Introduction
Hyperglycemia is a major risk factor for ischemic
stroke in the clinic and increases the risk of mortality and
morbidity in patients suffering from stroke (1). The Emerging Risk Factors
Collaboration showed that the adjusted hazard ratio for patients
with with diabetes was 2.27 (1.95-2.65) for ischemic stroke
(2). Animal studies have
confirmed that hyperglycemia enlarges cerebral infarction volume
and causes hemorrhagic transformation of the infarct area,
resulting in a poorer prognosis (3,4).
The mechanism of hyperglycemia aggravated ischemic brain injury may
involve lactic acidosis, oxidative stress and activation of cell
death pathways (5,6). Our previous studies have
demonstrated that imbalance of mitochondrial fission/fusion and
decreased mitochondrial biogenesis contribute to
hyperglycemia-enhanced ischemic brain damage (7-9).
The neurovascular unit (NVU), with the blood-brain
barrier (BBB) as its core, emphasizes the close link between brain
tissue and its blood supply (10). The BBB is a structural and
functional barrier between the central nervous system (CNS) and the
systemic blood circulation. It prevents harmful substances from
entering the brain parenchyma and regulates the transport of water,
ions and macromolecules, thus maintaining the stability of brain
homeostasis (11). The major
components of the BBB include vascular endothelial cells
interconnected with tight junctions (TJ), basement membrane (BM)
and astrocytic endfeet. TJs are located between adjacent vascular
endothelial cells, where they play a key role in limiting
paracellular permeability, thus regulating permeability of the BBB.
Three types of transmembrane proteins primarily constitute the TJ:
Claudin, occludin and zonula occluden-1 (ZO-1) (12). Claudin is the principal component
of the TJ complex and is involved in selective permeability and
cellular polarization (13). It
is considered to be the most important regulator of cerebrovascular
endothelial cell permeability (14). There are different subtypes of
claudin. Claudin-5 is the primary transmembrane protein that forms
the BBB (15). Miyamori et
al (16) observed that in
the late stage of cerebral ischemia/reperfusion (I/R), claudin-5
migrates from endothelial cells to surrounding astrocytes, which
disrupts the BBB. Occludin is considered as a staple of TJs between
blood endothelial cells. Occludin expression in cerebrovascular
endothelial cells is suppressed and integrity of the BBB is
compromised following cerebral I/R (17). This suggests that decreased
expression of occludin can be used as an indicator of BBB
disruption. ZO-1 is the first confirmed TJ adhesion protein
(18) and is used as a marker
for BBB damage. It connects transmembrane proteins (occludin and
claudins) with cytoskeletal proteins on the inner side of the cell
(such as actin), thus serving a key role in TJ (18). During cerebral
ischemia/reperfusion (I/R), expression of ZO-1 decreases
significantly, resulting in degradation of TJ, increased BBB
permeability and formation of vasogenic cerebral edema (19). BM is located at the basal side of
epithelial cells and is composed of laminin, collagen IV, nidogen
and heparan sulfate proteoglycans (20-22). The role of BM ranges from
providing structural support to modulating molecular signaling and
maintaining normal function of the BBB (23,24). Loss of BM occurs soon after the
onset of ischemia (25), or as
early as 10 min post-reperfusion in a middle cerebral artery
occlusion (MCAO) model (26).
Astrocytic endfeet cover almost the entire surface of
intraparenchymal capillaries in the adult brain and are involved in
the formation and maintenance of the BBB (27). In ischemic brain, astrocytes are
activated, reflected by increased numbers of dendrites and
enlargement of the cell body (28). Moreover, swelling of astrocytic
endfeet is one of the earliest morphological events following
ischemic stroke (29). Although
several studies have shown that hyperglycemia significantly
increases brain edema and BBB dysfunction, as shown by Evans blue
leakage following I/R injury (30-32), detailed morphological study of
the BBB has not previously been performed.
Microglia, macrophages, neutrophils and T
lymphocytes are the most well-studied immune cells in the CNS that
interact directly or indirectly with BBB components and affect the
integrity of the BBB following ischemia (33). Microglial cells, the resident
immune cells in the brain, are among the first responders to
ischemia, followed by neutrophil influx and infiltration of
peripheral macrophages, lymphocytes and dendritic cells (34,35). Activated microglia serve dual
role in the BBB and ischemic brain damage. On one hand, they
produce excessive cytokines and chemokines that overexpress cell
adhesion molecules and promote leukocyte infiltration (36). On the other hand, activated
microglia may be beneficial to the injured brain by phagocytizing
cell debris and inhibiting the inflammatory response (33). In addition, damage to the BBB
permits extravasation of neutrophils to the brain parenchyma, which
increases the release of deleterious inflammatory mediators.
Accumulation of neutrophils, along with proinflammatory mediators
and proteases, also potentiates junction disassembly, endothelial
malfunction, and extracellular matrix degradation, resulting in
irreversible BBB disruption (37). The effect of hyperglycemia on
activation of microglia and neutrophil infiltration is in need of
further investigation.
The aim of the present study was to investigate the
effect of preischemic hyperglycemia on key components of the BBB
following I/R injury in the rat brain. To this end, streptozotocin
(STZ)-induced hyperglycemic (HG) rats were subjected to 30 min
MCAO, followed by 1, 3 or 7 days reperfusion. Levels of TJ and
BM-associated proteins. as well as changes in astrocytes and
microglia and neutrophil infiltration, were assessed using
immunohistochemistry (IHC) and/or western blotting.
Materials and methods
Animals
A total of 95 male 8-week-old Sprague-Dawley rats
(weight, 200-230 g) were provided by the Experimental Animal Center
of Ningxia Medical University (Yinchuan China). The animals were
reared in a specific-pathogen-free environment with a 12-h
light/dark cycle and free access to water and food, the temperature
and humidity were controlled (23°C, 55% humidity). Animal
procedures were performed in strict accordance with the Chinese
Laboratory Animal Use Regulation and were approved by the
Institutional Animal Care and Use Committee of Ningxia Medical
University.
STZ-induced diabetic hyperglycemia
A total of 95 rats were fasted for 12 h and
intraperitoneally injected with STZ (60 mg/kg) freshly dissolved in
citrate buffer (pH 4.5) or an equal amount of solvent as a control.
Blood glucose levels were measured 3 days after STZ injection. Rats
with blood glucose >16.7 mM were placed in the HG group. The rat
body weight and blood glucose levels were measured once/week for 4
weeks Cerebral ischemia was induced after 4 weeks in STZ-induced
diabetic and citrate buffer-injected normoglycemic (NG) rats.
Animals in the NG and HG groups were further divided into Sham, 1-,
3- and 7 day I/R (I/R 1, 3 and 7 day) subgroups. Animal groups and
number of animals in each group are given in Table I.
 | Table IAnimal groups and numbers. |
Table I
Animal groups and numbers.
A, Normoglycemic
|
|---|
| Group | TTC staining TTC
staining | IHC | WB |
|---|
| Sham | 5 | 5 | 3 |
| I/R 1 day | 6 | 7 | 4 |
| I/R 3 days | 0 | 6 | 4 |
| I/R 7 days | 0 | 5 | 3 |
|
B, Hyperglycemic
|
| Group | TTC staining | IHC | WB |
|
| Sham | 0 | 5 | 4 |
| I/R 1 day | 5 | 9 | 4 |
| I/R 3 days | 0 | 6 | 5 |
| I/R 7 days | 0 | 5 | 4 |
I/R model
MCAO was induced for 30 min by inserting an
intraluminal filament through the right common carotid artery and
advancing to the internal carotid artery and the base of the middle
cerebral artery. Reperfusion was achieved by withdrawing the
filament after 30 min occlusion. Neurological defects were scored
at 1 day post-I/R according to the modified Neurological Severity
Score (mNSS), which primarily assesses sensory, motor, reflex,
balance and muscle tone (35).
The higher the score, the more serious the neurological deficit.
Normal rats have score 0, and mild, moderate, and severe injuries
scores are 1-6, 7-12 and 3-18, respectively. The deficit score was
assessed 3 times in each rat and the mean value was calculated. At
the predetermined experimental end-points, the rats (including the
sham group) were euthanized and the brain was extracted. The brain
samples used for morphological studies were fixed in 4%
paraformaldehyde for 4 h at room temperature and embedded in
paraffin. The brain samples used for western blot analysis were
frozen in liquid nitrogen and stored in a −80°C freezer. Brain
edema was evaluated by dry-wet weight ratio as follows: W/D
ratio=(wet weight-dry weight)/dry weight
2,3,5-Triphenyltetrazolium chloride (TTC)
& Evans blue staining
The cerebral infarct volume and integrity of the BBB
were assessed by TTC and Evans blue staining, respectively, as
previously described (38).
Briefly, Evans blue dye (2%; 4 ml/kg) was injected into the left
femoral vein and allowed to circulate for 60 sec before the rats
were euthanized with 1% pentobarbital (40 mg/kg) and perfused
transcardially with PBS for 60 sec at 37°C. The brains were
removed, washed with PBS and kept at −20°C for 30 min before
sectioning. Coronal sections (2 mm) were cut in a rat brain matrix
and stained with 2% TTC (Amresco, LLC) for 30 min at 37°C.
Following staining, the brain slices were fixed with 4%
paraformaldehyde for 10 min at 37°C to conserve the area stained by
TTC. The stained brain sections were digitally photographed using a
digital camera (PowerShot G7 X Mark II Canon, Inc.). The infarcted
area (white) and BBB disruption (blue) of each brain section was
measured using Image-Pro Plus 6.0 software (Media Cybernetics,
Inc.). The infarcted volume and the BBB disrupted area were
calculated according to the formula: 100% × (ipsilateral
volume-contralateral volume)/contralateral volume.
Immunohistochemistry (IHC)
IHC staining was performed as described previously
(9). Briefly, the
4-μm-thick brain sections were de-paraffinized, rehydrated
(in descending alcohol series) and washed (in PBS). Antigen
retrieval was performed using a high-temperature (120°C) and
high-pressure antigen repairing method in sodium citrate buffer (pH
6.0). The sections were then blocked with 3%
H2O2 and 10% normal goat serum (OriGene
Technologies, Inc.) in PBS for 30 min at 37°C. The sections were
incubated overnight at 4°C with primary antibodies against von
Willebrand Factor (vWF; 1:300; cat. no. ab6994; Abcam), claudin-5
(1:100; cat. no. ab15106; Abcam), ZO-1 (1:200; cat. no. ab216880;
Abcam), occludin (1:200; cat. no. ab216327; Abcam), laminin (1:200;
cat. no. ab11575; Abcam), collagen IV (1:400; cat. no. ab6586;
Abcam), glial fibrillary acidic protein (GFAP; 1:200; cat. no.
3670S; Cell Signaling Technology, Inc.) and anti-neutrophil
antibody (NIMP-R14; 1:50; cat. no. ab2557; Abcam). The sections
were rewarmed for 60 min at room temperature and then incubated
with horseradish peroxidase (HRP)-conjugated secondary antibody
(1:300; cat. no. TA130003, OriGene Technologies, Inc.) for 30 min
at 37°C. Color reaction was developed by 3,3′-diaminobenzidine
(DAB) (1:20) incubation for 5 min and counterstained with
hematoxylin for 1 min, both at 37°C.
For IgG staining, after being de-paraffinized,
rehydrated and blocked as aforementioned, the sections were
incubated with HRP-conjugated Affinipure Goat Anti-Mouse IgG (H+L;
1:2,000; cat. no. SA00001-1; ProteinTech Group, Inc.) for 2 h at
37°C. The sections were washed, and color reaction was developed by
DAB (1:20) incubation for 5 min and counterstained with hematoxylin
for 1 min at 37°C. The images were captured using a light
microscope (cat. no. VM1000; Motic Incorporation, Ltd.;
magnification, x40), and the IgG-positive areas were measured using
Image-Pro-Plus 6.0 software (Media Cybernetics, Inc.).
The immunofluorescent double labeling of vWF (1:200;
cat. no. ab6994; Abcam), with astrocyte marker GFAP (1:200; cat.
no. 3670S; Cell Signaling Technology, Inc.) and double labeling of
proliferating cell nuclear antigen (PCNA; 1:400; cat. no. ab29;
Abcam) with the microglia marker ionized calcium-binding adaptor
molecule (IBA1) (1:200; cat. no. ab5076; Abcam) were achieved by
incubating the two primary antibodies with brain sections
separately for 4 h at 37°C and then with a mixture of two different
fluorescence-conjugated secondary antibodies for 2 h at 37°C. The
specimens were mounted with Antifade Polyvinylpyrrolidone Mounting
Medium (Vector Informatik GmbH) containing DAPI and then examined
under a fluorescence confocal-scanning microscope (cat. no. FV1000;
Olympus Corporation; magnification, x400).
Morphological analysis
Following immunostaining, astrocyte and microglia
morphology were evaluated using software (ImageJ; version
2.0.0-rc-69/1.52p; National Institutes of Health). Analysis of
dendrite branching features, including number of endpoints, total
segment length and maximum branch length, was performed using the
plugin Analyze Skeleton (fiji.sc/AnalyzeSkeleton) (39). Briefly, images were converted to
8-bit gray type and filtered with Fast Fourier Transform bandpass
filter plugin (imagej.nih.gov/ij/plugins/fft-filter.html). The
brightness and contrast were adjusted to show cell morphology and
noise was removed. The images were then skeletonized and the number
of endpoints, total segment length and maximum branch length were
measured automatically using the Analyze Skeleton plugin. The area
of IBA1+ stained cells and circularity index (CI) were
measured using the plugin Shape Descriptors (40). The CI parameter was calculated by
the Shape Descriptors plugin as follows:
Area=[4p(area)/(perimeter)2] (41). The numbers of vWF+,
GFAP+ and IBA1+ cells were counted using
Image-Pro-Plus 6.0 software (Media Cybernetics, Inc.).
Western blotting
Following reperfusion, ipsilateral brain tissue was
homogenized in RIPA lysis buffer containing protease inhibitors
(Nanjing KeyGen Biotech Co., Ltd.). The protein concentration of
the supernatants (24,148.8 × g, 5 min, 4°C) was measured using a
BCA Protein Quantitation Assay (Nanjing KeyGen Biotech Co., Ltd.).
Equal amounts of protein (30 μg) were loaded onto 8-10%
sodium dodecyl sulfate-polyacrylamide gels. Following
electrophoresis, bands were transferred onto PVDF membranes. After
being blocked with 5% BSA (OriGene Technologies, Inc.) for 2 h at
37°C, the membranes were incubated overnight at 4°C using the
aforementioned primary antibodies against claudin-5, ZO-1,
occludin, laminin and collagen IV or TNF-α (1:1,000; cat. no.
17590-1-AP; ProteinTech Group, Inc.), IL-1β (1:500; cat. no.
ab200478; Abcam) or IL-6 (1:2,000; cat. no. 66146-1-Ig; ProteinTech
Group, Inc.). The membranes were washed and incubated with
HRP-conjugated secondary antibody (1:2,000; cat. no. SA00013-2;
ProteinTech Group, Inc.) for 2 h at room temperature. Visualization
was performed using ECL kit (cat. no. 34577; Thermo Fisher
Scientific, Inc.). The immunoblots were visualized using a
computerized image analysis system (Amersham Imager 600; Cytiva),
and the results were expressed as the ratio of corresponding
protein to β-actin (1:5,000; cat. no. SA00001-7L; ProteinTech
Group, Inc.).
Statistical analysis
Data are presented as the mean ± SEM (n=3). Data
were analyzed by one- or two-way ANOVA followed by post hoc Tukey's
test. All statistical analysis was performed using the SPSS 22.00
software (IBM Corp.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Hyperglycemia enhances neurological
deficit following cerebral I/R injury
The experimental paradigm is presented in Fig. 1A. The rat body weight and blood
glucose levels were measured once/week for 4 weeks following STZ
injection. Weight of NG animals increased from 215.7±31 to 331.2±20
g after 4 weeks; HG animals did not gain weight during the 4-week
period. As a result, the body weight of HG animals in weeks 2-4
were significantly lower (Fig.
1B). The blood glucose levels of HG animals elevated to
>16.7 mM at 1 week post-STZ injection and further increased to
22-34 mM in 2-4 weeks post-STZ injection. By contrast, the blood
glucose in NG animals stayed at baseline levels of 5-8 mM
(P<0.01 vs. HG; Fig. 1C).
Neurological function was assessed using the mNSS at 1 day
post-reperfusion (Fig. 1D). The
sham control animals had a mean score of 2. Cerebral ischemia in NG
animals increased the deficit score to 9 (P<0.01 vs. Sham) and
to 13.6 in HG ischemic animals (P<0.05 vs. NG).
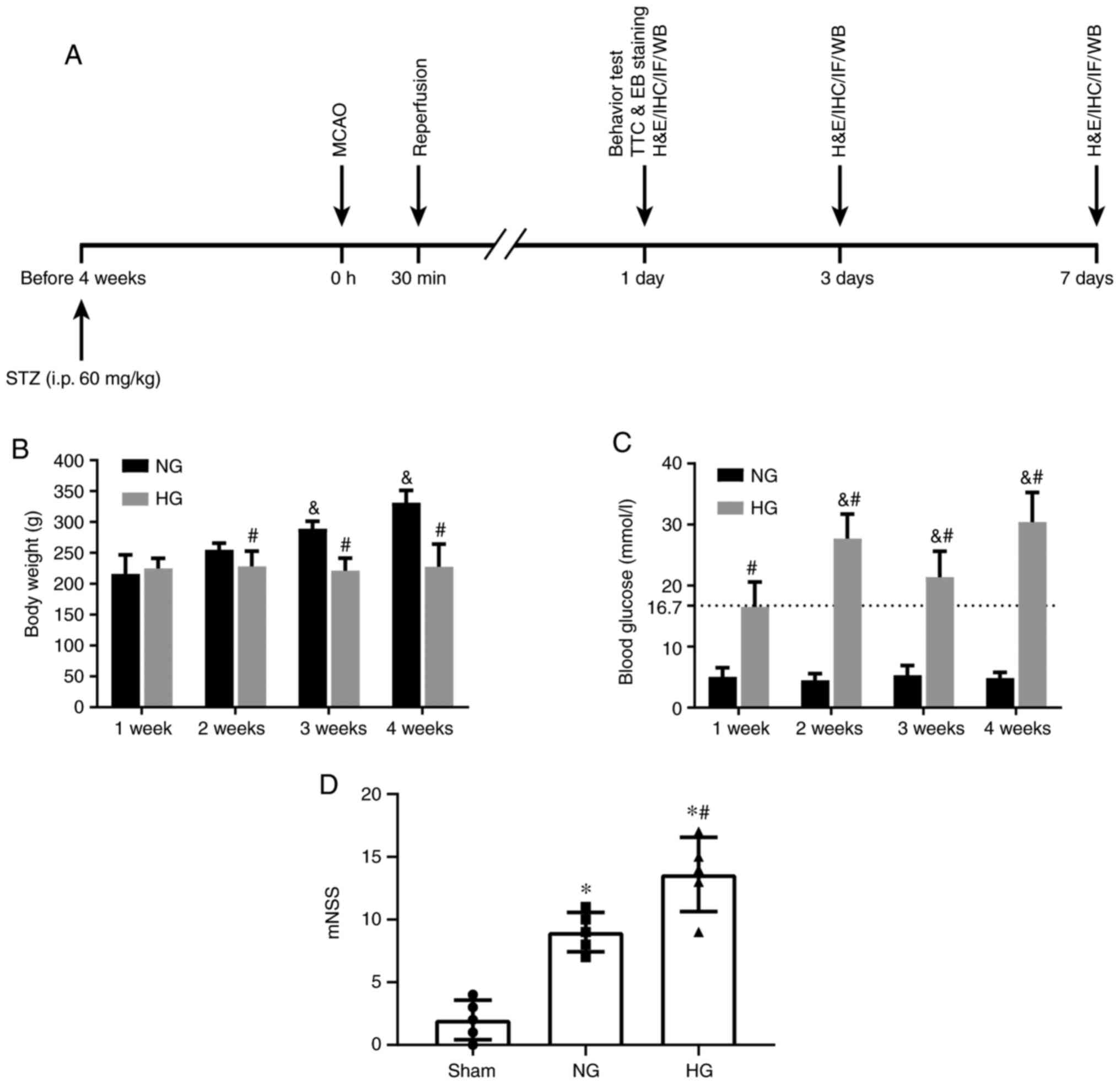 | Figure 1Schematic diagram and physiological
data (A) Schematic diagram of experimental design. Changes in (B)
body weight, (C) blood glucose level and (D) neurological deficit
following cerebral I/R in NG and HG animals. Hyperglycemia was
induced by STZ injection and neurological deficit was assessed
using the mNSS at 1 day post-reperfusion following 30 min MCAO.
n=5/group. &P<0.05 vs. 1 week,
*P<0.05 vs. Sham, #P<0.05 vs. NG. NG,
normoglycemic; HG, hyperglycemic; STZ, streptozotocin; MCAO, middle
cerebral artery occlusion; i.p., intraperitoneal; H&E,
hematoxylin and eosin; IHC, immunohistochemistry; IF,
immunofluorescence; WB, western blotting; mNSS, modified
neurological severity score; TTC, 2,3,5-triphenyltetrazolium
chloride; EB, Evans blue. |
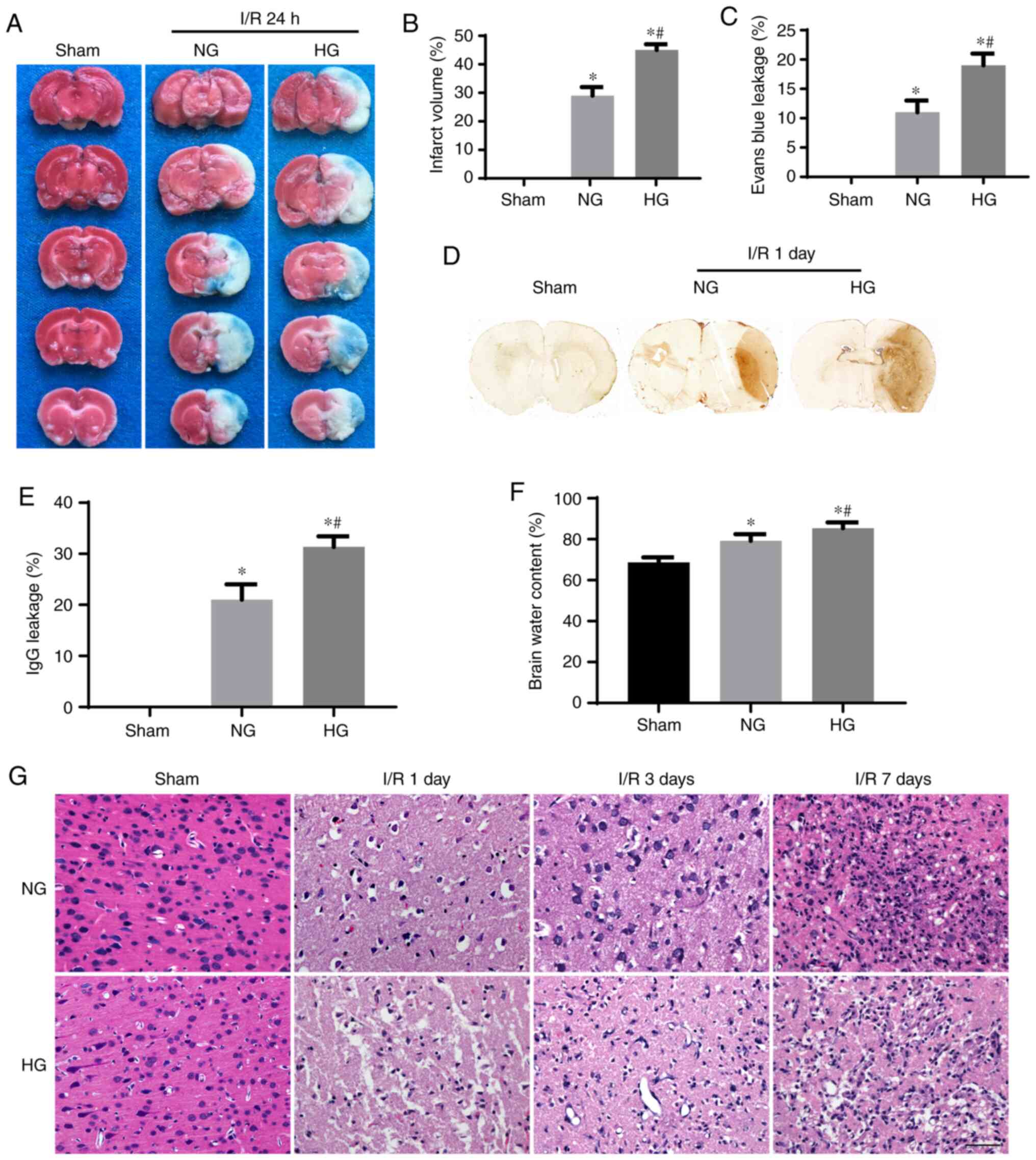
Hyperglycemia aggravates brain injury and
increases permeability of the BBB following I/R
TTC & Evans blue double staining were used to
evaluate the infarct volume and BBB disruption. The normal
non-ischemic brain tissue was stained pink while the ischemic
infarcted area remained white. When the permeability of the BBB
increased, Evans blue penetrated the BBB via blood vessels,
staining brain tissue blue. TTC staining revealed that ischemia in
NG animals resulted in 29% infarct volume at 1 day recovery. As
expected, preischemic hyperglycemia enlarged the infarct volume to
45% (Fig. 2A and B). The area of
Evans blue staining in NG animals was 11% leaking area; in HG
animals, this was increased to 19% (P<0.05 vs. NG), suggesting
HG exacerbated BBB leakage at 1 day reperfusion (Fig. 2A and C). In order to confirm that
HG increased the permeability of the BBB following I/R injury, IgG
staining was performed (Fig. 2D and
E). The results demonstrated that ischemia in NG animals led to
moderately increased IgG staining in the right middle cerebral
artery territory. In HG animals, the IgG-stained area was
significantly increased (P<0.01 vs. NG), confirming that
hyperglycemia damaged the integrity of the BBB. Evaluation of brain
edema by dry-wet weight ratio revealed that brain edema was mildly
and moderately increased in NG and HG animals, respectively.
The effect of HG on histopathology of brain tissue
at the frontal cortex was evaluated using hematoxylin and eosin
(H&E) staining. Sham group exhibited normal brain structure
with no evidence of tissue swelling, necrosis or other visible
abnormalities (Fig. 2G). No
abnormalities were observed in HG Sham animals, aside from an
increased number of microvessels compared with the NG Sham group.
At 1 day post-I/R, cerebral infarction, which is characterized by
brain edema, loosening of the matrix, pyknotic neurons and loss of
blood vessels (42), was
observed. These changes were significantly worsened in the HG group
compared with the NG group. At 3- and 7-days post-reperfusion,
glial cell proliferation and irregular neovascularization appeared.
Glial cell infiltration in HG animals was less than that in the NG
group.
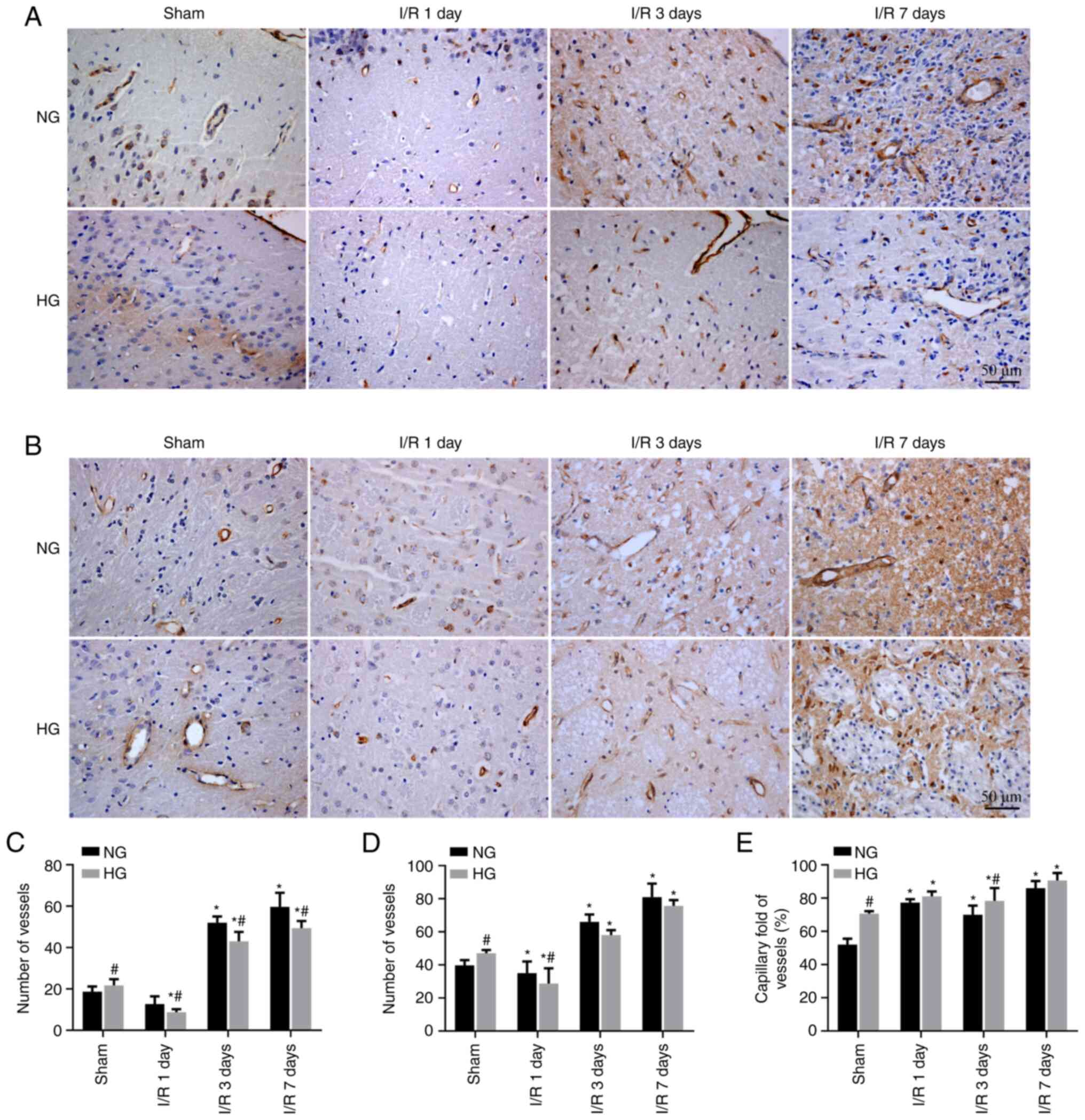
Effect of hyperglycemia on cerebral
vessel density following cerebral I/R injury
In order to observe the influence of HG ischemia on
cerebral blood vessels, vascular density was evaluated by labeling
vascular endothelial cells via vWF staining and counting the number
of vessels in NG- and HG-ischemic rats (Fig. 3). Consistent with the
observations in H&E-stained sections, the number of vessels in
both cortex and striatum regions significantly increased in the HG
Sham group compared with the NG Sham group. The number of blood
vessels initially decreased after 1 day and significantly increased
in both structures at 3 and 7 days post-I/R in NG animals.
Similarly, the number of blood vessels decreased at 1 day and
significantly increased in the cortex and striatum at 3 and 7 days
post-I/R in HG animals, but the extent of these increases were less
than those in NG animals (Fig.
3A-D).
Blood vessels with diameters <10 μm were
defined as capillaries and the ratio of capillaries to all vessels
was calculated. The ratio in the NG Sham group was 47.6%; in the HG
Sham group, this was increased to 70.2% (Fig. 3E). Following I/R injury, the
capillary ratio increased significantly at 1-7 days
post-reperfusion compared with sham groups but there was no
significant difference between the NG and HG groups.
TJ and BM are damaged by hyperglycemia
following I/R
TJs are the most important barrier structure of the
BBB. They are primarily composed of claudins, occludins and ZO-1
(43). These three proteins were
distributed continuously and uniformly among vascular endothelial
cells with a granular shape in NG and HG Sham groups. At 1 day
post-reperfusion, TJ proteins were distributed intermittently among
vascular endothelial cells with loss of continuity and decreased
staining intensity. These changes were more notable in the HG group
than in the NG group. At 3 and 7 days post-reperfusion, TJ proteins
recovered continuity of distribution around the vessels and
increased intensity of staining; this recovery was slower in HG
compared with NG animals (Fig.
4A-C).
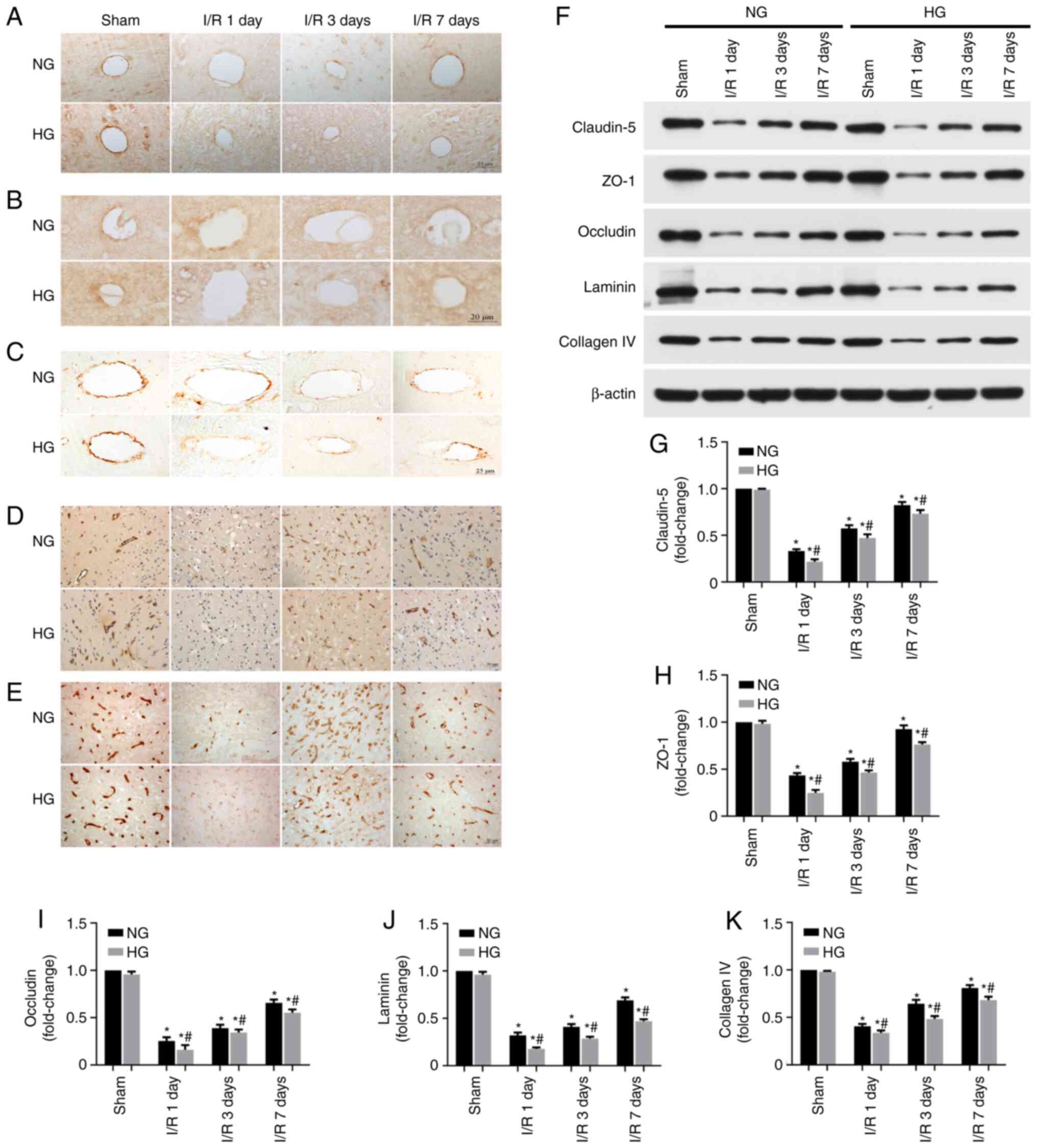 | Figure 4Immunoreactivity and levels of tight
junction and basement membrane proteins following cerebral I/R
injury. Representative photomicrographs showing (A) claudin-5, (B)
ZO-1, (C) occludin, (D) laminin and (E) collagen IV staining in the
ischemic cortical penumbra (n=5/group). (F) Representative western
blots and relative amounts of (G) claudin-5, (H) ZO-1, (I)
occludin, (J) laminin and (K) collagen IV (n=6/group).
*P<0.05 vs. Sham, #P<0.05 vs. NG. I/R,
ischemia/reperfusion; ZO-1, zonula occluden-1; NG, normoglycemic;
HG, hyperglycemic. |
Laminin and collagen IV constitute the network
skeleton of the BM (44). In the
Sham NG and HG groups, laminin and collagen IV were distributed in
the vessels and their shapes could be clearly seen (dark brown). At
1 day post-reperfusion, the two proteins were lost along with
infarct formation in brain tissue of both NG and HG animals, with
more pronounced changes observed in the HG animals. At 3 and 7 days
post-reperfusion, immunoreactivity of the BM protein was detected,
along with the neovascularization of cerebral vessels. The density
of laminin and collagen IV was weaker in HG compared with NG groups
(Fig. 4D and E).
The aforementioned observations were confirmed by
western blot analysis of the proteins. Similar to the observations
in IHC, the levels of these proteins decreased sharply at 1 day
post-reperfusion and increased gradually from 3 to 7 days
post-reperfusion. Compared with NG animals, the levels of all five
aforementioned proteins were lower in HG animals (Fig. 4F-K).
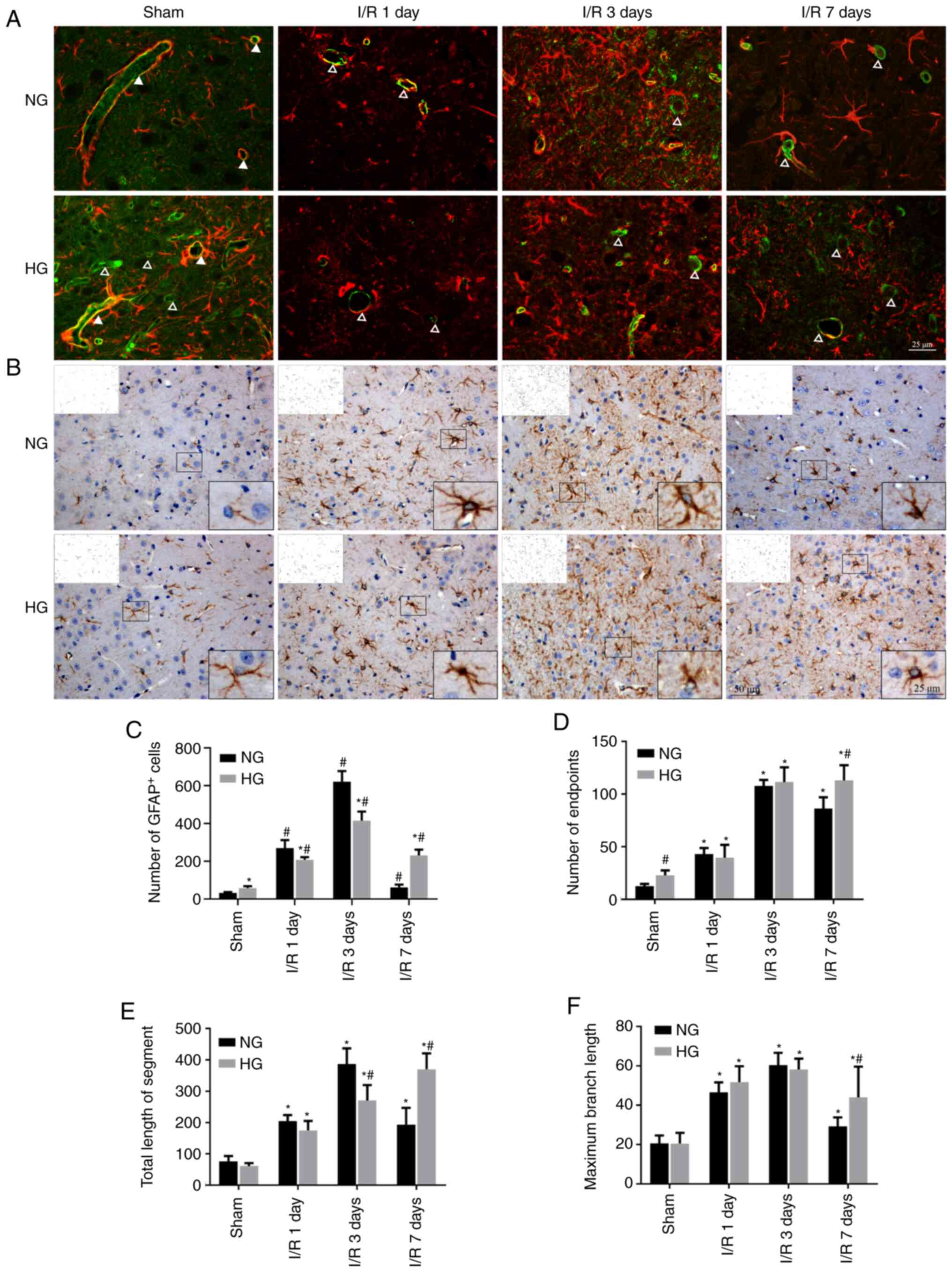
Damage to astrocytic endfeet and
astrocytic activation
The astrocytic endfoot is an important component of
the BBB (45). In the Sham HG
and NG groups, vWF-labeled vessels were tightly surrounded by
GFAP-stained astrocyte endfeet. This was observed in both large and
small vessels in the NG Sham group. At 1 day post-reperfusion, most
of the vessels lost astrocytic endfeet and only a few large vessels
were partially surrounded by astrocytes in the NG and HG groups. By
contrast, HG Sham group exhibited more blood vessels than the NG
Sham group. At 1 day recovery, both large and small vessels lost
astrocyte endfoot wrappings in HG animals (Fig. 5A).
IHC of astrocytes using anti-GFAP antibody
demonstrated that astrocytes had small cell bodies and fewer
dendrites in NG Sham group compared with the HG Sham group. Number
of astrocytes increased moderately after 1 day, peaked at 3 days
and slightly declined at 7 days in NG animals. Compared with NG
Sham, the astrocyte bodies enlarged and exhibited increased number
of dendritic endpoints, length of the dendrite segment and maximum
branch length of dendrites in NG I/R animals (Fig. 5B-F), suggesting that astrocytes
were activated by I/R in NG animals. Compared with NG animals, the
number of astrocytes increased slightly in the HG Sham group.
Following I/R, the number of astrocytes in NG groups increased to
lesser extent at days 1 and 3 but was higher at day 7 than those in
HG I/R at corresponding endpoints. The number of endpoints, total
length of branch segments and maximum branch length increased in HG
1 and 3 day groups but did not exceed those in NG animals, but
exhibited higher values at 7 days compared with NG (Fig. 5B-F). These data suggest that HG
activated astrocytes and HG-activated astrocytes persisted longer
than in NG animals following I/R.
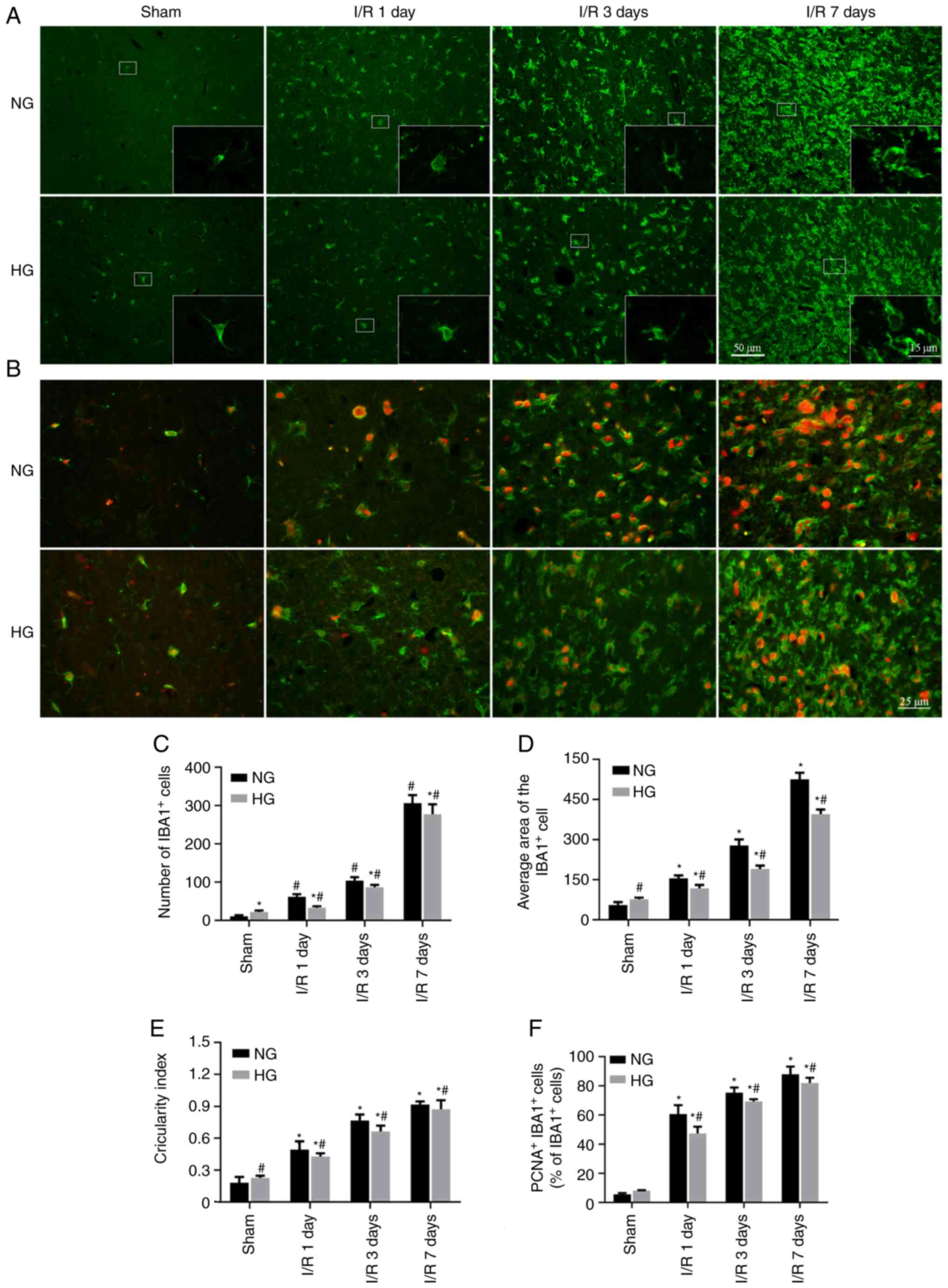
Inhibition of microglia by hyperglycemia
following cerebral I/R injury
As innate immune cells in the CNS, microglia remove
harmful substances that enter the brain parenchyma (41). Microglial activation was assessed
by labeling IBA1 [a marker of mature microglia (46)] coupled with morphological
analysis. The results (Fig. 6)
showed that in the NG Sham group, microglia remained in a survey
state, characterized by a ramified morphology with long branches
and small cellular body. However, in the HG Sham group, the number
of microglia cells increased and amoeboid morphology with a larger
cellular body and shorter branches were observed, indicating
activation of microglial cells by HG. Following I/R injury, the
number of IBA1-stained cells increased from days 1 to 7 in NG
animals, as did the average area of the microglial cells,
circularity index and newly generated microglial cells, reflected
by positive double labelling of IBA1 with cell proliferation marker
PCNA (Fig. 6A-F). The activated
microglia exhibited typical morphological characteristics, such as
thickening and retraction of dendrites and enlargement of cell body
to nearly circular shape (Fig.
6E). Compared with NG ischemic animals, the number and average
area of microglial cells, as well as the number of newly generated
microglial cells, decreased in the corresponding HG group,
suggesting that the activation of microglia in HG I/R group was as
strong as in the NG I/R counterparts (Fig. 6A-F).
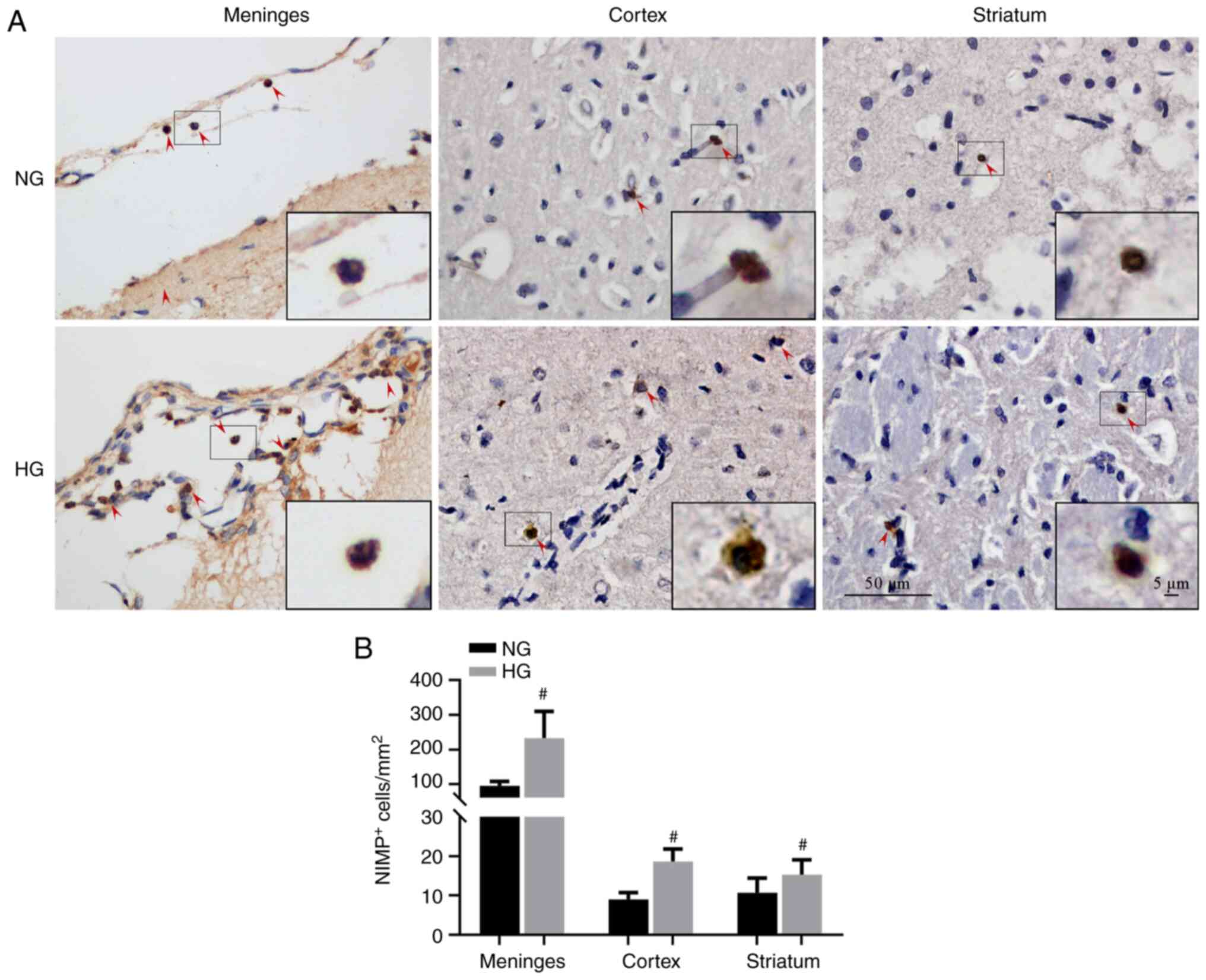
Neutrophil infiltration increases in the
meninges of HG I/R animals
Neutrophil infiltration damages the BBB, which
further increases neutrophil infiltration (47). Neutrophil infiltration was
investigated by labeling brain sections with neutrophil antibody
(anti-NIMP-R14) followed by IHC analysis of meningeal and brain
parenchyma (cortex and striatum) with respect to the number of
NIMP-R14+ neutrophils. NG I/R-increased neutrophil
infiltration primarily occurred in the meninges and HG I/R caused
further increases. In the brain parenchyma, only a small number of
NIMP-R14+ cells were detected and there was no
significant difference between the NG I/R and HG I/R groups
(Fig. 7A and B).
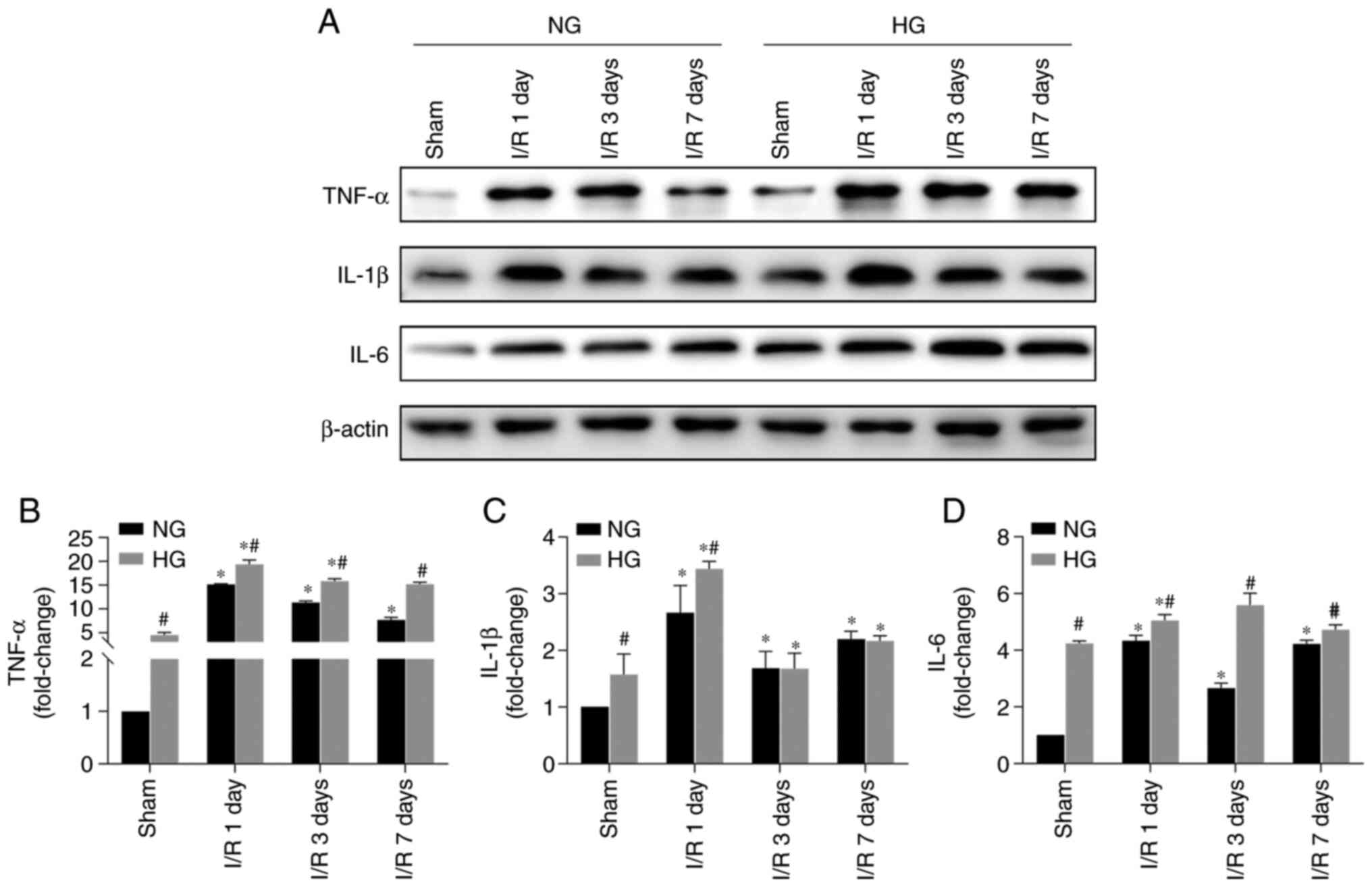
Inflammation increases in the penumbra of
HG I/R animals
In order to determine the change in
neuroinflammation, protein levels of pro-inflammatory factors
TNF-α, IL-1 β and IL-6 in the cortical penumbral tissue were
measured. Protein levels of these three factors were significantly
increased following I/R in NG animals, with peak levels at 1 day
post-I/R (Fig. 8). Compared with
NG I/R animals, the increases were further enhanced in the HG I/R
groups. In addition, animals in the HG sham groups also exhibited
increased levels of inflammatory markers, suggesting
hyperglycemia-alone caused stress to brain. This was consistent
with observations that astrocytes and microglia were activated in
the HG Sham group.
Discussion
Both animal and clinical studies have confirmed that
diabetes is an important independent risk factor for stroke
(48-51). Compared with non-diabetic
patients, people with diabetes are more likely to suffer stroke
(52). Furthermore, the size of
cerebral infarction, mortality and rate of disability are
significantly increased following stroke in patients with
hyperglycemia or diabetes (53,54). In the present study, HG animals
exhibited more severe cerebral edema, larger infarct volume and
higher neurological deficit scores than NG animals following 30 min
ischemia and 24 h reperfusion. The pathomorphological results
showed that hyperglycemia aggravated brain tissue damage; more
severe pan-necrosis was observed in the brain sections from HG
compared with those from from NG animals at days 1, 3 and 7
post-reperfusion. These results support the hypothesis that
hyperglycemia is an aggravating factor in cerebral I/R injury. In
addition, hyperglycemia-alone increased the number of small vessels
and capillaries, as observed by routine H&E and vascular
specific vWF staining. The percentage of capillaries increased in
HG Sham animals, suggesting hyperglycemia may increase vasogenesis
in normal healthy animals (55).
Over the past few decades, a number of clinical
trials have examined the efficacy of neuroprotective agents
(56,57), however, none of them improved
clinical recovery in patients following a stroke (58). Lo et al (10) proposed the concept of the NVU in
2003, which refers to an overall structural and functional unit
consisting of neurons, endothelial cells, glial cells with
pericytes, BM and extracellular matrix (10,59,60). As a core structure of the NVU,
the BBB not only prevents harmful substances entering the brain
parenchyma but also regulates endothelial-glial and glial
cell-neuron interaction. In order to determine the effects of
hyperglycemia on the BBB following I/R, the integrity of the BBB
was assessed using Evans blue and IgG leakage assays. HG I/R
significantly increased leakage of both Evans blue and IgG from
cerebral blood vessels to the brain parenchyma, suggesting that
hyperglycemia damaged the BBB and increased its permeability. This
finding is consistent with results reported by Kamada et al
(30), who demonstrated that
MMP-9 is activated following HG I/R injury.
TJs, primarily composed of claudin-5, occludin and
ZO-1 proteins, are key for maintaining structure and function of
the BBB (33). Claudin-5 is a
TJ-specific protein necessary for TJ formation and BBB function
(61). Occludin is a cell
polarity protein that primarily has the function of palisade and
barrier regulation. The palisade function is to divide the plasma
membrane of epithelial cells into functional zones (the lipid part
of the top and the protein part of the basal side) while preventing
mutual diffusion between the functional areas, which mediates
formation of cell polarity. The barrier regulation function is
selective to transmembrane materials (15,62-64). It regulates the transport of
small molecules between cells and maintains brain tissue
homeostasis (65). ZO-1 protein
is key to TJ stability and function (66-68). Dissociation of ZO-1 from the TJ
complex is associated with increased permeability of the BBB
(69). Since these three
proteins are the major structural proteins of the TJ, changes in
their expression result in alteration of BBB permeability and brain
edema (70,71). In the present study,
hyperglycemia notably suppressed protein levels and exacerbated
discontinuity of these TJ proteins around vessels following I/R
injury compared with NG animals, suggesting that
hyperglycemia-induced BBB leakage resulted from damage to TJ
proteins.
The BM is the second barrier structure of the BBB.
Located at the interface of the circulation system and the CNS, the
BM is well positioned to regulate BBB integrity under both
homeostatic and pathological conditions. The brain BM primarily
consists of laminin, collagen IV, nidogen and heparan sulfate
proteoglycans. Laminin and collagen IV are the two most abundant
components, which constitute the basic framework of the BM.
Cerebral ischemia damages BM components, including laminin and
collagen IV in gerbil, rat and primate models of cerebral I/R
injury, potentially due to activation of MMP-9 (20,22,44,72-74). In the present study,
hyperglycemia further decreased the levels of laminin and collagen
IV at 1, 3 and 7 days post-reperfusion compared with NG animals at
identical time points, suggesting that HG ischemia causes more
severe damage to the BM than NG ischemia. Suppression of TJ
proteins and damage to the BM by hyperglycemia may explain why
there is an increased transition from ischemic to hemorrhagic
stroke in animal stroke models and clinical patients with stroke
(75,76).
Astrocytes are the most abundant cell type in the
CNS (77). They support and
protect neurons by maintaining the homeostatic balance of the
neural microenvironment (78).
Astrocytes also interact with endothelial cells and support BBB
integrity via endfeet that encircle cerebral blood vessels
(27). In our previous study,
partial or complete absence of astrocyte endfeet around the blood
vessel wall was observed in diabetic animals following MCAO
(79). In the present study, the
loss of astrocytic endfeet following I/R injury was primarily
observed in the small cerebral blood vessels in the NG group, while
hyperglycemia exacerbated loss of endfeet to all types of cerebral
blood vessel. This variable damaging effect of HG and NG on endfeet
of blood vessels with different diameters may be one of the
mechanisms underlying the exacerbated damage induced by
hyperglycemia on the BBB. Several studies have shown that activated
astrocytes release trophic factors and extracellular matrix
molecules, which promote neuronal survival and plasticity following
ischemia (80-82).
In the present study, astrocytes were activated at
day 1 post-reperfusion following ischemia. In NG animals, the
activation peaked at 3 days post-I/R, and then tapered off at 7
days. Changes of astrocytes, such as activation, in HG animals
followed the same trend as seen in NG groups. However, the number
of GFAP+ cells and endpoints, total length of dendrite
segments and maximum branch length were significantly higher at 7
days in HG compared with NG animals. These results suggest that
hyperglycemia prolonged the duration of astrocytic activation. This
may facilitate tissue repair; however, continuously activated
astrocytes form glial scar tissue around the necrotic brain tissue
of the infarct, which provides a physical barrier against axonal
growth, thus preventing neurological recovery following stroke
(60,83).
Activation of microglial cells occurs in CNS
infectious disease and neurodegenerative disorders. The activation
of microglia has also been shown to regulate brain endothelial cell
proliferation (84). The present
results showed that cerebral I/R injury resulted in a
time-dependent activation of microglial cells in the brain, as
reflected by increases in number of IBA-1+ cells,
average area of the microglia and circularity index. I/R increased
microglial cell proliferation, as detected by double positive
labeling of IBA-1 and PCNA. Compared with NG animals, hyperglycemia
repressed activation and proliferation of microglial cells. This
may suppress the immune response and tissue repair in the
reperfusion stage.
The infiltration of neutrophils serves an important
role in mediating post-stroke damage in the brain. It causes
increased production of free radicals, exacerbation of
neuroinflammation and disruption of the BBB and hemorrhagic
transformation post-stroke (35). Activation of neutrophils via
lipopolysaccharide notably enhances BBB disruption in an animal
model of stroke (85). By
contrast, inhibition of neutrophils alleviates BBB leakage
following stroke and decreases the risk of hemorrhagic
transformation following thrombolysis (86). Consistent with these reports, the
present study demonstrated ~3-fold increases in numbers of of
neutrophils in the meninges of HG animals compared with the NG
group; no significant difference in the number of neutrophils in
the brain parenchyma or striatum was observed between groups.
Activation of microglia and infiltration of
neutrophils may elicit release of pro-inflammatory cytokines, which
contribute to neuronal damage following stroke (87). Increases in levels of TNF-α,
IL-1β and IL-6 were observed at 1 day post-I/R; these levels
decreased at 3 and 7 days post-I/R in NG animals. The changes in
expression levels of these inflammatory cytokines followed the same
trend in HG groups; however, the levels were higher than in the
corresponding NG groups. These results are consistent with the
increased infiltration of neutrophils in HG groups, suggesting that
hyperglycemia aggravated the ischemia-induced neuroinflammatory
response.
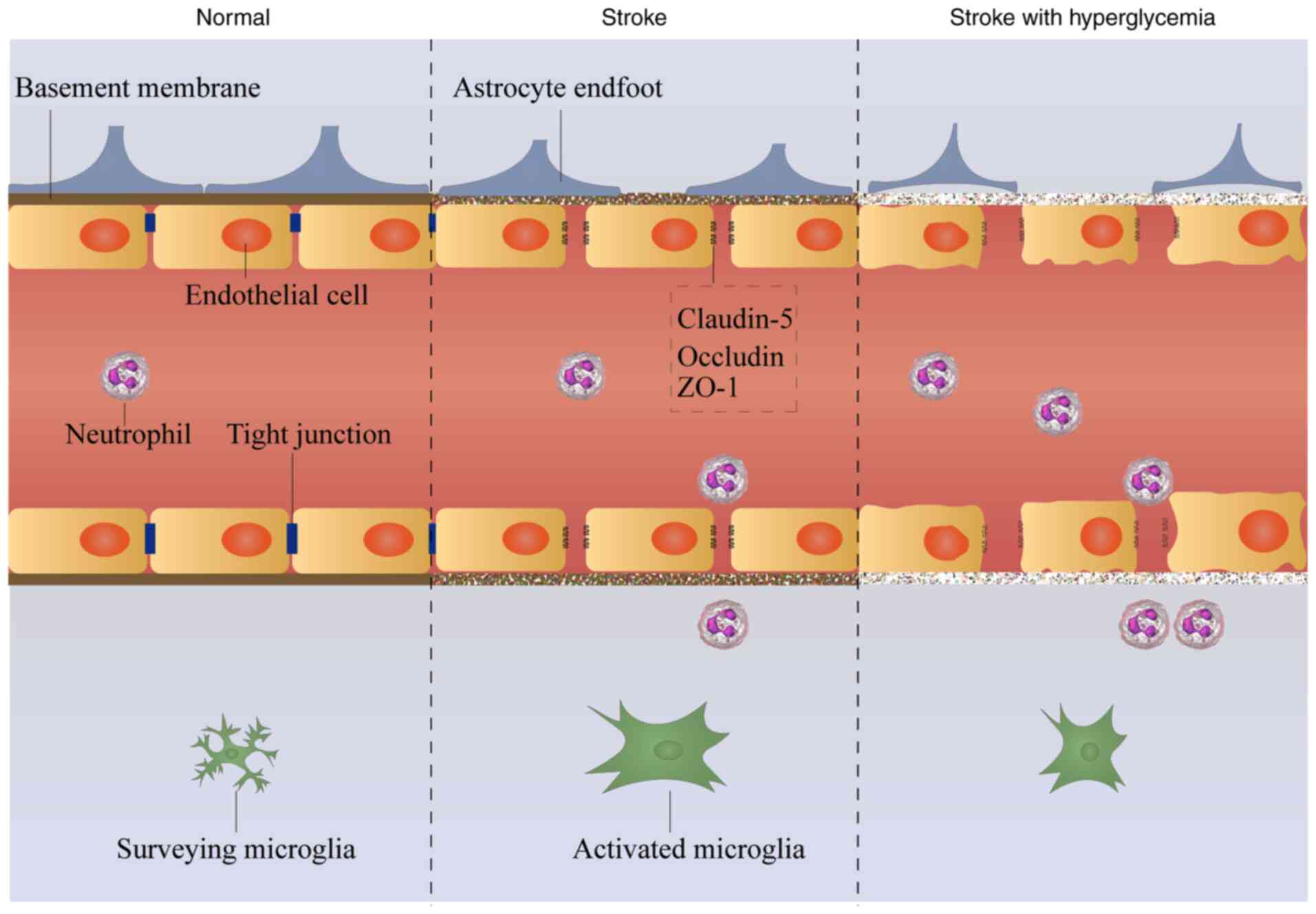
In conclusion, hyperglycemia increased BBB leakage,
suppressed levels of TJ and BM proteins and damaged astrocyte
endfeet that enclose cerebral vasculature following cerebral I/R
injury. Furthermore, hyperglycemia suppressed microglial activation
and proliferation, enhanced neutrophil infiltration into the brain
and increased levels of inflammatory cytokines. It was hypothesized
that hyperglycemia-exacerbated BBB damage is caused by damage to
TJs, BM and astrocytes, and is associated with increased
neuroinflammation (Fig. 9). The
present study aimed to determine the underlying mechanism of
aggravated BBB damage in diabetes. However, other aspects, such as
the association or cascade reaction between these structures
(endothelial cells, TJs, astrocytes, BM and microglia) and the role
of pericytes, an important barrier for the BBB (66,67), deserve further investigation.
Availability of data and materials
The datasets used and analyzed in the current study
are available from the corresponding authors on reasonable
request.
Authors' contributions
YZG conceived the study. YZG and JZZ performed data
analysis. LJ and JZZ acquired funding. LDD and AG performed the
experiments. JZZ was responsible for project administration. JWZ
established the experimental animal model. LJ supervised the study
and performed the MCAO model. TD and PAAL analyzed data and wrote
the original draft of the manuscript. JZZ and YZG confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Animal Care and Use Committee of Ningxia Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Mrs Laretha Davis
(North Carolina Central University, NC, USA) for editing and
proofreading.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 31960177 and 31780280) and the
Natural Science Foundation of Ningxia (grant no. 2018AAC03092).
References
|
1
|
Li W, Prakash R, Kelly-Cobbs AI, Ogbi S,
Kozak A, El-Remessy AB, Schreihofer DA, Fagan SC and Ergul A:
Adaptive cerebral neovascularization in a model of type 2 diabetes:
Relevance to focal cerebral ischemia. Diabetes. 59:228–235. 2010.
View Article : Google Scholar
|
|
2
|
Chen R, Ovbiagele B and Feng W: Diabetes
and stroke: Epidemiology, pathophysiology, pharmaceuticals and
outcomes. Am J Med Sci. 351:380–386. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li W, Qu Z, Prakash R, Chung C, Ma H, Hoda
MN, Fagan SC and Ergul A: Comparative analysis of the neurovascular
injury and functional outcomes in experimental stroke models in
diabetic Goto-Kakizaki rats. Brain Res. 1541:106–114. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu Q, Manaenko A, Bian H, Guo Z, Huang JL,
Guo ZN, Yang P, Tang J and Zhang JH: Hyperbaric oxygen reduces
infarction volume and hemorrhagic transformation through
ATP/NAD+/Sirt1 pathway in hyperglycemic middle cerebral
artery occlusion rats. Stroke. 48:1655–1664. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lanier WL, Hofer RE and Gallagher WJ:
Metabolism of glucose, glycogen, and high-energy phosphates during
transient forebrain ischemia in diabetic rats: Effect of insulin
treatment. Anesthesiology. 84:917–925. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Muranyi M, Fujioka M, He Q, Han A, Yong G,
Csiszar K and Li PA: Diabetes activates cell death pathway after
transient focal cerebral ischemia. Diabetes. 52:481–486. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu WJ, Jiang HF, Rehman FU, Zhang JW,
Chang Y, Jing L and Zhang JZ: Lycium barbarum polysaccharides
decrease hyperglycemia-aggravated ischemic brain injury through
maintaining mitochondrial fission and fusion balance. Int J Biol
Sci. 13:901–910. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu CJ, Guo YZ, Zhang Y, Yang L, Chang Y,
Zhang JW, Jing L and Zhang JZ: Coenzyme Q10 ameliorates cerebral
ischemia reperfusion injury in hyperglycemic rats. Pathol Res
Pract. 213:1191–1199. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang L, Ma YM, Shen XL, Fan YC, Zhang JZ,
Li PA and Jing L: The involvement of mitochondrial biogenesis in
selenium reduced Hyperglycemia-Aggravated cerebral ischemia injury.
Neurochem Res. 45:1888–1901. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lo EH, Dalkara T and Moskowitz MA:
Mechanisms, challenges and opportunities in stroke. Nat Rev
Neurosci. 4:399–415. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao Z, Nelson AR, Betsholtz C and
Zlokovic BV: Establishment and dysfunction of the Blood-Brain
barrier. Cell. 163:1064–1078. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Engelhardt B and Liebner S: Novel insights
into the development and maintenance of the blood-brain barrier.
Cell Tissue Res. 355:687–699. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Abdullahi W, Tripathi D and Ronaldson PT:
Blood-brain barrier dysfunction in ischemic stroke: Targeting tight
junctions and transporters for vascular protection. Am J Physiol
Cell Physiol. 315:C343–C356. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fang B, Zhang Y and Ma H: Effects of
MIR-122a on blood-spinal cord barrier after spinal cord
ischemia-reperfusion injury in rats. Chin Pharmacol Bull.
33:703–706. 2017.In Chinese.
|
|
15
|
Lv J, Hu W, Yang Z, Li T, Jiang S, Ma Z,
Chen F and Yang Y: Focusing on claudin-5: A promising candidate in
the regulation of BBB to treat ischemic stroke. Prog Neurobiol.
161:79–96. 2018. View Article : Google Scholar
|
|
16
|
Miyamori H, Takino T, Kobayashi Y, Tokai
H, Itoh Y, Seiki M and Sato H: Claudin promotes activation of
pro-matrix metalloproteinase-2 mediated by membrane-type matrix
metalloproteinases. J Biol Chem. 276:28204–28211. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang GS, Tian Y, Huang JY, Tao RR, Liao
MH, Lu YM, Ye WF, Wang R, Fukunaga K, Lou YJ and Han F: The
γ-secretase blocker DAPT reduces the permeability of the
blood-brain barrier by decreasing the ubiquitination and
degradation of occludin during permanent brain ischemia. CNS
Neurosci Ther. 19:53–60. 2013. View Article : Google Scholar
|
|
18
|
Fanning AS, Jameson BJ, Jesaitis LA and
Anderson JM: The tight junction protein ZO-1 establishes a link
between the transmembrane protein occludin and the actin
cytoskeleton. J Biol Chem. 273:29745–29753. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sandoval KE and Witt KA: Blood-brain
barrier tight junction permeability and ischemic stroke. Neurobiol
Dis. 32:200–219. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Paulsson M: Basement membrane proteins:
Structure, assembly, and cellular interactions. Crit Rev Biochem
Mol Biol. 27:93–127. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yurchenco PD, Amenta PS and Patton BL:
Basement membrane assembly, stability and activities observed
through a developmental lens. Matrix Biol. 22:521–538. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Martin GR and Timpl R: Laminin and other
basement membrane components. Annu Rev Cell Biol. 3:57–85. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hallmann R, Horn N, Selg M, Wendler O,
Pausch F and Sorokin LM: Expression and function of laminins in the
embryonic and mature vasculature. Physiol Rev. 85:979–1000. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Di Russo J, Hannocks MJ, Luik AL, Song J,
Zhang X, Yousif L, Aspite G, Hallmann R and Sorokin L: Vascular
laminins in physiology and pathology. Matrix Biol. 57-58:140–148.
2017. View Article : Google Scholar
|
|
25
|
Hamann GF, Burggraf D, Martens HK,
Liebetrau M, Jäger G, Wunderlich N, DeGeorgia M and Krieger DW:
Mild to moderate hypothermia prevents microvascular basal lamina
antigen loss in experimental focal cerebral ischemia. Stroke.
35:764–769. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yepes M, Sandkvist M, Wong MK, Coleman TA,
Smith E, Cohan SL and Lawrence DA: Neuroserpin reduces cerebral
infarct volume and protects neurons from ischemia-induced
apoptosis. Blood. 96:569–576. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Abbott NJ, Rönnbäck L and Hansson E:
Astrocyte-endothelial interactions at the blood-brain barrier. Nat
Rev Neurosci. 7:41–53. 2006. View Article : Google Scholar
|
|
28
|
Sofroniew MV and Vinters HV: Astrocytes:
Biology and pathology. Acta Neuropathol. 119:7–35. 2010. View Article : Google Scholar
|
|
29
|
Dodson RF, Chu LW, Welch KM and Achar VS:
Acute tissue response to cerebral ischemia in the gerbil. An
ultrastructural study. J Neurol Sci. 33:161–170. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kamada H, Yu F, Nito C and Chan PH:
Influence of hyperglycemia on oxidative stress and matrix
metalloproteinase-9 activation after focal cerebral
ischemia/reperfusion in rats: Relation to blood-brain barrier
dysfunction. Stroke. 38:1044–1049. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang S, Shao SY, Song XY, Xia CY, Yang
YN, Zhang PC and Chen NH: Protective effects of Forsythia suspense
extract with antioxidant and anti-inflammatory properties in a
model of rotenone induced neurotoxicity. Neurotoxicology. 52:72–83.
2016. View Article : Google Scholar
|
|
32
|
Zhang S, An Q, Wang T, Gao S and Zhou G:
Autophagy- and MMP-2/9-mediated reduction and redistribution of
ZO-1 contribute to Hyperglycemia-increased Blood-Brain barrier
permeability during early reperfusion in stroke. Neuroscience.
377:126–137. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jiang X, Andjelkovic AV, Zhu L, Yang T,
Bennett MVL, Chen J, Keep RF and Shi Y: Blood-brain barrier
dysfunction and recovery after ischemic stroke. Prog Neurobiol.
163-164:144–171. 2018. View Article : Google Scholar :
|
|
34
|
Gelderblom M, Leypoldt F, Steinbach K,
Behrens D, Choe CU, Siler DA, Arumugam TV, Orthey E, Gerloff C,
Tolosa E and Magnus T: Temporal and spatial dynamics of cerebral
immune cell accumulation in stroke. Stroke. 40:1849–1857. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jickling GC, Liu D, Ander BP, Stamova B,
Zhan X and Sharp FR: Targeting neutrophils in ischemic stroke:
Translational insights from experimental studies. J Cereb Blood
Flow Metab. 35:888–901. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
da Fonseca AC, Matias D, Garcia C, Amaral
R, Geraldo LH, Freitas C and Lima FR: The impact of microglial
activation on blood-brain barrier in brain diseases. Front Cell
Neurosci. 8:3622014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hu X, De Silva TM, Chen J and Faraci FM:
Cerebral vascular disease and neurovascular injury in ischemic
stroke. Circ Res. 120:449–471. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fan Q, Chen M, Zuo L, Shang X, Huang MZ,
Ciccarelli M, Raake P, Brinks H, Chuprun KJ, Dorn GW II, et al:
Myocardial ablation of G Protein-Coupled Receptor Kinase 2 (GRK2)
decreases Ischemia/Reperfusion injury through an Anti-Intrinsic
apoptotic pathway. PLoS One. 8:e662342013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Morrison HW and Filosa JA: A quantitative
spatiotemporal analysis of microglia morphology during ischemic
stroke and reperfusion. J Neuroinflammation. 10:42013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Thorsten W and Hans-Gerd L: IJBlob: An
ImageJ Library for Connected Component Analysis and Shape Analysis.
J Open Res Softw. 1:e62013. View Article : Google Scholar
|
|
41
|
Otxoa-de-Amezaga A, Miro-Mur F, Pedragosa
J, Gallizioli M, Justicia C, Gaja-Capdevila N, Ruíz-Jaen F,
Salas-Perdomo A, Bosch A, Calvo M, et al: Microglial cell loss
after ischemic stroke favors brain neutrophil accumulation. Acta
Neuropathol. 137:321–341. 2019. View Article : Google Scholar :
|
|
42
|
Tyson GW, Teasdale GM, Graham DI and
McCulloch J: Focal cerebral ischemia in the rat: Topography of
hemodynamic and histopathological changes. Ann Neurol. 15:559–567.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sweeney MD, Zhao Z, Montagne A, Nelson AR
and Zlokovic BV: Blood-Brain barrier: From physiology to disease
and back. Physiol Rev. 99. pp. 21–78. 2019, View Article : Google Scholar
|
|
44
|
Kang M and Yao Y: Basement membrane
changes in ischemic stroke. Stroke. 51:1344–1352. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kacem K, Lacombe P, Seylaz J and Bonvento
G: Structural organization of the perivascular astrocyte endfeet
and their relationship with the endothelial glucose transporter: A
confocal microscopy study. Glia. 23:1–10. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sasaki Y, Ohsawa K, Kanazawa H, Kohsaka S
and Imai Y: Iba1 is an actin-cross-linking protein in
macrophages/microglia. Biochem Biophys Res Commun. 286:292–297.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang C, Hawkins KE, Dore S and
Candelario-Jalil E: Neuroinflammatory mechanisms of blood-brain
barrier damage in ischemic stroke. Am J Physiol Cell Physiol.
316:C135–C153. 2019. View Article : Google Scholar :
|
|
48
|
Barrett-Connor E and Khaw KT: Diabetes
mellitus: An independent risk factor for stroke? Am J Epidemiol.
128:116–123. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Luitse MJ, Biessels GJ, Rutten GE and
Kappelle LJ: Diabetes, hyperglycaemia, and acute ischaemic stroke.
Lancet Neurol. 11:261–271. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ankolekar S, Rewell S, Howells DW and Bath
PM: The influence of stroke risk factors and comorbidities on
assessment of stroke therapies in humans and animals. Int J Stroke.
7:386–397. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
van Sloten TT, Sedaghat S, Carnethon MR,
Launer LJ and Stehouwer CDA: Cerebral microvascular complications
of type 2 diabetes: Stroke, cognitive dysfunction, and depression.
Lancet Diabetes Endocrinol. 8:325–336. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sander D and Kearney MT: Reducing the risk
of stroke in type 2 diabetes: Pathophysiological and therapeutic
perspectives. J Neurol. 256:1603–1619. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ergul A, Kelly-Cobbs A, Abdalla M and
Fagan SC: Cerebrovascular complications of diabetes: Focus on
stroke. Endocr Metab Immune Disord Drug Targets. 12:148–158. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Reeves MJ, Vaidya RS, Fonarow GC, Liang L,
Smith EE, Matulonis R, Olson DM and Schwamm LH: Quality of care and
outcomes in patients with diabetes hospitalized with ischemic
stroke: Findings from get with the Guidelines-Stroke. Stroke.
41:e409–e417. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Warner DS, Smith ML and Siesjö BK:
Ischemia in normo-and hyperglycemic rats: Effects on brain water
and electrolytes. Stroke. 18:464–471. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Godínez-Rubí M, Rojas-Mayorquín AE and
Ortuño-Sahagún D: Nitric oxide donors as neuroprotective agents
after an ischemic stroke-related inflammatory reaction. Oxid Med
Cell Longev. 2013:2973572013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fukuta T, Ishii T, Asai T, Sato A, Kikuchi
T, Shimizu K, Minamino T and Oku N: Treatment of stroke with
liposomal neuroprotective agents under cerebral ischemia
conditions. Eur J Pharm Biopharm. 97(Pt A): 1–7. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Birmingham K: Future of neuroprotective
drugs in doubt. Nat Med. 8:52002. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Guo S, Kim WJ, Lok J, Lee SR, Besancon E,
Luo BH, Stins MF, Wang X, Dedhar S and Lo EH: Neuroprotection via
matrix-trophic coupling between cerebral endothelial cells and
neurons. Proc Natl Acad Sci USA. 105:7582–7587. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Iadecola C: Neurovascular regulation in
the normal brain and in Alzheimer's disease. Nat Rev Neurosci.
5:347–360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jia W, Lu R, Martin TA and Jiang WG: The
role of claudin-5 in blood-brain barrier (BBB) and brain metastases
(review). Mol Med Rep. 9:779–785. 2014. View Article : Google Scholar
|
|
62
|
Findley MK and Koval M: Regulation and
roles for claudin-family tight junction proteins. IUBMB Life.
61:431–437. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Van Itallie CM and Anderson JM: Claudin
interactions in and out of the tight junction. Tissue Barriers. 1.
pp. e252472013, View Article : Google Scholar
|
|
64
|
Anderson JM and Van Itallie CM: Physiology
and function of the tight junction. Cold Spring Harb Perspect Biol.
1:a0025842009. View Article : Google Scholar :
|
|
65
|
Sladojevic N, Stamatovic SM, Keep RF,
Grailer JJ, Sarma JV, Ward PA and Andjelkovic AV: Inhibition of
junctional adhesion molecule-A/LFA interaction attenuates leukocyte
trafficking and inflammation in brain ischemia/reperfusion injury.
Neurobiol Dis. 67:57–70. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Abbruscato TJ, Lopez SP, Mark KS, Hawkins
BT and Davis TP: Nicotine and cotinine modulate cerebral
microvascular permeability and protein expression of ZO-1 through
nicotinic acetylcholine receptors expressed on brain endothelial
cells. J Pharm Sci. 91:2525–2538. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Fischer S, Wobben M, Marti HH, Renz D and
Schaper W: Hypoxia-Induced hyperpermeability in brain microvessel
endothelial cells involves VEGF-Mediated changes in the expression
of Zonula Occludens-1. Microvasc Res. 63:70–80. 2002. View Article : Google Scholar
|
|
68
|
Mark KS and Davis TP: Cerebral
microvascular changes in permeability and tight junctions induced
by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol.
282:H1485–H1494. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang H, Li T, Zhao L, Sun M, Jian Y, Liu
J, Zhang Y, Li Y, Dang M and Zhang G: Dynamic effects of ioversol
on the permeability of the blood-brain barrier and the expression
of ZO-1/occludin in rats. J Mol Neurosci. 68:295–303. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Bazzoni G and Dejana E: Endothelial
cell-to-cell junctions: Molecular organization and role in vascular
homeostasis. Physiol Rev. 84:869–901. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Huber JD, Egleton RD and Davis TP:
Molecular physiology and pathophysiology of tight junctions in the
blood-brain barrier. Trends Neurosci. 24:719–725. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Xu L, Nirwane A and Yao Y: Basement
membrane and blood-brain barrier. Stroke Vasc Neurol. 4:78–82.
2018. View Article : Google Scholar
|
|
73
|
Yao Y: Basement membrane and stroke. J
Cereb Blood Flow Metab. 39:3–19. 2019. View Article : Google Scholar :
|
|
74
|
Kwon I, Kim EH, del Zoppo GJ and Heo JH:
Ultrastructural and temporal changes of the microvascular basement
membrane and astrocyte interface following focal cerebral ischemia.
J Neurosci Res. 87:668–676. 2009. View Article : Google Scholar :
|
|
75
|
Kazmierski R, Michalak S, Wencel-Warot A
and Nowinski WL: Serum tight-junction proteins predict hemorrhagic
transformation in ischemic stroke patients. Neurology.
79:1677–1685. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Khatri R, McKinney AM, Swenson B and
Janardhan V: Blood-brain barrier, reperfusion injury, and
hemorrhagic transformation in acute ischemic stroke. Neurology.
79(13 Suppl 1): S52–S57. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Rodríguez-Arellano J, Parpura V, Zorec R
and Verkhratsky A: Astrocytes in physiological aging and
Alzheimer's disease. Neuroscience. 323:170–182. 2016. View Article : Google Scholar
|
|
78
|
Hansson E and Rönnbäck L: Astrocytes in
glutamate neurotransmission. FASEB J. 9:343–350. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Jing L, He Q, Zhang JZ and Li PA: Temporal
profile of astrocytes and changes of oligodendrocyte-based myelin
following middle cerebral artery occlusion in diabetic and
non-diabetic rats. Int J Biol Sci. 9:190–199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Anderson MA, Burda JE, Ren Y, Ao Y, O'Shea
TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ and Sofroniew MV:
Astrocyte scar formation aids central nervous system axon
regeneration. Nature. 532:195–200. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Overman JJ, Clarkson AN, Wanner IB,
Overman WT, Eckstein I, Maguire JL, Dinov ID, Toga AW and
Carmichael ST: A role for ephrin-A5 in axonal sprouting, recovery,
and activity-dependent plasticity after stroke. Proc Natl Acad Sci
USA. 109:E2230–E2239. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Liauw J, Hoang S, Choi M, Eroglu C, Choi
M, Sun GH, Percy M, Wildman-Tobriner B, Bliss T, Guzman RG, et al:
Thrombospondins 1 and 2 are necessary for synaptic plasticity and
functional recovery after stroke. J Cereb Blood Flow Metab.
28:1722–1732. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Fitch MT and Silver J: CNS injury, glial
scars, and inflammation: Inhibitory extracellular matrices and
regeneration failure. Exp Neurol. 209:294–301. 2008. View Article : Google Scholar
|
|
84
|
Bannister JV, Bellavite P, Davoli A,
Thornalley PJ and Rossi F: The generation of hydroxyl radicals
following superoxide production by neutrophil NADPH oxidase. FEBS
Lett. 150:300–302. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
McColl BW, Rothwell NJ and Allan SM:
Systemic inflammatory stimulus potentiates the acute phase and CXC
chemokine responses to experimental stroke and exacerbates brain
damage via interleukin-1- and neutrophil-dependent mechanisms. J
Neurosci. 27:4403–4412. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Gautier S, Ouk T, Petrault O, Caron J and
Bordet R: Neutrophils contribute to intracerebral haemorrhages
after treatment with recombinant tissue plasminogen activator
following cerebral ischaemia. Br J Pharmacol. 156:673–679. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Pan W and Kastin AJ: Tumor necrosis factor
and stroke: Role of the blood-brain barrier. Prog Neurobiol.
83:363–374. 2007. View Article : Google Scholar : PubMed/NCBI
|