Introduction
Bone homeostasis depends on an intricate coupling
between bone formation and resorption (1). Age-related bone loss is one main
cause of fractures in the elderly population, which is
characterized by reduced bone formation but persistent bone
resorption (2). Despite
considerable progress in research on the pathophysiology of
osteoporosis over the past several decades, the underlying
mechanisms of defective bone coupling in age-related bone loss
remain poorly understood.
Terminally differentiated and, matrix-embedded
osteocytes, which are regarded as the orchestrators of bone
remodeling, regulate the activities of osteoblasts and osteoclasts
(3). An important factor of the
osteocytic regulation of bone metabolism is sclerostin, a
Wnt/β-catenin antagonist (4).
Previous studies have reported that sclerostin can largely affect
bone homeostasis by decreasing bone formation but increasing bone
resorption (5,6). Plasma sclerostin levels increase
with age, and are associated with low bone mass and osteoporotic
fracture (7,8). Moreover, treatment with an
anti-sclerostin-neutralizing antibody has been revealed to restore
bone mass and strength in aged rats (9). Thus, considering these findings, it
can be inferred that elevated levels of sclerostin during aging,
may contribute to age-related bone loss.
Reactive oxygen species (ROS), which include
hydroxyl radicals, superoxides and hydrogen peroxides
(H2O2), are important secondary messengers
and serve essential roles in multiple physiological cellular
responses at their normal low levels (10). However, excessive intracellular
ROS result in oxidative stress (OS), which damages biomolecules,
including proteins, lipids, and nucleotides (11). Advanced oxidation protein
products (AOPPs) are cross-linked proteins enriched in dityrosine,
which are formed by the reaction of plasma proteins with
chlorinated compounds during OS (12,13). The accumulation of AOPPs occurs
in numerous redox-related diseases, such as chronic kidney
diseases, diabetes, inflammatory bowel diseases, rheumatoid
arthritis, postmenopausal osteoporosis, and spinal cord injury.
AOPPs are regarded as new biomarkers of OS (14-19). However, previous studies have
revealed that AOPPs are not only the byproducts of OS, but also
induce intracellular OS by triggering NADPH oxidases in various
cell types, and promote numerous diseases (14,16,19-22).
Our previous study reported that the level of AOPPs
in plasma increased with age and was negatively correlated with
bone mass in rats (23).
Furthermore, it was revealed that AOPPs accelerated the loss of
bone mass and microstructure degeneration in aged rats (24). However, the exact cellular basis
of age-related bone loss induced by AOPPs remains unknown. The
present study demonstrated that AOPPs aggravated age-related bone
loss by increasing the expression of sclerostin in osteocytes via
the downregulation of sirtuin 1 (Sirt1), and this process was
induced by NADPH oxidases dependent of ROS generation.
Materials and methods
Mice and reagents
Male C57Bl/6 mice (age, 12 months) were obtained
from Chengdu Dashuo Laboratory Animal Co., Ltd. The MLO-Y4
mouse-derived osteocytic cell line (derived from long bones of
mice) was purchased from PythonBio (http://www.pythonbiotech.com). Fetal bovine serum
(FBS), calf serum (CS), and α-minimum essential medium (α-MEM) were
purchased from Thermo Fisher Scientific, Inc. A Cell Counting Kit-8
(CCK-8) assay was purchased from Dojindo Molecular Technologies,
Inc. The ROS assay kit was purchased from Beyotime Institute of
Biotechnology. Primary antibodies against
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (product code
ab9485), sclerostin (product code ab63097), and Sirt1 (product code
ab12193) were purchased from Abcam. Goat anti-rabbit secondary
antibody conjugated with Cy3 (cat. no. SA00009-2) was purchased
from ProteinTech Group, Inc. Horseradish peroxidase
(HRP)-conjugated secondary anti-bodies (product code FD0155) and
protease inhibitor tablets were purchased from Fude Biological
Technology Co. Ltd. Apocynin, which is an inhibitor of nicotinamide
adenine dinucleotide phosphate (NADPH) oxidases,
N-acetyl-L-cysteine (NAC), which is an ROS scavenger, fluorescein
diacetate (FDA) and bovine serum albumin (BSA) were obtained from
Sigma-Aldrich; Merck KGaA. The Sirt1 activator, SRT3025, was
purchased from Selleck Chemicals.
Preparation of AOPPs-BSA and
concentration determination of AOPPs
AOPPs-BSA was prepared as previously described
(12,24). Briefly, 20 mg/ml BSA was
incubated (at 37°C for 30 min) with 40 mM hypochlorous acid (HOCl)
and diluted using phosphate-buffered saline (PBS, pH 7.4). To
remove free HOCl, the mixed solution was dialyzed in PBS at 4°C for
24 h. The controls used were BSA and PBS alone. Furthermore, a
Detoxi-Gel column (Pierce; Thermo Fisher Scientific, Inc.) was used
to remove contaminated endotoxins from all the samples. The
endotoxin levels in the obtained samples were measured using a
Limulus Amebocyte Lysate kit (Sigma; Merck KGaA) and a level of
<0.05 ng/mg was considered as acceptable. Content of AOPPs in
the sample was determined as previously described (12).
Animal treatment protocols
In total, 56 male C57Bl/6 mice (age, 12 months; body
weight, 33.5±3.1 g) were used. All mice were maintained at 23°C and
50-70% humidity with a 12-h light/dark cycle and were provided with
free access to food and water. All animal experiments were approved
(approval no. NFYY-2018-68) by the Laboratory Animal Care and Use
Committee of Nanfang Hospital, Southern Medical University
(Guangzhou, China).
The animal study consisted of two independent
experiments. For the AOPPs-BSA interventional experiment, 28 mice
matched by body weight were randomized into four groups (7 mice in
each group) and subjected to the following treatments: Group 1,
intraperitoneal (i.p.) injection of vehicle (PBS, pH 7.4); Group 2,
i.p. injection of native BSA at 50 mg/kg/day; Group 3, i.p.
injection of AOPPs-BSA at 50 mg/kg/day; and Group 4, i.p. injection
of AOPPs-BSA at 100 mg/kg/day. For the blocking experiment, the
remaining 28 mice were randomized into four groups and subjected to
treatments as follows: Group 1, i.p. injection of AOPPs-BSA alone
at 100 mg/kg/day; Group 2, i.p. injection of AOPPs-BSA at 100
mg/kg/day and apocynin at 100 mg/kg/day in drinking water; Group 3,
i.p. injection of AOPPs-BSA at 100 mg/kg/day and NAC at 100
mg/kg/day in drinking water; Group 4, i.p. injection of AOPPs-BSA
at 100 mg/kg/day and SRT3025 at 50 mg/kg/day in drinking water.
After a treatment duration of 16 weeks, mice in each group were
anesthetized using isoflurane (4% for induction, and 2.5% for
maintenance), and blood samples were collected from the
retroorbital vein (~500 µl blood was obtained from each
mouse). Mice were then immediately sacrificed by standard cervical
dislocation to avoid suffering. Subsequently, plasma was separated
and stored at -80°C. The tibia, femur and lumbar vertebra specimens
were also obtained for further assessments.
Micro-CT analysis
The trabecular microstructure of L4 vertebral bodies
and left proximal tibias were analyzed using micro-CT
(µCT80; SCANCO Medical AG) at a resolution of 10 µm.
The volume of interest (VOI) was defined as the entire trabecular
bone region in the L4 vertebral body, and the trabecular bone
region 100 slices downstream from the proximal growth plate of the
tibia. The trabecular microstructure morphometric parameters,
including bone volume over total volume (BV/TV), trabecular number
(Tb.N), trabecular thickness (Tb.Th), and trabecular separation
(Tb.Sp) were obtained via VOI analysis. All µCT analyses
were performed in accordance with the guidelines of the American
Society for Bone and Mineral Research (25).
Bone histological analysis and
immunohistochemical staining
Tibial bone samples were fixed in 2%
paraformaldehyde at 25°C for 24 h, decalcified in 0.5 M EDTA (pH
7.5) for 3 weeks, and then embedded in paraffin. Osteoblast number
and osteoclast area were measured on hematoxylin and eosin
(H&E)-stained sections (5 µm) and tartrate-resistant
acid phosphatase (TRAP)-stained sections (5 µm),
respectively. For H&E-staining, paraffin sections were stained
with hematoxylin for 2 min at 37°C, followed by eosin for 5 min at
37°C. For TRAP staining, paraffin sections were first reacted for
TRAP activity (37°C, 50 min) and then counterstained at 37°C for 5
min with light green SF yellowfish solution (Merck KGaA,).
For immunohistochemical staining, paraffin sections
(5 µm) were de-waxed, rehydrated, incubated in antigen
retrieval solution, and washed with Tris-buffered saline (TBS).
Endogenous superoxidase was then eliminated using 0.3%
H2O2. TBS containing 2% BSA was used to block
the non-specific antibody binding (37°C, 60 min). Then, sections
were incubated overnight at 4°C with primary antibodies against
sclerostin (product code ab63097; 1:50) and Sirt1 (product code
ab12193; 1:100; both from Abcam), followed by incubation (37°C, 60
min) with biotinylated secondary antibodies (1:2,500; cat. no.
ZB-2306; Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.).
Proteins were visualized under a light microscope using brown
pigments via a standard diaminobenzidine protocol. Subsequently,
slides were lightly counterstained with hematoxylin.
Serum biochemistry
Serum levels of N-terminal propeptide of type 1
procollagen (P1NP) and C-terminal crosslinked telopeptides of type
I collagen (CTX-I) were measured using ELISA kits (CSB-E12775m and
CSB-E12782m, respectively; Cusabio Technology LLC), following the
manufacturer's instructions. Serum levels of total superoxide
dismutase (t-SOD) and malondialdehyde (MDA) were quantified using
commercial reagent kits (cat. nos. A001-1-2 and A003-1-2,
respectively; Nanjing Jiancheng Bioengineering Institute).
Cell treatment and viability assay
MLO-Y4 cells were cultured in growth medium
containing α-MEM, 2.5% CS, 2.5% FBS, 100 U/ml penicillin, and 100
µg/ml streptomycin. The cells were incubated at 37°C in an
atmosphere of 5% CO2. The medium was changed every 2
days, and the cells were passaged after reaching 80%
confluence.
During the experiment, MLO-Y4 cells were treated
with various concentrations of AOPPs-BSA (from 0 to 200
µg/ml) for different durations (from 0 to 24 h), as well as
co-incubated with apocynin, NAC, or SRT3025, dissolved in dimethyl
sulfoxide (DMSO).
Subsequently, cell viability at different
concentrations of AOPPs-BSA (50,100, 200 and 400 µg/ml) was
qualitatively assessed using the fluorescein diacetate (FDA)
staining assay. After washing MLO-Y4 cells three times in PBS, 5
µg/ml FDA (dissolved in DMSO) was added, and the cells were
incubated at 37°C for 10 min. Images of the live cells were
captured using a model IX71 fluorescence microscope (Olympus
Corporation). To quantitatively determine the effect of AOPPs-BSA
on cell viability, a CCK-8 assay was performed following the
manufacturer's protocol. MLO-Y4 cells were washed three times using
PBS, and 10 µl CCK-8 solution was added to each well. Cells
were incubated at 37°C for 1 h and the absorbance was measured at
450 nm under a microplate reader (BioTek Instruments, Inc.).
Measurement of intracellular ROS
According to the manufacturer's instructions,
intracellular ROS levels were measured in MLO-Y4 cells using a ROS
assay kit (cat. no. S0033M; Beyotime Biotechnology) containing
2,7,-dichlorofluorescein diacetate (DCFH-DA) as the probe. MLO-Y4
cells under different treatment conditions (AOPPs-BSA of 0 to 200
µg/ml) were incubated with 10 µM DCFH-DA for 30 min
at 37°C. The fluorescence intensity was measured using a SpectraMax
M5 microplate reader (Molecular Devices, LLC), and the fluorescence
images were captured following ROS staining.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA of MLO-Y4 cells or femoral bone homogenate
was extracted using an RNA extraction kit (Takara Bio, Inc.) and
quantified using a NanoDrop 1000 spectrophotometer (Thermo Fisher
Scientific, Inc.). cDNA was obtained from total RNA (50 ng), using
a PrimeScript™ RT reagent kit (Takara Bio, Inc.). To quantify mRNA
expression, the obtained cDNA was amplified using TB
Green® Premix Ex Taq™ (TliRNaseH Plus; Takara Bio,
Inc.), and qPCR was performed using a LightCycler480 system (Roche
Diagnostics), following the manufacturer's instructions.
Thermocycling conditions were set as follows: Incubation at 95°C
for 30 sec, amplification at 95°C for 5 sec, 60°C for 30 sec for a
total of 40 cycles, 95°C for 5 sec, with a final extension at 60°C
for 60 sec. The expressions levels of Sclerostin (SOST) and
SIRT1 genes were measured, with GAPDH as the
reference gene. The 2−ΔΔCq method was used for the
measurement of gene expression (26). The primer sequences are listed in
Table I.
 | Table IPrimer sequences for real-time
PCR. |
Table I
Primer sequences for real-time
PCR.
| Gene name | Primers |
|---|
| SOST | F:
AGCCTTCAGGAATGATGCCAC |
| R:
CTTTGGCGTCATAGGGATGGT |
| SIRT1 | F:
GCTGACGACTTCGACGACG |
| R:
TCGGTCAACAGGAGGTTGTCT |
| GAPDH | F:
AGGTCGGTGTGAACGGATTTG |
| R:
TGTAGACCATGTAGTTGAGGTCA |
Western blotting
To obtain the total cell protein, cells were rinsed
with PBS at 4°C and lysed using a cell lysis buffer (Hangzhou Fude
Biological Technology Co., Ltd.). To collect the protein, the cell
lysate was centrifuged at a speed of 1,2000 × g for 30 min at 4°C.
The protein concentration was determined using a BCA protein assay
kit (Thermo Fisher Scientific, Inc.). Subsequently, the proteins
(20 µg/lane) were separated via 6-10% SDS-PAGE, and
transferred to PVDF membranes (EMD Millipore). Furthermore, the
membranes were blocked (60 min at 37°C) using 5% BSA in TBS
containing 0.1% Tween-20, and incubated overnight at 4°C with
primary antibodies. The primary antibodies were directed against
GAPDH (1:5,000 dilution), sclerostin (1:1,000 dilution) and Sirt1
(1:2,000 dilution). The proteins bound to the membrane were
detected ECL reagent (Bio-Rad Laboratories, Inc.) after incubation
with goat anti-rabbit IgG-HRP conjugated secondary antibody
(product code ab205718; 1:2,000 dilution; Abcam) at 37°C for 1 h.
The integrated density of all protein bands was analyzed using
ImageJ software (1.52v; National Institutes of Health).
Immunofluorescence staining
MLO-Y4 cells were washed three times using PBS and
fixed for 15 min at room temperature using 4% paraformaldehyde.
Following another three washes with PBS, the cells were
permeabilized for 20 min at room temperature, using 1% Triton
X-100/PBS solution. Then, the cells were blocked with 5% BSA in PBS
for 30 min at 37°C. The cells were incubated overnight at 4°C with
primary rabbit anti-mouse sclerostin antibody or Sirt1 antibody
(both 1:50 dilution). Primary antibodies were not added in the
negative control. The following day, the primary immunoreaction was
detected by incubation at 4°C for 12 h with a Cy3-conjugated goat
anti-rabbit secondary antibody (1:200 dilution). Finally, the cells
were counterstained using Fluoroshield mounting medium with DAPI
(37°C, 5 min). Images were captured using IX71 fluorescence
microscope (Olympus Corporation).
Statistical analysis
Data are presented as the mean ± SEM. All
statistical analyses were performed using the SPSS version 22.0
(IBM Corp.). For each independent in vitro experiment, ≥3
technical replicates were analyzed. Differences between groups
in vivo and in vitro were evaluated using one-way
ANOVA, followed by post hoc least significant difference (LSD)
test. For all statistical tests, P<0.05 was considered to
indicate a statistically significant difference.
Results
AOPPs accelerate bone loss by suppressing
bone formation but promoting bone resorption
The present study first measured the effect of AOPPs
on bone mass and microstructure in aging mice. Compared with PBS or
BSA, 50 mg/ml AOPPs-BSA significantly decreased the bone mineral
density (BMD) of L4 vertebral body, and this effect was more
notable in 100 mg/ml AOPPs-treated mice. However, no marked
difference was observed in femoral BMD of different groups.
(Fig. 1A and B). Micro-CT
analysis revealed that treatment with AOPPs-BSA significantly
aggravated the trabecular microstructure in aging mice. Compared
with PBS or BSA-treated mice, AOPPs-treated mice displayed a
significant decrease in BV/TV, Tb.N, and Tb.Th but an increase in
Tb.Sp in both L4 vertebral bodies and proximal tibias. Furthermore,
the degeneration of bone microstructure in the high-dose AOPPs-BSA
group was greater than that in the low-dose group. (Fig. 1C-L).
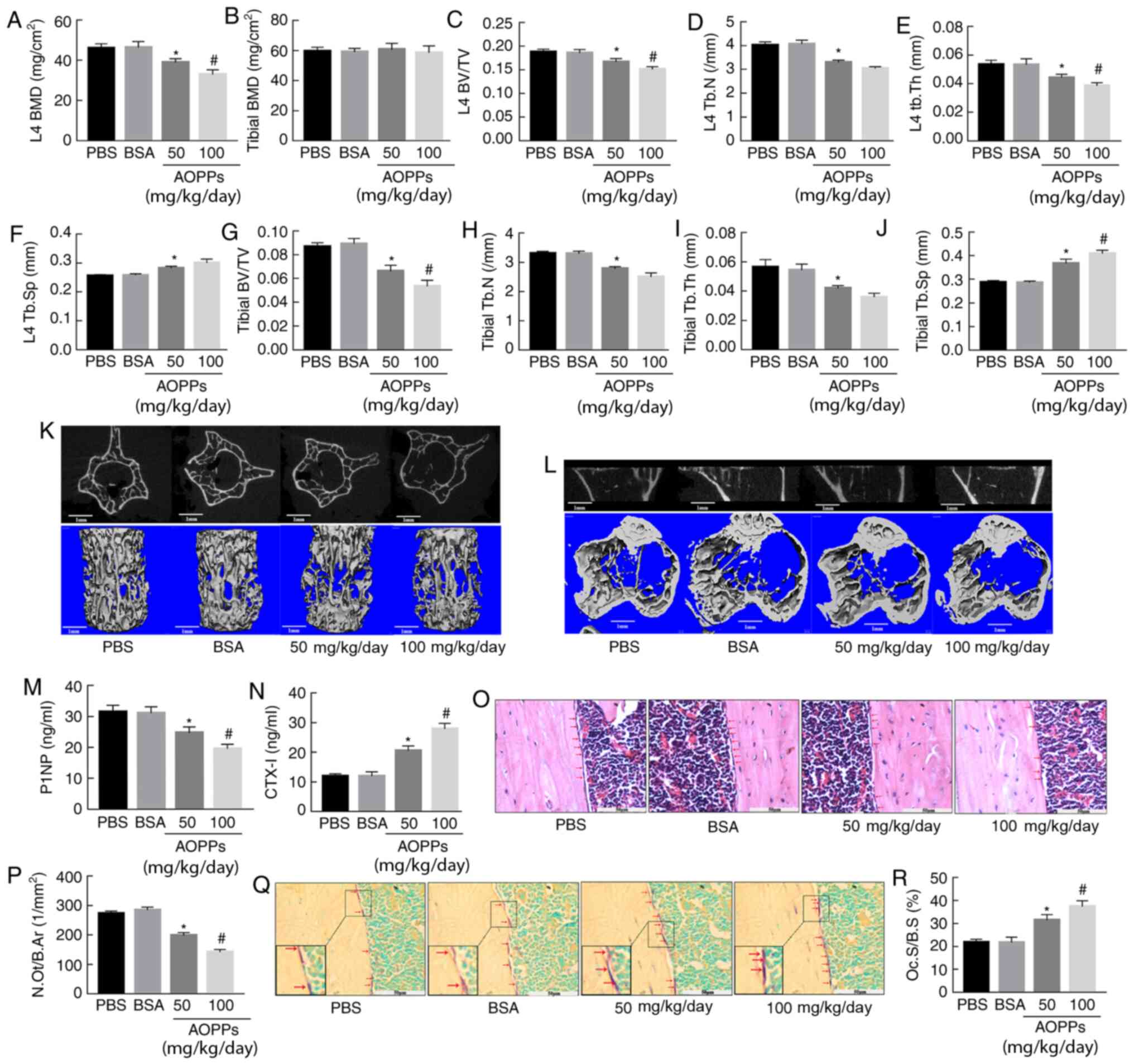 | Figure 1AOPPs accelerate bone loss by
suppressing bone formation but promoting bone resorption. (A and B)
AOPPs-BSA significantly decreased the BMD of the L4 vertebral body,
but did not significantly affect the femoral BMD. AOPPs-BSA
decreased the BV/TV, Tb.N, Tb.Th, but increased the Tb.Sp of both
(C-F) L4 vertebral bodies and (G-J) proximal tibias. Representative
middle cross or coronal sectional images and 3D reconstructed
images of (K) L4 vertebral bodies and (L) proximal tibias in
different groups. (M and N) ELISA results demonstrated that
treatment with AOPPs decreased the serum P1NP levels but increased
the CTX-I levels. (O and P) H&E and (Q and R) TRAP staining
identified that AOPPs decreased the number of osteoblasts (red
arrow) but increased the osteoclasts (red arrow) area (claret-red
area) in cortical diaphysis, respectively. Magnification, ×400. The
data are presented as the mean ± SEM. *P<0.05 vs. the
PBS or BSA group; #P<0.05 vs. the 50-mg/kg/day
AOPPs-BSA group; n=7. AOPPs, advanced oxidation protein products;
bovine serum albumin; BMD, bone mineral density; BV/TV, bone
volume/total volume; Tb.N, trabecular number; Tb.Th, trabecular
thickness; Tb.Sp, trabecular separation; P1NP, N-terminal
propeptide of type 1 procollagen; CTX-I, C-terminal crosslinked
telopeptides of type I collagen. |
To understand the effect of AOPPs on bone turnover
in aging mice, the serum level of a bone formation marker (P1NP),
and a bone resorption marker (CTX-1) were measured in mice of
different groups. Consistent with our previous study (24), the plasma P1NP concentration was
significantly decreased in mice treated with AOPPs-BSA, while the
CTX-1 concentration was increased (Fig. 1M and N). Along with this finding,
there was a significant decrease in the number of osteoblasts and
an increase in the osteoclast area observed in the cortical
diaphysis of mice treated with AOPPs-BSA, as evident by the H&E
and TRAP staining, respectively (Fig. 1O-R). These data indicated that
AOPPs accelerated bone deterioration in aging mice by suppressing
bone formation and promoting bone resorption.
AOPPs increase sclerostin expression, and
enhance OS and Sirt1 expression in mice
Sclerostin, a WNT/β-catenin inhibitor, is
predominantly secreted by osteocytes (5). This protein decreases the bone mass
by promoting bone resorption and suppressing bone formation
(5). In the present study, it
was revealed that AOPP-treated mice displayed decreased bone
forming activity but increased bone resorbing activity, which was
consistent with our previous study (24). These results indicated a probable
involvement of sclerostin in AOPP-induced bone loss. Given that
plasma sclerostin increases with age (27), the effect of AOPPs on sclerostin
expression was therefore examined in aging mice. As presented in
Fig. 2A, the SOST mRNA
expression in the AOPP-treated groups was higher compared with that
in PBS- or BSA-treated groups. An increased expression of
sclerostin protein in mice challenged with AOPPs was also observed
with immunohistochemical staining (Fig. 2B and C).
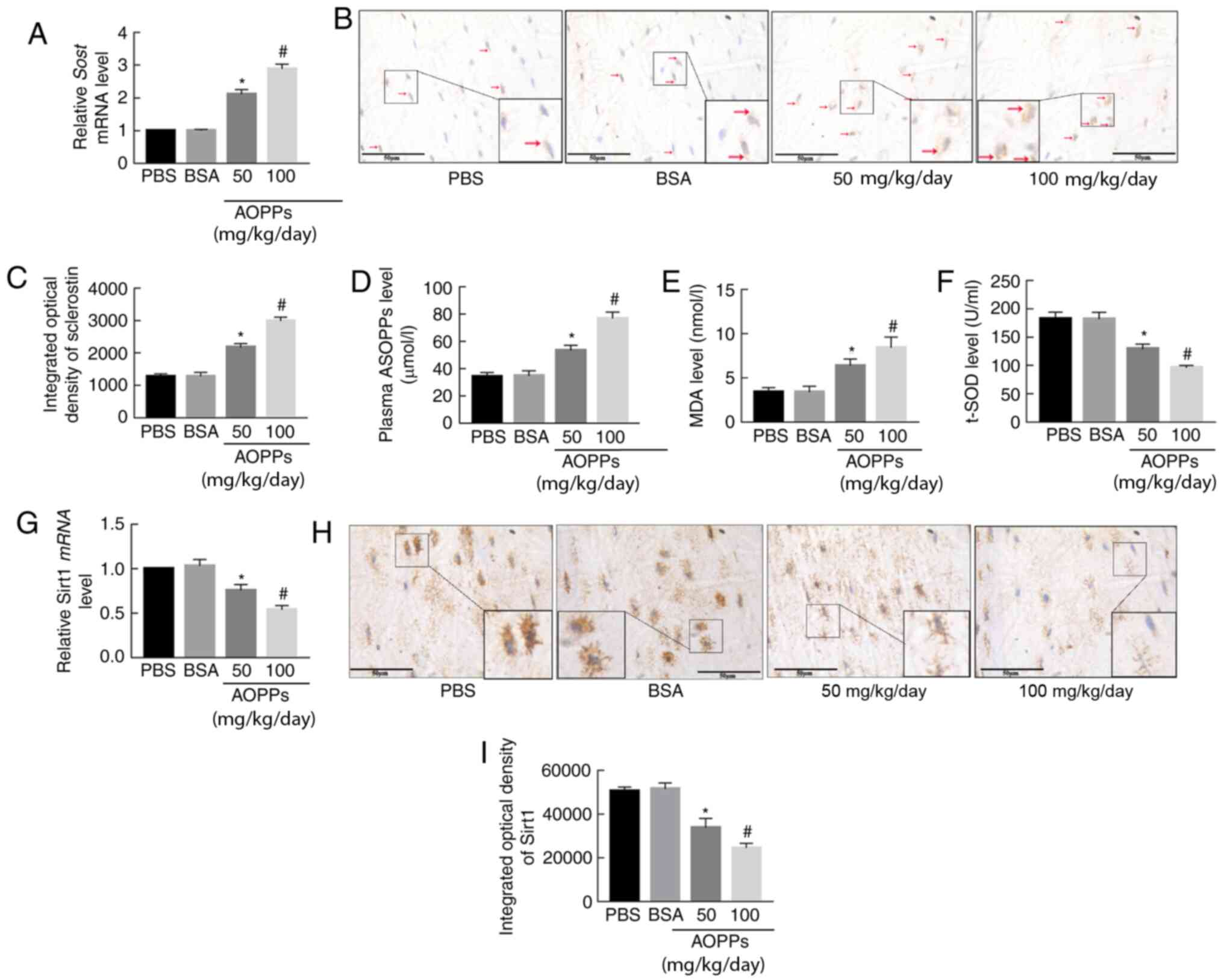 | Figure 2AOPPs increase sclerostin expression,
enhance oxidative stress and downregulate Sirt1 expression in aging
mice. (A) Reverse transcription-quantitative PCR results indicated
that AOPPs-BSA significantly increased the mRNA expression level of
SOST in femoral bone tissue. (B and C) Immunohistochemical
staining demonstrated that sclerostin protein expression (red
arrow) was significantly higher in AOPP-treated mice compared with
in PBS- or BSA-treated mice. AOPPs-BSA treatment increased (D)
serum levels of AOPPs and (E) MDA levels in femoral bone tissue,
but (F) decreased femoral t-SOD levels. AOPPs decreased (G)
SIRT1 mRNA and (H and I) protein expression levels in
femoral bone. Magnification, ×400. The data are presented as the
mean ± SEM. *P<0.05 vs. the PBS or BSA group;
#P<0.05 vs. the 50-mg/kg/day AOPPs-BSA group; n=7.
AOPPs, advanced oxidation protein products; Sirt1, sirtuin 1; BSA,
bovine serum albumin; SOST, sclerostin; PBS,
phosphate-buffered saline; MDA, malondialdehyde; t-SOD, total
superoxide dismutase. |
OS is regarded as a key promoting factor of
osteoporosis (28). Previous
studies have reported that AOPPs increase the OS status in a number
of animal models (16,21,22,29). The present study evaluated the
impact of AOPPs on the OS status of aging mice by testing the
plasma AOPPs, MDA and t-SOD levels in femoral bone. Compared with
mice treated with PBS or BSA, mice treated with AOPPs-BSA exhibited
higher AOPPs and MDA levels but lower t-SOD levels (Fig. 2D-F). These results indicated that
AOPPs enhanced OS in aging mice.
Sirt1 regulates sclerostin expression in osteocytes
(30), and under OS status, the
abundance and deacetylating activity of Sirt1 is compromised
(31). Therefore, it was
investigated whether there was a fluctuation of Sirt1 expression
under challenge of AOPPs. As was revealed by RT-qPCR and
immunohistochemical staining, Sirt1 mRNA and protein expression
levels were significantly decreased in AOPP-treated mice compared
with those in PBS- or BSA-treated mice (Fig. 2G-I).
Treatment with AOPPs increases sclerostin
expression in MLO-Y4 cells
To investigate the effect of AOPPs on the expression
of sclerostin in vitro, an osteocytic cell line, MLO-Y4, was
used. First, the cytotoxic effect of AOPPs-BSA on MLO-Y4 cells was
tested. As previously described (22,32), the viability of MLO-Y4 cells
treated with different doses of AOPPs, ranging between 50-400
µg/ml, was examined. FDA staining revealed that there was
only a slight decrease in the viability of cells treated with
50-200 µg/ml of AOPPs-BSA. However, cells treated with 400
µg/ml AOPPs displayed a sharp decrease in fluorescence
intensity, indicating a high cytotoxic effect of AOPPs-BSA at this
concentration (Fig. 3A).
Furthermore, a CCK-8 assay was performed to quantify the influence
of AOPPs-BSA on the viability of MLO-Y4 cells. Consistent with the
FDA staining, AOPPs-BSA up to 200 µg/ml did not markedly
affect cellular activity, while the absorbance value was noticeably
lower in the 400 µg/ml AOPP-treated group, suggesting
compromised cell viability in this group (Fig. 3B).
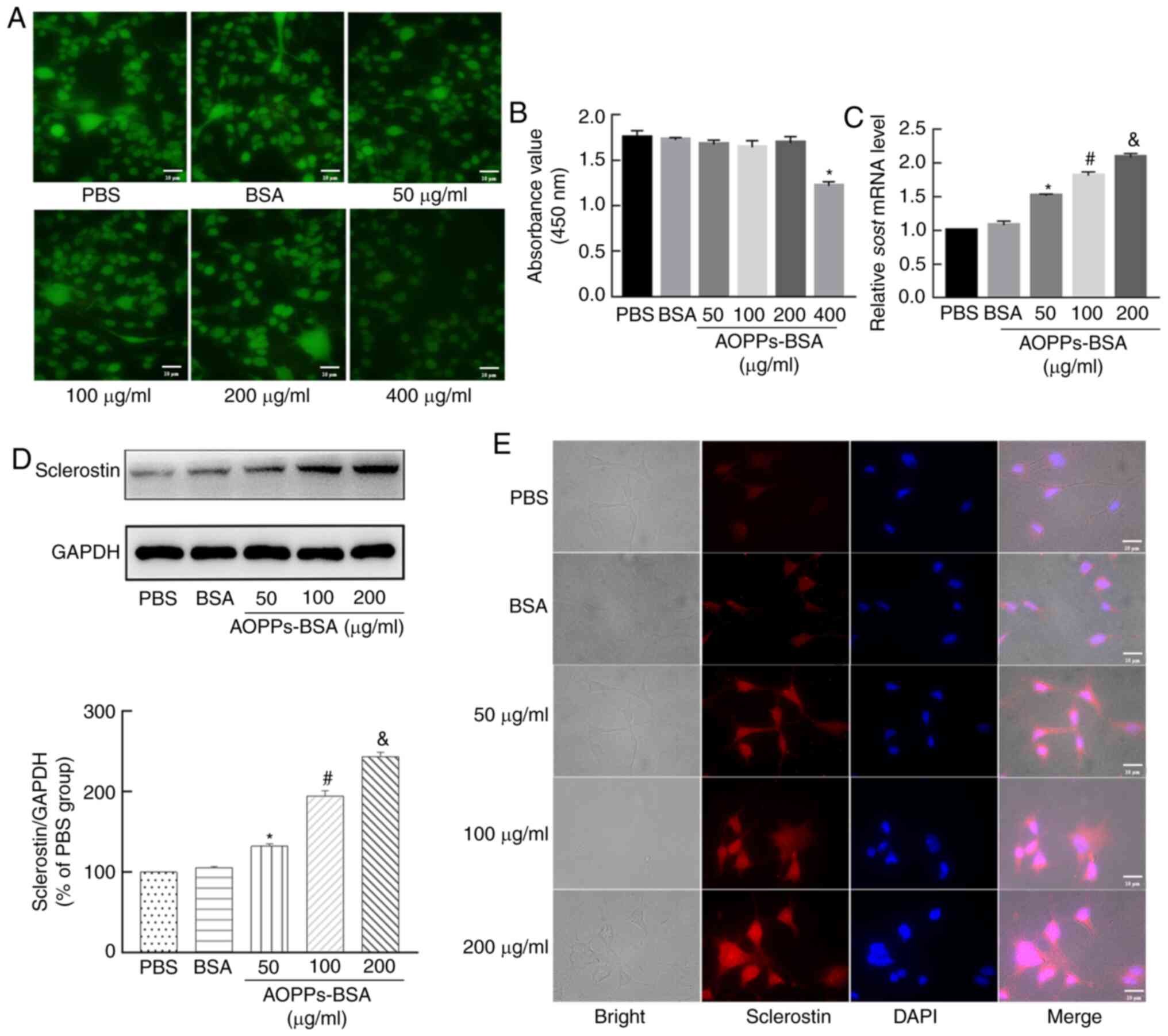 | Figure 3AOPPs increase sclerostin expression
in MLO-Y4 cells. (A) Compared with the PBS or BSA group, FDA
staining indicated there was only a slight decrease in cell
viability in the 50, 100 and 200 µg/ml AOPPs-BSA groups.
However, cell viability was significantly decreased in the
400-µg/ml AOPP-treated group. (B) The Cell Counting Kit-8
assay presented similar results. (C) Reverse
transcription-quantitative PCR results demonstrated that AOPPs
increased SOST mRNA expression in MLO-Y4 cells in a
concentration-dependent manner. (D) Western blotting and (E)
immunofluorescence staining revealed that AOPPs increased
sclerostin protein in MLO-Y4 cells. Magnification, ×400. The data
are presented as the mean ± SEM. *P<0.05 vs. the PBS
group; #P<0.05 vs. the 50-µg/ml group;
&P<0.05 vs. the 100-µg/ml group, n=3.
AOPPs, advanced oxidation protein products; PBS, phosphate-buffered
saline; BSA, bovine serum albumin; FDA, fluorescein diacetate;
SOST, sclerostin. |
To evaluate the effect of AOPPs on the expression of
sclerostin in MLO-Y4 cells, the cells were incubated for 24 h with
AOPPs-BSA (ranging between 50-200 µg/ml). According to
RT-qPCR analysis, the SOST mRNA expression was nearly
0.5-fold higher in the 50 µg/ml AOPPs-BSA group compared
with that in the PBS group. As the concentration of AOPPs-BSA
increased, the SOST mRNA expression levels were also
increased (Fig. 3C). However,
compared to the PBS group, SOST mRNA levels did not notably
change in the BSA-treated group. The results from the western blot
analysis and immunofluorescence staining further indicated that
treatment with AOPPs upregulated the expression of sclerostin in
MLO-Y4 cells in a dose-dependent manner (Fig. 3D and E).
AOPPs increase sclerostin expression in
MLO-Y4 cells by ROS-triggered downregulation of Sirt1
Previous, including the research group of the
present authors, have reported that AOPPs significantly increase
the cellular ROS levels in numerous pathological processes by
activating NADPH oxidases (21,22,33). Therefore, the present study
examined the ROS levels in MLO-Y4 cells treated with AOPPs.
Compared with PBS, BSA did not markedly affect DCFH fluorescence,
which represented the intracellular ROS levels. However, 50
µg/ml AOPPs-BSA notably increased the cellular fluorescence
intensity. In addition, the AOPPs-BSA-induced ROS effect was more
evident in the 100- and 200-µg/ml groups (Fig. 4A and B). To investigate whether
NADPH oxidases were involved in AOPP-induced ROS accumulation,
MLO-Y4 cells were co-incubated with apocynin, an NADPH oxidase
inhibitor. As presented in Fig.
4C, apocynin successfully decreased the ROS increase triggered
by AOPPs.
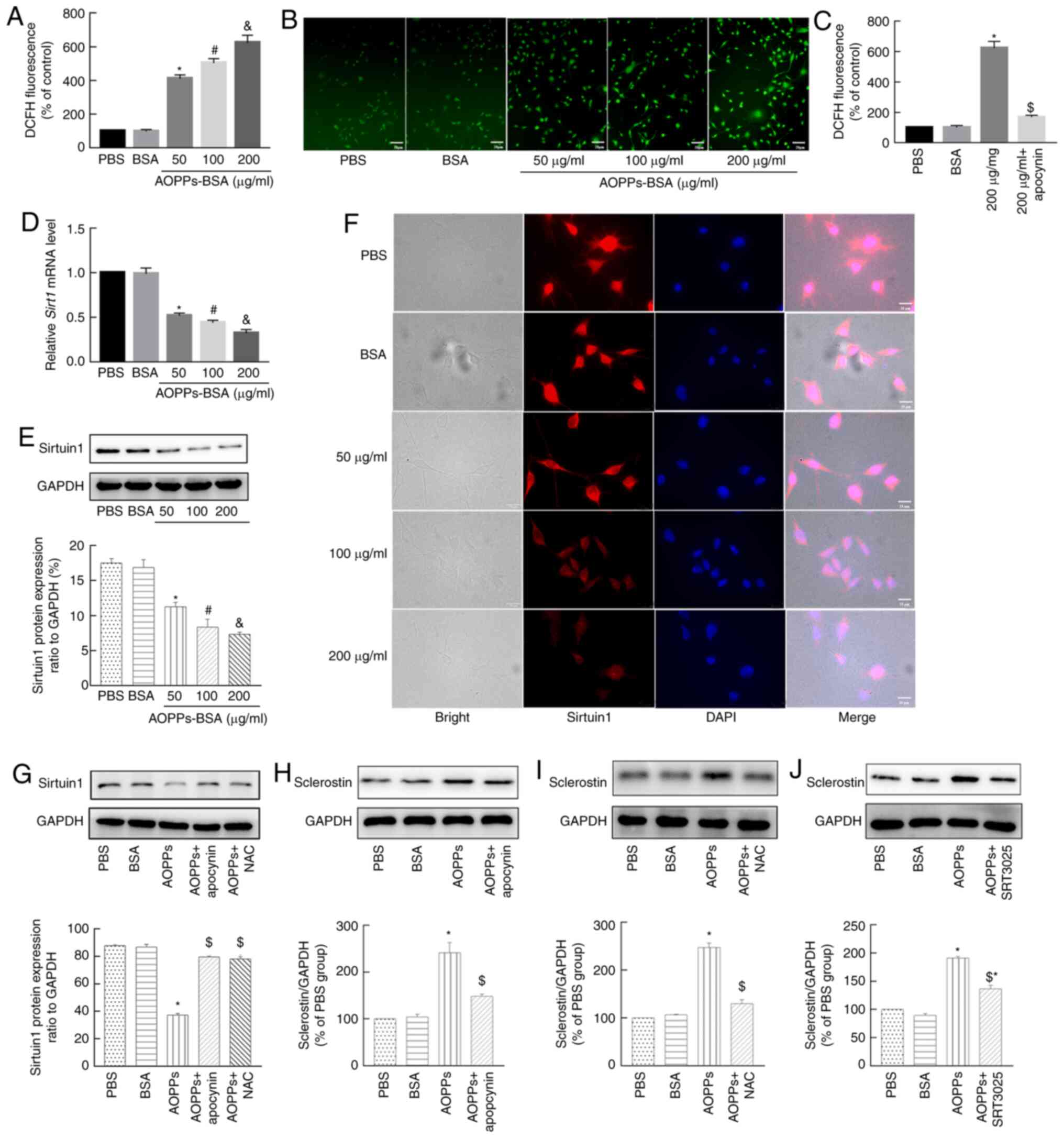 | Figure 4AOPPs increase sclerostin expression
in MLO-Y4 cells via ROS-triggered downregulation of Sirt1. (A and
B) AOPPs-BSA increased the intracellular ROS levels of MLO-Y4
cells, with a concentration-dependent effect. (C) Apocynin
successfully eliminated the AOPP-triggered ROS accumulation. (D)
SIRT1 mRNA expression was downregulated by treatment with
AOPPs, in a concentration-dependent manner. (E) This effect of
AOPPs was further confirmed at the protein level via (E) western
blotting and (F) immunofluorescence staining. (G) The AOPPs-induced
downregulation of Sirt1 was largely blocked by apocynin or NAC.
Western blotting results revealed that the AOPP-increased
sclerostin expression in MLO-Y4 cells was significantly suppressed
by (H) apocynin, (I) NAC, or (J) SRT3025. Magnification, ×400. The
data are presented as the mean ± SEM. *P<0.05 vs. the
PBS group; #P<0.05 vs. the 50-µg/ml group;
&P<0.05 vs. the 100-µg/ml group,
$P<0.05 vs. the 200-µg/ml group; n=3. AOPPs,
advanced oxidation protein products; ROS, reactive oxygen species;
Sirt1, sirtuin 1; BSA, bovine serum albumin; NAC,
N-acetyl-L-cysteine; PBS, phosphate-buffered saline. |
Evidence from previous studies has suggested that
Sirt1 directly decreases the expression of sclerostin in osteocytes
via epigenetic regulation (30,34,35). In addition, OS decreases Sirt1
expression in multiple diseases (31). Thus, it was investigated whether
AOPPs downregulated Sirt1 expression in MLO-Y4 cells. As revealed
in Fig. 4D, AOPPs significantly
decreased the mRNA expression levels of Sirt1 in a
dose-dependent manner. This effect of AOPPs was further confirmed
at the protein expression level, as detected via western blotting
(Fig. 4E) and immunofluorescence
staining (Fig. 4F). However,
when the cells were co-incubated with apocynin, or a ROS scavenger,
such as NAC, the aforementioned effect of AOPPs was largely blocked
(Fig. 4G). These results
indicated that AOPPs downregulated the expression level of Sirt1 in
MLO-Y4 cells by triggering NADPH oxidase-dependent ROS
accumulation.
To verify the roles of NADPH oxidases, ROS, and
Sirt1 in the AOPP-induced expression of sclerostin, MLO-Y4 cells
treated with AOPPs were co-incubated with apocynin, NAC, and a
Sirt1 activator, SRT3025. The western blotting results demonstrated
that the AOPP-induced expression of sclerostin was largely
abrogated by apocynin (Fig. 4H)
and NAC (Fig. 4I), whereas the
expression of this protein was significantly, but not completely,
diminished by SRT3025 (Fig. 4J).
These results indicated that the NADPH oxidase-dependent and
ROS-triggered downregulation of Sirt1 was involved in the
AOPP-induced upregulation of sclerostin.
Apocynin, NAC, and SRT-3025 alleviate
bone deterioration and decrease sclerostin expression in
AOPP-treated mice
To further confirm the mechanism of AOPP-induced
bone deterioration in vivo, AOPP-treated mice were
co-treated with apocynin, NAC, or SRT3025. The results of the BMD
measurement results identified that apocynin, NAC, and SRT3025
effectively ameliorated the loss of bone mass in L4 vertebral
bodies in mice treated with AOPPs-BSA (Fig. 5A). However, the BMD of the
femoral specimens was not markedly different among the various
groups (Fig. 5B). Apocynin, NAC,
and SRT3025 also largely alleviated the bone microstructural
degeneration induced by AOPPs. As indicated by the micro-CT
analyses results, the BV/TV, Tb.N and Tb.Th of L4 vertebral bodies
were notably higher in the AOPPs-BSA + apocynin, AOPPs-BSA + NAC or
AOPPs-BSA + SRT3025 groups compared with in the AOPPs-BSA group.
However, Tb.Sp in these three groups was reduced (Fig. 5C-G). Moreover, the same
microstructural changes were observed in the proximal tibias
(Fig. 5H-L).
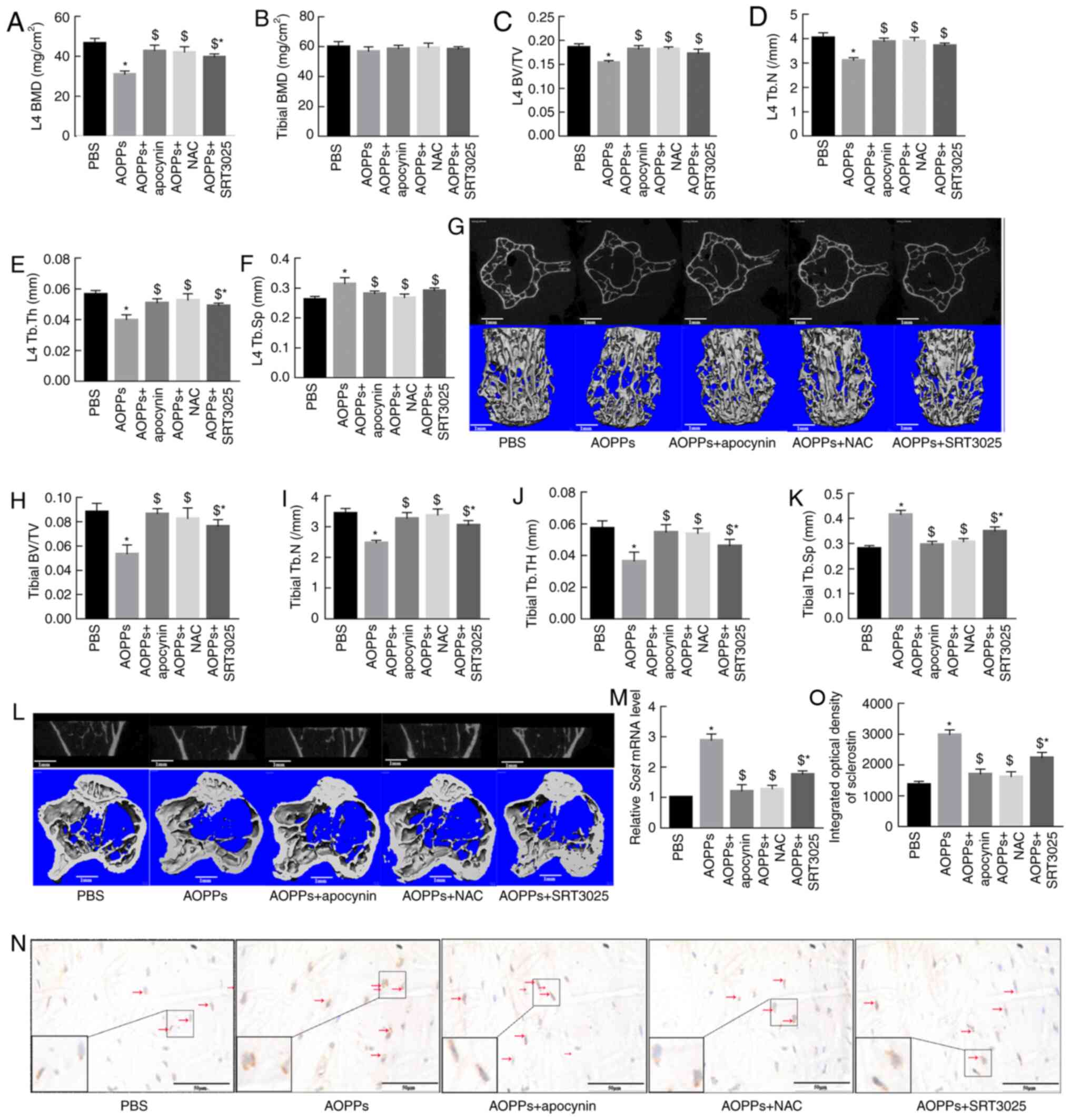 | Figure 5Apocynin, NAC and SRT3025 alleviate
bone deterioration and decrease sclerostin expression in
AOPP-treated mice. (A) Apocynin, NAC or SRT3025 effectively
ameliorated the bone mass loss of L4 vertebral bodies in
AOPP-treated mice. (B) The femoral BMD revealed no difference among
different groups. Apocynin, NAC or SRT3025 improved AOPP-induced
trabecular microstructural degeneration by increasing BV/TV, Tb.N
and Tb.Th but by decreasing Tb.Sp in both (C-F) L4 vertebral bodies
and (H-K) proximal tibias. Representative middle cross or coronal
sectional images and 3D reconstructed images of (G) L4 vertebral
bodies and (L) proximal tibias in different groups. (M) Reverse
transcription-quantitative PCR results demonstrated that the
AOPP-increased SOST mRNA expression was suppressed by
apocynin, NAC or SRT3025. (N and O) A similar effect was confirmed
at the protein level via immunohistochemical staining.
Magnification, ×400. The data are presented as the mean ± SEM.
*P<0.05 vs. the PBS group; $P<0.05 vs.
the 200-mg/kg/day AOPPs-BSA group; n=7. AOPPs, advanced oxidation
protein products; NAC, N-acetyl-L-cysteine; BMD, bone mineral
density; BV/TV, bone volume/total volume; Tb.N, trabecular number;
Tb.Th, trabecular thickness; Tb.Sp, trabecular separation;
SOST, sclerostin; PBS, phosphate-buffered saline. |
The effects of apocynin, NAC, and SRT3025 on the
expression of sclerostin in the cortical bone of AOPP-treated mice
were also evaluated. The RT-qPCR results demonstrated that the mRNA
expression levels of SOST were significantly lower in the
AOPPs-BSA + apocynin, AOPPs-BSA + NAC, and AOPPs + SRT3025 groups
compared with in the AOPPs-BSA group (Fig. 5M). This result was also confirmed
at the protein level via immunohistochemical staining (Fig. 5N and O).
Discussion
AOPPs are important proinflammatory mediators
involved in the pathological processes of diverse diseases
(14,22,36,37). Both animal and clinical
observational studies have revealed that the plasma levels of AOPPs
increase with age (23,38). However, whether AOPPs participate
in the process of age-related bone loss remains unknown. Our
previous study confirmed that AOPPs accelerated bone deterioration
in aged rats (24). Moreover,
the present study demonstrated that AOPPs aggravated the loss of
bone mass and the degeneration of bone microstructure by increasing
the expression level of sclerostin in osteocytes, which was
mediated by ROS-dependent downregulation of the Sirt1 pathway.
Osteocytes are the most abundant cells in bones,
accounting for 90-95% of all bone cells in the adult skeleton
(39). In the past decade,
accumulating data have been published regarding the biology and
function of osteocytes. Far from being 'passive embedded' cells,
osteocytes express a series of molecules that are involved in
multiple activities, especially in bone metabolism (40). Sclerostin is a cysteine-rich
protein that is predominantly expressed in osteocytes and is
largely considered as a negative regulator of bone formation
(5,41). Sclerostin inhibits the
Wnt/β-catenin signaling pathway by binding to a low-density
lipoprotein receptor-related protein 5/6 (42). While genetic inactivation or
mutation of the SOST gene, in humans and animals, results in
increased bone mass, its overexpression in animals leads to
osteopenia (43-45). In a 16-month-old, gonad-intact,
aged male rat model of age-related osteoporosis, 5 weeks of
treatment with an antibody against sclerostin effectively restored
the bone mass and strength (9).
The sclerostin monoclonal antibody, romosozumab, is now an optional
treatment for osteoporosis (46). Previous studies have also
reported that the plasma sclerostin levels increase with age and
are associated with low bone mass and osteoporotic fracture
(7,8). However, the mechanism of elevated
levels of sclerostin during aging are yet to be fully elucidated.
In the present study, it was identified that AOPPs upregulated the
expression level of sclerostin in osteocytes, both in vitro
and in vivo. This result was consistent with that of a
previous study (47). Given the
increased level of AOPPs with age, AOPP-induced sclerostin
expression may be an important contributor to age-related bone
loss.
OS is crucial in the progression of numerous
diseases, including osteoporosis (48). Patients with osteoporosis are
characterized by enhanced OS (28). Thus, factors that induce OS can
promote osteoporosis, and when osteoporosis is improved by
anti-osteoporotic treatment, the OS level also decreases (28). Previous researchers have reported
that AOPPs induce OS in multiple cell types, including neutrophils,
monocytes, vascular endothelial cells, mesangial cells,
osteoblasts, and hepatocytes, amongst others (21,22). Consistent with these findings,
the present study demonstrated that treatment with AOPPs increased
MDA levels but decreased the t-SOD levels in bone tissues of
AOPP-treated mice, and there was also an increase in the ROS levels
in AOPP-treated cells.
NADPH oxidases are one of the main contributors of
ROS. Previous studies have revealed that AOPPs induce OS mainly by
activating NADPH oxidases (16).
In accordance with this finding, the present study observed that
the NADPH oxidase inhibitor, apocynin, effectively eliminated
cellular ROS increased by AOPPs in MLO-Y4 cells.
It is important to elucidate how AOPPs influence
sclerostin expression in osteocytes. Sirt1 is the most widely
investigated member of the Sirt family, and serves important
protective roles in age-related diseases, including osteoporosis
(49,50). It has been revealed that both
natural and synthetic Sirt1 activator compounds restore bone loss
in animal models of osteoporosis (51). Furthermore, global or targeted
deletion of Sirt1 in bone cells results in a low bone mass
phenotype, while overexpression of Sirt1 protects against bone loss
(50). Clinical data have
suggested an association between low levels of Sirt1 and
osteoporotic fractures (52).
Thus, the molecular basis of Sirt1 in the regulation of bone
metabolism appears complicated. In recent years, several studies
have reported that Sirt1 increases bone mass by downregulating the
expression of sclerostin. Sirt1 directly and negatively regulates
the SOST gene by deacetylating histone 3 at the lysine 9
residue of the promoter (30,34,35). In the present study, treatment
with AOPPs decreased the protein expression level of Sirt1 in
MLO-Y4 cells, which was associated with increased sclerostin
expression. Furthermore, the Sirt1 activator, SRT-3025,
significantly weakened the aforementioned effects of AOPPs.
However, SRT-3025 did not completely abrogate the AOPP-induced
increase of sclerostin expression in MLO-Y4 cells, indicating that
AOPPs increase sclerostin expression using other pathways, such as
the c-Jun N-terminal kinase/p38 mitogen-activated protein kinase
activation (47).
How AOPPs decrease Sirt1 expression level remains
unknown. Previously published data suggest a relationship between
Sirt1 and OS. While it is widely accepted that Sirt1 regulates the
cellular OS burden and its toxicity, Sirt1 itself is also regulated
by OS (31). For instance, both
the abundance and deacetylating activity of Sirt1 decrease in
response to oxidants (53,54). Similar with these findings, the
present study demonstrated that AOPPs significantly suppressed
Sirt1 expression via the induction of OS in MLO-Y4 cells, and this
effect was effectively blocked by both the NADPH oxidase inhibitor
and ROS scavenger.
Since cortical and trabecular bone does not
degenerate at a same speed during aging (55), the present study is limited for
not detecting the microstructural changes in cortical bone.
Moreover, the sclerostin/OS/Sirt1 axis appears not to be the only
axis involved in AOPP-induced bone loss. AOPPs are documented to
inhibit proliferation and differentiation of osteoblasts via NF-κB
pathway (32), AOPPs also induce
pre-osteoblast apoptosis through a ROS-dependent, mitogen-activated
protein kinase-mediated intrinsic apoptosis pathway (22). The present study is also shorted
for not investigating those reported pathways.
In conclusion, the present study demonstrated that
AOPPs increase sclerostin expression in osteocytes via a NADPH
oxidase-dependent, ROS-triggered downregulation of the Sirt1
pathway. Therefore, the present data provide novel insights into
the understanding of the pathogenic basis of senile
osteoporosis.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article or can be obtained from the
corresponding author on reasonable request.
Authors' contributions
JC and JZ conceived and designed the study. JC and
QX provided administrative support. JZ and XL provided study
materials. JZ and QX collected analyzed and interpreted the data.
All authors wrote, read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Laboratory
Animal Care and Use Committee of Nanfang Hospital, Southern Medical
University (approval no. NFYY-2018-68).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 81772395) and the Jiangxi Provincial
Natural Science Foundation (grant no. 20202BAB216008).
References
|
1
|
Modinger Y, Loffler B, Huber-Lang M and
Ignatius A: Complement involvement in bone homeostasis and bone
disorders. Semin Immunol. 37:53–65. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Khosla S: Pathogenesis of age-related bone
loss in humans. J Gerontol A Biol Sci Med Sci. 68:1226–1235. 2013.
View Article : Google Scholar :
|
|
3
|
Bonewald LF: The amazing osteocyte. J Bone
Miner Res. 26:229–238. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Winkler DG, Sutherland MK, Geoghegan JC,
Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR,
Staehling-Hampton K, et al: Osteocyte control of bone formation via
sclerostin, a novel BMP antagonist. EMBO J. 22:6267–6276. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Delgado-Calle J, Sato AY and Bellido T:
Role and mechanism of action of sclerostin in bone. Bone. 96:29–37.
2017. View Article : Google Scholar :
|
|
6
|
Rauch F and Adachi R: Sclerostin: More
than a bone formation brake. Sci Transl Med. 8:330fs72016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roforth MM, Fujita K, McGregor UI, Kirmani
S, McCready LK, Peterson JM, Drake MT, Monroe DG and Khosla S:
Effects of age on bone mRNA levels of sclerostin and other genes
relevant to bone metabolism in humans. Bone. 59:1–6. 2014.
View Article : Google Scholar
|
|
8
|
Ardawi MS, Rouzi AA, Al-Sibiani SA,
Al-Senani NS, Qari MH and Mousa SA: High serum sclerostin predicts
the occurrence of osteoporotic fractures in postmenopausal women:
The Center of Excellence for Osteoporosis Research Study. J Bone
Miner Res. 27:2592–2602. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li X, Warmington KS, Niu QT, Asuncion FJ,
Barrero M, Grisanti M, Dwyer D, Stouch B, Thway TM, Stolina M, et
al: Inhibition of sclerostin by monoclonal antibody increases bone
formation, bone mass, and bone strength in aged male rats. J Bone
Miner Res. 25:2647–2656. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Finkel T: Signal transduction by reactive
oxygen species. J Cell Biol. 194:7–15. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schieber M and Chandel NS: ROS function in
redox signaling and oxidative stress. Curr Biol. 24:R453–R462.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Witko-Sarsat V, Friedlander M,
Capeillere-Blandin C, Nguyen-Khoa T, Nguyen AT, Zingraff J, Jungers
P and Descamps-Latscha B: Advanced oxidation protein products as a
novel marker of oxidative stress in uremia. Kidney Int.
49:1304–1313. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Capeillere-Blandin C, Gausson V,
Descamps-Latscha B and Witko-Sarsat V: Biochemical and
spectrophotometric significance of advanced oxidized protein
products. Biochim Biophys Acta. 1689:91–102. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Witko-Sarsat V, Friedlander M, Nguyen Khoa
T, Capeillere-Blandin C, Nguyen AT, Canteloup S, Dayer JM, Jungers
P, Drüeke T and Descamps-Latscha B: Advanced oxidation protein
products as novel mediators of inflammation and monocyte activation
in chronic renal failure. J Immunol. 161:2524–2532. 1998.PubMed/NCBI
|
|
15
|
Wu Q, Zhong ZM, Zhu SY, Liao CR, Pan Y,
Zeng JH, Zheng S, Ding RT, Lin QS, Ye Q, et al: Advanced oxidation
protein products induce chondrocyte apoptosis via receptor for
advanced glycation end products-mediated, redox-dependent intrinsic
apoptosis pathway. Apoptosis. 21:36–50. 2016. View Article : Google Scholar
|
|
16
|
Xie F, Sun S, Xu A, Zheng S, Xue M, Wu P,
Zeng JH and Bai L: Advanced oxidation protein products induce
intestine epithelial cell death through a redox-dependent, c-jun
N-terminal kinase and poly (ADP-ribose) polymerase-1-mediated
pathway. Cell Death Dis. 5:e10062014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu Q, Zhong ZM, Pan Y, Zeng JH, Zheng S,
Zhu SY and Chen JT: Advanced oxidation protein products as a novel
marker of oxidative stress in postmenopausal osteoporosis. Med Sci
Monit. 21:2428–2432. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kalousova M, Skrha J and Zima T: Advanced
glycation end-products and advanced oxidation protein products in
patients with diabetes mellitus. Physiol Res. 51:597–604. 2002.
|
|
19
|
Liu Z, Yao X, Jiang W, Li W, Zhu S, Liao
C, Zou L, Ding R and Chen J: Advanced oxidation protein products
induce microglia-mediated neuroinflammation via MAPKs-NF-κB
signaling pathway and pyroptosis after secondary spinal cord
injury. J Neuroinflammation. 17:902020. View Article : Google Scholar
|
|
20
|
Witko-Sarsat V, Gausson V, Nguyen AT,
Touam M, Drueke T, Santangelo F and Descamps-Latscha B:
AOPP-induced activation of human neutrophil and monocyte oxidative
metabolism: A potential target for N-acetylcysteine treatment in
dialysis patients. Kidney Int. 64:82–91. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun S, Xie F, Xu X, Cai Q, Zhang Q, Cui Z,
Zheng Y and Zhou J: Advanced oxidation protein products induce
S-phase arrest of hepatocytes via the ROS-dependent,
β-catenin-CDK2-mediated pathway. Redox Biol. 14:338–353. 2018.
View Article : Google Scholar
|
|
22
|
Zhu SY, Zhuang JS, Wu Q, Liu ZY, Liao CR,
Luo SG, Chen JT and Zhong ZM: Advanced oxidation protein products
induce pre-osteoblast apoptosis through a nicotinamide adenine
dinucleotide phosphate oxidase-dependent, mitogen-activated protein
kinases-mediated intrinsic apoptosis pathway. Aging Cell.
17:e127642018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang YB, Zhong ZM, Hou G, Jiang H and
Chen JT: Involvement of oxidative stress in age-related bone loss.
J Surg Res. 169:e37–42. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeng JH, Zhong ZM, Li XD, Wu Q, Zheng S,
Zhou J, Ye WB, Xie F, Wu XH, Huang ZP and Chen JT: Advanced
oxidation protein products accelerate bone deterioration in aged
rats. Exp Gerontol. 50:64–71. 2014. View Article : Google Scholar
|
|
25
|
Bouxsein ML, Boyd SK, Christiansen BA,
Guldberg RE, Jepsen KJ and Muller R: Guidelines for assessment of
bone microstructure in rodents using micro-computed tomography. J
Bone Miner Res. 25:1468–1486. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Method. 25:402–408. 2001.
View Article : Google Scholar
|
|
27
|
Coulson J, Bagley L, Barnouin Y, Bradburn
S, Butler-Browne G, Gapeyeva H, Hogrel JY, Maden-Wilkinson T, Maier
AB, Meskers C, et al: Circulating levels of dickkopf-1,
osteoprotegerin and sclerostin are higher in old compared with
young men and women and positively associated with whole-body bone
mineral density in older adults. Osteoporos Int. 28:2683–2689.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Manolagas SC: From estrogen-centric to
aging and oxidative stress: A revised perspective of the
pathogenesis of osteoporosis. Endocr Rev. 31:266–300. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shi XY, Hou FF, Niu HX, Wang GB, Xie D,
Guo ZJ, Zhou ZM, Yang F, Tian JW and Zhang X: Advanced oxidation
protein products promote inflammation in diabetic kidney through
activation of renal nicotinamide adenine dinucleotide phosphate
oxidase. Endocrinology. 149:1829–1839. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cohen-Kfir E, Artsi H, Levin A, Abramowitz
E, Bajayo A, Gurt I, Zhong L, D'Urso A, Toiber D, Mostoslavsky R
and Dresner-Pollak R: Sirt1 is a regulator of bone mass and a
repressor of Sost encoding for sclerostin, a bone formation
inhibitor. Endocrinology. 152:4514–4524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hwang JW, Yao H, Caito S, Sundar IK and
Rahman I: Redox regulation of SIRT1 in inflammation and cellular
senescence. Free Radic Biol Med. 61:95–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhong ZM, Bai L and Chen JT: Advanced
oxidation protein products inhibit proliferation and
differentiation of rat osteoblast-like cells via NF-kappaB pathway.
Cell Physiol Biochem. 24:105–114. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ding R, Jiang H, Sun B, Wu X, Li W, Zhu S,
Liao C, Zhong Z and Chen J: Advanced oxidation protein products
sensitized the transient receptor potential vanilloid 1 via NADPH
oxidase 1 and 4 to cause mechanical hyperalgesia. Redox Biol.
10:1–11. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Artsi H, Cohen-Kfir E, Gurt I, Shahar R,
Bajayo A, Kalish N, Bellido TM, Gabet Y and Dresner-Pollak R: The
Sirtuin1 activator SRT3025 down-regulates sclerostin and rescues
ovariectomy-induced bone loss and biomechanical deterioration in
female mice. Endocrinology. 155:3508–3515. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stegen S, Stockmans I, Moermans K,
Thienpont B, Maxwell PH, Carmeliet P and Carmeliet G: Osteocytic
oxygen sensing controls bone mass through epigenetic regulation of
sclerostin. Nat Commun. 9:25572018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu SX, Hou FF, Guo ZJ, Nagai R, Zhang WR,
Liu ZQ, Zhou ZM, Zhou M, Xie D, Wang GB and Zhang X: Advanced
oxidation protein products accelerate atherosclerosis through
promoting oxidative stress and inflammation. Arterioscler Thromb
Vasc Biol. 26:1156–1162. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li HY, Hou FF, Zhang X, Chen PY, Liu SX,
Feng JX, Liu ZQ, Shan YX, Wang GB, Zhou ZM, et al: Advanced
oxidation protein products accelerate renal fibrosis in a remnant
kidney model. J Am Soc Nephrol. 18:528–538. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Komosinska-Vassev K, Olczyk P,
Winsz-Szczotka K, Kuznik-Trocha K, Klimek K and Olczyk K: Age- and
gender-related alteration in plasma advanced oxidation protein
products (AOPP) and glycosaminoglycan (GAG) concentrations in
physiological ageing. Clin Chem Lab Med. 50:557–563. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bonewald LF: Osteocytes as dynamic
multifunctional cells. Ann N Y Acad Sci. 1116:281–290. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dallas SL, Prideaux M and Bonewald LF: The
osteocyte: An endocrine cell and more. Endocr Rev. 34:658–690.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
van Bezooijen RL, ten Dijke P, Papapoulos
SE and Lowik CW: SOST/sclerostin, an osteocyte-derived negative
regulator of bone formation. Cytokine Growth Factor Rev.
16:319–327. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li XF, Zhang Y, Kang H, Liu W, Liu P,
Zhang J, Harris SE and Wu D: Sclerostin binds to LRP5/6 and
antagonizes canonical Wnt signaling. J Biol Chem. 280:19883–19887.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Balemans W, Patel N, Ebeling M, Van Hul E,
Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ,
et al: Identification of a 52 kb deletion downstream of the SOST
gene in patients with van Buchem disease. J Med Genet. 39:91–97.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Balemans W, Ebeling M, Patel N, Van Hul E,
Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P,
et al: Increased bone density in sclerosteosis is due to the
deficiency of a novel secreted protein (SOST). Hum Mol Genet.
10:537–543. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang D, Park BM, Kang M, Nam H, Kim EJ,
Bae C and Lim SK: The systemic effects of sclerostin overexpression
using φC31 integrase in mice. Biochem Biophys Res Commun.
472:471–476. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cosman F, Crittenden DB, Adachi JD,
Binkley N, Czerwinski E, Ferrari S, Hofbauer LC, Lau E, Lewiecki
EM, Miyauchi A, et al: Romosozumab treatment in postmenopausal
women with osteoporosis. N Engl J Med. 375:1532–1543. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yu C, Huang D, Wang K, Lin B, Liu Y, Liu
S, Wu W and Zhang H: Advanced oxidation protein products induce
apoptosis, and upregulate sclerostin and RANKL expression, in
osteocytic MLO-Y4 cells via JNK/p38 MAPK activation. Mol Med Rep.
15:543–550. 2017. View Article : Google Scholar :
|
|
48
|
Finkel T and Holbrook NJ: Oxidants,
oxidative stress and the biology of ageing. Nature. 408:239–247.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Baur JA, Ungvari Z, Minor RK, Le Couteur
DG and de Cabo R: Are sirtuins viable targets for improving
healthspan and lifespan? Nat Rev Drug Discov. 11:443–461. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zainabadi K: Drugs targeting SIRT1, a new
generation of therapeutics for osteoporosis and other bone related
disorders? Pharmacol Res. 143:97–105. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kim HN, Han L, Iyer S, de Cabo R, Zhao H,
O'Brien CA, Manolagas SC and Almeida M: Sirtuin1 suppresses
osteoclastogenesis by deacetylating FoxOs. Mol Endocrinol.
29:1498–1509. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
El-Haj M, Gurt I, Cohen-Kfir E, Dixit V,
Artsi H, Kandel L, Yakubovsky O, Safran O and Dresner-Pollak R:
Reduced Sirtuin1 expression at the femoral neck in women who
sustained an osteoporotic hip fracture. Osteoporos Int.
27:2373–2378. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Caito S, Rajendrasozhan S, Cook S, Chung
S, Yao H, Friedman AE, Brookes PS and Rahman I: SIRT1 is a
redox-sensitive deacetylase that is post-translationally modified
by oxidants and carbonyl stress. Faseb J. 24:3145–3159. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Furukawa A, Tada-Oikawa S, Kawanishi S and
Oikawa S: H2O2 accelerates cellular
senescence by accumulation of acetylated p53 via decrease in the
function of SIRT1 by NAD+ depletion. Cell Physiol
Biochem. 20:45–54. 2007. View Article : Google Scholar
|
|
55
|
Farr JN and Khosla S: Skeletal changes
through the lifespan-from growth to senescence. Nat Rev Endocrinol.
11:513–521. 2015. View Article : Google Scholar : PubMed/NCBI
|














