–442. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gootenberg JS, Abudayyeh OO, Kellner MJ,
Joung J, Collins JJ and Zhang F: Multiplexed and portable nucleic
acid detection platform with Cas13, Cas12a, and Csm6. Science.
360:439–444. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Chen JS, Ma E, Harrington LB, Da Costa M,
Tian X, Palefsky JM and Doudna JA: CRISPR-Cas12a target binding
unleashes indiscriminate single-stranded DNase activity. Science.
360:436–439. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Chakrabarti R and Schutt CE: The
enhancement of PCR amplification by low molecular weight amides.
Nucleic Acids Res. 29:2377–2381. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kovárová M and Dráber P: New specificity
and yield enhancer of polymerase chain reactions. Nucleic Acids
Res. 28:E702000. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Chester N and Marshak DR: Dimethyl
sulfoxide-mediated primer Tm reduction: A method for analyzing the
role of renaturation temperature in the polymerase chain reaction.
Anal Biochem. 209:284–290. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Sarkar G, Kapelner S and Sommer SS:
Formamide can dramatically improve the specificity of PCR. Nucleic
Acids Res. 18:74651990. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Yasukawa K, Konishi A and Inouye K:
Effects of organic solvents on the reverse transcription reaction
catalyzed by reverse transcriptases from avian myeloblastosis virus
and Moloney murine leukemia virus. Biosci Biotechnol Biochem.
74:1925–1930. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Ahokas H and Erkkilä MJ: Interference of
PCR amplification by the polyamines, spermine and spermidine. PCR
Methods Appl. 3:65–68. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Roperch JP, Benzekri K, Mansour H and
Incitti R: Improved amplification efficiency on stool samples by
addition of spermi-dine and its use for non-invasive detection of
colorectal cancer. BMC Biotechnol. 15:412015. View Article : Google Scholar
|
|
93
|
Kikuchi A, Sawamura T, Kawase N, Kitajima
Y, Yoshida T, Daimaru O, Nakakita T and Itoh S: Utility of
spermidine in PCR amplification of stool samples. Biochem Genet.
48:428–432. 2010. View Article : Google Scholar : PubMed/NCBI
|