Introduction
Nasopharyngeal carcinoma (NPC) is highly prevalent
in Southern China and Southeast Asia (1,2).
Advanced NPC is often associated with resistance to conventional
radiotherapy and chemotherapy (3,4).
A combination chemotherapy regimen with different pharmacological
mechanism drugs could be an effective strategy to enhance the
therapeutic effect (5,6). In the present study, proteasome
inhibitor was combined with paclitaxel and their efficacy was
evaluated in the treatment of NPC.
Bortezomib (PS341) is an inhibitor of the 26S
proteasome, used in the treatment of multiple myeloma, as well as
in clinical trials for solid tumors as a single agent or in
combination with other drugs (7-9).
Proteasome inhibition causes rapid accumulation of ubiquitinated
proteins and perturbation of protein metabolism, leading to
initiation of apoptosis by the endoplasmic reticulum to overcome
irreversible cellular damage (10,11). The ubiquitin-proteasome system is
a non-lysosomal protein degradation pathway, which regulates
numerous eukaryotic cell signaling pathways, including the cell
cycle (12). In the cell cycle
pathway, two families of E3 ubiquitin ligases, anaphase promoting
complex or cyclosome (APC/C) and Skp1/CUL1/F-box protein, are
responsible for periodic proteolysis of cell cycle regulators,
ensuring regulated cell cycle progression (13).
On the other hand, paclitaxel is a member of the
taxane class of drugs, used as an anti-microtubule agent in the
treatment of cancer, including NPC (14). Binding of paclitaxel to β-tubulin
dimers inhibits the hydrolysis of GTP, resulting in microtubule
stabilization and loss of dynamics (15). Dysfunction of microtubules causes
incorrect spindle-chromosome attachment and prolonged mitotic
arrest, resulting in mitotic catastrophe, and initiation of cell
death to prevent transmission of erroneous genetic information
(16,17).
Mitotic catastrophe refers to cell death associated
with inappropriate mitosis or irreversible cell cycle arrest
(18). Cells that undergo
mitotic catastrophe are morphologically distinguishable, since
nuclear envelopes form around individual or groups of chromosomes,
resulting in large nonviable cells with multiple micronuclei
(19,20). Mitotic catastrophe has been
proposed to be a tumor suppression mechanism, while evasion of
mitotic catastrophe constitutes one of the mechanisms of cancer
development (21). Cells
typically undergo prolonged mitotic arrest when treated with
antimitotic drugs, and the cell fate is determined by assessing the
slow or sudden degradation of cyclin B1 resulting in mitotic
slippage or activation of cell death pathways, respectively
(22). The activation of cell
death pathways leads to accumulation of death activators or loss of
death inhibitors (23).
Additionally, mitotic cell death results from intracellular signals
breaching the death threshold before achieving the mitotic slippage
threshold (24).
The CDK1/cyclin B1 complex is essential for cell
cycle progression in eukaryotes. Cyclin B1 is a regulatory subunit,
while CDK1 is a catalytic subunit (25). CDK1 phosphorylates a number of
mitotic substrates, which are involved in mitotic spindle
generation, chromosome condensation, nuclear envelope breakdown and
mitotic death (26).
Accumulation of the CDK1-cyclin B1 proteins is critical for the
activation of CDK1 and entry of mitosis (27). In addition to cyclin B1 binding,
CDK1 activation requires the phosphorylation at T161 of its
activation segment and the dephosphorylation of Y15 and T14
residues (28). After mitosis,
destruction of cyclin B1 provides a mechanism to rapidly inactivate
CDK1 and exit mitosis, and CDK1/cyclin B1 catalyzes its own
destruction by stimulating the activity of APC/CCDC20
(29). When cells undergo
inappropriate mitosis or irreversible cell cycle arrest after
treatment with antimitotic drugs, the mitotic cells also require
the sudden degradation of CDK1/cyclin B1 and the breaching of the
death threshold (30,31).
In the present study examined the combination
chemotherapy regimen, and revealed that proteasome inhibitor PS341
markedly decreased the number of rounded cells in
paclitaxel-treated NPC. Accordingly, the present study aimed to
investigate the effect of proteasome inhibitor in attenuating the
lethality of paclitaxel against NPC cells and to explore its
related molecular mechanism.
Materials and methods
Cell culture and drugs
The 5-8F and 6-10B NPC cell lines (Cancer Center of
Sun Yat-sen University, Guangzhou, China) used in the present study
were cultured in DMEM (Biological Industries) with 10% FBS
(Biological Industries), penicillin (100 U/ml; Biological
Industries) and streptomycin (100 µg/ml; Biological
Industries) in a humidified incubator with 5% CO2 at
37°C. Paclitaxel, PS341, MG132, Ro3306 and cycloheximide were
purchased from MedChemExpress. Flutax1 was purchased from R&D
Systems, Inc. The cells were cultured with paclitaxel (400 nM),
flutax1 (3 µM), PS341 (30 nM), MG132 (700 nM), Ro3306 (5
µM) or cycloheximide (1 µg/ml) in a humidified
incubator with 5% CO2 at 37°C for 24 or 48 h during the
test. In addition, the sets and procedure of the cycloheximide
assay (Fig. S1) were as
follows: Two sets were cycloheximide-treated cells and
non-cycloheximide-treated cells, and each set contained four
groups: NC (0.5%, v/v, DMSO), PTX (400 nM), PS341 (30 nM) and PTX
(400 nM)-PS341 (30 nM). Cycloheximide-treated cells were cultured
as groups for 16 h, then cycloheximide (1 µg/ml) was added
for a further 8-h treatment, and cells were harvested at 24 h.
While non-cycloheximide-treated cells were cultured as groups and
harvested at 24 h. The used drug concentrations were tested against
5-8F and 6-10B cells in the present study based on the following
principles: Paclitaxel and flutax1 were used at a lethal
concentration, and PS341, MG132, Ro3306 and cycloheximide were used
at low toxicity concentrations or nontoxic concentrations, ensuring
cell proliferation inhibition and death were mainly caused by taxol
analogues. After cell attachment, taxol-analogues and PS341, MG132
or Ro3306 were added simultaneously.
Cell Counting Kit-8 (CCK-8) assay
To examine the effect on cell proliferation, 15,000
cells per well were seeded in a 96-well plate in DMEM with 10% FBS
(n=3). After cell adhesion, the medium was replaced with a fresh
complete medium containing drugs at the corresponding
concentrations as follow: Negative control (NC; 0.5%, v/v, DMSO),
PTX (400 nM), PS341 (30 nM), PTX (400 nM)-PS341 (30 nM), MG132 (700
nM), PTX (400 nM)-MG132 (700 nM), PTX (400 nM)-PS341 (0, 5, 10, 25,
50 or 100 nM), PTX (400 nM)-MG132 (0, 50, 100, 200, 400 or 700 nM),
Ro3306 (5 µM) or PTX (400 nM)-Ro3306 (5 µM). A total
of 10 µl CCK-8 reagent (Shanghai Yeasen Biotechnology Co.,
Ltd.) was added to each well every 24 h for 2 days. The cells were
incubated for 1 h and then absorbance at 450 nm was measured using
a microplate reader (ELx800; BioTek Instruments, Inc.).
Western blotting
To examine the related protein expression changes,
5×105 cells per well were seeded in a 12-well plate with
DMEM with 10% FBS. After cell culture in a humidified incubator
with 5% CO2 at 37°C for cell adhesion, the medium was
replaced with a fresh complete medium containing drugs at the
corresponding concentrations as follows: Negative control (NC;
0.5%, v/v, DMSO), PTX (400 nM), PS341 (30 nM), PTX (400 nM)-PS341
(30 nM), MG132 (700 nM), PTX (400 nM)-MG132 (700 nM), Ro3306 (5
µM) or PTX (400 nM)-Ro3306 (5 µM). After 24 or 48 h,
cells were harvested and lysed in Cell lysis buffer for Western and
IP (Beyotime Institute of Biotechnology). The BCA Protein Assay kit
(Beyotime Institute of Biotechnology) was selected for protein
concentration determination. A total of 20 µg total proteins
were loaded per lane and resolved by 10% SDS-PAGE, and then
transferred onto a 0.45-µm PVDF membrane (MilliporeSigma),
except p21 Waf1/Cip1 which was transferred onto a 0.22-µm
PVDF membrane (MilliporeSigma). The membrane was blocked in PBS
with 0.05% Tween-20 (PBST) with 5% w/v nonfat dry milk at room
temperature for 1 h, then washed three times in PBST for 2 min each
at room temperature, followed by incubation with primary antibody
in PBST with gentle agitation at 4°C overnight. The membrane was
washed three times for 10 min each in PBST and then incubated with
an HRP-conjugated secondary antibody at room temperature for 60 min
in PBST. After three washes for 5 min each in PBST, the signal was
visualized using an ECL detection reagent (Thermo Fisher
Scientific, Inc.) and imaged. Ubiquitin antibody (cat. no. 3933;
dilution, 1:1,000), CDK1 antibody (cat. no. 77055; dilution,
1:1,000), phosphorylated (p-)CDK1(Thr14) antibody (cat. no. 2543;
dilution, 1:1,000), p-CDK1(Thr161) antibody (cat. no. 9114;
dilution, 1:1,000), p21 Waf1/Cip1 antibody (cat. no. 2947;
dilution, 1:1,000), MCL1 apoptosis regulator, BCL2 family member
(MCL1) antibody (cat. no. 94296; dilution, 1:1,000), caspase-9
antibody (for detection of procaspase-9 and cleaved caspase-9; cat.
no. 9502; dilution, 1:1,000), poly (ADP-ribose) polymerase (PARP)
antibody (for detection of full-length PARP and cleaved PARP; cat.
no. 9532; dilution, 1:1,000) were purchased from Cell Signaling
Technology, Inc. Cyclin D1 (cat. no. AF1183; dilution, 1:1,000),
cyclin E1 (cat. no. AF2491; dilution, 1:1,000), cyclin A2 (cat. no.
AF2524; dilution, 1:1,000), cyclin B1 (cat. no. AF6627; dilution,
1:1,000), aurora A (cat. no. AF1708; dilution, 1:1,000), aurora B
(cat. no. AF1930; dilution, 1:1,000) and GAPDH (cat. no. AF0006;
dilution, 1:2,000) primary antibodies were purchased from Beyotime
Institute of Biotechnology. GAPDH was used as a control. The
secondary antibodies, including anti-rabbit IgG, HRP-linked
antibody (cat. no. 7074; dilution, 1:2,000) and anti-mouse IgG,
HRP-linked antibody (cat. no. 7076; dilution, 1:2,000), were
purchased from Cell Signaling Technology, Inc. Grayscale
semi-quantifications of bands was performed using ImageJ software
(version 1.42q; National Institutes of Health). GAPDH was used as a
loading control for total protein, the grayscale values of each
protein band were normalized to GAPDH within groups, and relative
protein levels were normalized to the control group.
Cell microscopic observation
To examine the paclitaxel-induced cell morphology
changes, 3×105 cells per well were seeded in a 24-well
plate with DMEM with 10% FBS. After cell culture in a humidified
incubator with 5% CO2 at 37°C for cell adhesion, the
medium was replaced with a fresh complete medium containing drugs
at the corresponding concentrations as follows: NC (0.5% v/v DMSO),
PTX (400 nM), PS341 (30 nM), PTX (400 nM)-PS341 (30 nM), MG132 (700
nM), PTX (400 nM)-MG132 (700 nM). After culture in a humidified
incubator with 5% CO2 at 37°C for 24 h, the cells were
observed under a Leica DM IL LED inverted light microscope (Leica
Microsystems, Inc.) and images were captured. The ratio of rounded
cells to total cells in five random fields of vision
(magnification, ×200) was counted. To examine the
paclitaxel-induced nuclear condensation in NPC cells, Hoechst 33342
Staining Solution for Live Cells 100X (cat. no. C1028; Beyotime
Institute of Biotechnology) was used to stain the nucleus. After
removing the medium and rinsing with PBS, nuclei were stained with
200 µl diluted Hoechst 33342 (dilution, 1:100) per well for
10 min at 37°C, rinsed three times with PBS and observed under a
fluorescence microscope (Leica Microsystems, Inc.). Merging of the
images was performed using ImageJ software (version 1.42q; National
Institutes of Health). To examine the taxol-induced microtubule
changes, fluorescently labeled taxol flutax1 (R&D Systems,
Inc.) was used for tracing taxol in NPC cells, while the
transformation of microtubules in flutax1-treated NPC cells was
observed using Tubulin-Tracker Red (cat. no. C1050; Beyotime
Institute of Biotechnology). After culture with flutax1 (3
µM), flutax1 (3 µM)-PS341 (30 nM) and flutax1 (3
µM) -MG132 (700 nM) in a humidified incubator with 5%
CO2 at 37°C for 24 h, cells were rinsed with PBS two
times and fixed with 3.7% paraformaldehyde (Sigma-Aldrich; Merck
KGaA) in PBS for 20 min at room temperature. Subsequently, cells
were washed three times with PBS containing 0.1% Triton X-100 for 5
min each time at room temperature. Tubulin-Tracker Red was diluted
with PBS containing 3% BSA (Beyotime Institute of Biotechnology)
and 0.1% Triton X-100 (Beyotime Institute of Biotechnology), and
200 µl Tubulin-Tracker Red (dilution, 1:100) was added to
each well for 60 min at room temperature, followed by
counterstaining with 200 µl diluted Hoechst 33342 (dilution,
1:100) per well for 10 min at 37°C. After three washes with PBS for
5 min each at room temperature, related results were visualized
with a fluorescence microscope (Lionheart; BioTek Instruments,
Inc.).
Cell cycle detection
To investigate the cell cycle distribution,
2.5×106 cells per well were seeded in a 6-well plate
with DMEM with 10% FBS (n=3). After cell adhesion, the medium was
replaced with fresh complete medium containing drugs at the
corresponding concentrations as follows: NC (0.5% v/v DMSO), PTX
(400 nM), PS341 (30 nM), PTX (400 nM)-PS341 (30 nM), MG132 (700
nM), PTX (400 nM)-MG132 (700 nM), Ro3306 (5 µM) and PTX (400
nM)-Ro3306 (5 µM). After culture in a humidified incubator
with 5% CO2 at 37°C for 24 h, the cells were harvested
and fixed in 75% alcohol at 4°C. After 24 h, 75% alcohol was
removed by centrifugation at 500 × g at 4°C for 5 min, followed by
two washes with cold PBS. Cells were stained using propidium iodide
(PI final concentration, 50 µg/ml; Beyotime Institute of
Biotechnology) and RNase treatment (RNase final concentration, 100
µg/ml; Beyotime Institute of Biotechnology), and incubated
for 20 min at 37°C in the dark. The cell cycle distribution was
determined by evaluating DNA content using a BD Accuri C6 Flow
Cytometer (BD Biosciences), and the data were analyzed using the BD
Accuri C6 Software (version 1.0.264.21; BD Biosciences). The NC
group was set as the control when comparing SubG1 phase
change, PTX (400 nM) group was set as the control when comparing
restored G1 phase and decreased G2/M
phase.
Cell apoptosis detection
The cell apoptosis assay was performed using the
Annexin V-FITC/PI Apoptosis Detection kit (Shanghai Yeasen
Biotechnology Co., Ltd.) according to the manufacturer's protocol.
Briefly, 1.5×106 cells per well were seeded in a 6-well
plate with DMEM with 10% FBS (n=3). After cell adhesion, the medium
was replaced with fresh complete medium containing drugs at the
corresponding concentrations as follows: NC (0.5% v/v DMSO), PTX
(400 nM), PS341 (30 nM), PTX (400 nM)-PS341 (30 nM), MG132 (700
nM), PTX (400 nM)-MG132 (700 nM), Ro3306 (5 µM) and PTX (400
nM)-Ro3306 (5 µM). After culture in a humidified incubator
with 5% CO2 at 37°C for 48 h, the cells were harvested
by centrifugation at 500 × g at 4°C for 5 min and washed twice with
cold PBS, then resuspended in 1X binding buffer at a concentration
of 5×105 cells/ml. Cells (100 µl) were
resuspended with 5 µl Annexin V-FITC and 10 µl PI.
After incubation for 15 min at room temperature in the dark, 400
µl 1X Binding Buffer was added to each tube. The proportion
of apoptotic cells was measured using a BD Accuri C6 Flow Cytometer
(BD Biosciences) within 1 h, and data were analyzed using the BD
Accuri C6 Software (version 1.0.264.21; BD Biosciences). Annexin
V-FITC and PI double-positive cells were considered to be late
apoptotic cells, while Annexin V-FITC positive and PI-negative
cells were considered to be early apoptotic cells. The fold changes
in the percentage of apoptotic cells were calculated, and each
group was normalized to the NC group. The NC group was set as the
control when comparing fold change of paclitaxel-induced apoptosis,
and the PTX (400 nM) group was set as the control when comparing
fold change of proteasome inhibitors or Ro3306 decreased
paclitaxel-induced apoptosis. All fold change data were normalized
to the NC group.
Statistical analysis
All experiments were performed at least in
triplicate and data are presented as the mean ± SD. Data were
analyzed using one-way ANOVA or two-way ANOVA with multiple
comparison as Tukey's post hoc test or Dunnett's post hoc test as
appropriate. Figures were generated and statistical analysis was
performed using GraphPad Prism 8.3.0 (GraphPad Software, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
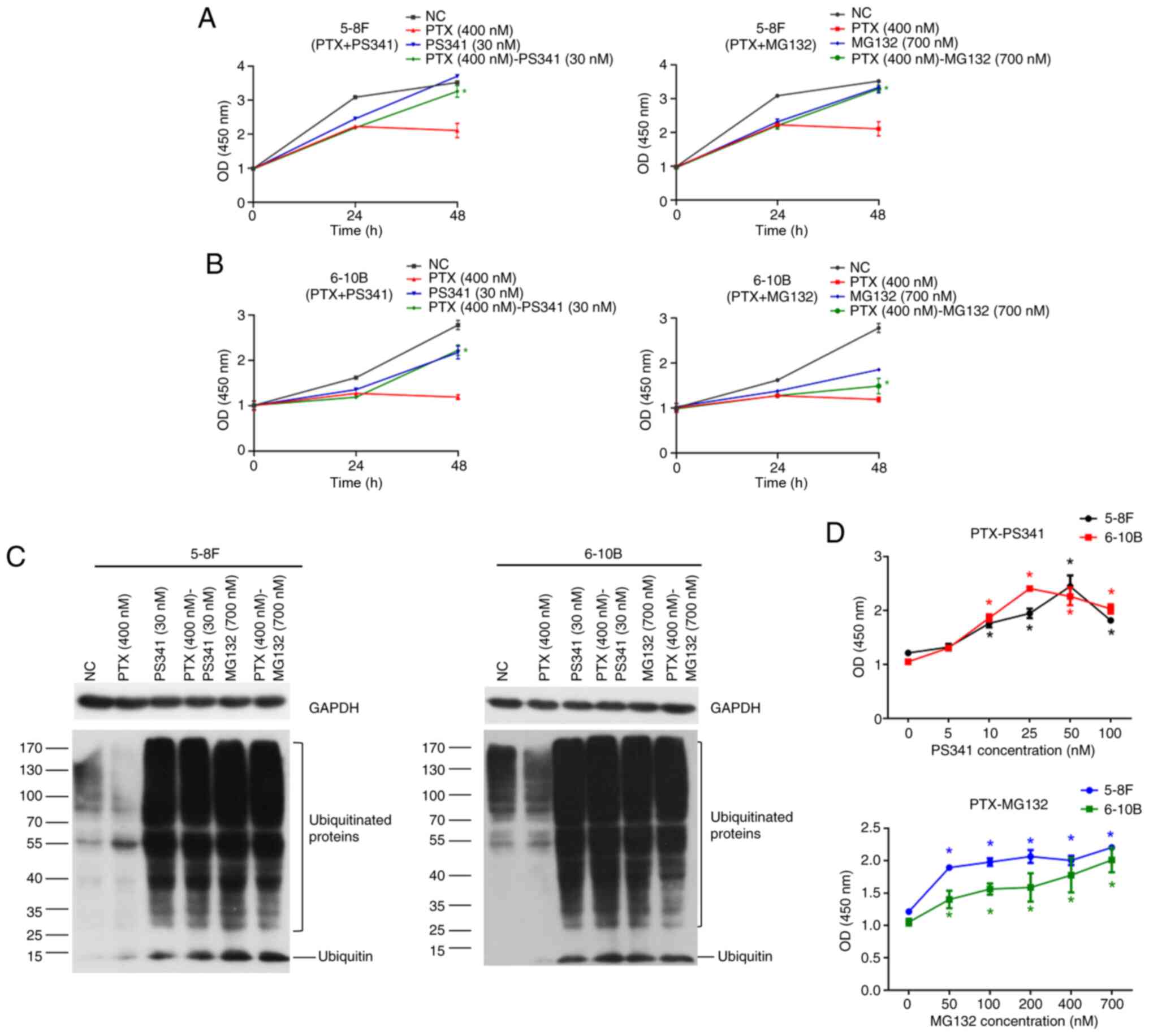
Proteasome inhibitors reduce the
paclitaxel-specific inhibition of NPC cell proliferation
A CCK-8 assay was performed to determine the
inhibition of NPC cell proliferation by proteasome blockers. The
present results demonstrated that 5-8F cells and 6-10B cells were
still growing after culture with proteasome inhibitor PS341 (30 nM)
or MG132 (700 nM) after 48 h. Additionally, both inhibitors reduced
the inhibitory effect of paclitaxel on NPC cell proliferation after
48 h, while paclitaxel (400 nM) was toxic to 5-8F and 6-10B cells
based on the decreased absorbance values at 48 h (Fig. 1A and B). Accumulation of
ubiquitinated-proteins indicated compromised proteasome functions
following treatment with PS341 (30 nM) and MG132 (700 nM) after 24
h (Fig. 1C). In addition, the
linear plot of CCK-8 results revealed increasing absorbance values
at 450 nm with the gradual increase in concentrations of proteasome
inhibitors compared with the negative control group (PTX 400 nM).
PS341 (10-100 nM) and MG132 (50-700 nM) attenuated the inhibitory
effect of paclitaxel (400 nM) on cell proliferation after 48 h
(Fig. 1D). Therefore, proteasome
inhibition was associated with reduced lethality of paclitaxel.
Proteasome inhibitors reduce
paclitaxel-specific morphological changes in NPC cells
Paclitaxel-induced mitotic catastrophe often leads
to changes in cell morphology and chromatin condensation after 24 h
(31). The present findings
demonstrated that proteasome inhibitors markedly reduced the
paclitaxel-specific morphological changes and multiple micronuclei
of NPC cells after 24 h (Fig.
2A-D). Representative images (magnification, ×40; top rows in
Fig. 2A and B) revealed that
most of the paclitaxel-treated cells were rounded; however, rounded
cells decreased when the cells were treated with paclitaxel and
proteasome inhibitors (PS341 or MG132) synchronously. The ratio of
rounded cells and total cells in five random fields of vision
(magnification, ×200; bottom row in Fig. 2A and B) was determined. The
results of statistical analyses indicated that proteasome
inhibitors significantly decreased paclitaxel-induced rounded
cells. These data demonstrated that the proteasome inhibitors aided
the NPC cells in evading mitotic catastrophe after 24 h, speculated
that more NPC cells survived with spindle-chromosome attachment
errors represented higher chromosome mutation risk.
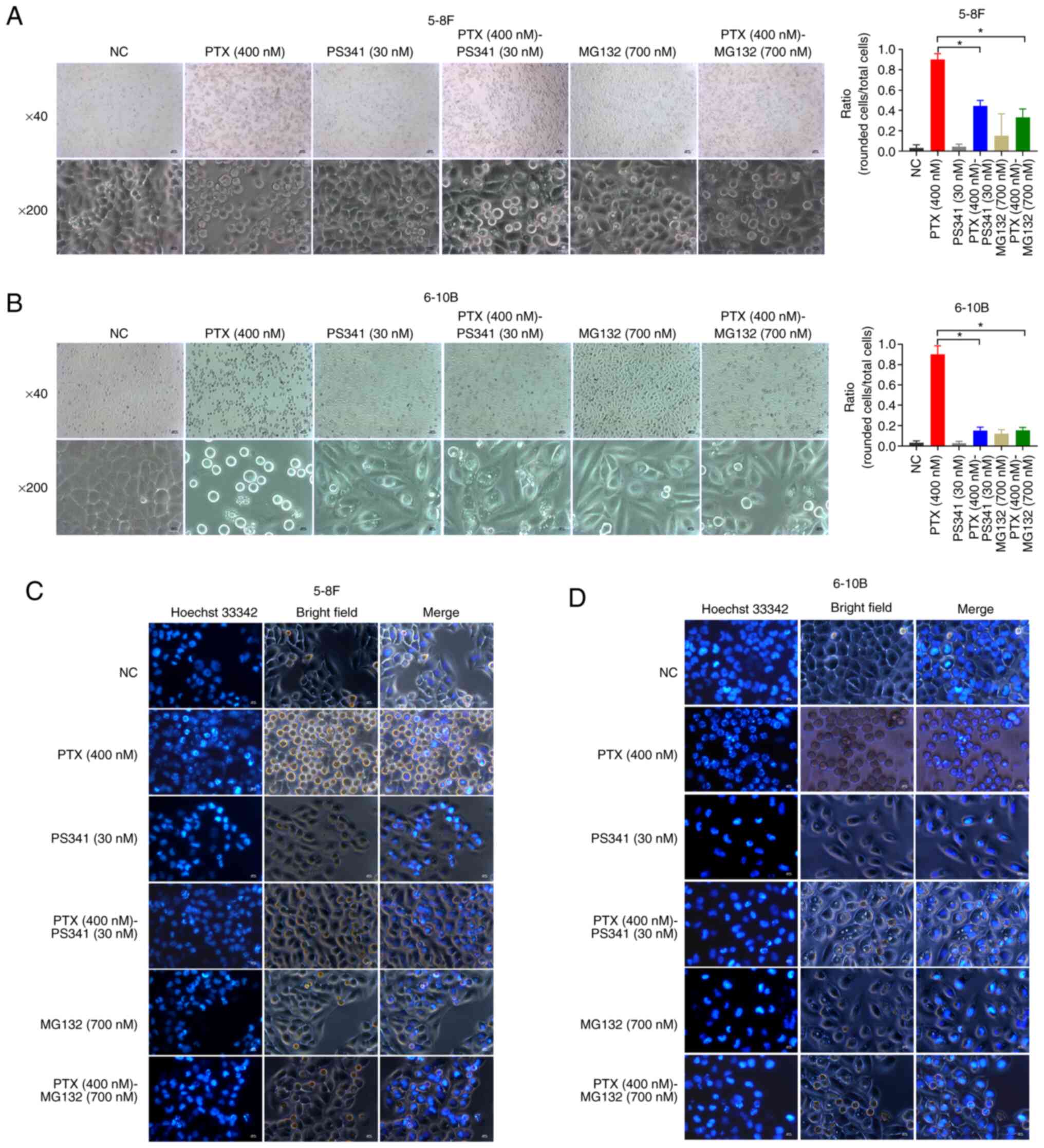 | Figure 2Proteasome inhibitors reduce
paclitaxel-induced morphological changes and nuclear condensation
in NPC cells. Proteasome inhibitors (PS341 and MG132) significantly
reduced the paclitaxel-induced morphological changes of (A) 5-8F
and (B) 6-10B cells. The data are presented as the mean ± SD (n=5)
and were analyzed using one-way ANOVA followed by Tukey's post hoc
test. *P<0.05. Magnification, ×40; scale bar, 500
µm (top row). Magnification, ×200; scale bar, 100 µm
(bottom row). Proteasome inhibitors reduced nuclear condensation in
rounded (C) 5-8F and (D) 6-10B cells. Magnification, ×200; scale
bar, 100 µm. NC, negative control; NPC, nasopharyngeal
carcinoma; PS341, bortezomib; PTX, paclitaxel. |
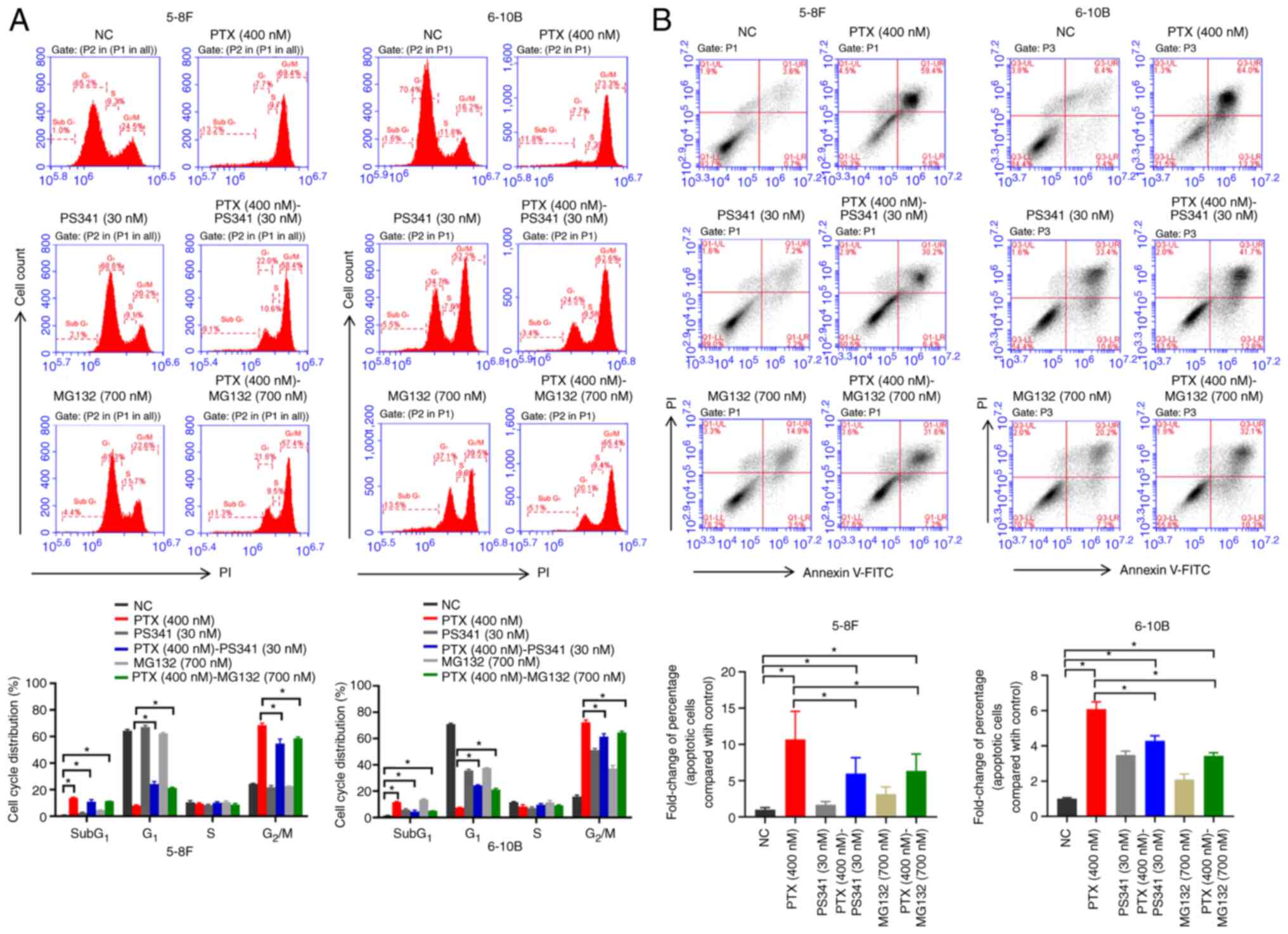
Proteasome inhibitors decrease the
paclitaxel-induced cell cycle arrest in NPC cells
To determine the cell cycle profile in the
drug-treated NPC cells after 24 h, the DNA content was evaluated
using flow cytometry. Compared with NC groups, addition of
paclitaxel increased the percentage of Sub G1 phase
cells in the PTX (400 nM), PTX (400 nM)-PS341 (30 nM) and PTX (400
nM)-MG132 (700 nM) groups. Paclitaxel blocked almost all NPC cells
at the G2/M phase; however, when the cells were treated
with paclitaxel and proteasome inhibitors (PS341 or MG132)
synchronously, the G1 phase was significantly restored
and G2/M phase was significantly decreased compared with
the paclitaxel-treated group (Fig.
3A). These results combined with the CCK-8 assay data (Fig. 1A and B) suggested that proteasome
inhibitors could help NPC cells in escaping paclitaxel-induced
mitotic arrest and proliferation inhibition.
Proteasome inhibitors reduce
paclitaxel-induced cell death in NPC cells
Apoptosis is the main outcome of mitotic
catastrophe-induced cell death (32). An Annexin V-FITC/PI assay was
performed to determine the effect of paclitaxel coupled with
proteasome inhibitors on cell apoptosis after 48 h. Compared with
the NC groups, addition of paclitaxel in the PTX (400 nM), PTX (400
nM)-PS341 (30 nM) and PTX (400 nM)-MG132 (700 nM) groups increased
the percentage of apoptotic cells. When the cells were treated with
paclitaxel and proteasome inhibitors (PS341 or MG132)
synchronously, the percentage of apoptotic cells was significantly
decreased compared with that in the paclitaxel-treated group. The
data demonstrated a marked reduction of paclitaxel-induced
apoptosis in NPC cells by the proteasome inhibitors (Fig. 3B).
Proteasome inhibitors lead to an enlarged
microtubule cytoskeleton system and freed nuclear condensation of
NPC cells treated with flutax1
Furthermore, the present study used fluorescently
labeled taxol flutax1 (green) to treat NPC cells, tubulin-tracker
(red) to label microtubules, and hoechst 33342 (blue) to stain DNA
to extract more data from microtubule stabilized cells. In the
Flutax1 (3 µM) group, most of the flutax1-treated NPC cells
(green) presented multiple micronuclei (blue) after 24 h, and
pyknotic microtubules (red) wrapped with these condensed genetic
materials (Fig. 4A; first and
fourth row). Condensed microtubules and chromatin indicated that
the mitotic spindle failed in separating chromosomes to their
respective poles, and most NPC cells were stuck in the mitosis
phase for taxol-treatment (33).
Notably, a combination of flutax1 and proteasome inhibitors (PS341
or MG132) led to an enlarged microtubule cytoskeleton system and
fewer multiple micronuclei in NPC cells, and also fewer NPC cells
were stuck in the mitosis phase (Fig. 4A).
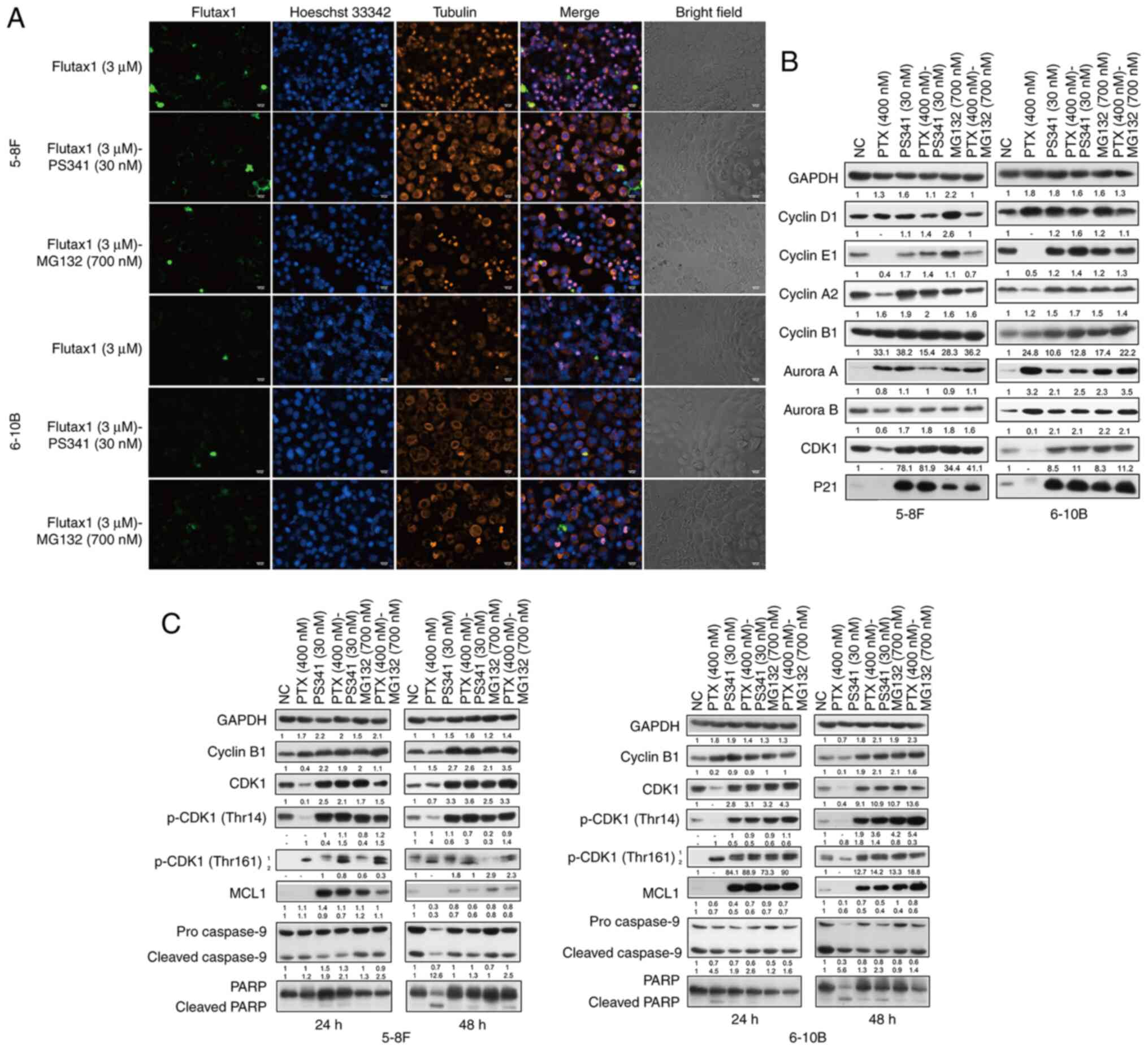 | Figure 4Proteasome inhibitors perturb
CDK1/cyclin B1 functions in regulating the NPC cell cycle. (A)
Proteasome inhibitor-treated NPC cells were freed from nuclear
condensation induced by flutax1 and had an enlarged microtubule
cytoskeleton system. Magnification, ×200; scale bar, 100 µm.
(B) NPC cells treated with the proteasome inhibitors presented a
different cell cycle proteins profile compared with the
paclitaxel-treated cells at 24 h, particularly in terms of CDK1 and
cyclin B1. (C) Proteasome inhibitors altered the expression profile
of CDK1/cyclin B1 and decreased paclitaxel-induced apoptosis
through MCL1 accumulation and caspase-9 stabilization at 24 and 48
h. Representative western blotting results from three independent
experiments. Grayscale semi-quantification of bands was performed
using ImageJ software. Control group was normalized to 1. Data are
presented as fold change in protein expression relative to the
control group. No protein detected was marked as '-'. MCL1, MCL1
apoptosis regulator, BCL2 family member; NC, negative control; NPC,
nasopharyngeal carcinoma; p-, phosphorylated; PARP, poly
(ADP-ribose) polymerase; PS341, bortezomib; PTX, paclitaxel. |
Proteasome inhibitors alter the
expression pattern of the CDK1/cyclin B1 protein in NPC cells
There are four main types of cyclin in human cells,
cyclin D1 (protein exists throughout the cell cycle, triggers cell
from G0 to G1 and G1 to S phase),
cyclin E1 (protein mainly exists during G1/S phase,
preparing for DNA replication in S phase), cyclin A2 (protein
mainly exists during G2/M transition, for the activation
of DNA replication in S phase) and cyclin B1 (protein mainly exists
during G2/M phase, for the assembly of mitotic spindle
and mitosis promotion) (34,35). In the present study,
paclitaxel-treated NPC cells were blocked at M phase after 24 h.
Compared with the NC group of 5-8F and 6-10B cells in Fig. 4B, the expression profile of
cyclin-related proteins in the PTX (400 nM) group exhibited mitotic
phase-specific features, such as low expression levels of cyclin E1
(during G1/S phase) and cyclin A2 (during
G2/M transition), and high expression levels of aurora A
and cyclin B1. Additionally, there was a reduction of CDK1
expression in the PTX (400 nM) group. On the other hand, the
combination of paclitaxel and proteasome inhibitors restored cyclin
E1 and cyclin A2 expression in the PTX (400 nM)-PS341 (30 nM) and
PTX (400 nM)-MG132 (700 nM) groups. The inhibition of proteasomes
led to high expression levels of both CDK1 and cyclin B1 in
proteasome inhibitor-treated groups [PS341 (30 nM), PTX (400
nM)-PS341 (30 nM), MG132 (700 nM) and PTX (400 nM)-MG132 (700 nM)]
compared with the NC group (Fig.
4B). The contrast of the CDK1/cyclin B1 expression pattern
between the PTX (400 nM) group (high cyclin B1 and low CDK1
expression compared with NC group) and proteasome inhibitor-treated
groups (high cyclin B1 and high CDK1 expression compared with NC
group) suggested that proteasome inhibitor-induced alterations in
the CDK1/cyclin B1 expression pattern could be essential for
escaping paclitaxel-induced mitotic arrest.
Proteasome inhibitors reduce
paclitaxel-initiated cell death via CDK1/cyclin B1 signaling
Considering the onset time of paclitaxel-initiated
cell death and the key role of CDK1/cyclin B1 in mitosis
regulation, the present study examined the expression profile of
CDK1/cyclin B1 and its related proteins at 24 and 48 h. In the
paclitaxel-treated NPC cells, CDK1 was activated (compared with NC
groups, T161 phosphorylation was increased and T14 phosphorylation
was decreased) at 24 h, while cyclin B1 proteolysis and the
caspase-9/PARP apoptosis cascade were activated at 48 h. In the
present study, p-CDK1 T161 separated into two bands: Band 1
reflected T161 phosphorylation combination with inhibitory
phosphorylation of either T14 or Y15, whereas band 2 reflected the
active single T161 phosphorylated form of CDK1. In the proteasome
inhibitor-treated NPC cells, PS341 (30 nM), PTX (400 nM)-PS341 (30
nM), MG132 (700 nM) and PTX (400 nM)-MG132 (700 nM) groups
sustained the high expression levels of CDK1/cyclin B1 proteins,
and multi-band T161 phosphorylation status of CDK1. Multisite
phosphorylation of CDK1 prevents its premature activation and
demonstrates that the cells are distributed in a different phase of
the cell cycle (28).
Additionally, compared with paclitaxel-treated cells in the PTX
(400 nM) group, there was persistent high CDK1/cyclin B1 protein
expression with MCL1 accumulation, less cleavage of caspase-9 and
PARP in the PS341 (30 nM), PTX (400 nM)-PS341 (30 nM), MG132 (700
nM) and PTX (400 nM)-MG132 (700 nM) groups (Fig. 4C). Following culture with
cycloheximide for 8 h, PS341 was also associated with accumulation
of cyclin B1 and MCL1 proteins in 5-8F and 6-10B cells at 24 h
(Fig. S1).
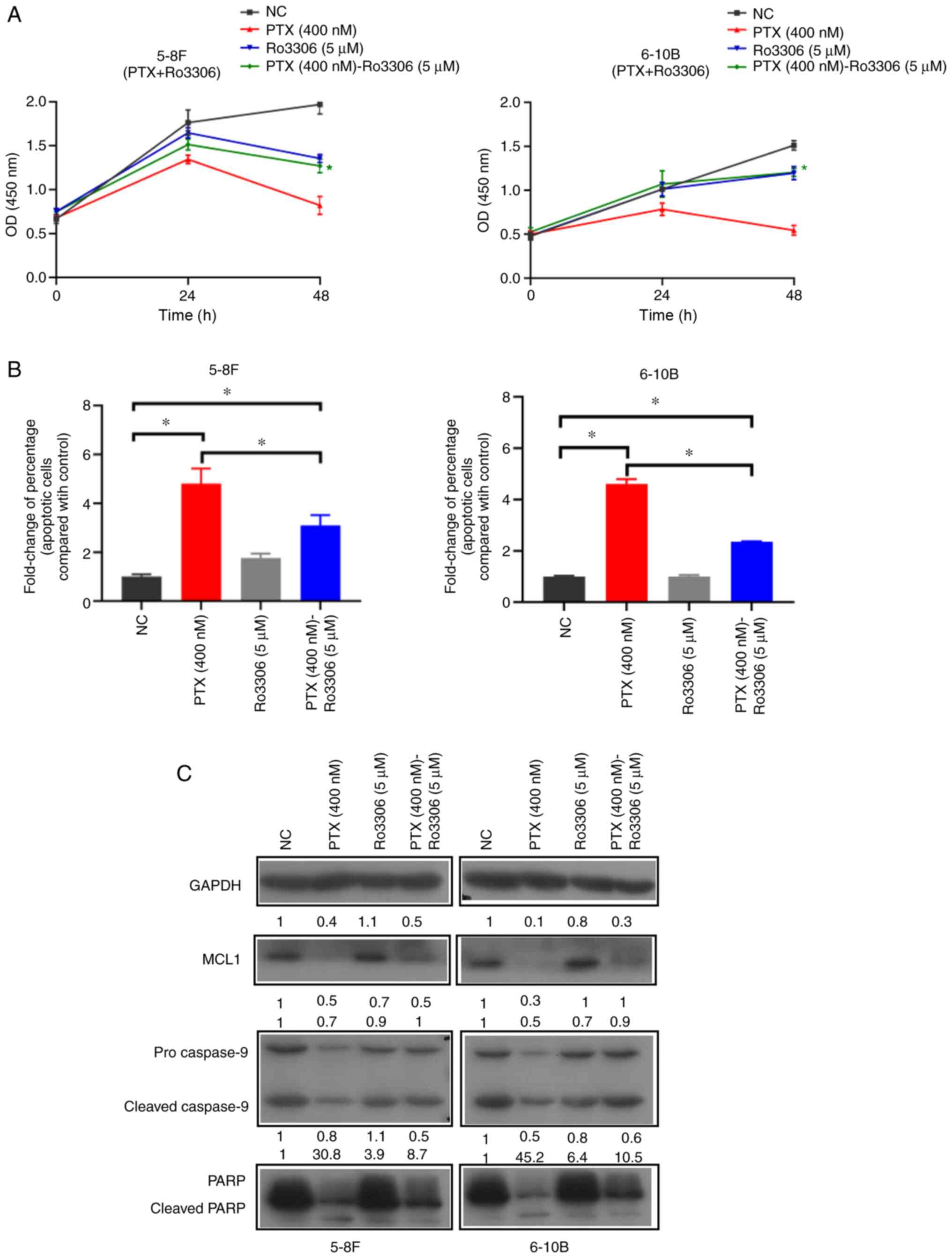
Inhibition of CDK1 by Ro3306 decreases
the paclitaxel-induced mitotic catastrophe
Ro3306, a potent and selective inhibitor of CDK1
(36), also decreased
paclitaxel-induced mitotic catastrophe and cell death in NPC cells
in the present study. When the cells were treated with paclitaxel
and Ro3306, compared with the PTX (400 nM) group, the optical
density results of the CCK-8 assay at 48 h were significantly
increased in the PTX (400 nM)-Ro3306 (5 µM) group (Fig. 5A), there was a decreased
paclitaxel-specific inhibition of cell proliferation. In apoptotic
cell detection, compared with the NC group, the addition of
paclitaxel in the PTX (400 nM) and PTX (400 nM)-Ro3306 (5
µM) groups increased the percentage of apoptotic cells
(Figs. 5B and S2). When the cells were treated with
paclitaxel and Ro3306, compared with PTX (400 nM) group, the
paclitaxel-induced apoptosis in the PTX (400 nM)-Ro3306 (5
µM) group was decreased (Figs. 5B and S2). In addition, western blotting
results indicated that Ro3306 decreased the paclitaxel-induced
apoptosis signaling via the MCL1/caspase-9/PARP signaling pathway
(Fig. 5C). Therefore, loss of
paclitaxel lethality may be due to the inhibition of CDK1.
Discussion
A combination of drugs with different mechanisms
presents an alternative and effective strategy in enhancing cancer
therapeutic effects. For instance, in paclitaxel-resistant ovarian
cancer, PS341 restores paclitaxel-induced cell death by alteration
of microtubule dynamics and hedgehog signaling (37). Furthermore, in H-ras-dependent
paclitaxel-resistant epithelial solid tumors, PS341 blocks
Bcl-2-like protein 11 (BIM) degradation and restores BIM-dependent
apoptosis associated with paclitaxel treatment (38). Therefore, the combination of
paclitaxel with PS341 appears to be a promising tumor
chemotherapeutic alternative in overcoming paclitaxel-resistance.
While a previous clinical trial associated the use of PS341 with
striking toxicity and limited clinical benefit in solid tumors
(39-42), the drug is recommended in
refractory advanced solid tumors and unresectable stage III
non-small-cell lung cancer (43-46). Therefore, more trials are
required to further dissect the effects of the use of PS341 with
paclitaxel.
In antimitotic drug-induced prolonged mitotic arrest
of cells, CDK1/cyclin B1 can be regarded as a hinge between
survival and death (47).
Stabilization and persistent activation of CDK1/cyclin B1 maintains
cells in the mitosis phase (48). Rapid degradation of cyclin B1 and
inactivation of CDK1 promote cell death pathways with accumulation
of death activators and loss of death inhibitors (49). For example, caspase-9 and MCL1
are typical apoptosis and anti-apoptosis proteins, respectively,
regulated by CDK1 (50,51). CDK1 phosphorylates caspase-9 at
an inhibitory site to prevent caspase-9 cleavage, while
phosphorylation of MCL1 at the T92 site triggers its degradation
(50,51). During paclitaxel-induced mitotic
arrest, rapid degradation of cyclin B1 activates death signals,
such as MCL1 hydrolysis and the cleavage of caspase-9 (52).
The present results demonstrated that nontoxic
concentrations of proteasome inhibitors and CDK1 inhibitor RO3306
decreased the paclitaxel-induced mitotic death in NPC cells. It was
hypothesized that the perturbed CDK1/cyclin B1 balance in mitosis
could be defining the reduced mitotic death. Additionally, when
restraining CDK1/cyclin B1 destruction via blockage of the
ubiquitin-proteasome pathway, the side-effect of proteasome
inhibition could further lead to accumulation of
p21Waf1/Cip1 and MCL1 (53,54). P21Waf1/Cip1 can
interact with CDK1 and inhibits its function (55). Accumulation of MCL1 was
associated with higher anti-apoptotic signaling (52), and this side-effect of proteasome
inhibitors may have enhanced the suppression in CDK1/cyclin
B1-triggered cell death. Therefore, it was hypothesized that the
mechanism of proteasome inhibitors in disturbing CDK1 function is
more complex and stronger than that of RO3306. Whereas the
CDK1/cyclin B1 catalytic activity defines the mitosis-promoting
factor, data on its role in mitosis remains insufficient.
Additionally, data on when and how rapidly CDK1/cyclin B1 is
activated in mammalian cells, as well as reorganization of the cell
as it enters and exits mitosis is limited (56,57).
The present study demonstrated that NPC cells
treated with nontoxic doses of proteasome inhibitors block
proliferation without CDK1/cyclin B1 extinction. Combined with
previous data (58,59), it was suggested that CDK1/cyclin
B1 could be more closely associated with the regulation of
apoptosis compared with cell reproduction. CDK1/cyclin B1 functions
as a rate-limiting regulator but is not essential for mitotic entry
and progression in mammalian somatic cells (60). It was hypothesized that the
side-effect of proteasome inhibitors could be vital in cell
cycle-related research, especially in cell cycle regulation and
anti-mitotic drug resistance.
In conclusion, the present study demonstrated that
the abnormal accumulation of CDK1/cyclin B1 protein could be
mediating the suppressed effects of paclitaxel when used in
combination with proteasome inhibitors. Therefore, increased
caution is required when using a combination of paclitaxel with
proteasome or CDK1 inhibitors in the treatment of NPC.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LH, XP, HZ and BJ performed the experiments. XL and
MJ analyzed the data. JH conducted literature search and data
interpretation. BJ drafted the initial manuscript. BJ and JH
confirm the authenticity of all the raw data. BJ designed and
supervised the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 81802952), the Natural Science
Foundation of Hunan Province (grant no. 2020JJ5608), the Scientific
Research Funds of Health Commission of Hunan Province (grant no.
B2019141), and the Science and Technology Program Foundation of
Changsha (grant no. kq1901017).
References
|
1
|
Chen YP, Chan ATC, Le QT, Blanchard P, Sun
Y and Ma J: Nasopharyngeal carcinoma. Lancet. 394:64–80. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ji MF, Sheng W, Cheng WM, Ng MH, Wu BH, Yu
X, Wei KR, Li FG, Lian SF, Wang PP, et al: Incidence and mortality
of nasopharyngeal carcinoma: Interim analysis of a cluster
randomized controlled screening trial (PRO-NPC-001) in southern
China. Ann Oncol. 30:1630–1637. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vasan N, Baselga J and Hyman DM: A view on
drug resistance in cancer. Nature. 575:299–309. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kaidar-Person O, Gil Z and Billan S:
Precision medicine in head and neck cancer. Drug Resist Updat.
40:13–16. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Madani Tonekaboni SA, Soltan Ghoraie L,
Manem VSK and Haibe-Kains B: Predictive approaches for drug
combination discovery in cancer. Brief Bioinform. 19:263–276. 2018.
View Article : Google Scholar :
|
|
6
|
Wong AS, Soo RA, Lu JJ, Loh KS, Tan KS,
Hsieh WS, Shakespeare TP, Chua ET, Lim HL and Goh BC: Paclitaxel,
5-fluorouracil and hydroxyurea concurrent with radiation in locally
advanced nasopharyngeal carcinoma. Ann Oncol. 17:1152–1157. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Scott K, Hayden PJ, Will A, Wheatley K and
Coyne I: Bortezomib for the treatment of multiple myeloma. Cochrane
Database Syst Rev. 4:CD0108162016.PubMed/NCBI
|
|
8
|
Davies AM, Lara PN Jr, Mack PC and Gandara
DR: Incorporating bortezomib into the treatment of lung cancer.
Clin Cancer Res. 13:s4647–4651. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang IT, Dhungel B, Shrestha R, Bridle
KR, Crawford DHG, Jayachandran A and Steel JC: Spotlight on
Bortezomib: Potential in the treatment of hepatocellular carcinoma.
Expert Opin Investig Drugs. 28:7–18. 2019. View Article : Google Scholar
|
|
10
|
Ri M: Endoplasmic-reticulum stress
pathway-associated mechanisms of action of proteasome inhibitors in
multiple myeloma. Int J Hematol. 104:273–280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Manasanch EE and Orlowski RZ: Proteasome
inhibitors in cancer therapy. Nat Rev Clin Oncol. 14:417–433. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hochstrasser M: Ubiquitin, proteasomes,
and the regulation of intracellular protein degradation. Curr Opin
Cell Biol. 7:215–223. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Skaar JR and Pagano M: Control of cell
growth by the SCF and APC/C ubiquitin ligases. Curr Opin Cell Biol.
21:816–824. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Joerger M: Treatment regimens of classical
and newer taxanes. Cancer Chemother Pharmacol. 77:221–233. 2016.
View Article : Google Scholar
|
|
15
|
Alushin GM, Lander GC, Kellogg EH, Zhang
R, Baker D and Nogales E: High-resolution microtubule structures
reveal the structural transitions in alphabeta-tubulin upon GTP
hydrolysis. Cell. 157:1117–1129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Weaver BA: How Taxol/paclitaxel kills
cancer cells. Mol Biol Cell. 25:2677–2681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi X and Sun X: Regulation of paclitaxel
activity by microtubule-associated proteins in cancer chemotherapy.
Cancer Chemother Pharmacol. 80:909–917. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vitale I, Galluzzi L, Castedo M and
Kroemer G: Mitotic catastrophe: A mechanism for avoiding genomic
instability. Nat Rev Mol Cell Biol. 12:385–392. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roninson IB, Broude EV and Chang BD: If
not apoptosis, then what? Treatment-induced senescence and mitotic
catastrophe in tumor cells. Drug Resist Updat. 4:303–313. 2001.
View Article : Google Scholar
|
|
20
|
Castedo M, Perfettini JL, Roumier T,
Andreau K, Medema R and Kroemer G: Cell death by mitotic
catastrophe: A molecular definition. Oncogene. 23:2825–2837. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Denisenko TV, Sorokina IV, Gogvadze V and
Zhivotovsky B: Mitotic catastrophe and cancer drug resistance: A
link that must to be broken. Drug Resist Updat. 24:1–12. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shi J and Mitchison TJ: Cell death
response to anti-mitotic drug treatment in cell culture, mouse
tumor model and the clinic. Endocr Relat Cancer. 24:T83–T96. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brito DA and Rieder CL: Mitotic checkpoint
slippage in humans occurs via cyclin B destruction in the presence
of an active checkpoint. Curr Biol. 16:1194–1200. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mc Gee MM: Targeting the mitotic
catastrophe signaling pathway in cancer. Mediators Inflamm.
2015:1462822015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fung TK and Poon RY: A roller coaster ride
with the mitotic cyclins. Semin Cell Dev Biol. 16:335–342. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kalous J, Jansova D and Susor A: Role of
cyclin-dependent kinase 1 in translational regulation in the
M-Phase. Cells. 9:15682020. View Article : Google Scholar :
|
|
27
|
Yang J, Bardes ES, Moore JD, Brennan J,
Powers MA and Kornbluth S: Control of cyclin B1 localization
through regulated binding of the nuclear export factor CRM1. Genes
Dev. 12:2131–2143. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Coulonval K, Kooken H and Roger PP:
Coupling of T161 and T14 phosphorylations protects cyclin B-CDK1
from premature activation. Mol Biol Cell. 22:3971–3985. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lindqvist A, Rodriguez-Bravo V and Medema
RH: The decision to enter mitosis: Feedback and redundancy in the
mitotic entry network. J Cell Biol. 185:193–202. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sinha D, Duijf PHG and Khanna KK: Mitotic
slippage: An old tale with a new twist. Cell cycle. 18:7–15. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mascaraque M, Delgado-Wicke P, Damian A,
Lucena SR, Carrasco E and Juarranz A: Mitotic catastrophe induced
in HeLa tumor cells by photodynamic therapy with
methyl-aminolevulinate. Int J Mol Sci. 20:12292019. View Article : Google Scholar :
|
|
32
|
Vakifahmetoglu H, Olsson M and Zhivotovsky
B: Death through a tragedy: Mitotic catastrophe. Cell Death Differ.
15:1153–1162. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Orth JD, Kohler RH, Foijer F, Sorger PK,
Weissleder R and Mitchison TJ: Analysis of mitosis and antimitotic
drug responses in tumors by in vivo microscopy and single-cell
pharmacodynamics. Cancer Res. 71:4608–4616. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jackman M, Kubota Y, den Elzen N, Hagting
A and Pines J: Cyclin A- and cyclin E-Cdk complexes shuttle between
the nucleus and the cytoplasm. Mol Biol Cell. 13:1030–1045. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Harashima H, Dissmeyer N and Schnittger A:
Cell cycle control across the eukaryotic kingdom. Trends Cell Biol.
23:345–356. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vassilev LT, Tovar C, Chen S, Knezevic D,
Zhao X, Sun H, Heimbrook DC and Chen L: Selective small-molecule
inhibitor reveals critical mitotic functions of human CDK1. Proc
Natl Acad Sci USA. 103:10660–10665. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Steg AD, Burke MR, Amm HM, Katre AA,
Dobbin ZC, Jeong DH and Landen CN: Proteasome inhibition reverses
hedgehog inhibitor and taxane resistance in ovarian cancer.
Oncotarget. 5:7065–7080. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tan TT, Degenhardt K, Nelson DA, Beau B,
Nieves-Neira W, Bouillet P, Villunger A, Adams JM and White E: Key
roles of BIM-driven apoptosis in epithelial tumors and rational
chemotherapy. Cancer Cell. 7:227–238. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Edelman MJ, Burrows W, Krasna MJ, Bedor M,
Smith R and Suntharalingam M: Phase I trial of
carboplatin/paclitaxel/bortezomib and concurrent radiotherapy
followed by surgical resection in Stage III non-small cell lung
cancer. Lung cancer. 68:84–88. 2010. View Article : Google Scholar
|
|
40
|
Jatoi A, Dakhil SR, Foster NR, Ma C,
Rowland KM Jr, Moore DF Jr, Jaslowski AJ, Thomas SP, Hauge MD,
Flynn PJ, et al: Bortezomib, paclitaxel, and carboplatin as a
first-line regimen for patients with metastatic esophageal,
gastric, and gastroesophageal cancer: Phase II results from the
North Central Cancer Treatment Group (N044B). J Thorac Oncol.
3:516–520. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Croghan GA, Suman VJ, Maples WJ, Albertini
M, Linette G, Flaherty L, Eckardt J, Ma C, Markovic SN and
Erlichman C: A study of paclitaxel, carboplatin, and bortezomib in
the treatment of metastatic malignant melanoma: A phase 2
consortium study. Cancer. 116:3463–3468. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cresta S, Sessa C, Catapano CV, Gallerani
E, Passalacqua D, Rinaldi A, Bertoni F, Vigano L, Maur M, Capri G,
et al: Phase I study of bortezomib with weekly paclitaxel in
patients with advanced solid tumours. Eur J Cancer. 44:1829–1834.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mehnert JM, Tan AR, Moss R, Poplin E,
Stein MN, Sovak M, Levinson K, Lin H, Kane M, Gounder M, et al:
Rationally designed treatment for solid tumors with MAPK pathway
activation: A phase I study of paclitaxel and bortezomib using an
adaptive dose-finding approach. Mol Cancer Ther. 10:1509–1519.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ramaswamy B, Bekaii-Saab T, Schaaf LJ,
Lesinski GB, Lucas DM, Young DC, Ruppert AS, Byrd JC, Culler K,
Wilkins D, et al: A dose-finding and pharmacodynamic study of
bortezomib in combination with weekly paclitaxel in patients with
advanced solid tumors. Cancer Chemother Pharmacol. 66:151–158.
2010. View Article : Google Scholar
|
|
45
|
Zhao Y, Foster NR, Meyers JP, Thomas SP,
Northfelt DW, Rowland KM Jr, Mattar BI, Johnson DB, Molina JR,
Mandrekar SJ, et al: A phase I/II study of bortezomib in
combination with paclitaxel, carboplatin, and concurrent thoracic
radiation therapy for non-small-cell lung cancer: North Central
Cancer Treatment Group (NCCTG)-N0321. J Thorac Oncol. 10:172–180.
2015. View Article : Google Scholar :
|
|
46
|
Ma C, Mandrekar SJ, Alberts SR, Croghan
GA, Jatoi A, Reid JM, Hanson LJ, Bruzek L, Tan AD, Pitot HC, et al:
A phase I and pharmacologic study of sequences of the proteasome
inhibitor, bortezomib (PS-341, Velcade), in combination with
paclitaxel and carboplatin in patients with advanced malignancies.
Cancer Chemother Pharmacol. 59:207–215. 2007. View Article : Google Scholar
|
|
47
|
Castedo M, Perfettini JL, Roumier T and
Kroemer G: Cyclin-dependent kinase-1: Linking apoptosis to cell
cycle and mitotic catastrophe. Cell Death Differ. 9:1287–1293.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sakurikar N, Eichhorn JM and Chambers TC:
Cyclin-dependent kinase-1 (Cdk1)/cyclin B1 dictates cell fate after
mitotic arrest via phosphoregulation of antiapoptotic Bcl-2
proteins. J Biol Chem. 287:39193–39204. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hou Y, Allan LA and Clarke PR:
Phosphorylation of XIAP by CDK1-cyclin-B1 controls mitotic cell
death. J Cell Sci. 130:502–511. 2017.
|
|
50
|
Harley ME, Allan LA, Sanderson HS and
Clarke PR: Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its
Cdc20-dependent destruction during mitotic arrest. The EMBO J.
29:2407–2420. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Allan LA and Clarke PR: Phosphorylation of
caspase-9 by CDK1/cyclin B1 protects mitotic cells against
apoptosis. Mol Cell. 26:301–310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Clarke PR and Allan LA: Destruction's our
delight: Controlling apoptosis during mitotic arrest. Cell Cycle.
9:4035–4036. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lu Z and Hunter T: Ubiquitylation and
proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2)
CDK inhibitors. Cell Cycle. 9:2342–2352. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Millman SE and Pagano M: MCL1 meets its
end during mitotic arrest. EMBO Rep. 12:384–385. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kreis NN, Louwen F and Yuan J: Less
understood issues: p21(Cip1) in mitosis and its therapeutic
potential. Oncogene. 34:1758–1767. 2015. View Article : Google Scholar
|
|
56
|
Gavet O and Pines J: Progressive
activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell.
18:533–543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Rata S, Suarez Peredo Rodriguez MF, Joseph
S, Peter N, Echegaray Iturra F, Yang F, Madzvamuse A, Ruppert JG,
Samejima K, Platani M, et al: Two interlinked bistable switches
govern mitotic control in mammalian cells. Curr Biol. 28:3824–3832
e3826. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Diril MK, Ratnacaram CK, Padmakumar VC, Du
T, Wasser M, Coppola V, Tessarollo L and Kaldis P: Cyclin-dependent
kinase 1 (Cdk1) is essential for cell division and suppression of
DNA re-replication but not for liver regeneration. Proc Natl Acad
Sci USA. 109:3826–3831. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Saito M, Mulati M, Talib SZ, Kaldis P,
Takeda S, Okawa A and Inose H: The indispensable role of
cyclin-dependent kinase 1 in skeletal development. Sci Rep.
6:206222016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Soni DV, Sramkoski RM, Lam M, Stefan T and
Jacobberger JW: Cyclin B1 is rate limiting but not essential for
mitotic entry and progression in mammalian somatic cells. Cell
Cycle. 7:1285–1300. 2008. View Article : Google Scholar : PubMed/NCBI
|