Introduction
Pulmonary hypertension (PH) is a chronic and
progressive disease with multiple etiologies and a high mortality
rate. At the Sixth World Symposium on Pulmonary Hypertension, the
hemodynamic definition of PH was proposed as a mean pulmonary
artery pressure (mPAP) >20 mmHg (1). Based on the etiology and treatment
modalities, the World Health Organization (WHO) has defined 5
groups of PH. Group 1 pulmonary arterial hypertension (PAH)
encompasses diverse diseases that result in similar pathological
changes within the pulmonary vasculature, which is a small subset
of pulmonary hypertensive syndromes (WHO categories 2-5) (2,3).
PAH is characterized by the pathological vascular remodeling of
arterial vessels and elevated pulmonary artery pressure, leading to
pressure overload of the right ventricle (RV) and eventually, to
heart failure.
Although the exact mechanisms responsible for the
development of PAH remain unclear, several molecular and cellular
abnormalities appear to play important roles. For example,
pulmonary artery endothelial cell (PAEC) dysfunction, local
inflammation, and the abnormal proliferation of pulmonary artery
smooth muscle cells (PASMCs) underlie the pathological changes of
PAH (4). PAECs are recognized as
a major regulator of pulmonary vascular function, and their
dysfunction leads to the imbalanced production of endogenous
vasoconstrictors (serotonin and endothelin) and vasodilators
(nitric oxide and prostacyclin) (5). In addition, in vitro analyses
have demonstrated that the exposure of PAECs to hypoxia causes the
synthesis and release of inflammatory cytokines, such as tumor
necrosis factor-α (TNF-α) and interleukin (IL)-1β (6). These changes underpin the abnormal
proliferation of PASMCs, and the abnormal vasoconstriction and
remodeling of pulmonary vessels. Evidence from animal models and
studies on patients with PAH suggests that inflammation may
contribute to pulmonary vascular remodeling (7,8).
The number of inflammatory cells (T and B lymphocytes,
macro-phages) and the level of cytokines (TNF-α, IL-1β, IL-6, IL-8)
are increased in PAH, and have been shown to participate in the
initiation and progression of PAH by directly modulating the
proliferation and migration of pulmonary vascular cells (9,10).
In addition, the emergence of highly proliferative and
apoptosis-resistant PASMCs acts as a hallmark of pulmonary vessel
wall thickening and vascular remodeling (11), and there is evidence to indicate
that the improvement of vascular remodeling by normalizing the
abnormal proliferation of PASMCs is an effective strategy for the
treatment of PAH (12,13). Taken together, attenuating PAEC
dysfunction, inhibiting inflammation and reducing the abnormal
proliferation of PASMCs are promising approaches for the prevention
and treatment of PAH.
Among the several existing experimental models of
PAH, the monocrotaline (MCT) model is perhaps the one that has most
contributed to the understanding of PAH pathophysiology (14). MCT is derived from the plant,
Crotalaria spectabilis. A single dose of MCT (usually 60
mg/kg, intraperitoneally or subcutaneously) reliably causes PAH in
rats within 3 to 4 weeks (15).
The MCT model has the advantage of mimicking several key aspects of
human PAH, such as pulmonary vascular remodeling, RV failure, PASMC
proliferation, PAEC dysfunction and the increased expression of
inflammatory cytokines (14).
Astragalus membranaceus, the main ingredient
of the majority of Chinese herbal antidiabetic formulas, exerts
protective effects on the cardiovascular system, the immune system
and the nervous system (16,17). Astragalus membranaceus
contains saponins, polysaccharides and flavonoids. Astragaloside IV
(ASIV,
3-O-β-D-xylopyranosyl-6-O-β-D-glucopyranosylcyl-cloastragenol)
is a purified small molecular saponin included in Astragalus
membranaceus that has a wide range of pharmacological
properties, such as antioxidant, anti-inflammatory, antidiabetic,
antitumor, immunoregulatory and antiviral activities (18-20). ASIV has been shown to exert
protective effects against cardiovascular disease and pulmonary
fibrosis (21-23). These findings suggest that ASIV
may exert a therapeutic effect against PAH.
Several classes of drugs are currently used
clinically in the treatment of PAH; however, their usage is greatly
compromised by either limited effectiveness or unwanted
side-effects. This reality highlights the urgent need for the
development of novel pharmaceutical candidates for PAH. Screening
and identifying bioactive compounds from herbs that exert
therapeutic effects on the cardiovascular system is a promising
approach. Therefore, in the present study, the possible protective
effects of ASIV on PAH were evaluated, and the relevant mechanisms
were investigated.
Materials and methods
Reagents and antibodies
ASIV was purchased from Shanghai Yuanye
Bio-Technology Co., Ltd. (HPLC >98%; cat. no. C14J9Q65734).
High-glucose Dulbecco's modified Eagle's medium (DMEM) and fetal
bovine serum (FBS) were purchased from HyClone Laboratories, Inc.
Endothelial cell medium (ECM) and endothelial cell growth
supplement (ECGS) were purchased from ScienCell Research
Laboratories. MCT, dimethyl sulfoxide (DMSO), tribromoethanol, and
hematoxylin and eosin were purchased from Sigma-Aldrich; Merck
KGaA. Rabbit monoclonal anti-hypoxia-inducible factor (HIF)-1α
(#14179), rabbit monoclonal anti-phospho-ERK1/2 (p-ERK1/2; #4377),
rabbit polyclonal anti-total ERK1/2 (#9102), mouse monoclonal
anti-p27 (#3698), rabbit monoclonal anti-p21 (#2947), and mouse
monoclonal anti-Bcl-2 (#15071) were purchased from Cell Signaling
Technology, Inc. Mouse monoclonal anti-Bax 6A7 (sc-23959), mouse
monoclonal anti-vascular endothelial growth factor (VEGF; sc-7269),
mouse monoclonal anti-α-smooth muscle actin (SMA; sc-53142) and
mouse monoclonal anti-proliferating cell nuclear antigen (PCNA;
sc-56) were purchased from Santa Cruz Biotechnology, Inc. Rabbit
monoclonal anti-cleaved caspase-3 (AC033), mouse monoclonal
anti-cleaved caspase-9 (AC062), mouse monoclonal anti-GAPDH
(AF0006), horseradish peroxidase-conjugated goat anti-rabbit IgG
(A0208), goat anti-mouse IgG (A0216) and
3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT;
ST316) were purchased from Beyotime Institute of Biotechnology.
Animal experiments
Male Sprague-Dawley rats, 8 weeks old weighing
200-230 g, were obtained from the Animal Center of Qiqihar Medical
University. The protocol for the present study was approved by the
Qiqihar Medical University Institutional Review Board (no.
QMU-AECC-2018-27). The rats were housed in a temperature- and
humidity-controlled environment with 12-h light/dark cycles. Food
and water were available ad libitum. The experiments
conformed to the National Institutes of Health guidelines
concerning the care and use of laboratory animals, and all animal
procedures were approved by the Animal Care and Use Committee of
the Qiqihar Medical University. The rats were randomly assigned to
4 groups (8 rats per group) as follows: The control group, the MCT
group, the MCT + 10 mg/kg/dahy ASIV (ASIV10) group, and the MCT +
30 mg/kg/day ASIV (ASIV30) group. To establish MCT-induced PAH, the
rats were administered a single intraperitoneal injection of MCT
(60 mg/kg), while the control group received the same volume of
saline. MCT was dissolved in 1 N HCl, diluted in sterile saline and
adjusted to pH 7.4 with 1 N NaOH. ASIV was initially dissolved in
DMSO as a stock solution and further diluted in saline immediately
prior to use; the final DMSO concentration was 0.5%. Within hours
of the MCT injection, there were signs of pulmonary vascular
endothelial damage, but without an increase in pulmonary artery
pressure. By 2 weeks, pulmonary artery pressure began to increase,
as previously described (24). At
2 days following the MCT administration, ASIV or the vehicle (0.5%
DMSO in saline) were administered intraperitoneally once a day for
21 days.
Hemodynamic experiments
Rats were anesthetized by an intraperitoneal
injection of tribromoethanol (250 mg/kg), and a polyethylene (PE)
catheter was inserted into the RV through the right jugular vein.
Then, the RV systolic pressure (RVSP) was measured using a force
transducer and recorded via PowerLab Software (ADInstruments).
After hemodynamic measurements, the right carotid artery was
inserted with a polyethylene catheter to collect blood samples for
further biochemical assay. The rats were then euthanized by
CO2 exposure (CO2 displacement rate
equivalent to 20% of the chamber volume/min; these experiments were
performed in 2019) and cervical dislocation, and the lung and heart
tissues were collected for following analyses. To assess the
hypertrophy degree of the RV, the hearts were divided into the RV
and left ventricle plus septum (LV + S). The weight ratio of RV/(LV
+ S), known as the Fulton index, was calculated to indicate RV
hypertrophy.
Morphological investigation
The lungs were dissected into 3-mm-thick slices and
soaked in 4% neutral-buffered formalin. The lung slices were
subjected to paraffin embed-ding and sectioned into
4-µm-thick sections. The lung sections were then dewaxed in
xylene, hydrated with graded ethanol and stained with hematoxylin
and eosin. To assess pulmonary artery structural remodeling, the
percentage medial wall thickness (WT%) = (outside diameter-inside
diameter)/(outside diameter) ×100 and the percentage medial wall
area (WA%) = (medial wall area)/(total vessel area) ×100 were
calculated, as previously described (25).
Immunohistochemical staining
Lung sections were dewaxed in xylene and hydrated
with graded ethanol, and antigens were retrieved. Unspecific
protein binding was blocked with 5% bovine serum albumin for 30 min
at room temperature. After rinsing with phosphate-buffered saline,
the lung sections were incubated overnight with anti-α-SMA antibody
(1:500) or anti-PCNA antibody (1:1,000) at 4°C. The sections were
then incubated with a biotinylated anti-mouse IgG antibody diluted
at 1:500 for 1 h at room temperature. Immunoreactivity was
visualized using diaminobenzidine. The sections were then
counterstained with hematoxylin for 5 min at room temperature.
Quantitative immunohistochemical assessments of α-SMA and PCNA were
performed as previously described (25).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from each sample using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and
subjected to reverse transcription using the PrimeScript™ RT
reagent kit (Takara Bio, Inc.). RT-qPCR analysis was performed
using the Applied LightCycler 2.0 detection system (Roche Applied
Science) and the SYBR-Green I reagent (Takara Bio, Inc.) following
the manufacturers' instructions. The relative expression levels of
TNF-α and IL-1β were calculated using β-actin as an internal
control by the 2−ΔΔCq method (26). The sequences of the primers were
as follows: TNF-α forward, 5′-ATG AGC ACA GAA AGC ATG ATC-3′ and
reverse, 5′-TAC AGG CTT GTC ACT CGA ATT-3′; IL-1β forward, 5′-TAC
CTA TGT CTT GCC CGT GGA G-3′ and reverse, 5′-ATC ATC CCA CGA GTCACA
GAG G-3′; and β-actin forward, 5′-CTG TGC CCA TCT ATG AGG GT-3′ and
reverse, 5′-CAG TGA GGC CAG GAT AGA GC-3′.
Cell culture and hypoxia in vitro
Primary human PAECs (HPAECs) and human PASMCs
(HPASMCs) were obtained from ScienCell Research Laboratories. The
HPAECs were cultured in ECM (5% FBS, 1% ECGS), the HPASMCs were
cultured in DMEM supplemented with 10% FBS, and the cells used in
the experiments were between passages 3 and 8. The cells were
divided into 6 groups as follows: The normoxia, hypoxia, hypoxia +
10 µM ASIV, hypoxia + 20 µM ASIV, hypoxia + 40
µM ASIV, and hypoxia + 80 µM ASIV groups, and then
cultured either under normoxic (21% O2 and 5%
CO2) or hypoxic (2% O2 and 5% CO2)
conditions for 24 h.
HPASMC proliferation analysis
The proliferation of HPASMCs was assessed by MTT
assay. The HPASMCs in each group were placed in 96-well plates
(5,000 cells per well) and then cultured under either normoxic or
hypoxic conditions. Following treatment, an MTT solution was added
to each well at a 5 mg/ml concentration, and the plates were
incubated at 37°C for 4 h. DMSO was then added. The optical density
(OD) of the samples was measured at 570 nm in a microplate
spectrophotometer (BioTek Instruments, Inc.).
Detection of apoptosis
Lung tissue sections or HPASMC apoptosis was
evaluated by TUNEL staining using the terminal deoxyribonucleotidyl
transferase-mediated dUTP nick end-labeling (TUNEL) Apoptosis Assay
kit (Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. In brief, lung tissue sections were
dewaxed in xylene and dehydrated with graded ethanol, and incubated
with 3% H2O2 at room temperature for 10 min.
The sections were then incubated with proteinase K at room
temperature for 30 min. The sections were then incubated with the
TUNEL reaction mixture for 1 h at 37°C in the dark. Apoptotic cells
were visualized with diaminobenzidine, which exhibited a brown
color. The sections were then counterstained with hematoxylin for 5
min at room temperature. In each tissue section, 3 pulmonary
arteries at ×200 magnification were randomly selected, and the
percentage of positive cells was quantified using Image Pro Plus
software (Media Cybernetics, Inc.). For in vitro analysis,
HPASMCs were fixed in 4% paraformaldehyde for 30 min at room
temperature. Subsequently, the cells were treated with 0.3% Triton
X-100 for 5 min at room temperature, and then incubated with the
TUNEL reaction mixture for 1 h at 37°C in the dark. Localized green
fluorescence of apoptotic FITC-labeled TUNEL-positive cells was
imaged using a fluorescence microscope (Carl Zeiss), and images of
4 random and non-overlapping fields were selected from each well of
12-well plates at ×400 magnification for analysis.
Assay of TNF-α and IL-1β
Blood samples from each rat were centrifuged at
1,500 × g for 20 min at 4°C, and serum was separated. The sera and
supernatants of HPAEC culture media were collected. TNF-α and IL-1β
concentrations in the serum or supernatants were measured using
enzyme-linked immunosorbent assay (ELISA) kits (Shanghai Jianglai
Industrial Co., Ltd.) according to the manufacturer's
instructions.
Western blot analysis
Cellular proteins were extracted using RIPA lysis
buffer containing protease and phosphatase inhibitors (Beyotime
Institute of Biotechnology). The protein concentrations were
determined with a BCA protein assay kit (Beyotime Institute of
Biotechnology). Equal amounts of protein (30 µg) were
separated on 10% sodium dodecyl sulfate polyacrylamide gels and
transferred to polyvinylidene fluoride membranes. After blocking
with 5% (W/V) non-fat milk at room temperature for 2 h, the
membranes were incubated with primary antibodies against HIF-1α
(1:1,000), p-ERK1/2 (1:1,000), total ERK1/2 (1:2,000), cleaved
caspase-3 (1:1,000), cleaved caspase-9 (1:1,000), p27 (1:1,000),
p21 (1:1,000), Bcl-2 (1:1,000), Bax (1:1,000), VEGF (1:1,000) and
GAPDH (1:2,000) at 4°C overnight. The membranes were then incubated
with corresponding horseradish peroxidase-conjugated secondary
antibodies at room temperature for 1 h. Immunoreactive proteins
were detected by enhanced chemiluminescence (ECL) solution
(Beyotime Institute of Biotechnology), and densitometric analysis
was performed with the use of a Gel Imaging System 4.2 (Tanon,
Tanon Science & Technology Co., Ltd.).
Statistical analyses
All data are presented as the means ± SEM.
Statistical analysis was performed using SPSS 11.5 (SPSS, Inc.).
Statistical comparisons were performed by one-way analysis of
variance (ANOVA) followed by the Holm-Sidak post hoc test. A
significant difference was accepted at P<0.05.
Results
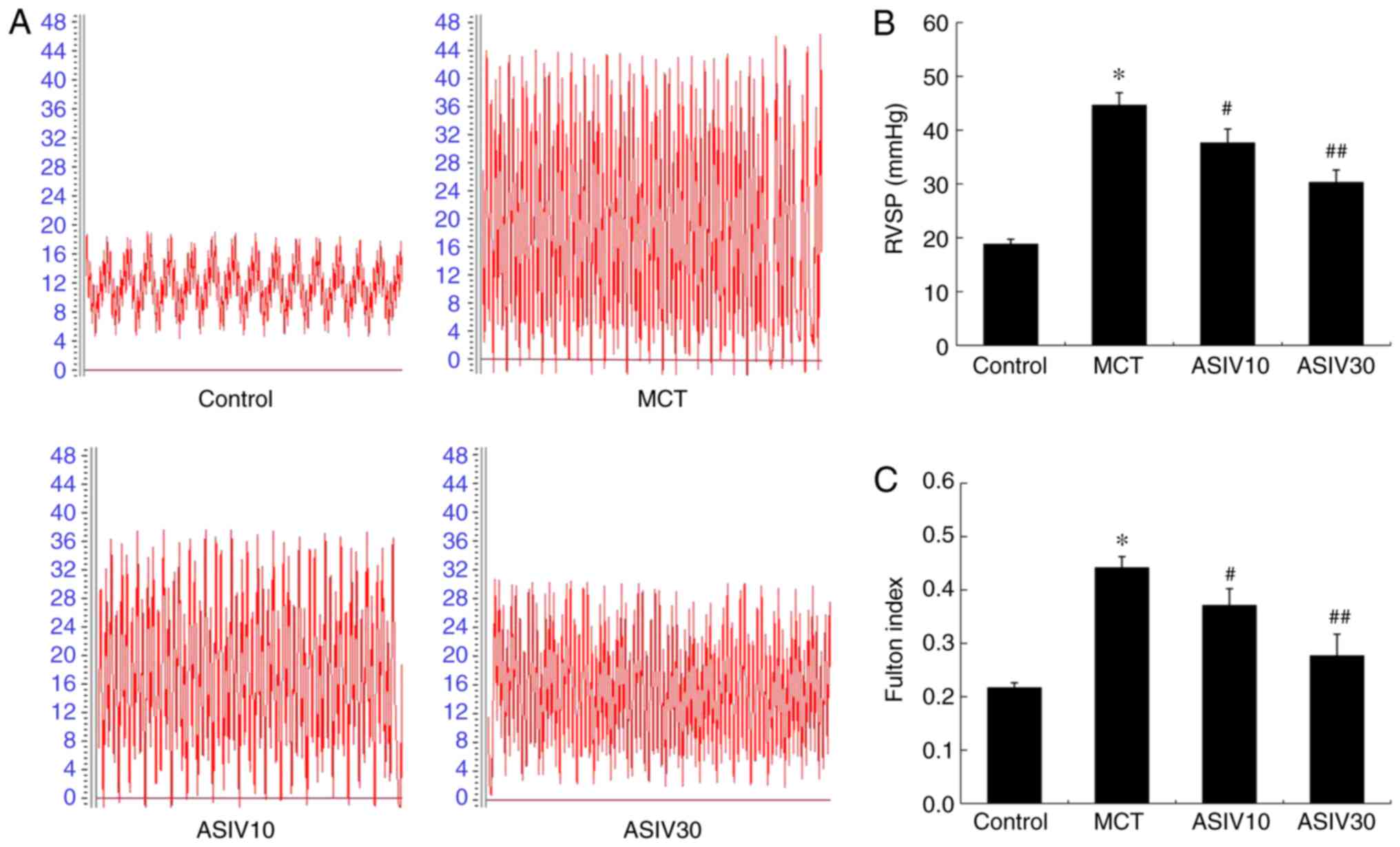
ASIV attenuates pulmonary arterial
pressure and pulmonary artery structural remodeling
The rats in the MCT group exhibited a higher RVSP
than those of the control group, indicating the establishment of
PAH. However, treatment with both doses of ASIV (10 and 30
mg/kg/day) prevented this pathophysiological change (Fig. 1A and B). The Fulton index in the
MCT group was markedly elevated compared with that in the control
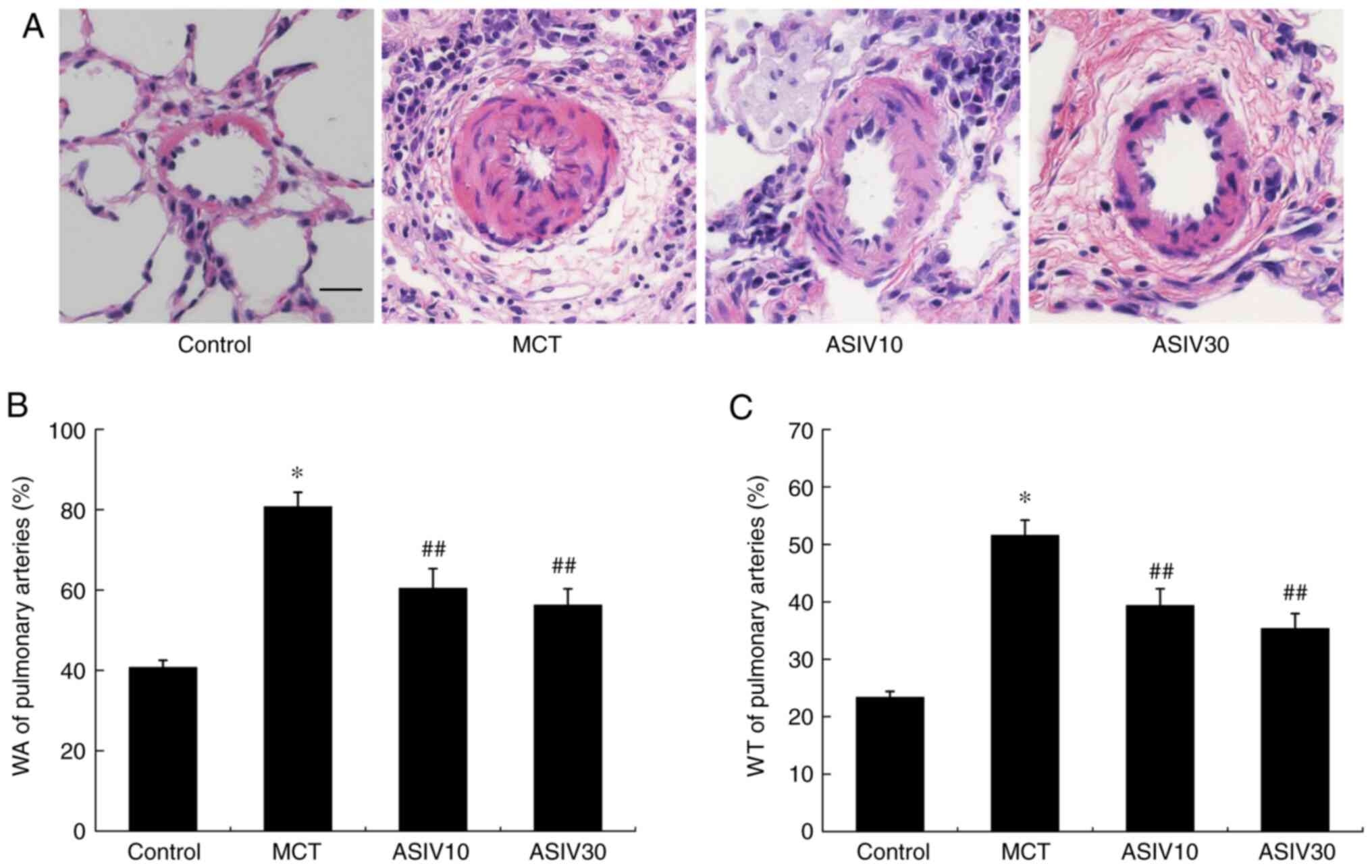
group, which was also normalized by treatment with ASIV (Fig. 1C). Furthermore, as shown in
Fig. 2, the MCT significantly
elevated WA and WT%, while treatment with 10 and 30 mg/kg/day ASIV
attenuated these pathological changes, marking the improvement of
pulmonary artery structural remodeling.
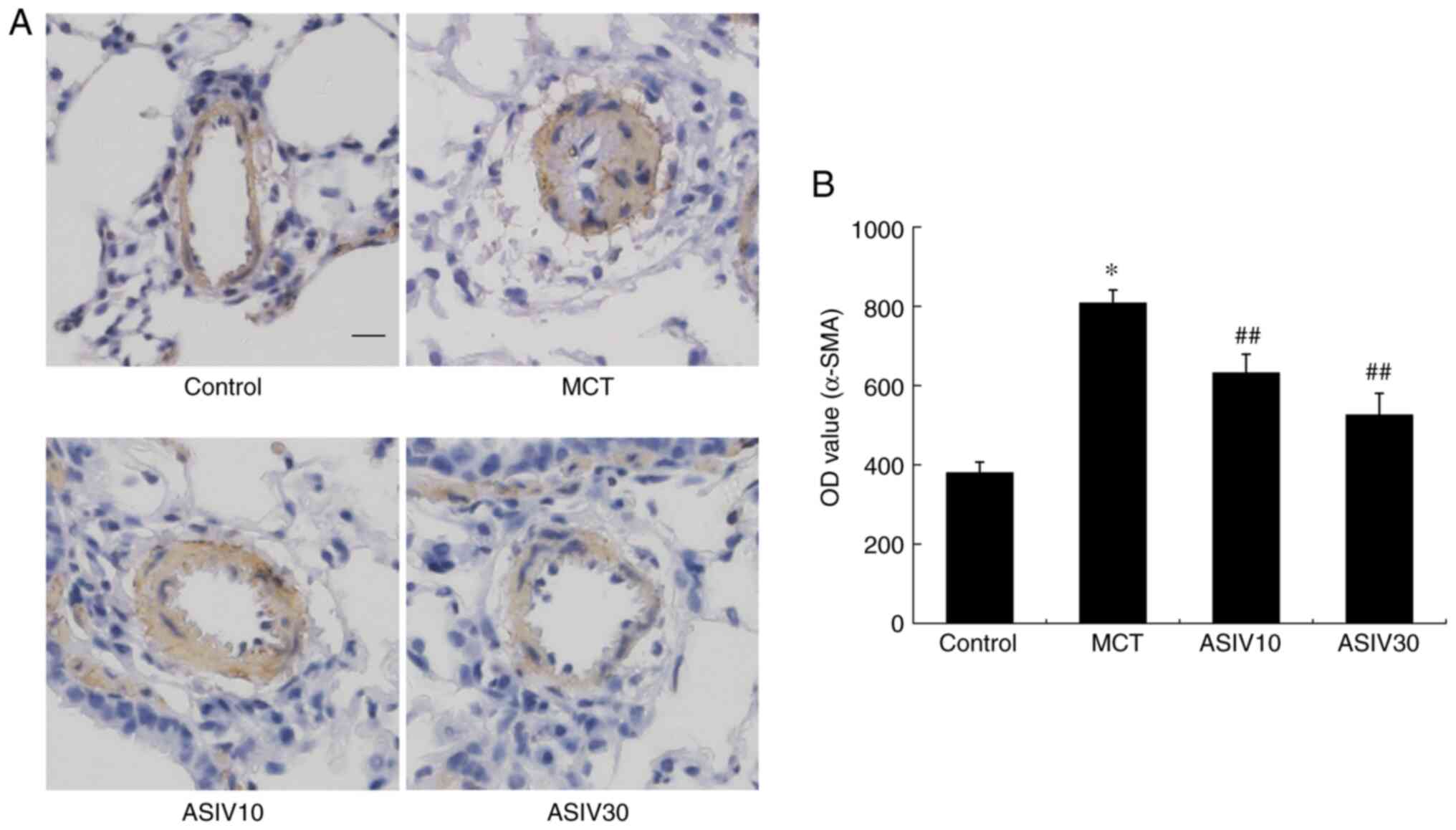
ASIV attenuates the increased expression
of α-SMA
In the present study, the levels of α-SMA reflected
the hyperplastic smooth muscularization of pulmonary arteries
(Fig. 3). The integrated OD value
of α-SMA in the MCT group was elevated compared with that in the
control group. However, both doses of ASIV inhibited the elevation
of the OD value of α-SMA.
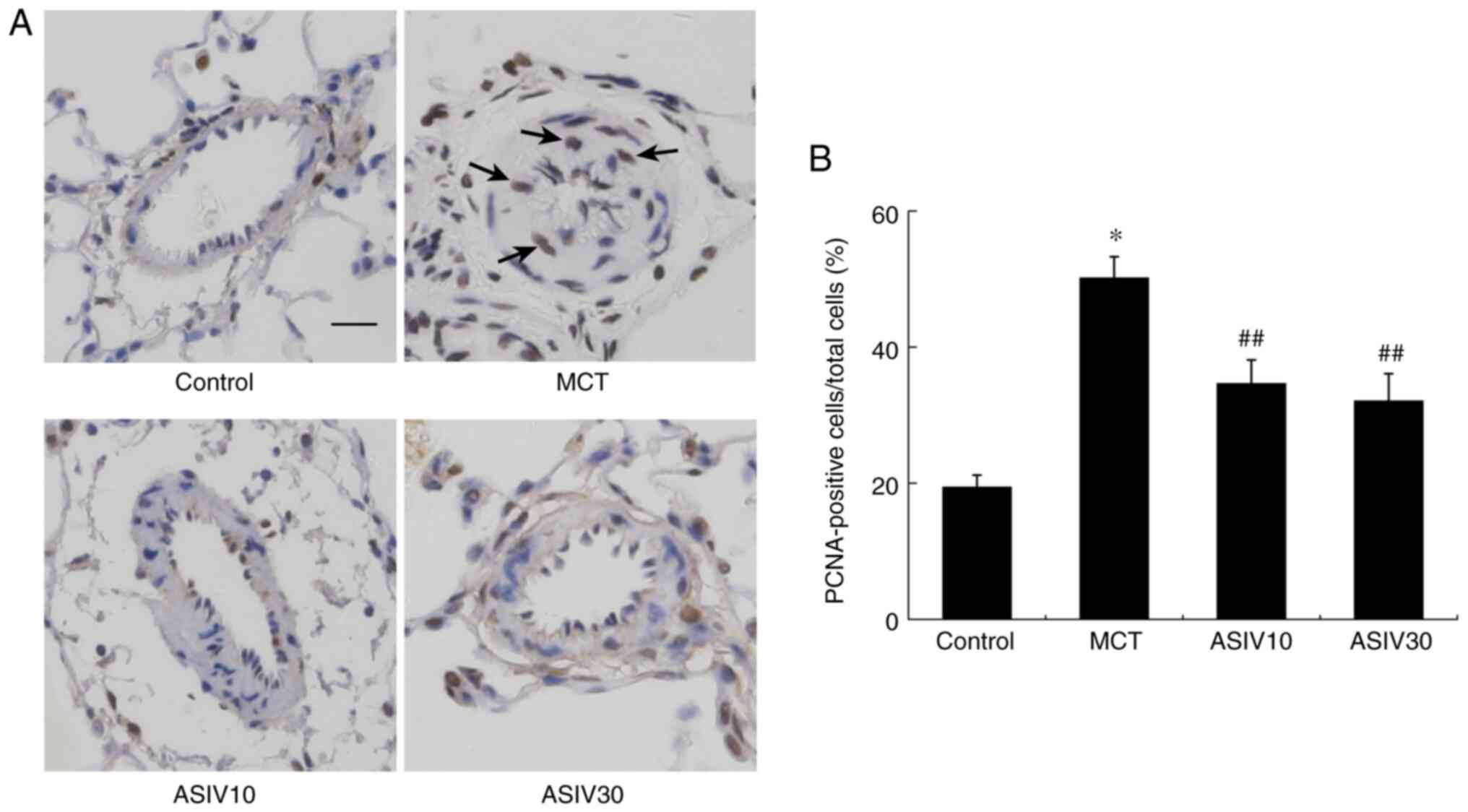
ASIV attenuates PASMC proliferation
To evaluate the role of ASIV in PASMC proliferation,
PCNA expression was measured in the medial wall of pulmonary
arteries. As shown in Fig. 4, the
numbers of PCNA-positive cells in the rats of the MCT group were
significantly increased compared with those of the control animals.
However, treatment with both doses of ASIV reduced the abnormal
PASMC proliferation. Additionally, in the in vitro
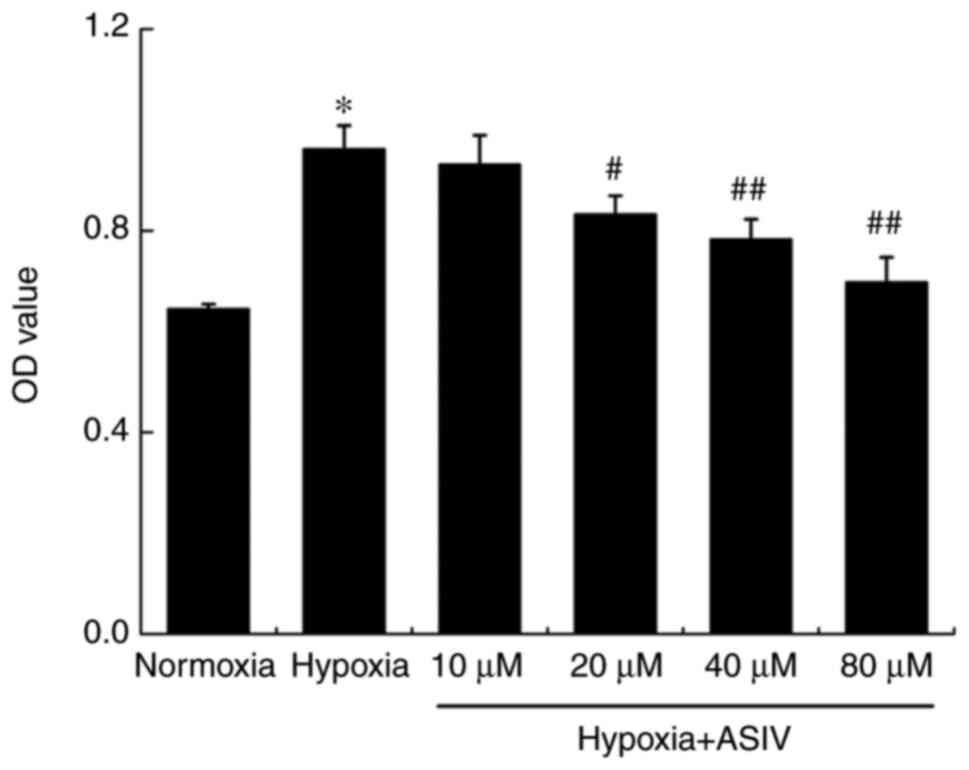
experiments, the results of MTT assay revealed that ASIV inhibited
the hypoxia-induced HPASMC proliferation, and the effects were
concentration-dependent (Fig. 5).
Moreover, the results of western blot analysis found that ASIV also
reversed the enhancement of HIF-1α and p-ERK1/2 protein expression,
and the decreases in p27 and p21 levels in the HPASMCs induced by
hypoxia (Fig. 6).
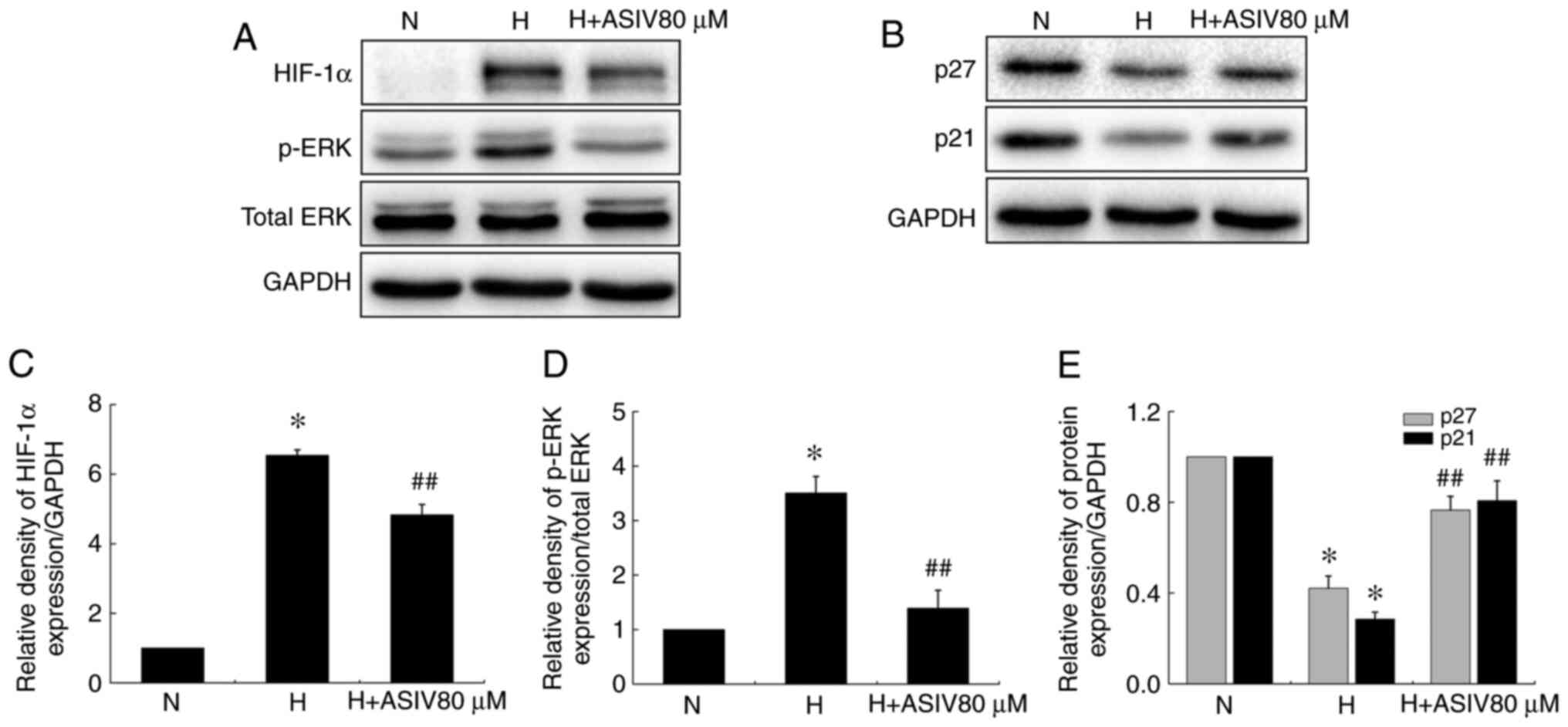 | Figure 6ASIV decreases HIF-1α and p-ERK1/2
protein levels, and increases p27 and p21 protein levels in
HPASMCs. (A and B) Western blot analysis of HIF-1α, p-ERK1/2, p27
and p21 protein levels in HPASMCs under normoxic or hypoxic
conditions. (C-E) HIF-1α, p-ERK1/2, p27 and p21 protein levels,
respectively (relative to normoxia group). Values are means ± SEM
of 3 independent experiments. *P<0.01 compared with
the normoxia group; ##P<0.01, compared with the
hypoxia group. ASIV, astragaloside IV; HPASMCs, human pulmonary
artery smooth muscle cells; N, normoxia; H, hypoxia. |
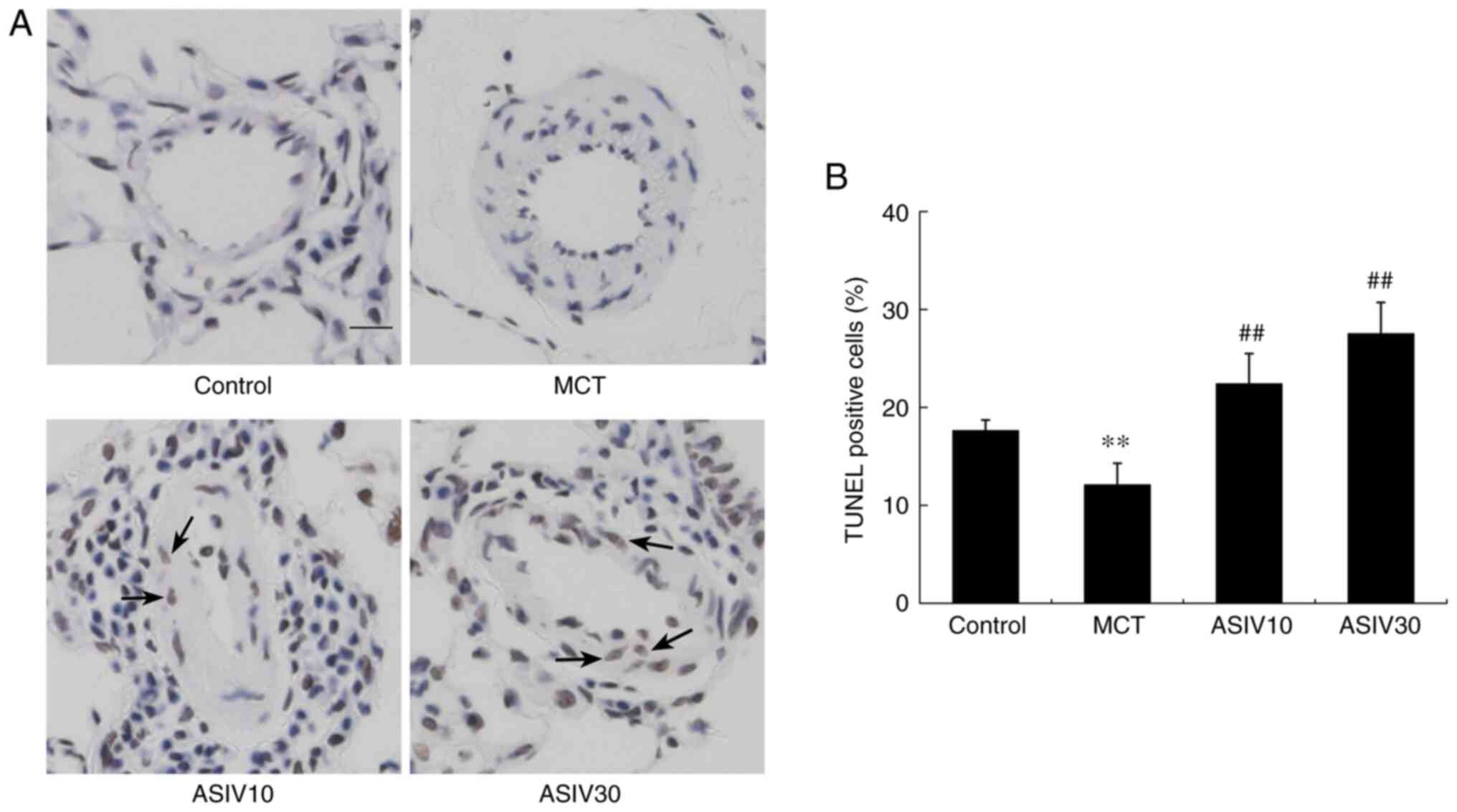
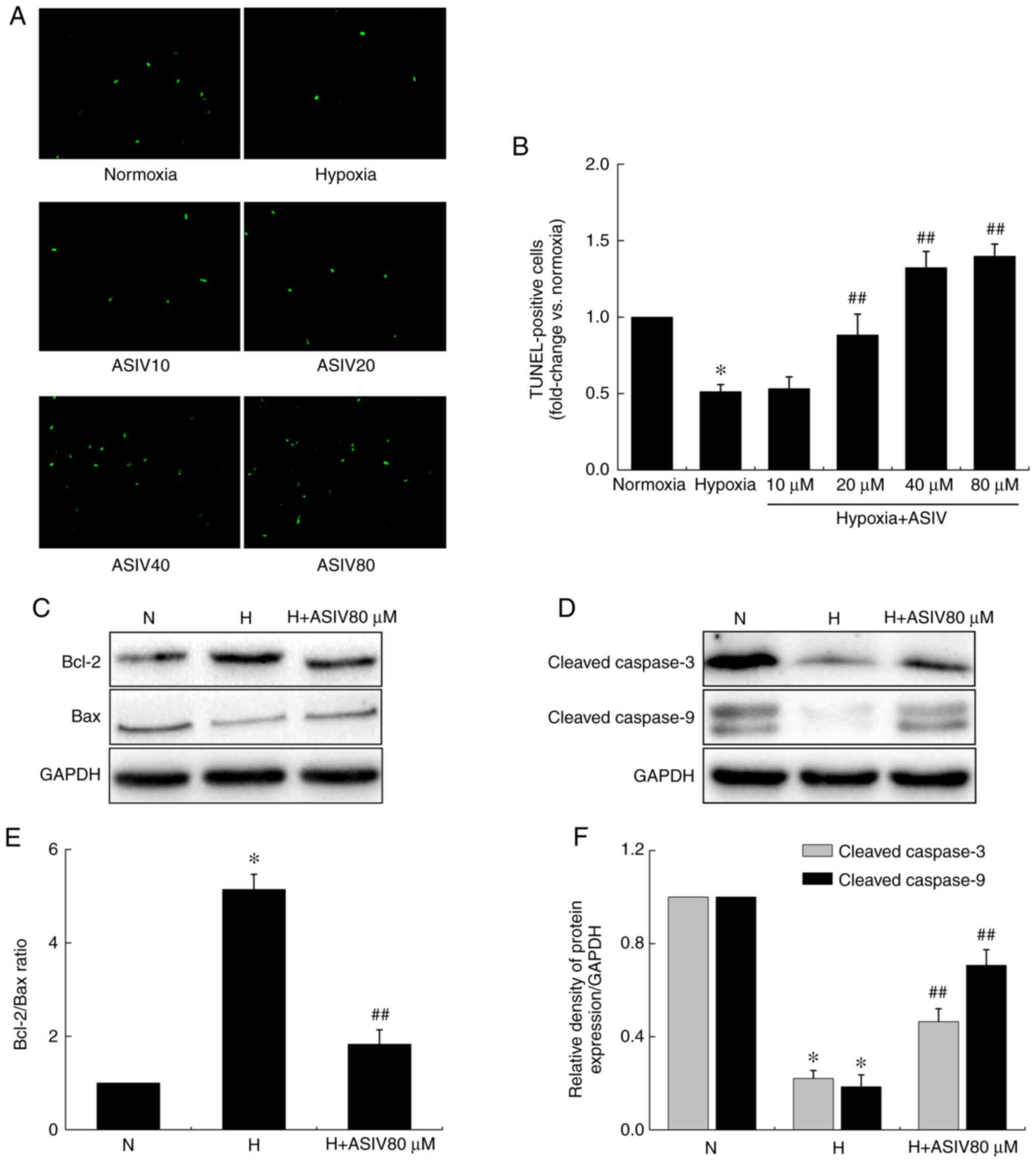
ASIV contributes to PASMC apoptosis
To determine the effects of ASIV on the apoptotic
resistance of PASMCs, a TUNEL assay was performed. As shown in
Fig. 7, disrupted apoptosis was
observed in the MCT group, while both concentrations of ASIV
significantly increased PASMC apoptosis. In the in vitro
experiments, apoptotic HPASMCs were illustrated by the green
fluorescence of the free labeled 3′-OH termini (Fig. 8A and B). The results revealed that
the percentage of apoptotic cells under hypoxic conditions was
markedly decreased compared with that under normoxic conditions,
while hypoxia-induced apoptotic resistance was reversed by ASIV. In
addition, the levels of apoptosis-related proteins in the HPASMCs
were detected by western blot analysis (Fig. 8C-F). Hypoxia markedly decreased
the levels of the pro-apoptotic proteins, Bax, cleaved caspase-9
and cleaved caspase-3, while it increased the expression of the
anti-apoptotic protein, Bcl-2, in HPASMCs. However, ASIV treatment
normalized these alterations.
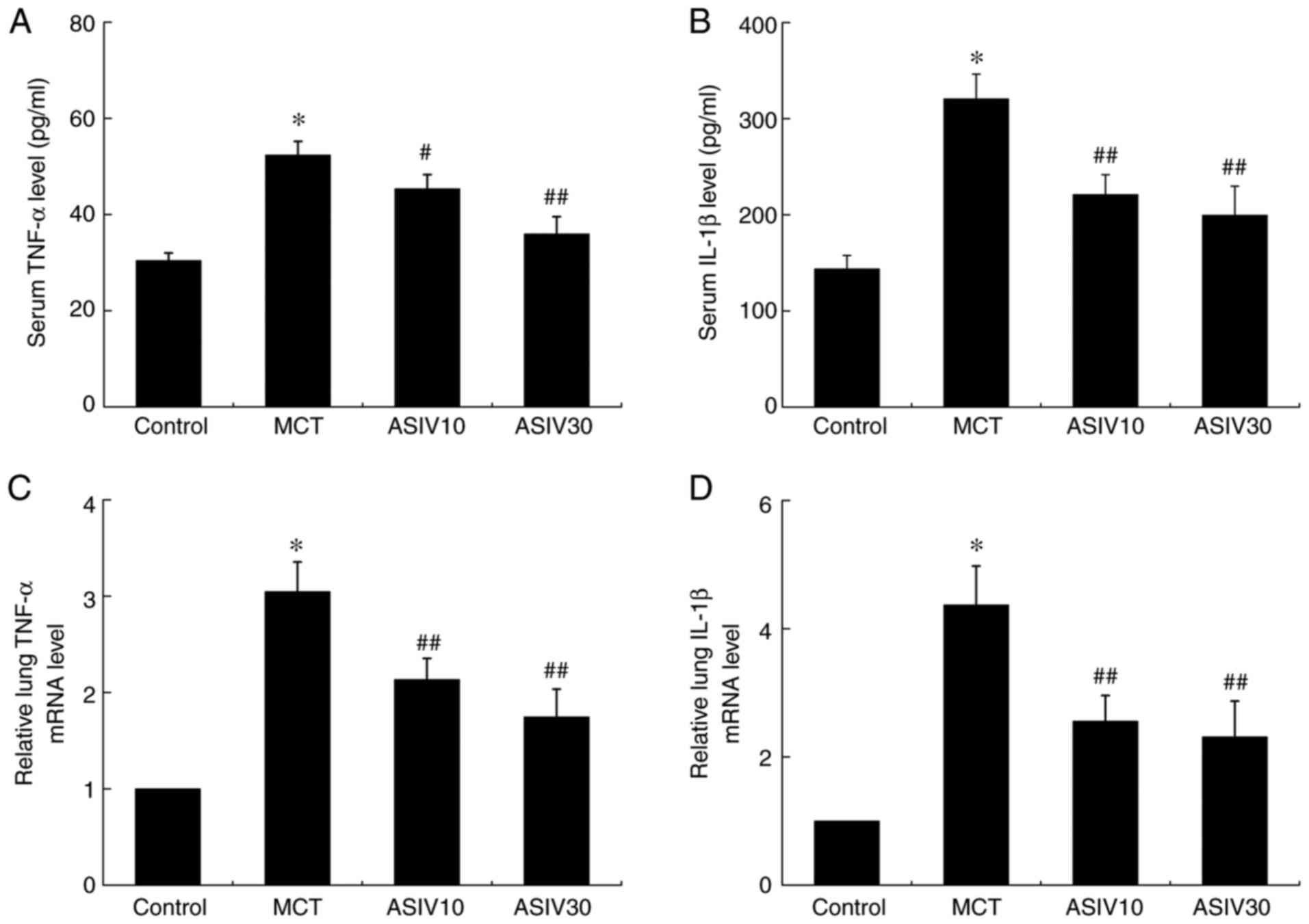
ASIV prevents the elevation of TNF-α and
IL-1β levels
To determine the anti-inflammatory effects of ASIV,
TNF-α and IL-1β levels were analyzed in serum (Fig. 9A and B). The serum levels of TNF-α
and IL-1β were elevated in the MCT group, while both doses of ASIV
suppressed these elevations. Similarly, RT-qPCR analysis revealed
that the mRNA expression of TNF-α and IL-1β in lung tissues was
upregulated in the MCT group compared with the control group, which
was also normalized by both doses of ASIV (Fig. 9C and D).
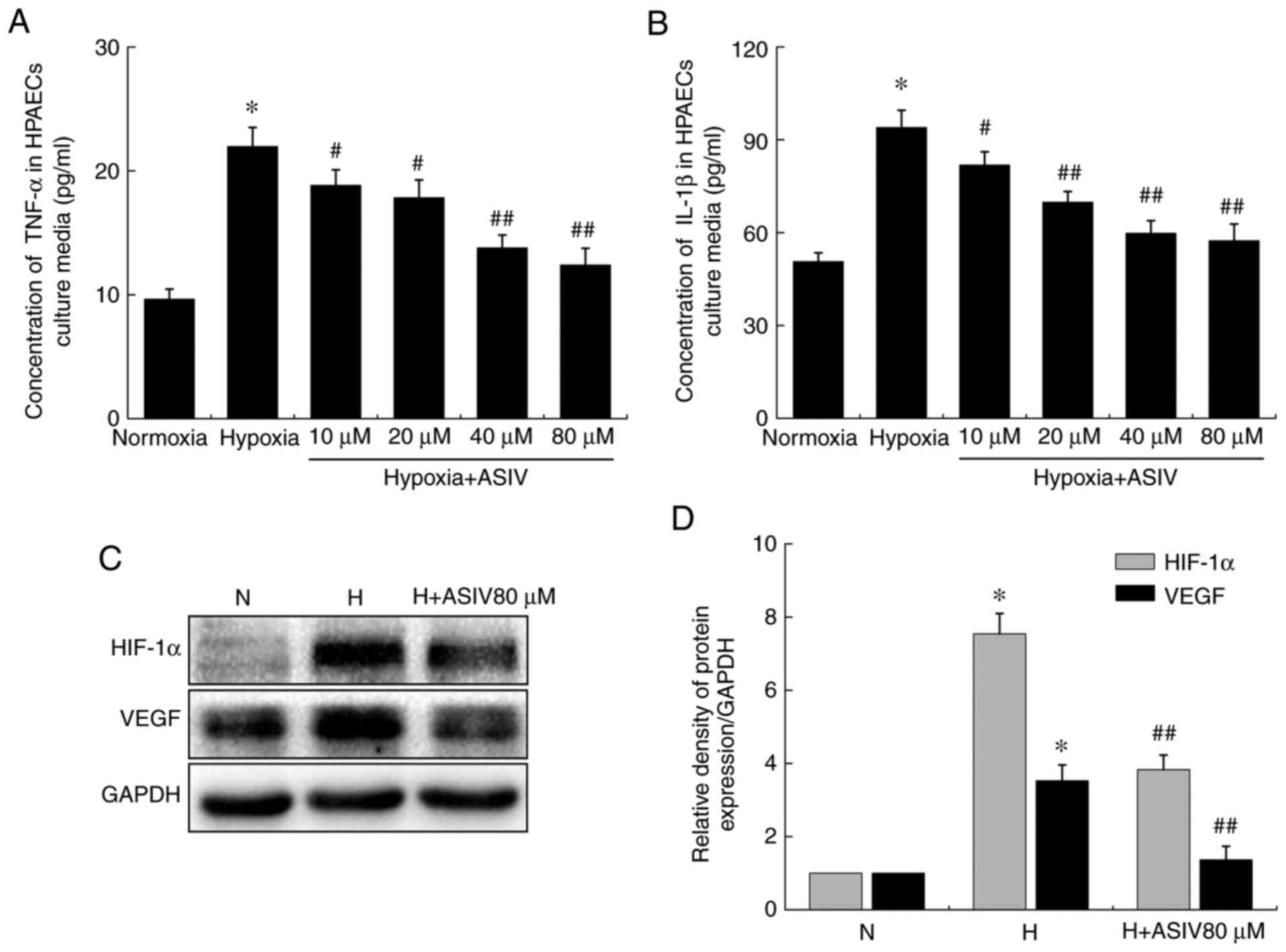
ASIV improves HPAEC dysfunction
Hypoxia increased the concentrations of TNF-α and
IL-1β in the supernatants of the HPAEC culture media compared with
normoxia. However, ASIV treatment prevented these changes in a
concentration-dependent manner (Fig.
10A and B). In addition, western blot analysis revealed that
hypoxia also increased the HIF-1α and VEGF protein levels in
HPAECs, which were reversed by ASIV treatment (Fig. 10C and D).
Discussion
In the present study, the rats in the MCT group
exhibited significant pulmonary artery remodeling, an increased
pulmonary artery pressure and right ventricular hypertrophy.
However, these pathophysiological changes were improved by both
doses of ASIV. Additionally, ASIV suppressed the elevation of TNF-α
and IL-1β secretion in serum and their gene expression in lung
tissues induced by MCT. Furthermore, in the in vivo and
in vitro experiments, ASIV normalized the abnormal
proliferation and apoptosis resistance of PASMCs and PAEC
dysfunction. These results indicate that ASIV exerts protective
effects against PAH induced by MCT. Pulmonary function tests could
permit accurate, reproducible assessment of the functional state of
the respiratory system. It would be of interest to evaluate whether
drugs exert protective effects on respiratory system. In the
present study, the pulmonary functions of the animals were not
examined. Thus, whether ASIV exerts protective effects on the
respiratory system warrants further investigation.
The pathogenesis of MCT-induced PAH is characterized
by persistent inflammation. A single MCT dose universally elevates
the numbers of inflammatory cells and increases the expression of
inflammatory cytokines in the lungs (14). For example, TNF-α and IL-1β have
been found to be closely associated with the accumulation of
extracellular matrix proteins in PAH lesions and poor clinical
outcomes in some patients with PAH. In addition, serum levels of
TNF-α and IL-1β are higher in patients with PAH than in normal
controls, and serve as biomarkers of disease progression (7,27).
TNF-α elevates pulmonary arterial reactivity and inhibits the
vasodilating action of prostacyclin via the downregulation of
prostacyclin synthase mRNA expression (28). In addition, the overexpression of
TNF-α in alveolar type II cells leads to an increased lung volume,
alveolar enlargement and increased pulmonary artery pressure
(29), while blocking TNF-α and
IL-1β signaling can ameliorate pulmonary inflammation, pulmonary
hemodynamics, pulmonary vascular remodeling and right ventricular
hypertrophy (30-32). ASIV attenuates some
inflammation-associated cardiopulmonary diseases, such as
obesity-related hypertension and cigarette smoke-induced pulmonary
inflammation by decreasing the levels of TNF-α and IL-1β (33,34). Consistent with these findings, in
the present study, ASIV suppressed the inflammatory response in a
rat model of PAH, suggesting that the anti-inflammatory action of
ASIV underlies its therapeutic effects on MCT-induced PAH.
PASMCs in patients with PAH exhibit high
proliferative and apoptosis-resistant properties similar to some
cancer cells, which appear to be associated with the thickening and
narrowing of pulmonary arteries and increased pulmonary arterial
pressure, hence constituting a promising target for the treatment
of PAH. In the present study, immunohistochemical analysis with an
anti-PCNA antibody and TUNEL assay revealed that ASIV suppressed
the proliferation and promoted the apoptosis of PASMCs in the
medial wall of the pulmonary arteries. The results of the MTT assay
and TUNEL assay in vitro also supported these findings.
HIF-1α, as an oxygen-sensitive transcription factor, is involved in
the crosstalk among several hypoxia-related signaling pathways and
promotes pulmonary artery structural remodeling by increasing
proliferation and suppressing apoptosis (35). The loss of HIF-1α in PASMCs
significantly suppresses the remodeling of pulmonary arteries and
the associated pulmonary hypertension (36). In addition, the MAPK/ERK and
PI3K/Akt signaling pathways have been recognized to mediate a wide
range of functions, including proliferation, growth and survival.
As regards the association between HIF-1α, ERK1/2 and Akt
signaling, it has been reported that the overexpression of HIF-1α
can lead to the activation of various signaling molecules,
including ERK1/2 and Akt (37).
In the presents study, in the in vitro experiments, HIF-1α
protein expression was detected in HPASMCs. The results indicated
that ASIV reversed the enhancement of HIF-1α protein expression in
HPASMCs induced by hypoxia. Subsequently, p-ERK1/2 and p-Akt
protein expression was detected. Of note, ASIV reversed the
enhancement of p-ERK1/2 protein expression in HPASMCs induced by
hypoxia, but not p-Akt protein expression (data not shown). p27 and
p21 are cyclin-dependent kinase inhibitors that play a key role in
the regulation of cell cycle progression. Recent studies have
indicated that they exert inhibitory effects on PASMC proliferation
(38,39). It has also been reported that the
suppression of proliferative signaling, such as ERK1/2 can inhibit
the degradation of p27 and p21 (40). The results of the present study
demonstrated that ASIV upregulated the protein expression of p27
and p21 in HPASMCs. Apoptosis is the process of normal programmed
cell death and involves a large number of molecules and pathways,
including Bcl-2 family proteins and caspase signaling. The protein
expression ratio of Bcl-2/Bax is important in the
mitochondria-dependent apoptotic pathway and is associated with
higher and lower apoptotic thresholds. In the present study, to
determine whether ASIV-induced PASMC apoptosis is associated with
the Bax/Bcl-2 and mitochondrial apoptosis pathways, the protein
expression of Bcl-2, Bax, cleaved caspase-9 and cleaved caspase-3
was detected in HPASMCs. The results revealed that ASIV upregulated
the protein expression of Bax, cleaved caspase-9 and cleaved
caspase-3 in HPASMCs, but downregulated Bcl-2 protein expression.
Collectively, these findings indicate that ASIV can reduce PASMC
proliferation and apoptotic resistance.
In the pulmonary vasculature, PAECs are located as
the innermost layer of the blood vessel and are the main barrier to
the movement of molecules through the vascular wall. In PAH, the
dysfunction of PAECs leads to the impairment of
endothelial-dependent vasodilatation, a decrease in anticoagulant
properties, elevated levels of adhesion molecules, an enhancement
of reactive oxygen species production and the release of different
inflammatory cytokines (41). In
addition, the dysfunction of PAECs contributes to the increased
angiogenesis that underlies plexiform lesion formation (9). As markers of angiogenesis, HIF-1α
and VEGF are highly expressed in endothelial cells of plexiform
lesions in patients with PAH (42). In the present study, it was
observed that ASIV significantly reduced the increased HIF-1α and
VEGF protein levels in HPAECs induced by hypoxia. Furthermore, ASIV
also suppressed the hypoxia-induced release of TNF-α and IL-1β in
HPAECs.
In conclusion, the present study demonstrated that
the administration of ASIV improved the pathological changes in
pulmonary artery structural remodeling, pulmonary arterial pressure
and right ventricular hypertrophy in a rat model of MCT PAH. The
therapeutic effects of ASIV were associated with the improvement of
the inflammatory response, PAEC dysfunction, and abnormal PASMC
proliferation and apoptotic resistance. These findings provide
indicate that ASIV exerts protective effects against PAH, and may
thus have potential to be developed into a promising
pharmacological candidate for the treatment of PAH. However, the
precise molecular mechanisms of action ASIV against PAH require
further investigation. In future experiments, the authors aim to
establish the Sugen 5416/hypoxia mouse model of PAH to further
confirm the possible protective effects of ASIV against PAH and
explore the precise molecular mechanisms.
Funding
The present study was supported by the Science
Research Foundation of Education Department of Heilongjiang
Province (grant no. 2018-KYYWF-0115), China; the Heilongjiang
Postdoctoral Science Foundation (grant no. LBH-Z17213), China; the
Science Research Foundation of Qiqihar Medical University (grant
no. QY2016M-02), China; and the National Research Foundation of
Korea (grant no. 2018R1A5A2025272), Korea.
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article or are available from
the corresponding author on reasonable request.
Authors' contributions
HJ, JL, RZ and SCK designed the study. HJ, YJ, LG,
YM, XL and LS conducted the experiments. HJ and ZZ performed the
statistical analysis. JL, HJ, RZ and SCK wrote the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Animal
Care and Use Committee of the Qiqihar Medical University and
conducted in accordance with the National Institutes of Health
guidelines concerning the care and use of laboratory animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Simonneau G, Montani D, Celermajer DS,
Denton CP, Gatzoulis MA, Krowka M, Williams PG and Souza R:
Haemodynamic definitions and updated clinical classification of
pulmonary hypertension. Eur Respir J. 53:18019132019. View Article : Google Scholar :
|
|
2
|
Sahay S: Evaluation and classification of
pulmonary arterial hypertension. J Thorac Dis. 11(Suppl 14):
S1789–S1799. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Archer SL, Weir EK and Wilkins MR: Basic
science of pulmonary arterial hypertension for clinicians: New
concepts and experimental therapies. Circulation. 121:2045–2066.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guignabert C and Dorfmuller P: Pathology
and pathobiology of pulmonary hypertension. Semin Respir Crit Care
Med. 34:551–559. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pullamsetti SS, Schermuly R, Ghofrani A,
Weissmann N, Grimminger F and Seeger W: Novel and emerging
therapies for pulmonary hypertension. Am J Respir Crit Care Med.
189:394–400. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pugliese SC, Poth JM, Fini MA, Olschewski
A, El Kasmi KC and Stenmark KR: The role of inflammation in hypoxic
pulmonary hypertension: From cellular mechanisms to clinical
phenotypes. Am J Physiol Lung Cell Mol Physiol. 308:L229–L252.
2015. View Article : Google Scholar :
|
|
7
|
Rabinovitch M, Guignabert C, Humbert M and
Nicolls MR: Inflammation and immunity in the pathogenesis of
pulmonary arterial hypertension. Circ Res. 115:165–175. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mushaben EM, Hershey GK, Pauciulo MW,
Nichols WC and Le Cras TD: Chronic allergic inflammation causes
vascular remodeling and pulmonary hypertension in BMPR2 hypomorph
and wild-type mice. PLoS One. 7:e324682012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vaillancourt M, Ruffenach G, Meloche J and
Bonnet S: Adaptation and remodelling of the pulmonary circulation
in pulmonary hypertension. Can J Cardiol. 31:407–415. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Freund-Michel V, Cardoso Dos Santos M,
Guignabert C, Montani D, Phan C, Coste F, Tu L, Dubois M, Girerd B,
Courtois A, et al: Role of nerve growth factor in development and
persistence of experimental pulmonary hypertension. Am J Respir
Crit Care Med. 192:342–355. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jin H, Liu M, Zhang X, Pan J, Han J, Wang
Y, Lei H, Ding Y and Yuan Y: Grape seed procyanidin extract
attenuates hypoxic pulmonary hypertension by inhibiting oxidative
stress and pulmonary arterial smooth muscle cells proliferation. J
Nutr Biochem. 36:81–88. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thenappan T, Ormiston ML, Ryan JJ and
Archer SL: Pulmonary arterial hypertension: Pathogenesis and
clinical management. BMJ. 360:j54922018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li MX, Jiang DQ and Wang Y, Chen QZ, Ma
YJ, Yu SS and Wang Y: Signal mechanisms of vascular remodeling in
the development of pulmonary arterial hypertension. J Cardiovasc
Pharmacol. 67:182–190. 2016. View Article : Google Scholar
|
|
14
|
Nogueira-Ferreira R, Vitorino R, Ferreira
R and Henriques-Coelho T: Exploring the monocrotaline animal model
for the study of pulmonary arterial hypertension: A network
approach. Pulm Pharmacol Ther. 35:8–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xiong PY, Potus F, Chan W and Archer SL:
Models and molecular mechanisms of world health organization group
2 to 4 pulmonary hypertension. Hypertension. 71:34–55. 2018.
View Article : Google Scholar
|
|
16
|
Fu J, Wang Z, Huang L, Zheng S, Wang D,
Chen S, Zhang H and Yang S: Review of the botanical
characteristics, phytochemistry, and pharmacology of Astragalus
membranaceus (Huangqi). Phytother Res. 28:1275–1283. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li X, Qu L, Dong Y, Han L, Liu E, Fang S,
Zhang Y and Wang T: A review of recent research progress on the
astragalus genus. Molecules. 19:18850–18880. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li L, Hou X, Xu R, Liu C and Tu M:
Research review on the pharmacological effects of astragaloside IV.
Fundam Clin Pharmacol. 31:17–36. 2017. View Article : Google Scholar
|
|
19
|
Qiu L, Yin G, Cheng L, Fan Y, Xiao W, Yu
G, Xing M, Jia R, Sun R, Ma X, et al: Astragaloside IV ameliorates
acute pancreatitis in rats by inhibiting the activation of nuclear
factor-κB. Int J Mol Med. 35:625–636. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu X, Zhang J, Wang S, Qiu J and Yu C:
Astragaloside IV attenuates the H2O2-induced apoptosis of neuronal
cells by inhibiting α-synuclein expression via the p38 MAPK
pathway. Int J Mol Med. 40:1772–1780. 2017.PubMed/NCBI
|
|
21
|
Wang J, Zhou Y, Wu S, Huang K, Thapa S,
Tao L, Wang J, Shen Y, Wang J, Xue Y and Ji K: Astragaloside IV
attenuated 3,4-benzopyrene-induced abdominal aortic aneurysm by
ameliorating macrophage-mediated inflammation. Front Pharmacol.
9:4962018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu Y, Li S, Wu H, Bian Z, Xu J, Gu C, Chen
X and Yang D: Beneficial effects of astragaloside IV against
angio-tensin II-induced mitochondrial dysfunction in rat vascular
smooth muscle cells. Int J Mol Med. 36:1223–1232. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qian W, Cai X, Qian Q, Zhang W and Wang D:
Astragaloside IV modulates TGF-β1-dependent epithelial-mesenchymal
transition in bleomycin-induced pulmonary fibrosis. J Cell Mol Med.
22:4354–4365. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
West J and Hemnes A: Experimental and
transgenic models of pulmonary hypertension. Compr Physiol.
1:769–782. 2011.PubMed/NCBI
|
|
25
|
Jin H, Wang Y, Zhou L, Liu L, Zhang P,
Deng W and Yuan Y: Melatonin attenuates hypoxic pulmonary
hypertension by inhibiting the inflammation and the proliferation
of pulmonary arterial smooth muscle cells. J Pineal Res.
57:442–450. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
27
|
Matura LA, Ventetuolo CE, Palevsky HI,
Lederer DJ, Horn EM, Mathai SC, Pinder D, Archer-Chicko C, Bagiella
E, Roberts KE, et al: Interleukin-6 and tumor necrosis factor-α are
associated with quality of life-related symptoms in pulmonary
arterial hypertension. Ann Am Thorac Soc. 12:370–375. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Itoh A, Nishihira J, Makita H, Miyamoto K,
Yamaguchi E and Nishimura M: Effects of IL-1beta, TNF-alpha, and
macrophage migration inhibitory factor on prostacyclin synthesis in
rat pulmonary artery smooth muscle cells. Respirology. 8:467–472.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fujita M, Shannon JM, Irvin CG, Fagan KA,
Cool C, Augustin A and Mason RJ: Overexpression of tumor necrosis
factor-alpha produces an increase in lung volumes and pulmonary
hypertension. Am J Physiol Lung Cell Mol Physiol. 280:L39–L49.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Groth A, Vrugt B, Brock M, Speich R,
Ulrich S and Huber LC: Inflammatory cytokines in pulmonary
hypertension. Respir Res. 15:472014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Q, Zuo XR, Wang YY, Xie WP, Wang H
and Zhang M: Monocrotaline-induced pulmonary arterial hypertension
is attenuated by TNF-α antagonists via the suppression of TNF-α
expression and NF-κB pathway in rats. Vascul Pharmacol. 58:71–77.
2013. View Article : Google Scholar
|
|
32
|
Campos M and Schiopu E: Pulmonary arterial
hypertension in adult-onset still's disease: Rapid response to
anakinra. Case Rep Rheumatol. 2012:5376132012.PubMed/NCBI
|
|
33
|
Jiang P, Ma D, Wang X, Wang Y, Bi Y, Yang
J, Wang X and Li X: Astragaloside IV prevents obesity-associated
hypertension by improving pro-inflammatory reaction and leptin
resistance. Mol Cells. 41:244–255. 2018.PubMed/NCBI
|
|
34
|
Meiqian Z, Leying Z and Chang C:
Astragaloside IV inhibits cigarette smoke-induced pulmonary
inflammation in mice. Inflammation. 41:1671–1680. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Paulin R and Michelakis ED: The metabolic
theory of pulmonary arterial hypertension. Circ Res. 115:148–164.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ball MK, Waypa GB, Mungai PT, Nielsen JM,
Czech L, Dudley VJ, Beussink L, Dettman RW, Berkelhamer SK,
Steinhorn RH, et al: Regulation of hypoxia-induced pulmonary
hypertension by vascular smooth muscle hypoxia-inducible factor-1α.
Am J Respir Crit Care Med. 189:314–324. 2014. View Article : Google Scholar :
|
|
37
|
Wang G, Wang JJ, Fu XL, Guang R and To ST:
Advances in the targeting of HIF-1α and future therapeutic
strategies for glioblastoma multiforme (Review). Oncol Rep.
37:657–670. 2017. View Article : Google Scholar
|
|
38
|
Fouty BW, Grimison B, Fagan KA, Le Cras
TD, Harral JW, Hoedt-Miller M, Sclafani RA and Rodman DM: p27(Kip1)
is important in modulating pulmonary artery smooth muscle cell
proliferation. Am J Respir Cell Mol Biol. 25:652–658. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mizuno S, Kadowaki M, Demura Y, Ameshima
S, Miyamori I and Ishizaki T: p42/44 mitogen-activated protein
kinase regulated by p53 and nitric oxide in human pulmonary
arterial smooth muscle cells. Am J Respir Cell Mol Biol.
31:184–192. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ashok C, Owais S, Srijyothi L, Selvam M,
Ponne S and Baluchamy S: A feedback regulation of CREB activation
through the CUL4A and ERK signaling. Med Oncol. 36:202019.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Humbert M, Guignabert C, Bonnet S,
Dorfmüller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti
SS, Schermuly RT, Stenmark KR and Rabinovitch M: Pathology and
pathobiology of pulmonary hypertension: State of the art and
research perspectives. Eur Respir J. 53:18018872019. View Article : Google Scholar :
|
|
42
|
Budhiraja R, Tuder RM and Hassoun PM:
Endothelial dysfunction in pulmonary hypertension. Circulation.
109:159–165. 2004. View Article : Google Scholar : PubMed/NCBI
|