Introduction
Pulmonary arterial hypertension (PAH) is a chronic
and progressive cardiovascular disease with a high mortality rate.
It is mainly characterized by pulmonary vascular remodeling,
endothelial cell (EC) dysfunction, apoptosis and inflammation.
These features increase pulmonary vascular resistance and
subsequent pulmonary arterial pressure, causing right heart failure
and mortality (1-3). Despite notable improvements being
made in therapeutic strategies for PAH, the prognosis of patients
with PAH remains unsatisfactory (4). Therefore, a novel signaling pathway
related to the pathophysiology of PAH urgently needs to be
explored.
Peroxisome proliferator-activated receptor γ (PPARγ)
is a nuclear hormone receptor and transcription factor that
regulates multiple genes in cardiovascular homeostasis (5). Increasing evidence indicates that
PPARγ is a potent, protective regulator of PAH (6) and pulmonary artery ECs (PAECs)
(7,8). In addition, a previous study proved
that PPARγ activation alleviated hypoxia-induced pulmonary arterial
remodeling and collagen deposition in hypoxia-induced PAH (9). However, the role of PPARγ in EC
dysfunction and inflammation in hypoxia-exposed human PAECs
(HPAECs) has yet to be fully elucidated.
MicroRNAs (miRNAs or miRs) are a conserved class of
small non-coding RNAs that can regulate gene expression at the
post-transcriptional level (10). Recent studies have indicated that
abundant miRNAs participate in the pathogenesis of PAH (11-14). miR-27b has been shown to suppress
endothelial cell proliferation and migration in Kawasaki disease
(15). In addition, miR-27b has
been shown to regulate heat shock protein 90 (Hsp90)/endothelial
nitric oxide synthase (eNOS) signaling and nitric oxide production
by targeting PPARγ (16). Thus,
it was hypothesized that miR-27b is involved in hypoxia-induced
HPAEC dysfunction and inflammation.
Fibroblast growth factor 21 (FGF21) is a member of
the FGF superfamily, which is mainly expressed in the liver,
pancreas, testis and brown adipose tissue. As a hormone, FGF21
regulates a variety of pharmacological effects, including blood
glucose and lipid metabolism (17). A previous study demonstrated that
FGF21 attenuated hypoxia-induced dysfunction and apoptosis in
HPAECs by alleviating endoplasmic reticulum stress (18). Yu et al (19) suggested that FGF21 inhibited
macrophage-mediated inflammation by activating nuclear factor
erythroid 2-related factor 2 (Nrf2) and suppressing the nuclear
factor (NF)-κB signaling pathway. Moreover, a relevant study
demonstrated that FGF21 attenuated hypoxia-induced pulmonary
hypertension (HPH) by upregulating PPARγ expression and suppressing
the expression of inflammatory cytokines (20). Another study reported that FGF21
inhibited miRNA-33 expression to alleviate inflammation and thus
prevent atherosclerosis (21),
indicating that miRNAs may play an important role in the regulatory
effects of FGF21 in several diseases. Therefore, the present study
established a model of hypoxia-induced HPAEC to investigate whether
FGF21 can alleviate PAH through the miR-27b-mediated PPARγ
pathway.
Materials and methods
Reagents and antibodies
Pioglitazone (a PPARγ agonist) was obtained from
Sigma-Aldrich; Merck KGaA. FGF21 was obtained from PeproTech, Inc.
Rabbit antibodies against PPARγ (cat. no. ab178860) and NF-κB p65
(cat. no. ab32536) were purchased from Abcam). Rabbit antibodies
against p-NF-κB (cat. no. 3033T) and β-actin (cat. no. 4970S) were
purchased from Cell Signaling Technology, Inc. A horseradish
peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG)
antibody (cat. no. BL003A) was obtained from Biosharp Life
Sciences. Alexa Fluor 488-conjugated donkey anti-rabbit IgG (H+L;
cat. no. PC02A) was obtained from Shanghai Boyun BioTech Co., Ltd.
Alexa Fluor 594 AffiniPure goat anti-rabbit IgG (H+L; cat. no.
33112ES60) was purchased from Shanghai Yeasen Biotechnology Co.,
Ltd. Endothelial cell medium (ECM; 1001), fetal bovine serum (FBS)
and endothelial cell growth supplement (ECGS) were purchased from
ScienCell Research Laboratories, Inc. Phosphate-buffered saline was
purchased from HyClone; Cytiva. SuperSignal (R) West Femto Maximum
Sensitivity Substrate, radio immunoprecipitation assay (RIPA)
buffer, protease and phosphatase inhibitor mini tablets, and the
bicinchoninic acid protein assay kit were purchased from Pierce;
Thermo Fisher Scientific, Inc. The antifade-4,
6-diamidino-2-phenylindole (DAPI) was purchased from Beijing
Solarbio Science & Technology Co., Ltd. The Cell Counting Kit-8
(CCK-8) was purchased from Dojindo Molecular Technologies, Inc. The
ELISA kit was purchased from Shanghai Boyun BioTech Co., Ltd.
Cells and cell culture
Primary HPAECs (cat. no. 3100) were obtained from
ScienCell Research Laboratories, Inc. For the use of primary cells,
the present study was approved by the Ethics Committee of the First
Affiliated Hospital of Wenzhou Medical University (Wenzhou, China;
approval no. 2020-232). They were cultured in ECM supplemented with
5% FBS, 1% ECGS, 100 µ/ml penicillin and 100 µg/ml
streptomycin. After reaching 80-90% confluency, the cells were
treated with 0.25% trypsin-ethylene diamine tetraacetic acid (EDTA)
for further passaging. Confluent cultures of cells were used in the
experiments. For the analysis of the miR-27b/PPARγ axis, the HPAECs
were divided into the following groups: i) N, normoxia + negative
control (NC) group; ii) H, hypoxia + NC group; iii) H + I, hypoxia
+ miR-27b inhibitor (100 nM) group; iv) H + I + siPPARγ, hypoxia +
miR-27b inhibitor + siPPARγ; v) H + Pio: Hypoxia + pioglitazone
(PPARγ agonist, 6.25 µmol/l) + NC group. For the analysis of
the FGF21/miR-27b/PPARγ axis, the cells were divided into the
following groups: i) N, normoxia + NC group; ii) H, hypoxia + NC
group; iii) H + F, hypoxia + FGF21 (50 ng/ml) + NC group; and iv) H
+ F + m, hypoxia + FGF21 + miR-27b mimic (50 nM) group. The
hypoxia-exposed HPAECs were treated as aforementioned for 24 h. The
cells in the normoxia group were cultured in a normal incubator
(37°C with 21% O2, 5% CO2, and 74% N) for 24
h, while the cells in the hypoxia group were kept in a hypoxia
incubator (37°C with 5% CO2, 2% O2, and 90%
N2) for 24 h, with these conditions based on the results
of previous studies (18,22).
Cell transfection
The small interfering RNAs (siRNAs) targeting PPARγ
(si-PPARγ) with the negative control (si-NC), miRNAs for miR-27b
overexpression (miR-27b mimics), respective NC (mimics NC), miR-27b
knockdown (miR-27b inhibitors) and respective NC (inhibitors NC)
were designed and synthesized by Guangzhou RiboBio Co., Ltd. The
Ribo FECT. CP transfection kit (Guangzhou RiboBio Co., Ltd.) was
used to transfect the siPPARγ (50 nM), si-NC (50 nM), inhibitor
(100 nM), inhibitors NC (100 nM), mimic (50 nM) and mimics NC (50
nM) into the cells following the manufacturer's protocol. Following
transfection for 24 h, the cells were used in further
experiments.
Western blot analysis
Following treatment, the HPAECs were lysed with
ice-cold RIPA lysis buffer containing phenylmethylsulfonyl fluoride
(PMSF) for 30 min. The lysates were then centrifuged at 16,000 × g
for 30 min at 4°C, and the supernatant was collected. The protein
concentrations were determined using a Pierce bicinchoninic acid
(BCA) protein assay kit (Thermo Fisher Scientific, Inc.). Equal
amounts of protein (20 µg) were then separated by 10% sodium
dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE),
transferred onto polyvinylidene fluoride membranes, blocked with 5%
skimmed milk for 1 h at room temperature, and incubated overnight
with specific primary rabbit antibodies against PPARγ (1:1,000),
p-NF-κB (1:1,000), NF-κB p65 (1:1,000) and β-actin (1:1,000) at 4°C
overnight and then incubated with the HRP-labeled secondary
antibody (1:10,000) at room temperature for 1 h. β-actin was used
as an endogenous control. Super Signal West Fem to Maximum
Sensitivity substrate (cat. no. 3409; Shanghai Boyun BioTech Co.,
Ltd.) was used as the visualization reagent. The optical density of
the western blots was quantified using Quantity One 4.6.2 software
(Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the treated HPAECs using
TRIzol reagent (Sangon Biotech). Reverse transcription was
conducted using the Takara PrimeScript kit (cat. no. RR036A; Takara
Bio, Inc.) at 37°C for 15 min. Subsequently, qPCR was performed
using the Real-Time PCR System (cat. no. A25742; Thermo Fisher
Scientific, Inc.) following the manufacturers' instructions. The
PCR amplification reaction were as follows: 95°C for 10 min, 95°C
for 15 sec, 62°C for 30 sec, and 72°C for 30 sec. Relative
quantification was calculated using the 2−ΔΔCq method,
as previously described (23).
U6 and β-actin were set as internal controls. The primers used for
PCR were as follows: miR-27b forward, 5′-GCG CGT TCA CAG TGG CTA
AG-3′ and reverse, 5′-AGT GCA GGG TCC GAG GTA TT-3′; interleukin
(IL)-1β forward, 5′-CCC TAA ACA GAT GAA GTG CTC CTT-3′ and reverse,
5′-GTA GCT GGA TGC CGC CAT-3′; IL-6 forward, 5′-AAC CTG AAC CTT CCA
AAG ATG G-3′ and reverse, 5′-TCT GGC TTG TTC CTC ACT ACT-3′; tumor
necrosis factor (TNF)-α forward, 5′-CCT CTC TCT AAT CAG CCC TCT
G-3′ and reverse, 5′-GAG GAC CTG GGA GTA GAT GAG-3′; U6 forward,
5′-AGA GAA GAT TAG CAT GGC CCC TG-3′ and reverse, 5′-ATC CAG TGC
AGG GTC CGA GG-3′; and β-actin forward, 5′-CTG GAA CGG TGA AGG TGA
CA-3′ and reverse, 5′-AAG GGA CTT CCT GTA ACA ATG CA-3′.
Bioinformatics analysis and
dual-luciferase assay
TargetScan (www.targetscan.org) were utilized to predict the
binding site between the targeting gene and miR-27b. The
3′-untranslated region (3′-UTR) of PPAR mRNA with the
putative/mutant miR-27b-binding site was cloned into the
pmiR-RB-Report vector (Guangzhou RiboBio Co., Ltd.). Renilla
luciferase (Rluc) acted as a reporter and Firefly luciferase (Luc)
acted as a control. 293T cells (American Type Culture Collection
Co., Ltd.) grown in 96-well plates were co-transfected with the
vector (2 µg) and miR-27b mimic (50 nM) using a ribo FECTTM
CP transfection kit (Guangzhou RiboBio Co., Ltd.). Following
incubation (37°C with 21% O2, 5% CO2, and 74%
N) for 48 h, Renilla and Firefly luciferase activities were
detected using a dual-luciferase reporter assay system (Promega
Corporation).
Immunofluorescence (IF)
IF assays were conducted to determine the protein
expression levels of PPARγ and NF-κB. HPAECs were seeded at a
density of 1.0×105 cells per well of a 6-well plate in 2
ml of growth medium. Following stimulation, HPAECs were fixed with
4% paraformaldehyde for 30 min at 37°C and permeabilized using 0.1%
Triton X-100 for 10 min. The cells were then blocked using 5%
bovine serum albumin for 30 min at 37°C and immunostained using
anti-PPARγ (1:200) and anti-NF-κB p65 (1:200) antibodies overnight
at 4°C, followed incubation at 24°C in the dark for 1 h with 1:200
Alexa Fluor 488 conjugated donkey anti-rabbit IgG (H+L) and Alexa
Fluor 594 AffiniPure goat anti-rabbit IgG (H+L). For mounting, DAPI
was added to the coverslips at 24°C for 5-10 min. Images were
acquired using a fluorescence microscope (Leica DMi8; Leica
Microsystems, Inc.). Quantitative analysis was performed using
ImageJ analysis software (v 1.51, National Institutes of Health).
Each condition was repeated three times per experiment.
Cell viability assay
CCK-8 assay was performed to evaluate the viability
of the HPAECs. HPAECs were seeded in 96-well plates at a density of
1×104 cells/well. Following pre-incubation in complete
medium at 37°C in the presence of 21% O2 and 5%
CO2 for 12-24 h, HPAECs were treated with FGF21, miR-27b
mimics, miR-27b inhibitor, siPPARγ and pioglitazone prior to
exposure to hypoxia. Following 24 h of exposure to hypoxia, the
HPAECs were treated with 10 µl/well CCK-8 reagent at 37°C
for 1 h, and the results were examined every 30 min. The absorbance
of each well was measured using a microplate reader
(SoftMax®Pro 6 Software) at the wavelength of 450
nm.
Transwell migration chamber assay
Cell migration assay was performed to determine the
migration of HPAECs. In the present study, 24-well Transwell system
(5 µm; Corning Inc.) was used for migration assays.
Subsequently, 600 µl ECM containing 5% FBS were added to the
lower chamber to establish a chemical attractant.t HPAECs were
suspended in ECM without serum, and 100 µl
(2×104/100 µl) of cell suspension was seeded to
the upper chambers of the Transwell plates. Following incubation at
37°C for 24 h, the cells in the upper chamber were gently wiped off
using a cotton swab. The migrated cells were then fixed with 4%
paraformaldehyde for 20 min, followed by staining with crystal
violet for 20 min at 24°C. Finally, the migrated cells were counted
under a microscope (Leica DMi8, Leica Microsystems, Inc.) by
randomly selecting six fields for every sample.
ELISA for the measurement of endothelin-1
(ET-1), TNF-α and IL-1β levels
The levels of accumulated ET-1, TNF-α and IL-1β in
the culture medium were determined using ELISA kits (ET-1, cat. no.
BP-E10711; IL-1β, cat. no. BP-E10081; IL-6, cat. no. BP-E10140;
TNF-α, cat. no. BP-E10110) following the manufacturer's
protocol.
Statistical analysis
GraphPad Prism (v7.0; GraphPad Software, Inc.) was
used for the statistical analysis of the experimental data. All
data are presented as the mean ± SD of at least three independent
experiments. The differences among multiple groups were analyzed
using one-way ANOVA with repeated measures followed by Tukey's
post-hoc test, while differences between two groups were analyzed
using an unpaired Student's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
PPARγ expression is downregulated and
miR-27b expression is upregulated in HPAECs exposed to hypoxia
HPAECs were cultured under hypoxic conditions for 24
h to examine the changes in PPARγ and miR-27b expression in
response to hypoxia. The results of western blot analysis
demonstrated that PPARγ expression was significantly decreased in
the H group compared with the N group (Fig. 1A). miR-27b expression measured by
RT-qPCR was increased in the H group compared with the N group
(Fig. 1B). These results
indicated that hypoxia decreased PPARγ expression and increased
miR-27b expression in HPAECs.
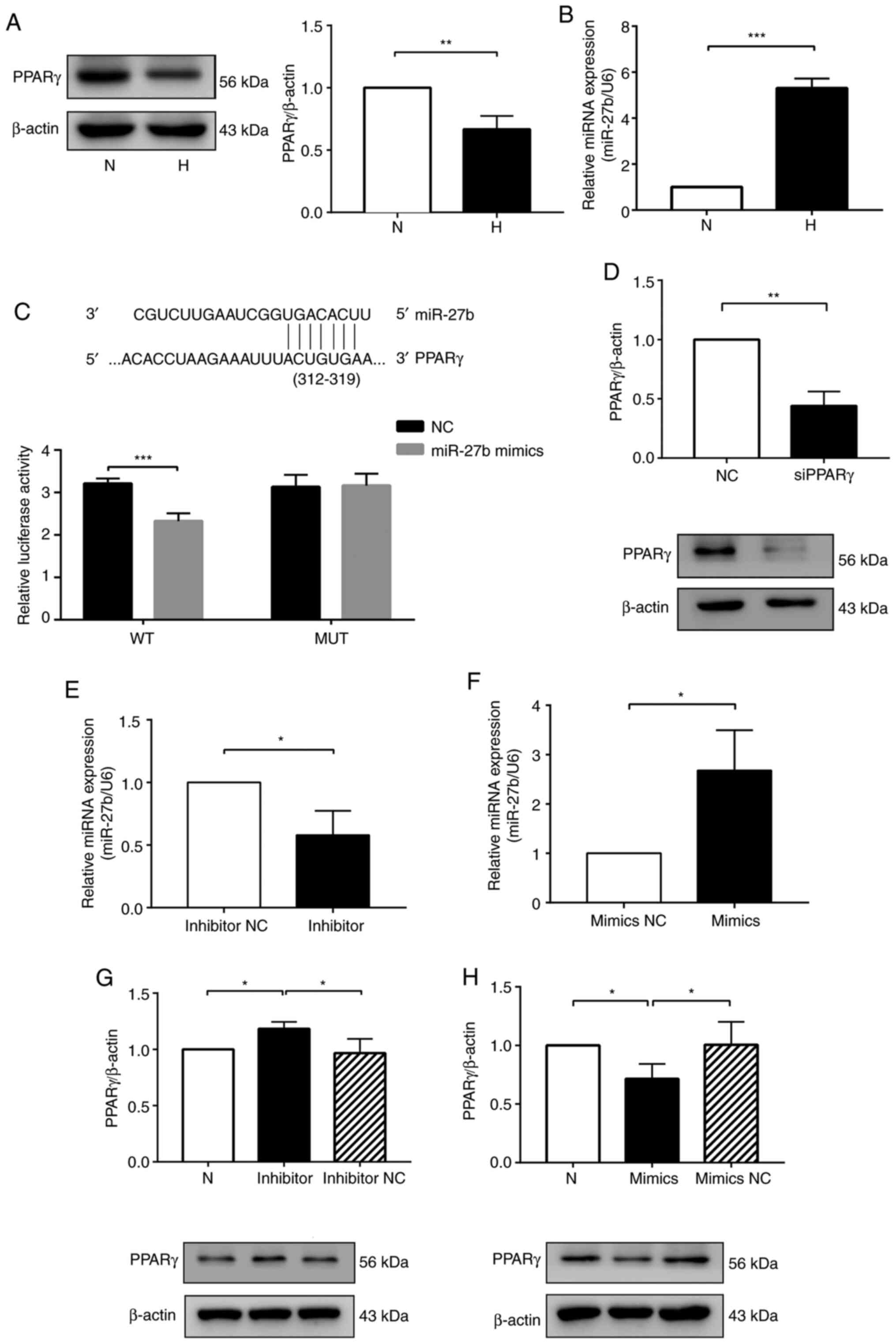 | Figure 1miR-27b suppresses PPARγ expression
by targeting the PPARγ gene. (A) Protein expression levels of PPARγ
were determined by western blot analysis. (B) Expression of miR-27b
was determined by RT-qPCR. (C) Predicted binding sites of miR-27b
matching the 3′-UTR of PPARγ. The luciferase activity was decreased
following treatment with a combination of miR-27b mimic and
PPARγ-3′-UTR-WT, suggesting that miR-27b regulated PPARγ. (D)
Western blot analysis to determine PPARγ siRNA efficiency. (E and
F) HPAECs were transfected with inhibitors NC or miR-27b
inhibitors, mimics NC or miR-27b mimics, and at 24 h following
transfection, the expression of miR-27b was determined by RT-qPCR.
(G and H) Western blot analysis of PPARγ protein levels in the
different groups. β-actin was used as a loading control. The
measurement data are presented as the mean ± standard deviation and
analyzed by one-way analysis of variance. The experiment was
performed in triplicate. *P<0.05;
**P<0.01; ***P<0.001, vs. the
respective control. RT-qPCR, reverse transcription-quantitative
polymerase chain reaction; WT, wild-type; MUT, mutant-type; UTR,
untranslated region; NC, negative control; N, normoxia + NC group;
H, hypoxia + NC group; PPARγ, peroxisome proliferator-activated
receptor γ; HPAECs, human pulmonary arterial endothelial cells. |
miR-27b supresses PPARγ expression by
targeting the PPARγ gene
Based on a bioinformatics database TargetScan,
miR-27b was predicted to target PPARγ mRNA (Fig. 1C). Therefore, a luciferase
reporter gene assay was performed to examine the direct interaction
between PPARγ and miR-27b. The results revealed that miR-27b mimic
bound to the PPARγ 3′-UTR compared with miR-27b mimics NC + PPARγ
3′-UTR [NC + wild-type (WT)], markedly inhibiting the fluorescence
expression of PPARγ. By contrast, the fluorescence level was
unaffected when the cells were co-transfected with the MUT-PPARγ
3′-UTR vector and miR-27b mimic (Fig. 1C). These data thus indicated that
miR-27b directly targeted PPARγ by binding to its 3′-UTR.
Subsequently, the present study explored the
regulatory effects of miR-27b on PPARγ. PPARγ siRNA was used to
downregulate the expression of PPARγ (Fig. 1D). The knockdown of miR-27b was
achieved by transfecting the HPAECs with miR-27b inhibitor, and
transfection with miR-27b inhibitor markedly reduced miR-27b
expression in the HPAECs compared with the inhibitor NC group
(Fig. 1E). Moreover, the
downregulation of miR-27b markedly promoted the PPARγ protein
expression levels in the hypoxia-exposed HPAECs compared with the
inhibitor NC group (Fig. 1G).
The overexpression of miR-27b was achieved by transfecting HPAECs
with miR-27b mimics, and transfection with miR-27b mimics
significantly upregulated miR-27b expression in the HPAECs compared
with the mimics NC group (Fig.
1F). Simultaneously, the decrease in the PPARγ protein
expression levels was detected in the HPAECs overexpressing miR-27b
(Fig. 1H).
Furthermore, the results of immunofluorescence
staining indicated that the fluorescence intensity of PPARγ was
markedly downregulated in the H group compared with the N group.
However, the fluorescence intensity of PPARγ was significantly
upregulated in the H + I and H + Pio groups compared with the H
group. The fluorescence intensity of PPARγ was decreased in the H +
I + siPPARγ group compared with the H + I group (Fig. 2A and B). Simultaneously, the
results of western blot analysis demonstrated that the protein
expression of PPARγ was markedly decreased in the H group compared
with the N group. Compared with the H group, the H + I and H + Pio
groups exhibited a higher PPARγ level, whereas an opposite trend
was observed in the H + I + siPPARγ group (Fig. 2C). Overall, these results
illustrated that miR-27b inhibited PPARγ expression via targeting
the PPARγ gene.
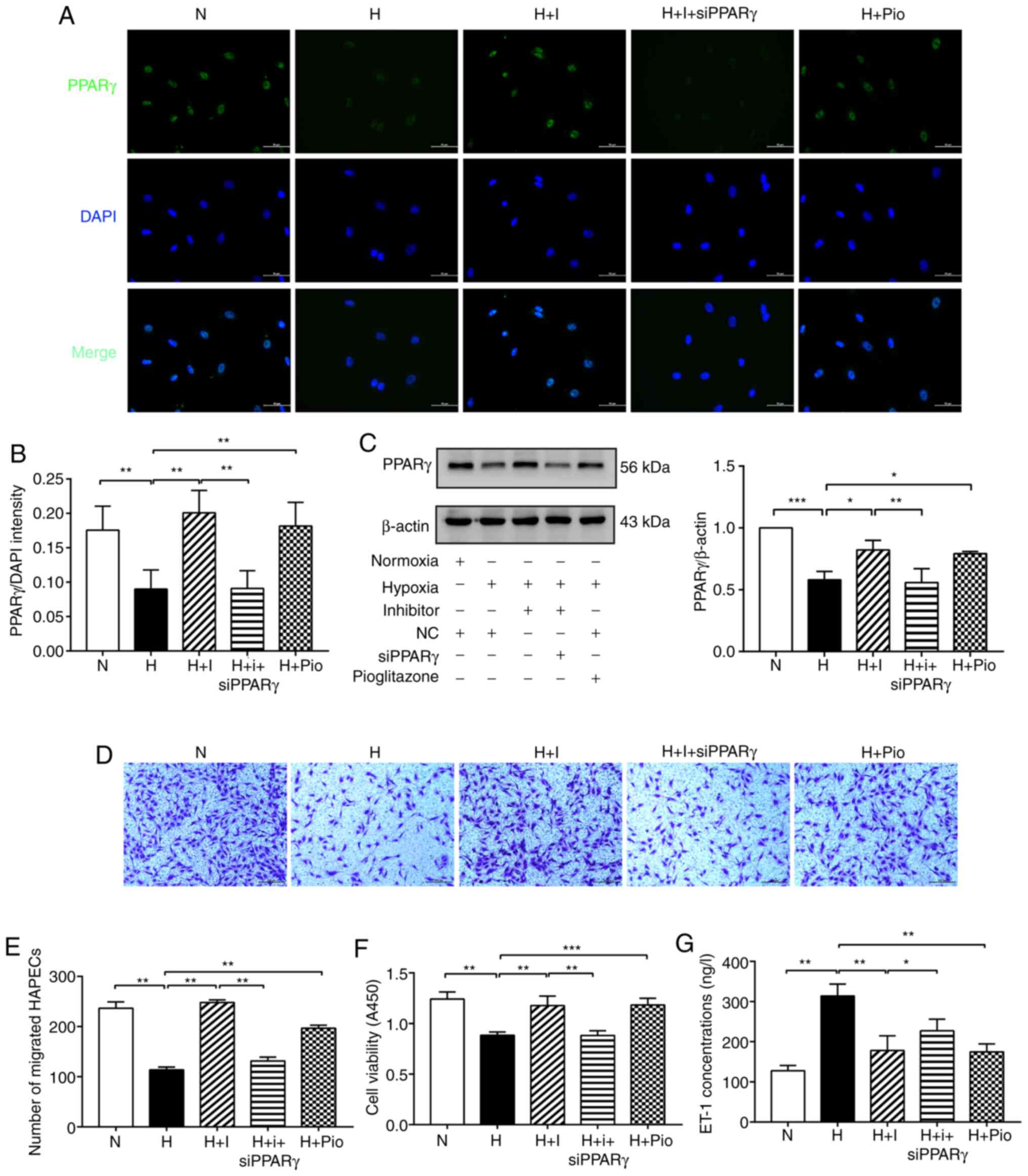 | Figure 2miR-27b aggravates dysfunction of
hypoxia-exposed HPAECs by targeting PPARγ. (A and B) Expression of
PPARγ (green) was detected by immunofluorescence staining, and the
fluorescence intensity of PPARγ was calculated; DAPI was used to
stain cell nuclei (blue). Scale bars, 50 µm (magnification,
×400). (C) Protein levels of of PPARγ was determined by western
blot analysis. β-actin was used as a loading control. (D and E)
Cell migration was assessed using Transwell migration chambers.
Scale bars, 200 µm (magnification, ×100). (F) Cell viability
was analyzed by CCK-8 assay. (G) The concentration of ET-1 was
assayed by ELISA. The measurement data are presented as mean ±
standard deviation and analyzed by one-way analysis of variance.
The experiment was performed in triplicate. *P<0.05;
**P<0.01; ***P<0.001, vs. the
respective control. NC, negative control; N, normoxia + NC group;
H, hypoxia + NC group; H + I, hypoxia + miR-27b inhibitor (100 nM)
group;) H + I + siPPARγ, hypoxia + miR-27b inhibitor + siPPARγ; H +
Pio: Hypoxia + pioglitazone (PPARγ agonist, 6.25 µmol/l) +
NC group; DAPI, 4,6-diamidino-2-phenylindole; CCK-8, cell counting
kit 8; ELISA, enzyme-linked immunosorbent assay; ET-1,
endothelin-1; PPARγ, peroxisome proliferator-activated receptor γ;
HPAECs, human pulmonary arterial endothelial cells. |
miR-27b aggravates hypoxia-induced HPAEC
dysfunction by targeting PPARγ
Transwell assay was used to determine relative cell
numbers and viability so as to assess hypoxia-induced HPAEC cell
migratory ability and its regulation by miR-27b. The migratory
ability of the HPAECs was decreased in the H group compared with
the N group, and was markedly increased in the H + I and H + Pio
groups compared with the H group, suggesting that the
downregulation of miR-27b and the overexpression of PPARγ had a
similar function in repairing the cell migratory ability. However,
a significant reduction in the number of migrated cells was
observed in the H + I + siPPARγ group compared with the H + I group
(Fig. 2D and E). Subsequently,
the present study examined the effects of miR-27b on cell viability
by CCK-8 assay. As regards the results of cell viability shown in
Fig. 2F, the absorbance was
reduced in the H group compared with the N group. The absorbance
was markedly enhanced in the H + I and H + Pio groups compared with
the H group. However, this change was markedly diminished in the H
+ I + siPPARγ group.
ET-1 is a marker of endothelial dysfunction. The
secretion of ET-1 was increased by hypoxia in the H group compared
with the N group. ET-1 secretion was significantly decreased in the
H + I and H + Pio groups compared with the H group; however, the
secretion of ET-1 was upregulated in the H + I + siPPARγ group
(Fig. 2G). Hence, these findings
suggested that miR-27b exacerbated hypoxia-induced HPAEC
dysfunction through the PPARγ gene.
miR-27b enhances hypoxia-induced HPAEC
inflammation by targeting PPARγ
The relative levels of p-NF-κB and NF-κB expression
in the different groups of HPAECs were determined by western blot
analysis to elucidate the molecular mechanisms underlying the
effects of miR-27b on PAH-related inflammation. The results
revealed that the ratio of p-NF-κB/NF-κB was markedly increased in
the H group compared with the N group. The ratio of p-NF-κB/NF-κB
was decreased in the H + I and H + Pio groups compared with the H
group; however, an adverse trend was observed in the H + I +
siPPARγ group (Fig. 3A). The
nuclear translocation of NF-κB was then examined by
immunofluorescence. The results revealed that hypoxia enhanced the
translocation of NF-κB protein into the nucleus, whereas this
translocation was inhibited in the H + I and H + Pio groups.
However, this inhibitory effect was abolished in the H + I +
siPPARγ group (Fig. 3B and
C).
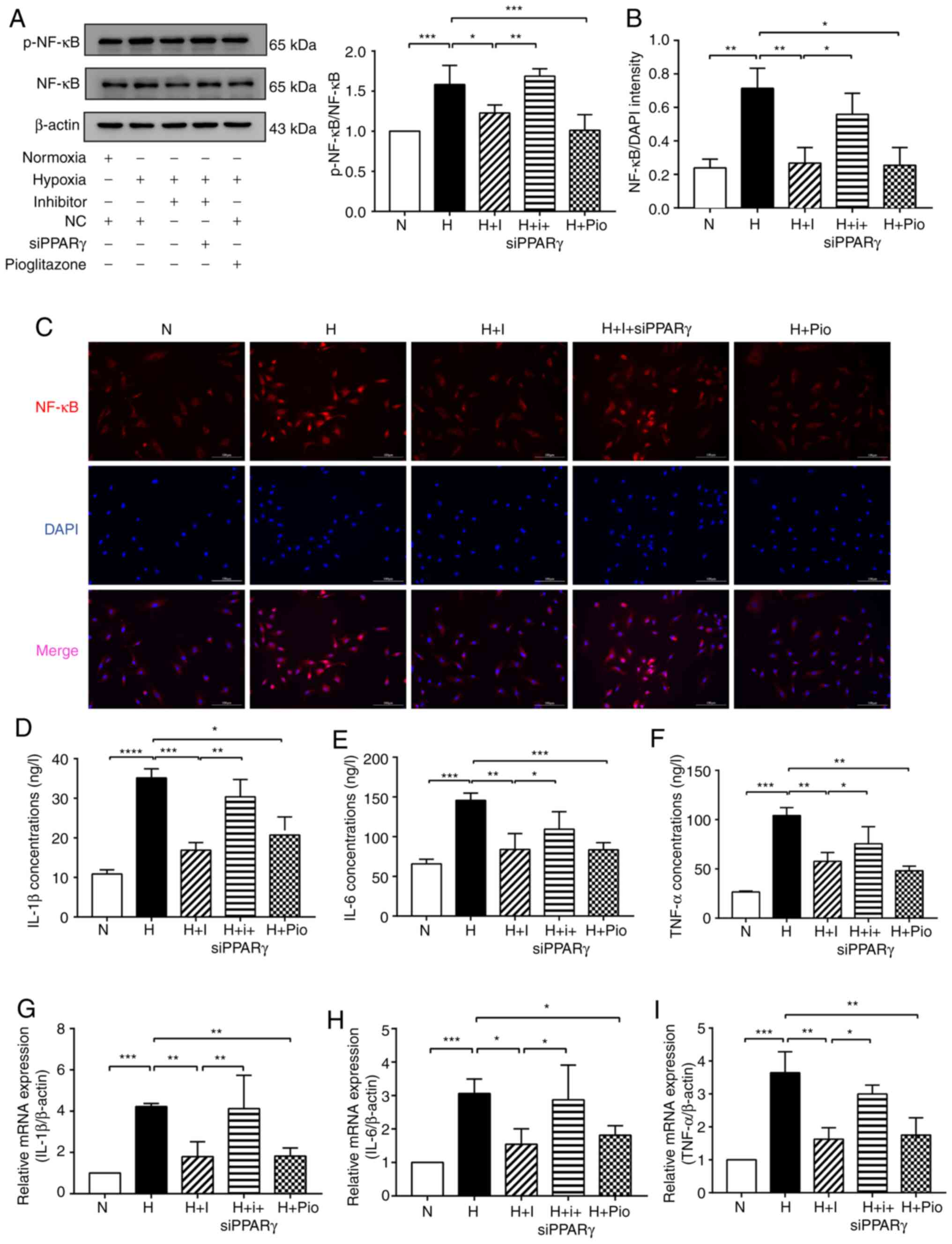 | Figure 3miR-27b pomotes hypoxia-induced HPAEC
inflammation by targeting PPARγ. (A) Protein levels of p-NF-κB and
NF-κB were detected by western blot analysis. β-actin was used as a
loading control. (B and C) Immunofluorescence staining was used to
detect NF-κB (red) entering the nucleus. DAPI was used to stain
cell nuclei (blue). Scale bars, 100 µm (magnification,
×200). (D-F) The expression levels of IL-1β, IL-6 and TNF-α were
analyzed using ELISA. (G-I) The expression levels of IL-1β, IL-6
and TNF-α were detected by RT-qPCR. The measurement data are
presented as mean ± standard deviation and analyzed using one-way
analysis of variance. The experiment was performed in triplicate.
*P<0.05; **P<0.01;
***P<0.001, vs. the respective control. NC, negative
control; N, normoxia + NC group; H, hypoxia + NC group; H + I,
hypoxia + miR-27b inhibitor (100 nM) group;) H + I + siPPARγ,
hypoxia + miR-27b inhibitor + siPPARγ; H + Pio: Hypoxia +
pioglitazone (PPARγ agonist, 6.25 µmol/l) + NC group; DAPI,
4,6-diamidino-2-phenylindole; ELISA, enzyme-linked immunosorbent
assay. RT-qPCR, reverse transcription-quantitative polymerase chain
reaction; PPARγ, peroxisome proliferator-activated receptor γ;
HPAECs, human pulmonary arterial endothelial cells. |
Subsequently, the levels of inflammatory factors,
such as IL-1β, IL-6 and TNF-α, as secreted cytokines, were examined
by ELISA, as this may better reflect their expression in the
medium. The results indicated that markedly higher levels of IL-1β,
IL-6 and TNF-α were found in the culture medium of HPAECs exposed
to hypoxia. The levels of IL-1β, IL-6 and TNF-α were significantly
decreased in the H + I and H + Pio groups, whereas this decreasing
tendency was reversed in the H + I + siPPARγ (Fig. 3D-F). Similarly, RT-qPCR analysis
revealed that the mRNA expression levels of IL-1β, IL-6 and TNF-α
in the hypoxia-exposed HPAECs were upregulated in the H group
compared with the N group, which was also normalized in the H + I
and H + Pio groups. However, the IL-1β, IL-6 and TNF-α expression
levels were decreased in the H + I + siPPARγ group (Fig. 3G-I). Taken together, these data
suggested that miR-27b promoted hypoxia-induced HPAEC inflammation
by targeting PPARγ.
FGF21 attenuates HPAEC dysfunction by
inhibiting the miR-27b/PPARγ pathway
The present study then examined the effect of FGF21
on the miR-27/PPARγ axis. Firstly, it was found that the expression
of FGF21 was decreased in the H group compared with the N group
(Fig. 4A), which indicated that
FGF21 may play a protective role in HPH. The results of RT-qPCR
indicated that the expression of miR-27b was decreased in the
hypoxia-exposed HPAECs following treatment with FGF21 (Fig. 4B). Moreover, the results of
immunofluorescence staining revealed that FGF21 significantly
increased the fluorescence intensity of PPARγ, which was
significantly decreased in the F + m group. (Fig. 4C and D). Consistent with the
aforementioned results, western blot analysis revealed that the
expression of PPARγ was significantly increased in the H + F group
compared with the H group. However, the agonistic effect of FGF21
on PPARγ expression was markedly abolished in the H + F + m group
(Fig. 4E). These results
indicated that FGF21 reversed the hypoxia-mediated upregulation of
miR-27b, which attenuated the downregulated expression of PPARγ
induced by miR-27b overexpression.
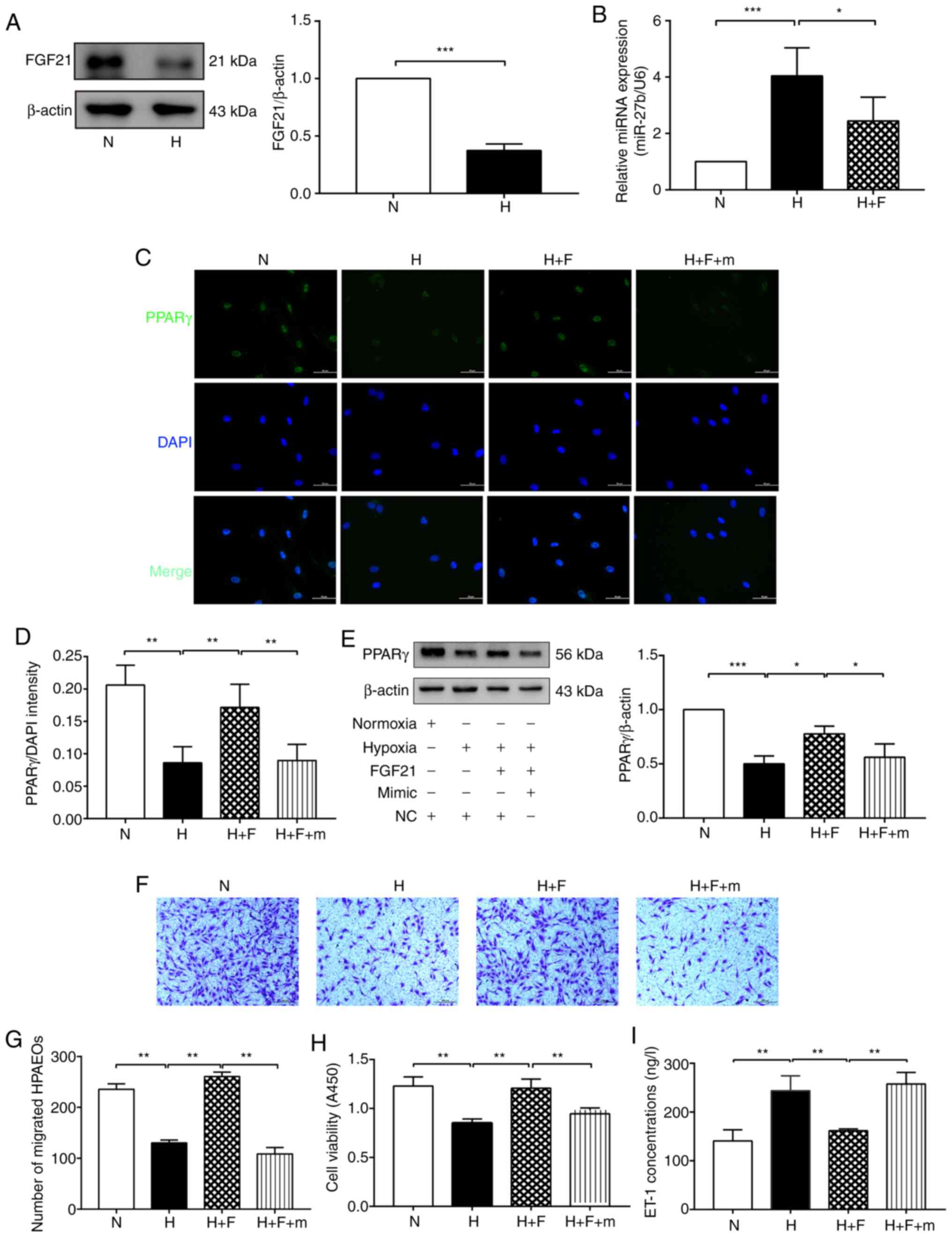 | Figure 4FGF21 attenuates hypoxia-induced
dysfunction of HPAECs through the miR-27b/PPARγ axis. (A) Protein
levels of of FGF21 was determined by western blot analysis. β-actin
was used as a loading control. (B) RT-qPCR was used to determine
the expression of miR-27b. (C and D) The expression of PPARγ
(green) was detected by immunofluorescence staining, and the
fluorescence intensity of PPARγ was calculated. DAPI was used to
stain cell nuclei (blue). Scale bars, 50 µm (magnification,
×400). (E) Protein levels of of PPARγ was determined by western
blot analysis. β-actin was used as a loading control (F and G) Cell
migration was assessed using Transwell migration chambers. Scale
bars, 200 µm (magnification, ×100). (H) Cell viability was
analyzed by CCK-8 assay. (I) The concentration of ET-1 was assayed
using ELISA. The measurement data were presented as mean ± standard
deviation and analyzed using one-way analysis of variance. The
experiment was performed in triplicate. *P<0.05;
**P<0.01; ***P<0.001, vs. the
respective control. N, normoxia + NC group;) H, hypoxia + NC group;
H + F, hypoxia + FGF21 (50 ng/ml) + NC group; H + F + m, hypoxia +
FGF21 + miR-27b mimic (50 nM) group; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; DAPI,
4,6-diamidino-2-phenylindole; CCK-8, cell counting kit-8; ELISA,
enzyme-linked immunosorbent assay; ET-1, endothelin-1; NC, negative
control; PPARγ, peroxisome proliferator-activated receptor γ;
HPAECs, human pulmonary arterial endothelial cells; FGF21,
fibroblast growth factor 21. |
Furthermore, CCK-8 and Transwell assays, as well as
ELISA were used to examine the effect of FGF21 on the dysfunction
of hypoxia-exposed HPAECs via the miR-27b/PPARγ axis. FGF21
markedly promoted cell viability and migration under hypoxic
conditions; however, this change was abolished in the H + F + m
group (Fig. 4F-H). As regards
ET-1 secretion, the ET-1 level was markedly reduced following FGF21
treatment, whereas the inhibitory effects of FGF21 on ET-1
secretion were reversed in the H + F + m group (Fig. 4I). The aforementioned data
illustrated that FGF21 inhibited the expression of miR-27b, thereby
promoting PPARγ expression and consequently alleviating
hypoxia-induced HPAEC dysfunction.
FGF21 alleviates hypoxia-induced
inflammation in HPAECs through the miR-27b/PPARγ axis
HPAECs were treated with FGF21 prior to exposure to
hypoxia to further examine the functional antagonistic effect of
FGF21 on hypoxia-induced inflammation. The results of western blot
analysis indicated that the ratio of p-NF-κB/NF-κB was markedly
decreased in the H + F group compared with the H group. However,
this change was reversed in the H + F + m group (Fig. 5A). The results of
immunofluorescence staining also revealed that FGF21 inhibited the
translocation of NF-κB protein into the nucleus in the
hypoxia-exposed HPAECs. However, the inhibitory effects of FGF21 on
NF-κB nuclear translocation were abolished in the H + F + m group
(Fig. 5B and C). The results of
ELISA revealed that FGF21 treatment substantially inhibited the
expression of IL-1β, IL-6 and TNF-α in the culture medium of
hypoxia-exposed HPAECs, whereas this phenomenon was significantly
reversed in the H + F + m group (Fig. 5D-F). Similarly, RT-qPCR analysis
revealed that FGF21 reduced the mRNA expression of TNF-α and IL-1β
in the hypoxia-exposed HPAECs, whereas this effect was abolished in
the H + F + m group (Fig. 5G-I).
Collectively, these results demonstrated that FGF21 reversed
hypoxia-induced miR-27b upregulation, which attenuated the
miR-27b-mediated reduction in PPARγ expression and thereby
activated PPARγ to attenuate HPAEC inflammation under hypoxic
conditions.
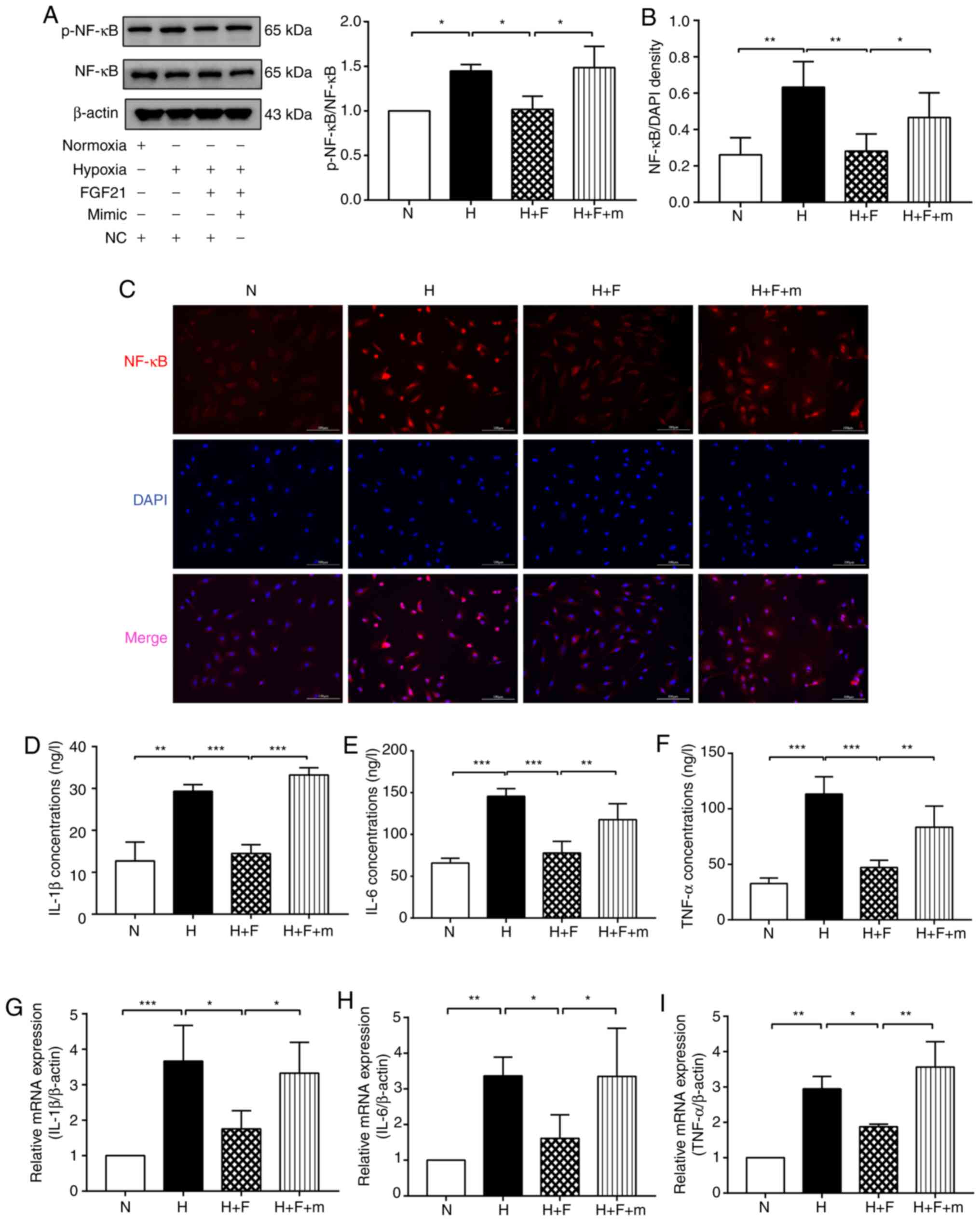 | Figure 5FGF21 suppresses the NF-κB signaling
pathway and the inflammatory response in hypoxia-exposed HPAECs via
the miR-27b/PPARγ pathway. (A) Protein levels of p-NF-κB and NF-κB
were detected by western blot analysis. β-actin was used as a
loading control. (B and C) Immunofluorescence staining was used to
detect NF-κB (red) entering the nucleus. DAPI was used to stain
cell nuclei (blue). Scale bars, 100 µm (magnification, 200).
(D-F) The expression levels of IL-1β, IL-6 and TNF-α were analyzed
using ELISA. (G-I) The expression levels of IL-1β, IL-6 and TNF-α
were detected by RT-qPCR. The measurement data are presented as the
mean ± standard deviation and analyzed by one-way analysis of
variance. The experiment was performed in triplicate.
*P<0.05; **P<0.01;
***P<0.001, vs. the respective control. N, normoxia +
NC group;) H, hypoxia + NC group; H + F, hypoxia + FGF21 (50 ng/ml)
+ NC group; H + F + m, hypoxia + FGF21 + miR-27b mimic (50 nM)
group; RT-qPCR, reverse transcription-quantitative polymerase chain
reaction; DAPI, 4,6-diamidino-2-phenylindole; ELISA, enzyme-linked
immunosorbent assay; NC, negative control; PPARγ, peroxisome
proliferator-activated receptor γ; HPAECs, human pulmonary arterial
endothelial cells; FGF21, fibroblast growth factor 21. |
Discussion
PAH is a vascular disorder with a high morbidity and
mortality due to the limited treatment methods available. Recent
studies have indicated that PAH os closely related to endothelial
dysfunction and inflammation (24,25).
Previously, PPARγ was reported to reduce ET-1 and
endothelial dysfunction in sickle cell disease-associated PH
(SCD-PH) (26). In another
study, in a rat model of SU5416/hypoxia (SuHx), oral treatment with
the PPARγ agonist, pioglitazone, completely reversed severe PAH and
vascular remodeling and prevented right ventricular failure
(27). Moreover, PPARγ has been
demonstrated to suppress inflammatory cytokine levels in
hypoxia-exposed pulmonary artery smooth muscle cells (20). Recently, hypoxia-induced PH was
found to be associated with the reductions in PPARγ expression, and
the activation of PPARγ attenuated increased pulmonary arterial
pressure and right ventricular hypertrophy in hypoxia-exposed mice
(9). The aforementioned findings
suggest the protective role of PPARγ in PAH. The present study
extended these findings by providing evidence that hypoxia
decreased PPARγ expression and aggravated HPAEC dysfunction by
reducing cell viability and migration ability, and promoting the
secretion of ET-1. Moreover, the results indicated that hypoxia
activated the NF-κB signaling pathway and increased the expression
of inflammatory cytokines. However, all these pathological
processes were reversed by treatment with the PPARγ agonist,
pioglitazone. These results indicated that PPARγ attenuated
endothelial dysfunction and inflammation in hypoxia-exposed
HPAECs.
Recently, miR-27b has been suggested to be a key
biomarker in various diseases (28-30). miR-27b expression was increased
in hypoxia-exposed HPAECs in the present study, indicating that
miR-27b was involved in the mechanisms of PAH development. To date,
several studies have provided evidence that miR-27b participates in
the regulation of inflammatory reactions. For example, Huang et
al (31) demonstrated that
the inhibition of miR-27b attenuated LPS-induced acute lung injure
(ALI) via downregulating the NF-κB signaling pathway. In accordance
with previous findings (16),
the present study found that the downregulated expression of
miR-27b enhanced the viability of HPAECs, improved the cell
migratory ability and decreased the secretion of ET-1 under hypoxic
conditions. The present study found that the downregulated
expression of miR-27b ameliorated hypoxia-induced inflammation by
inhibiting the expression of NF-κB and inflammatory factors, which
was in accordance with previous results (31). A previous study revealed that
anti-miR-27b attenuated cardiac hypertrophy and dysfunction in a
mouse model of transverse aortic constriction by targeting PPARγ
(32). Notably, the present
study found that the inhibition of miR-27b reversed the decreased
expression of PPARγ in hypoxia-exposed HPAECs by targeting its
3′-UTR. Simultaneously, the effect of miR-27b inhibition on
hypoxia-exposed HPAECs was abolished on by transfection with
siPPARγ. Taken together, these data indicated that miR-27b promoted
hypoxia-exposed HPAEC dysfunction and inflammation via targeting
PPARγ.
FGF21, as an important endocrine regulator, has
recently been reported to prevent angiotensin II-induced
hypertension and vascular dysfunction in mice (33). Previous research has indicated
the important roles of FGFs in defining and regulating the
functions of some endocrine-relevant tissues and organs, as well as
modulating various metabolic processes (34). Apart from its effects on energy
metabolism, FGF21 has been shown to exert protective effects
against hypertension and vascular dysfunction (35). In the present study, miR-27b was
downregulated in hypoxia-exposed HPAECs following FGF21 treatment,
suggesting that FGF21 prevented PAH by regulating miR-27b. Based on
the aforementioned findings, HPAECs were treated with FGF21 prior
to hypoxia exposure to further verify whether FGF21 can protect
HPAECs against hypoxia-induced dysfunction via the miR-27b/PPARγ
axis. The present study indicated that FGF21 restored EC
dysfunction and inhibited the activation of the NF-κB pathway and
inflammatory cytokine levels in the hypoxic environment. However,
the overexpression of miR-27b blocked the protective effects of
FGF21 on the hypoxia-exposed HPAECs. These findings elucidated that
FGF21 ameliorated hypoxia-induced HPAEC dysfunction and
inflammation via decreasing miR-27b expression, thereby enhancing
PPARγ expression. The data further suggested that FGF21 prevented
PAH by promoting PPARγ expression.
In conclusion, the findings of the present study
suggested that miR-27b exacerbated hypoxia-exposed HPAEC
dysfunction and inflammation by inversely regulating PPARγ.
However, FGF21 abolished the effect of miR-27b on hypoxia-exposed
HPAECs, mainly by inhibiting miR-27b expression, thereby promoting
the expression of PPARγ. Due to limited conditions, the role of the
FGF21/miR-27b/PPARγ axis in vivo remains unclear and the
mechanisms through which FGF21 regulates miR-27b warrants further
investigation. Moreover, the effect of the miR-27b/PPARγ axis on
the apoptosis of hypoxia-exposed HPAECs needs to be further
explored in future studies. Overall, the present study provides
theoretical evidence for the basis of further clinical research for
the treatment of PAH.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DY and QH designed and performed the experiments. QH
analyzed the data and wrote the manuscript. XH, LW and YL designed
the experiments and assisted in the drafting of the manuscript. JS,
LC and JW performed the experiments and collected the data. GC and
JL collected the data and performed the analysis. DY and QH confirm
the authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the First Affiliated Hospital of Wenzhou Medical
University (Wenzhou, China) (approval no. 2020-232).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Project of Health
Department of Zhejiang Province of China (grant no. 2017171805),
the Natural Science Foundation Grants of Zhejiang Province (grant
no. Y17H010028), and the Chinese National Natural Science
Foundation Grants (grant no. 81873411).
References
|
1
|
Thompson AAR and Lawrie A: Targeting
vascular remodeling to treat pulmonary arterial hypertension.
Trends Mol Med. 23:31–45. 2017. View Article : Google Scholar
|
|
2
|
Lan NSH, Massam BD, Kulkarni SS and Lang
CC: Pulmonary arterial hypertension: Pathophysiology and treatment.
Diseases. 6:382018. View Article : Google Scholar :
|
|
3
|
Rafikova O, Al Ghouleh I and Rafikov R:
Focus on early events: Pathogenesis of pulmonary arterial
hypertension development. Antioxid Redox Signal. 31:933–953. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Baptista R, Meireles J, Agapito A, Castro
G, da Silva AM, Shiang T, Gonçalves F, Robalo-Martins S,
Nunes-Diogo A and Reis A: Pulmonary hypertension in Portugal: First
data from a nationwide registry. Biomed Res Int. 2013:4895742013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pelham CJ, Ketsawatsomkron P, Groh S,
Grobe JL, de Lange WJ, Ibeawuchi SR, Keen HL, Weatherford ET,
Faraci FM and Sigmund CD: Cullin-3 regulates vascular smooth muscle
function and arterial blood pressure via PPARγ and RhoA/Rho-kinase.
Cell Metab. 16:462–472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hansmann G and Zamanian RT: PPARgamma
activation: A potential treatment for pulmonary hypertension. Sci
Transl Med. 1:12ps142009. View Article : Google Scholar
|
|
7
|
Diebold I, Hennigs JK, Miyagawa K, Li CG,
Nickel NP, Kaschwich M, Cao A, Wang L, Reddy S, Chen PI, et al:
BMPR2 preserves mitochondrial function and DNA during reoxygenation
to promote endothelial cell survival and reverse pulmonary
hypertension. Cell Metab. 21:596–608. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Archer SL, Marsboom G, Kim GH, Zhang HJ,
Toth PT, Svensson EC, Dyck JR, Gomberg-Maitland M, Thébaud B,
Husain AN, et al: Epigenetic attenuation of mitochondrial
superoxide dismutase 2 in pulmonary arterial hypertension: A basis
for excessive cell proliferation and a new therapeutic target.
Circulation. 121:2661–2671. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cai G, Liu J, Wang M, Su L, Cai M, Huang
K, Li X, Li M, Wang L and Huang X: Mutual promotion of FGF21 and
PPARγ attenuates hypoxia-induced pulmonary hypertension. Exp Biol
Med (Maywood). 244:252–261. 2019. View Article : Google Scholar :
|
|
10
|
Small EM and Olson EN: Pervasive roles of
microRNAs in cardiovascular biology. Nature. 469:336–342. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tan H, Yao H, Lie Z, Chen G, Lin S and
Zhang Y: MicroRNA-30a-5p promotes proliferation and inhibits
apoptosis of human pulmonary artery endothelial cells under hypoxia
by targeting YKL-40. Mol Med Rep. 20:236–244. 2019.PubMed/NCBI
|
|
12
|
Zhao H, Guo Y, Sun Y, Zhang N and Wang X:
miR-181a/b-5p ameliorates inflammatory response in
monocrotaline-induced pulmonary arterial hypertension by targeting
endocan. J Cell Physiol. 235:4422–4433. 2020. View Article : Google Scholar
|
|
13
|
Zhao M, Chen N, Li X, Lin L and Chen X:
MiR-19a modulates hypoxia-mediated cell proliferation and migration
via repressing PTEN in human pulmonary arterial smooth muscle. Life
Sci. 239:1169282019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mondejar-Parreño G, Callejo M, Barreira B,
Morales-Cano D, Esquivel-Ruiz S, Filice M, Moreno L, Cogolludo A
and Perez-Vizcaino F: miR-1 induces endothelial dysfunction in rat
pulmonary arteries. J Physiol Biochem. 75:519–529. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rong X, Ge D, Shen D, Chen X, Wang X,
Zhang L, Jia C, Zeng J, He Y, Qiu H, et al: miR-27b suppresses
endothelial cell proliferation and migration by targeting Smad7 in
kawasaki disease. Cell Physiol Biochem. 48:1804–1814. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bi R, Bao C, Jiang L, Liu H, Yang Y, Mei J
and Ding F: MicroRNA-27b plays a role in pulmonary arterial
hypertension by modulating peroxisome proliferator-activated
receptor γ dependent Hsp90-eNOS signaling and nitric oxide
production. Biochem Biophys Res Commun. 460:469–475. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Geng L, Lam KSL and Xu A: The therapeutic
potential of FGF21 in metabolic diseases: From bench to clinic. Nat
Rev Endocrinol. 16:654–667. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen A, Liu J, Zhu J, Wang X, Xu Z, Cui Z,
Yao D, Huang Z, Xu M, Chen M, et al: FGF21 attenuates
hypoxia-induced dysfunction and apoptosis in HPAECs through
alleviating endoplasmic reticulum stress. Int J Mol Med.
42:1684–1694. 2018.PubMed/NCBI
|
|
19
|
Yu Y, He J, Li S, Song L, Guo X, Yao W,
Zou D, Gao X, Liu Y, Bai F, et al: Fibroblast growth factor 21
(FGF21) inhibits macrophage-mediated inflammation by activating
Nrf2 and suppressing the NF-κB signaling pathway. Int
Immunopharmacol. 38:144–152. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu J, Cai G, Li M, Fan S, Yao B, Ping W,
Huang Z, Cai H, Dai Y, Wang L and Huang X: Fibroblast growth factor
21 attenuates hypoxia-induced pulmonary hypertension by
upregulating PPARγ expression and suppressing inflammatory cytokine
levels. Biochem Biophys Res Commun. 504:478–484. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo Y, Luo F, Yi Y and Xu D: Fibroblast
growth factor 21 potentially inhibits microRNA-33 expression to
affect macrophage actions. Lipids Health Dis. 15:2082016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang X, Mao W, Zhang T, Wang M, Wang X,
Li Y, Zhang L, Yao D, Cai X and Wang L: Baicalin promotes apoptosis
and inhibits proliferation and migration of hypoxia-induced
pulmonary artery smooth muscle cells by up-regulating A2a receptor
via the SDF-1/CXCR4 signaling pathway. BMC Complement Altern Med.
18:3302018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Cao Y, Zhang X, Wang L, Yang Q, Ma Q, Xu
J, Wang J, Kovacs L, Ayon RJ, Liu Z, et al: PFKFB3-mediated
endothelial glycolysis promotes pulmonary hypertension. Proc Natl
Acad Sci USA. 116:13394–13403. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ban Y, Liu Y, Li Y, Zhang Y, Xiao L, Gu Y,
Chen S, Zhao B, Chen C and Wang N: S-nitrosation impairs KLF4
activity and instigates endothelial dysfunction in pulmonary
arterial hypertension. Redox Biol. 21:1010992019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kang BY, Park K, Kleinhenz JM, Murphy TC,
Sutliff RL, Archer D and Hart CM: Peroxisome proliferator-activated
receptor γ regulates the V-Ets avian erythroblastosis virus E26
oncogene homolog 1/microRNA-27a axis to reduce endothelin-1 and
endothelial dysfunction in the sickle cell mouse lung. Am J Respir
Cell Mol Biol. 56:131–144. 2017. View Article : Google Scholar :
|
|
27
|
Legchenko E, Chouvarine P, Borchert P,
Fernandez-Gonzalez A, Snay E, Meier M, Maegel L, Mitsialis SA,
Rog-Zielinska EA, Kourembanas S, et al: PPARγ agonist pioglitazone
reverses pulmonary hypertension and prevents right heart failure
via fatty acid oxidation. Sci Transl Med. 10:eaao03032018.
View Article : Google Scholar
|
|
28
|
Hannafon BN, Cai A, Calloway CL, Xu YF,
Zhang R, Fung KM and Ding WQ: miR-23b and miR-27b are oncogenic
microRNAs in breast cancer: Evidence from a CRISPR/Cas9 deletion
study. BMC Cancer. 19:6422019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang X, Chen J, Liao Y, Huang L, Wen C,
Lin M, Li W, Zhu Y, Wu X, Iwamoto A, et al: MiR-27b-3p promotes
migration and invasion in colorectal cancer cells by targeting
HOXA10. Biosci Rep. 39:BSR201910872019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen J, Wang Y, Du G, Zhang W, Cao T, Shi
L, Wang Y, Mi J and Tang G: Down-regulation of miRNA-27b-3p
suppresses keratinocytes apoptosis in oral lichen planus. J Cell
Mol Med. 23:4326–4337. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang Y, Huang L, Zhu G, Pei Z and Zhang
W: Downregulated microRNA-27b attenuates lipopolysaccharide-induced
acute lung injury via activation of NF-E2-related factor 2 and
inhibition of nuclear factor κB signaling pathway. J Cell Physiol.
234:6023–6032. 2019. View Article : Google Scholar
|
|
32
|
Wang J, Song Y, Zhang Y, Xiao H, Sun Q,
Hou N, Guo S, Wang Y, Fan K, Zhan D, et al: Cardiomyocyte
overexpression of miR-27b induces cardiac hypertrophy and
dysfunction in mice. Cell Res. 22:516–527. 2012. View Article : Google Scholar :
|
|
33
|
Pan X, Shao Y, Wu F, Wang Y, Xiong R,
Zheng J, Tian H, Wang B, Wang Y, Zhang Y, et al: FGF21 prevents
angiotensin II-induced hypertension and vascular dysfunction by
activation of ACE2/angiotensin-(1-7) axis in mice. Cell Metab.
27:1323–1337.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kharitonenkov A, Shiyanova TL, Koester A,
Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers
JS, Owens RA, et al: FGF-21 as a novel metabolic regulator. J Clin
Invest. 115:1627–1635. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ruan CC, Kong LR, Chen XH, Ma Y, Pan XX,
Zhang ZB and Gao PJ: A2A receptor activation attenuates
hypertensive cardiac remodeling via promoting brown adipose
tissue-derived FGF21. Cell Metab. 28:476–489.e5. 2018. View Article : Google Scholar
|














