Introduction
Atherosclerosis (AS) is a chronic inflammatory
reaction of the arterial walls in response to vascular endothelial
cell injury. AS pathogenesis involves endothelial cell injury,
lipid accumulation in the vascular wall, monocyte adhesion and
transformation, the release of inflammatory factors and finally,
the proliferation and migration of smooth muscle cells, which
eventually leads to AS development (1). AS is the main pathological basis of
cardiovascular diseases, which are associated with high morbidity
and mortality rates (2,3). Significant progress has been made
concerning the treatment options of patients with AS; however, the
majority of therapeutics used are often associated with chronic
side-effects (4). Therefore, it
is necessary to elucidate the molecular mechanisms of AS in order
to identify novel diagnostic and therapeutic modalities, that can
more treat AS more efficiently.
Krüppel-like factor 5 (KLF5) is a protein, encoded
in humans by the KLF5 gene, and belongs to the Krüppel-like factor
subfamily of zinc finger proteins. KLF5 can regulate the expression
of a number of downstream target genes, including cyclin D1, cyclin
B1, fibroblast growth factor-binding protein and other coding genes
(5). KLF5 is expressed in a wide
variety of cells, including vascular smooth muscle cells,
lipocytes, neurons and white blood cells, and its expression is
particularly high in intestinal epithelial cells (6). Therefore, KLF5 is involved in the
regulation of inflammatory stress response and intestinal
development, which is caused by cardiovascular remodeling in
embryonic development (5). KLF5
promotes angiogenesis through the upregulation of vascular
endothelial growth factor (VEGFA), myosin heavy chain kinase (MHC),
myosin light chain kinase (MLCK), calponin, smooth muscle actin
(SMA) and transgelin (SM22-a), thereby increasing the proliferation
and migration of vascular smooth muscle cells (7-9).
MicroRNA (miRNA/miR)-152 prevents AS progression and reduces
β-catenin expression through the downregulation of KLF5 (10). KLF5 promotes the proliferation of
vascular smooth muscle cells and subsequently promotes the
formation of atherosclerotic plaques (11). However, the role of KLF5 in
endothelial cell damage caused by AS, at least to the best of our
knowledge, has not yet been studied.
Long non-coding RNAs (lncRNAs) are a group of
transcripts of >200 nucleotides in length, which lack protein
coding potential (12). lncRNAs
may play an important role in the treatment of AS. lncRNA
non-coding RNA activated by DNA damage (NORAD) expression has been
observed to be increased in human umbilical vein endothelial cells
(HUVECs). This is induced by oxidized low-density lipoprotein
(OX-LDL) and NORAD knockdown and has been shown to function as a
promoter of OX-LDL-induced HUVEC injury and AS (13). In a previous study, lncRNA
forkhead box C2-antisense RNA 1 (FOXC2-AS1) expression was found to
be markedly increased in patients with AS, and FOXC2-AS1
overexpression promoted the proliferation and inhibited apoptosis
of vascular smooth muscle cells (VSMCs) (14). lncRNA CDKN2B antisense RNA 1
expression has been noted to be elevated in human atherosclerotic
plaques and OX-LDL-stimulated HUVECs, and can promote cell
proliferation and migration by sponging miR-399-5p (15).
The present study aimed to explore the important
role of the KLF5/p53 regulated carcinoma associated Stat3
activating long intergenic non-protein coding transcript
(LINC00346)/miR-148a-3p axis in AS. It was found that the
transcription factor, KLF5, promoted LINC00346 transcription. In
addition, the role of LINC00346 in AS progression was confirmed and
the positive feedback loop of KLF5/LINC00346/miR-148a-3p in AS was
revealed, providing a potential therapeutic target for AS.
Materials and methods
Serum samples
A total of nine patients with AS and nine healthy
volunteers were recruited from Changzhou Second People's Hospital
(Jiangsu, China) between July, 2019 and October, 2019. The
inclusion criteria were as follows: i) An age 25-75 years; ii)
patients exhibiting hypertension, coronary heart disease and
hyperlipidemia; and iii) patients provided informed consent for
participation. The exclusion criteria were a history of stroke,
transient ischemic attack, coronary instability, congestive heart
failure, chronic or acute inflammatory conditions, cancer and
recent intracranial hemorrhage. From the health check-up center,
nine healthy donor control samples, (25-75 years) were selected as
the control group.
Approval for the study was obtained from the Ethics
Committee of Changzhou No. 2 People's Hospital, Affiliated Nanjing
Medical University. Informed consent and relevant clinical
information were obtained from all participants. Blood samples (5
ml) were collected from all participants and were centrifuged at
3,000 × g for 10 min at 4°C. Serum samples were then stored at
−80°C.
Cell culture and transfection
HUVECs were obtained from the American Type Culture
Collection (ATCC). HUVECs were routinely cultured in DMEM (Thermo
Fisher Scientific, Inc.) containing 15% FBS (Gibco; Thermo Fisher
Scientific, Inc.) in an incubator under humidity conditions of 5%
CO2 and 37°C, and cells in logarithmic growth phase were
used. HUVECs were treated with 100 μmol/l OX-LDL for 24 h.
The short hairpin RNA (shRNA)-negative control (NC),
shRNA-KLF5-1/2, shRNA-LINC00346-1/2 were obtained from Shanghai
GenePharma Co., Ltd. A KLF5 overexpression plasmid, pcDNA3.1-KLF5
(OV-KLF5), was commercially constructed by Shanghai GenePharma Co.,
Ltd., and an empty pcDNA 3.1 vector (OV-NC) was used as the
control. For miRNA transfection, miR-148-5p mimic, mimic-NC were
obtained from Sangon Biotech Co., Ltd. OX-LDL-stimulated HUVECs
(2nd generation) were transfected with shRNA-NC (500 μM),
shRNA-KLF5-1/2 (500 μM), OV-NC (500 μM), OV-KLF5 (500
μM), shRNA-LINC00346-1/2 (500 μM), miR-NC (500
μM) and miR-148-5p mimic (500 μM) using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
After 48 h of transfection, the cells were harvested for downstream
assays.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the serum samples and
HUVECs in each group using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions, and 2 μg RNA per sample were reverse
transcribed into cDNA by First-Stand cDNA Synthesis Super Mix
(TransGen Biotech Co., Ltd.). Each gene was amplified by qPCR. The
following thermocycling conditions were used: The reaction
conditions were 95°C pre-denaturation for 30 sec, 95°C denaturation
for 5 sec, 60°C annealing for 30 sec, 72°C extension for 30 sec,
and 40 cycles were repeated. The primer sequences of all genes were
as follows: KLF5 forward, 5′-ACG CTT GGC CTA TAA CTT GGT-3′ and
reverse, 5′-TGG AGG AAG CTG AGG TGTCA-3′; LINC00346 forward, 5′-AGC
TTG AAT GGC GTT GGA ACC TAT AG-3′ and reverse, 5′-ATA GTC CCT TCC
TCG AAT CCT AGT-3′; GAPDH forward, 5′-AAG GTG AAG GTC GGA GTC A-3′
and reverse, 5′-GGA AGA TGG TGA TGG GAT TT-3′; miR-148a-3p forward,
5′-TCA GTG CAC TAC AGA ACT TTG T-3′ and reverse, 5′-GTC ACC CCT GTT
TCT GGC AC-3′; U6 forward, 5′-CTC GCT TCG GCA GCA CA-3′ and
reverse, 5′-AAC GCT TCA CGA ATT TGC GT-3′. GAPDH was used as the
endogenous control of KLF5 and LINC00346, and U6 was used as the
endogenous control of miR-148a-3p. The relative expression of KLF5,
LINC00346 and miR-148a-3p was calculated using the
2−ΔΔCq method (16).
Western blot analysis
HUVECs in each group were digested and collected by
trypsin. Subsequently, total protein was extracted from the cells
in each group with the addition of cell lysis solution and
centrifugation at 3,000 × g at 4°C for 10 min. The quantity
measurement of the total isolated protein was performed using the
BCA method. Protein samples were denatured in a boiling water bath
for 5 min. Denatured proteins were obtained and 80 μg
protein per lane was separated via 12% SDS-PAGE electrophoresis.
The PVDF membrane was then blocked with 5% skimmed milk powder on a
shaking table at room temperature for 2 h, and then incubated with
anti-KLF5 (dilution, 1:1,000; ab137676), phosphorylated
(p-)endothelial nitric oxide synthase (eNOS; dilution, 1:1,000;
ab184154), eNOS (dilution, 1:1,000; ab76198) and GAPDH (dilution,
1:25,00; ab9485) (all from Abcam) antibodies overnight at 4°C.
Following primary antibody incubation, the membranes were incubated
with horseradish peroxidase-conjugated IgG secondary antibody
(dilution, 1:2,000; ab6721; Abcam) for 1 h at room temperature.
Protein bands were visualized using ECL reagent (Thermo Fisher
Scientific, Inc.) and densitometric analysis was performed using
Quantity One software (Version 4.6.6; Bio-Rad Laboratories,
Inc.).
ELISA
HUVECs at the logarithmic growth stage were
inoculated into a 10-cm culture dish at an appropriate density.
After the indicated treatments, the HUVECs, together with the
culture medium, were centrifuged at 3,000 × g for 5 min at 4°C, in
order to obtain the supernatant. The contents of TNF-α, IL-1β and
IL-6 in the supernatant were determined according to the
instructions of TNF-α (cat. no. PT518), IL-1β (cat. no. PI305) and
IL-6 (cat. no. PI330) ELISA kits (Beyotime Institute of
Biotechnology).
Detection of nitric oxide (NO)
levels
HUVECs were digested to prepare a 1×108/l
cell suspension, which was inoculated into a 24-well plate.
According to the experimental groups, the corresponding treatment
was performed. The cell culture supernatant was obtained to detect
NO levels according to the instructions provided with the NO kit
(cat. no. S0021S; Beyotime Institute of Biotechnology).
Chromatin immunoprecipitation (ChIP)-PCR
assay
HUVECs were fixed at room temperature with
formaldehyde for 10 min, washed with PBS, and the collected cells
were placed in an ultrasonic water bath. Ultrasonic treatment was
then carried out. The long strand DNA of the gene was broken into
200-1,000 bp DNA fragments by ultrasonic treatment, which was
centrifuged at 16,000 × g for 15 min at room temperature to obtain
the supernatant. A total of 20 μl supernatant was removed as
the input. KLF5 antibody (dilution, 2.5-5 μg/106
cells; ab277773, Abcam) and corresponding IgG antibody (A7016;
Beyotime Institute of Biotechnology) were added into the remaining
supernatant, which was incubated overnight at 4°C. Magnetic beads
were added the following day for 2 h at room temperature. Following
centrifugation at 16,000 × g for 2 min at 4°C, the precipitate was
eluted step by step with low-salt buffer solution and high-salt
buffer solution, respectively, and NaCl solution was then added
followed by incubation at 65°C overnight to remove chromatin
fixation. EDTA, Tris-HCl and protease were added followed by
incubation at 65°C for 1 h. Finally, phenol-chloroform method was
used to extract the 50 μl purified product for PCR
detection.
Luciferase reporter assay
HUVECs were transfected with LINC00346 (full)-L and
OV-NC/OV-KLF5, LINC00346 (site2)-L and OV-KLF5 using Lipofectamine
2000®. The relative luciferase activity was detected
using a Dual Luciferase Reporter assay system (Promega Corporation)
to confirm the binding between KLF5 and LINC00346.
Wild-type (WT)-LINC00346, mutant (Mut)-LINC00346,
WT-KLF5 or Mut-KLF5 were co-transfected into HUVECs with
miR-148a-3p mimic or miR-NC using Lipofectamine 2000®
and the luciferase activity was detected to confirm the binding
between LINC00346/KLF5 and miR-148a-3p after transfection for 48 h.
Relative luciferase activity was normalized to Renilla
luciferase activity (control).
Statistical analysis
SPSS 22.0 software (IBM Corp.) was used for the
statistical analysis and the data are presented as the mean ± SD.
One-way ANOVA was used for the statistical comparisons between
groups, followed by Tukey's post hoc test for multiple comparisons
between groups. An independent samples unpaired Student's t-test
was used for comparisons between two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
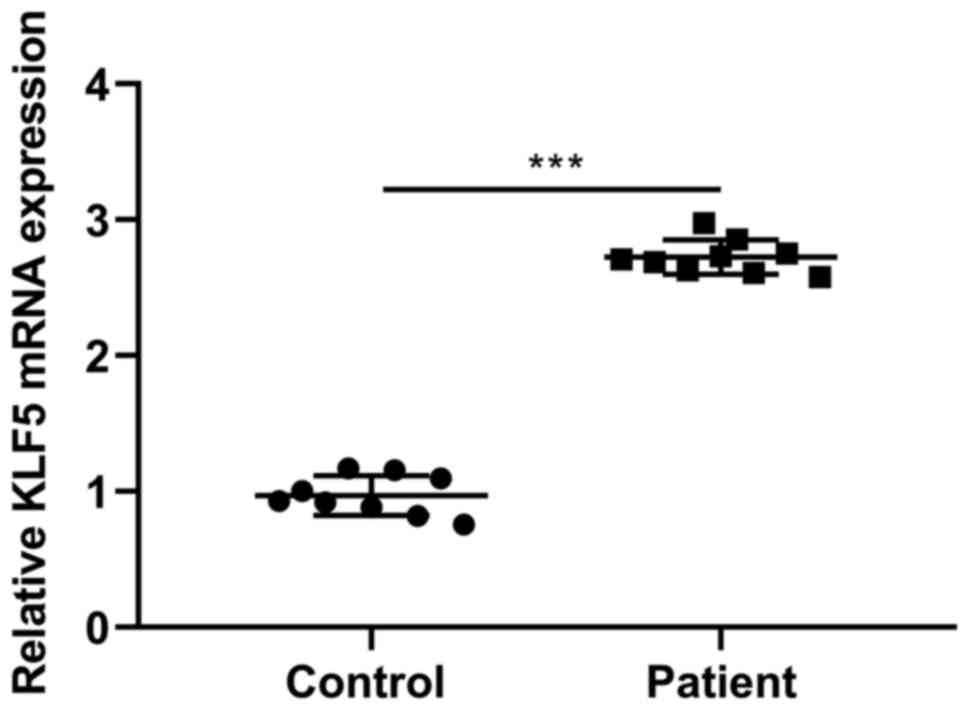
KLF5 expression in AS patient serum
As shown in Fig.
1, KLF5 expression levels were increased in serum from patients
with AS in comparison with the respective healthy donor expression
levels.
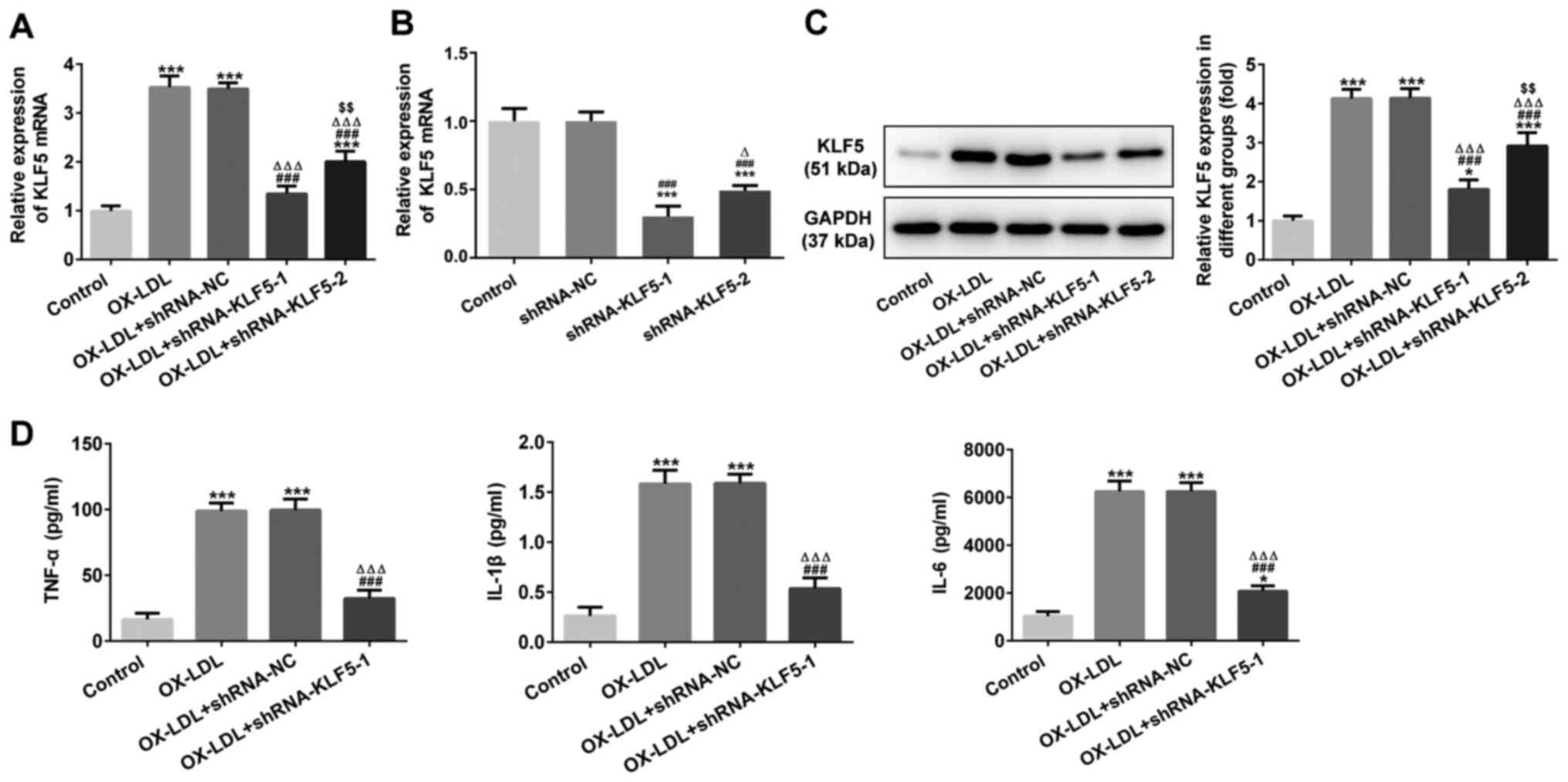
KLF5 interference inhibits OX-LDL-induced
inflammatory factor expression in HUVECs
KLF5 mRNA expression was increased in
OX-LDL-stimulated HUVECs and was decreased when the
OX-LDL-stimulated HUVEC cells were transfected with shRNA-KLF5-1/2.
KLF5 mRNA expression was decreased further in the OX-LDL-stimulated
HUVECs transfected with shRNA-KLF5-1 in comparison with HUVECs
transfected with shRNA-KLF5-2 (Fig.
2A). KLF5 mRNA expression was also decreased further in HUVECs
transfected with shRNA-KLF5-1 as compared with HUVECs transfected
with shRNA-KLF5-2 (Fig. 2B).
Changes in KLF5 protein expression in these groups were similar to
those obtained for KLF5 mRNA expression (Fig. 2C). Therefore, shRNA-KLF5-1 was
selected for use in subsequent experiments. The TNF-α, IL-1β and
IL-6 levels in OX-LDL-stimulated HUVECs were upregulated and were
subsequently suppressed by KLF5 interference (Fig. 2D).
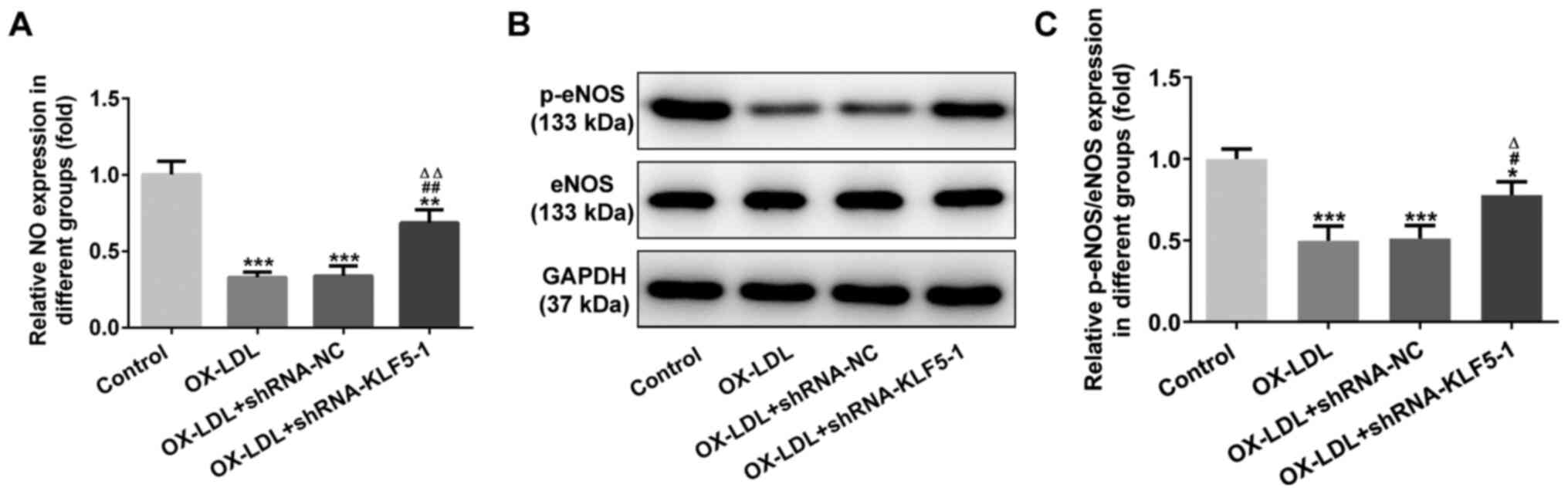
KLF5 interference inhibits OX-LDL-induced
injury to HUVECs
NO expression was decreased in the OX-LDL-stimulated
HUVECs, while KLF5 interference increased NO expression (Fig. 3A). p-eNOS/eNOS expression in
OX-LDL-stimulated HUVECs was decreased. This effect was reversed by
KLF5 interference (Fig. 3B and
C).
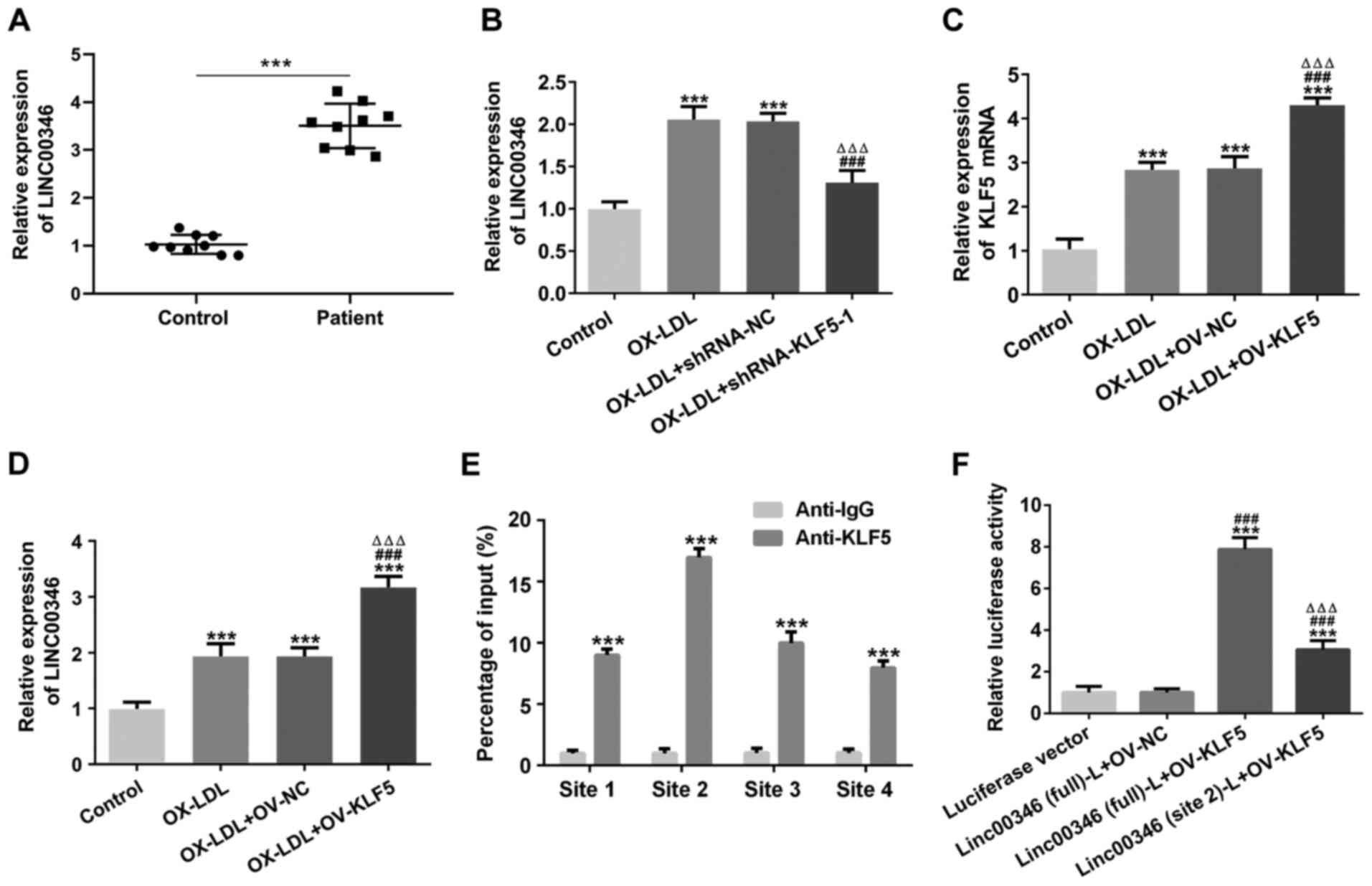
Transcription factor KLF5 promotes
LINC00346 transcription
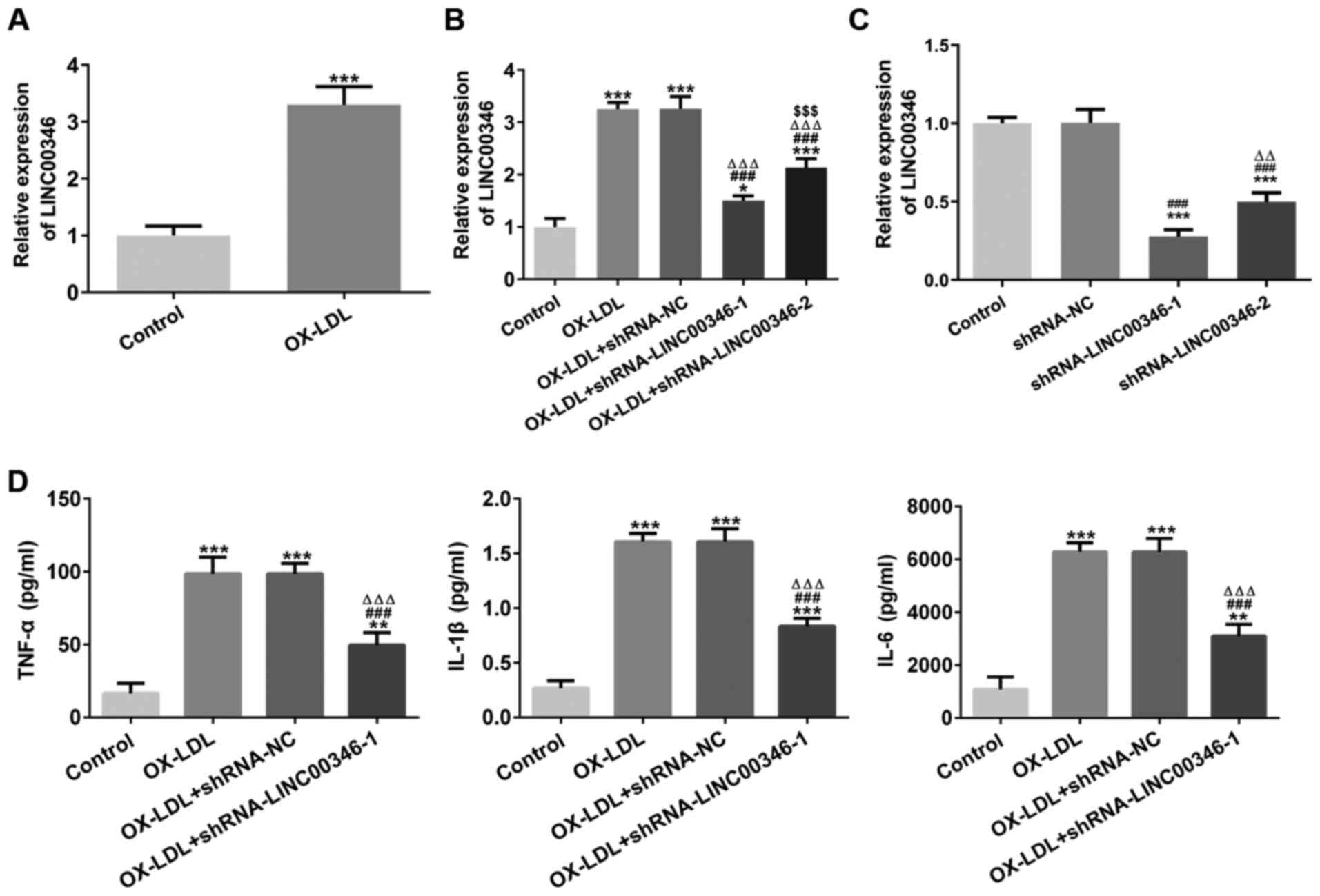
LINC00346 expression was increased in the serum of
patients with AS (Fig. 4A).
LINC00346 expression was increased in OX-LDL-stimulated HUVECs and
was subsequently suppressed by KLF5 interference (Fig. 4B). KLF5 expression was increased
in OX-LDL-stimulated HUVECs, and was further promoted by KLF5
overexpression (Fig. 4C). KLF5
overexpression further increased LINC00346 expression in
OX-LDL-stimulated HUVECs (Fig.
4D). The results presented in Fig. 4E illustrate that KLF5 can bind to
the four promoter regions of LINC00346. Mutated site2 LINC00346 and
full LINC00346 were respectively co-transfected into HUVECs with
OV-KLF5 plasmid and the results revealed that KLF5 (site2) could
bind to LINC00346 (Fig. 4F).
LINC00346 interference inhibits the
expression of inflammatory factors in HUVECs, induced by
OX-LDL
LINC00346 expression was increased in
OX-LDL-stimulated HUVECs (Fig.
5A) and LINC00346 expression was decreased in OX-LDL-stimulated
HUVECs transfected with shRNA-LINC00346-1/2. LINC00346 expression
was decreased to a greater extent in shRNA-LINC00346-1-transfected
OX-LDL-stimulated HUVEC cells in comparison with the
shRNA-LINC00346-2-transfected cells (Fig. 5B); LINC00346 expression was
further downregulated in shRNA-LINC00346-1-transfected HUVECs in
comparison with the shRNA-LINC00346-2-transfected cells (Fig. 5C); thus, shRNA-LINC00346-1 was
selected for use in subsequent experiments. OX-LDL induction
upregulated the TNF-α, IL-1β and IL-6 levels, which were then
decreased by LINC00346 interference (Fig. 5D).
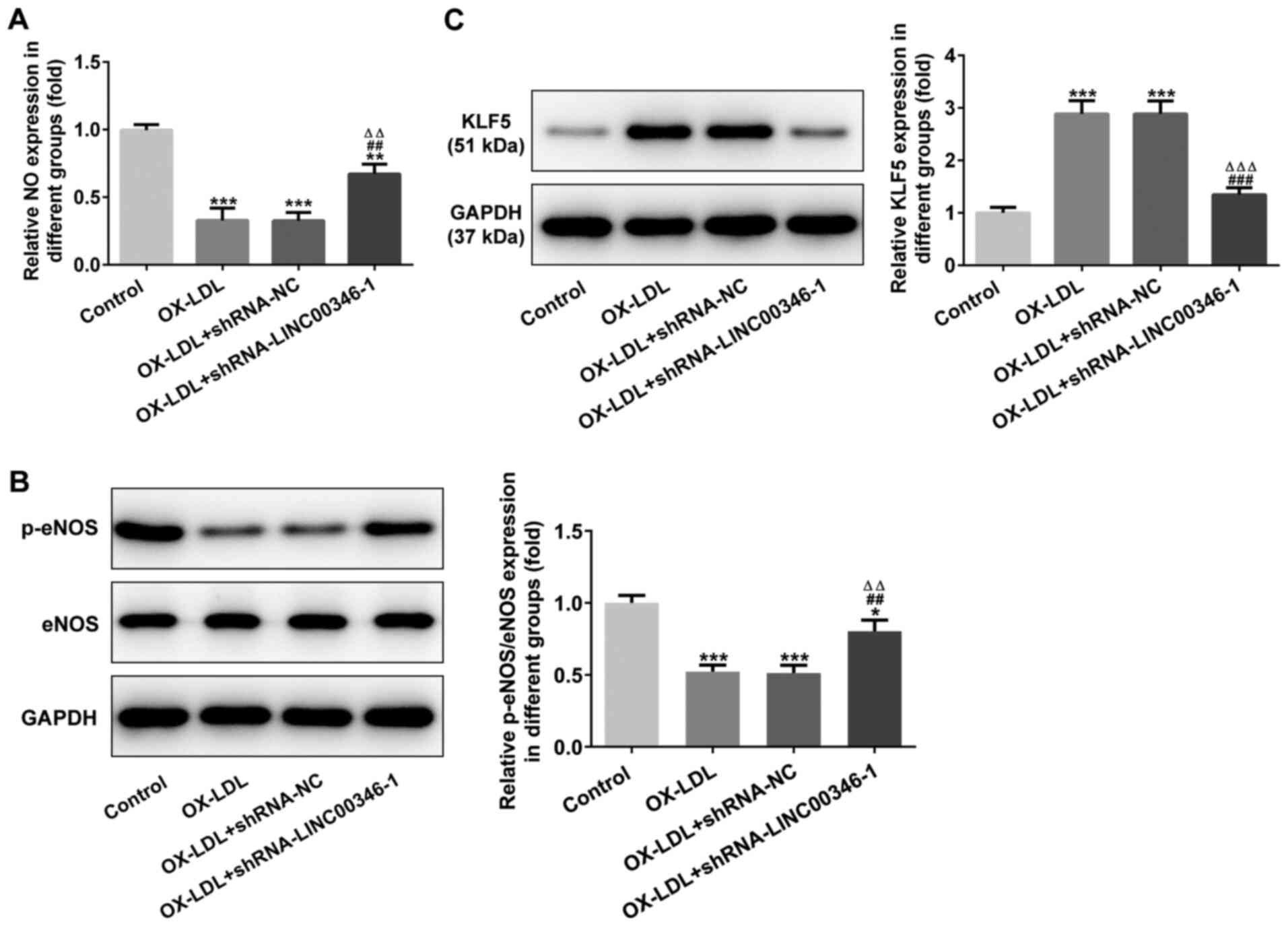
LINC00346 interference inhibits
OX-LDL-induced injury and KLF5 expression in HUVECs
NO expression was decreased in OX-LDL-stimulated
HUVECs and was promoted by LINC00346 interference (Fig. 6A). p-eNOS/eNOS expression was
downregulated in OX-LDL-stimulated HUVECs, while LINC00346
interference increased p-eNOS/eNOS expression (Fig. 6B). KLF5 expression was also
increased in OX-LDL-stimulated HUVECs, which was suppressed by
LINC00346 interference (Fig.
6C).
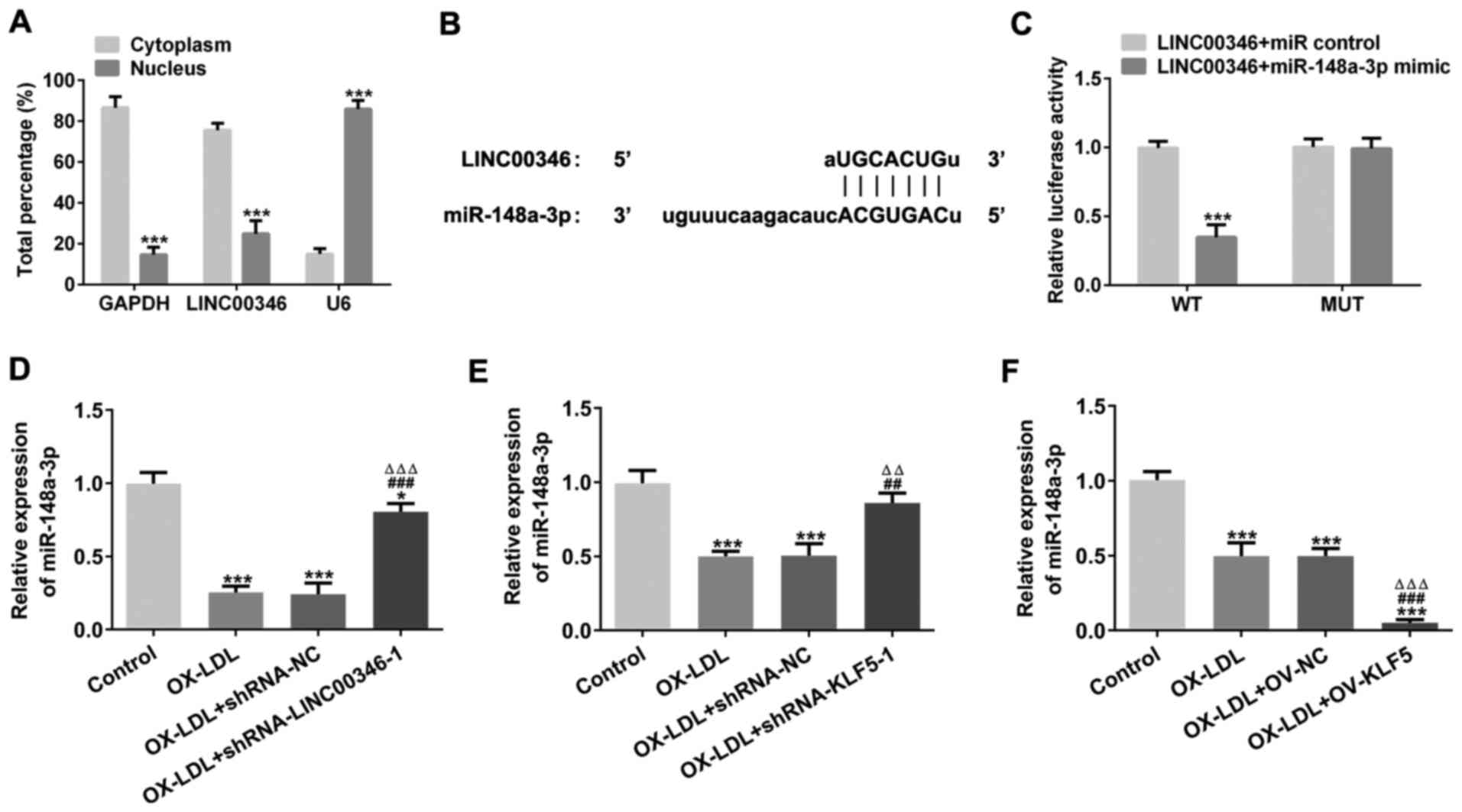
LINC00346 functions as a sponge for
miR-148a-3p
LINC00346 expression in the cytoplasm was increased
compared with that in the nucleus (Fig. 7A). The binding sites between
LINC00346 and miR-148a-3p shown in Fig. 7B and C confirmed that LINC00346
could bind to miR-148a-3p. miR-148a-3p expression was decreased in
OX-LDL-stimulated HUVECs. LINC00346 interference or KLF5
interference promoted miR-148a-3p expression, while KLF5
overexpression suppressed miR-148a-3p expression in
OX-LDL-stimulated HUVECs (Fig.
7D-F).
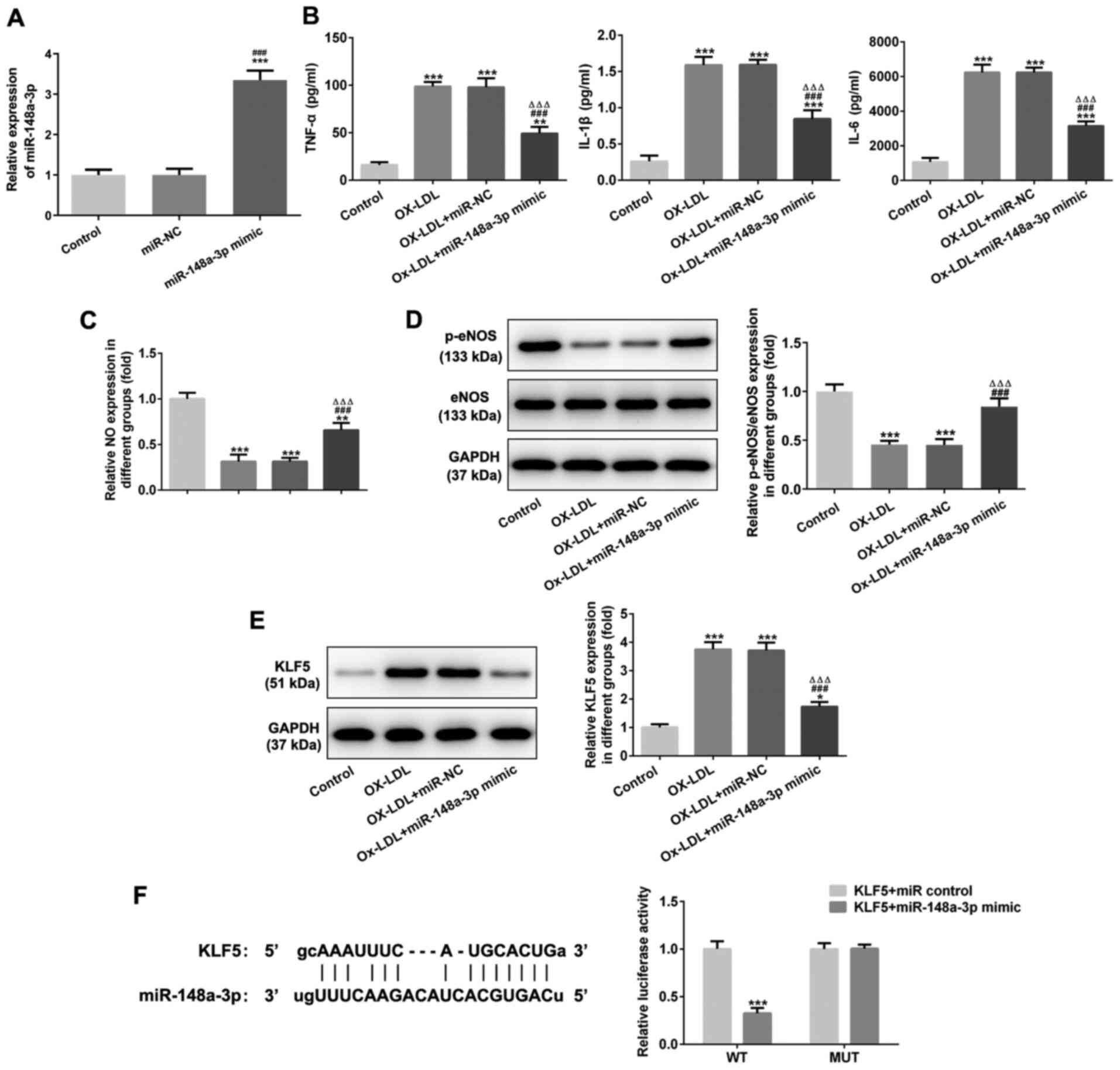
miR-148a-3p overexpression inhibits
OX-LDL-induced injury and OX-LDL-induced inflammatory factor
expression in HUVEC cells and miR-148a-3p targets KLF5
miR-148a-3p expression was increased in HUVECs
transfected with miR-148a-3p mimic (Fig. 8A). miR-148a-3p overexpression
decreased the TNF-α, IL-1β and IL-6 levels (Fig. 8B) and increased NO expression in
OX-LDL-stimulated HUVECs (Fig.
8C). miR-148a-3p overexpression increased the expression of
p-eNOS/eNOS (Fig. 8D) and
inhibited the expression of KLF5 in OX-LDL-stimulated HUVECs
(Fig. 8E). As shown in Fig. 8F, binding sites were predicted
between miR-148a-3p and KLF5, and dual luciferase reporter assay
confirmed that miR-148a-3p could bind with KLF5.
Discussion
AS is a chronic inflammatory disease, in which
lipids and cholesterol accumulate in large and medium-sized
arteries and develop into fibrous plaques (17). The pathogenesis of AS is complex,
and involves an inflammatory reaction, abnormal cholesterol levels
and activation of the damage response (18,19). After decades of research, the
inflammatory response has been found to play an important role in
the occurrence and development of AS, and even in the later stage
of plaque formation (18,20).
The dysfunction of vascular endothelial cells occurs in the
impaired area of the arterial vascular system (21,22). The earliest pathological change
that can be detected in AS lesions is the focal infiltration of
circulating lipoprotein particles into the subendothelial layer
following physicochemical modification (23). Vascular lumen obstruction may be
caused by erosion of the surface intima (24), which may be triggered by
endothelial cell apoptosis, local endothelial denudation and
thrombosis (25). The
aforementioned studies demonstrated that vascular endothelial cell
dysfunction is an important factor leading to AS progression and
adverse outcomes. HUVEC cells are often used as the cellular model
of AS in previous studies (26-28). In the present study, OX-LDL
induced inflammation and injury, and KLF5 interference inhibited
the inflammatory response and improved the function of HUVECs
induced by OX-LDL.
There is increasing evidence to indicate that
lncRNAs can regulate vascular remodeling, lipid metabolism and the
inflammatory response (29-31). A previous study indicated that
KLF5 transcription factor, promoted the transcription of LINC00346
(32). Current research on
LINC00346 focuses on its role in multiple tumors. LINC00346 is
highly expressed in gastric cancer and is considered a tumor
inducer in vitro and in vivo (32). In pancreatic cancer, LINC00346
expression has been found to be increased in tumor tissue samples,
while the knockdown of LINC00346 suppresses pancreatic cancer cell
proliferation, migration, invasion and tumor formation (33). LINC00346 has been shown to be
upregulated in tissues and hepatocellular carcinoma cell lines, and
LINC00346 may promote the viability, proliferation, migration and
invasion of hepatocellular carcinoma cells (34). LINC00346 has been observed to be
overexpressed in colorectal cancer tissues and cell lines, and the
inhibition of LINC00346 impairs the proliferative, migratory, and
invasive abilities of colorectal cancer cells (35). The present study explored the
role of LINC00346 in AS. LINC00346 expression was increased in
OX-LDL-stimulated HUVECs, and LINC00346 interference resulted in
the suppression of OX-LDL-induced inflammation, decreased of
OX-LDL-induced injury and the downregulation of KLF5 expression in
HUVECs.
In recent years, studies have demonstrated that
miRNAs are involved in AS lesions, and AS lesions also cause miRNA
changes in vivo. miRNAs are more than clinically detectable
lesion indicators and may also represent possible novel future
therapeutic targets. A recent study on miRNA expression profiles in
AS demonstrated that miR-21 expression was found to be
significantly increased (36).
AS-related inflammation has been found to be inhibited by the
expression of miR-146 in macrophages, endothelial cells and
hematopoietic cells (37-39).
miR-126 is expressed in vascular endothelial cells participating in
the inflammatory process of AS, inhibits inflammatory factors,
promotes the autophagy of endothelial cells, and thus plays an
anti-AS role in maintaining vascular integrity (40,41). In the present study, it was
confirmed that LINC00346 could bind to miR-148a-3p, and that it
could also bind to KLF5. miR-148a-3p expression has been shown to
be highly expressed in patients with AS in comparison with healthy
individuals, and miR-148a-3p overexpression promotes proliferation
and migration, whereas on the other hand it suppresses endothelial
cell apoptosis (42).
Furthermore, miR-148a-3p overexpression inhibited OX-LDL-induced
inflammatory factors expression and injury in HUVECs; the
corresponding findings of the present study were consistent with
those of the aforementioned previous study.
In conclusion, the present study demonstrated that
the expression of KLF5 and LINC00346 was increased in AS patient
serum. Additionally, KLF5 interference inhibited inflammation and
attenuated injury to HUVECs stimulated with OX-LDL, which was also
suppressed by LINC00346 interference. In addition, OX-LDL-induced
inflammatory factor expression and injury in HUVECs were also
impaired by miR-148a-3p overexpression. The
KLF5/LINC00346/miR-148a-3p axis may therefore enhance current
understanding of AS pathogenesis and may represent potential
targets for the development of novel therapeutics for
cardiovascular diseases.
However, there are also some limitations to the
present study. OX-LDL through lectin-type oxidized LDL receptor 1
(LOX-1), may cause an increase in leukocyte adhesion molecules, the
activation of apoptotic pathways and an increase in reactive oxygen
species in endothelial cells and cause endothelial dysfunction
(43). OX-LDL and LOX-1, in
combination, play a role in the pathogenesis of atherosclerosis.
The authors aim to explore the association between
KLF5/LINC00346/miR-148a-3p loop and LOX-1 in future studies. In
addition, the effects of disease duration, serum lipids such as
cholesterol and medication on the expression of KLF5 were not
investigated in the present study; thus, this is also a future
research objective. Furthermore, more advanced techniques are
required, in order to elucidate further the possibility of KLF5
originating also from blood cells.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FW and YJ conceived and designed the study. FW, JG,
SH and CZ performed the experiments. FW, ZS, YS and YX analyzed the
data. FW wrote the manuscript. YJ revised the manuscript. All
authors gave read and approved the final manuscript. FW, JG and SH
are responsible for confirming the authenticity of the raw
data.
Ethics approval and consent to
participate
Approval for the study was obtained from the Ethics
Committee of Changzhou No. 2 People's Hospital, Affiliated Nanjing
Medical University. Informed consent and relevant clinical
information were obtained from all participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nature. 473:317–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Beaufrère H, Vet DM, Cray C, Ammersbach M
and Tully TN Jr: Association of plasma lipid levels with
atherosclerosis prevalence in psittaciformes. J Avian Med Surg.
28:225–231. 2014. View
Article : Google Scholar
|
|
3
|
Herrington W, Lacey B, Sherliker P,
Armitage J and Lewington S: Epidemiology of atherosclerosis and the
potential to reduce the global burden of atherothrombotic disease.
Circ Res. 118:535–546. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dou Y, Chen Y, Zhang X, Xu X, Chen Y, Guo
J, Zhang D, Wang R, Li X and Zhang J: Non-proinflammatory and
responsive nanoplatforms for targeted treatment of atherosclerosis.
Biomaterials. 143:93–108. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gao Y, Ding Y, Chen H, Chen H and Zhou J:
Targeting Krüppel-like factor 5 (KLF5) for cancer therapy. Curr Top
Med Chem. 15:699–713. 2015. View Article : Google Scholar
|
|
6
|
Bell SM, Zhang L, Xu Y, Besnard V, Wert
SE, Shroyer N and Whitsett JA: Kruppel-like factor 5 controls
villus formation and initiation of cytodifferentiation in the
embryonic intestinal epithelium. Dev Biol. 375:128–139. 2013.
View Article : Google Scholar :
|
|
7
|
Ha JM, Yun SJ, Jin SY, Lee HS, Kim SJ,
Shin HK and Bae SS: Regulation of vascular smooth muscle phenotype
by cross-regulation of krüppel-like factors. Korean J Physiol
Pharmacol. 21:37–44. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao Y, Wu K, Chen Y, Zhou J, Du C, Shi Q,
Xu S, Jia J, Tang X, Li F, et al: Beyond proliferation: KLF5
promotes angiogenesis of bladder cancer through directly regulating
VEGFA transcription. Oncotarget. 6:43791–43805. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Owens GK: Regulation of differentiation of
vascular smooth muscle cells. Physiol Rev. 75:487–517. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang W, Zhang Y, Wang L, Li J, Li Y, Yang
X and Wu Y: MircroRNA-152 prevents the malignant progression of
atherosclerosis via down-regulation of KLF5. Biomed Pharmacother.
109:2409–2414. 2019. View Article : Google Scholar
|
|
11
|
Zhang YN, Xie BD, Sun L, Chen W, Jiang SL,
Liu W, Bian F, Tian H and Li RK: Phenotypic switching of vascular
smooth muscle cells in the 'normal region' of aorta from
atherosclerosis patients is regulated by miR-145. J Cell Mol Med.
20:1049–1061. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Song X, Li Z and Liu B: Long
non-coding RNAs in coronary atherosclerosis. Life Sci. 211:189–197.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bian W, Jing X, Yang Z, Shi Z, Chen R, Xu
A, Wang N, Jiang J, Yang C, Zhang D, et al: Downregulation of
LncRNA NORAD promotes Ox-LDL-induced vascular endothelial cell
injury and atherosclerosis. Aging (Albany NY). 12:6385–6400. 2020.
View Article : Google Scholar
|
|
14
|
Wang YQ, Xu ZM, Wang XL, Zheng JK, Du Q,
Yang JX and Zhang HC: LncRNA FOXC2-AS1 regulated proliferation and
apoptosis of vascular smooth muscle cell through targeting
miR-1253/FOXF1 axis in atherosclerosis. Eur Rev Med Pharmacol Sci.
24:3302–3314. 2020.PubMed/NCBI
|
|
15
|
Huang T, Zhao HY, Zhang XB, Gao XL, Peng
WP, Zhou Y, Zhao WH and Yang HF: LncRNA ANRIL regulates cell
proliferation and migration via sponging miR-339-5p and regulating
FRS2 expression in atherosclerosis. Eur Rev Med Pharmacol Sci.
24:1956–1969. 2020.PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
17
|
Libby P, Buring JE, Badimon L, Hansson GK,
Deanfield J, Bittencourt MS, Tokgözoğlu L and Lewis EF:
Atherosclerosis. Nat Rev Dis Primers. 5:562019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Geovanini GR and Libby P: Atherosclerosis
and inflammation: Overview and updates. Clin Sci (Lond).
132:1243–1252. 2018. View Article : Google Scholar
|
|
19
|
Wang HH, Garruti G, Liu M, Portincasa P
and Wang DQ: Cholesterol and lipoprotein metabolism and
atherosclerosis: Recent advances in reverse cholesterol transport.
Ann Hepatol. 16(Suppl 1: s3-105): S27–S42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Taleb S: Inflammation in atherosclerosis.
Arch Cardiovasc Dis. 109:708–715. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stary HC: Natural history and histological
classification of atherosclerotic lesions: An update. Arterioscler
Thromb Vasc Biol. 20:1177–1178. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Virmani R, Kolodgie FD, Burke AP, Farb A
and Schwartz SM: Lessons from sudden coronary death: A
comprehensive morphological classification scheme for
atherosclerotic lesions. Arterioscler Thromb Vasc Biol.
20:1262–1275. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Simionescu N, Vasile E, Lupu F, Popescu G
and Simionescu M: Prelesional events in atherogenesis. Accumulation
of extracellular cholesterol-rich liposomes in the arterial intima
and cardiac valves of the hyperlipidemic rabbit. Am J Pathol.
123:109–125. 1986.PubMed/NCBI
|
|
24
|
Libby P: Mechanisms of acute coronary
syndromes. N Engl J Med. 369:883–884. 2013.PubMed/NCBI
|
|
25
|
Quillard T, Araújo HA, Franck G, Shvartz
E, Sukhova G and Libby P: TLR2 and neutrophils potentiate
endothelial stress, apoptosis and detachment: Implications for
superficial erosion. Eur Heart J. 36:1394–1404. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wen Y, Chun Y, Lian ZQ, Yong ZW, Lan YM,
Huan L, Xi CY, Juan LS, Qing ZW, Jia C and Ji ZH:
circRNA-0006896-miR1264-DNMT1 axis plays an important role in
carotid plaque destabilization by regulating the behavior of
endothelial cells in atherosclerosis. Mol Med Rep. 23:3112021.
View Article : Google Scholar :
|
|
27
|
Qian X, Wang H, Wang Y, Chen J, Guo X and
Deng H: Enhanced autophagy in GAB1-Deficient vascular endothelial
cells is responsible for atherosclerosis progression. Front
Physiol. 11:5593962021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu H, Ma S, Sun L, Gao J and Zhao C:
TGF-β1 upregulates the expression of lncRNA-ATB to promote
atherosclerosis. Mol Med Rep. 19:4222–4228. 2019.PubMed/NCBI
|
|
29
|
Deng L, Bradshaw AC and Baker AH: Role of
noncoding RNA in vascular remodelling. Curr Opin Lipidol.
27:439–448. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen Z: Progress and prospects of long
noncoding RNAs in lipid homeostasis. Mol Metab. 5:164–170. 2015.
View Article : Google Scholar
|
|
31
|
Hu YW, Zhao JY, Li SF, Huang JL, Qiu YR,
Ma X, Wu SG, Chen ZP, Hu YR, Yang JY, et al:
RP5-833A20.1/miR-382-5p/ NFIA-dependent signal transduction pathway
contributes to the regulation of cholesterol homeostasis and
inflammatory reaction. Arterioscler Thromb Vasc Biol. 35:87–101.
2015. View Article : Google Scholar
|
|
32
|
Xu TP, Ma P, Wang WY, Shuai Y, Wang YF, Yu
T, Xia R and Shu YQ: KLF5 and MYC modulated LINC00346 contributes
to gastric cancer progression through acting as a competing
endogeous RNA and indicates poor outcome. Cell Death Differ.
26:2179–2193. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Peng WX, He RZ, Zhang Z, Yang L and Mo YY:
LINC00346 promotes pancreatic cancer progression through the
CTCF-mediated Myc transcription. Oncogene. 38:6770–6780. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang N and Chen X: A positive feedback
loop involving the LINC00346/β-catenin/MYC axis promotes
hepatocellular carcinoma development. Cell Oncol (Dordr).
43:137–153. 2020. View Article : Google Scholar
|
|
35
|
Tong WH, Mu JF and Zhang SP: LINC00346
accelerates the malignant progression of colorectal cancer via
competitively binding to miRNA-101-5p/MMP9. Eur Rev Med Pharmacol
Sci. 24:6639–6646. 2020.PubMed/NCBI
|
|
36
|
Pordzik J, Pisarz K, De Rosa S, Jones AD,
Eyileten C, Indolfi C, Malek L and Postula M: The potential role of
platelet-related microRNAs in the development of cardiovascular
events in high-risk populations, including diabetic patients: A
review. Front Endocrinol (Lausanne). 9:742018. View Article : Google Scholar
|
|
37
|
Li Z, Wang S, Zhao W, Sun Z, Yan H and Zhu
J: Oxidized low-density lipoprotein upregulates microRNA-146a via
JNK and NF-κB signaling. Mol Med Rep. 13:1709–1716. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cheng HS, Sivachandran N, Lau A, Boudreau
E, Zhao JL, Baltimore D, Delgado-Olguin P, Cybulsky MI and Fish JE:
MicroRNA-146 represses endothelial activation by inhibiting
pro-inflammatory pathways. EMBO Mol Med. 5:1017–1034. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Del Monte A, Ar royo AB, Andrés-Manzano
MJ, García-Barberá N, Caleprico MS, Vicente V, Roldan V,
Gonzalez-Conejero R, Martínez C and Andrés V: MiR-146a deficiency
in hematopoietic cells is not involved in the development of
atherosclerosis. PLoS One. 13:e01989322018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chistiakov DA, Orekhov AN and Bobryshev
YV: The role of miR-126 in embryonic angiogenesis, adult vascular
homeostasis, and vascular repair and its alterations in
atherosclerotic disease. J Mol Cell Cardiol. 97:47–55. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tang F and Yang TL: MicroRNA-126
alleviates endothelial cells injury in atherosclerosis by restoring
autophagic flux via inhibiting of PI3K/Akt/mTOR pathway. Biochem
Biophys Res Commun. 495:1482–1489. 2018. View Article : Google Scholar
|
|
42
|
Shang L, Quan A, Sun H, Xu Y, Sun G and
Cao P: MicroRNA-148a-3p promotes survival and migration of
endothelial cells isolated from Apoe deficient mice through
restricting circular RNA 0003575. Gene. 711:1439482019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kattoor AJ, Kanuri SH and Mehta JL: Role
of Ox-LDL and LOX-1 in atherogenesis. Curr Med Chem. 26:1693–1700.
2019. View Article : Google Scholar
|