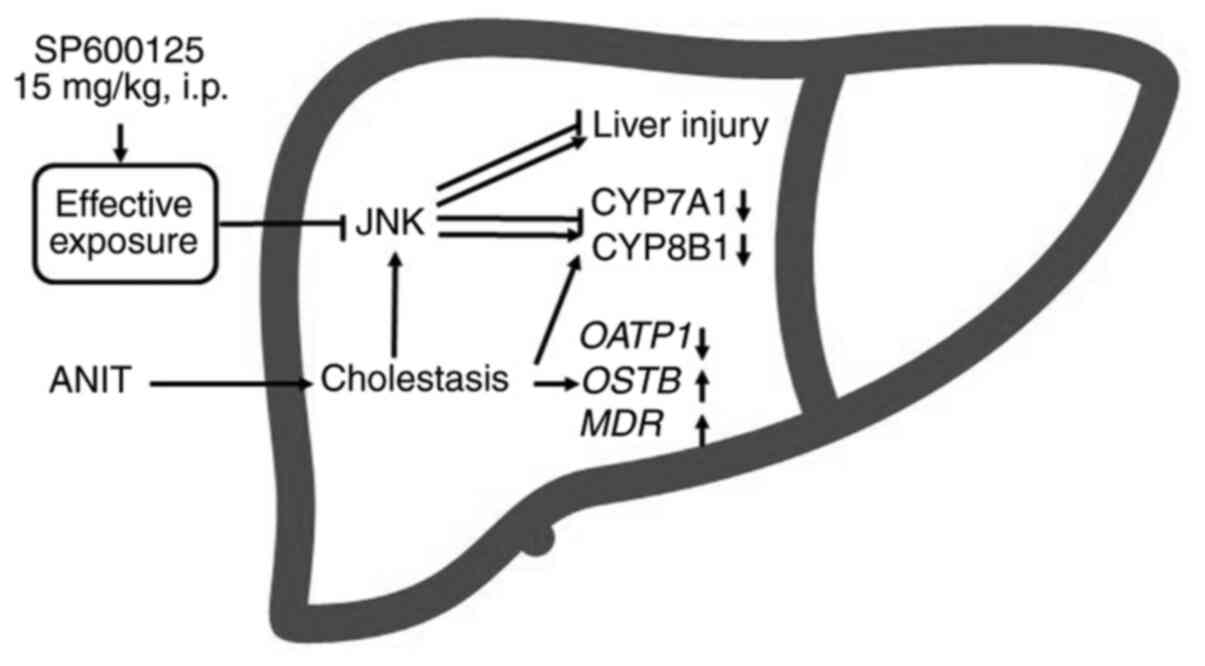
Introduction
c-Jun-N-terminal kinase (JNK) is a member of the
mitogen-activated protein kinase (MAPK) family and includes 3
isoforms in mammals, namely JNK1, JNK2 and JNK3 (1). JNK1 and JNK2 are expressed in almost
all cells, whereas JNK3 is mainly expressed in the brain, heart,
and testis (2). The JNK pathway
may be activated by cytokines, pathogens, toxins, drugs and
metabolic changes (3). JNK
activation is involved in cell death, differentiation,
proliferation, and tumorigenesis in hepatocytes (4). In hepatic macrophages and hepatic
stellate cells, JNK was revealed to contribute to liver
inflammation and fibrosis (5).
The JNK pathway also regulates inflammation, and insulin
resistance, which are associated with hepatic diseases such as
non-alcoholic steatosis, fibrosis, and hepatocellular carcinoma
(3).
SP600125 is widely used in biochemical studies as a
selective JNK inhibitor (6) and
it can block the activation of JNK to modulate the expression of
inflammatory factors (6-8). Additionally, this inhibitor has been
used in numerous cellular models to investigate the underlying
pathophysiological and pharmacological mechanisms. Incubation with
bile acid significantly reduced L02 cell viability, and this effect
was reduced by pre-treatment with 20 µM SP600125, which
involved downregulation of IL-1β and inhibition of JNK
phosphorylation (9). In MC3T3-E1
cells, 25 and 50 µM SP600125 reduced osteoblast apoptosis in
an LPS-induced model and restored differentiation of osteoblasts
(10).
In a rat model, administration of 30 mg/kg SP600125
by intraperitoneal (i.p.) injection once per day for 8 weeks
suppressed autophagy and attenuated insulin resistance by
inhibiting JNK signalling (11).
In C57BL/6 mice, a single dose of 10 and 30 mg/kg SP600125 by i.p.
injection inhibited 3-chloro-1,2-propanediol esters-induced
apoptosis by downregulating the JNK/p53 pathway in tubular cells
(12). In a type 1 diabetic mouse
model induced by streptozocin, 5 mg/kg SP600125 by intragastric
(i.g.) injection every other day for 3 months inhibited cardiac
fibrosis, oxidative stress, endoplasmic reticulum stress, and cell
death by inhibiting the JNK pathway (13,14). However, the optimal administration
regimen of SP600125 inhibiting the JNK pathway and its exposure
profile have yet to be determined.
Cholestasis is caused by disrupted structure and
impaired function of the hepatobiliary system, which occurs in a
number of clinical disorders (15). Patients with cholestasis may
even-tually develop sepsis, immune depression, cardiovascular,
hepatic and renal failure (16).
Cholestatic liver injury is closely associated with several
inflammatory pathways (17,18). It was reported that the synthesis
and excretion of bile acids increased the levels of TNF-α and IL-1β
in hepatic Kupffer cells (19).
In addition, cytokines, cholic acid and deoxycholic acid have been
revealed to activate the JNK/c-Jun pathway (20,21). In cellular models, the JNK pathway
was also found to regulate bile acid metabolism (22,23). Thus, α-naphthylisothiocyanate
(ANIT)-induced cholestatsis is a suitable model for investigating
the inhibitory effect of SP600125 on the JNK pathway.
In the present study, ANIT-induced cholestatic liver
injury was used to evaluate the inhibitory effect of SP600125
administered by i.p. and i.g. injections, on the JNK pathway. The
data were revealed to be informative in designing biomedical
experiments where SP600125 may be used.
Materials and methods
Chemicals and reagents
ANIT was purchased from Sigma-Aldrich; Merck KGaA.
Alkaline phosphatase (ALP), total bile acid (TBA), alanine
aminotransferase (ALT), and aspartate aminotransferase (AST) assay
kits were purchased from Ningbo Ruiyuan Biotechnology Co., Ltd.
TRIzol® and the reverse transcription kit were purchased
from Invitrogen (Thermo Fisher Scientific, Inc). The LightCycle 480
SYBR Green I Master Mix was obtained from Roche Diagnostics.
Antibodies against phosphorylated (p)-MAPK kinase 4 (MKK4)
(1:1,000; product no. 4514) and total (t)-MKK4 (1:1,000; product
no. 9152), t-JNK (1:1,000; product no. 9252) and p-JNK (1:1,000;
product no. 9912), p-c-Jun (1:1,000; product no. 2361) and t-c-Jun
(1:1,000; product no. 9165), were obtained from Cell Signalling
Technology, Inc. Primary antibodies against CYP7A1 (1:1,000;
product code ab65596), CYP8B1(1:1,000; product code ab191910),
GAPDH (1:10,000; product code ab181602) were acquired from Abcam.
Secondary antibody IgG H&L (HRP) was also obtained from Abcam
(1:2,000; product code ab205718). All the other chemicals were of
the highest grade available from commercial sources.
Animals and treatment
A total of 50 mice were maintained under a 12-h
light/dark cycle with free access to water and a commercial diet.
The SPF room housing the mouse cages was set at 23±1°C with a
relative humidity of 60-70%. All the mice were allowed to acclimate
for 7 days before the experiments. The procedures performed were
approved by the Animal Care and Use Committee of Ningbo University.
Twenty of the mice were assigned into 4 groups (n=5/group) as
follows: Negative control (Con), ANIT/positive control (ANIT),
ANIT/SP600125-i.p. (A-SP-i.p.) and ANIT/SP600125-i.g. (A-SP-i.g.).
A single dose of 15 mg/kg SP600125 dissolved in corn oil was
administered by i.p. or i.g. injections to pre-treat the mice. At
30 min after SP600125 administration, a single dose of 75 mg/kg
ANIT in corn oil was injected to induce cholestasis. At 48 h after
the treatment, the mice were weighed and then killed by
asphyxiation after blood collection. To perform euthanasia, the
mouse cage was moved into a trans-parent polycarbonate chamber.
Compressed CO2 gas (purity >99%) was introduced at a
rate of 30% chamber volume per min using a CO2-specific
regulator. Death was confirmed by observing absence of respiratory
movement and faded eye colour. The chamber was cleaned thoroughly
to remove any potential risk for euthanasia. Liver tissues and the
gallbladder were harvested for calculation of liver and gallbladder
indices. Half of the isolated liver tissues were cut and fixed in
10% neutral buffered formalin at room temperature for 24 h. The
remaining liver tissues were immediately frozen and stored under at
−80°C.
Biochemical analysis and
histopathological assessment
The Multiskan GO (Thermo Fisher Scientific, Inc.)
was used to measure the TBA, ALP, ALT and AST levels in the serum
according to the manufacturer's instructions.
The liver tissues were dehydrated through serial
concentrations of alcohol (70, 80, 90 and 100%) and xylene,
followed by paraffin embedding at 60 C and cooled to −20 C. The
paraffin block was cut into 4-µm sections which were then
stained with hematoxylin for 3 min and eosin for 2 min at room
temperature. A BX51 light microscope (Olympus Corporation) at ×40
and ×400 magnification and ten serial sections per preparation were
used to randomly capture histopathological images.
Quantitative PCR (qPCR) and western blot
(WB) analysis
Processing of liver samples for the qPCR and WB
analysis followed previously published procedures with slight
modifications (18).
Specifically, a 5-µl PCR system was designed for the 384
plate. For qPCR analysis, the primers (Table SI) were also obtained from a
public database (http://mouseprim-erdepot.nci.nih.gov). This system
configuration followed the protocols provided by the manufacturer.
Melting curves were used to assess primer specificity. 18S rRNA was
used as an internal standard to calculate the relative
transcription level for each run.
Concentration-time profile of
SP600125
In a second pilot experiment, 30 mice were divided
into 6 groups (n=5/group), as follows: SP600125-i.p.-A (SP-i.p.-A),
SP600125-i.p.-B (SP-i.p.-B), SP600125-i.p.-C (SP-i.p.-C),
SP600125-i.g.-A (SP-i.g.-A) SP600125-i.g.-B (SP-i.g.-B) and
SP600125-i.g.-C (SP-i.g.-C). A single dose of 15 mg/kg SP600125 was
administered by i.p. and i.g. injections. Blood (30 µl) was
collected from the tail vein for exposure assessment. The blood
collection was performed at 0.3, 1.5, 3, 6 and 14 h following
SP600125 treatment in the SP-i.p.-A and SP-i.g.-A groups.
Similarly, the blood collection in the SP-i.p.-B and SP-i.g.-B
groups was performed at 0.5, 2, 8 and 18 h. For the SP-i.p.-C and
SP-i.g.-C groups, blood was collected at 1, 4, 10 and 24 h after
SP600125 treatment. The determination followed previously published
procedures (18).
Statistical analysis
All data were expressed as the mean ± SD. GraphPad
Prism (version 7.0; GraphPad Software, Inc.) for Windows was used
for the data analysis. Two-samples t-test was used to test the
difference between two groups, and Bonferroni's correction was
performed for all P-values. The Cmax and AUC were
calculated using Drug and Statistics (Version 3.0; Mathematical
Pharmacology Professional Committee of China, Shanghai, China)
(24).
Results
Cholestatic liver injury is
differentially inhibited
The levels of the biochemical markers ALP, TBA, ALT
and AST in the serum were all markedly increased in the ANIT group
compared with those in the control group (Fig. 1; all P<0.0001). The serum TBA
levels in the A-SP-i.p. and A-SP-i.g. groups were similar to those
in the ANIT group (Fig. 1A). In
the A-SP-i.p. group, but not in the A-SP-i.g. group, the ALP, ALT
and AST levels in serum were decreased compared with those in the
ANIT group (Fig. 1B-D; all
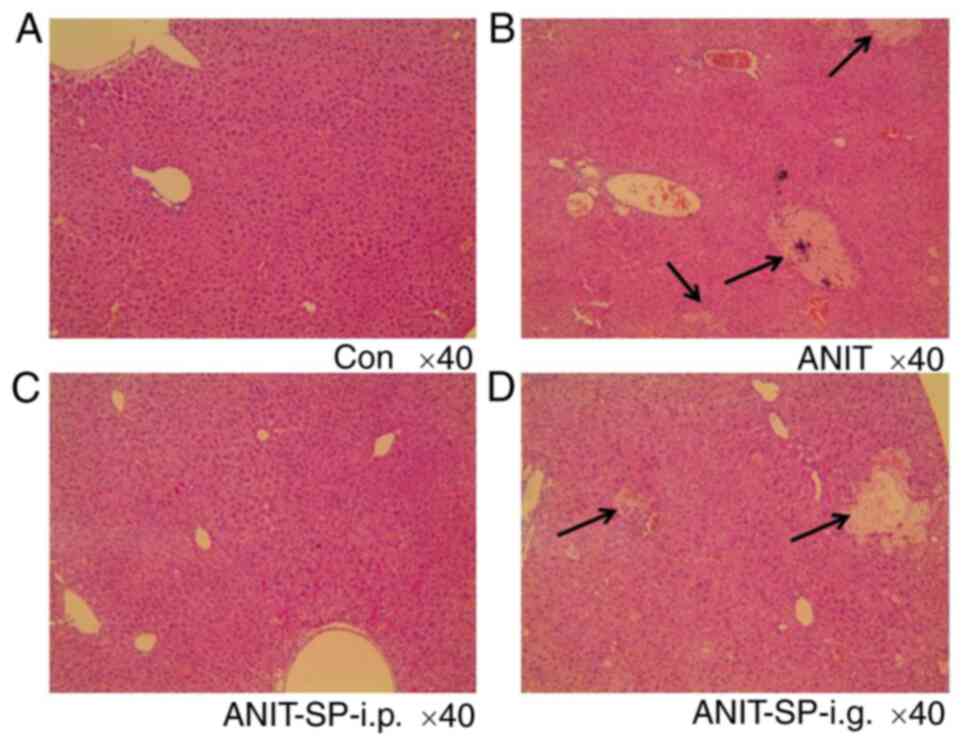
P<0.0001). The hepatic pathological analysis in the control
group revealed characteristic features (Fig. 2A). Loss of cellular boundaries,
degenerative changes and marked necrosis were identified in the
ANIT group (Fig. 2B). The
A-SP-i.p. group, but not the A-SP-i.g. group, exhibited no
significant difference compared with the control group (Fig. 2C and D). Thus, pre-treatment with
SP600125 at a dose of 15 mg/kg by i.p. injection reversed the liver
injury induced by ANIT in mice, although the serum TBA level did
not improve. However, the aforementioned protective effect was not
observed in the mice pre-treated with SP600125 at 15 mg/kg by i.g.
injection.
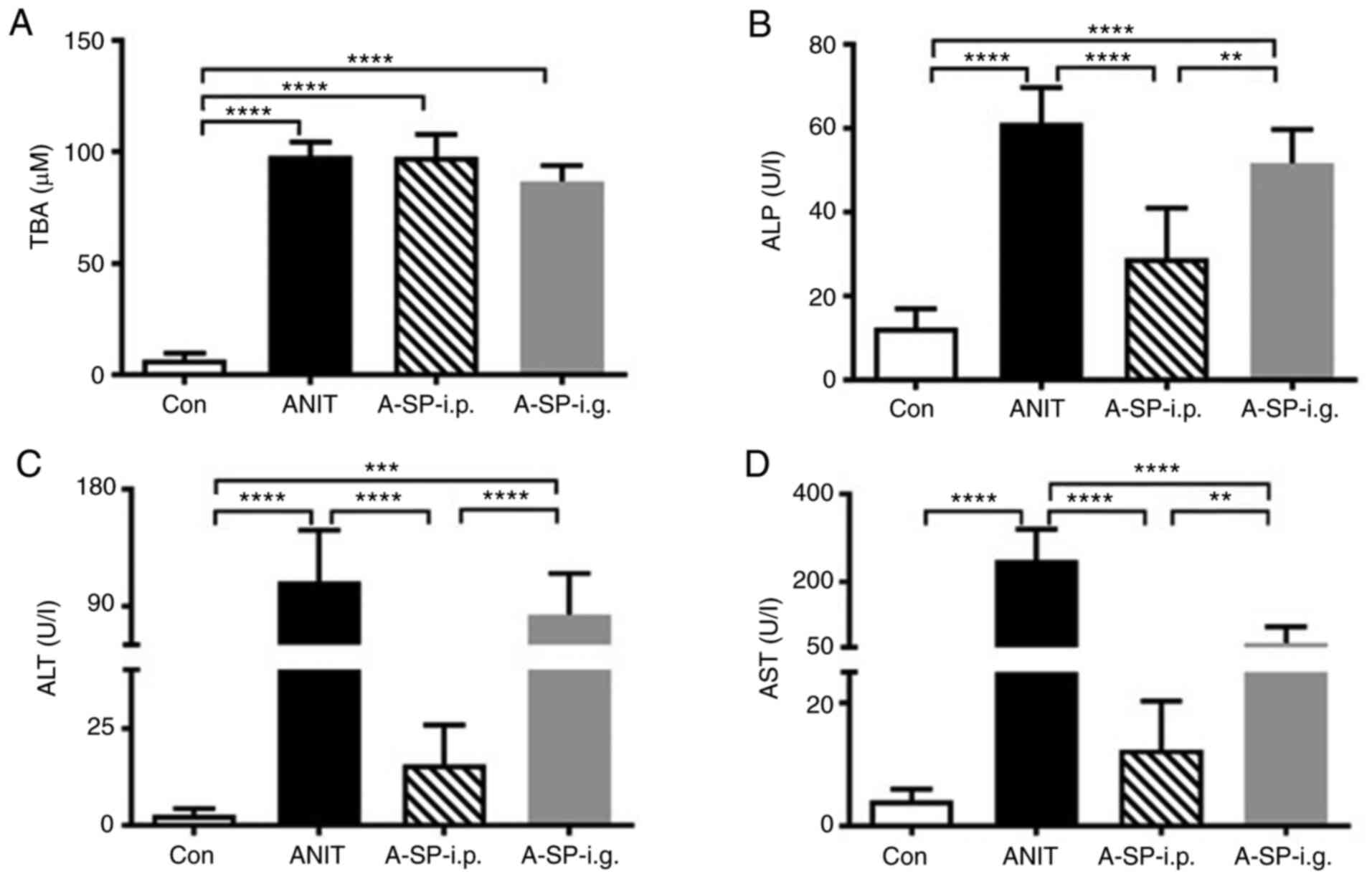 | Figure 1Biochemical markers indicating the
inhibitive effect of SP600125 against intrahepatic cholestasis. (A)
TBA, (B) ALP, (C) ALT and (D) AST in the mouse serum of four groups
48 h after ANIT administration. The data are expressed as the mean
± SD. (n=5). **P<0.01, ***P<0.001 and
****P<0.0001. TBA, total bile acid; ALP, alkaline
phosphatase; ALT, alanine aminotransferase; AST, aspartate
aminotransferase; ANIT, α-naphthylisothiocyanate; A-SP-i.p.,
ANIT/SP600125-i.p.; A-SP-i.g., ANIT/SP600125-i.g.; i.p,
intraperitoneal; i.g., intragastric; Con, negative control. |
Adaptation of bile acid metabolism and
transport
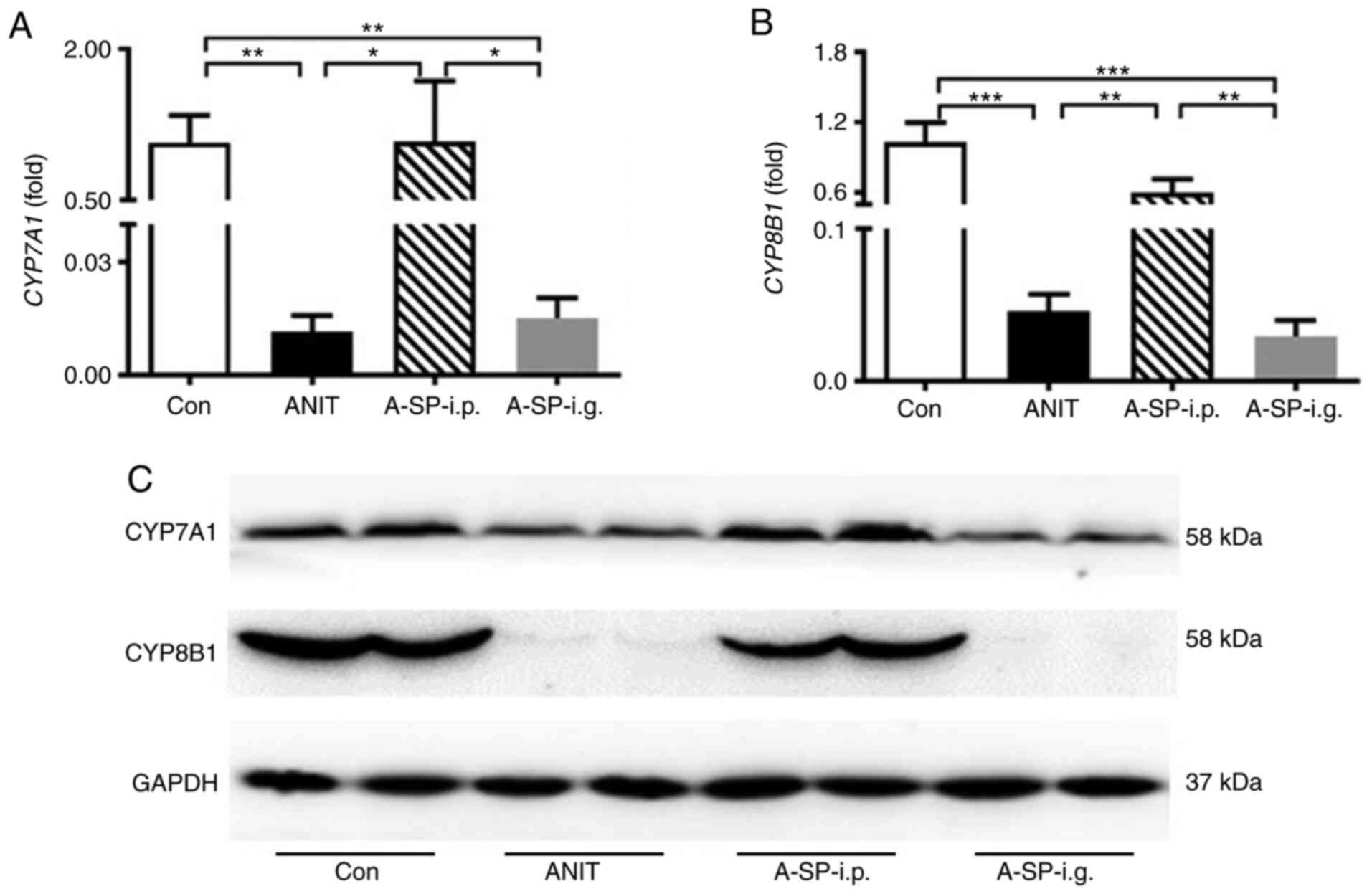
The rate-limiting enzymes CYP7A1 and CYP8B1 play an
important role in bile acid homeostasis (25). ANIT challenge decreased the
CYP7A1 and CYP8B1 mRNA levels in the ANIT group
compared with those in the control group (Fig. 3A and B; P<0.01 and P<0.001).
In the A-SP-i.p. group, but not in the A-SP-i.g. group, both
CYP7A1 and CYP8B1 mRNA levels were increased compared
with those in the ANIT group (Fig. 3A
and B; P<0.05 and P<0.01). In the A-SP-i.g. group,
CYP7A1 and CYP8B1 mRNA levels were lower compared
with those in the A-SP-i.p. group, as the adaptation remained
strong (P<0.05 and P<0.01). In the WB analysis, the protein
levels of CYP7A1 and CYP8B1 in the A-SP-i.g. group were decreased
compared with those in the control group, similar to those in the
ANIT group. In the A-SP-i.p. group, these levels were almost
identical to those in the control group (Fig. 3C).
Organic anion-transporting polypeptide 1 (OATP1),
heteromeric organic solute transporter β subunit (OSTB), multidrug
resistance 2 (MDR2) and multidrug resistance 1A (MDR1A) are
involved in bile acid efflux (25). OATP1 was decreased, and
OSTB, MDR2, MDR1A were all increased by ANIT
treatment. However, no difference of these transporters was
observed among the A-SP-i.p., A-SP-i.g. and ANIT groups. The
changes in bile acid transporters were similar in the three groups
challenged by ANIT, which was mainly considered to be adaptive
responses (Fig. 4A-D). Other
genes involved in bile acid metabolism were not obviously affected
(data not shown).
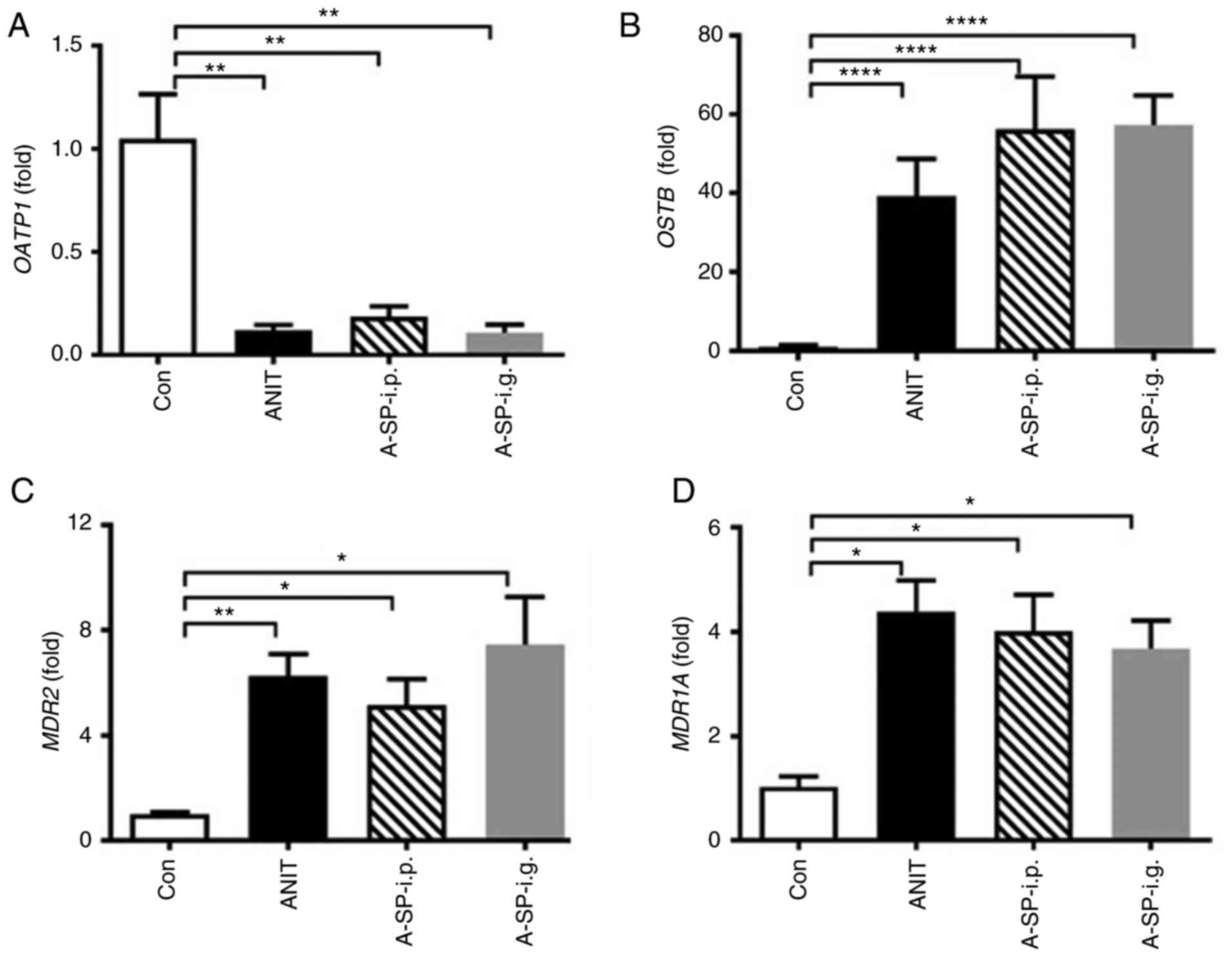 | Figure 4Adaptation of genes involved in bile
acid efflux transport. (A) OATP1, (B) OSTB, (C)
MDR2 and (D) MDR1A mRNA levels in four groups 48 h
after the ANIT administration. mRNAs levels in the vehicle-treated
control mice were set as 1 and the results are expressed as the
mean ± SD. (n=5). *P<0.05, **P<0.01 and
****P<0.0001. OATP1, organic
anion-transporting polypeptide 1; OSTB, organic solute
transporter β subunit; MDR2, multidrug resistance 2;
MDR1A, multidrug resistance 1A; Con, negative control; ANIT,
α-naphthylisothiocyanate; A-SP-i.p., ANIT/SP600125-i.p.; A-SP-i.g.,
ANIT/SP600125-i.g.; i.p, intraperitoneal; i.g., intragastric. |
Differential inflammation responses
between the two administration routes
The mRNA expression levels of IL-6,
IL-1β and ICAM-1 was revealed to be significantly
increased following ANIT treatment (Fig. 5A-C, P<0.05, P<0.001 and
P<0.01). IL-6 and IL-1β mRNA levels were decreased
in the A-SP-i.p. and A-SP-i.g. groups, but the decrease was greater
in the A-SP-i.p. group compared with the ANIT group. The protective
inflammation factor IL-10 was upregulated in the ANIT and
A-SP-i.g. groups compared with the control group, which was not
observed in the A-SP-i.p. group (Fig.
5D). In terms of apoptosis, the BCL2 and P53 mRNA
levels were increased following administration of ANIT at 75 mg/kg,
but both were suppressed by pretreatment with SP600125, without a
significant difference between the two administration routes
(Fig. 5E and F). These data
indicated that inflammation developed in the mice challenged with
ANIT, but it was differentially blocked by i.p. and i.g.
administration of SP600126.
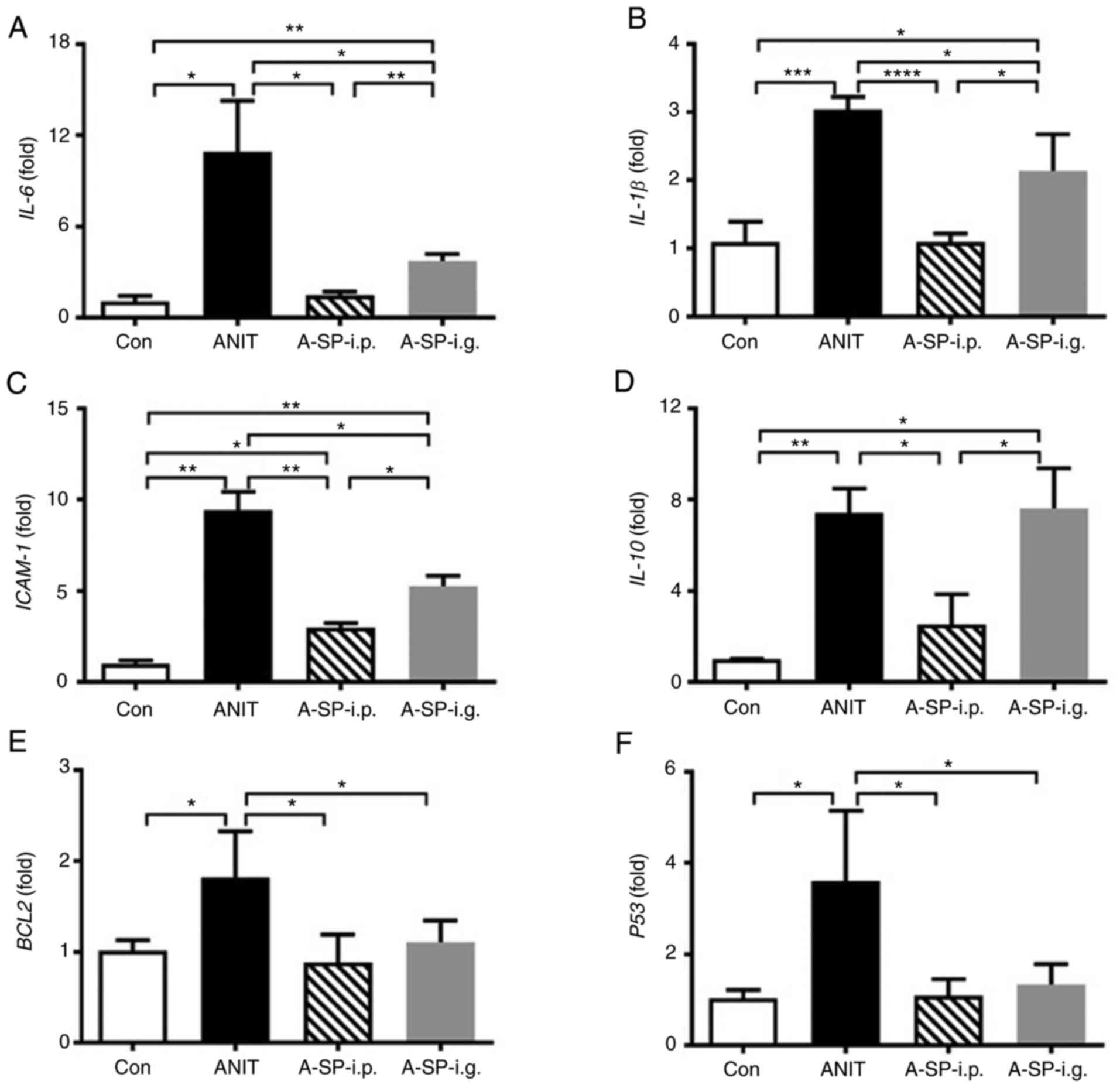 | Figure 5Levels of mRNAs encoding
inflammation-related factors and apoptosis genes. (A) IL-6,
(B) IL-1β, (C) ICAM-1, (D) IL-10, (E)
BCL2 and (F) P53 mRNA levels in four groups 48 h
after the ANIT administration. The mRNA levels were measured by
qPCR and normalized by 18S rRNA. mRNA levels in the
vehicle-treated control mice were set as 1 and the results are
expressed as the mean ± SD. n=5. *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001. Con, negative control; ANIT,
α-naphthylisothiocyanate; A-SP-i.p., ANIT/SP600125-i.p.; A-SP-i.g.,
ANIT/SP600125-i.g.; i.p, intraperitoneal; i.g., intragastric. |
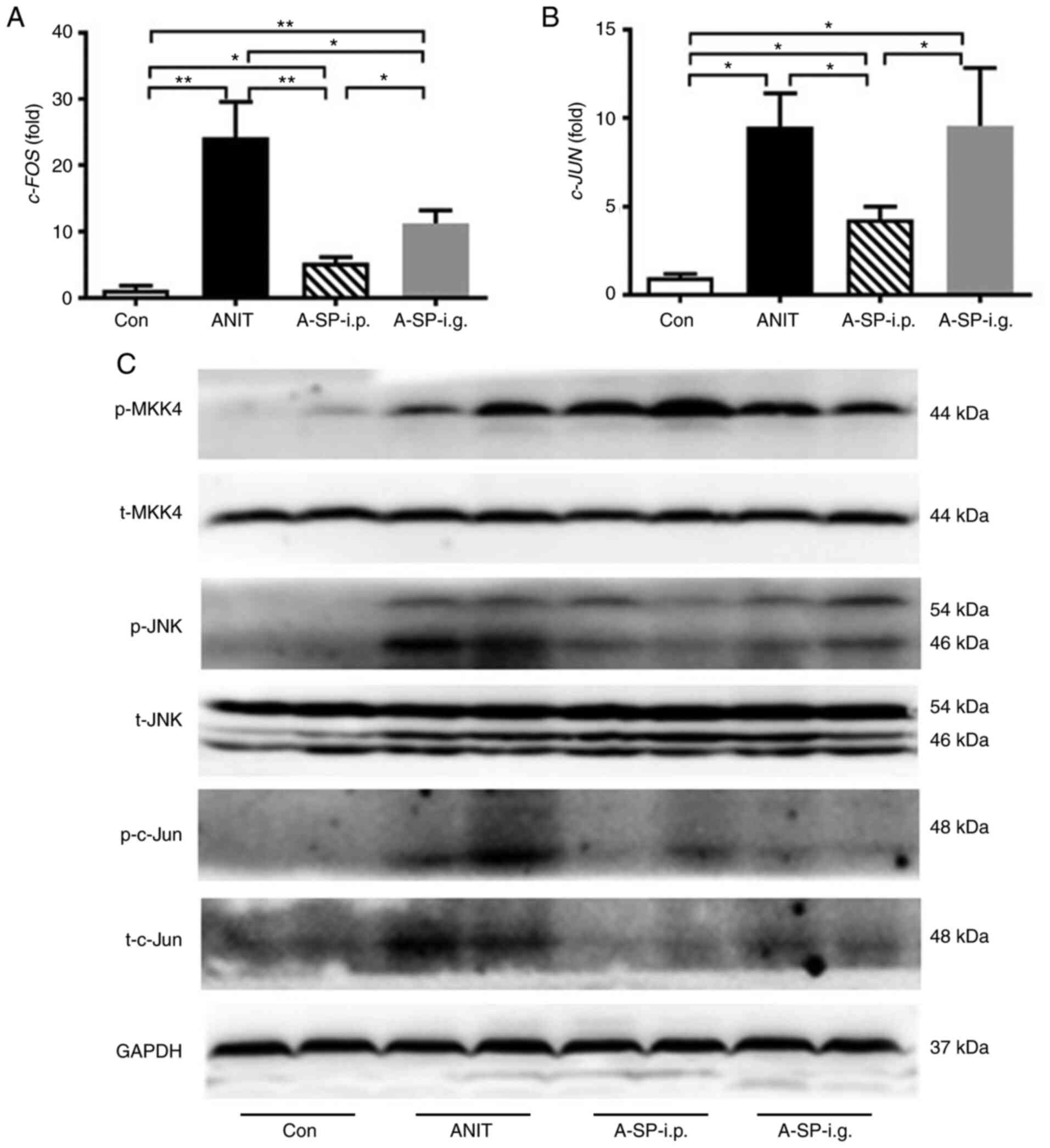
SP600125 i.p. inhibits JNK signaling
The c-JUN and c-FOS mRNA levels were
increased by ANIT. Compared with the ANIT group, the c-JUN
mRNA was decreased in the A-SP-i.p. group (P<0.05), but not in
the A-SP-i.g. group (Fig. 6A).
The c-FOS mRNA was decreased in both the A-SP-i.p. and
A-SP-i.g. groups, and this decrease was greater in the A-SP-i.p.
group (Fig. 6B, P<0.01 and
P<0.05). The WB analysis revealed that the p-MKK4 was activated
in the ANIT, A-SP-i.p. and A-SP-i.g. groups and p-JNK was activated
in the ANIT group compared with the control group. This pathway was
strongly inhibited in the A-SP-i.p. group, but only partly
inhibited in the A-SP-i.g. group. Activation of p-c-Jun was
observed in the ANIT group, but it was markedly inhibited by
SP600125 in the A-SP-i.p. group, in which liver injury was not
observed (Fig. 6C). Thus,
SP600125 acted against ANIT-induced cholestatic liver injury
following i.p. administration. The aforementioned protective effect
and differential inflammation regulation were attributed to the
inhibition of JNK signalling following i.p. administration.
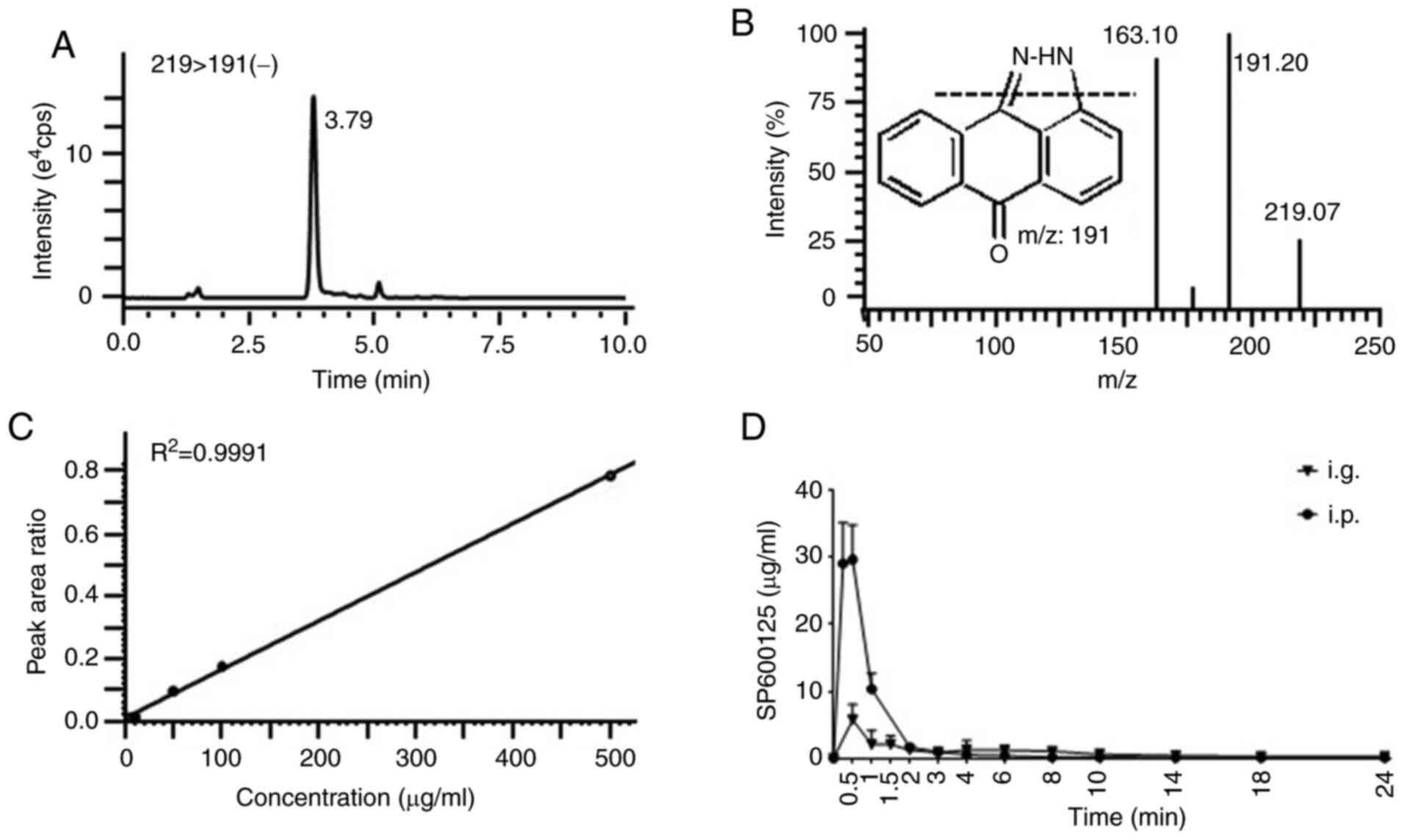
SP600125 exposure assessment
The multiple reaction monitoring mode was used to
detect the negative ion transition m/z 191/219 for SP600125
(Fig. 7A and B). The calibration
curve demonstrated a good linear association
(R2>0.99) with satisfac-tory chromatographic
performance (Fig. 7C). The mean
plasma concentration-time profiles after a single administration of
SP600125 (15 mg/kg i.p. and i.g.) are presented in Fig. 7D. The AUC0-24 of
SP600125 was 31.05±2.53 and 19.82±4.57 µg h/ml, and the
Cmax was 29.01±6.64 and 5.75±1.06 µg/ml by i.p.
and i.g. administration, respectively. Thus, the exposure level of
SP600125 in the ANIT-SP-i.p. group was significantly higher
compared with that in the ANIT-SP-i.g. group treated with
SP600125.
Discussion
Cholic acid, deoxycholic acid and typical
pro-inflammatory cytokines can activate JNK signalling (21,26). The JNK pathway has been reported
to regulate the bile acid metabolism (21,23,27). This pathway was activated in
cholestatic mice challenged by ANIT and its activation profile was
revealed to be correlated with cholestasis and liver injury
(26). In the present study, a
cholestatic liver injury model was induced by ANIT in mice, and the
successful construction of the model was confirmed by serum
biochemistry. Obvious loss of cellular boundaries, degenerative
changes and marked necrosis were observed in the ANIT group. As
anticipated, JNK signalling was activated when cholestasis was
induced by ANIT.
The balance among synthesis, uptake and export of
bile acids contributes to bile acid homeostasis (25). Bile acids are endogenous ligands
for the farnesoid X receptor (FXR), which decreases their synthesis
by the SHP-mediated suppression of the genes encoding CYP7A1 and
CYP8B1 (27). The activated JNK
downregulated the expression of CYP7A1 and CYP8B1 in primary rat
hepatocytes (23). In human
hepatocytes, FXR induced the expression of fibroblast growth factor
19, a secreted protein that suppresses CYP7A1 through a
JNK-dependent pathway (28). In
the present study, CYP7A1 and CYP8B1 were
downregulated in the cholestatic groups induced by ANIT, while they
were not altered in the A-SP-i.p. group, in which JNK signalling
was inhibited. Therefore, the downregulation of CYP7A1 and
CYP8B1 in the ANIT and A-SP-i.g. groups was the result of
both adaptation and JNK-dependent inhibition. CYP7A1 and
CYP8B1 levels were not affected in the A-SP-i.p. group, in
which cholestasis was also induced and the JNK pathway was
inhibited. This indicated the dependence of FXR-mediated adaptation
on JNK activation, which was in agreement with previously reported
findings (29). These data
suggested that i.p. administration strongly inhibited JNK
signalling and blocked adaptation of synthesis (Fig. 8).
The basolateral uptake transporters, OATP1
transports bile acids from the portal blood into hepatocytes; The
basolateral efflux transporter MDR is involved in export of bile
acids into the blood and OSTB exports bile acids into canaliculi
(25). Bile acids activate FXR,
which in turn accelerates bile acid export by inducing BSEP and
OSTβ expression, and decreases bile acid uptake by suppressing
sodium taurocholate co-transporting polypeptide and OATP expression
(30). In the present study, the
changes in the bile acid transporters OATP1, OSTB,
MDR1A and MDR2 were similar among the three groups
challenged by ANIT, which were considered as adaptive responses.
This indicated that the FXR-mediated transport adaptation was
independent of JNK activation, which was different from the
synthesis adaptation indicated above.
In the present study, JNK signalling was activated
in the ANIT group, but only partially inhibited in the SP-A-i.g.
group, as indicated by the increase in p-MKK4, p-JNK and c-Jun in
the WB analysis. This serial activation triggered liver injury,
which was substantially attenuated when JNK signalling was more
markedly inhibited in the SP-A-i.p. group compared with the
SP-A-i.g. group. In the present study, SP600125 protected against
cholestatic liver injury, as indicated by the regulation of bile
acid metabolism, normalization of serum ALT, AST, and ALP levels
and histomorphology in the A-SP-i.p. group. Thus, the
administration route was found to be crucial for the inhibitory
effect of SP600125 on the JNK pathway, which contributed to
inhibition of the cholestatic liver injury.
As a JNK inhibitor, SP600125 has been widely used to
investigate pathophysiological and pharmacological mechanisms
(31). Its concentration used in
cellular experiments was usually between 25 and 50 µM
(5.5-11 µg/ml). In in vivo models of i.g. and i.p.
administration, the dose range was 5-30 mg/kg (32,33). Unfortunately, no studies to date
have explored the plasma exposure profile of SP600125. In the
present study, the exposure of SP600125 by i.p. injection was
significantly higher compared with that by i.g. injection and
produced very different exposure profiles. The AUC0–24
(31.05 vs. 19.82 µg h/ml) and Cmax (29.01 vs.
5.75 µg/ml) were statistically significantly different
between the two administration routes. The small Tmax
(0.5-1 h) indicated the high potential of SP600125 to be absorbed
and distributed. Thus, the in vivo exposure level of
SP600125 was close to the concentration of SP600125 used in in
vitro models. In the ANIT-SP-i.p. group, the duration of an
SP600125 concentration >5.5 µg/ml was markedly longer
compared with that in the ANIT-SP-i.g. group. In cellular models,
SP600125 as a competitive inhibitor inhibited the phosphorylation
of c-Jun in a dose-dependent manner (33). It may be reasonably inferred that
SP600125 at a high concentration binds more JNK and inhibits its
signalling. Therefore, the administration route of SP600125
determined its exposure level, and thereby the inhibition of JNK
signalling (Fig. 8).
Collectively, the findings of the present study
indicated that an effective dose-exposure level was crucial for the
inhibitory effect of SP600125 on the JNK pathway, and i.p.
administration was more effective compared with i.g.
administration. This optimized regimen of SP600125 may lead to an
improved design for future in vivo investigations. However,
the dose-effect relationship in i.p. administration remains to be
investigated for strongest inhibition of JNK signalling without
toxic action.
Supplementary Data
Acknowledgments
Not applicable.
Funding
The present study was supported by the Ningbo
Natural Science Foundation (grant nos. 2018A610253 and
2018A610384), the Ningbo Public Welfare Project (grant no.
202002N3160), the Zhejiang Public Welfare Technology Research
Program (grant no. LGD19H070001), the Zhejiang Provincial Natural
Science Foundation of China (grant no. LY20H030001) and the K.C.
Wong Magna Fund of Ningbo University.
Availability of data and materials
The datasets used during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
XZ, MD, JY and AL participated in the research
design, conducted experiments, performed data analysis and wrote
the manuscript. GX conducted experiments and performed data
analysis. LC performed data analysis and contributed new reagents
or analytic tools. All authors read and approved the final version
of the manuscript to be published.
Ethics approval and consent to
participate
All experiments were approved by the Animal Care and
Use Committee of Ningbo University.
Patient consent for publication
Not applicable.
Competing interests
Not applicable.
References
|
1
|
Gupta S, Barrett T, Whitmarsh AJ, Cavanagh
J, Sluss HK, Derijard B and Davis RJ: Selective interaction of JNK
protein kinase isoforms with transcription factors. EMBO J.
15:2760–2770. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Seki E, Brenner DA and Karin M: A liver
full of JNK: Signaling in regulation of cell function and disease
pathogenesis, and clinical approaches. Gastroenterology.
143:307–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brenner C, Galluzzi L, Kepp O and Kroemer
G: Decoding cell death signals in liver inflammation. J Hepatol.
59:583–594. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kallunki T, Deng T, Hibi M and Karin M:
c-jun Can recruit JNK to phosphorylate dimerization partners via
specific docking interactions. Cell. 87:929–939. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Williams AS, Issa R, Leung SY, Nath P,
Ferguson GD, Bennett BL, Adcock IM and Chung KF: Attenuation of
ozone-induced airway inflammation and hyper-responsiveness by c-Jun
NH2 terminal kinase inhibitor SP600125. J Pharmacol Exp Ther.
322:351–359. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nath P, Eynott P, Leung SY, Adcock IM,
Bennett BL and Chung KF: Potential role of c-Jun NH2-terminal
kinase in allergic airway inflammation and remodelling: Effects of
SP600125. Eur J Pharmacol. 506:273–283. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Assi K, Pillai R, Gomez-Munoz A, Owen D
and Salh B: The specific JNK inhibitor SP600125 targets tumour
necrosis factor-alpha production and epithelial cell apoptosis in
acute murine colitis. Immunology. 118:112–121. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Y, Mei X, Yuan J, Lai X and Xu D:
Taurine zinc solid dispersions enhance bile-incubated L02 cell
viability and improve liver function by inhibiting ERK2 and JNK
phosphory-lation during cholestasis. Toxicology. 366-367:10–19.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guo C, Wang SL, Xu ST, Wang JG and Song
GH: SP600125 reduces lipopolysaccharide-induced apoptosis and
restores the early-stage differentiation of osteoblasts inhibited
by LPS through the MAPK pathway in MC3T3-E1 cells. Int J Mol Med.
35:1427–1434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yan H, Gao Y and Zhang Y: Inhibition of
JNK suppresses autophagy and attenuates insulin resistance in a rat
model of nonalcoholic fatty liver disease. Mol Med Rep. 15:180–186.
2017. View Article : Google Scholar :
|
|
12
|
Liu M, Huang G, Wang TT, Sun X and Yu LL:
3-MCPD 1-Palmitate induced tubular cell apoptosis in vivo via
JNK/p53 pathways. Toxicol Sci. 151:181–192. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu Y, Wang Y, Miao X, Zhou S, Tan Y,
Liang G, Zheng Y, Liu Q, Sun J and Cai L: Inhibition of JNK by
compound C66 prevents pathological changes of the aorta in
STZ-induced diabetes. J Cell Mol Med. 18:1203–1212. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Y, Zhou S, Sun W, McClung K, Pan Y,
Liang G, Tan Y, Zhao Y, Liu Q, Sun J and Cai L: Inhibition of JNK
by novel curcumin analog C66 prevents diabetic cardiomyopathy with
a preservation of cardiac metallothionein expression. Am J Physiol
Endocrinol Metab. 306:E1239–E1247. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hirschfield GM, Heathcote EJ and Gershwin
ME: Pathogenesis of cholestatic liver disease and therapeutic
approaches. Gastroenterology. 139:1481–1496. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hofmann AF: Cholestatic liver disease:
Pathophysiology and therapeutic options. Liver. 22(Suppl 2):
S14–S19. 2002. View Article : Google Scholar
|
|
17
|
Huang YH, Chuang JH, Yang YL, Huang CC, Wu
CL and Chen CL: Cholestasis downregulate hepcidin expression
through inhibiting IL-6-induced phosphorylation of signal
transducer and activator of transcription 3 signaling. Lab Invest.
89:1128–1139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tan Z, Liu A, Luo M, Yin X, Song D, Dai M,
Li P, Chu Z, Zou Z, Ma M, et al: Geniposide inhibits
alpha-naphthylisothio-cyanate-induced intrahepatic cholestasis: The
downregulation of STAT3 and NF[Formula: See text]B signaling plays
an important role. Am J Chin Med. 44:721–736. 2016. View Article : Google Scholar
|
|
19
|
Miyake JH, Wang SL and Davis RA: Bile acid
induction of cytokine expression by macrophages correlates with
repression of hepatic cholesterol 7alpha-hydroxylase. J Biol Chem.
275:21805–21808. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Higuchi H, Grambihler A, Canbay A, Bronk
SF and Gores GJ: Bile acids up-regulate death receptor
5/TRAIL-receptor 2 expression via a c-Jun N-terminal
kinase-dependent pathway involving Sp1. J Biol Chem. 279:51–60.
2004. View Article : Google Scholar
|
|
21
|
Li D, Zimmerman TL, Thevananther S, Lee
HY, Kurie JM and Karpen SJ: Interleukin-1 beta-mediated suppression
of RXR: RAR transactivation of the Ntcp promoter is JNK-dependent.
J Biol Chem. 277:31416–31422. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li T, Jahan A and Chiang JY: Bile acids
and cytokines inhibit the human cholesterol 7 alpha-hydroxylase
gene via the JNK/c-jun pathway in human liver cells. Hepatology.
43:1202–1210. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gupta S, Stravitz RT, Dent P and Hylemon
PB: Down-regulation of cholesterol 7alpha-hydroxylase (CYP7A1) gene
expression by bile acids in primary rat hepatocytes is mediated by
the c-Jun N-terminal kinase pathway. J Biol Chem. 276:15816–15822.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guo R, Wu H, Yu X, Xu M, Zhang X, Tang L
and Wang Z: Simultaneous determination of seven anthraquinone
aglycones of crude and processed semen cassiae extracts in rat
plasma by UPLC-MS/MS and its application to a comparative
pharmacokinetic study. Molecules. 22:18032017. View Article : Google Scholar
|
|
25
|
Chiang JY: Bile acid metabolism and
signaling. Compr Physiol. 3:1191–1212. 2013.PubMed/NCBI
|
|
26
|
Dai M, Yang J, Xie M, Lin J, Luo M, Hua H,
Xu G, Lin H, Song D, Cheng Y, et al: Inhibition of JNK signalling
mediates PPARα-dependent protection against intrahepatic
cholestasis by fenofibrate. Br J Pharmacol. 174:3000–3017. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gonzalez FJ, Jiang C, Xie C and Patterson
AD: Intestinal farnesoid X receptor signaling modulates metabolic
disease. Dig Dis. 35:178–184. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Holt JA, Luo G, Billin AN, Bisi J, McNeill
YY, Kozarsky KF, Donahee M, Wang DY, Mansfield TA, Kliewer SA, et
al: Definition of a novel growth factor-dependent signal cascade
for the suppression of bile acid biosynthesis. Genes Dev.
17:1581–1591. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Inagaki T, Choi M, Moschetta A, Peng L,
Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA,
et al: Fibroblast growth factor 15 functions as an enterohepatic
signal to regulate bile acid homeostasis. Cell Metab. 2:217–225.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dawson PA: Role of the intestinal bile
acid transporters in bile acid and drug disposition. Handb Exp
Pharmacol. 169–203. 2011. View Article : Google Scholar
|
|
31
|
Wu Q, Wu W, Jacevic V, Franca TCC, Wang X
and Kuca K: Selective inhibitors for JNK signalling: A potential
targeted therapy in cancer. J Enzyme Inhib Med Chem. 35:574–583.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hanajiri K, Mitsui H, Maruyama T,
Hashimoto N, Sata M and Omata M: Echographic detection of
diethylnitrosamine-induced liver tumors in rats and the effect of
the intratumoral injection of an inhibitor of c-Jun N-terminal
kinase. J Gastroenterol Hepatol. 24:866–871. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bennett BL, Sasaki DT, Murray BW, O'Leary
EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et
al: SP600125, an anthrapyrazolone inhibitor of Jun N-terminal
kinase. Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|