Introduction
Long-QT syndrome (LQTS) is a hereditary cardiac ion
channel disease with 17 clinical subtypes characterized by
potentially fatal arrhythmia and sudden cardiac death (1). Long QT syndrome type 2 (LQT2) is
caused by a human-ether-a-go-go-related gene (HERG) mutation
(2). HERG is located at 7q35-36
and encodes the cardiac Kv11.1 potassium channel, which rapidly
activates the delayed rectifier potassium current (IKr)
(3). The HERG channel is an
essential cardiac ion channel whose mutations can cause partial or
complete reduction of IKr current and delayed ventricular
repolarization (4). Four
possible mechanisms have been identified for the loss of HERG
channel function caused by HERG gene mutations: Channel protein
synthesis defects, trafficking barriers after protein synthesis,
gate control defects and current conduction defects (5). A previous study has reported that a
heterozygous missense mutation (A561V) linked to LQT2, syncope and
epilepsy was identified in the S5/pore region of HERG protein
(6). HERG-A561V mutation causes
the HERG channel protein to change conformation and remain in the
endoplasmic reticulum (ER), preventing it from maturing and
trafficking to the plasma membrane where it can serve a role
(7).
The ER quality control (ERQC) system, which mediates
folding and trafficking of channel proteins in the ER, is
fundamental to folding newly synthesized proteins (8). The ERQC system monitors HERG and
other nascent proteins and facilitates their exit from the ER for
the following processing stage (9). The calnexin (CNX)/calreticulin
(CRT) cycle, composed of lectin-like molecular chaperones CNX and
CRT, is an essential part of the ERQC system that depends on
modifying protein glycosylation and sugar chain structures in the
ER (10). The CNX/CRT cycle is
one of the important monitoring mechanisms for protein folding and
assembly (11). The disulfide
bond isomerase ER protein 57 (ERP57), involved in the CNX/CRT
cycle, catalyzes the oxidation and isomerization of disulfide bonds
in glycoproteins and binds to CNX/CRT glycoproteins (12,13). The formation of transient
disulfide bonds assists protein folding (14). ERP57, a prominent multifunctional
member of the protein disulfide isomerase (PDI) family, is detected
at various levels in multiple cellular locations, including the ER,
nucleus, cytoplasm, mitochondria and plasma membrane (15). ERP57 has 505 amino acids and
consists of four domains: A-b-b′-a′, N-terminal signal sequence and
Gln-Glu-Asp-Leu C-terminal ER retention/search motif (16). Both the a and a′ domains have
active sites similar to thioredoxin, and each region has a
redox-active Cys-Gly-His-Cys catalytic sequence (15). The b and b′ domains contain
binding sites for CRT and CNX (17). The catalytically inactive central
domains, b and b′, serve a vital role in the specific functionality
of ERP57 to bind proteins and assist in their folding, and contain
binding sites for CNX and CRT (18).
At present, no study has described how to influence
the trafficking of mutant HERG proteins by regulating key molecular
chaperones in the CNX/CRT cycle. Our previous study verified that
the two chaperone proteins CNX and CRT arrest the export of mutant
HERG from the ER and empower it to refold into the correct native
conformation and thus serve a role in preventing trafficking and
degrading defective HERG mutant protein (19). The present study further
evaluated the roles of CNX, CRT and ERP57 in trafficking the
defective HERG-A561V mutant protein.
Materials and methods
Cells, cDNA and cell culture
Experiments were performed using the 293 cell line
obtained from The Cell Bank of Type Culture Collection of The
Chinese Academy of Sciences. Wild-type (WT)-HERG was expressed
using pcDNA3 vector (Invitrogen; Thermo Fisher Scientific, Inc.).
A561V was produced by site-directed mutagenesis of WT cDNA and then
subcloned into WT pcDNA3 vector at the BstEII/XhoI
restriction sites. The WT, heterozygous and mutant cell lines were
constructed by transiently transfecting pcDNA3-WT,
pcDNA3-WT/pcDNA3-A561V and pcDNA3-A561V, respectively. At 24 h
after expression of the aforementioned plasmid, pcDNA3-Vector
(negative control), pcDNA3-CRT (CALR; NM_004343) and pcDNA3-ERP57
(PDIA3; NM_005313) were transfected to overexpress CRT and ERP57.
pL-short hairpin RNA-Vector (negative control) and pL-short hairpin
RNA-CRT (CALR-RNAi:5′-GAT CCC ctC TGT GAG ACT CGA GAA CTT CTC GAG
AAG TTC TCG AGT CTC ACA GATT TT T-3′) were transfected to reduce
CRT expression. Domain deletion was implemented by transfecting
pcDNA3-ERP57-b domain deletion [PDIA3, NM_005313(del244-357aa)], p
cDNA 3 -E R P 57-b′ domain deletion [ PDIA3,
NM_005313(del135-240aa)] or pcDNA3-ERP57-bb′ domain deletion
[PDIA3, NM_005313(del135-357aa)] (all plasmids were obtained from
Shanghai GeneChem Co., Ltd.) into the heterozygous cells.
TransIT-2020 (Mirus Bio, LLC) diluted in Opti-MEM I reducing serum
medium (Gibco; Thermo Fisher Scientific, Inc.) was used to
transfect a total of 2.5 µg of plasmid into the cells at
room temperature (23±2°C) for 48 h, according to the manufacturer's
instructions. After 48 h, the transfected cells were used for
experiments. All cells were cultured in high glucose DMEM (Gibco;
Thermo Fisher Scientific, Inc.) containing 10% FBS (Bovogen
Biologicals Pty, Ltd.), and kept in a humid 5% CO2
incubator at 37°C.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from the treated 293 cells
using TRIzol reagent (Sangon Biotech Co., Ltd.). Reverse
transcription was conducted using TransScript®
All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (One-Step
gDNA Removal) (cat. no. AT341-01; TransGen Biotech Co., Ltd.) at
42°C for 15 min and heating at 85°C for 5 sec. Subsequently, qPCR
was performed using the SYBR-Green Real-Time PCR kit (cat. no.
TSE202; TsingKe Biological Technology) according to the
manufacturer's protocols. The PCR amplification reaction was as
follows: 95°C for 1 min, followed by 45 cycles of 95°C for 10 sec
and 60°C for 60 sec. Relative quantification was calculated using
the 2−ΔΔCq method, with GAPDH being used to normalize
mRNA expression (20). The
primers used for PCR were as follows: GAPDH forward, 5′-GGT GTG AAC
CAT GAG AAG TAT GA-3′ and reverse, 5′-GAG TCC TTC CAC GAT ACC AAA
G-3′; HERG forward, 5′-AGG ACA AGT ATG TGA CGG CG-3′ and reverse,
5′-AGG GAG CCA ATG AGC ATG AC-3′; ERP57 forward, 5′-GGA GGA GTT CTC
GCG TGA TG-3′ and reverse, 5′-CAG GCC CAT CAT TGC TCT CT-3′; and
CRT forward, 5′-GGC AGA TCG ACA ACC CAG AT-3′ and reverse, 5′-GAT
GGT GCC AGA CTT GAC CT-3′.
Western blotting
293 cells expressing HERG in 35-mm diameter culture
dishes were harvested for analysis at 48 h after transient
transfection. Resuspended cells were washed with ice-cold PBS. Cell
pellets were solubilized in ice-cold RIPA buffer (Beijing Solarbio
Science & Technology Co., Ltd.) containing the protease
inhibitor PMSF (Beijing Solarbio Science & Technology Co.,
Ltd.), incubated on ice for 30 min and centrifuged at 13,800 × g
for 15 min at 4°C to pellet detergent-insoluble cell debris. The
supernatant was stored at -80°C, and the proteins contents were
quantified using the BCA protein assay kit (Thermo Fisher
Scientific, Inc.). Subsequently, 30 µg/lane of protein was
separated by 6% SDS-PAGE, and then transferred to PVDF membranes
(MilliporeSigma). Membranes were blocked for 1 h at room
temperature with blocking solution (5% non-fat dry milk powder and
0.2% Tween-20 in TBS). The membranes were incubated with rabbit
polyclonal anti-HERG (1:400; cat. no. APC-109; Alomone Labs), mouse
monoclonal anti-CNX (1:2,000; cat. no. ab92573; Abcam),
anti-β-tubulin (reference protein in the cytoplasm; 1:2,000; cat.
no. K200059M; Beijing Solarbio Science & Technology Co., Ltd.),
anti-GAPDH (reference protein for the whole cell; 1:3,000; cat. no.
K200057M; Beijing Solarbio Science & Technology Co., Ltd.),
anti-Na/K ATPase (reference protein for cell membranes; 1:10,000;
cat. no. ab254025; Abcam), anti-ERP57 (1:1,000; cat. no. ab13506;
Abcam) and anti-CRT (1:2,000; cat. no. ab22683; Abcam) antibodies
at 4°C overnight. After three washes with TBS with 0.3% Tween-20,
the membrane was probed with a goat anti-mouse antibody
HRP-conjugated secondary antibody (1:2,000; cat. no. SE131; Beijing
Solarbio Science & Technology Co., Ltd.) or goat anti-rabbit
antibody (1:2,000; cat. no. SE134; Beijing Solarbio Science &
Technology Co., Ltd.) for 1 h at room temperature. Western blots
were visualized using WesternBright ECL chemiluminescent substrate
(Advansta, Inc.) according to manufacturer's protocol using an
ImageQuant LAS 500 imager (General Electric). Band densities were
quantitated using ImageJ (v1.51; National Institutes of
Health).
Co-immunoprecipitation of CNX, CRT and
ERP57 with immature HERG
293 cells expressing HERG in 6-well cell culture
plates were harvested at 48 h after transient transfection as
aforementioned. Subsequently, the IP/CoIP kit (cat. no. abs955;
Absin (Shanghai) Biotechnology Co., Ltd.) was used. After
centrifugation at 14,000 × g for 10 min at 4°C, the supernatant was
the cell division product. Subsequently, beads (5 µl Protein
A and 5 µl Protein G) were added to 500 µl
(containing 200-1,000 µg total protein) cell lysate.
CNX-HERG, CRT-HERG and ERP57-HERG complexes were immunoprecipitated
by incubation with 2 µg antibody against CNX (cat. no.
ab92573; Abcam), CRT (cat. no. ab22683; Abcam) and ERP57 (cat. no.
ab13506; Abcam), respectively, at 4°C overnight. Furthermore, 5
µl Protein A and 5 µl Protein G were added and mixed
gently at 4°C for 1-3 h, then the precipitate was washed with 0.5
ml 1X Wash buffer (from the IP/CoIP kit), centrifuged at 12,000 × g
for 1 min at 4°C, and the precipitate was retained. Subsequently,
20-40 µl 1X SDS was added to the precipitate, and the sample
was heated to 100°C for 5 min. This was followed by analysis by
western blotting and band densities were quantitated using
ImageJ.
Immunofluorescence and confocal
imaging
293 cells were seeded at a density of
1.0×105 cells per well of a 6-well plate coverslips and
transiently transfected with pcDNA3-WT, pcDNA3-A561V and
pcDNA3-WT/A561V plasmids. After 24 h of incubation, pcDNA3-CRT and
pcDNA3-ERP57 were transiently transfected into the cells, which
were cultured for an additional 48 h. The cells were fixed with 4%
paraformaldehyde for 30 min at 37°C, permeabilized with 0.5% Triton
X-100 for 10 min and blocked with 5% goat serum (all from Beijing
Solarbio Science & Technology Co., Ltd.) at room temperature
for 1 h. Cells were labeled with rabbit polyclonal anti-HERG
antibody (1:200; cat. no. APC-109; Alomone Labs) at 4°C overnight
and then incubated with FITC-conjugated goat anti-rabbit IgG
immunofluorescent secondary antibody (1:1,000; cat. no. SA00013-2;
ProteinTech Group, Inc.) and phalloidin (1:1,000; cat. no.
AC18L022; Shanghai Life iLab Bio Technology Co., Ltd.) at room
temperature for 1 h. Signals were captured using a Leica TCS SP8
confocal laser scanning microscope (Leica Microsystems, Inc.).
Whole-cell patch-clamp recordings
293 cells were collected 48 h after transfection
(DNA plasmid only) to examine differences in tail currents of WT,
A561V and WT/A561V and the effect of overexpression of ERP57 and
CRT on WT/A561V using the patch-clamp technique. A pipette with an
end resistance of 2-5 MΩ, when filled with the internal solution,
was used to record membrane currents in a whole-cell recording
configuration, as described in previous studies (21,22). The electrodes were connected to
an Axopatch 700B amplifier (Molecular Devices, LLC), and currents
were analog filtered at a frequency of 2 kHz and digitized by an
analog-to-digital converter (DigiData1440A; Molecular Devices,
LLC). For this experiment, pCLAMP version 10.3 software (Molecular
Devices, LLC) was used to edit the stimulation program, record the
current, and analyze and measure the raw data. Excel 2016
(Microsoft Corporation) and Origin7.5 (OriginLab) software were
used to perform statistics and map the original data, activate the
current and tail current, and calculate current density according
to battery capacitance, thereby eliminating the impact of battery
size on data.
Statistical analysis
GraphPad Prism 7.0 software (GraphPad Software,
Inc.) was used to perform statistical analysis. All experiments
were performed at least in triplicate and data are presented as the
mean ± SD. Differences between two groups were analyzed using an
unpaired Student's t-test and differences among three or more
groups were analyzed using one-way ANOVA followed by Tukey's post
hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
HERG-A561V mutation upregulates
CNX/CRT/ERP57 expression
To improve the understanding of maturation and
trafficking of HERG protein in cells expressing WT, mutant and
heterozygous HERG, pcDNA3-WT, pcDNA3-A561V or
pcDNA3-WT/A561Vplasmids were transiently transfected into 293 cells
and the successful transfection was demonstrated by RT-qPCR
(Fig. 1A). At 48 h
post-transfection, the protein was extracted from cells for western
blot analysis, and the protein expression levels of CNX, CRT and
ERP57 were detected. As shown in (Fig. 1E-H) the expression levels of
CNX/CRT/ERP57 were significantly increased in the A561V and
WT/A561V groups (P<0.05). WT-transfected cells exhibited two
protein bands for HERG, of which the upper band at 155 kDa
represents a mature, fully glycosylated glycoprotein form of HERG,
which is transported to the cell membrane surface. The lower band
at 135 kDa represents a core-glycosylated immature form of HERG,
and the band at 132 kDa is the precursor of immature form of HERG
protein, all located in the ER. As shown in (Fig. 1B-D) immature forms of HERG (135
kDa) were significantly increased in the A561V and WT/A561V groups
compared with the WT group (P<0.05). Furthermore, the mature
form expression of HERG protein (155 kDa) was significantly reduced
in the A561V group compared with the WT group (P<0.05), and 155
kDa HERG protein expression was not increased in the WT/A561V group
compared with the WT group (P>0.05). These results indicated
that co-expression of WT and A561V will not lead to complete loss
of HERG function related to the negative dominant effect of the
A561V mutation site and molecular chaperone CNX/ERP57/CRT serves a
role in this process.
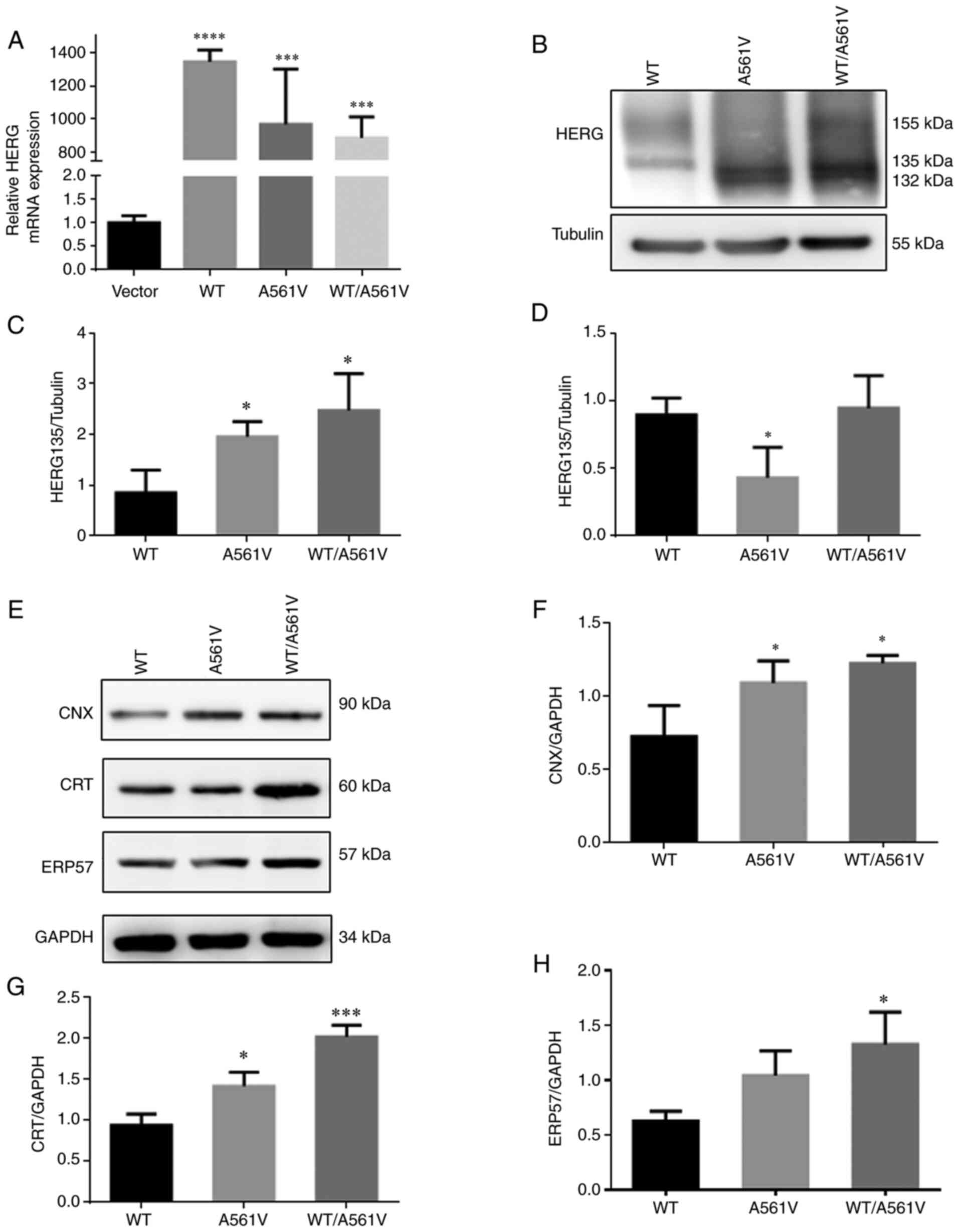 | Figure 1HERG-A561V mutation upregulates
CNX/CRT/ERP57 expression. (A) Relative HERG mRNA expression in
cells transfected with WT, A561V and WT/A561V.
***P<0.001, ****P<0.0001 vs. Vector.
(B) HERG protein expression in cells transfected with WT, A561V and
WT/A561V were detected by western blotting. (C) Optical density
analysis of immature (135 kDa) forms of HERG protein.
*P<0.05 vs. WT. (D) Optical density analysis of
mature (155 kDa) forms of HERG protein. *P<0.05 vs.
WT. (E) CNX/CRT/ERP57 protein expression in cells transfected with
WT, A561V and WT/A561V were detected by western blotting. (F)
Optical density analysis of CNX protein expression.
*P<0.05 vs. WT. (G) Optical density analysis of CRT
protein expression. *P<0.05, ***P<0.001
vs. WT. (H) Optical density analysis of ERP57 protein expression.
*P<0.05 vs. WT. Data are presented as the mean ± SD;
n=3 in each group. One-way ANOVA was used to analyze the data. CNX,
calnexin; CRT, calreticulin; ERP57, endoplasmic reticulum protein
57; HERG, human ether-a-go-go-related gene; WT, wild-type HERG
group; A561V, A561V-HERG mutation group; WT/A561V, heterozygous
HERG group. |
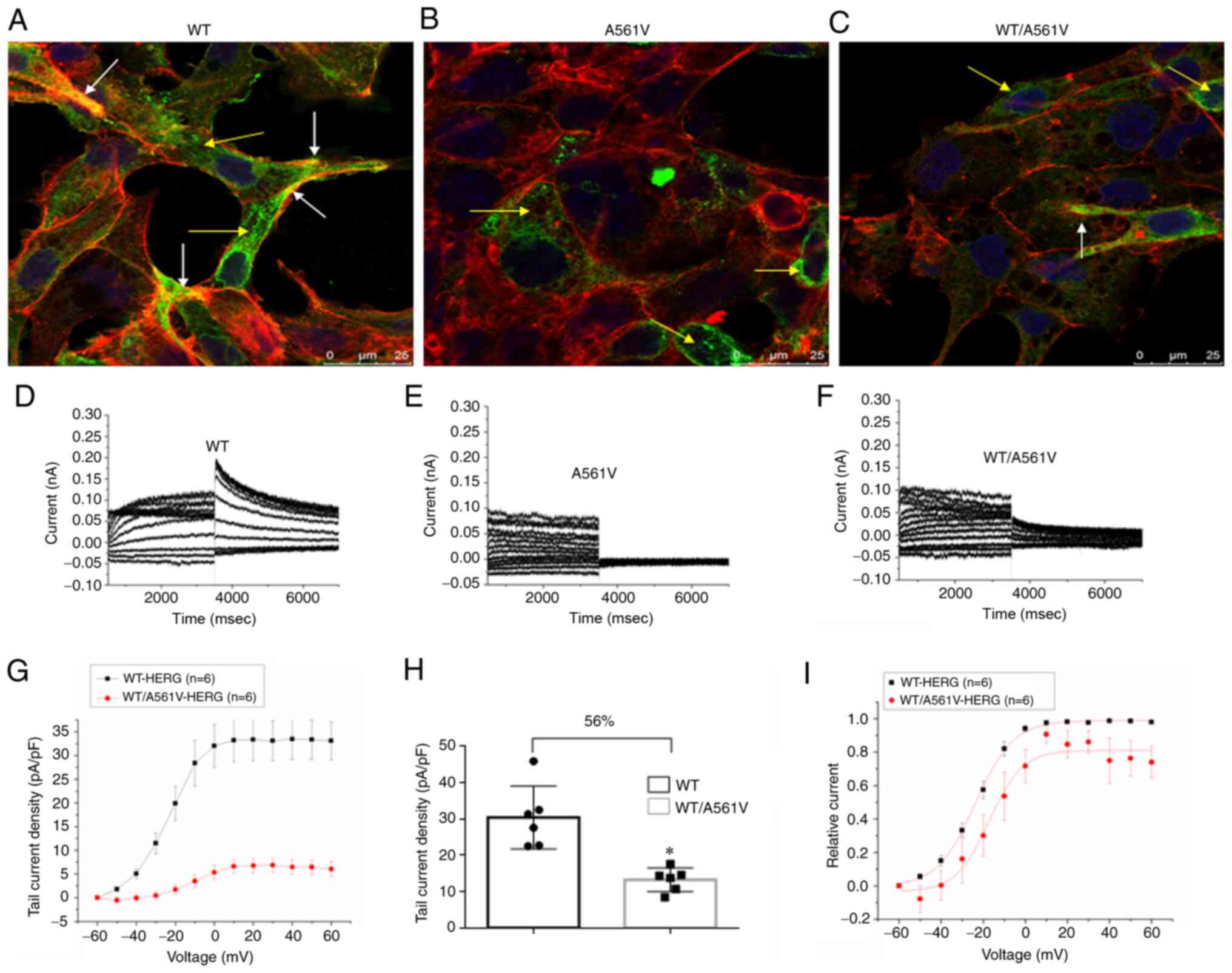
Tail current of WT/A561V is suppressed
but it does not disappear completely
The localization and protein trafficking of HERG
protein in cells expressing WT, A561V and WT/A561V were examined by
immunostaining and confocal imaging (Fig. 2A-C). The confocal image clearly
revealed that WT protein was expressed on the cell surface (white
arrows) and in the cytoplasm (yellow arrows). By contrast, A561V
and WT/A561V proteins were mainly expressed in the cytoplasm.
Specifically, the mutant HERG protein was mainly retained in the
ER. No tail current was observed in A561V cells but this was
present in WT/A561V cells. The tail current density in WT/A561V
cells (13.30±3.23 pA/pF) was 56% lower than that of WT cells
(30.49±8.67 pA/pF; n= 6; P<0.05) (Fig. 2H). In Fig. 2I, the normalized data of the tail
currents were plotted against the test potential and fitted to a
Boltzmann function. The half-maximal activation voltage (V1/2) in
WT/A561V cells was-17.19709±2.22513 mV, whereas that in WT cells
was-23.76787±0.4569 mV (n=6; P>0.05). A similar trend was seen
in the slope factor k values of WT/A561V cells (7.87613±1.95185 mV)
compared with WT cells (9.02434±0.39344 mV) (n=6; P>0.05).
Therefore, there was no significant difference in activation phase
properties of WT and WT/A561V protein channels. As shown, the
heterozygous channel did not completely lose its function. If
normal transportation can be partially restored after refolding,
the heterozygous channel may still serve a certain compensatory
role. Therefore, it was concluded that the mutant HERG protein
could not be transported to the membrane to perform its function.
HERG function did not completely disappear in heterozygous cells,
and some HERG proteins were still transported to the cell
membrane.
Mutual binding ability of A561V and
WT/A561V proteins with molecular chaperones CNX/ERP57/CRT is
increased compared with that of the WT group
To clarify the role of key molecular chaperones CNX,
CRT and ERP57 in mediating A561V folding in the CNX/CRT cycle, the
present study used immunoprecipitation to detect HERG protein
binding to molecular chaperones CNX, CRT and ERP57 in cells
expressing WT, A561V and WT/A561V. The physical association of the
HERG channel with chaperones was determined by immunoprecipitation
with anti-CNX, anti-CRT and anti-ERP57 antibodies followed by
western blot analysis with anti-HERG, anti-CNX, anti-CRT and
anti-ERP57 antibodies (Fig. 3).
In A561V and WT/A561V cells, CNX/CRT/ERP57 and immature forms of
HERG protein binding increased (P<0.05). Simultaneously, the
present study also revealed that, in WT/A561V cells, ERP57 binding
to CRT protein was significantly enhanced compared with the WT
group (P<0.05). bb′ domain of ERP57 is a key domain that
binds to HERG and CRT proteins. As sshown in Fig. 4A-D, b and b ′ are key
domains required for ERP57 to bind to HERG and CRT proteins and
promote proper protein folding. After the modified ERP57 was
expressed in WT/A561V cells, ERP57 in the bb′ domain deletion group
was identified to exhibit enhanced binding to HERG and CRT proteins
compared with the Vector group (P<0.05), and there was no
significant difference in binding among the remaining groups
(P>0.05).
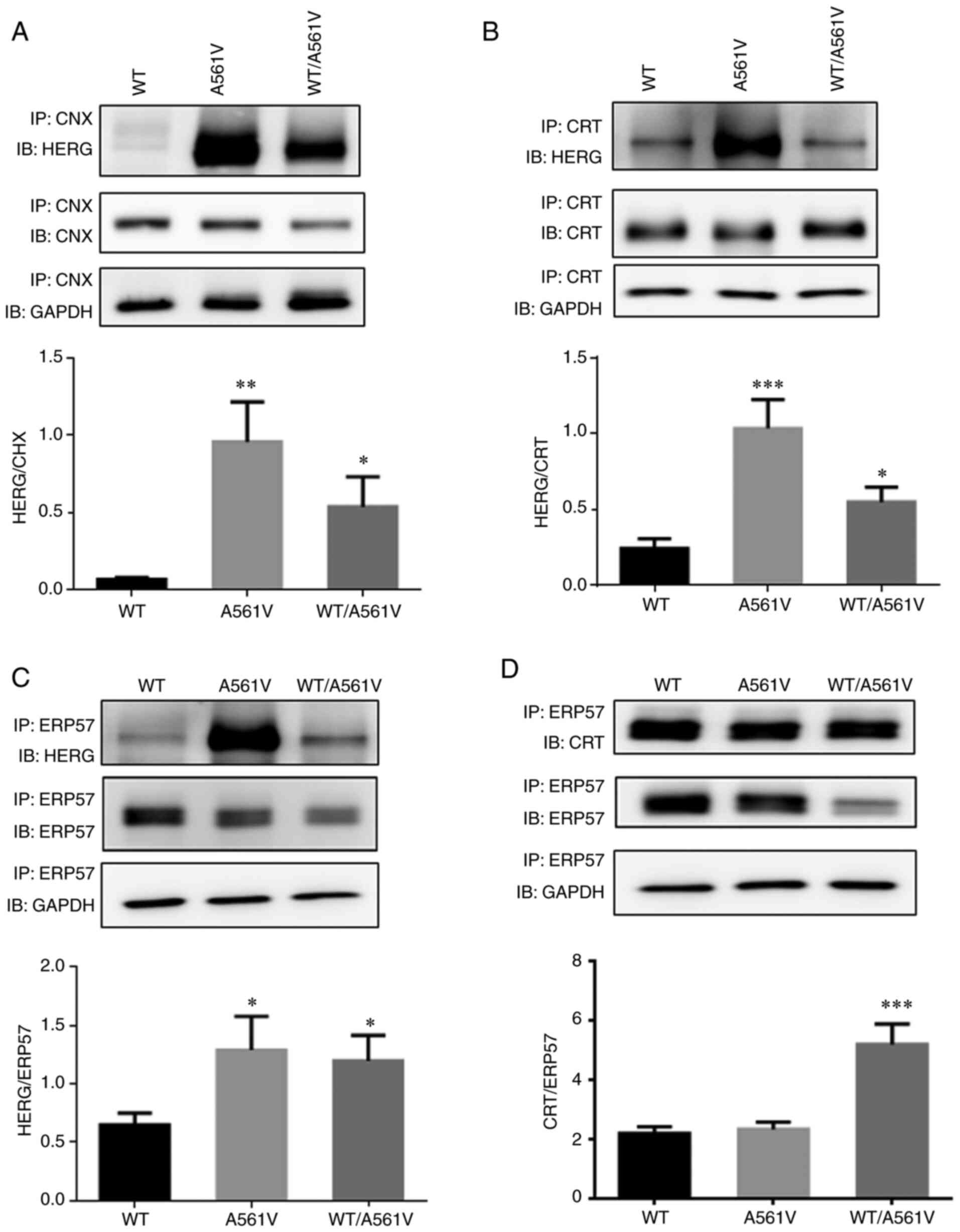 | Figure 3Mutual binding of A561V and WT/A561V
proteins with molecular chaperones CNX/ERP57/CRT was significantly
increased compared with the WT group. (A) HERG and CNX binding and
optical density analysis. *P<0.05,
**P<0.01 vs. WT. (B) HERG and CRT binding and optical
density analysis. *P<0.05, ***P<0.001
vs. WT. (C) HERG and ERP57 binding and optical density analysis.
*P<0.05 vs. WT. (D) CRT and ERP57 binding and optical
density analysis. ***P<0.001 vs. WT. Data are
presented as the mean ± SD; n=3 in each group. One-way ANOVA was
used to analyze the data. CNX, calnexin; CRT, calreticulin; ERP57,
endoplasmic reticulum protein 57; HERG, human ether-a-go-go-related
gene; IB, antibody used to blot the membrane; IP, lysate after
incubation with antibody and pulled down using beads; WT, wild-type
HERG group; A561V, A561V-HERG mutation group; WT/A561V,
heterozygous HERG group. |
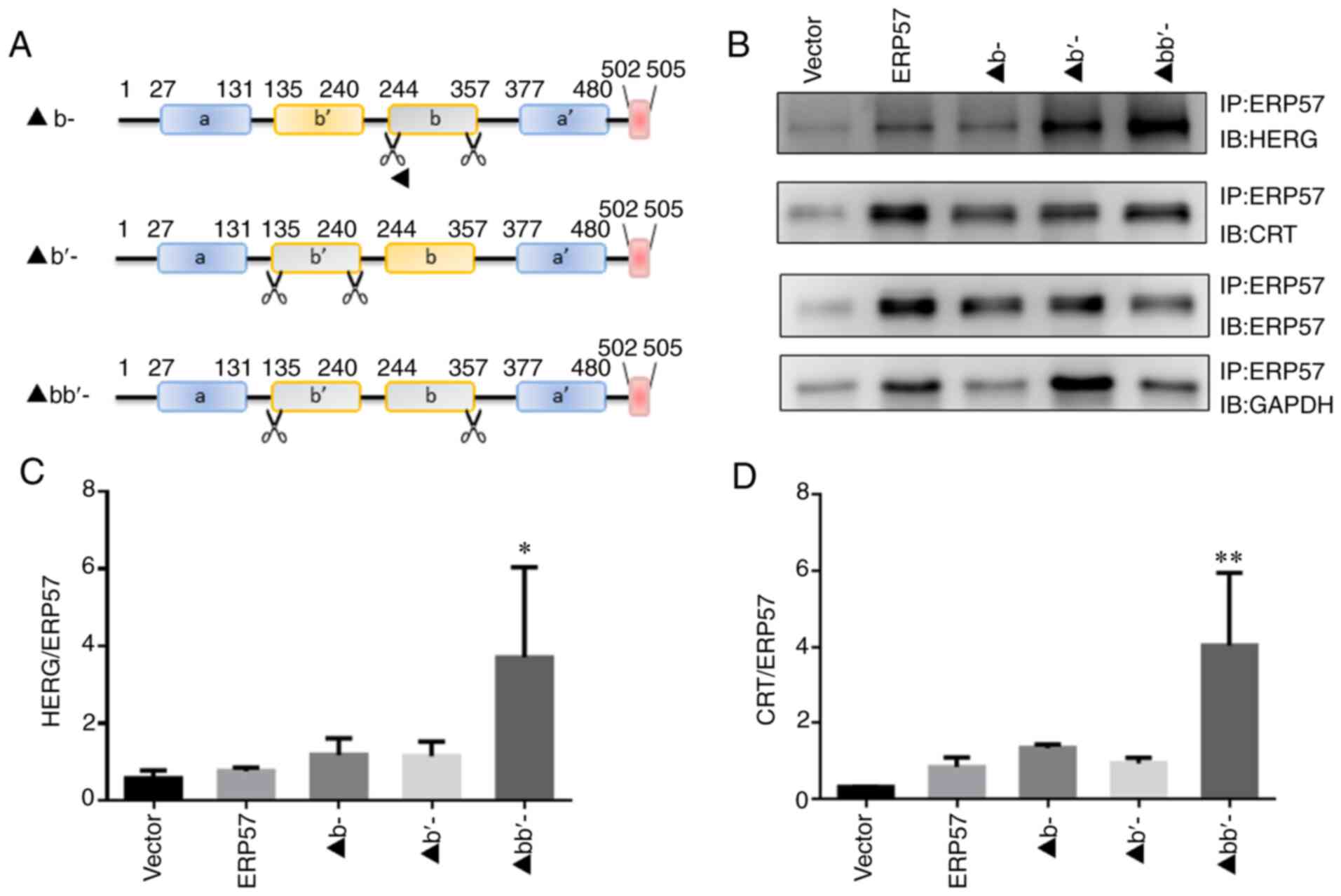 | Figure 4bb′ domain of ERP57 is a key domain
that binds to HERG and CRT proteins. (A) Specific location of ERP57
domain deletion. (B) After modified ERP57 was expressed in WT/A561V
cells, ERP57 binding to HERG and CRT was detected. Optical density
analysis of (C) HERG and (D) CRT binding to modified ERP57,
including ERP57 lacking its b and b′ domains. Data are presented as
the mean ± SD; n=3 in each group. One-way ANOVA was used to analyze
the data. *P<0.05, **P<0.01 vs. vector
group. b-, ERP57 b-domain deletion group; ▲b′-, ERP57 b′-domain
deletion group; ▲bb′-, ERP57 bb′-domain deletion group; CRT,
calreticulin; ERP57, endoplasmic reticulum protein 57; HERG, human
ether-a-go-go-related gene; IB, antibody used to blot the membrane;
IP, lysate after incubation with antibody and pulled down using
beads; WT, wild-type. |
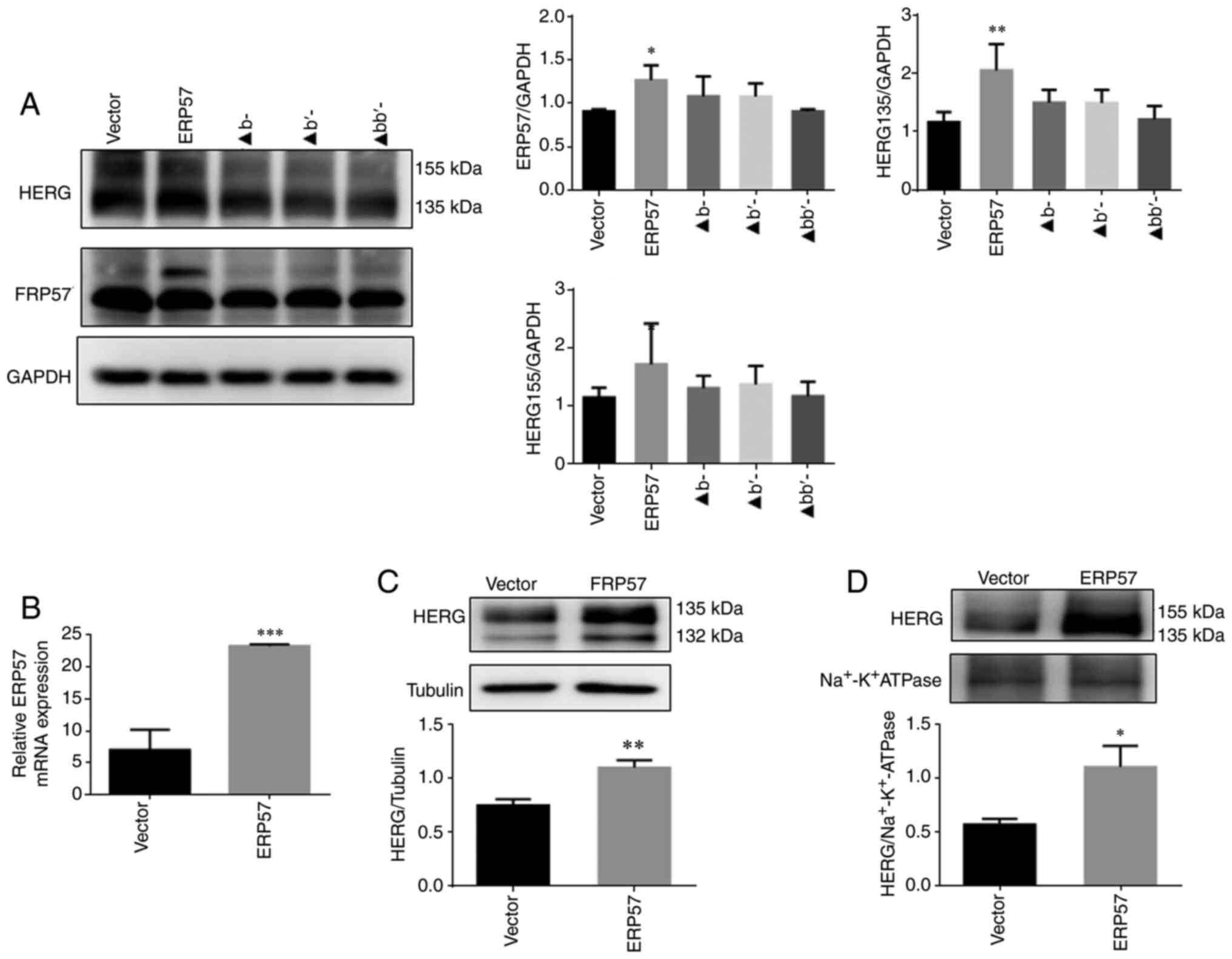
Overexpression of ERP57 can promote
folding and trafficking of WT/A561V mutant protein
In the WT/A561V cell group (Fig. 5A), the immature form of HERG
protein in the ERP57 overexpression group was increased compared
with that in other groups (P<0.05), indicating that ERP57
overexpression can promote the correct folding of protein. ERP57
expression, as measured by RT-qPCR (Fig. 5B), was increased compared with
the Vector group (P<0.001). After plasma membrane separation
(Fig. 5C and D), western
blotting revealed that not only immature form expression of HERG
protein in the cytoplasm increased (P<0.05) but the expression
levels of mature form of HERG protein on the membrane were also
increased (P<0.05).
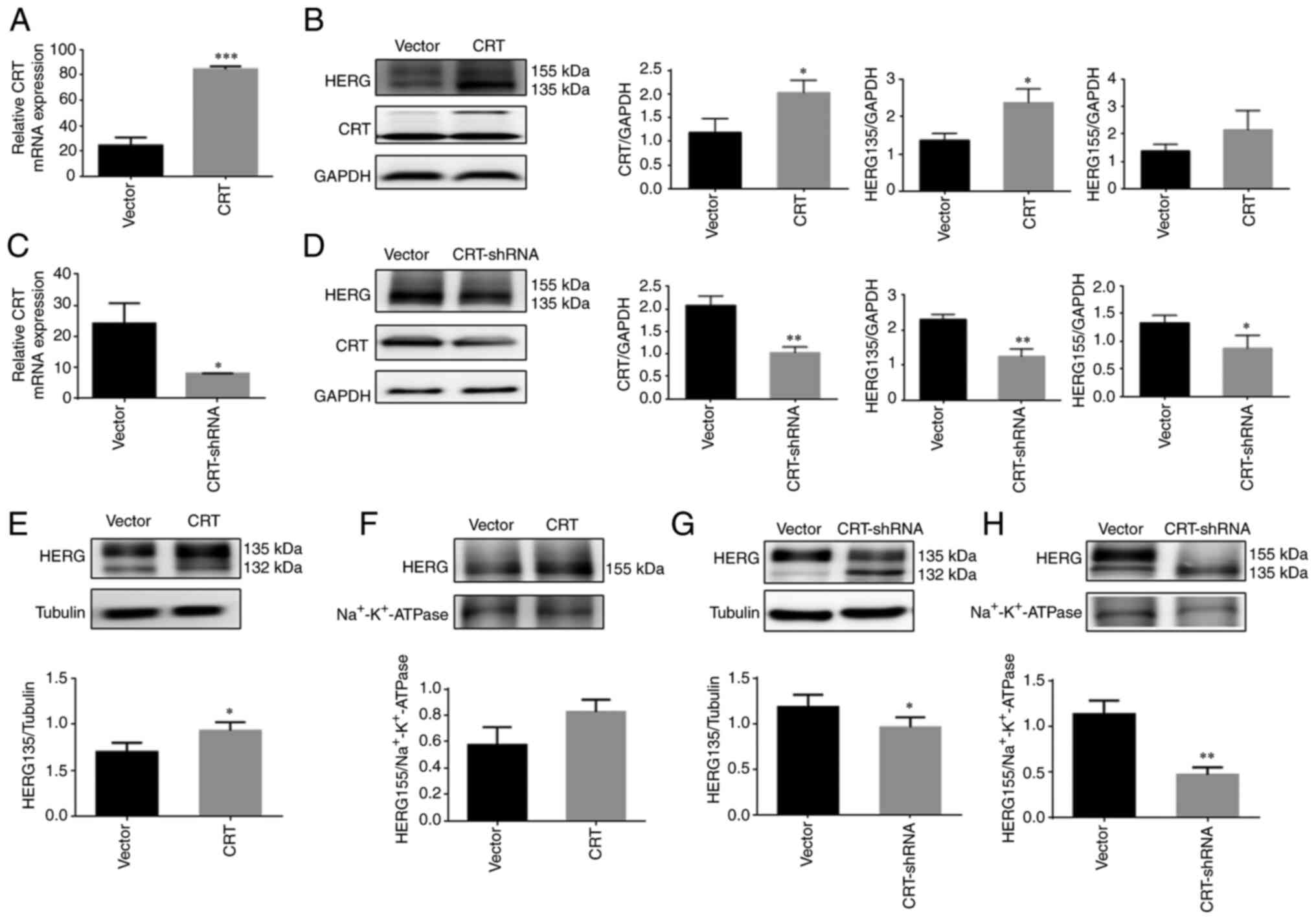
Overexpression of CRT can help the
folding of WT/A561V mutant protein but knockdown of CRT cannot
Relative CRT mRNA expression, as measured by
RT-qPCR, was increased compared with the Vector group (P<0.001;
Fig. 6A). In WT/A561V cells, the
expression levels of the immature form of HERG in the
CRT-overexpressing group were significantly increased (P<0.05),
and the mature form of HERG protein did not increase significantly
(P>0.05; Fig. 6B).
Additionally, relative CRT mRNA expression (Fig. 6C) was decreased compared with the
Vector group (P<0.05). The expression levels of immature and
mature forms of HERG in the knockdown of CRT protein group were
significantly lower than those in the control group (P<0.05;
Fig. 6D). After plasma membrane
separation, as shown in Fig. 6E and
F, the HERG protein expression in the cytoplasm was
significantly increased in the CRT overexpression group
(P<0.05), and that on the membrane did not increase
significantly in the CRT overexpression group (P>0.05). As shown
in Fig. 6G and H, HERG protein
expression in the cytoplasm was significantly reduced in the
knockdown of CRT group (P<0.05), and that on the membrane was
significantly reduced in the knockdown of CRT group (P<0.05).
The present study revealed that CRT overexpression promoted the
correct folding and trafficking of the immature form of HERG.
Furthermore, knockdown of CRT reduced the expression of both the
mature and immature forms of HERG protein, and failed to serve a
role in the correct folding and trafficking of WT/A561V
protein.
Overexpression of CRT and ERP57 can
increase the tail current density of WT/A561V
Confocal images clearly demonstrated that
overexpression of CRT and ERP57 increased HERG protein localization
in the plasma membrane (Fig.
7A-C). As shown in Fig. 7H,
the tail current amplitude of the CRT overexpression group
(19.48±7.48 pA/pF) increased by 46% compared with the heterozygous
control group (13.30±3.23 pA/pF; n=6; P<0.05), and the tail
current amplitude of the ERP57 overexpression group (20.41±4.07
pA/pF) increased by 53% compared with the heterozygous control
group (13.30±3.23 pA/pF; n=6; P<0.05). In Fig. 7I, the normalized data of the tail
currents were plotted against the test potential and fitted to a
Boltzmann function. The half-maximal activation voltage (V1/2) in
WT/A561V cells was -17.19709±2.22513 mV, whereas that in
WT/A561V+CRT cells was -18.57577±1.16886 mV (n=6; P>0.05), and
that in WT/A561V cells was -17.19709±2.22513 mV, whereas that in
WT/A561V+ERP57 cells was -21.63619±1.42333 mV (n=6; P>0.05). A
similar trend was seen in the slope factor k values of WT/A561V
cells (7.87613±1.95185 mV) compared with WT/A561V+CRT cells
(9.45329±1.02986 mV; n=6; P>0.05), and WT/A561V cells
(7.87613±1.95185 mV) compared with WT/A561V+ERP57 cells
(10.86673±1.2318 mV; n=6; P>0.05). Therefore, there was no
significant difference in activation phase properties among the CRT
overexpression, ERP57 overexpression and WT/A561V groups. The
heterozygous channel did not completely lose its function. If it
can partially resume normal transport after its regulation, it can
still perform a certain compensatory function. These results
suggested that overexpression of key molecular chaperones,
including CRT and ERP57, could correct the function of the WT/A561V
channel.
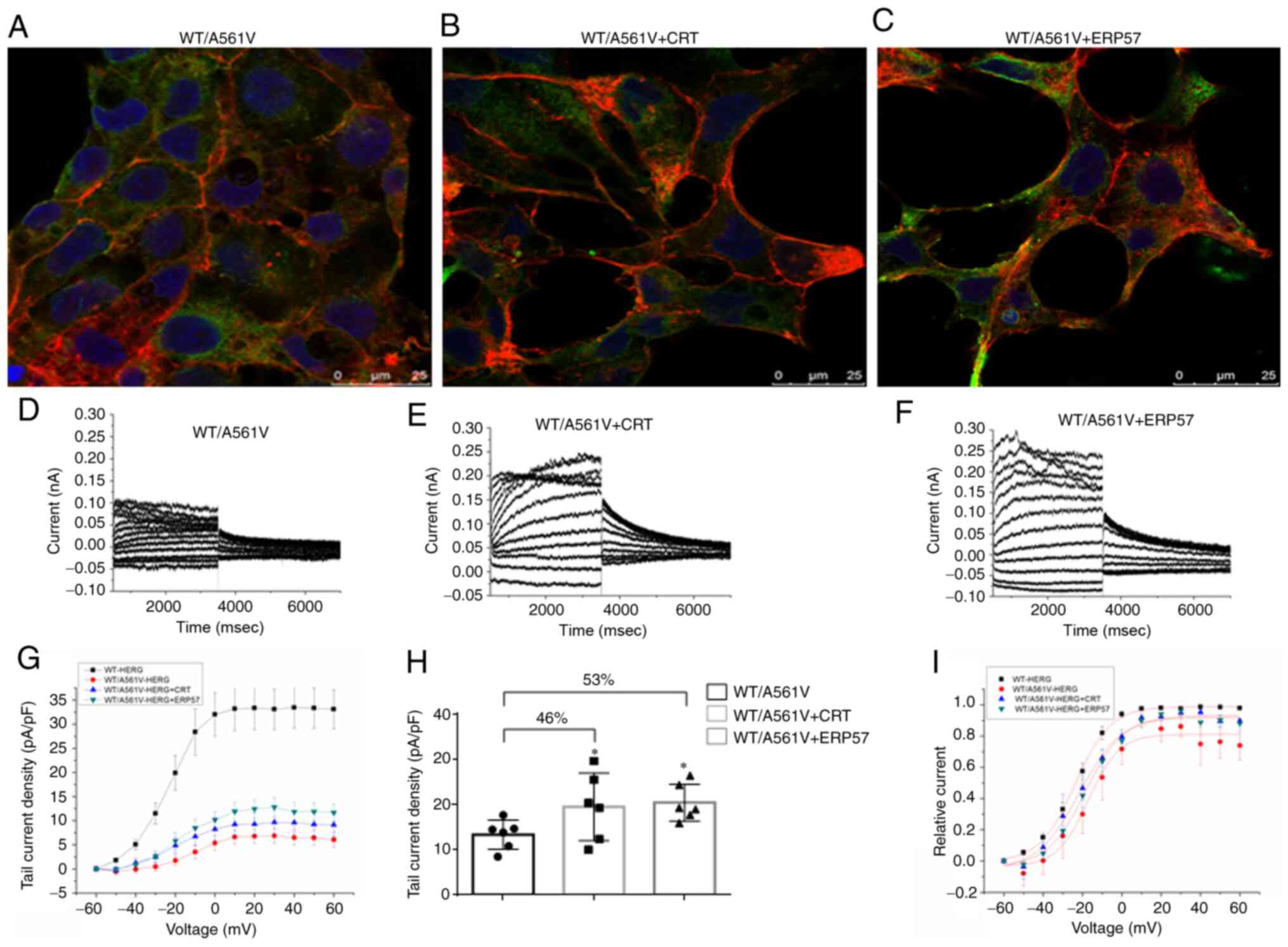 | Figure 7Overexpression of CRT and ERP57 can
increase the tail current density of WT/A561V. (A) Confocal imaging
of WT/A561V channel in 293 cells (60X confocal special oil lens).
(B) Confocal imaging of WT/A561V channel in 293 cells
overexpressing CRT protein (60X confocal special oil lens). (C)
Confocal imaging of WT/A561V channel in 293 cells overexpressing
ERP57 protein (60X confocal special oil lens). Co-staining with
anti-HERG antibody (green), DAPI-stained nuclei (blue) and
phalloidin (red). (D) Tail current amplitudes of WT/A561V group.
(E) Tail current amplitudes of CRT overexpression group. (F) Tail
current amplitudes of ERP57 overexpression group. (G) Current
density-voltage relationship for tail currents amplitudes of WT,
WT/A561V, overexpression of CRT and overexpression of ERP57 groups.
(H) Statistical graph of tail current amplitudes. The tail current
amplitudes of the CRT group were increased by 46% compared with the
WT/A561V group (n=6; *P<0.05). The tail current
amplitudes of the ERP57 group were increased by 53% compared with
the WT/A561V group (n=6; *P<0.05). (I) Amplitudes of
tail currents of WT, WT/A561V, WT/A561V+CRT, WT/A561V+ERP57 channel
plotted as a function of the test potential and fitted to a
Boltzmann function (n=6). CRT, calreticulin; ERP57, endoplasmic
reticulum protein 57; HERG, human ether-a-go-go-related gene; WT,
wild-type. |
Discussion
The HERG channel protein is a glycosylated protein
composed of four α subunits (3).
The channel protein-peptide chain enters the ER after ribosome
synthesis and undergoes core glycosylation at position N598 to form
a 135 kDa precursor (23). The
precursor is then transported to the Golgi apparatus and undergoes
complex glycosylation to form a 155 kDa mature channel protein
inserted into the cell membrane (24). Compared with WT HERG, represented
on a western blot by a 155-kDa band of mature HERG channel protein,
the mutant HERG protein, represented on a western blot by a 135-kDa
immature HERG protein band, is prone to misfolding and is retained
in the ER (25). The mutant
channel protein will combine with the WT protein to form a hybrid
channel, producing two protein bands of 155 and 135 kDa on a
western blot, and stay in the ER, thereby reducing the expression
of the WT protein in the cell membrane (19). These mutant proteins, especially
heterozygous channels, are not completely non-functional (5). If their normal transport can be
partially restored by overexpression of related chaperones, the
mutant protein can still perform a compensatory function (26). In other words, the upstream
regulatory mechanism of HERG channel abnormalities is the ERQC
system, which mediates channel protein folding and transport in the
ER, and serves a vital role in changes in ion channel function
(27). The aforementioned
viewpoints were verified in the present study.
The CNX/CRT cycle is an essential part of the ERQC
system and one of the critical mechanisms for monitoring protein
folding and assembly (17). The
disulfide bond isomerase ERP57 can form transient disulfide bonds
with CNX/CRT-bound glycoproteins to facilitate protein folding
(13,14), and the b and b′ domains of ERP57
protein contain the binding sites for CRT and CNX. UDP-glucose:
Glycoprotein glucosyltransferase 1 (UGGT1) is an important enzyme
in the circulatory system and can specifically recognize
incompletely folded proteins and catalyze their glycosylation,
which is called the ER folding induction device (28,29). Once the glycoprotein is
misfolded, Man9GlcNAc2 will add glucose under UGGT1 catalysis, and
the protein will re-enter the CNX/CRT cycle for refolding (30). If the protein still fails to fold
correctly, to avoid excessive accumulation of misfolded protein in
the ER, it will be released from the CNX/CRT cycle under the action
of ER mannosidase I, and the mannosidase, which enhances ER
degradation enzyme-like protein is combined and degraded through
ER-associated degradation (31-34). The specific process is shown in
Fig. 8.
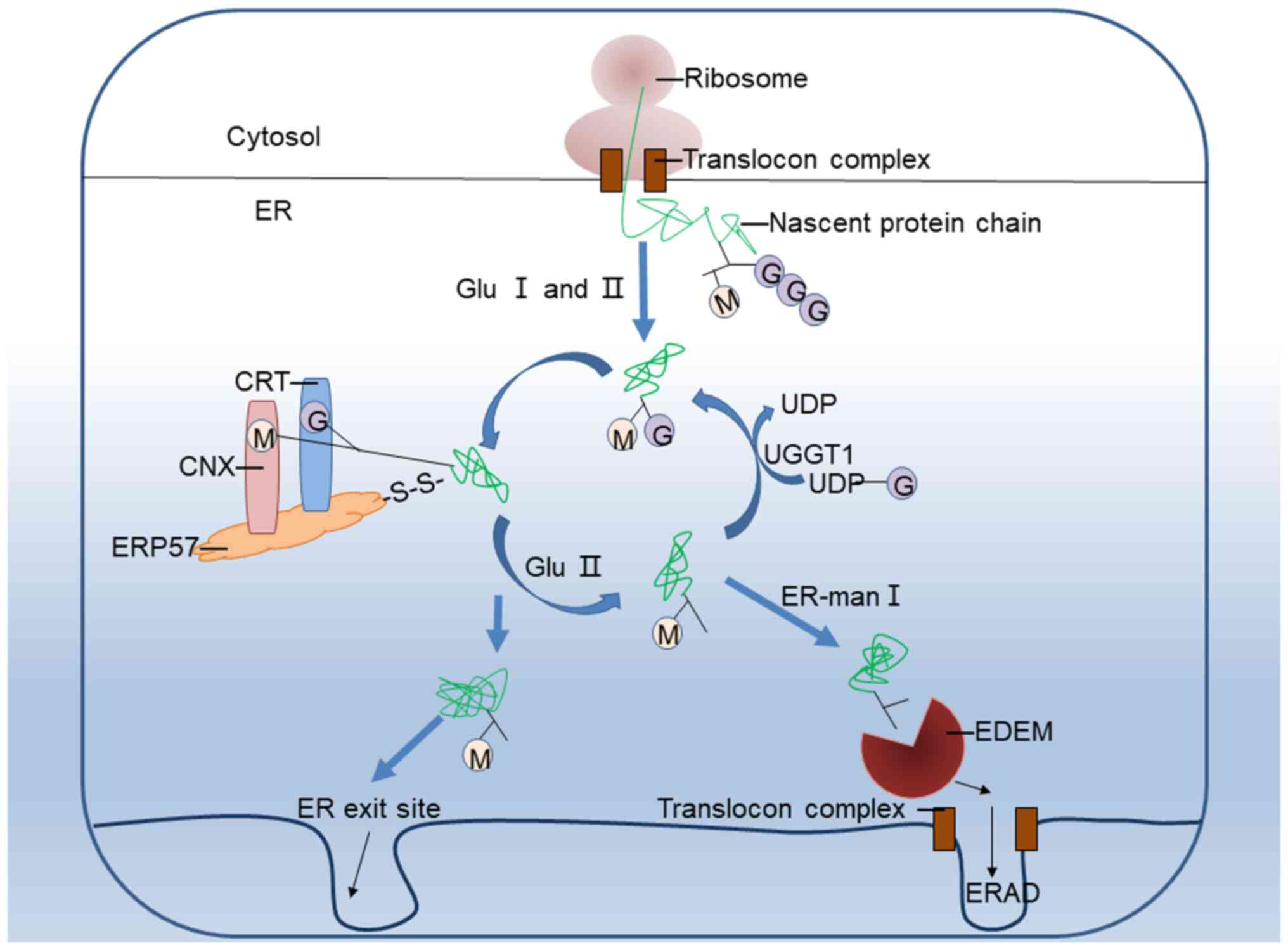 | Figure 8CNX/CRT cycle is an important part of
the ERQC system, and its key proteins are CNX, CRT, ERP57, UGGT1,
Glu II, ER-man I and EDEM, all of which are involved in the process
of protein folding and transport. CNX, calnexin; CRT, calreticulin;
ERP57, endoplasmic reticulum protein 57; ER, endoplasmic reticulum;
ERQC, ER quality control; ERAD, ER-associated degradation; GluI/II,
glucosidases I/II; UGGT1, UDP-glucose: Glycoprotein
glucosyltransferase 1; ER-man I, endoplasmic reticulum-mannosidase
I; EDEM, ER degradation-enhancing α-mannosidase-like protein. |
To study the role of the chaperone proteins CNX, CRT
and ERP57 in the process of A561V trafficking and defects, the
present study examined the expression levels of HERG protein and
CNX/CRT/ERP57 in WT, A561V and WT/A561V cells. The data revealed
that immature HERG protein expression was significantly increased
in A561V and WT/A561V cells compared with its expression in WT
cells and the expression of molecular chaperones CNX and CRT was
also significantly increased compared with their expression in WT
cells, and ERP57 was significantly increased in WT/A561V cells
compared with WT cells. This result indicated that molecular
chaperones CNX, CRT and ERP57 serve a role in the A561V transport
process. Furthermore, the CNX/CRT chaperone role in the A561V
mutant was consistent with the results obtained for E637K-HERG and
G572R-HERG mutants (24).
To the best of our knowledge, the present study
demonstrated for the first time that folding and maturation of
A561V were controlled by cytoplasmic chaperone ERP57 and verified
the key domains of ERP57 protein and its specific mechanism of
action. Currently, ERP57 protein is primarily used in tumor
research. ERP57 is abnormally dysregulated in a number of cancer
types, including cervical cancer and laryngeal cancer, and abnormal
ERP57 expression has been evaluated as a clinical prognostic
indicator (35). Upregulation or
downregulation of ERP57 may be associated with poor prognosis
(36). Furthermore, CRT protein,
the key molecular chaperone in the CNX/CRT protein cycle, is one of
the main calcium-binding proteins in the ER (37). In addition to coordinating
membrane surface and exocrine proteins in the ER, CRT can also help
the folding and trafficking of misfolded proteins retained in the
ER (38). However, a recent
study has revealed that CRT is not only present in the ER but also
localizes to the cell membrane and is secreted to the outside of
the cell to perform its specific functions (39). In the future, it will be
necessary to conduct experiments to determine whether these
functions are also involved in folding and transportation of HERG
protein.
Accumulating misfolded proteins is a major feature
of pathology of numerous neurodegenerative diseases, including
frontotemporal dementia, amyotrophic lateral sclerosis, Parkinson's
disease, Alzheimer's Mer's disease, Huntington's disease and
Creutzfeldt-Jakob disease (16).
ER stress and activation of unfolded protein response after
misfolding and/or nascent protein accumulation are common features
in the cytopathology of these diseases, which is the pathology of
accumulating mutant proteins or misfolded proteins in the ER caused
by mutations in HERG gene features (40). However, the cumulative effect of
activating the unfolded protein response depends on integrating
cell survival time and the duration of apoptosis signals (41). Activation of the unfolded protein
response will increase the recruitment of PDI family members to
promote protein folding or trigger cell apoptosis (42). In Creutzfeldt-Jakob disease,
ERP57 upregulation is considered a defense response against
toxicity of misfolded prion virus protein (43). However, characterization of the
role of ERP57 in mediating the steady-state levels of WT and prion
virus-associated disease related mutant proteins suggests that
ERP57 deficiency may cause prion virus protein folding but may not
contribute to resistance to ER stress sensitivity (44). The ability to inhibit mutant
huntingtin-mediated toxicity by inhibiting ERP57 and/or PDI
highlights that PDI can promote apoptosis by stimulating
mitochondrial outer membrane permeability (MOMP) to stimulate
neurodegenerative diseases with abnormal cytoplasmic protein
aggregation characteristics (45). In the present study, upregulation
of ERP57 promoted the correct folding and transportation of
misfolded and unfolded proteins. ERP57 may be upregulated during ER
stress as part of a protective response to promote the folding of
aggregated proteins and restore cellular protein stability.
Simultaneously, excessive ERP57 can stimulate cell apoptosis
through MOMP. Notably, ERP57 is a selective folding enzyme that
only interacts with a part of potential protein aggregates
(46). It is unclear whether
ERP57 can protect HERG mutant protein or other aggregation-prone
proteins involved in misfolding in vivo, or if ERP57 can
directly rescue the mutant conformation of HERG protein.
Investigation of this will reveal the potential of ERP57 as a
possible therapeutic target for LQTS caused by mutations in the
HERG gene.
The present study detected no IKr current in
A561V cells in which A561V was transported from the ER to the cell
membrane. This result may be due to mutations in the channel pore
area and channel opening, preventing potassium ions from passing
through the channel. The function of the WT/A561V heterozygous
channel was not completely abolished. An IKr current was
detected; however, it was weaker than the tail current of the WT
HERG channel, so it would be important to correct the IKr
current of the heterozygous HERG channel. For this reason, the
expression of molecular chaperones was adjusted to interfere with
folding and transportation of the WT/A561V channel protein. It was
revealed that knockdown of CRT protein did not promote HERG protein
transport to the membrane, while CRT and ERP57 overexpression
promoted the correct folding and transport of the protein.
Knockdown of CRT can lead to non-apoptotic cell death, leading to
complete cell disintegration (47), consistent with reduced expression
levels of mature and immature HERG protein forms after knockdown of
CRT protein. In the overexpression group, the amounts of immature
(135 kDa) and mature forms (155 kDa) of HERG protein were increased
to varying degrees in the cytoplasm and membrane. To verify the
impact of overexpression of CRT and ERP57 in the WT/A561V cell
model, whole-cell patch-clamp technology was used to detect the
IKr current in cells from each group, indicating that
overexpression of CRT and ERP57 further corrected the WT/A561V
trafficking defect.
The present study clarified the specific role of the
CNX/CRT cycle in mediating the folding and transport of WT and
mutant HERG and analyzed the regulatory roles of key factors CNX,
CRT and ERP57 in binding HERG protein and HERG channel function. It
was revealed that these factors can change the transport process of
proteins, especially heterozygous mutant proteins, by improving
HERG channel protein transport and increasing HERG channel protein
function, suggesting novel avenues for developing treatments for
LQTS from the perspective of correcting protein transport defects.
The present study demonstrated that the three chaperone proteins,
CNX, CRT and ERP57, block the mutant protein in the ER and refold
it into its correct conformation, which serves a crucial role in
trafficking the A561V mutant protein. Additionally, overexpression
of CRT and ERP57 promoted the correct transport of WT/A561V and
restored its function. This potential therapeutic method may also
provide ideas for treating other clinically relevant protein
trafficking diseases.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XH, XY and JL conceived and designed the
experiments. YW, YB and ZZ performed the experiments, YW and YB
analyzed the data. XH and JL contributed reagents and materials.
YW, XH and JL wrote the paper. YW and JL confirm the authenticity
of all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Abbreviations:
|
CNX
|
calnexin
|
|
CRT
|
calreticulin
|
|
ERP57
|
endoplasmic reticulum protein 57
|
|
ER
|
endoplasmic reticulum
|
|
ERQC
|
ER quality control
|
|
HERG
|
human-ether-a-go-go-related gene
|
|
IKr
|
rapidly activating delayed rectifier
K-current
|
|
LQTS
|
long QT syndrome
|
|
LQT2
|
long QT syndrome type 2
|
|
UGGT1
|
UDP-glucose: glycoprotein
glucosyltransferase 1
|
References
|
1
|
Wallace E, Howard L, Liu M, O'Brien T,
Ward D, Shen S and Prendiville T: Long QT syndrome: Genetics and
future perspective. Pediatr Cardiol. 40:1419–1430. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kanner SA, Jain A and Colecraft HM:
Development of a high-throughput flow cytometry assay to monitor
defective trafficking and rescue of long QT2 mutant hERG channels.
Front Physiol. 9:3972018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Butler A, Helliwell MV, Zhang Y, Hancox JC
and Dempsey CE: An update on the structure of hERG. Front
Pharmacol. 10:15722020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Perissinotti L, Guo J, Kudaibergenova M,
Lees-Miller J, Ol'khovich M, Sharapova A, Perlovich GL, Muruve DA,
Gerull B, Noskov SY and Duff HJ: The pore-lipid interface: Role of
amino-acid determinants of lipophilic access by ivabradine to the
hERG1 pore domain. Mol Pharmacol. 96:259–271. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang KP, Yang BF and Li BX: Translational
toxicology and rescue strategies of the hERG channel dysfunction:
Biochemical and molecular mechanistic aspects. Acta Pharmacol Sin.
35:1473–1484. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li G, Shi R, Wu J, Han W, Zhang A, Cheng
G, Xue X and Sun C: Association of the hERG mutation with long-QT
syndrome type 2, syncope and epilepsy. Mol Med Rep. 13:2467–2475.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Foo B, Williamson B, Young JC, Lukacs G
and Shrier A: hERG quality control and the long QT syndrome. J
Physiol. 594:2469–2481. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Araki K and Nagata K: Protein folding and
quality control in the ER. Cold Spring Harb Perspect Biol.
3:a0075262011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marti L, Lia A, Reca IB, Roversi P,
Santino A and Zitzmann N: In planta preliminary screening of ER
glycoprotein folding quality control (ERQC) modulators. Int J Mol
Sci. 19:21352018. View Article : Google Scholar :
|
|
10
|
Lamothe SM, Hulbert M, Guo J, Li W, Yang T
and Zhang S: Glycosylation stabilizes hERG channels on the plasma
membrane by decreasing proteolytic susceptibility. FASEB J.
32:1933–1943. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Patel C, Saad H, Shenkman M and
Lederkremer GZ: Oxidoreductases in glycoprotein glycosylation,
folding, and ERAD. Cells. 9:21382020. View Article : Google Scholar :
|
|
12
|
Tannous A, Pisoni GB, Hebert DN and
Molinari M: N-linked sugar-regulated protein folding and quality
control in the ER. Semin Cell Dev Biol. 41:79–89. 2015. View Article : Google Scholar :
|
|
13
|
Foo B, Barbier C, Guo K, Vasantharuban J,
Lukacs GL and Shrier A: Mutation-specific peripheral and ER quality
control of hERG channel cell-surface expression. Sci Rep.
9:60662019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi YQ, Yan M, Liu LR, Zhang X, Wang X,
Geng HZ, Zhao X and Li BX: High glucose represses hERG K+ channel
expression through trafficking inhibition. Cell Physiol Biochem.
37:284–296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hettinghouse A, Liu R and Liu CJ:
Multifunctional molecule ERp57: From cancer to neurodegenerative
diseases. Pharmacol Ther. 181:34–48. 2018. View Article : Google Scholar
|
|
16
|
Song D, Liu H, Wu J, Gao X, Hao J and Fan
D: Insights into the role of ERp57 in cancer. J Cancer.
12:2456–2464. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kozlov G and Gehring K: Calnexin
cycle-structural features of the ER chaperone system. FEBS J.
287:4322–4340. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pollock S, Kozlov G, Pelletier MF, Trempe
JF, Jansen G, Sitnikov D, Bergeron JJ, Gehring K, Ekiel I and
Thomas DY: Specific interaction of ERp57 and calnexin determined by
NMR spectroscopy and an ER two-hybrid system. EMBO J. 23:1020–1029.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Y, Shen T, Fang P, Zhou J, Lou K, Cen
Z, Qian H, Zhou J, Liu N and Lian J: The role and mechanism of
chaperones calnexin/calreticulin in which ALLN selectively rescues
the trafficking defective of HERG-A561V mutation. Biosci Rep. Sep
7–2018.Epub ahead of print.
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Smith JL, Anderson CL, Burgess DE, Elayi
CS, January CT and Delisle BP: Molecular pathogenesis of long QT
syndrome type 2. J Arrhythm. 32:373–380. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jenewein T, Kanner SA, Bauer D, Hertel B,
Colecraft HM, Moroni A, Thiel G and Kauferstein S: The mutation
L69P in the PAS domain of the hERG potassium channel results in
LQTS by trafficking deficiency. Channels (Austin). 14:163–174.
2020. View Article : Google Scholar
|
|
23
|
Zhan G, Wang F, Ding YQ, Li XH, Li YX,
Zhao ZR, Li JX, Liu Y, Zhao X, Yan CC and Li BX: Rutaecarpine
targets hERG channels and participates in regulating
electrophysiological properties leading to ventricular arrhythmia.
J Cell Mol Med. 25:4938–4949. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Y, Huang X, Zhou J, Yang X, Li D, Mao
H, Sun HH, Liu N and Lian J: Trafficking-deficient G572R-hERG and
E637K-hERG activate stress and clearance pathways in endoplasmic
reticulum. PLoS One. 7:e298852012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ono M, Burgess DE, Schroder EA, Elayi CS,
Anderson CL, January CT, Sun B, Immadisetty K, Kekenes-Huskey PM
and Delisle BP: Long QT syndrome type 2: Emerging strategies for
correcting class 2 KCNH2 (hERG) mutations and identifying new
patients. Biomolecules. 10:11442020. View Article : Google Scholar :
|
|
26
|
Anderson CL, Delisle BP, Anson BD, Kilby
JA, Will ML, Tester DJ, Gong Q, Zhou Z, Ackerman MJ and January CT:
Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2
(trafficking-deficient) mechanism. Circulation. 113:365–373. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Du H, Zheng C, Aslam M, Xie X, Wang W,
Yang Y and Liu X: Endoplasmic reticulum-mediated protein quality
control and endoplasmic reticulum-associated degradation pathway
explain the reduction of N-glycoprotein level under the lead
stress. Front Plant Sci. 11:5985522021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ito Y, Takeda Y, Seko A, Izumi M and
Kajihara Y: Functional analysis of endoplasmic reticulum
glucosyltransferase (UGGT): Synthetic chemistry's initiative in
glycobiology. Semin Cell Dev Biol. 41:90–98. 2015. View Article : Google Scholar
|
|
29
|
Tannous A, Patel N, Tamura T and Hebert
DN: Reglucosylation by UDP-glucose: Glycoprotein
glucosyltransferase 1 delays glycoprotein secretion but not
degradation. Mol Biol Cell. 26:390–405. 2015. View Article : Google Scholar :
|
|
30
|
Ferris SP, Jaber NS, Molinari M, Arvan P
and Kaufman RJ: UDP-glucose: Glycoprotein glucosyltransferase
(UGGT1) promotes substrate solubility in the endoplasmic reticulum.
Mol Biol Cell. 24:2597–2608. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guerriero CJ and Brodsky JL: The delicate
balance between secreted protein folding and endoplasmic
reticulum-associated degradation in human physiology. Physiol Rev.
92:537–576. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shenkman M, Ron E, Yehuda R, Benyair R,
Khalaila I and Lederkremer GZ: Mannosidase activity of EDEM1 and
EDEM2 depends on an unfolded state of their glycoprotein
substrates. Commun Biol. 1:1722018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Benyair R, Ogen-Shtern N, Mazkereth N,
Shai B, Ehrlich M and Lederkremer GZ: Mammalian ER mannosidase I
resides in quality control vesicles, where it encounters its
glycoprotein substrates. Mol Biol Cell. 26:172–184. 2015.
View Article : Google Scholar :
|
|
34
|
Ogen-Shtern N, Avezov E, Shenkman M,
Benyair R and Lederkremer GZ: Mannosidase IA is in quality control
vesicles and participates in glycoprotein targeting to ERAD. J Mol
Biol. 428:3194–3205. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chung H, Cho H, Perry C, Song J, Ylaya K,
Lee H and Kim JH: Downregulation of ERp57 expression is associated
with poor prognosis in early-stage cervical cancer. Biomarkers.
18:573–579. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Choe MH, Min JW, Jeon HB, Cho DH, Oh JS,
Lee HG, Hwang SG, An S, Han YH and Kim JS: ERp57 modulates STAT3
activity in radioresistant laryngeal cancer cells and serves as a
prognostic marker for laryngeal cancer. Oncotarget. 6:2654–2666.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kozlov G, Pocanschi CL, Rosenauer A,
Bastos-Aristizabal S, Gorelik A, Williams DB and Gehring K:
Structural basis of carbohydrate recognition by calreticulin. J
Biol Chem. 285:38612–38620. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kiuchi T, Izumi M, Mukogawa Y, Shimada A,
Okamoto R, Seko A, Sakono M, Takeda Y, Ito Y and Kajihara Y:
Monitoring of glycoprotein quality control system with a series of
chemically synthesized homogeneous native and misfolded
glycoproteins. J Am Chem Soc. 140:17499–17507. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ito Y, Kajihara Y and Takeda Y:
Chemical-synthesis-based approach to glycoprotein functions in the
endoplasmic reticulum. Chemistry. 26:15461–15470. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ghemrawi R and Khair M: Endoplasmic
reticulum stress and unfolded protein response in neurodegenerative
diseases. Int J Mol Sci. 21:61272020. View Article : Google Scholar :
|
|
41
|
Wang SB, Shi Q, Xu Y, Xie WL, Zhang J,
Tian C, Guo Y, Wang K, Zhang BY, Chen C, et al: Protein disulfide
isomerase regulates endoplasmic reticulum stress and the apoptotic
process during prion infection and PrP mutant-induced cytotoxicity.
PLoS One. 7:e382212012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Perri E, Parakh S and Atkin J: Protein
disulphide isomerases: Emerging roles of PDI and ERp57 in the
nervous system and as therapeutic targets for ALS. Expert Opin Ther
Targets. 21:37–49. 2017. View Article : Google Scholar
|
|
43
|
Turano C, Gaucci E, Grillo C and
Chichiarelli S: ERp57/GRP58: A protein with multiple functions.
Cell Mol Biol Lett. 16:539–563. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sepulveda M, Rozas P, Hetz C and Medinas
DB: ERp57 as a novel cellular factor controlling prion protein
biosynthesis: Therapeutic potential of protein disulfide
isomerases. Prion. 10:50–56. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hoffstrom BG, Kaplan A, Letso R, Schmid
RS, Turmel GJ, Lo DC and Stockwell BR: Inhibitors of protein
disulfide isomerase suppress apoptosis induced by misfolded
proteins. Nat Chem Biol. 6:900–906. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Grek C and Townsend DM: Protein disulfide
isomerase superfamily in disease and the regulation of apoptosis.
Endoplasmic Reticulum Stress Dis. 1:4–17. 2014.PubMed/NCBI
|
|
47
|
Han A, Li C, Zahed T, Wong M, Smith I,
Hoedel K, Green D and Boiko AD: Calreticulin is a critical cell
survival factor in malignant neoplasms. PLoS Biol. 17:e30004022019.
View Article : Google Scholar : PubMed/NCBI
|