Introduction
Hepatic fibrosis is a wound-healing response that
requires a range of cell types and mediators to encapsulate the
injury (1). Activation of
resident hepatic stellate cells (HSCs), the primary source of
extracellular matrix (ECM) proteins, into proliferative,
contractile and fibrogenic cells serves a pivotal role in fibrosis
(2). Among the numerous
fibrogenic factors involved in the activation of HSCs, one of the
most important is transforming growth factor β1 (TGF-β1), which can
affect HSC growth and differentiation, as well as the production of
ECM proteins (3). The discovery
of HSC activation induced by TGF-β1 remains meaningful in
understanding the basis of hepatic fibrogenesis.
Autophagy is an essential catabolic degradation
process, in which cellular proteins, organelles and invading
microbes are engulfed by double-membraned autophagosomes and are
degraded in the lysosomes (4).
Autophagy serves dual roles dependent on the conditions, promoting
survival or promoting cell death (4). Recently, autophagy has been
implicated in a variety of liver diseases (5,6).
The selective breakdown of lipids stored in lipid droplets by
autophagy is termed lipophagy (7). Although originally described in
hepatocytes, lipophagy may metabolize lipids in all cells,
including HSCs (7,8). Thus, autophagy regulates hepatic
metabolism under healthy conditions and various chronic liver
diseases, such as fibrosis (9,10).
A previous study showed that autophagy flux is increased during
activation of HSCs, and autophagy stimulates the loss of lipid
droplets (11). This provides
cellular energy to promote the survival and activation of HSCs
(12). Chloroquine and
3-methyladenine (3-MA), inhibitors of autophagy, may improve carbon
tetrachloride (CCl4)-induced liver fibrosis in
vivo (13,14). The mechanism involved in this
process includes inhibition of autophagy pathways and inhibition of
HSC activation (13,14). However, another study has shown
that inducing autophagy promotes the degradation of collagen and
reverses hepatic fibrosis (15).
The complex function of autophagy in the regulation of fibrosis may
differ between various tissues or cells.
Studies have reported that autophagy is involved in
regulating the fibrosis of human subconjunctival fibroblasts,
tubular epithelial kidney cells, primary human lung fibroblast
cells, human atrial myofibroblasts and human peritoneal mesothelial
cells induced by TGF-β1 (16-20). However, the effect of TGF-β1 on
autophagy in HSC is unclear. In the present study, the relationship
and mechanism between autophagy and TGF-β1-induced fibrogenesis in
JS1 cells, a mouse immortalized HSC line, were assessed.
Materials and methods
CCl4-induced liver
fibrosis
A total of 32 C57BL/6 male mice, aged 6-8 weeks,
were purchased from Shanghai Laboratory Animal Resources, Chinese
Academy of Sciences (cat. no. SCXK 2007-0005). All mice were housed
in specific pathogen-free sterile rooms, with a 12-h light/dark
cycle, at 22±2°C and 40-60% humidity, with free access to water and
food in the Laboratory Animal Center, Shanghai University of
Traditional Chinese Medicine. The present study was performed in
accordance with the Guide for the Care and Use of Laboratory
Animals of the National Institutes of Health. All experimental
protocols were approved by Shuguang Hospital Affiliated to Shanghai
University of Traditional Chinese Medicine Animal Care and Ethics
committee. All mice were randomly divided into control (n=11) and
experimental groups (n=21). The experimental group was further
subdivided into a chronic injury group (n=13) and an acute hepatic
injury group (n=8). CCl4 (Sigma-Aldrich; Merck KGaA) was
intraperitoneally injected to induce hepatic fibrosis or liver
injury. In the chronic injury group, the mice were injected with
12% CCl4 (diluted in olive oil) three times per week for
6 weeks at a dose of 2 ml/kg body weight. In the acute hepatic
injury group, the mice were injected with a single dose of 0.5%
CCl4 at 5 ml/kg. The control mice were injected with
saline at the same dosing schedule as the experimental subgroups.
All mice were euthanized by intraperitoneal injection of
pentobarbital sodium (200 mg/kg) after 6 weeks of treatment.
Subsequently, liver tissues were removed and immediately
cryopreserved in liquid nitrogen for subsequent experiments.
Cells and cell culture
Primary HSCs were isolated from normal and
CCl4-induced acute hepatic injured C57BL/6 mice through
enzymatic digestion and 11.5% Nycodenz density gradient
centrifugation with modifications (21). Once adherent, the cells were
collected for subsequent experiments. The mouse immortalized HSC
cell line, JS1, was kindly provided by Professor Scott L. Friedman
(Mt. Sinai School of Medicine, USA). JS1 cells were cultured in
DMEM (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10%
FBS (Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin at 5% CO2 and 37°C. The medium
was changed to media supplemented with 0.5% FBS for 12 h prior to
treatment. Recombinant human TGF-β1 (PeproTech, Inc.) was added to
the medium for different durations, according to the design of the
experiments. JS1 cells were pre-treated with 10 µM PD98059
(cat. no. ab120234; Abcam), 10 µM SB203580 (cat. no.
ab120162; Abcam) or 10 µM SP600125 (cat. no. ab120065;
Abcam) for 1 h, followed by treatment with or without 10 ng/ml
TGF-β1 for varying durations (2, 4, 8 or 12 h). Each in
vitro experiment was repeated three times.
Measurement JS1 cell viability
Cells were plated in 96-well plates and incubated
with 0.5, 1, 5 or 10 mM 3-MA (cat. no. M9281; Sigma-Aldrich; Merck
KGaA), 10, 50 or 100 µM chloroquine (CQ) diphosphate salt
(cat. no. C6628; Sigma-Aldrich; Merck KGaA) or 100 ng/ml rapamycin
(cat. no. S1842; Beyotime Institute of Biotechnology) with or
without TGF-β1 (10 ng/ml) for 12 or 24 h. Subsequently, Cell
viability was assessed using an MTT Cell Proliferation and
Cytotoxicity Assay kit (cat. no. C0009; Beyotime Institute of
Biotechnology) according to the manufacturer's protocol. The
supernatant was removed after cell treatment, and 90 µl
fresh culture medium and 10 µl MTT solution were added for
further culture for 4 h at 37°C. The cell supernatant was then
discarded, and 100 µl formazan was added with gentle shaking
for 10 min. The absorbance was recorded at a wavelength of 490 nm
with a microplate reader (SpectraMax M5; Molecular Devices
LLC).
Fluorescence staining
Lipid droplets in primary HSCs were stained using 4
µM BODIPY 581/591 (cat. no. D3861; Invitrogen; Thermo Fisher
Scientific, Inc.) after fixing the cells with 4% paraformaldehyde
for 30 min at room temperature. Immunofluorescence staining of
liver tissue was performed using 7-mm sections sliced using a
cryostat machine (CM1850; Leica Microsystems, Inc.) at −20°C. The
sections were fixed with 100% acetone for 30 min at 4°C and blocked
with 5% BSA (cat. no. ST025; Beyotime Institute of Biotechnology)
for 30 min at room temperature. Subsequently, two primary
antibodies were used for staining: Mouse anti-α-smooth muscle actin
(α-SMA; 1:100; cat. no. AA123; Beyotime Institute of Biotechnology)
and rabbit anti- microtubule-associated protein 1 light chain 3B
(LC3B; 1:100; cat. no. L7543; Sigma-Aldrich; Merck KGaA) overnight
at 4°C. The sections were subsequently exposed to the following
secondary antibodies (Beyotime Institute of Biotechnology):
Cy3-labeled goat anti-mouse antibody (cat. no. A0521; 1:100;
Beyotime Institute of Biotechnology) or FITC-labeled goat
anti-rabbit antibody (cat. no. A0562; 1:100; Beyotime Institute of
Biotechnology) for 1 h at 37°C. Representative images were captured
using confocal microscopy at ×100 or ×200 magnification (FV10C-PSU;
Olympus Corporation). ImageJ 6.0 (National Institutes of Health)
was used for quantification of fluorescence intensity and the
number of fluorescent cells.
Transmission electron microscopy
Cells were fixed with 2.5% glutaraldehyde followed
by 1% osmic acid successively for 2 h at 4°C. The samples were
dehydrated with gradient ethanol and embedded with Epon for 12 h at
45°C and then for 36 h at 60°C. Ultra-thin sections were stained
with uranyl acetate for 30 min and lead citrate for 10 min at room
temperature. The ultrastructure was observed using a transmission
electron microscope (Tecnai-12 Biotwin; Philips Healthcare).
Lentiviral autophagy-related gene 7
(Atg7) short hairpin RNA (shAtg7) and RFP-GFP-LC3B
transduction
shAtg7 expression plasmids were kindly provided by
Professor Mark J. Czaja (Albert Einstein College of Medicine, USA).
High-titer lenti-viral stocks were produced by calcium
phosphate-mediated transfection of the modified transfer vectors
and the packaging vectors pMDLg-pRRE, pRSV-Rev and pMD2.VSVG into
293T cells (Cell Bank of the Chinese Academy of Sciences).
Supernatants were harvested over 36 to 48 h, titered by plaque
assay and used at a multiplicity of infection (MOI) of 50 to infect
JS1 cells. Empty lentiviral vectors were used as the control. JS1
cells were cultured at a density of 1×105 cells/ml
overnight. The medium was then replaced with serum-free medium and
JS1 cells were infected with lentiviral shAtg7 or empty vectors
with an MOI of 50 for 24 h at 37°C. Optimal knockdown and
inhibition of autophagy was observed 5 days after transduction,
which was the time point used for the subsequent experiments. JS1
cells were transfected with RFP-GFP-LC3B viral particles using a
Premo Autophagy Tandem Sensor RFP-GFP-LC3B kit (Invitrogen; Thermo
Fisher Scientific, Inc.) with an MOI of 30 for 24 h at 37°C, and
subsequently treated with 10 ng/ml TGF-β1 alone or in combination
with 50 µM CQ for 24 h. Treated cells were fixed with 4%
paraformaldehyde for 30 min at room temperature and autophagic flux
was observed using a confocal microscope (FV10C-PSU; Olympus
Corporation).
Western blotting
Lysates of the liver or HSCs were prepared for
electrophoresis as previously described (22). After resolving, protein
concentration was measured using a bicinchoninic acid protein assay
kit (cat. no. P0012; Beyotime Institute of Biotechnology). Proteins
were separated by SDS-PAGE (5% stacking gel, 10 or 15% separating
gel; 30 µg protein in 15 µl loaded per lane) and
subsequently transferred to a PVDF membrane. The membrane was
blocked using blocking buffer (cat. no. 927-40000; LI-COR
Biosciences) for 1 h at room temperature and incubated with primary
antibody overnight at 4°C. Subsequently, the membrane was incubated
with a fluorescently tagged secondary antibody in the dark at room
temperature for 45 min and read at a wavelength of 800 nm (780 nm
excitation, 820 nm detection) or 700 nm (680 nm excitation, 720 nm
detection) with an Odyssey Infrared Imaging system (Odyssey; LI-COR
Biosciences) according to the manufacturer's instructions. ImageJ
6.0 (National Institutes of Health) was used for quantification.
The antibodies used were as follows: Rabbit anti-α-SMA (cat. no.
1184-1; Epitomics; Abcam), rabbit anti-collagen type I (Col.I; cat.
no. ab292; Abcam), rabbit anti-Atg7 (cat. no. ab133528; Abcam),
rabbit anti-LC3B (cat. no. L7543; Sigma-Aldrich; Merck KGaA),
rabbit anti-p62 (cat. no. 5114; Cell Signaling Technology, Inc.),
rabbit anti-cleaved-caspase3 (cat. no. 9664; Cell Signaling
Technology, Inc.), rabbit anti-Beclin-1 (cat. no. 3495; Cell
Signaling Technology, Inc.), rabbit anti-JNK (cat. no. 9258; Cell
Signaling Technology, Inc.), rabbit anti-phosphorylated (p)-JNK
(cat. no. 4671; Cell Signaling Technology, Inc.), rabbit anti-ERK
(cat. no. 4695; Cell Signaling Technology, Inc.), rabbit anti-p-ERK
(cat. no. 4370; Cell Signaling Technology, Inc.), rabbit anti-p38
MAPK (cat. no. 8690; Cell Signaling Technology, Inc.), rabbit
anti-p-p38 MAPK (cat. no. 4511; Cell Signaling Technology, Inc.),
all at 1:500 dilution and mouse anti-GAPDH (cat. no. KC-5G4;
1:10,000; KangChen BioTech Co., Ltd.). The secondary antibodies
used were as follows: IRDye 680RD goat anti-rabbit (cat. no.
926-68071; LI-COR Biosciences) and IRDye 800CW donkey anti-mouse
(cat. no. 926-32212; LI-COR Biosciences), all 1:10,000
dilution.
Reverse transcription-quantitative (RT-q)
PCR
RT-qPCR was performed as previously described to
determine the gene expression levels in the cultured cells
(23). Total RNA was extracted
from the cells using a Nucleic Acid Purification kit (cat. no.
NPK-201; Toyobo Life Science), and cDNA was synthesized using a
RevertAid First Strand cDNA Synthesis kit (cat. no. K1622; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. Total cDNA was mixed with PCR master mix (cat. no.
DRR041A; Takara Bio, Inc.), and pre-designed primers. The following
primer pairs (synthesized by Sangon Biotech co., Ltd.) were used
for the qPCR: α-SMA forward, 5′-ACT ACT GCC GAG CGT GAG ATT G-3′
and reverse, 5′-CGT CAG GCA GTT CGT AGC TCT T-3′; Col.I, forward,
5′-GGA CCT CCG GCT CCT GCT CCT C-3′ and reverse, 5′-GCA TTG CAC GTC
ATC GCA CAC -3′; beclin 1 forward, 5′-CTT ACC ACA GCC CAG GCG AA-3′
and reverse, 5′-AGA TGC CTC CCC GAT CAG AG-3′; Atg5 forward, 5′-TAT
CAG ACC ACG ACG GAG CG-3′ and reverse, 5′-CTG GCT CCT CTT CTC TCC
ATC TTC -3′; Bcl-2 forward, 5′-TGT GGA GAG CGT CAA CAG GG-3′ and
reverse, 5′-AGA CAG CCA GGA GAA ATC AAA CAG A-3′; p21 forward,
5′-ATG TCC AAT CCT GGT GAT GTC C-3′ and reverse, 5′-AAG TCA AAG TTC
CAC CGT TCT CG-3′; p53 forward, 5′-CGC CGA CCT ATC CTT ACC ATC
AT-3′ and reverse, 5′-CTC CCA GGG CAG GCA CAA AC-3′ and GAPDH
forward, 5′-AAG GTC ATC CAT GAC AAC TTT GGC -3′ and reverse, 5′-ACA
GTC TTC TGG GTG GCA GTG AT-3′. GAPDH was used as the internal
reference gene. The thermocycling conditions used for the qPCR were
as follows: Initial denaturation for 1 min at 95°C; followed by 40
cycles of 15 sec at 95°C and 30 sec at 60°C. Amplification was
performed on a ViiA7 system (Thermo Fisher Scientific, Inc.). Gene
expression was normalized to that of GAPDH using the
2−ΔΔCq method (24).
Flow cytometry analysis of apoptosis
JS1 cells were incubated with TGF-β1 (10 ng/ml) for
12 h and pre-treated with or without 10 µM SB203580 (cat.
no. ab120162; Abcam) for 1 h. In the absence or presence of the
previously mentioned agents, JS1 cells were labeled with PE Annexin
V and 7-amino-actino-mycin using a PE Annexin V Apoptosis Detection
kit according to the manufacturer's protocol (BD Biosciences) and
analyzed using flow cytometry (DxFLEX; Beckman Coulter, Inc.). Flow
cytometry data were analyzed using CytExpert software (version 2.0;
Beckman Coulter, Inc.).
Statistical analysis
Data were collected in triplicate from at least
three separate cell cultures. Results are expressed as the mean ±
SEM of three repeats. Differences amongst groups were compared
using a one-way ANOVA followed by Tukey's post hoc test, or
otherwise a t-test. SPSS 17.0 (SPSS, Inc.) was used to perform
statistical analysis. P<0.05 was considered to indicate a
statistically significant difference.
Results
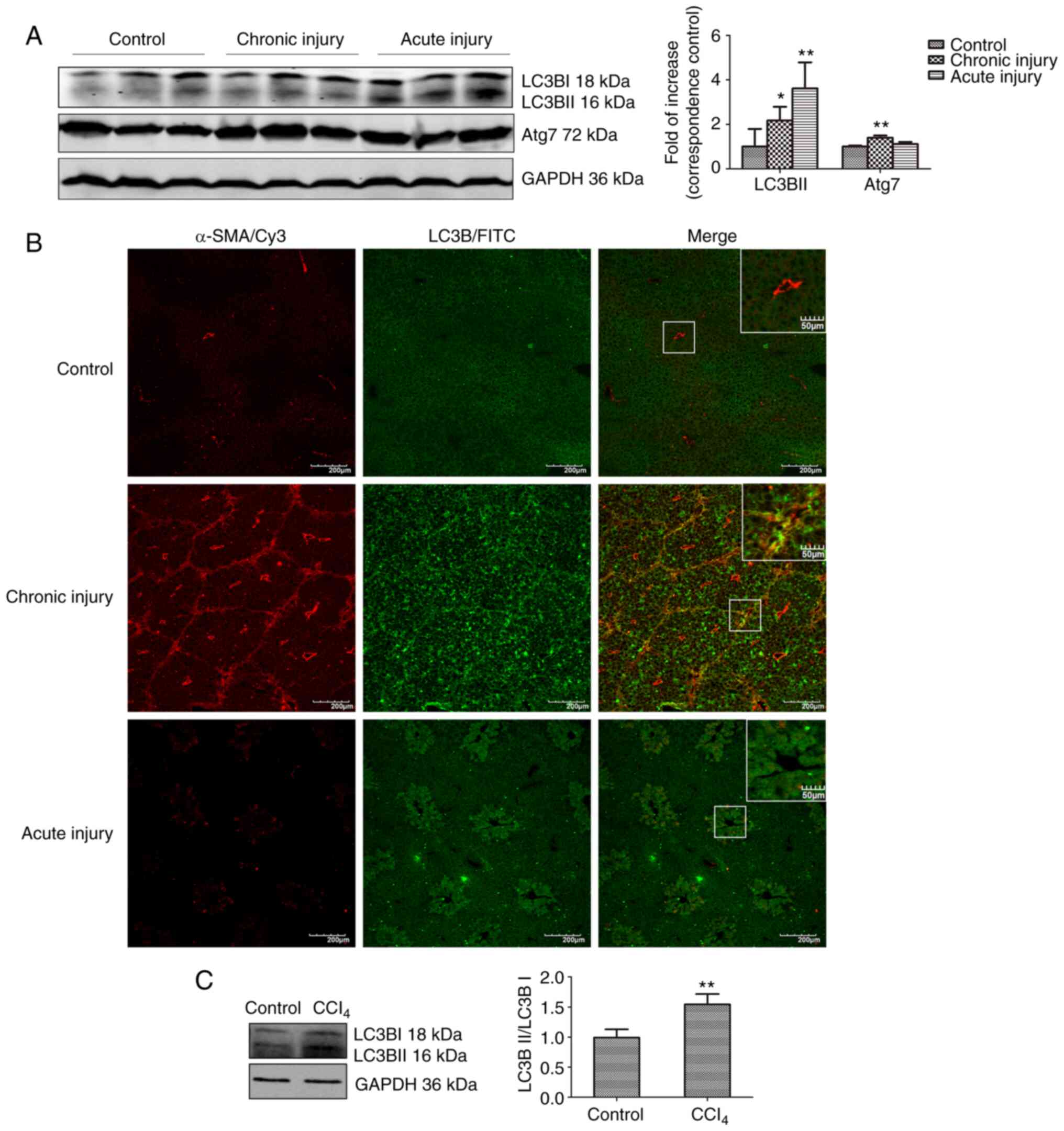
Autophagy is increased in liver tissues
and HSCs following CCL4-induced injury
The expression of LC3BII and Atg7 was assessed using
western blotting to determine the extent of autophagy in liver
samples of mice with acute and chronic liver injury induced by
CCl4. The results showed that LC3BII expression was
significantly higher in both hepatic injury models compared with
the control (Fig. 1A). The
expression of Atg7, another autophagy-related protein, which
promotes the conjugation of LC3-I to phosphatidylethanolamine to
form LC3-II, was significantly increased in the chronic injury
group, and only slightly increased in the acute injury model
(Fig. 1A).
Confocal microscopy was used to determine the
relationship between HSCs and autophagy, and to monitor the
co-localization of α-SMA (a marker of HSC activation) and LC3B (a
marker of autophagy). Autophagy and activation of HSC occurred
simultaneously in fibrotic liver tissue. As shown in Fig. 1B, marked co-localization between
α-SMA and LC3B was observed in the chronic injury tissue. However,
α-SMA expression was only observed around the vessels, whereas LC3B
expression was hardly observed, and there was no co-localization in
the normal liver tissue. Primary mouse HSCs were isolated from the
acute injury liver tissue to show that autophagy was increased
following activation of HSCs in vivo. The expression of
LC3BII in HSCs from injured liver tissues was significantly higher
compared with the control (Fig.
1C). These findings are consistent with those of previous
reports (13,14), showing that hepatic fibrosis is
accompanied by autophagy.
TGF-β1 simultaneously induces activation
and autophagy in JS1 cells
One of the most characteristic features of HSC
activation is the loss of lipid droplets (25). As shown in Fig. 2A, TGF-β1 significantly promoted
the loss of lipid droplets in primary HSCs. Consistent with
previous studies, autophagy regulated hepatic metabolism (7,8),
and stimulated the loss of lipid droplets to provide energy for the
activation of HSCs (12).
Subsequently, whether there was an association between
TGF-β1-induced activation and autophagy in HSC was assessed.
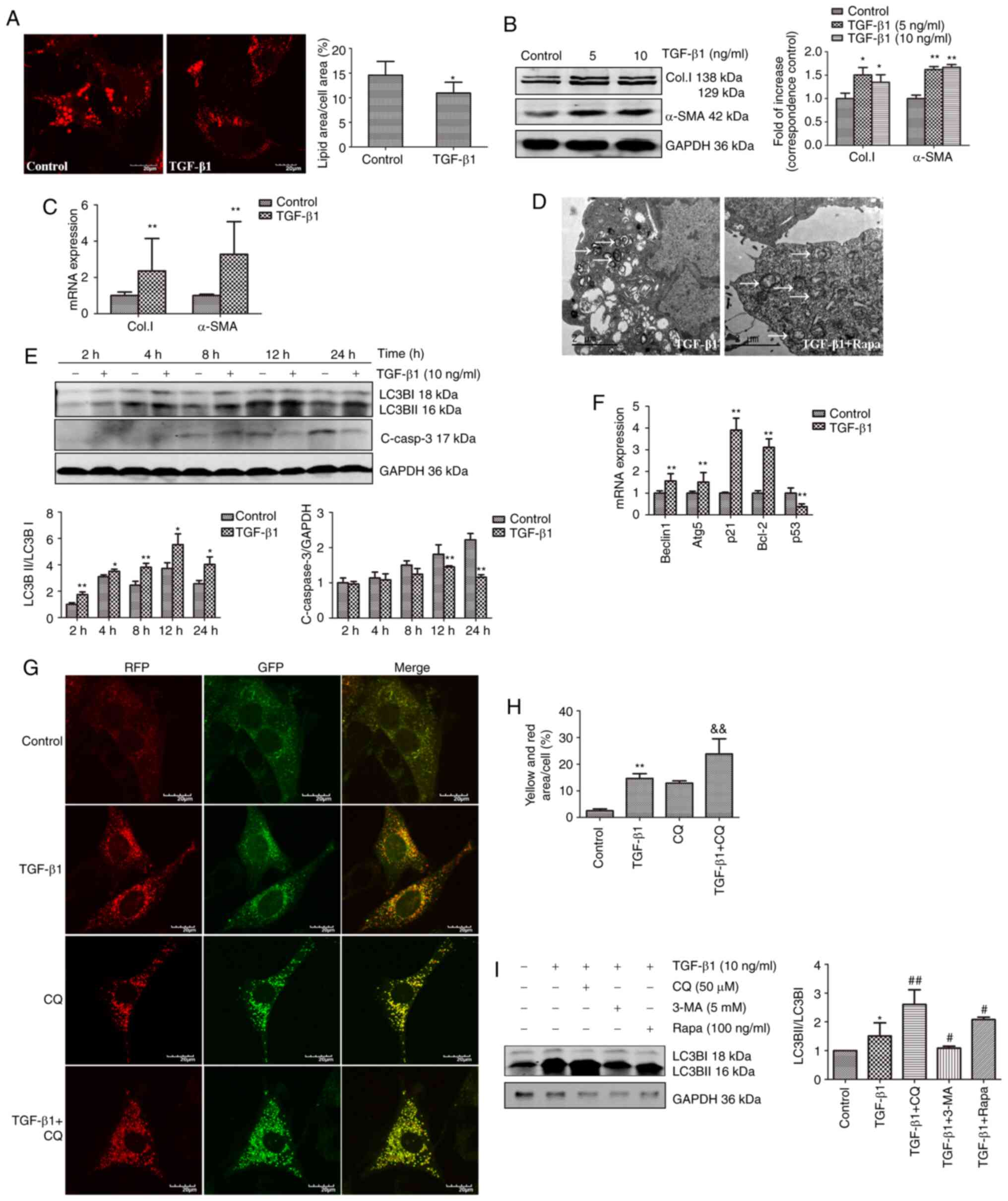 | Figure 2TGF-β1 simultaneously induces
activation and autophagy in JS1 cells. (A) On day 5 after isolation
of primary HSCs, the cells were fixed, and lipid droplets were
stained using BODIPY581/591 after treatment with 10 ng/ml TGF-β1
for 24 h. The quantity of droplets per cells was quantified. Scale
bar, 20 µm. *P<0.05 vs. control. (B) JS1 cells
were treated with TGF-β1 (5 or 10 ng/ml) for 24 h. Western blotting
was performed to assess the expression of fibrosis-associated
markers α-SMA and Col.I. (C) mRNA expression levels of α-SMA and
Col.I in JS1 cells were determined using reverse transcription-
quantitative PCR following treatment with 10 ng/ml TGF-β1 for 24 h.
(D) JS1 cells were either treated with 10 ng/ml TGF-β1 either alone
or in combination with 100 ng/ml rapamycin for 2 h. Cells were
fixed and subsequently imaged using transmission electron
microscopy. Autophagosomes (indicated by the white arrows) were
clearly visible in the cytoplasm and were more abundant in cells
treated with both TGF-β1 and rapamycin. Scale bar, 2 µm. (E)
Protein expression levels of LC3BII and cleaved-caspase-3 were
detected by western blotting after incubation of JS1 cells with 10
ng/ml TGF-β1 for 2, 4, 8, 12 or 24 h. (F) mRNA expression levels of
Beclin-1, Atg5, p21, Bcl-2 and p53 in JS1 cells incubated with 10
ng/ml TGF-β1 for 24 h. (G and H) JS1 cells were transfected with an
RFP-GFP-LC3B virus vectors for 24 h, and subsequently treated with
10 ng/ml TGF-β1 alone or in combination with 50 µM CQ for 24
h. Cells were imaged using a confocal microscope. A total of 10
different fields (10 cells per field) were randomly selected and
manually counted. The percentage of cells with yellow
(colocalization of GFP and RFP) and red puncta (autophagosomes and
lysosomes merged with GFP quenched) were compared with the control
and treatment with CQ groups alone. TGF-β1 significantly increased
the number of autophagosomes and autolysosomes. Scale bar, 20
µm. **P<0.01 vs. control; &&P<0.01
vs. CQ. (I) JS1 cells were incubated with 10 ng/ml TGF-β1 for 12 h,
and subsequently pretreated with 5 mM 3-MA, 50 µm CQ for 2
h, or 100 ng/ml rapamycin for 2 h. LC3BII was detected using
western blotting. Protein ratios were used to quantify fold change
relative to the control. The mRNA expression levels were expressed
as the fold-change relative to control. *P<0.05 and
**P<0.01 vs. control; #P<0.05 and
##P<0.01 vs. TGF-β1. Data are presented as the mean ±
the standard error of the mean. LC3B, microtubule-associated
protein 1 light chain 3B; α-SMA, α-smooth muscle actin; HSC,
hepatic stellate cell; Col.I, collagen type I; TGF-β1, transforming
growth factor-β1; CQ, chloroquine; 3-MA, 3-methyladenine; C-Casp3,
cleaved caspase-3; Rapa, rapamycin. |
JS1 cells were used to study TGF-β1-induced fibrosis
and autophagy. TGF-β1 significantly increased the expression of the
fibrosis markers α-SMA and Col.I in these cells compared with
controls (Fig. 2B and C).
Autophagic bodies were clearly observed in JS1 cells treated with
10 ng/ml TGF-β1 for 2 h through transmission electron microscopy.
More autophagic bodies were observed when cells were co-treated
with 10 ng/ml TGF-β1 and 100 ng/ml rapamycin (Fig. 2D). Moreover, compared with
controls, 10 ng/ml TGF-β1 significantly induced the expression of
LC3BII (Fig. 2E) and
significantly upregulated the mRNA expression levels the of
autophagy-related genes, Atg5 and Beclin-1 (Fig. 2F). These results also indicated
that TGF-β1 may induce autophagy in JS1 cells. LC3BII expression
was significantly increased 12 h after treatment with TGF-β1.
Conversely, the expression of cleaved-caspase 3 was significantly
inhibited at 12 and 24 h after treatment with TGF-β1. These results
suggested that treatment with TGF-β1 protected JS1 against
apoptosis. The results of PCR analysis are shown in Fig. 2F. The results showed that compared
with controls, the mRNA expression levels of p21 and Bcl-2
(anti-apoptosis genes) were significantly upregulated, whereas the
levels of p53 (pro-apoptotic gene) was downregulated. These results
indicate a negative association between the induction of autophagy
and suppression of apoptosis following treatment with TGF-β1 in JS1
cells, which may be attributed to crosstalk between autophagy and
apoptosis (26).
JS1 cells were transfected with a viral vector
containing RFP-GFP-LC3B to confirm that treatment with TGF-β1
increased the expression of LC3BII, rather than leading to its
accumulation by blocking the turnover of LC3BII. The
immunofluorescence data indicated a significant increase in
punctate LC3BII staining in JS1 cells treated with TGF-β1. Under
physiological conditions, autophagic activity proceeded at a very
low level, leading to yellow puncta being diffusely distributed in
the cytoplasm. In addition, treatment with TGF-β1 increased
immunofluorescence staining of autophagosomes (yellow) and
autolysosomes (red) compared with the control (Fig. 2G and H).
Treatment with CQ inhibited autophagy by suppressing
the fusion of autophagosomes and lysosomes, further confirming that
TGF-β1 induced autophagy. As shown in Fig. 2G, JS1 cells treated with CQ
demonstrated only bright yellow fluorescent puncta. This was
attributed to blockage of the maturation of autophagosomes into
autolysosomes, thus the GFP signal could not be quenched by
lysosomal fusion. The presence of yellow puncta was more prominent
in the cytoplasm when co-treated with TGF-β1 and CQ, suggesting
that TGF-β1 increased autophagosome synthesis. Another autophagy
inhibitor, 3-MA, and the autophagy inducer rapamycin were used to
further assess the presence of autophagy flux. These data
demonstrated that compared with controls, pretreatment with 3-MA
significantly inhibited TGF-β1-induced LC3BII expression, whereas
treatment with CQ and rapamycin significantly increased the
expression of LC3BII in JS1 cells (Fig. 2I). Collectively, these data
supported the notion that TGF-β1 induces autophagy in JS1
cells.
Inhibition of autophagy attenuates the
activation of JS1 cells induced by TGF-β1
It was hypothesized that autophagy was involved in
TGF-β1-induced activation of HSCs. Therefore, blocking autophagy
may attenuate the activation of HSCs. The effects of JS1 cell
stimulation by TGF-β1 in the presence of autophagy inhibitors were
assessed. The results suggested that treatment with 3-MA or CQ
significantly inhibited TGF-β1-induced activation of JS1 cells. As
shown in Fig. 3A, 3-MA or CQ
inhibited TGF-β1-induced proliferation. Moreover, CQ significantly
inhibited TGF-β1-induced expression (both at the mRNA and protein
expression level) of α-SMA and Col.I in JS1 cells (Fig. 3C and D). However, 3-MA only
suppressed the expression of α-SMA mRNA and protein of Col.I
(Fig. 3B and D). This may due to
3-MA not only blocking class III PI3K, but also class I PI3K.
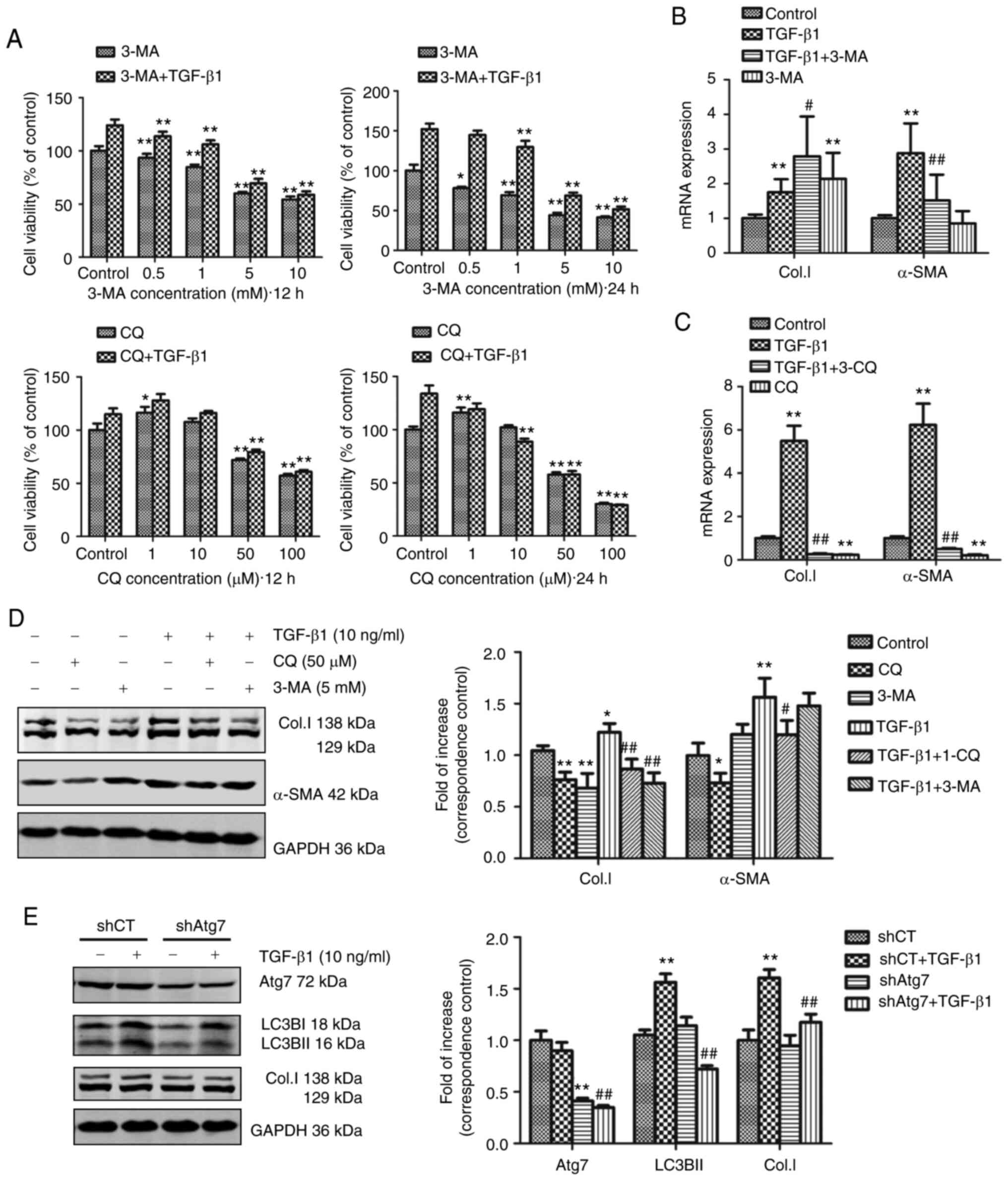 | Figure 3Inhibition of autophagy either by
treatment with pharmacological inhibitors or shAtg7 reduces
TGF-β1-induced activation of JS1 cells. (A) Cell viability was
determined using an MTT assay. JS1 cells were incubated with 3-MA
(0.5, 1, 5 or 10 mM) or CQ (10, 50 or 100 µM) with or
without 10 ng/ml TGF-β1 for 12 or 24 h. Absorbance values at each
concentration were obtained by comparison with their respective
controls. *P<0.05 and **P<0.01 vs.
corresponding control. JS1 cells were incubated with 10 ng/ml
TGF-β1 combined with (B) 50 µM CQ or (C) 5 mM 3-MA for 24 h.
The mRNA expression levels of α-SMA and Col.I were detected through
reverse transcription-quantitative PCR and expressed as the fold
change relative to the control. **P<0.01 vs. control;
#P<0.05 and ##P<0.01 vs. TGF-β1. (D)
JS1 cells were incubated with 10 ng/ml TGF-β1 in combination with
50 µM CQ or 5 mM 3-MA for 12 h. The protein expression
levels of α-SMA and Col.I were detected through western blotting.
Protein ratios were used to quantify fold change relative to
control. *P<0.05 and **P<0.01 vs.
control; #P<0.05 and ##P<0.01 vs.
TGF-β1. (E) JS1 cells were transduced with shAtg7 lentiviral vector
for 5 days, and subsequently incubated with 10 ng/ml TGF-β1 for 24
h. Western blotting was used to detect the expression of Atg7, LC3B
and Col.I in JS1 cells. Protein ratios were used to quantify fold
change relative to control. **P<0.01 vs. shCT;
##P<0.01 vs. shCT + TGF-β1. Data are presented as the
mean ± the standard error of the mean. LC3B, microtubule-associated
protein 1 light chain 3B; α-SMA, α-smooth muscle actin; Col.I,
collagen type I; TGF-β1, transforming growth factor-β1; CQ,
chloroquine; 3-MA, 3-methyladenine; shRNA, small hairpin RNA; CT,
control. |
As previously described (27), Atg7 is a protein-coding gene which
promotes the conjugation of LC3-I to phosphati-dylethanolamine to
form LC3-II. shAtg7 lentiviral vector was used to interfere with
the formation of the essential autophagy gene product, Atg7. The
results suggested that JS1 cells infected with shAtg7 lentiviral
vector significantly reduced the protein expression levels of Atg7.
Following treatment with TGF-β1, LC3BII and Col.I expression was
significantly inhibited compared with the corresponding empty
vector control (Fig. 3E).
Therefore, inhibition of autophagy may reduce TGF-β1-induced
activation of JS1 cells, suggesting that TGF-β1-induced autophagy
might be required for fibrosis in HSCs.
TGF-β1 induces autophagy via activation
of ERK and JNK of the MAPK signaling pathway
The mechanism involved in autophagy was next
investigated. The MAPK signaling pathway has been shown to be
associated with cell proliferation, differentiation, migration,
senescence, autophagy and apoptosis (28). In addition, previous studies have
shown that TGF-β1-induced activation of HSCs is associated with the
MAPK signaling pathway (22,29,30). The results of the present study
showed that TGF-β1 simultaneously induced autophagy and fibrosis in
HSCs, and that inhibition of autophagy is associated with a
concurrent reduction in TGF-β1-induced fibrosis. Thus, it was
hypothesized that the MAPK pathway (including the ERK, JNK, and p38
MAPK cascades) may be involved in TGF-β1-induced autophagy.
The effects of JS1 cell stimulation using TGF-β1 in
the presence of the ERK inhibitor PD98059, p38 MAPK inhibitor
SB203580 and JNK inhibitor SP600125 were assessed to investigate
the role of the MAPK signaling pathways in TGF-β1-induced
autophagy. Subsequently, expression of Col.I, α-SMA and autophagy
markers were examined using western blotting or PCR. As shown in
Fig. 4A, the ERK inhibitor
PD98059 abrogated ERK phosphorylation in the presence of TGF-β1
after 2 and 12 h. To investigate this further, the effects of
PD98059 on autophagy and fibrosis were determined. TGF-β1 treatment
resulted in a significant increase in the expression of the
autophagy markers LC3BII, beclin-1 and Atg5, and the fibrosis
markers Col.I and α-SMA. These effects were partially reversed by
treatment with PD98059 (Fig. 4B and
C). As shown in Fig. 4D and
E, the JNK inhibitor SP600125 reversed JNK phosphorylation and
significantly decreased the expression of autophagy and fibrosis
markers in the presence of TGF-β1. In summary, TGF-β1-induced
autophagy and fibrosis were reduced following inhibition of the ERK
and JNK signaling pathways.
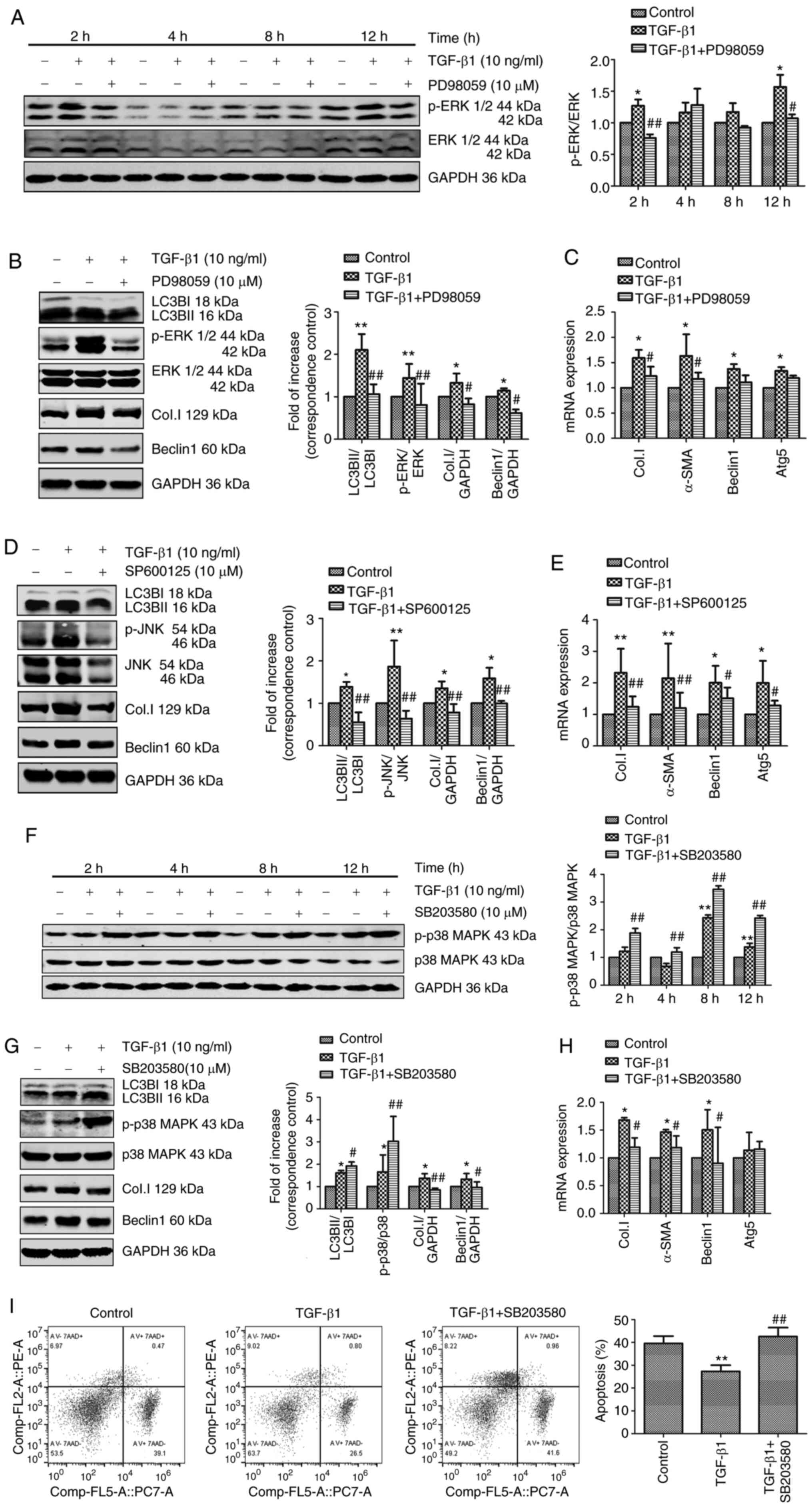 | Figure 4TGF-β1 induces autophagy via
activation of the ERK and JNK signaling pathways. (A) JS1 cells
were pretreated with or without 10 µM PD98059 for 1 h and
incubated with 10 ng/ml TGF-β1 for 2, 4, 8 and 12 h. The levels of
p-ERK and ERK were detected through western blotting. JS1 cells
were pretreated with or without 10 µM PD98059 for 1 h and
incubated with 10 µM TGF-β1 for 12 h. (B) Protein expression
levels of LC3BII, p-ERK, ERK, Col.I and Beclin-1 were detected
through western blotting. (C) mRNA expression of Col.I, α-SMA,
Beclin-1 and Atg5 were detected using RT-qPCR. JS1 cells were
pretreated with or without 10 µM SP600125 for 1 h and
incubated with 10 ng/ml TGF-β1 for 12 h. (D) The levels of LC3BII,
JNK, p-JNK, Col.I and Beclin-1 were detected using western
blotting. (E) The mRNA expression levels of Col.I, α-SMA, Beclin-1
and Atg5 were detected using RT-qPCR. (F) JS1 cells were pretreated
with or without 10 µM SB203580 for 1 h and incubated with 10
ng/ml TGF-β1 for 2, 4, 8 and 12 h. The levels of p-p38 MAPK and p38
MAPK were detected through western blotting. JS1 cells were
pretreated with or without 10 µM SB203580 for 1 h and
incubated with 10 ng/ml TGF-β1 for 12 h. (G) The protein expression
levels of LC3BII, p-p38 MAPK, p38 MAPK, Col.I and Beclin-1 were
detected through western blotting. (H) The mRNA expression levels
of Col.I, α-SMA, Beclin-1 and Atg5 were detected using RT-qPCR. (I)
The proportion of apoptotic cells was assessed by flow cytometry
using PE Annexin V and 7-AAD staining. Protein ratios were used to
quantify fold-change relative to the control. The mRNA expression
levels were expressed as the fold change relative to the control.
*P<0.05 and **P<0.01 vs. control;
#P<0.05 and ##P<0.01 vs. TGF-β1. Data
are presented as the mean ± the standard error of the mean. TGF-β1,
transforming growth factor-β1; p, phosphorylated; LC3B,
microtubule-associated protein 1 light chain 3B; RT-qPCR, reverse
transcription-quantitative PCR; α-SMA, α-smooth muscle actin;
Col.I, collagen type I; 7AAD, 7-actinomycin D. |
Treatment with the p38 MAPK inhibitor SB203580
resulted in phosphorylation of p38 MAPK (Fig. 4F) and significantly increased the
expression of LC3BII in JS1 cells (Fig. 4G). However, the expression of
fibrosis markers and Beclin-1 induced by TGF-β1 was significantly
decreased following SB203580 treatment (Fig. 4G and H). Furthermore, whether the
p38 MAPK signaling pathway regulated apoptosis rather than
autophagy to participate in TGF-β1-induced activation of HSCs was
assessed. The rate of apoptosis was assessed using flow cytometry
(Fig. 4I). Treatment with TGF-β1
resulted in a significant decrease in apoptosis, which was reversed
through pretreatment with SB203580. This finding supported the
hypothesis that the p38 MAPK signaling pathway regulated
TGF-β1-induced activation of JS1 cells to inhibit apoptosis. In
conclusion, these findings supported the notion that, unlike the
p38 MAPK signaling cascade, ERK and JNK may be responsible for
TGF-β1-induced autophagy.
Discussion
In the present study, it was shown that TGF-β1
simultaneously induced autophagy and fibrosis in a mouse HSC line.
Moreover, inhibition of autophagy, either through the use of
pharmacological inhibitors (CQ and 3-MA) or shAtg7, reduced
TGF-β1-induced fibrosis. These results showed that TGF-β1 was able
to induce autophagy in HSCs, thereby further showing that
TGF-β1-induced the activation of HSCs, and this mechanism was shown
to be associated with the ERK and JNK signaling pathway.
Autophagy is a cellular pathway through which
proteins and organelles are delivered to lysosomes for degradation,
allowing for turnover of cell components and providing energy and
macromolecular precursors (31).
Basal level autophagy occurs in all types of cells; however,
uncontrolled autophagy leads to programmed cell death (4). This duality of outcomes also
manifests in the treatment of liver fibrosis. For example, oroxylin
A (32) and caffeic acid
phenethyl ester (33) attenuated
liver fibrosis via activation of the autophagy pathway, whereas
trolline (34) and quercetin
(35) ameliorated liver fibrosis
by suppressing autophagy.
The complex function of autophagy in promoting or
preventing liver fibrosis may be dependent on different cells and
different stages of the disease. Hernández-Gea et al
(12) reported that autophagy
stimulated the loss of lipid droplets, which provides cellular
energy to promote survival and activation of HSCs. Lodder et
al (36) found that
atg5−/− mice were more susceptible to liver fibrosis, as
shown by enhanced matrix and fibrogenic cell accumulation. Hepatic
myofibro-blasts exposed to the conditioned medium of macrophages
from atg5−/− mice showed increased profibrogenic gene
expression. However, Hong et al (37) showed that activation of HSCs was
inhibited in ATG2A-deficient LX2 cells. Ruart et al
(38) recently suggested that the
selective potentiation of autophagy in liver sinusoidal endothelial
cells during the early stages of liver disease may be an attractive
approach to modify the disease course and prevent the progression
of fibrosis.
TGF-β1 is a key regulator of different cellular
processes and regulates both autophagy and fibrosis in numerous
types of tissues. The effect of TGF-β1 on autophagy has been
previously reported in several fibrotic diseases, and its role may
differ between various tissues or cells (19,39-43). A number of studies have shown that
TGF-β1 induces autophagy. In cardiac fibrosis, TGF-β1 increased
autophagic activation and promoted fibrosis in primary human atrial
myofibroblasts (19). Moreover,
in renal fibrosis, TGF-β1 induced autophagy and protected
glomerular mesangial cells via activation of the Akt pathway
(42,43). However, other studies reported
contrasting results. In pulmonary fibrosis, autophagy was inhibited
by TGF-β1 in human lung fibroblast cells, and the expression of
α-SMA and fibronectin was enhanced (39,40).
The results of the present study showed that the
effect induced by TGF-β1 was attenuated following inhibition of
autophagy through treatment with pharmacological inhibitors (CQ and
3-MA) or shAtg7. CQ inhibits autophagy by suppressing the fusion of
autophagosomes and lysosomes (44). Therefore, although the progression
of autophagy was inhibited, autophagosome accumulation still
occurred. 3-MA is an inhibitor of PI3K and has been widely used as
an inhibitor of autophagy, based on its inhibitory effect on class
III PI3K, which is essential for the induction of autophagy
(45). However, 3-MA also blocks
class I PI3K, which exhibits the opposite effect to that of class
III PI3K (14). This may explain
the upregulation of Col.I mRNA expression levels in JS1 cells
incubated with 3-MA (5 mM) for 24 h. A similar result was reported
in mouse embryonic fibro-blasts and primary HSCs (45). Finally, the results of the present
study revealed that TGF-β1 induced autophagy in HSCs, and autophagy
is involved in TGF-β1-induced activation of HSCs. Although these
results are consistent with a previous study (26), contrasting results have also been
reported. Thomes et al (46) found that exposure of primary rat
HSCs to TGF-β (5 ng/ml) increased the levels of fibrogenic markers,
but simultaneously decreased the rate of synthesis of
autophagosomes. These conflicting results may suggest that the role
of TGF-β in autophagy is related to the level and duration of its
activation.
The mechanism involved in the processes assessed
were also determined. The MAPK signal transduction pathways are
amongst the most widespread mechanisms involved in the regulation
of eukaryotic cells (47). As
shown previously, MAPKs can be activated by TGF-β1 and regulate
autophagy (48-52). In the present study, the role of
the most extensively studied MAPK pathways (ERK, p38 and JNK) in
TGF-β1-induced autophagy were assessed. The ERK inhibitor, PD98059,
and JNK inhibitor, SP600125, were able to attenuate TGF-β1-induced
autophagy and fibrosis in JS1 cells. Although the p38 MAPK
inhibitor SB203580 decreased the activation of JS1 cells, an
opposing effect on the autophagy pathway was observed. SB203580
resulted in phosphorylation of p38 MAPK and the induction of
autophagy. Further investigation showed that activation of p38 MAPK
may exert a protective effect, preventing apoptosis and cell death.
According to these results, TGF-β1 may induce autophagy through the
activation of the ERK and JNK pathways and inhibit apoptosis via
the p38 MAPK pathway in HSCs.
MAPK pathways, in particular p38 MAPK, serve dual
roles in autophagy, and the role exhibited may depend upon the
nature of the stimulus, and the level and duration of MAPK pathway
activation. Webber (51), Webber
and Tooze (52) reported that
p38α MAPK negatively regulates the levels of autophagy, and
activation of p38 may exhibit a protective role against autophagy.
In contrast, inhibition of p38 with SB203580 results in an
induction of autophagy, and prolonged blockade leads to cell death
with autophagic features. Moreover, activation of JNK contributes
to autophagy and cell death. These findings are in agreement with
the results of the present study.
In conclusion, the present study showed that
autophagy is involved in TGF-β1-induced activation of HSCs.
Furthermore, TGF-β1 may partly induce autophagy to promote HSC
activation via the ERK and JNK pathways. These results provide a
novel framework for understanding the mechanism of TGF-β1 and its
involvement in the activation of HSCs. Additionally, the present
study highlighted autophagy as a putative novel therapeutic target
for the attenuation of fibrosis. However, crosstalk occurs between
the ERK, JNK and p38 MAPK pathways, as well as autophagy and
apoptosis. Thus, further research is warranted to investigate the
molecular mechanisms of MAPK signaling, and its involvement in the
regulation of autophagy.
Funding
The present study was supported by the General
Program of the National Natural Science Foundation of China (grant
no. 81373859).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LX conceived and designed the study, contributed
reagents and materials. JZ and NJ performed research and analyzed
data. JZ wrote the manuscript. JP analyzed and interpreted the data
and revised the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The experimental protocols used in the present study
were approved by the Shuguang Hospital Affiliated to Shanghai
University of Traditional Chinese Medicine Animal Care and Ethics
committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
We would like to thank Professor Scott L. Friedman
(Icahn School of Medicine at Mount Sinai, USA) for providing the
mouse immortalized HSC cell line, JS1. We would like to thank
Professor Mark J. Czaja (Albert Einstein College of Medicine, USA)
for providing shAtg7 expression plasmids.
References
|
1
|
Lee UE and Friedman SL: Mechanisms of
hepatic fibrogenesis. Best Pract Res Clin Gastroenterol.
25:195–206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsuchida T and Friedman SL: Mechanisms of
hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol.
14:397–411. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Higashi T, Friedman SL and Hoshida Y:
Hepatic stellate cells as key target in liver fibrosis. Adv Drug
Deliv Rev. 121:27–42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tseng YJ, Dong L, Liu YF, Xu N, Ma W, Weng
SQ, Janssen HLA and Wu SD: Role of autophagy in chronic liver
inflammation and fibrosis. Curr Protein Pept Sci. 20:817–822. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chao X and Ding WX: Role and mechanisms of
autophagy in alcohol-induced liver injury. Adv Pharmacol.
85:109–131. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cingolani F and Czaja MJ: Regulation and
functions of autophagic lipolysis. Trends Endocrinol Metab.
27:696–705. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carmona-Gutierrez D, Zimmermann A and
Madeo F: A molecular mechanism for lipophagy regulation in the
liver. Hepatology. 61:1781–1783. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mao YQ and Fan XM: Autophagy: A new
therapeutic target for liver fibrosis. World J Hepatol.
7:1982–1986. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Song Y, Zhao Y, Wang F, Tao L, Xiao J and
Yang C: Autophagy in hepatic fibrosis. Biomed Res Int.
2014:4362422014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thoen LF, Guimarães EL, Dollé L, Mannaerts
I, Najimi M, Sokal E and van Grunsven LA: A role for autophagy
during hepatic stellate cell activation. J Hepatol. 55:1353–1360.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hernández-Gea V, Ghiassi-Nejad Z,
Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ and Friedman SL:
Autophagy releases lipid that promotes fibrogenesis by activated
hepatic stellate cells in mice and in human tissues.
Gastroenterology. 142:938–946. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
He W, Wang B, Yang J, Zhuang Y, Wang L,
Huang X and Chen J: Chloroquine improved carbon
tetrachloride-induced liver fibrosis through its inhibition of the
activation of hepatic stellate cells: Role of autophagy. Biol Pharm
Bull. 37:1505–1509. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang B, Yang H, Fan Y, Yang Y, Cao W, Jia
Y, Cao Y, Sun K, Pang Z and Du H: 3-Methyladenine ameliorates liver
fibrosis through autophagy regulated by the NF-κB signaling
pathways on hepatic stellate cell. Oncotarget. 8:107603–107611.
2017. View Article : Google Scholar
|
|
15
|
Bridle KR, Popa C, Morgan ML, Sobbe AL,
Clouston AD, Fletcher LM and Crawford DH: Rapamycin inhibits
hepatic fibrosis in rats by attenuating multiple profibrogenic
pathways. Liver Transpl. 15:1315–1324. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu N, Chen L, Yan D, Zhou M, Shao C, Lu Y,
Yao Q, Sun H and Fu Y: Trehalose attenuates TGF-β1-induced fibrosis
of hSCFs by activating autophagy. Mol Cell Biochem. 470:175–188.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nam SA, Kim WY, Kim JW, Park SH, Kim HL,
Lee MS, Komatsu M, Ha H, Lim JH, Park CW, et al: Autophagy
attenuates tubulointerstital fibrosis through regulating
transforming growth factor-β and NLRP3 inflammasome signaling
pathway. Cell Death Dis. 10:782019. View Article : Google Scholar
|
|
18
|
Ghavami S, Yeganeh B, Zeki AA, Shojaei S,
Kenyon NJ, Ott S, Samali A, Patterson J, Alizadeh J, Alizadeh J, et
al: Autophagy and the unfolded protein response promote profibrotic
effects of TGF-β1 in human lung fibroblasts. Am J Physiol Lung Cell
Mol Physiol. 314:L493–L504. 2018. View Article : Google Scholar
|
|
19
|
Ghavami S, Cunnington RH, Gupta S, Yeganeh
B, Filomeno KL, Freed DH, Chen S, Klonisch T, Halayko AJ, Ambrose
E, et al: Autophagy is a regulator of TGF-β1-induced fibrogenesis
in primary human atrial myofibroblasts. Cell Death Dis.
6:e16962015. View Article : Google Scholar
|
|
20
|
Wu J, Xing C, Zhang L, Mao H, Chen X,
Liang M, Wang F, Ren H, Cui H, Jiang A, et al: Autophagy promotes
fibrosis and apoptosis in the peritoneum during long-term
peritoneal dialysis. J Cell Mol Med. 22:1190–1201. 2018.
|
|
21
|
Guo J, Loke J, Zheng F, Hong F, Yea S,
Fukata M, Tarocchi M, Abar OT, Huang H, Sninsky JJ and Friedman SL:
Functional linkage of cirrhosis-predictive single nucleotide
polymorphisms of Toll-like receptor 4 to hepatic stellate cell
responses. Hepatology. 49:960–968. 2009. View Article : Google Scholar
|
|
22
|
Lv Z, Song Y, Xue D, Zhang W, Cheng Y and
Xu L: Effect of salvianolic-acid B on inhibiting MAPK signaling
induced by transforming growth factor-β1 in activated rat hepatic
stellate cells. J Ethnopharmacol. 132:384–392. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu CH, Hu YY, Wang XL, Liu P and Xu LM:
Effects of salvianolic acid-A on NIH/3T3 fibroblast proliferation,
collagen synthesis and gene expression. World J Gastroenterol.
6:361–364. 2000. View Article : Google Scholar
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Blaner WS, O'Byrne SM, Wongsiriroj N,
Kluwe J, D'Ambrosio DM, Jiang H, Schwabe RF, Hillman EM, Piantedosi
R and Libien J: Hepatic stellate cell lipid droplets: A specialized
lipid droplet for retinoid storage. Biochim Biophys Acta.
1791:467–473. 2009. View Article : Google Scholar :
|
|
26
|
Fu MY, He YJ, Lv X, Liu ZH, Shen Y, Ye GR,
Deng YM and Shu JC: Transforming growth factor-β1 reduces apoptosis
via autophagy activation in hepatic stellate cells. Mol Med Rep.
10:1282–1288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mizushima N, Yoshimori T and Ohsumi Y: The
role of Atg proteins in autophagosome formation. Annu Rev Cell Dev
Biol. 27:107–132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun Y, Liu WZ, Liu T, Feng X, Yang N and
Zhou HF: Signaling pathway of MAPK/ERK in cell proliferation,
differentiation, migration, senescence and apoptosis. J Recept
Signal Transduct Res. 35:600–604. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lv Z and Xu L: Salvianolic acid B inhibits
ERK and p38 MAPK signaling in TGF-β1-stimulated human hepatic
stellate cell line (LX-2) via distinct pathways. Evid Based
Complement Alternat Med. 2012:9601282012. View Article : Google Scholar
|
|
30
|
Zhang W, Ping J, Zhou Y, Chen G and Xu L:
Salvianolic acid B inhibits activation of human primary hepatic
stellate cells through downregulation of the myocyte enhancer
factor 2 signaling pathway. Front Pharmacol. 10:3222019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Levy JMM, Towers CG and Thorburn A:
Targeting autophagy in cancer. Nat Rev Cancer. 17:528–542. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen W, Zhang Z, Yao Z, Wang L, Zhang F,
Shao J, Chen A and Zheng S: Activation of autophagy is required for
oroxylin A to alleviate carbon tetrachloride-induced liver fibrosis
and hepatic stellate cell activation. Int Immunopharmacol.
56:148–155. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang N, Dang S, Shi J, Wu F, Li M, Zhang
X, Li Y, Jia X and Zhai S: Caffeic acid phenethyl ester attenuates
liver fibrosis via inhibition of TGF-β1/Smad3 pathway and induction
of autophagy pathway. Biochem Biophys Res Commun. 486:22–28. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bai F, Huang Q, Nie J, Lu S, Lu C, Zhu X,
Wang Y, Zhuo L, Lu Z and Lin X: Trolline ameliorates liver fibrosis
by inhibiting the NF-κB pathway, promoting HSC apoptosis and
suppressing autophagy. Cell Physiol Biochem. 44:436–446. 2017.
View Article : Google Scholar
|
|
35
|
Wu L, Zhang Q, Mo W, Feng J, Li S, Li J,
Liu T, Xu S, Wang W, Lu X, et al: Quercetin prevents hepatic
fibrosis by inhibiting hepatic stellate cell activation and
reducing autophagy via the TGF-β1/Smads and PI3K/Akt pathways. Sci
Rep. 7:92892017. View Article : Google Scholar
|
|
36
|
Lodder J, Denaës T, Chobert MN, Wan J,
El-Benna J, Pawlotsky JM, Lotersztajn S and Teixeira-Clerc F:
Macrophage autophagy protects against liver fibrosis in mice.
Autophagy. 11:1280–1292. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hong Y, Li S, Wang J and Li Y: Author
correction: In vitro inhibition of hepatic stellate cell activation
by the autophagy-related lipid droplet protein ATG2A. Sci Rep.
8:145692018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ruart M, Chavarria L, Campreciós G,
Suárez-Herrera N, Montironi C, Guixé-Muntet S, Bosch J, Friedman
SL, Garcia-Pagán JC and Hernández-Gea V: Impaired endothelial
autophagy promotes liver fibrosis by aggravating the oxidative
stress response during acute liver injury. J Hepatol. 70:458–469.
2019. View Article : Google Scholar :
|
|
39
|
Patel AS, Lin L, Geyer A, Haspel JA, An
CH, Cao J, Rosas IO and Morse D: Autophagy in idiopathic pulmonary
fibrosis. PLoS One. 7:e413942012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Araya J, Kojima J, Takasaka N, Ito S,
Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi
M, et al: Insufficient autophagy in idiopathic pulmonary fibrosis.
Am J Physiol Lung Cell Mol Physiol. 304:L56–L69. 2013. View Article : Google Scholar
|
|
41
|
Ding Y, Kim JK, Kim SI, Na HJ, Jun SY, Lee
SJ and Choi ME: TGF-{beta}1 protects against mesangial cell
apoptosis via induction of autophagy. J Biol Chem. 285:37909–37919.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sureshbabu A, Muhsin SA and Choi ME: TGF-β
signaling in the kidney: Profibrotic and protective effects. Am J
Physiol Renal Physiol. 310:F596–F606. 2016. View Article : Google Scholar
|
|
43
|
Ding Y and Choi ME: Regulation of
autophagy by TGF-β: Emerging role in kidney fibrosis. Semin
Nephrol. 34:62–71. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wu YT, Tan HL, Shui G, Bauvy C, Huang Q,
Wenk MR, Ong CN, Codogno P and Shen HM: Dual role of
3-methyladenine in modulation of autophagy via different temporal
patterns of inhibition on class I and III phosphoinositide
3-kinase. J Biol Chem. 285:10850–10861. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Thomes PG, Brandon-Warner E, Li T, Donohue
TM Jr and Schrum LW: Reverb agonist and TGF-β similarly affect
autophagy but differentially regulate hepatic stellate cell
fibro-genic phenotype. Int J Biochem Cell Biol. 81:137–147. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Krishna M and Narang H: The complexity of
mitogen-activated protein kinases (MAPKs) made simple. Cell Mol
Life Sci. 65:3525–3544. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhou YY, Li Y, Jiang WQ and Zhou LF:
MAPK/JNK signal-ling: A potential autophagy regulation pathway.
Biosci Rep. 35:e001992015. View Article : Google Scholar
|
|
49
|
Cagnol S and Chambard JC: ERK and cell
death: Mechanisms of ERK-induced cell death-apoptosis, autophagy
and senescence. FEBS J. 277:2–21. 2010. View Article : Google Scholar
|
|
50
|
Xu XF, Liu F, Xin JQ, Fan JW, Wu N, Zhu
LJ, Duan LF, Li YY and Zhang H: Respective roles of the
mitogen-activated protein kinase (MAPK) family members in
pancreatic stellate cell activation induced by transforming growth
factor-β1 (TGF-β1). Biochem Biophys Res Commun. 501:365–373. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Webber JL: Regulation of autophagy by
p38alpha MAPK. Autophagy. 6:292–293. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Webber JL and Tooze SA: Coordinated
regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP.
EMBO J. 29:27–40. 2010. View Article : Google Scholar
|