Introduction
Glioblastoma (GBM) is a devastating disease that has
an extremely poor prognosis. The 5-year survival rate of patients
with GBM is <3% and the 5-year mortality rate ranks third
amongst all malignant tumors (1-3).
The high mortality rate is due to the invasive nature of GBM, as
tumors can spread locally within the brain or disseminate to other
sites within the body (4). There
is a critical need for the better understanding of the molecular
mechanisms that drive the invasive phenotype of GBM so that
improved therapeutic approaches can be developed. Moreover, the
identification of natural drugs with efficacy in the treatment of
GBM have a high potential to lead to the development of novel
therapies that may benefit a large number of patients.
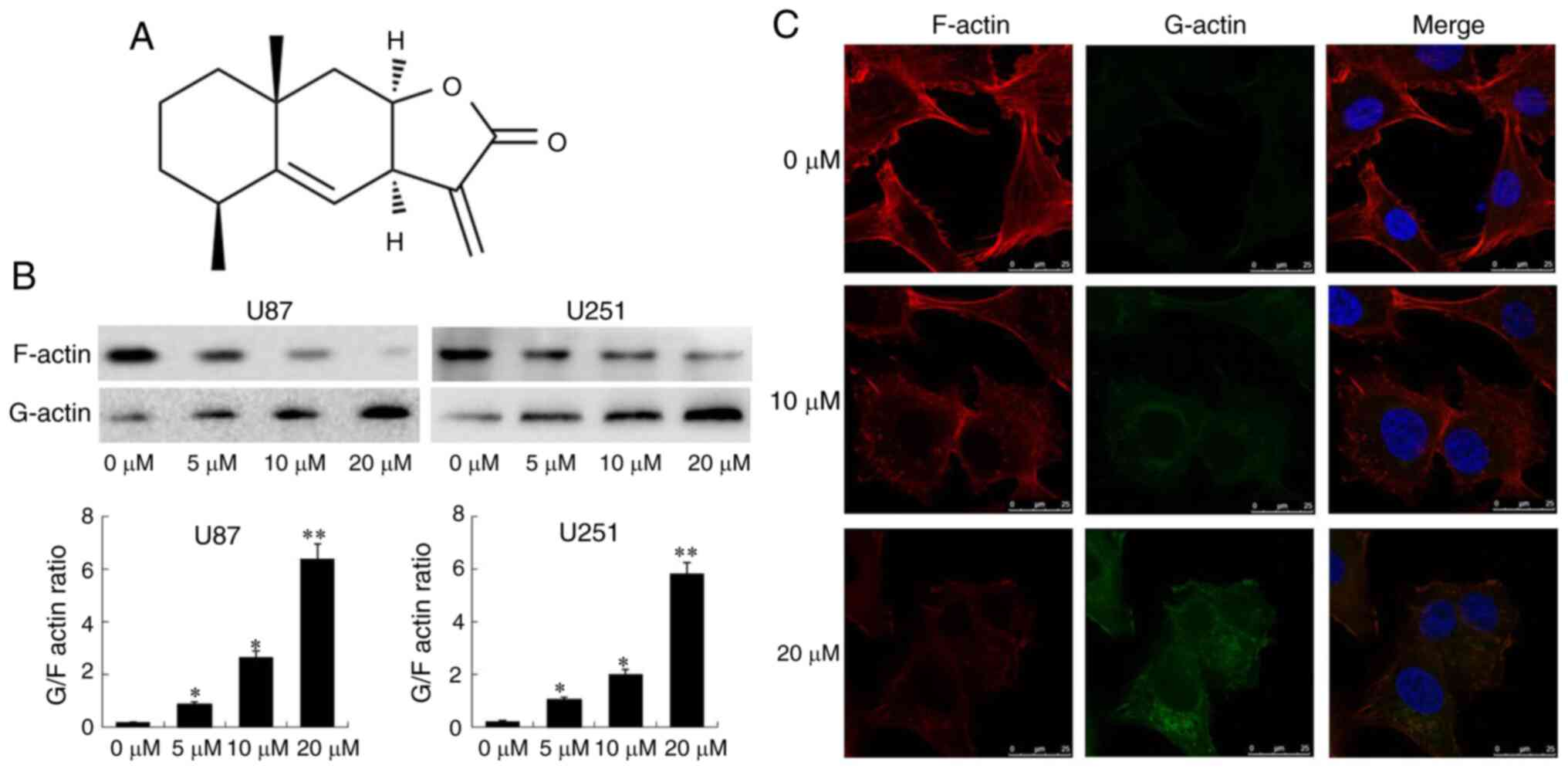
Alantolactone (ATL; chemical structure shown in
Fig. 1A) is a semi-terpene
lactone compound that is isolated from the roots of Inula
helenium. ATL has potent biological activities that include
anti-inflammatory, antitumor, anti-parasitic and hepatoprotective
properties (5). Previously, the
authors demonstrated that ATL can pass through the blood-brain
barrier to inhibit the growth and metastatic phenotype of GBM and
also induce apoptosis (6).
However, the underlying molecular mechanisms of these effects and
the regulatory targets activated by ATL remain to be fully
characterized.
Cell migration is a critical hallmark of tumor
invasion and metastasis (7). A
detailed investigation of the mechanisms of cell migration has
revealed that cell polarity, the dynamic regulation of
depolymerization, the polymerization of microfilaments and
microtubules, and changes in signal transduction during migration
are all related to the recombination of the actin cytoskeleton
(8).
Actin is a protein that is divided into a polymer
form of F-actin (fibrous) and a monomer form of G-actin
(spherical). The formation of pseudopodia is dependent on the
polymerization and depolymerization of actin. Cofilin is a binding
protein of actin and represents a family of actin depolymerization
factors that dominates the migration of cells. Cofilin induces the
depolymerization of fibrous F-actin into spherical G-actin, leading
to the remodeling of the cytoskeleton. This process affects cell
migration and occurs during embryonic development, wound repair and
tumor cell invasion and metastasis (9).
LIMK is a serine/threonine-protein kinase that
includes 2 members, LIMK1 and LIMK2. These proteins catalyze the
phosphorylation of cofilin as upstream molecules that regulate
cofilin. LIMK inactivates cofilin by phosphorylating the serine
residue at the 3rd position of cofilin protein. It also mediates
actin polymerization to form a pseudopodoid structure and can
regulate cofilin activity through changes in the enzyme activity of
LIMK to control the migration and invasion properties of tumor
cells (10). Studies have found
that LIMK is involved in the progression, migration and invasion of
various malignant tumors, including ovarian, breast and colon
cancer cells (11-13). It has also been demonstrated that
the translocation of activated cofilin to the mitochondria is an
early marker of apoptosis. Cofilin translocates to the outer
membrane of the mitochondria and releases cytochrome c into
the cytoplasm to induce apoptosis (14).
Based on these previous findings, it was
hypothesized that ATL may activate cofilin through the targeted
inhibition of LIMK enzyme activity. This may inhibit cell migration
and invasion and also induce apoptosis. In the present study, in
vitro and in vivo experiments were performed to
investigate the molecular mechanisms of ATL in modulating the
migration, invasion and apoptosis of GBM cells. The data of the
present study support the further development of ATL as a potential
therapy for the treatment of GBM.
Materials and methods
Chemicals and antibodies
ATL (≥98.5% purity) was prepared by the laboratory
at Dalian Medical University. Extraction and purification were
performed via stepwise elution in a solvent system containing
hexane:ethyl acetate:methanol:water at volumetric ratios of 5:3:1:2
(v/v). The concentration of the ATL stock solution was 100
µmol/l. The stock solution was dissolved in dimethyl
sulfoxide (DMSO) and stored at −20°C for further experimental use.
The final concentration of DMSO was <0.1% when ATL was applied
to the cells. The LIM kinase Inhibitor I (LIMKi 3) was purchased
from MedChem Express. Antibodies against cofilin (sc-376476), LIMK1
(sc-28370)/2 (sc-365414) and phospho-LIMK1/2 (Thr508/505)
(sc-28409) were purchased from Santa Cruz Biotechnology, Inc.
Antibodies against phospho-cofilin (Ser 3) (cat. no. 3311),
cytochrome c oxidase (Cox) IV (cat. no. 4844), slingshot
homolog (SSH; cat. no. 13578), testis associated actin remod- eling
kinase (cat. no. 4655, TESK), GAPDH (cat. no. 2118), β-actin (cat.
no. 4970), cleaved caspase-3 (cat. no. 9664)/9 (cat. no.
20750)/poly(ADP-ribose) polymerase (cat. no. 5625, PARP) were
purchased from Cell Signaling Technology, Inc. Matrix
metalloproteinase (MMP)-2 (cat. no. 66366-1-Ig)/9 (cat. no.
10375-2-AP) and cytochrome c (cat. no. 10993-1-AP)
antibodies were purchased from Proteintech Group, Inc. and actin
(cat. no. A4700) antibody was from Sigma-Aldrich; Merck KGaA.
Cells and cell culture
The U87MG (cat. no. HTB-14) and U251 cell lines were
obtained from the American Type Culture Collection (ATCC). All
cells were maintained in DMEM supplemented with 10% fetal bovine
serum (FBS) and maintained at 37°C in a humidified atmosphere
containing 5% CO2.
The U87MG cell line is a glioblastoma of unknown
origin. The authenticity of the cell line was verified through the
genomic short tandem repeat profile by Shanghai Biowing
Biotechnology Co. Ltd. and the cell line were confirmed to be free
of mycoplasma using a Mycoplasma Detection kit-Quick Test (Biotool,
LLC).
Wound healing migration assay
The cells were allowed to grow to become fully
confluent in 6-well plates. A line of cells was then scraped away
in each well using a pipette tip after 6 h of serum starvation. The
cells were then washed twice to remove detached cells. Fresh medium
containing different drug treatments (DMSO; ATL, 10 µM;
LIMKi 3, 5 µM; and ATL + LIMKi 3) were added to the
scratched monolayers. Images were acquired using a Leica DM 14000B
microscope following 24 h of incubation at 37°C. The migrating
cells were observed from 3 randomly chosen fields of view and
quantified by manual counting. The percentage inhibition of
migration was expressed as relative to the untreated cells
(100%).
Transwell invasion assay
Liquefied matrix glue was mixed with DMEM (1:3
ratio) and kept on ice. A total of 80 µl of the mixture was
applied to the cell membrane, and incubated at 37°C for 30 min to
solidify the matrix glue. Cells were trypsinized and resuspended in
culture medium without fetal bovine serum. Subsequently, 100
µl of cell suspension (density 5×105 cells/ml)
were added to the upper layer of the chamber, which were pre-coated
with Matrigel (BD Biosciences) and 700 µl of DMEM medium
containing fetal bovine serum was added to the lower layer. The
upper and lower layers of each group contained the same
concentration of cells in each of the treatment groups. Following
incubation for 24 h at 37°C, cotton swabs were used to remove the
residual matrix glue and cell suspension in the upper chamber. The
chamber was then fixed in methanol for 15 min. After removing the
chamber and allowing the membrane to air dry, the chamber was
stained with 1% crystal violet (Sigma-Aldrich; Merck KGaA) for 20
min at room temperature and washed 3 times with PBS. The invading
cells were detected after staining under the membrane of the
chamber were photographed under a Leica DM 14000B microscope (Leica
Microsystems GmbH).
Flow cytometry analysis
Cells were seeds in 6-cm culture dishes and treated
with the different drugs (DMSO; ATL, 10 µM; LIMKi 3, 5
µM; and ATL + LIMKi 3) for 24 h. The cells were then washed
with PBS twice and trypsinized. The cells were collected by
centrifugation (400 × g, 5 min) at 4°C. The cells were then washed
twice in PBS and centrifuged (400 × g, 5 min) at 4°C. Cells were
resuspended in 500 µl of binding buffer and 5 µl of
Annexin V-FITC were added to the samples. The samples were mixed
and 5 µl of propidium iodide added before being incubated
for 15 min at room temperature in the dark. Cells were then
analyzed using a C6 flow cytometer (BD FACS Accuri C6; BD
Biosciences).
G-actin/F-actin assay
G-actin and F-actin were detected using the
G-actin/F-actin in vivo detection kit (Cytoskeleton, Inc.)
according to the manufacturer's instructions. Following 48 h of
treatment with various concentrations of ATL (0, 5, 10 and 20
µM), the cells were lysed with LAS2 buffer (including lysis
solution, F-actin stabilization buffer, ATP stock solution and
protease inhibitor cocktail stock solution; Cytoskeleton, Inc.) at
37°C for 1 h. The cells were isolated by centrifugation at 800 × g
for 5 min at 4°C. The cell lysates were then centrifuged at 100,000
× g for 1 h at 4°C and F-actin in the precipitate and G-actin in
the supernatant were precipitated. Samples were mixed 5 times with
SDS sample buffer and western blot analysis was performed using an
anti-actin antibody. Grayscale analysis was performed using
ImageJ2x Software (Rawak Software Inc.).
Western blot analysis and
immunoprecipitation
Whole-cell, cytoplasmic, nuclear and mitochondrial
cytoplasmic proteins from each of the treatment groups were
extracted using a corresponding extraction kit. The total protein
extraction kit (cat. no. BC3790), cytoplasmic protein extraction
kit (cat. no. BC3740), nuclear protein extraction kit (cat. no.
R0050) and mitochondrial protein extraction kit (cat. no. SM0020)
were purchased from Solarbio Life Sciences, Inc. Equal quantities
of protein (40 µg/lane) were separated by electrophoresis on
a 7.5-12% sodium dodecyl sulfate polyacrylamide, and which were
transferred to PVP membranes and detected by specific antibodies
targeting the proteins of interest. The membranes were blocked with
5% non-fat milk for 2 h at 25°C. The membranes were incubated with
primary antibodies as follows: Cofilin (1:1,000), p-cofilin
(1:1,000), LIMK1/2 (1:1,000), p-LIMK1/2 (1:1,000), SSH (1:1,000),
TESK (1:1,000), MMP-2/9 (1:1,000), cytochrome c (1:1,000)
and cleaved caspase-3/9/PARP (1:1,000) overnight at 4°C, followed
by incubation with anti-rabbit HRP secondary anti-body (1:20,000,
cat. no. 7074, Cell Signaling Technology, Inc.), or anti-mouse
HRP-conjugated secondary antibody (1:20,000, cat. no. 7076, Cell
Signaling Technology, Inc.) for 2 h at room temperature. β-actin
(1:5,000), GAPDH (1:3,000) or Cox IV (1:3,000) served as the
loading controls. Proteins were visualized by exposure to Chem-Doc
(Bio-Rad Laboratories, Inc.). The concentration of proteins was
determined using a BCA protein detection kit (Beyotime Institute of
Biotechnology). All experiments were performed at least in
triplicate.
For immunoprecipitation, the cells were dissolved in
1% NP-40 buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1% Noindet P-40,
10% glycerol, 1 mM PMSF, 10 µg/ml aprotinin, 10 µg/ml
leupeptone and 1 mM Na3V4]. The same amount
of protein was then incubated with the primary antibody on a
shaking table at 4°C. Immune complexes were collected with protein
G agarose beads (Santa Cruz Biotechnology Co., Ltd.) and washed
several times in lysis buffer. The samples were boiled and western
blot analysis was performed as described above.
Immunofluorescence
Cells from each of the treatment groups were grown
on chamber slides, collected by centrifugation (400 × g, 5 min) at
4°C, resuspended gently in pre-warmed (37°C) staining solution
containing 200 nM MitoTracker Red CMXRos (cat. no. M7512, Molecular
Probes; Thermo Fisher Scienific, Inc.) for 1 h at 37°C, and washed
twice with RPMI-1640 medium. This was followed by fixation with
3.7% of methanol-free formaldehyde for 15 min, and permeabilization
with 0.1% Triton X-100 for 10 min. Slides were blocked with 1% BSA
in PBS for 30 min, then incubated with anti-cofilin (1:50) primary
antibody at 4°C overnight, followed by the secondary Alexa
488-conjugated goat anti-mouse antibody (1:300, cat. no. R37120,
Molecular Probes; Thermo Fisher Scienific, Inc.) for 1 h at room
temperature. Cells were incubated with 50 nM MitoTracker Green FM
(cat. no. M7514, Molecular Probes; Thermo Fisher Scienific, Inc.)
following fixation. The fluorescent staining of globular and
filamentous actin was performed using Fluorescent Deoxyribonuclease
I Conjugates (1:500, cat. no. D12371, Molecular Probes; Thermo
Fisher Scienific, Inc.) and Fluorescent phallotoxins (1:40, cat.
no. R415, Molecular Probes; Thermo Fisher Scienific, Inc.) at 4°C
overnight, followed by the secondary Alexa 488- conjugated goat
anti-mouse antibody (1:300, cat. no. R37120, Molecular Probes;
Thermo Fisher Scienific, Inc.) for 1 h at room temperature. The
stained samples were mounted with 4′,6-diamidino-2-phenylindole
(DAPI) at room temperature for 5 min to counterstain the cell
nuclei. Following 5 additional 5-min washes in PBS, the samples
were examined under a Leica DM 14000B confocal microscope (Leica
Microsystems GmbH).
Enzyme-linked immunosorbent assay
The LIMK enzyme was detected using a cell-based
fluorometric ELISA kit (ImmunoWay Biotechnology Co., Ltd.)
according to the manufacturer's instructions. Cells were seeded
onto 96-well plates. Following 48 h of treatment with various
concentrations of ATL (0, 5, 10 and 20 µM), the cells were
fixed by removing the cell growth culture medium, followed by
rinsing twice with PBS, and a final incubation with 100 µl
of 4% formaldehyde in PBS for adherent cells. Incubation was
performed for approximately 30 min at room temperature. The
formaldehyde solution was removed and the cells were rinsed 3 times
with wash buffer. The final wash buffer was then removed, and 100
µl of Quench buffer was added followed by incubation for 25
min at room temperature; the plates were sealed and covered with
parafilm. The Quench buffer was then removed and the cells were
rinsed 3 times for 5 min each with 200 µl of wash buffer on
a shaker. The wash buffer was removed and 100 µl of Blocking
Buffer were add followed by incubation for 1 h at room temperature.
After blocking, the plates were washed 3 times with wash buffer for
5 min each wash. Subsequently, 50 µl of Primary Antibody
Mixture (1:100 LIMK1/2; 1:100 GAPDH) were added into each relevant
well on the 96-well plate. The plates were incubated overnight at
4°C. The plates were sealed with parafilm or incubated in a
humid-box in refrigerator, ensuring that the plates were plated at
an even level. The Primary Antibody Mixture was then removed, and
the wells were washed 3 times for 5 min each with 200 µl of
wash buffer with gentle shaking on a shaker. The wash buffer was
removed, and 50 µl of Secondary Antibody Mixture
(DyLight®649-conjugated anti-rabbit IgG; FITC-conjugated
anti-mouse IgG) were then added to each well. The plates were
covered and sealed with parafilm, with gentle shaking on a shaker
for 1 h at the room temperature. The Secondary Antibody Mixture was
then removed and the cells were washed 3 times for 5 min each time
with 200 µl of wash buffer. Subsequently, 50 µl of 1X
PBS were added to each well on the 96-well plate. The plates were
then read at an excitation and emission wavelength: 654/673
(DyLight®649) and 495/521 (FITC) and kept in the dark.
The fluorescence intensity ratio was calculated between
DyLight®649 vs. FITC, comparing the ratio before and
after treatment.
Animal experiments
U87MG cells were trypsinized and resuspended in PBS
at a density of 1×107/ml. Under aseptic conditions, 100
µl of cell suspension was subcutaneously injected near the
axillary fossa of male nude mice (BALB/c nu/nu, 4 weeks old,
weighing 18-19 g, 25 mice in total. The mice were maintained in a
specific pathogen-free grade animal facility on a 12 h light/dark
cycles at 25±2°C. The mice were provided with free access to
sterilized food and water and were allowed to acclimatize for 7
days before start of the experiment. All procedures were performed
at the SPF Laboratory Animal Center of Dalian Medical University,
Dalian, China).
Following injection, the weights of the mice were
recorded daily, and the longest and shortest diameters of the
tumors were measured using a Vernier caliper. When the tumors
reached a size of 3×4 mm, the mice were randomly divided into 3
experimental groups with 5 mice in each group. In group A, the
animals were injected with 100 µl PBS; in group B, the
animals were treated with a low dose of ATL (10 mg/kg in PBS
containing 33% propylene glycol, 100 µl); and group C, the
animals were treated with a high dose of ATL (20 mg/kg in PBS
containing 33% propylene glycol, 100 µl). The doses used
were selected with reference to previous research (15). Treatments were delivered by
intraperitoneal injection once a day for 15 days. All experimental
animals were sacrificed and tumor tissues were collected for
immunohistochemical staining and western blot analysis. All
procedures were in performed in line with the National Institutes
of Health Guidelines for the Care and Use of Laboratory Animals
(National Institutes of Health, Bethesda, MD, USA). The
experimental protocol was approved by the Animal Care and Ethics
Committee of Dalian Medical University.
Statistical analysis
A Student's t-test (two-tailed), a t-test with
Welch's correction, and an F-test were used to statistically
analyze the datasets using GraphPad Prism 6.0 software (GraphPad
Software, Inc.). The concrete methods of t-test analysis in the
study were as follows: The data of 2 groups for comparison were
analyzed using an F-test firstly (homogeneity test of variance).
When the value of the F-test was >0.05, the value of the t-test
was obtained according to the heteroscedasticity double sample
test. When the value of the F-test was <0.05, the value of the
t-test was obtained according to the heteroscedasticity double
sample test. A value from the t-test <0.05 indicated significant
differences between the 2 experimental groups and a value of the
t-test >0.05 indicated no statistically significant differences
between the 2 experimental groups. Two-way analysis of variance
(ANOVA) followed by a Bonferroni's test for multiple comparisons
were performed to analyze the data involving multiple groups. The
data are represented as the means ± SD of at least 3 independent
experiments. P-values <0.05 were considered to indicate a
statistically significant difference. SPSS 18.0 software (SPSS
Inc.) was used for all of the statistical analyses.
Results
ATL induces the dephosphorylation of
Cofilin and regulates the ratio of G/F-actin
A previous study by the authors (6) demonstrated that ATL significantly
reduced the migration and invasiveness of GBM cells. Moreover,
previous research has indicated that the G/F-actin ratio is an
indicator of actin dynamics and is responsible for regulating cell
migration and invasion (16).
Cofilin is a binding protein of actin that functions function of
cofilin is to regulate the polymerization and depolymerization of
actin. Phosphorylated cofilin is inactive which hinders the process
of F-actin depolymerization into G-actin, and disrupts the
formation of invasive pseudopodia to enhance the migration and
invasion of tumor cells (11,17).
In the present study, G-actin was first separated
from F-actin using a G/F-actin separation kit. The results of
western blot analysis revealed that ATL significantly increased the
expression of G-actin and reduced the expression of F-actin
(Fig. 1B). Similarly, it was
also confirmed that ATL significantly upregulated the expression of
G-actin and downregulated the expression of F-actin in a
concentration-dependent manner, as shown by immunofluorescence
microscopy confocal experiments (Fig. 1C).
Subsequently, whole-cell protein, as well as
cytoplasmic and mitochondrial proteins were isolated from the U87MG
and U251 cells and were subjected to western blot analysis
(Fig. 2A-D). In the whole-cell
extract, it was found that ATL significantly inhibited the
expression of p-cofilin in a concentration-dependent manner, whilst
the total level of cofilin was not markedly altered (Fig. 2A). These results indicated that
ATL can induce the dephosphorylation and activation of cofilin and
depolymerize F-actin into G-actin, suggesting a potential role of
ATL in inhibiting the migration and invasion of GBM cells.
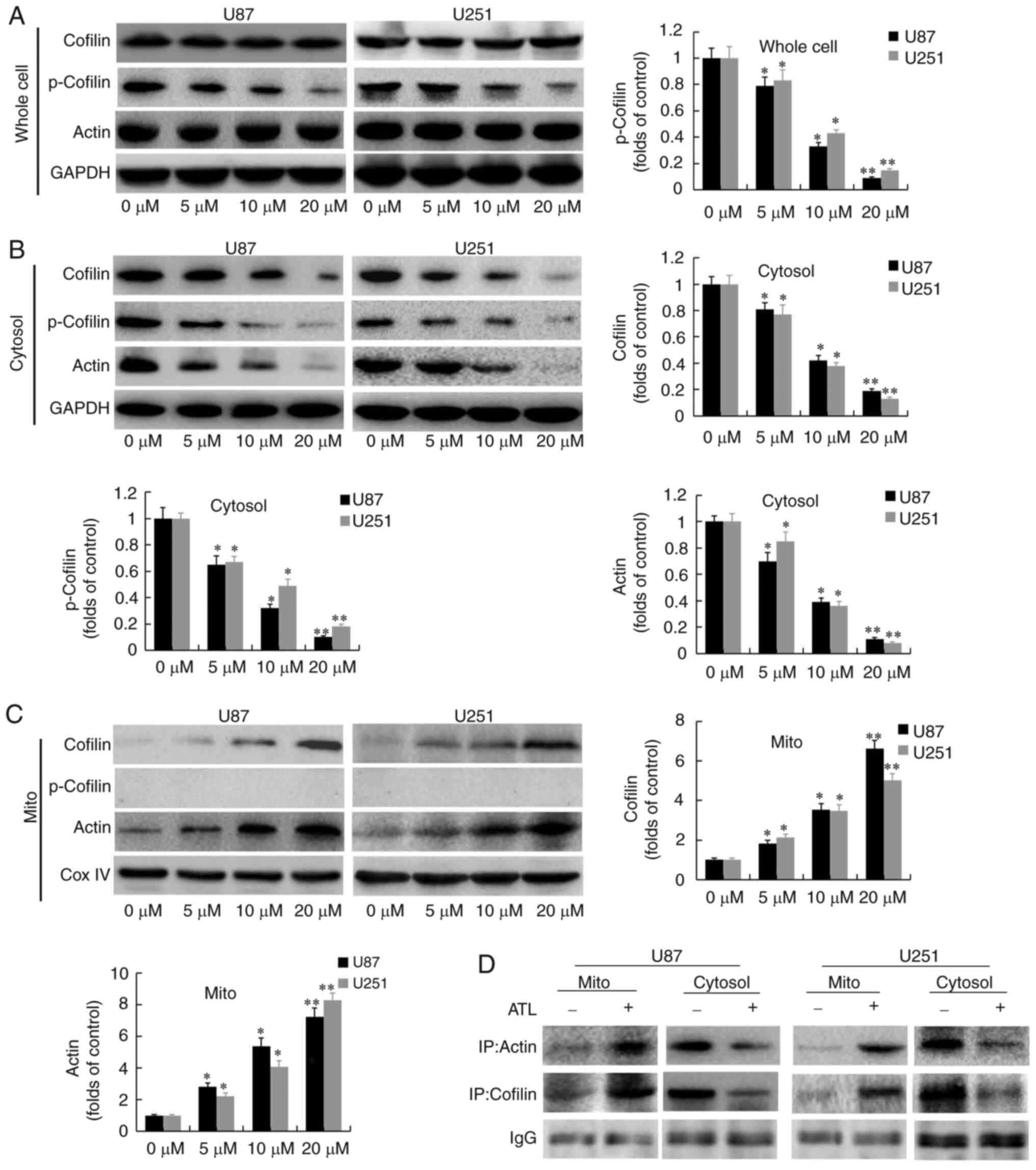 | Figure 2ATL induces cofilin and G-actin
co-translocation to the mitochondria. After U87MG and U251 cells
were treated with various concentrations of ATL (0-20 µM)
for 48 h, whole-cell, cytosolic and mitochondrial lysates were
prepared. (A) Western blot analysis was performed using antibodies
against cofilin, p-cofilin, actin and GAPDH. (B) The cytosolic
fractions were subjected to western blot analysis using antibodies
against cofilin, p-cofilin, actin and GAPDH. (C) The mitochondrial
fractions were subjected to western blot analysis using antibodies
against cofilin, p-cofilin, actin and Cox IV. (D) Cytosolic and
mitochondrial fractions of control and ATL-treated cells were
prepared and subjected to immunoprecipitation using an anti-cofilin
antibody, followed by western blot analysis. *P<0.05
and **P<0.01 vs. the DMSO group. ATL, alantolactone;
Cox IV, cytochrome c oxidase. |
ATL induces cofilin and G-actin
co-translocation to the mitochondria
The authors have previously demonstrated that ATL
induces the release of cytochrome c from the mitochondria
into the cytoplasm through endogenous pathways to initiate the
caspase cascade pathway and induce apoptosis in GBM cells (6). Previous research has found that
cofilin can translocate to the mitochondria to increase the
permeability transition pores, release cytochrome c and
initiate the apoptotic cascade (18). In the present study, it was found
that the expression of cofilin in the cytoplasm was significantly
decreased (Fig. 2B) and the
expression in the mitochondria was significantly increased
(Fig. 2C). The expression of
Cofilin in whole cells was not significantly changed (Fig. 2A) after ATL treatment. These
observations indicate that ATL can promote the transfer of cofilin
from the cytoplasm to the mitochondria.
It was also found that the expression of p-cofilin
(Ser 3) protein was significantly decreased in whole cells and in
the cytoplasm (Fig. 2B), whilst
its expression was not detected in the mitochondria (Fig. 2C). These results confirm that
cofilin may be activated by dephosphorylation at the Ser 3 site and
only activated cofilin may be translocated into the mitochondria,
whilst p-Cofilin cannot undergo mitochondrial translocation.
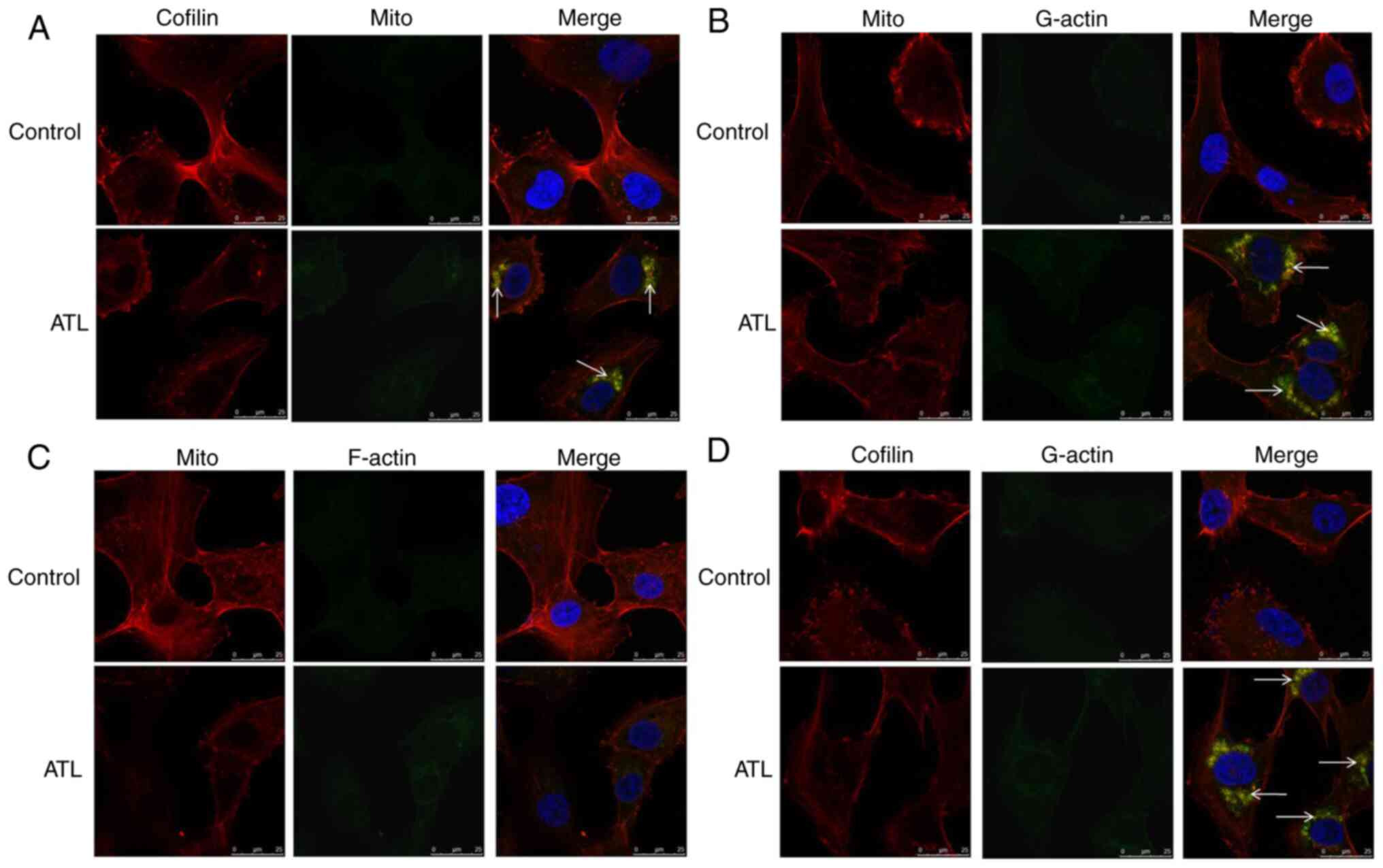
Cofilin (red light) and mitochondria (green light) were stained
with specific fluorescence probes observed under a laser confocal
microscope (Fig. 3A). Following
treatment with ATL, the fluorescence of cofilin and the
mitochondria overlapped (red light and green light overlapped into
yellow light), further confirming that ATL induced the
translocation of cofilin to the mitochondria.
Following treatment with ATL, the expression of
actin was upregulated in the mitochondria (Fig. 2C) and downregulated in the
cytoplasm (Fig. 2B) in a
concentration-dependent manner. The expression of actin at the
whole cell protein level was not significantly altered (Fig. 2A). These data suggest that ATL
can significantly increase the ratio of G-actin to F-actin; thus,
it was hypothesized that G-actin is transferred to the
mitochondria. Subsequently, specific fluorescence staining
technology was used to observe the localization associations
between G-actin, F-actin and the mitochondria under a laser
confocal microscope to examine the hypothesis. The results revealed
that the fluorescence of G-actin and the mitochondria overlapped
(yellow light, Fig. 3B) whilst
the co-localization between F-actin and the mitochondrial did not
occur (Fig. 3C), confirming the
hypothesis.
We speculate that the ATL-induced translocation of
Cofilin to the mitochondria causes apoptosis and may also be
related to actin translocation to mitochondria. We conducted
immunoprecipitation experiments to detect the levels of correlation
between Cofilin and actin. As shown in Fig. 2D, we found that the direct
binding of Cofilin and actin in mitochondrial components was
significantly increased after treated with ATL. Also, Cofilin (red
light) and G-actin (green light) were found to be co-localized and
had overlapping fluorescence (yellow light) detected by confocal
observation (Fig. 3D). These
results confirm that ATL induces cofilin and G-actin to
co-translocate to the mitochondria. These proteins interact with
each other and mediate the mitochondrial/cytochrome c
pathway and to initiate the caspase cascade signaling pathway,
leading to apoptosis.
ATL inhibits the activity of the LIMK
enzyme
Studies have demonstrated that the activity of
cofilin is mainly regulated by phosphorylation and
dephosphorylation, whilst the third serine site of Cofilin is only
one phosphorylation site (19).
When the serine 3 site is phosphorylated, cofilin loses its
activity and F-actin will accumulate due to blocked
depolymerization. By contrast, Cofilin will regain the function of
depolymerizing F-actin after the serine 3 site is dephosphorylated.
The enzymes that catalyze cofilin phosphorylation are LIMK and
TESK, whilst the enzymes that can remove cofilin phosphorylation
include SSH (20).
To further explore the target of ATL in regulating
cofilin activity, the expression of phosphorylase and
dephosphorylase that catalyze cofilin were detected. As shown in
Fig. 4A, ATL significantly
inhibited the expression of p-LIMK1/2 in a concentration-dependent
manner, whilst the total levels of LIMK1 and LIMK2, TESK and SHH
were not markedly altered. Furthermore, a significant downward
trend of p-LIMK1/2 protein was detected by ELISA (Fig. 4B). Based on these data, it was
hypothesized that ATL may activate cofilin by inducing the
dephosphorylation of LIMK, inhibiting the activity of LIMK enzyme
and blocking the phosphorylation of cofilin.
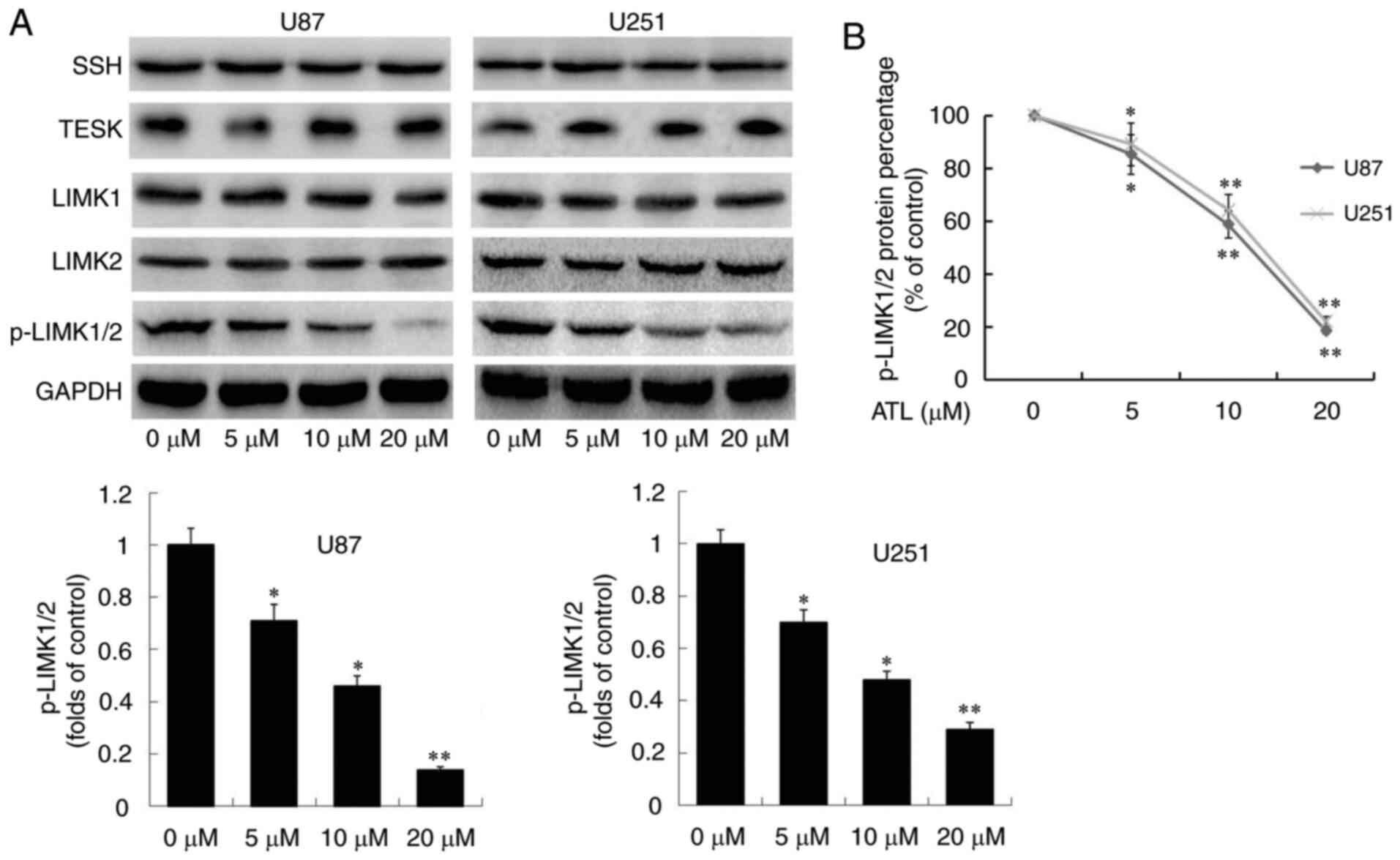 | Figure 4ATL inhibits the activity of LIMK
enzyme. At 48 h following treatment of U87MG and U251 cells with
ATL, (A) the protein levels of the SHH, TESK, LIMK1, LIMK2,
p-LIMK1/2 and GAPDH were detected by western blot analysis, and (B)
the percentage p-LIMK1/2 protein was detected by ELISA.
*P<0.05 and **P<0.01 vs. the DMSO
group. ATL, alantolactone; LIMK, LIM kinase; SSH, Sonic hedgehog;
TESK, testis associated actin remodelling kinase. |
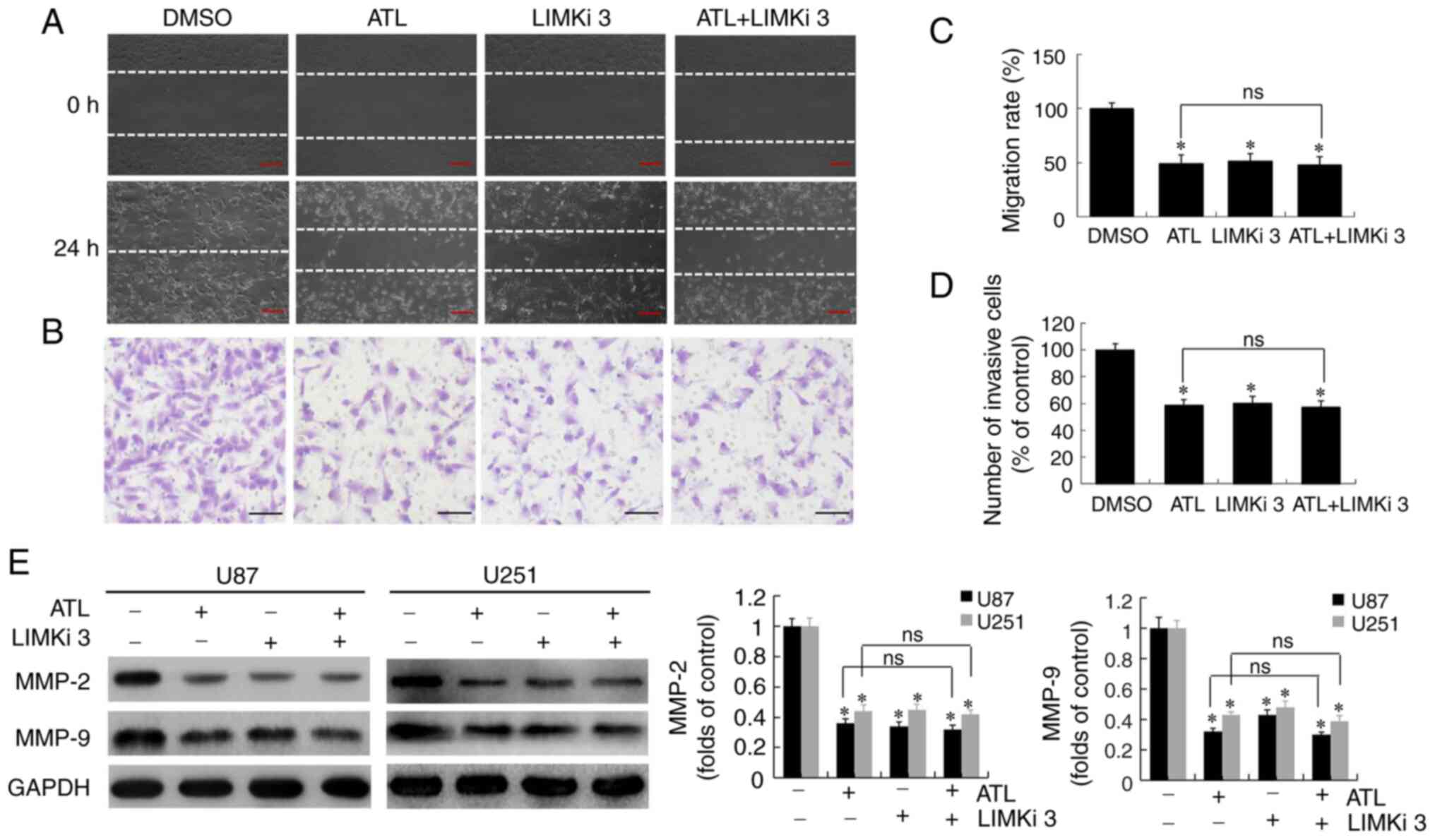
ATL inhibits the migratory ability and
invasiveness of GBM by targeting LIMK enzyme activity
In a previous study, the authors demonstrated that
ATL suppressed the migratory ability and invasiveness of GBM cells
and also downregulated the expression of MMP-2 and MMP-9 (6). Thus, in the present study,
experiments were conducted to verify whether ATL targeting LIMK
enzyme activity during the activation of cofilin would inhibit the
migration and invasiveness, and induce the apoptosis of GBM cells.
U87MG cells were pre-treated with LIMKi 3 (5 µM) for 8 h,
and then ATL (10 µM) was added at the corresponding time
points to detect migration and invasion. LIMKi 3 is a specific
selective LIMK inhibitor. The results of the wound scratch
(Fig. 5A and C) and invasion
assays (Fig. 5B and D) revealed
significant differences between the ATL, LIMKi 3 and ATL + LIMKi 3
groups compared to the control group (P<0.05). No significant
differences were observed between the ATL and ATL + LIMKi 3 group
(P>0.05). The above-mentioned results indicated that ATL,
similar to LIMKi, specifically inhibited LIMK enzyme activity to
suppress the migratory ability and invasiveness of GBM cells.
The U87MG and U25 cells were then treated in the
same manner and the protein expression of MMP-2 and MMP-9 was
detected by western blot analysis. The results revealed (Fig. 5E) that the expression of these 2
proteins was differed significantly between the ATL, LIMKi 3 and
ATL + LIMKi 3 groups, and the control group. No significant
difference was observed between the ATL and ATL + LIMKi groups.
These data suggest that ATL may play an anti-migratory and
anti-invasive role in GBM by inhibiting LIMK enzyme activity,
activating cofilin, and downregulating MMP-2 and MMP-9 protein
expression.
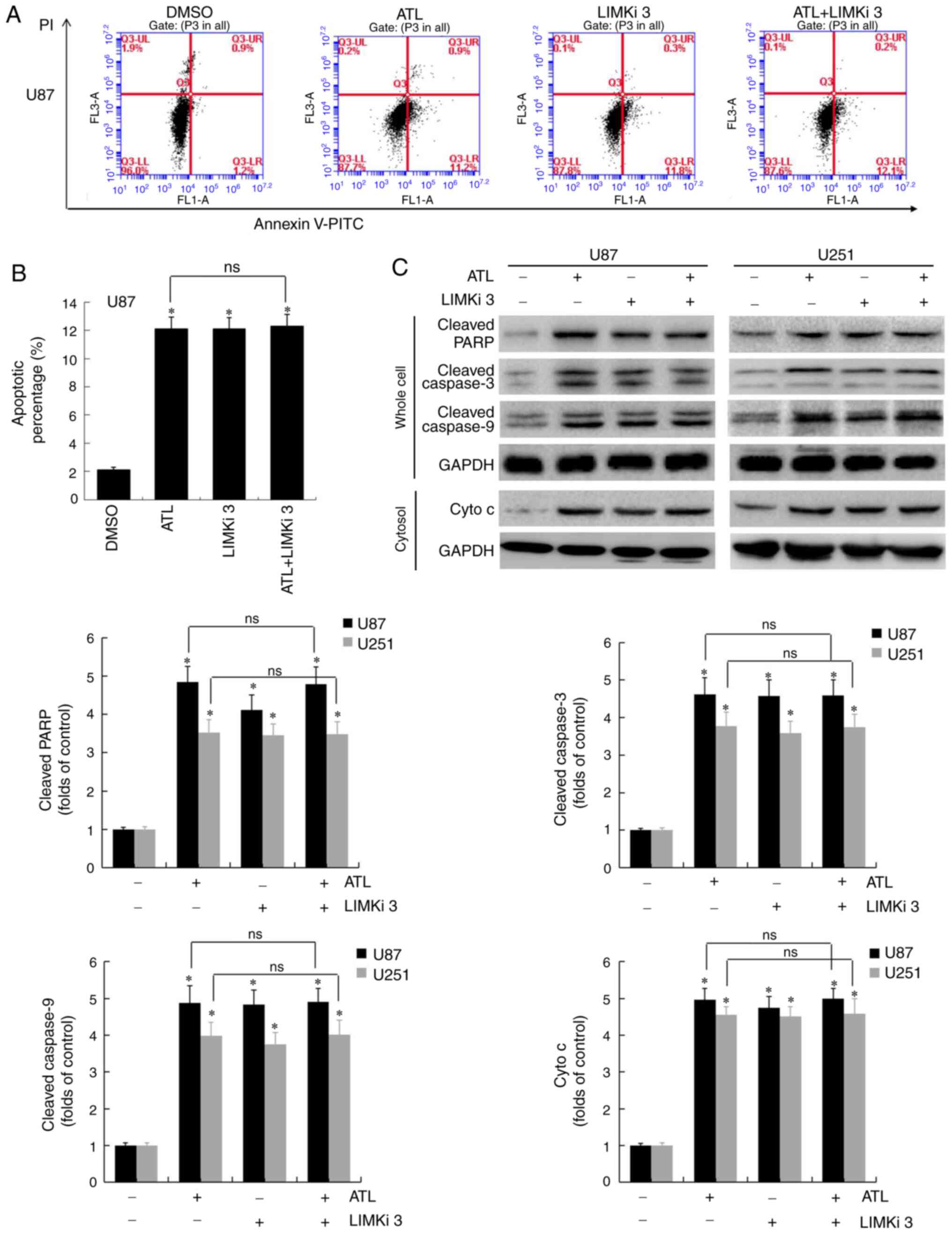
ATL induces the apoptosis of GBM cells by
targeting LIMK enzyme activity
Previously, it was confirmed that ATL induces the
release of cytochrome c from the mitochondria into the
cytoplasm through endogenous pathways to initiate the caspase
cascade pathway, and induces the apoptosis of GBM cells (6). In the present study, U87MG cells
were pre-treated with LIMKi 3 for 8 h and ATL was then applied (10
µM) for 24 h before detecting apoptosis by flow cytometry.
As shown in Fig. 6A and B,
compared to the control group, the number of apoptotic cells in the
ATL, LIMKi 3 and ATL + LIMKi 3 groups increased significantly
(P<0.05). No significant difference in the levels of apoptosis
was observed between the ATL and ATL + LIMKi 3 groups
(P>0.05).
The expression of key proteins regulating apoptosis
was then detected by western blot analysis. As shown in Fig. 6C, the expression levels of
cleaved caspase-3, cleaved caspase-9, cleaved PARP and cytochrome
c were significantly increased in the ATL, LIMKi 3 and ATL +
LIMKi 3 groups compared to the control group (P<0.05). No
significant differences in the expression of apoptosis-associated
proteins were detected between the ATL and ATL + LIMKi 3 groups
(P>0.05). These results confirm that ATL can activate cofilin by
targeting LIMK enzyme activity. In addition, cofilin and G-actin
co-translocate to the mitochondria, causing mitochondrial damage
and the release of cytochrome c from the mitochondria to the
cytoplasm, which acts to initiate the caspase signaling pathway to
induce apoptosis.
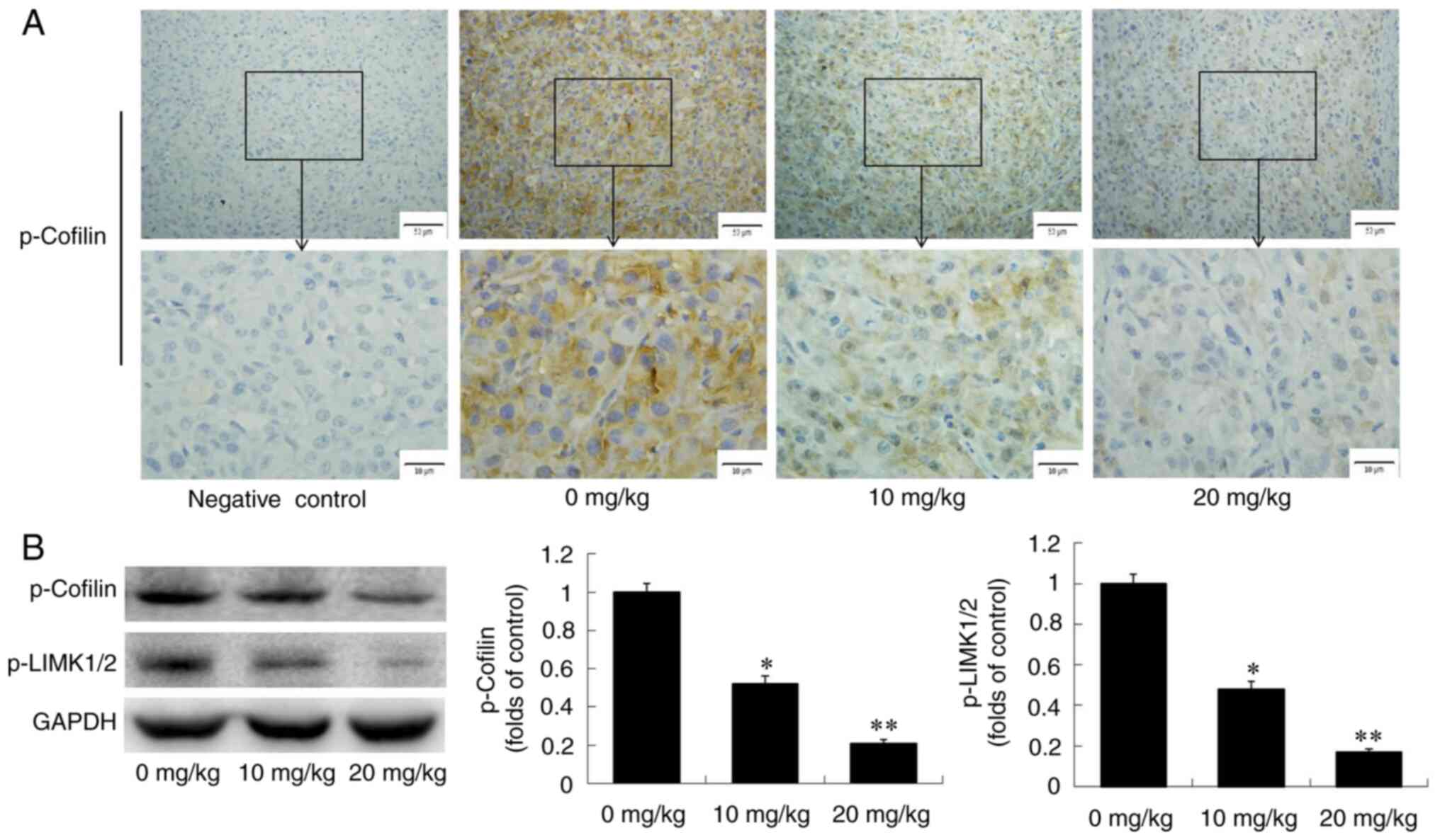
ATL inhibits the expression of p-cofilin
and p-LIMK1/2 in a heterotopic xenograft tumor model
To verify the results from the cell experiments,
in vivo experiments by were conducted establishing a GBM
xenograft model. Previously, it was confirmed that ATL can inhibit
the growth of transplanted tumors in nude mice in a dose-dependent
manner (6). In the present
study, transplanted tumor tissues from each of the experimental
groups were first examined by immunohistochemical analysis. The
results revealed that ATL inhibited the expression of p-cofilin in
a dose-dependent manner (Fig.
7A). Western blot analysis also revealed that ATL significantly
downregulated the expression levels of p-cofilin and p-LIMK1/2 in
tumor tissues in vivo (Fig.
7B).
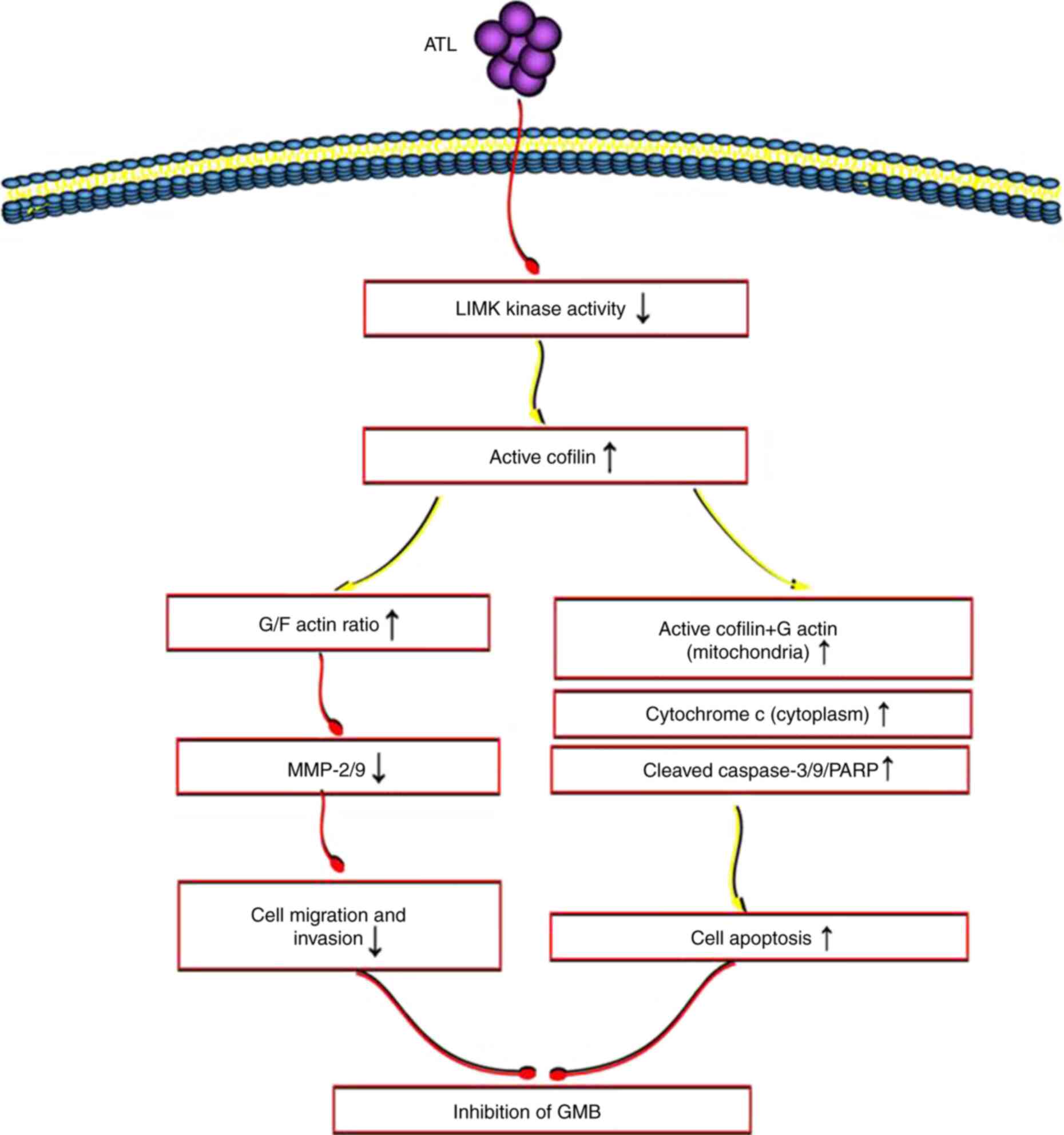
Molecular mechanisms responsible for the
inhibitory effects of ATL on the GBM metastatic phenotype and the
induction of apoptosis
As demonstrated by the above-mentioned results, ATL
may activate cofilin through the targeted inhibition of LIMK enzyme
activity. It can upregulate the ratio of G/F actin, and inhibit the
migration and invasion of GBM cells. Activated cofilin and G-actin
can be co-transferred to the mitochondria to initiate the
mitochondrial/cytochrome c pathway and induce apoptosis
(Fig. 8).
Discussion
GBM is the most common aggressive brain tumor and
has an extremely poor prognosis as current standard of care
treatments lack efficacy. There is an urgent need for the
development of novel therapeutics that are effective against the
disease. With advances in targeted therapies, it is particularly
important to explore new targets for the diagnosis and treatment of
GBM. ATL is a small molecule natural compound that has a variety of
pharmacological activities including antitumor effects (21-23). In a previous study by the authors
(6), it was found that ATL
inhibited the proliferation, growth, migration and invasion of GBM
cells, and induced apoptosis. The present study aimed to
systematically examine the specific molecular mechanism and
regulatory targets of ATL in GBM.
The migration and invasion of tumor cells is a
complex process (24) that
consists of 4 basic steps. The process begins with the separation
of cancer cells from tumor entities. The second step involves
adhesion to extracellular matrix followed by the activation of
matrix metalloproteinases (typically MMP-2, MMP-9) to degrade the
extracellular matrix and the final involves complete cell movement
and contraction. The migratory movement of cells is the first core
step in which actin and its regulatory proteins play a central role
(25). Actin can be divided into
2 forms, fibrous F-actin (polymer) and spherical G-actin (monomer).
These forms exist in the form of dynamic equilibrium by
depolymerizing F-actin into G-actin and polymerizing G-actin into
F-actin. Actin is closely related to tumor cell movement, vascular
invasion and metastasis. Tumor cells break the dynamic balance of
actin by blocking the depolymerization of F-actin. This causes the
accumulation of F-actin that induces the formation of invasive
pseudopodia of tumor cells under the guidance of chemotactic
signals and promotes the migration, invasion and metastasis of
tumor cells (26).
Actin depolymerizing factors are a group of proteins
that can bind to actin. Cofilin is an important family member with
a molecular weight of 21 kDa and is expressed in all eukaryotic
cells. Cofilin and contains sites that can bind to actin at both
ends of protein N and C termini (20). Previous research has demonstrated
that cofilin plays a key role in regulating the speed and direction
of tumor cell migration and in mediating adhesion to the
extracellular matrix during invasion and metastasis (27). Activated cofilin can combine with
actin to depolymerize F-actin in the multimeric form into G-actin
in monomer form, increasing the conversion of actin. Therefore,
cofilin is a key molecule that regulates dynamic changes in the
actin cytoskeleton (28). The
activation or inactivation of cofilin is closely related to the
invasion and metastasis of tumor cells.
The n-terminal serine 3 site of cofilin is the only
phosphorylation site in its structure. The phosphorylation and
dephosphorylation of cofilin are closely related to the assembly,
decomposition and cleavage of actin (9). Once the Ser 3 site is
phosphorylated, cofilin will lose its activity and cannot combine
with F-actin to prevent F-actin from depolymerizing into G-actin
and induce tumor cell migration, invasion and metastasis (29).
It has been demonstrated that the enzymes that
catalyze cofilin phosphorylation are LIMK and TESK. The
phospholipase that removes cofilin phosphorylation is SSH. LIMK is
a key enzyme that regulates cofilin (19). LIMK is a serine/threonine protein
kinase that exists in eukaryotes. It has 3 highly related family
members, LIMK1 and LIMK2. Phosphorylated LIMK is an activated form
and can specifically catalyze the phosphorylation of the Ser 3 site
of cofilin that inactivates cofilin. The change of cofilin activity
regulated by LIMK is directly related to the invasion, movement and
metastasis of tumor cells (30).
Previously, it was confirmed that ATL can suppress
the migratory ability and invasiveness of GBM cells and
downregulates the expression of MMP-2 and MMP-9 (6). However, in the present study, it
was found that the expression of p-cofilin was significantly
decreased, and the ratio of G/F-actin was significantly increased
by ATL. It was found that ATL specifically downregulated the
expression of p-LIMK1/2 in the detection of cofilin phosphorylation
and dephosphorylation enzyme. The protein expression of MMP-2/MMP-9
and the characterization of migration and invasion were determined
following treatment with LIMKi (a positive inhibitor of the LIMK
enzyme). It was confirmed that ATL inhibited migration and invasion
by inhibiting LIMK enzyme activity and activating cofilin. These
changes resulted in an increased ratio of G/F-actin. In addition,
it was verified that ATL significantly reduced the expression of
p-LIMK1/2 and p-cofilin in vivo using a xenograft tumor
model established in nude mice.
Apoptosis is the process of programmed cell death
and the unlimited growth of tumor cells results from the inhibition
of apoptosis. Studies have found that ATL can induce the apoptosis
of liver, colon, lung and leukemia cells (31-34). The occurrence of apoptosis is
mainly divided into the endogenous and exogenous pathways, of which
the endogenous pathway is also known as the
mitochondrial-cytochrome c pathway (35). In a previous study, the authors
confirmed that ATL can promote the transmission of cytochrome
c from the mitochondria to the cytoplasm and initiate the
caspase cascade to induce the apoptosis of GBM cells (6).
The mechanism through which ATL regulates the
transmission of cytochrome c by permeability changes caused
by mitochondrial damage remains largely unknown. Studies have found
that the mitochondrial translocation of cofilin is an early step in
the induction of apoptosis. Moreover, it has been confirmed that
only activated cofilin (dephosphorylated state) can be transferred
to the mitochondria and that this transfer is enabled by combining
the carboxyl-terminal sequence of the target signal with the
amino-terminal of the mitochondria (36). A previous study found that
following the activation of cofilin dephosphorylation Ser 3 site,
activated Cofilin translocated to the outer mitochondrial membrane
to open the permeability transition pore and induced the release of
cytochrome c to the cytoplasm, leading to cell apoptosis
(37). In the present study, it
was found that following treatment with ATL, p-Cofilin was not
detected in the mitochondria. The expression of active cofilin was
significantly increased in the mitochondria and significantly
decreased in the cytoplasm. In addition, the co-localization of
cofilin in the mitochondria was further confirmed, indicating that
ATL can induce active cofilin to translocate to the
mitochondria.
As previously demonstrated, the mitochondrial
translocation of cofilin is highly dependent on the mitochondrial
translocation of actin (38).
However, the mechanism of interaction between cofilin and actin
translocation remains unclear. In the present study, it was
demonstrated that ATL upregulated actin in the mitochondria, whilst
actin expression in the cytoplasm decreased. Moreover, only G-actin
was colocalized in the mitochondria, whilst F-actin was not. It was
found that Cofilin and actin exhibited an obvious co-precipitation
in the mitochondria following treatment with ATL, which only
minimally occurred in the cytoplasm. It was also confirmed that
only G-actin co-localized with cofilin.
It was hypothesized that ATL can promote cofilin and
G-actin co-translocation to the mitochondrial outer membrane to
initiate the endogenous apoptosis pathway. To examine this
hypothesis, LIMKi was used to detect the changes in the number of
apoptotic cells and the expression of key proteins in the
cytochrome c and caspase cascade pathways. The results
verified that ATL at least partially induced apoptosis by
inhibiting LIMK enzyme activity, activating cofilin, inducing the
co-translocation of activated cofilin and G-actin to the
mitochondria. These changes result in the release of cytochrome
c and initiate the caspase signaling pathway.
In conclusion, the present study found that ATL may
activate cofilin through the targeted inhibition of LIMK enzyme
activity. ATL can upregulate the ratio of G/F-actin, and inhibit
the migration and invasion of GBM cells. Activated cofilin and
G-actin can be co-transferred to the mitochondria to initiate the
mitochondrial/cytochrome c pathway to induce apoptosis
(Fig. 8). Combined with previous
findings (6), it was confirmed
that ATL acts on multiple pathways and has multiple targets through
which it exerts its anticancer effects on GBM. The data of the
present study highlight the therapeutic potential of ATL as a
natural product in the treatment of GBM and support its further
development for clinical applications.
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
LJZhang, XY and XW initiated the study and designed
the experiments. XW, SZ, TR and XYL performed the experiments. XW,
LJZhao and LFY analyzed the data. XW, SZ, TR and XYL prepared the
figures. XW, SZ and TR wrote the manuscript. All authors reviewed
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All procedures were in performed in line with the
National Institutes of Health Guidelines for the Care and Use of
Laboratory Animals (National Institutes of Health, Bethesda,
Maryland, USA). The experimental protocol was approved by the
Animal Care and Ethics Committee of Dalian Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by grants from the Liaoning
Provincial Natural Science Foundation of China (grant nos.
2019-BS-05, 2019-ZD-1009, 20180550761, 20180550976 and
2017010889-301), the Science and Technology Innovation Fund Project
of Dalian (grant no. 2019J13SN105), and the Health Commission
Foundation of Dalian (grant nos. 17Z1007 and 1911032).
References
|
1
|
Bryukhovetskiy I, Ponomarenko A, Lyakhova
I, Zaitsev S, Zayats Y, Korneyko M, Eliseikina M, Mischenko P,
Shevchenko V, Shanker Sharma H, et al: Personalized regulation of
glioblastoma cancer stem cells based on biomedical technologies:
From theory to experiment (Review). Int J Mol Med. 42:691–702.
2018.PubMed/NCBI
|
|
2
|
Jian S, Chen L, Minxue L, Hongmin C,
Ronghua T, Xiaoxuan F, Binbin Z and Shiwen G: Tanshinone I induces
apoptosis and protective autophagy in human glioblastoma cells via
a reactive oxygen species-dependent pathway. Int J Mol Med.
45:983–992. 2020.PubMed/NCBI
|
|
3
|
Hwang TW, Kim DH, Kim DB, Jang TW, Kim GH,
Moon M, Yoon KA, Choi DE, Park JH and Kim JJ: Synergistic
anticancer effect of acteoside and temozolomide-based glioblastoma
chemotherapy. Int J Mol Med. 43:1478–1486. 2019.PubMed/NCBI
|
|
4
|
Butowski NA, Sneed PK and Chang SM:
Diagnosis and treatment of recurrent high-grade astrocytoma. J Clin
Oncol. 24:1273–1280. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cantrell CL, Abate L, Fronczek FR,
Franzblau SG, Quijano L and Fischer NH: Antimycobacterial
eudesmanolides from Inula helenium and Rudbeckia subtomentosa.
Planta Med. 65:351–355. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang X, Yu Z, Wang C, Cheng W, Tian X, Huo
X, Wang Y, Sun C, Feng L, Xing J, et al: Alantolactone, a natural
sesquiterpene lactone, has potent antitumor activity against
glioblastoma by targeting IKKβ kinase activity and interrupting
NF-κB/COX-2-mediated signaling cascades. J Exp Clin Cancer Res.
36:932017. View Article : Google Scholar
|
|
7
|
Lauffenburger D and Horwitz A: Cell
migration: A physically integrated molecular process. Cell.
84:359–369. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mounier N and Arrigo AP: Actin
cytoskeleton and small heat shock proteins: How do they interact?
Cell Stress Chaperones. 7:167–176. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang T, DerMardirossian C and Bokoch G:
Cofilin phosphatases and regulation of actin dynamics. Curr Opin
Cell Biol. 18:26–31. 2006. View Article : Google Scholar
|
|
10
|
Zhang W, Gan N and Zhou J:
Immunohistochemical investigation of the correlation between LIM
kinase 1 expression and development and progression of human
ovarian carcinoma. J Int Med Res. 40:1067–1073. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou Y, Su J, Shi L, Liao Q and Su Q: DADS
downregulates the Rac1-ROCK1/PAK1-LIMK1-ADF/cofilin signaling
pathway, inhibiting cell migration and invasion. Oncol Rep.
29:605–612. 2013. View Article : Google Scholar
|
|
12
|
Li R, Doherty J, Antonipillai J, Chen S,
Devlin M, Visser K, Baell J, Street I, Anderson RL and Bernard O:
LIM kinase inhibition reduces breast cancer growth and invasiveness
but systemic inhibition does not reduce metastasis in mice. Clin
Exp Metastasis. 30:483–495. 2013. View Article : Google Scholar
|
|
13
|
Aggelou H, Chadla P, Nikou S, Karteri S,
Maroulis I, Kalofonos H, Papadaki H and Bravou V: LIMK/cofilin
pathway and slingshot are implicated in human colorectal cancer
progression and chemoresistance. Virchows Arch. 472:727–737. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Karbowski M and Youle RJ: Dynamics of
mitochondrial morphology in healthy cells and during apoptosis.
Cell Death Differ. 10:870–880. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khan M, Yi F, Rasul A, Li T, Wang N, Gao
H, Gao R and Ma T: Alantolactone induces apoptosis in glioblastoma
cells via GSH depletion, ROS generation, and mitochondrial
dysfunction. IUBMB life. 64:783–794. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen H, Bernstein B and Bamburg J:
Regulating actin-filament dynamics in vivo. Trends Biochem Sci.
25:19–23. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsai NM, Chen YL, Lee CC, Lin PC, Cheng
YL, Chang WL, Lin SZ and Harn HJ: The natural compound
n-butylidene- phthalide derived from angelica sinensis inhibits
malignant brain tumor growth in vitro and in vivo. J Neurochem.
99:1251–1262. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li GB, Cheng Q, Liu L, Zhou T, Shan CY, Hu
XY, Zhou J, Liu EH, Li P and Gao N: Mitochondrial translocation of
cofilin is required for allyl isothiocyanate-mediated cell death
via ROCK1/PTEN/PI3K signaling pathway. Cell Commun Signal.
11:502013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Van Troys M, Huyck L, Leyman S, Dhaese S,
Vandekerkhove J and Ampe C: Ins and outs of ADF/cofilin activity
and regulation. Eur J Cell Biol. 87:649–667. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bernstein BW and Bamburg JR: ADF/cofilin:
A functional node in cell biology. Trends Cell Biol. 20:187–195.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lei JC, Yu JQ, Yin Y, Liu YW and Zou GL:
Alantolactone induces activation of apoptosis in human hepatoma
cell. Food Chem Toxicol. 50:3313–3319. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mi XG, Song ZB, Wu P, Zhang YW, Sun LG,
Bao YL, Zhang Y, Zheng LH, Sun Y, Yu CL, et al: Alantolactone
induces cell apoptosis partially through down-regulation of
testes-specific protease 50 expression. Toxicol Lett. 224:349–355.
2014. View Article : Google Scholar
|
|
23
|
Shi Y, Bao YL, Wu Y, Yu CL, Huang YX, Sun
Y, Zheng LH and Li YX: Alantolactone inhibits cell proliferation by
interrupting the interaction between Cripto-1 and activin receptor
type II A in activin signaling pathway. J Biomol Screen.
16:525–535. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ridley AJ, Schwartz MA, Burridge K, Firtel
RA, Ginsberg MH, Borisy G, Parsons JT and Horwitz AR: Cell
migration: Integrating signals from front to back. Science.
302:1704–1709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Campellone KG and Welch MD: A nucleator
arms race: Cellular control of actin assembly. Nat Rev Mol Cell
Biol. 11:237–251. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hitchcock-Degregori SE: Chemotaxis:
Cofilin in the driver's seat. Curr Biol. 16:R1030–R1032. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee MH, Kundu JK, Chae JI and Shim JH:
Targeting ROCK/LIMK/cofilin signaling pathway in cancer. Arch Pharm
Res. 42:481–491. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Berger K and Moeller MJ: Cofilin-1 in the
podocyte: A molecular switch for actin dynamics. Int Urol Nephrol.
43:273–275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang W, Mouneimne G, Sidani M, Wyckoff J,
Chen X, Makris A, Goswami S, Bresnick AR and Condeelis JS: The
activity status of cofilin is directly related to invasion,
intravasation, and metastasis of mammary tumors. J Cell Biol.
173:395–404. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pandey D, Goyal P and Siess W:
Lysophosphatidic acid stimulation of platelets rapidly induces
Ca2+-dependent dephosphorylation of cofilin that is
independent of dense granule secretion and aggregation. Blood Cells
Mol Dis. 38:269–279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang C, Yang J, Sun M, Yan J, Meng X and
Ma T: Alantolactone inhibits growth of K562/adriamycin cells by
downregulating Bcr/Abl and P-glycoprotein expression. IUBMB Life.
65:435–444. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lei JC, Yu JQ, Yin Y, Liu YW and Zou GL:
Alantolactone induces activation of apoptosis in human hepatoma
cells. Food Chem Toxicol. 50:3313–3319. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ding Y, Wang H, Niu J, Luo M, Gou Y, Miao
L, Zou Z and Cheng Y: Induction of ROS overload by alantolactone
prompts oxidative DNA damage and apoptosis in colorectal cancer
cells. Int J Mol Sci. 17:5582016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pal HC, Sehar I, Bhushan S, Gupta BD and
Saxena AK: Activation of caspases and poly (ADP-ribose) polymerase
cleavage to induce apoptosis in leukemia HL-60 cells by Inula
racemosa. Toxicol In Vitro. 24:1599–1609. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dalla Via L, García-Argáez A,
Martínez-Vázquez M, Grancara S, Martinis P and Toninello A:
Mitochondrial permeability transition as target of anticancer
drugs. Curr Pharm Des. 20:223–244. 2014. View Article : Google Scholar
|
|
36
|
Kanellos G and Frame MC: Cellular
functions of the ADF/cofilin family at a glance. J Cell Sci.
129:3211–3218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chua BT, Volbracht C, Tan KO, Li R, Yu VC
and Li P: Mitochondrial translocation of cofilin is an early step
in apoptosis induction. Nat Cell Biol. 5:1083–1089. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rehklau K, Gurniak CB, Conrad M, Friauf E,
Ott M and Rust MB: ADF/cofilin proteins translocate to mitochondria
during apoptosis but are not generally required for cell death
signaling. Cell Death Differ. 19:958–967. 2012. View Article : Google Scholar :
|