Introduction
Macroautophagy (hereafter referred to as autophagy)
is an intracellular catabolic pathway essential for maintaining
cellular homeostasis (1,2). Autophagy is activated under
nutrient-starved conditions to induce bulk turnover of cytoplasmic
contents, including proteins and organelles, whereas basal
autophagy occurs under nutrient-rich conditions and is important
for the quality control of intracellular proteins and organelles
(3,4). Autophagy is initiated by the
formation of isolation membranes (also known as phagophores), which
expand to form double-membrane autophagosomes containing
cytoplasmic cargo, and then subsequently fuse with lysosomes to
form single-membrane degradative compartments, autolysosomes
(5-7). The low internal pH and acidic
enzymes, such as cathepsins (CTSs), degrade the autolysosomal
content to generate simple molecules, including amino acids, which
are then recycled back to the cytoplasm for reuse (8,9).
The rate at which this tightly regulated multistep dynamic process
occurs inside the cells is referred to as autophagic flux. As
autophagy is a fundamental control system that maintains the
homeostasis of cells and organisms, disturbances in autophagy have
been implicated in various human disorders, including
neurodegenerative diseases and cancer (10,11).
Lysosomes are membrane-enclosed cytoplasmic
organelles that mediate the degradation of various biological
macromolecules, including proteins, and the adaptation of cell
metabolism to environmental cues (12). Lysosomes are not static
organelles, but highly dynamic structures that move around the
cytoplasm to change their size and shape or to undergo fusion or
fission (13,14). In autophagic flux, lysosomes fuse
with autophagosomes to form autolysosomes and are reformed by
fission from autolysosomes, a process called autophagic lysosome
reformation (ALR) (9,15). mTORC1, a key regulator of
autophagy, localizes on lysosomes and senses cellular nutrients
(16-18). When the nutrients are limited,
mTORC1 is deactivated, which induces autophagy. Subsequently, the
release of free amino acids from autolysosomes upon autophagy
induction reactivates mTORC1, triggering ALR (15,19). In addition, actomyosin dynamics
also serve essential roles in each step of the autophagic process,
including biogenesis of autophagosomes, autophagosome-lysosome
fusion and ALR (20,21).
Cyclin G-associated kinase (GAK) is a ubiquitously
expressed serine/threonine-protein kinase that primarily localizes
to the perinuclear trans-Golgi and the plasma membrane regions
(22,23). GAK, which was initially
identified as a direct partner of cyclin G (24), has been associated with a diverse
range of biological processes, including the maintenance of
centrosome maturation and cell cycle progression (25-28), clathrin-mediated endocytosis
(22,23,29-31), epidermal growth factor receptor
(EGFR) signaling (32) and
lysosomal enzyme sorting (33).
In addition, GAK has been reported as a binding partner of
leucine-rich repeat kinase 2 (LRRK2) (34), which has previously been reported
to control autophagy in several systems (35-37). Regarding human disorders, GAK is
overexpressed in osteosarcoma cells and tissues (38), and a single nucleotide
polymorphism (SNP) rs1564282 in the intronic region of GAK is
associated with susceptibility to Parkinson's disease (PD)
(39-41).
The relationship between GAK and clathrin-mediated
endocytosis has been extensively studied, but the relationship
between GAK and the autophagy-lysosome system is not completely
understood. To determine whether GAK serves an essential role in
the autophagy-lysosome system, the present study performed targeted
disruption of GAK in non-small cell lung cancer (NSCLC) A549 cells,
analyzed the fluctuation of autophagic flux and examined the
underlying molecular mechanism.
Materials and methods
Reagents
Bafilomycin A1 (Baf), puromycin,
blasticidin S and Hanks' balanced salt solution (HBSS) were
purchased from FUJIFILM Wako Pure Chemical Corporation. Rapamycin
was obtained from LC Laboratories. Hydroxychloroquine (HCQ) was
obtained from Cayman Chemical Company. The GAK inhibitor (GAKi;
cat. no. 538770), Y-27632 (cat. no. 688000) and cycloheximide were
purchased from Calbiochem (Merck KGaA). DAPI was obtained from
Sigma-Aldrich (Merck KGaA). Protease inhibitor cocktail and
phosphatase inhibitor cocktail were obtained from Nacalai Tesque,
Inc. Baf and rapamycin were dissolved in DMSO and used at a
concentration of 10 nM, and the same amount of DMSO was used for
negative control. HCQ was dissolved in water and used at a
concentration of 50 µM. GAKi was dissolved in DMSO and used
at the indicated concentrations (3, 10 or 30 µM), and the
same amount of DMSO was used for negative control. Y-27632 was
dissolved in water and used at a concentration of 10 µM.
Cycloheximide was dissolved in DMSO and used at a concentration of
30 µM. Cells treated with Baf (8 or 24 h), rapamycin (24 h),
HCQ (8 or 24 h), GAKi (8 or 12 h), Y-27632 (8 h) or cycloheximide
(3, 9 or 24 h) at 37°C with 5% CO2 for an indicated
period of time.
Antibodies
The following primary antibodies were used in the
present study: Anti-GAK (cat. no. M057-3; Medical & Biological
Laboratories Co., Ltd.), anti-phosphorylated (p)-mTOR (cat. no.
5536), anti-mTOR (cat. no. 2983), anti-p-ribosomal protein S6
kinase (S6K; cat. no. 9234), anti-S6K (cat. no. 2708), anti-p-S6
(cat. no. 4858), anti-S6 (cat. no. 2217;), anti-p-AMPK (cat. no.
2535), anti-AMPK (cat. no. 5832), anti-LC3B for immunocytochemistry
(cat. no. 2775), anti-p62/sequestosome 1 (SQSTM1; cat. no. 7695),
anti-CTSB (cat. no. 31718), and anti-CTSD (cat. no. 2284),
anti-ROCK1 (cat. no. 4035; all from Cell Signaling Technology,
Inc.), anti-lysosomal associated membrane protein (LAMP)1 (cat. no.
sc-20011), anti-LAMP2 (cat. no. sc-18822), anti-β-actin (cat. no.
sc-47778), anti-GAPDH (cat. no. sc-32233; all from Santa Cruz
Biotechnology, Inc.), anti-LC3B for immunoblotting (cat. no.
NB600-1384; Novus Biologicals, LLC), anti-FLAG (cat. no. F1804;
Sigma-Aldrich; Merck KGaA) and anti-V5 (R960-25; Invitrogen; Thermo
Fisher Scientific, Inc.). Horseradish peroxidase-conjugated
secondary antibodies against mouse IgG (cat. no. 115-035-003) and
rabbit IgG (cat. no. 711-035-152) were purchased from Jackson
ImmunoResearch Laboratories, Inc. The Alexa Fluor 555-conjugated
secondary antibody against mouse IgG (cat. no. A21424) and Alexa
Fluor 488-conjugated secondary anti-body against rabbit IgG (cat.
no. A11034) were obtained from Invitrogen (Thermo Fisher
Scientific, Inc.).
Cell culture
The human NSCLC cell lines (A549 and H596), human
pancreatic cancer cell line (PANC-1) and human liver cancer cell
line (Hep-G2) were purchased from American Type Culture Collection.
Cells were cultured in RPMI-1640 (Sigma-Aldrich; Merck KGaA)
supplemented with 10% heat-inactivated FBS (Gibco; Thermo Fisher
Scientific, Inc.) at 37°C with 5% CO2. Stable
A549/GFP-LC3-mCherry-LC3ΔG cells were cultured under the same
conditions as A549 cells with the addition of puromycin (2
µg/ml). RPMI medium without serum and glutamine (RPMI-Gln)
was prepared by dissolving RPMI-1640 without glutamine and sodium
bicarbonate (cat. no. 05918; Nissui Pharmaceutical Co., Ltd.) in
water, followed by autoclaving and then adding sterilized sodium
bicarbonate. 293T cells were cultured in DMEM (Sigma-Aldrich; Merck
KGaA) supplemented with 10% FBS at 37°C with 5% CO2.
CRISPR/Cas9 genome editing
Target sequences for CRISPR interference (42) were designed using CRISPR direct
(http://crispr.dbcls.jp/) provided by the Database
Center for Life Science. The target sequences for human GAK were
GCA GTC GGC GCT CGA CTT CT (exon 1; N-terminus) or GAG GCC CCC TTT
CGT GCG ACA (exon 5; kinase domain). Subsequently, two
complementary oligonucleotides with BpiI restriction sites
for guide RNAs (gRNAs) were synthesized at Eurofins Genomics and
cloned into the pSpCas9(BB)-2A-Puro (PX459) vector (cat. no. 48139;
Addgene, Inc.), deposited by the Feng Zhang Laboratory (43). A549 cells (2×106
cells/100 mm dish) were transfected with pX459-gRNA (10 µg)
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Following culture at 37°C with 5% CO2 overnight, cells
were treated with 2 µg/ml puromycin for 2 days. Surviving
cells were diluted in growth medium (RPMI-1640 supplemented with
10% heat-inactivated FBS) to prepare a cell suspension (5
cells/ml). Then, 100 µl cell suspension was added to each
well of a 96-well plate. The expression of GAK in the expanded
colonies was detected by immunoblotting, described in the
immunoblotting section, using an anti-GAK antibody to select
GAK-depleted colonies. The genome sequences of the edited locus in
selected colonies were confirmed by Sanger DNA sequencing performed
at Eurofins Genomics, which demonstrated that the expected mutation
and frameshifting were present in each exon of GAK (Fig. S1). After cloning and expansion,
A549/GAK-knockout (KO) cells were cultured under the same
conditions as A549 cells and used for subsequent experiments.
Establishment of stable cell lines
The stable cell line was generated by transfecting
A549/GAK-KO cells (2×106 cells/100 mm dish) with plasmid
DNA (pMRX-IP-GFP-LC3-mCherry-LC3ΔG; 10 µg) using
Lipofectamine 2000 according to the manufacturer's instructions and
cultured at 37°C with 5% CO2 overnight. As previously
described (44), the
pMRX-IP-GFP-LC3-mCherry-LC3ΔG plasmid used in the present study was
recombined with the pMRX-IP-GFP-LC3-RFP-LC3ΔG plasmid, which was a
kind gift from Dr N. Mizushima (University of Tokyo, Japan)
(45). After selecting the
transfected cells using puromycin (1 µg/ml) from the day
after transfection, single clones of the cells were isolated and
the expression of transfected genes was confirmed by
immunoblotting. After cloning and expansion,
A549/GAK-KO/GFP-LC3-mCherry-LC3ΔG cells were cultured under the
same conditions as A549 cells with the addition of 1 µg/ml
puromycin. Since A549/GAK-KO cells are more sensitive to puromycin
than A549 cells, puromycin was used at a concentration of 1
µg/ml.
Transduction of lentiviral vectors
To construct the lentiviral expression vector,
pLentiN-GAK, GAK cDNA was amplified by PCR. Total RNA was isolated
from A549 cells using the NucleoSpin RNA kit (cat. no. U0955C;
Takara Bio, Inc.) according to the manufacturer's protocol. cDNA
was synthesized from the total RNA using the SuperScript III
First-Strand Synthesis system (cat. no. 18080-051; Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. cDNA for GAK was amplified using PrimeSTAR HS DNA
Polymerase (cat. no. R010A; Takara Bio, Inc.) according to the
manufacturer's protocol. The primers used for amplification were as
follows: forward, 5′-CAC CGG TAC CCA CCA TGT CGC TGC TGC AGT CGG
CGC TC-3′ and reverse, 5′-AGC TGA ATT CTC ACT TGT CGT CGT CAT CCT
TGT AGT CGA AGA GGG GCC GGG AGC CCT G-3′. The thermocycling
conditions were as follows: 98°C for 1 min; followed by 30 cycles
of 98°C for 10 sec, 60°C for 5 sec and 72°C for 4 min; and 72°C for
5 min. During PCR amplification, FLAG-tag was fused at the
C-terminus of GAK. The amplified GAK cDNA was subcloned into the
pcDNA6 mammalian expression vector (Invitrogen; Thermo Fisher
Scientific, Inc.). The cloned sequences were verified by Sanger
sequencing at Eurofins Genomics (Fig. S2). The DNA inserts encoding GAK
were then subcloned into the pLentiN lentiviral expression vector,
which was a kind gift from Dr Karl Munger (cat. no. 37444; Addgene,
Inc.) using restriction enzymes, SpeI and EcoRI
(46). To construct the
lentiviral short hairpin RNA (shRNA) vectors, pLKO.1-Rho-associated
protein kinase (ROCK)1, oligonucleotides for shRNAs targeting
ROCK1 on appropriate sites and a non-targeting (NT) control
oligonucleotide were synthesized (Table SI). The oligonucleotides were
phosphorylated, annealed and inserted into the pLKO.1 puro
lentiviral shRNA vector, which was a kind gift from Dr Bob Weinberg
(cat. no. 8453; Addgene, Inc.) (47). The production and transduction of
lentiviral particles were performed as previously described
(48). Briefly, 293T cells
(1×107 cells/100 mm dish) were cotransfected with the
constructed plasmid (pLentiN-GAK, pLentiN-empty, pLKO.1-ROCK1 or
pLKO.1-NT; all 4 µg), pMD2.G (a gift from Dr Didier Trono;
cat. no. 12259; Addgene, Inc.; 2 µg) and psPAX2 (a gift from
Dr Didier Trono; cat. no. 12260; Addgene, Inc.; 2 µg) using
PEI-MAX (cat. no. 24765-1, Polysciences, Inc.; 24 µg) and
cultured at 37°C with 5% CO2. At 2 days
post-transfection, the cultured supernatant containing lentivirus
was harvested and filtrated. The titer of lentiviral particles was
not determined. A549 and A549/GAK-KO cells were plated
(2×105 cells/well) in 6-well plate for 24 h before
transduction and infected with the prepared lentivirus (150
µl) at 37°C with 5% CO2 overnight, and selected
using blasticidin S (10 µg/ml for A549 cells; 5 µg/ml
for A549/GAK-KO cells) or puromycin (2 µg/ml for A549 cells;
1 µg/ml for A549/GAK-KO cells) from the day after infection.
In the present study, the following cell lines were established:
A549/GAK-KO/GAK-rescued, A549/GAK-KO/sham-rescued,
A549/GAK-overexpression (OE), A549/sham-OE, A549/GAK-KO/shROCK1#1,
A549/GAK-KO/shROCK1#2 and A549/GAK-KO/shNT cells.
A549/GAK-KO/sham-rescued and A549/sham-OE cells were established
using pLentiN-empty. The established cell lines were cultured under
the same conditions as A549 cells, except for the supplementation
of blasticidin S for GAK-rescued cells or puromycin for
ROCK1-knockdown cells.
Immunoblotting
Total cellular proteins were extracted from A549 and
A549/GAK-KO cells under the following conditions at 37°C with 5%
CO2: i) Basal-fed condition (RPMI-1640 supplemented with
10% FBS); ii) starved condition using RPMI-Gln (2, 4, 6 or 8 h);
iii) basal-fed condition with 30 µM cycloheximide (3, 9 or
24 h); iv) starved condition using HBSS (2 h); v) refed condition
(starvation using HBSS or RPMI-Gln for 1 h, followed by fed
conditions for 1 h) using lysis buffer containing 50 mM Tris-HCl
(pH 8.0), 150 mM NaCl, 1.0% Nonidet P-40, 0.5% sodium deoxycholate,
0.1% SDS, and protease and phosphatase inhibitor cocktails. Each
sample was sonicated on ice for 20 pulses (0.5 sec on/off) to
solubilize the aggregated proteins using a Branson 450D Sonifier
(Emerson Electric Co.). Protein concentrations were measured using
the BCA Protein Assay kit (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Equal amounts (10
µg) of proteins were resolved by SDS-PAGE (10% gel or 5-20%
gradient gel) and transferred onto Immobilon-P PVDF membranes
(Merck KGaA). Following blocking with 5% non-fat milk for 1 h at
room temperature, the membranes were probed with primary antibodies
at 4°C overnight. Primary antibodies used for immunoblotting were
as follows: Anti-LC3B (1:2,000), anti-GAK (1:500), anti-LAMP1
(1:1,000), anti-LAMP2 (1:1,000), anti-p-mTOR (1:1,000), anti-mTOR
(1:1,000), anti-p-AMPK (1:1,000), anti-AMPK (1:1,000), anti-p-S6K
(1:1,000), anti-S6K (1:1,000), anti-p-S6 (1:1,000), anti-S6
(1:1,000), anti-CTSB (1:1,000), anti-CTSD (1:1,000), anti-SQSTM1
(1:1,000), anti-ROCK1 (1:1,000), anti-FLAG (1:1,000), anti-V5
(1:5,000), anti-GAPDH (1:1,000), and anti-β-actin (1:1,000). These
membranes were incubated with horseradish peroxidase-conjugated
secondary antibodies (anti-mouse IgG or anti-rabbit IgG; both
1:2,500) for 1 h at room temperature. Immunoreactive proteins were
detected using Immobilon Western HRP substrate detection reagents
(Merck KGaA). Densitometry analysis was performed using a WSE-6300
Luminograph III molecular imager (ATTO Corporation) and CSAnalyzer4
software version 2.3.1 (ATTO Corporation).
Immunocytochemistry
A549, A549/GAK-KO and A549/GAK-rescued cells were
seeded (4×104 cells/well) onto glass coverslips in a
24-well culture plate and incubated for 48 h. After washing with
PBS, cells were fixed with 100% methanol at −30°C for 15 min. After
washing with TBST (0.05% Tween-20), cells were blocked with 10%
newborn calf serum (Gibco; Thermo Fisher Scientific, Inc.) in TBST
at room temperature for 1 h, followed by incubation with primary
antibodies in TBST containing 1.5% newborn calf serum and 0.1% BSA
(Nacalai Tesque, Inc.) at 4°C overnight. Primary antibodies used
for immunocytochemistry were follows: Anti-LC3B (1:100) and
anti-LAMP2 (1:100). After washing with TBST, cells were incubated
with Alexa Fluor 488-conjugated anti-rabbit IgG and/or Alexa Fluor
555-conjugated anti-mouse IgG (both 1:1,000) along with DAPI at
37°C for 1 h. Cells were washed again with TBST then mounted in
ProLong Diamond Antifade Mountant (Invitrogen; Thermo Fisher
Scientific, Inc.). Stained cells were visualized using a Zeiss
LSM700 confocal laser scanning fluorescence microscope (Zeiss GmbH)
equipped with PlanApochromat 63x/1.4 or 100x/1.4 oil DIC (Zeiss
GmbH). All images were acquired and processed using ZEN 2012
software (version 8.1.0.484; Zeiss GmbH). Object-based fluorescence
intensity was measured using ImageJ software (version 1.50i;
National Institutes of Health). The size of lysosomes was measured
using ZEN 2012 software. The number of autophagosomes was
automatically recognized based on size and intensity thresholds
using the Spot function in Imaris ×64 software (version 9.1.0;
Oxford Instruments Bitplane) (49).
Assessment of autophagic flux using
GFP-LC3-mCheery-LC3ΔG system
A549/GFP-LC3-mCherry-LC3ΔG and
A549/GAK-KO/GFP-LC3-mCherry-LC3ΔG cells were plated
(8×103 cells/well) in a 96-well plate for 24 h before
treatment. Fluorescence intensities derived from GFP-LC3 and
mCherry-LC3ΔG were monitored during the 24 h-culture at 37°C with
5% CO2 using an IncuCyte ZOOM cell imaging system (Essen
BioScience) under the following conditions: i) Basal-fed condition
(RPMI-1640 supplemented with 10% FBS); ii) basal-fed condition with
10 nM Baf; iii) basal-fed condition with 10 nM rapamycin; iv)
basal-fed condition with DMSO; v) starved condition using HBSS; vi)
starved condition using RPMI-Gln; vii) starved condition using HBSS
with GAKi (3, 10 or 30 µM); and viii) starved condition
using RPMI-Gln with GAKi (3, 10 or 30 µM). Autophagic flux
was presented as alterations in the relative intensities of
GFP/mCherry calculated using Microsoft Excel version 16.51
(Microsoft Corporation) from the measured fluorescence intensities
of GFP and mCherry.
LysoTracker/LysoSensor staining
A549, A549/GAK-KO and A549/GAK-rescued cells were
seeded (4×104 cells/well) into glass-bottom cell culture
dishes (Greiner Bio-One International GmbH). After a 48-h culture,
cells were incubated under basal-fed (RPMI-1640 supplemented with
10% FBS) or starvation conditions (RPMI-Gln) for 8 h at 37°C with
5% CO2 and then incubated for 1 h with 50 nM LysoTracker
Red DND-99 (cat. no. L7528; Invitrogen; Thermo Fisher Scientific,
Inc.) or 1 µM LysoSensor Green DND-189 (cat. no. L7535;
Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C with 5%
CO2. Stained cells were visualized using an LSM700
confocal laser scanning fluorescence microscope equipped with
PlanApochromat 63x/1.4 oil DIC. All images were acquired and
processed using ZEN 2012 software. The object-based fluorescence
intensity was measured using ImageJ software.
Transmission electron microscopy
A549 and A549/GAK-KO cells were plated
(2×105 cells/well) in 6-well plate for 24 h before
treatment and further cultured for 8 h at 37°C with 5%
CO2 under basal-fed (RPMI-1640 supplemented with 10%
FBS), starved condition (RPMI-Gln) or basal-fed with 50 µM
HCQ conditions. Then, the cells were fixed with 2.5% glutaraldehyde
in 0.1 M phosphate buffer (pH 7.3) for 1 h at 4°C. The samples were
further fixed in 1% osmium tetroxide for 1 h at 4°C, dehydrated in
a graded ethanol series (30-100%) and embedded in Quetol 812 epoxy
resin (Nisshin EM) for 2 days at 60°C. Ultrathin sections (70 nm)
were cut using an Ultracut J microtome. The sections were stained
with uranium acetate (Merck KGaA) for 15 min at room temperature in
a dark box. After washing with water, the sections were further
stained with lead nitrate for 10 min at room temperature and
subjected to electron microscopy using a transmission electron
microscope (JEM-1400Flash; JEOL, Ltd.).
Reverse transcription-quantitative PCR
(qPCR)
Total RNA was extracted from A549 and A549/GAK-KO
cells using the NucleoSpin RNA kit (Takara Bio, Inc.). Total RNA
was reverse transcribed into cDNA using the PrimeScript RT Master
Mix kit (Takara Bio, Inc.) according to the manufacturer's
instructions. Subsequently, qPCR was performed using the SYBR
Premix Ex Taq II Tli RNase H Plus kit (Takara Bio, Inc.). The
sequences of primers used for qPCR are listed in Table SI. qPCR was performed using a
Thermal Cycler Dice Real-Time System TP800 (Takara Bio, Inc.) and
the following thermocycling conditions: Initial cDNA denaturation
at 95°C for 30 sec; followed by 45 cycles of denaturation at 95°C
for 5 sec; and simultaneous annealing and extension at 60°C for 30
sec. The data were analyzed using Thermal Cycler Dice Real-Time
System Software (Takara Bio, Inc.). To determine relative gene
expression, the 2−ΔΔCq method was used (50). mRNA expression levels were
normalized to the internal reference gene GAPDH.
Immunoprecipitation
The cDNA of ROCK1 was amplified in the same manner
as GAK and cloned into the pcDNA6 vector. The primers used for
amplification were as follows: forward, 5′-CAC CGC GGC CGC CAC CAT
GTC GAC TGG GGA CAG TTT TG-3′ and reverse, 5′-AGC TGG CGC GCC CGC
TAG GTT TGT TTG GGG CAA GC-3′. The expression vectors (4 µg
each) for GAK-FLAG and ROCK1-V5 were transfected into 293T cells
(6×106 cells/dish) using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) and cultured at 37°C
with 5% CO2. At 48 h post-transfection, cells were lysed
in 500 µl lysis buffer [50 mM Tris-HCl (pH 8.0), containing
150 mM NaCl and 1.0% NP-40] containing a protease inhibitor
cocktail. Immunoprecipitation was performed on transfected cell
lysates (100 µl each) using anti-FLAG M2 magnetic beads
(cat. no. M8823; Sigma-Aldrich; Merck KGaA; 10 µl). The
beads were washed three times with lysis buffer using magnetic
separation. Immunoprecipitates were eluted with lysis buffer
containing 100 µg/ml 3xFLAG peptide (cat. no. F4799;
Sigma-Aldrich; Merck KGaA) and recovered using magnetic separation.
Immunoprecipitates or total cell lysates were analyzed by
immunoblotting using anti-FLAG (1:1,000) and anti-V5 (1:5,000)
primary antibodies according to the aforementioned protocol.
Statistical analysis
All data are presented as the mean ± standard
deviation from at least three independent experiments. The unpaired
two-tailed Student's t-test or one-way ANOVA followed by Dunnett's
or Bonferroni's post hoc test were used to analyze the data using
Microsoft Excel version 16.51 (Microsoft Corporation) with the
add-in software, Excel Statistical Program File ystat2008
(Igakutosho-shuppan, Ltd.). One-way ANOVA followed by Tukey's post
hoc test and two-way ANOVA followed by Bonferroni's post hoc test
were used to analyze the data using SPSS version 27 software (IBM
Corp.). P<0.05 was considered to indicate a statistically
significant difference.
Results
Targeted disruption of GAK stagnates the
autophagic flux and causes LC3B-II accumulation
To analyze the role of GAK in the autophagy-lysosome
system, GAK-KO A549-derived cell lines were established (Fig. 1A). GAK was completely knocked out
in clones 1-1 and 2-1, but not completely in clone 1-2. The
expression of LC3B-II, an autophagosome marker protein, was
significantly enhanced in all three clones compared with that in
wild-type cells (Fig. 1A and B),
whereas the expression of LC3B mRNA in GAK-KO cells was similar to
that in wild-type cells (Fig.
1C). Therefore, in the following experiments, clone 1-1 was
primarily used for the GAK-KO cells, and clones 1-2 and 2-1 were
used as needed. The significant enhancement of LC3B-puncta
formation in GAK-KO cells compared with that in wild-type cells was
detected by immunofluorescence analysis (Fig. 1D-F).
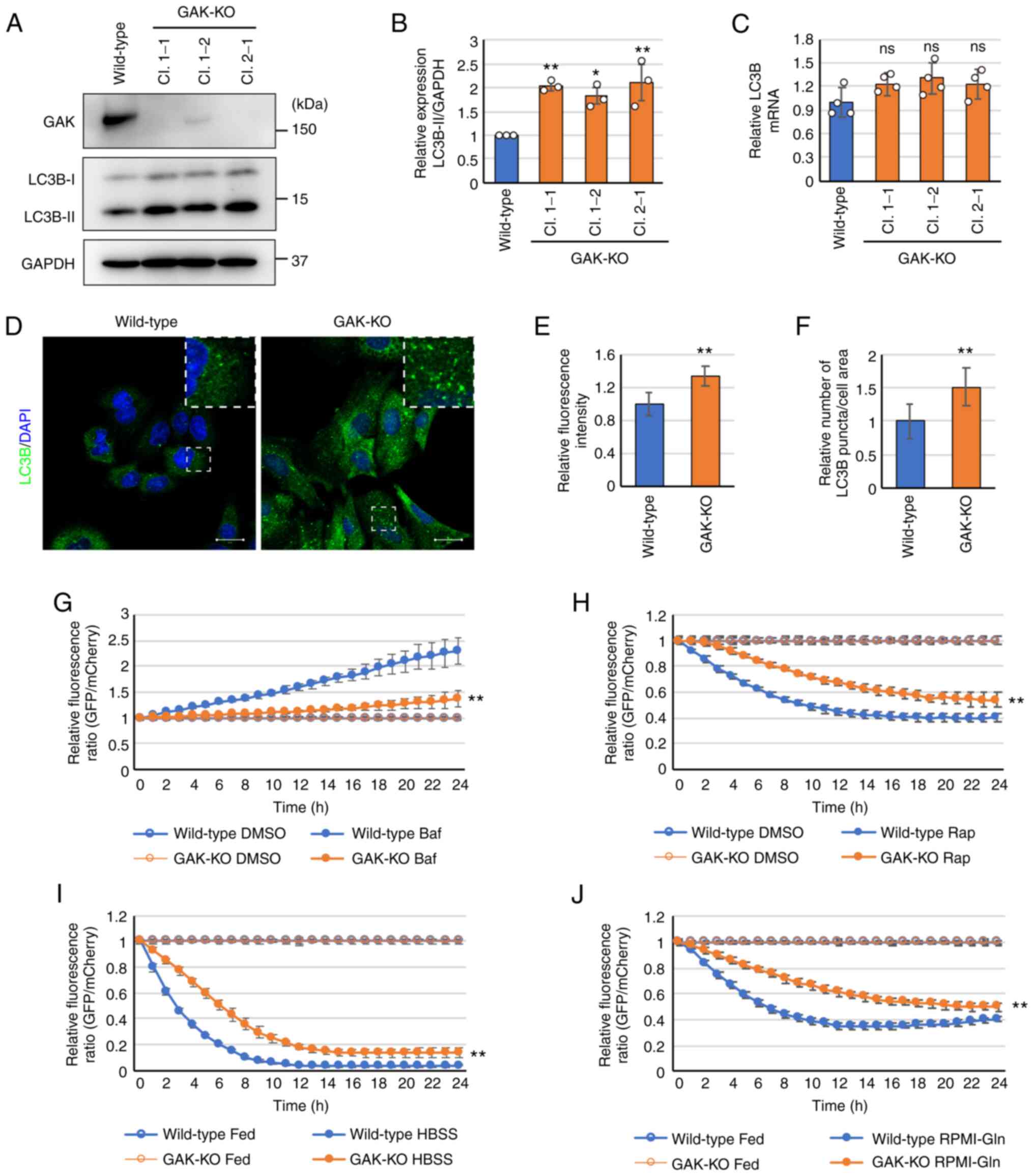 | Figure 1GAK disruption induces LC3B-II
accumulation and stagnates autophagic flux. (A) Immunoblot analysis
of GAK and LC3B protein expression levels in wild-type and
CRISPR/Cas9-mediated GAK-KO (clones 1-1, 1-2 and 2-1) A549 cells.
(B) Semi-quantification of relative LC3B-II protein expression
levels in wild-type and GAK-KO cells. Data were analyzed using
one-way ANOVA followed by Dunnett's post hoc test. Data are
presented as the mean ± SD (n=3). *P<0.05 and
**P<0.01 vs. wild-type. (C) Reverse
transcription-quantitative PCR analysis of relative mRNA expression
levels of LC3B in wild-type and GAK-KO cells under basal-fed
conditions. GAPDH was used as an internal control. Data were
analyzed using one-way ANOVA followed by Dunnett's post hoc test.
Data are presented as the mean ± SD (n=4). (D) Immunofluorescence
analysis of LC3B (green) in wild-type and GAK-KO cells. The dashed
boxed regions are shown at a high magnification (×3) in the inset.
Scale bar, 20 µm. (E) Relative mean fluorescence intensity
of LC3B in the cell area. Data were analyzed using an unpaired
two-tailed Student's t-test. Data are presented as the mean ± SD
(wild-type n=52; GAK-KO, n=20). **P<0.01 vs.
wild-type. (F) Relative number of LC3B puncta per cell area. Data
were analyzed using an unpaired two-tailed Student's t-test. Data
are presented as the mean ± SD (wild-type, n=40; GAK-KO, n=25).
**P<0.01 vs. wild-type. (G and H)
A549/GFP-LC3-mCherry-LC3ΔG and A549/GAK-KO/GFP-LC3-mCherry-LC3ΔG
cells were treated with (G) 10 nM Baf or (H) 10 nM rapamycin. The
fluorescence intensities derived from GFP-LC3 and mCherry-LC3ΔG
were monitored over 24 h. Autophagic flux was determined as
alterations in the relative intensities of GFP/mCherry, using
DMSO-treated groups as controls. Data were analyzed using two-way
ANOVA followed by Bonferroni's post hoc test. Data are presented as
the mean ± SD (n=8). **P<0.01 vs. wild-type. (I and
J) A549/GFP-LC3-mCherry-LC3ΔG and A549/GAK-KO/GFP-LC3-mCherry-LC3ΔG
cells were cultured under starvation conditions using (I) HBSS or
(J) RPMI-Gln. The fluorescence intensities derived from GFP-LC3 and
mCherry-LC3ΔG were monitored over 24 h. Autophagic flux was
determined as alterations in the relative intensities of
GFP/mCherry, using basal-fed condition groups as controls. Data
were analyzed using two-way ANOVA followed by Bonferroni's post hoc
test. Data are presented as the mean ± SD (n=8).
**P<0.01 vs. wild-type. Data are presented as the
mean ± SD. GAK, cyclin G-associated kinase; KO, knockout; Baf,
bafilomycin A1; Rap, rapamycin; HBSS, Hanks' balanced
salt solution; Gln, glutamine; ns, not significant. |
To analyze autophagic flux, a
GFP-LC3-mCherry-LC3ΔG-based system was used (44,45). Brief ly, GFP-LC3-mCherry-LC3ΔG is
cleaved into equimolar amounts of GFP-LC3 and mCherry-LC3ΔG.
GFP-LC3 is degraded by autophagy, whereas mCherry-LC3ΔG remains in
the cytosol and serves as an internal control. Therefore,
autophagic flux can be estimated by calculating the GFP/mCherry
signal ratio. In the present study, GAK-KO cells showed
significantly higher GFP/mCherry ratios under basal-fed conditions
compared with those in wild-type cells, suggesting that autophagic
flux levels were lower in GAK-KO cells compared with those in
wild-type cells (Fig. S3A and
B). Baf suppressed autophagic flux, as indicated by an
increased GFP/mCherry ratio, and cytoplasmic accumulation of
GFP-LC3 was more prominent in wild-type cells compared with that in
GAK-KO cells (Figs. 1G and
S3C). This result suggested
that the additional inhibitory effect of the autophagy inhibitor
was decreased in GAK-KO cells because the autophagic flux was
already inhibited in the steady-state. By contrast, as indicated by
a reduced GFP/mCherry ratio, rapamycin induced autophagic flux, and
the elimination rates of GFP-LC3 were more prominent in wild-type
cells compared with those in GAK-KO cells (Figs. 1H and S3D). Similarly, starvation conditions
(HBSS or RPMI-Gln medium) induced autophagic flux, and the
elimination rates of GFP-LC3 were more prominent in wild-type cells
compared with those in GAK-KO cells (Figs. 1I, J, S3E and S3F). Collectively, these
results suggested that GAK disruption decelerated autophagic flux
under basal-fed conditions and reduced the acceleration of
autophagic flux under autophagy-inducing conditions, such as
starvation.
To analyze the autophagy-related signaling pathways,
starvation-refeeding experiments were performed. Both in wild-type
and GAK-KO cells, starvation significantly decreased the
phosphorylation of mTOR (Ser2448), S6K (Thr389) and S6 (Ser235/236)
and induced the phosphorylation of AMPK (Thr172) compared with that
in the fed group, indicating that the mTOR signaling pathway was
repressed by starvation. Conversely, refeeding significantly
induced the phosphorylation of mTOR, S6K and S6 and decreased the
phosphorylation of AMPK compared with that in the starved group,
indicating that the mTOR signaling pathway was reactivated by
refeeding (Fig. S4). Therefore,
GAK disruption did not impair starvation and refeeding-induced
fluctuations in mTOR signaling. Collectively, these results
suggested that the loss of GAK stagnated autophagic flux,
but displayed limited effects on the signals that induce
autophagosome formation, as well as the formation of
autophagosomes.
Impact of GAK disruption on the
morphology and function of lysosomes
Transmission electron microscopy (TEM) revealed a
notable accumulation of cytoplasmic vesicles in GAK-KO cells
(Fig. 2A). To characterize the
cytoplasmic vesicles, immunofluorescence analysis was performed.
The size distribution of LAMP2-positive vesicles, including
lysosomes and autolysosomes, in GAK-KO cells was larger compared
with that in wild-type cells (Fig.
2B and C). The protein expression levels of LAMP1 and LAMP2 in
GAK-KO cells were significantly higher compared with those in
wild-type cells (Fig. S5A and
B), whereas the expression levels of LAMP1 and LAMP2 mRNA in
GAK-KO cells were similar to those in wild-type cells (Fig. S5C). These data indicated that
the de novo synthesis of lysosome-related proteins was not
upregulated; however, lysosome-related proteins accumulated in
GAK-KO cells under basal-fed conditions. The enlargement of
LAMP2-positive vesicles in GAK-KO cells was more prominent under
starvation conditions (Fig.
2D-F). Transduction of GAK-KO cells with a GAK-encoding
lentivirus canceled the GAK-KO cell-specific phenotype, enlarged
LAMP2-positive vesicles (Fig.
2D-F). Additionally, it was shown that the number of
LAMP2-positive vesicles in GAK-KO cells was significantly lower
compared with that in wild-type cells, and that transduction of
GAK-KO cells with a GAK-encoding lentivirus restored the number of
LAMP2-positive vesicles (Fig. 2D, E
and G). Although the expression of lysosome-related genes was
not decreased (Fig. S5), the
number of lysosomes decreased, suggesting that lysosomal
reformation was suppressed in GAK-KO cells.
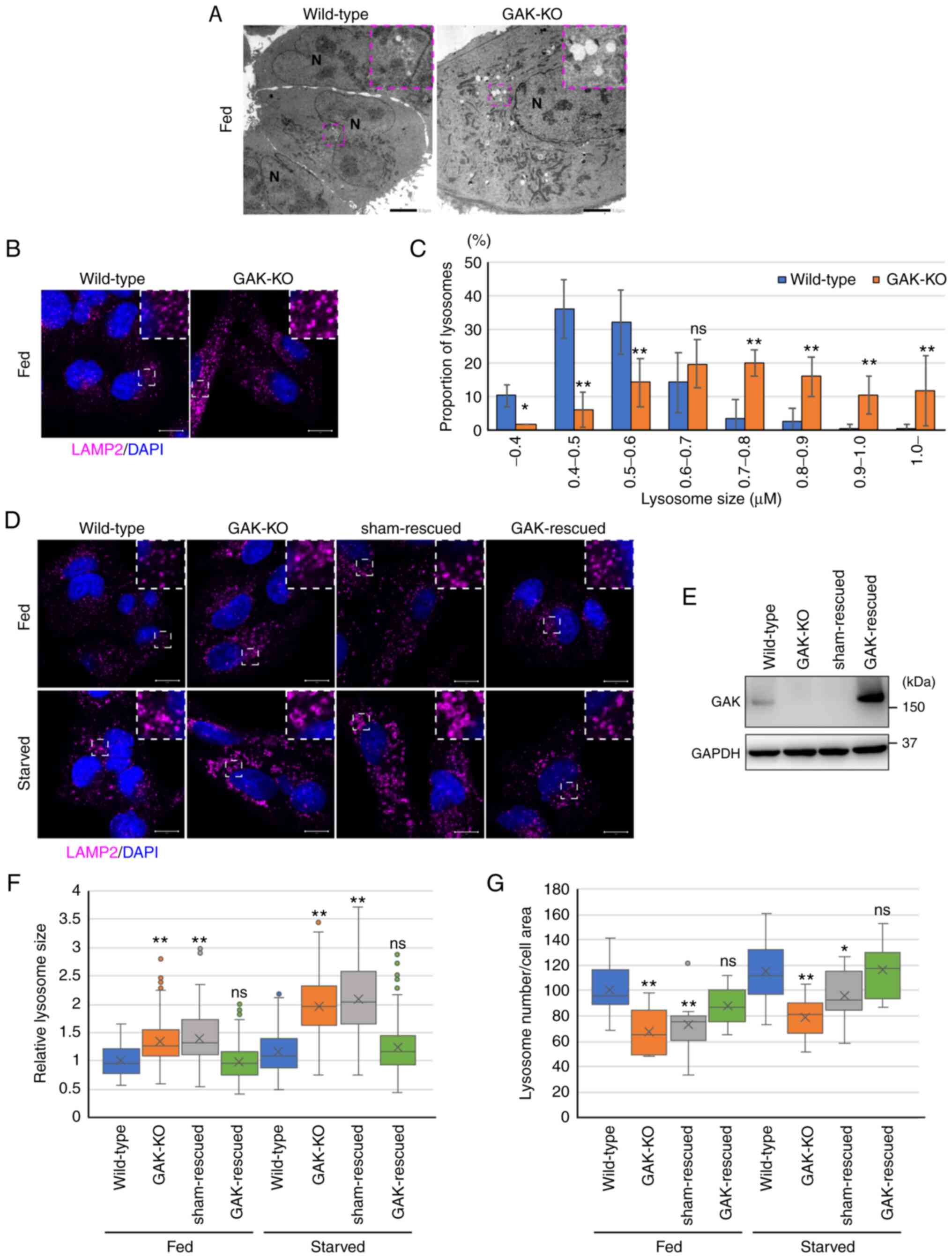 | Figure 2GAK disruption causes the
formation of enlarged LAMP2-positive vesicles. (A) Representative
transmission electron microscopy images of wild-type and GAK-KO
cells under basal-fed conditions. Dashed boxed regions are shown at
a high magnification (×3) in the inset. Scale bar, 5 µm. (B)
Immunofluorescence analysis of LAMP2 (magenta) in wild-type and
GAK-KO cells under basal-fed conditions. Dashed boxed regions are
shown at a high magnification (×3) in the inset. Scale bar, 10
µm. (C) Lysosomal size in wild-type and GAK-KO cells under
basal-fed conditions was quantified using ZEN 2012 software. Data
were analyzed using two-way ANOVA followed by Bonferroni's post hoc
test. Data are presented as the mean ± SD (wild-type, n=204;
GAK-KO, n=295). *P<0.05 and **P<0.01
vs. wild-type. (D) Immunofluorescence analysis of LAMP2 (magenta)
in wild-type, GAK-KO, sham-rescued and GAK-rescued cells under
basal-fed or starvation (RPMI-Gln for 8 h) conditions. Dashed boxed
regions are shown at a high magnification (×3) in the inset. Scale
bar, 10 µm. (E) GAK expression in wild-type, GAK-KO,
sham-rescued and GAK-rescued cells was determined by
immunoblotting. (F) Lysosomal size in wild-type, GAK-KO,
sham-rescued and GAK-rescued cells was quantified using ZEN 2012
software. Data were analyzed using one-way ANOVA followed by
Dunnett's post hoc test. The box extends from the lower to the
upper quartile, the middle line indicates the median, the X
indicates the mean and the whiskers represent the minimum to
maximum values, except for outliers, which are indicated by dots
(wild-type/fed, n=212; GAK-KO/fed, n=240; sham-rescued/fed, n=140;
GAK-rescued/fed, n=277; wild-type/starved, n=195; GAK-KO/starved,
n=112; sham-rescued/starved, n=172; GAK-rescued/starved, n=180).
**P<0.01 vs. wild-type. (G) Number of lysosomes in
wild-type, GAK-KO, sham-rescued and GAK-rescued cells was
quantified using Imaris. Data were analyzed using one-way ANOVA
followed by Dunnett's post hoc test. The box extends from the lower
to the upper quartile, the middle line indicates the median, the X
indicates the mean and the whiskers represent the minimum to
maximum values, except for outliers, which are indicated by dots
(wild-type/fed, n=25; GAK-KO/fed, n=10; sham-rescued/fed, n=9;
GAK-rescued/fed, n=12; wild-type/starved, n=21; GAK-KO/starved,
n=15; sham-rescued/starved, n=10; GAK-rescued/starved, n=16).
*P<0.05 and **P<0.01 vs. wild-type.
GAK, cyclin G-associated kinase; KO, knockout; LAMP, lysosomal
associated membrane protein; ns, not significant; N, nucleus; Gln,
glutamine. |
The enlargement of lysosomes/autolysosomes is
accompanied by acidification defects that promote the accumulation
of undegradable cargo in these organelles (14). To assess lysosomal function in
GAK-KO cells, GAK-KO cells were stained with LysoTracker dye, which
is usually trapped in acidified compartments, such as lysosomes.
Decreased staining with LysoTracker dye was detected in GAK-KO
cells compared with that in wild-type cells under basal-fed
conditions, indicating the elevation of lysosomal pH in GAK-KO
cells (Fig. 3A and B). However,
under starvation conditions, the intensity of LysoTracker staining
in GAK-KO cells was at the same level as that in wild-type cells
(Fig. 3A and B). Staining with
the LysoSensor dye displayed similar results to the LysoTracker dye
(Fig. S6A and B). The
maturation rates of CTSB in GAK-KO cells were significantly lower
compared with those in wild-type cells under both basal-fed and
starvation conditions (Fig. 3C and
D). However, mature CTSD expression levels were similar between
wild-type and GAK-KO cells (Fig. 3C
and D). In addition, the degradation rates of p62/SQSTM1, an
autophagy substrate, were also similar between wild-type and GAK-KO
cells (Fig. 3E and F). The TEM
images revealed a significant accumulation of cytoplasmic vesicles
in GAK-KO cells, but the vesicles contained little undigested cargo
(Fig. 2A). Collectively, these
results indicated that the loss of GAK altered the intra-lysosomal
conditions, but essentially maintained the hydrolytic capacity in
lysosomes and/or autolysosomes, suggesting that the alteration of
intra-lysosomal conditions may not be a major cause of the
stagnation of the autophagic flux.
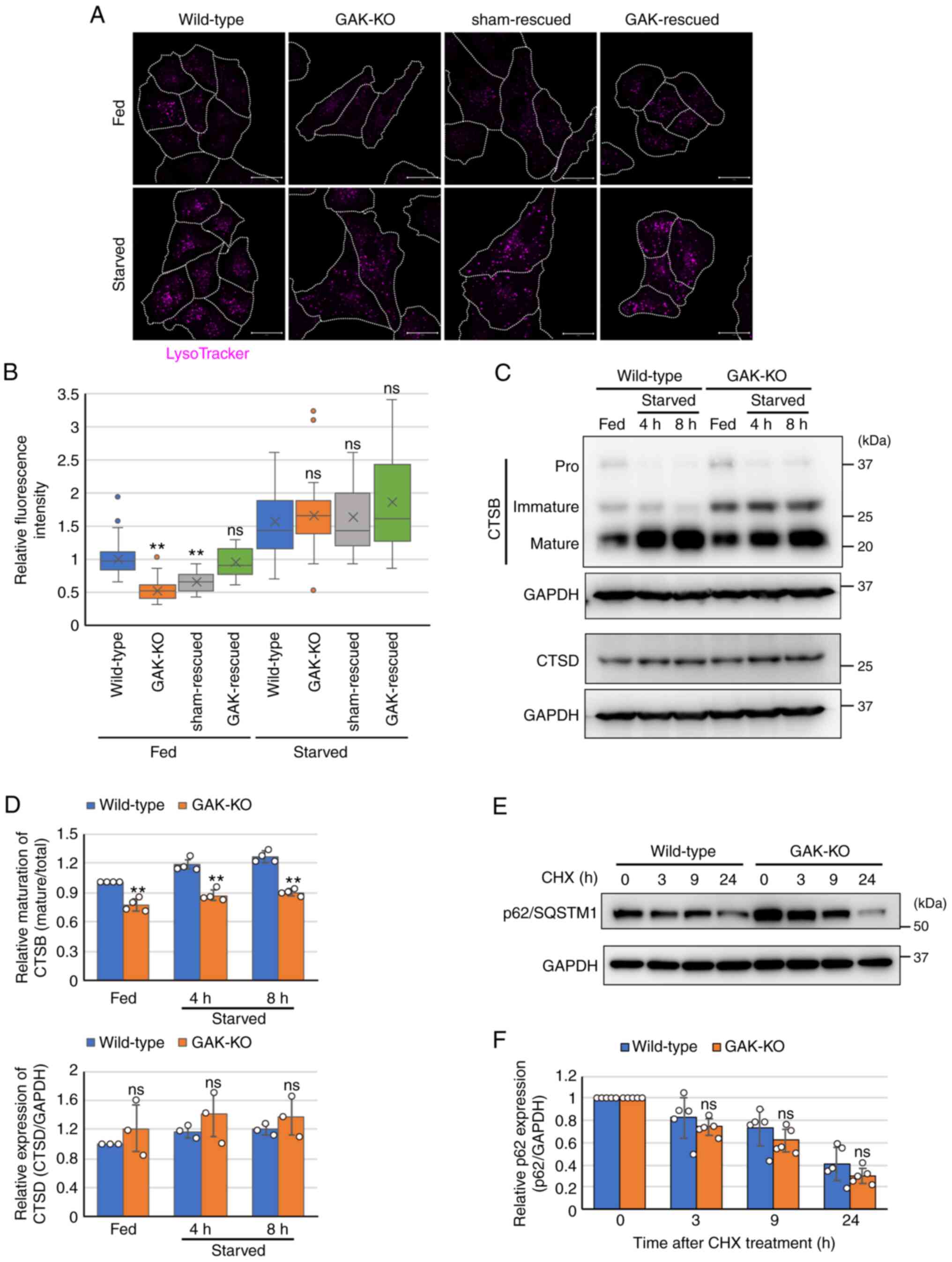 | Figure 3GAK disruption alters the
intra-lysosomal conditions. (A) LysoTracker staining in wild-type,
GAK-KO, sham-rescued and GAK-rescued cells under basal-fed or
starvation conditions (RPMI-Gln for 8 h). Scale bar, 20 µm.
(B) Relative mean fluorescence intensities of LysoTracker in
wild-type, GAK-KO, sham-rescued and GAK-rescued cells. Data were
analyzed using one-way ANOVA followed by Dunnett's post hoc test.
The box extends from the lower to the upper quartile, the middle
line indicates the median, the X indicates the mean and the
whiskers represent the minimum to maximum values, except for
outliers, which are indicated by dots (wild-type/fed, n=63;
GAK-KO/fed, n=42; sham-rescued/fed, n=20; GAK-rescued/fed, n=18;
wild-type/starved, n=52; GAK-KO/starved, n=40;
sham-rescued/starved, n=15; GAK-rescued/starved, n=19).
**P<0.01 vs. wild-type. (C) Immunoblot analysis of
CTSB and CTSD expression in wild-type and GAK-KO cells under
basal-fed or starvation conditions (RPMI-Gln for 4 or 8 h). The
membrane for CTSB was stripped and reprobed for GAPDH as the
loading control. (D) Semi-quantification of the relative maturation
levels of CTSB and relative expression levels of mature CTSD in
wild-type and GAK-KO cells. Ratios of mature CTSB/total CTSB and
CTSD/GAPDH are presented. Data were analyzed using two-way ANOVA
followed by Bonferroni's post hoc test. Data are presented as the
mean ± SD (n=4). **P<0.01 vs. wild-type. (E) Cell
lysates were prepared from wild-type and GAK-KO cells after 30
µM cycloheximide treatment under basal-fed conditions. The
expression of p62/SQSTM1 was analyzed by immunoblotting. (F)
Semi-quantification of the relative expression levels of p62 in
wild-type and GAK-KO cells. Data were analyzed using two-way ANOVA
followed by Bonferroni's post hoc test. Data are presented as the
mean ± SD (n=5). GAK, cyclin G-associated kinase; KO, knockout;
CTSD, cathepsin D; Gln, glutamine; CTSB, cathepsin B; SQSTM1,
sequestosome 1; CHX, cycloheximide; ns, not significant. |
GAK disruption stagnates
autophagosome-lysosome fusion and induces the accumulation of
autophagosomes and autolysosomes
Subsequently, the formation of autophagosomes and
autolysosomes under starvation conditions was assessed. The
accumulation of enlarged LC3B-positive puncta was detected in
GAK-KO cells at 4-24 h after starvation (Figs. 4A, 4B, S7A and S7B). In addition, enlarged
LC3B-positive puncta and enlarged LAMP2-positive vesicles were
observed side by side in GAK-KO cells (Fig. 4A). Transduction of GAK-KO cells
with a GAK-encoding lentivirus canceled the accumulation of
enlarged LC3B-positive puncta and the enlargement of LAMP2-positive
vesicles (Fig. 4A and B). In
GAK-OE cells, similar to wild-type cells, a small number of
enlarged LC3B-positive puncta were detected at 24 h after
starvation, but these were not observed at 10 h after starvation
(Fig. S7C-E). The TEM results
verified the accumulation of enlarged autophagosomes and
autolysosomes in GAK-KO cells under starvation conditions (Fig. 4C). These observations indicated
that the enlarged LAMP2-positive vesicles observed under starvation
conditions (Figs. 2D and
4A) were primarily
autolysosomes.
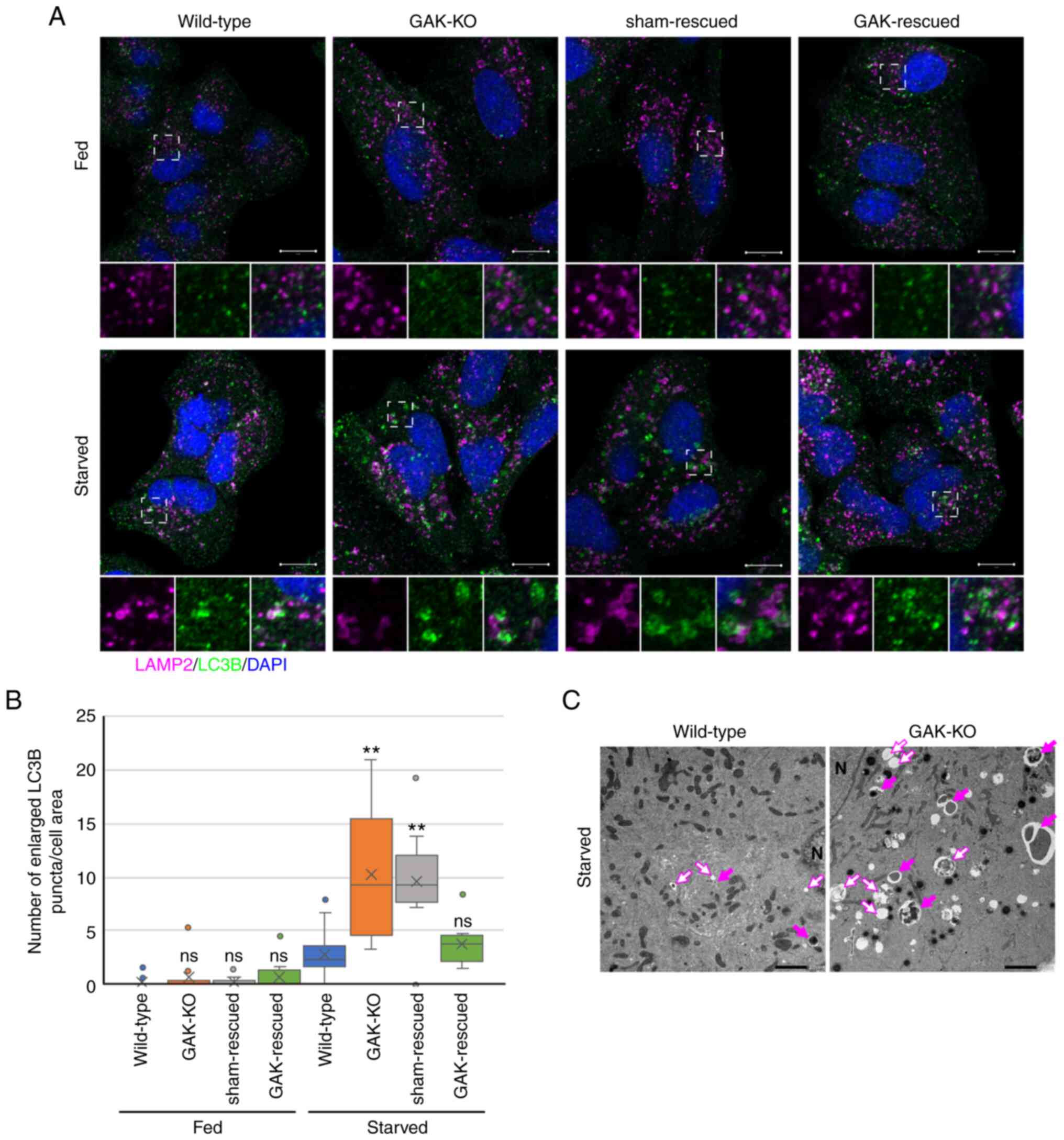 | Figure 4GAK disruption induces the
accumulation of autophagosomes and autolysosomes under starvation
conditions. (A) Immunofluorescence analysis of LAMP2 (magenta) and
LC3B (green) in wild-type, GAK-KO, sham-rescued and GAK-rescued
cells under basal-fed or starvation conditions (RPMI-Gln for 8 h).
The dashed boxed regions are shown at a high magnification (×3) at
the bottom. Scale bar, 10 µm. (B) Number of enlarged
LC3B-positive puncta in wild-type, GAK-KO, sham-rescued and
GAK-rescued cells was quantified using Imaris. Data were analyzed
using one-way ANOVA followed by Dunnett's post hoc test. The box
extends from the lower to the upper quartile, the middle line
indicates the median, the X indicates the mean and the whiskers
represent the minimum to maximum values, except for outliers, which
are indicated by dots (wild-type/fed, n=18; GAK-KO/fed, n=10;
sham-rescued/fed, n=9; GAK-rescued/fed, n=11; wild-type/starved,
n=18; GAK-KO/starved, n=14; sham-rescued/starved, n=9;
GAK-rescued/starved, n=10). **P<0.01 vs. wild-type.
(C) Representative transmission electron microscopy images of
wild-type and GAK-KO cells under starvation conditions (RPMI-Gln
for 8 h). Open arrows indicate autolysosomes and closed arrows
indicate autophagosomes. N, nucleus; Scale bar, 2 µm. GAK,
cyclin G-associated kinase; LAMP, lysosomal associated membrane
protein; KO, knockout; Gln, glutamine; ns, not significant. |
Chemical inhibition of GAK kinase activity also
induced the accumulation of enlarged LC3B-positive puncta and
LAMP2-positive vesicles in a dose-dependent manner under starvation
conditions (Fig. 5A-C). GAK-KO
cells showed an increase in lysosomal size even under basal-fed
conditions (Fig. 2D and F). By
contrast, wild-type cells in the presence of the GAKi showed an
increase in lysosomal size under starvation conditions, but no
increase under basal-fed conditions (Fig. 5A and C), suggesting that the
increase in lysosomal size involves more than just the kinase
activity of GAK, at least under basal-fed conditions. In addition
to A549 cells, H596, PANC-1, and Hep-G2 cells also showed enlarged
LC3B-positive puncta in the presence of the GAKi under starvation
conditions (Fig. S8). As
observed in GAK-KO cells, the GAKi reduced autophagic flux, which
was demonstrated by increased elimination rates of GFP-LC3 in a
GFP-LC3-mCherry-LC3ΔG-based system, in wild-type cells under
starvation conditions (Figs. 5D,
5E, S9A and S9B). Wild-type
cells in the presence of 30 µM GAKi showed a more
considerable decrease in autophagic flux compared with that
observed in GAK-KO cells (Figs.
1I, 1J, 5D and 5E), suggesting that high concentrations
of the GAKi may affect proteins other than GAK.
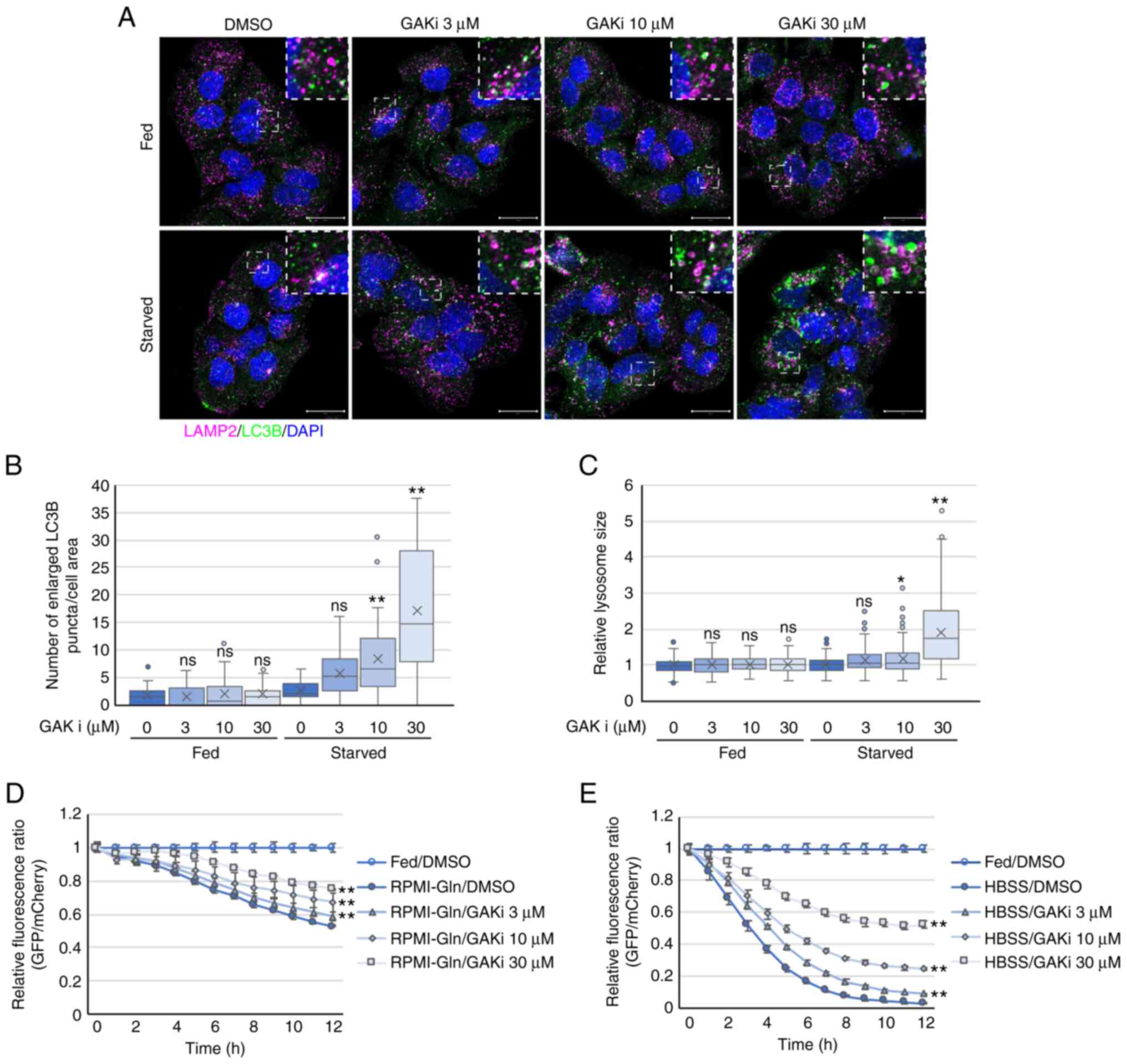 | Figure 5Chemical inhibition of GAK induces
the accumulation of autophagosomes and autolysosomes, and stagnates
autophagic flux. (A) Immunofluorescence analysis of LAMP2 (magenta)
and LC3B (green) in wild-type cells treated with the indicated
concentrations of GAKi under basal-fed or starvation conditions
(RPMI-Gln for 8 h). The dashed boxed regions are shown at a high
magnification (×3) in the inset. Scale bar, 20 µm. (B)
Relative number of enlarged LC3B-positive puncta was quantified
using Imaris. Data were analyzed using one-way ANOVA followed by
Dunnett's post hoc test. The box extends from the lower to the
upper quartile, the middle line indicates the median, the X
indicates the mean and the whiskers represent the minimum to
maximum values, except for outliers, which are indicated by dots
(GAKi 0 µM/fed, n=29; 3 µM/fed, n=32; 10
µM/fed, n=32; 30 µM/fed, n=31; 0 µM/starved,
n=32; 3 µM/starved, n=27; 10 µM/starved, n=35; 30
µM/starved, n=55). **P<0.01 vs. 0 µM.
(C) Relative lysosomal sizes were quantified using ZEN 2012
software. Data were analyzed using one-way ANOVA followed by
Dunnett's post hoc test. The box extends from the lower to the
upper quartile, the middle line indicates the median, the X
indicates the mean and the whiskers represent the minimum to
maximum values, except for outliers, which are indicated by dots
(n=150). *P<0.05 and **P<0.01 vs. 0
µM. (D and E) A549/GFP-LC3-mCherry-LC3ΔG cells were treated
with the indicated concentrations of GAKi under starvation
conditions using (D) RPMI-Gln or (E) HBSS. The fluorescence
intensities derived from GFP-LC3 and mCherry-LC3ΔG were monitored
over 12 h. Autophagic flux was determined as alterations in the
relative intensities of GFP/mCherry, using the basal-fed condition
group as controls. Data were analyzed using one-way ANOVA followed
by Dunnett's post hoc test. Data are presented as the mean ± SD
(n=8). **P<0.01 vs. RPMI-Gln/DMSO or HBSS/DMSO. GAK,
cyclin G-associated kinase; LAMP, lysosomal associated membrane
protein; GAKi, GAK inhibitor; Gln, glutamine; HBSS, Hanks' balanced
salt solution; ns, not significant. |
To facilitate the analysis of
autophagosome-lysosomal fusion, HCQ, which blocks the late phase of
autophagy by increasing the lysosomal pH (51-53), was used. After 8 h of treatment
with HCQ, most of the LC3B-positive puncta were enclosed within the
LAMP2-positive vesicles in wild-type and GAK-rescued cells
(Fig. 6A, B and E). By contrast,
in GAK-KO cells, most of the LC3B-positive puncta were in contact
with the LAMP2-positive vesicles but were not enclosed within them
(Fig. 6A, B, and E). However,
after 24 h of treatment with HCQ, most of the LC3B-positive puncta
were enclosed within the LAMP2-positive vesicles in GAK-KO and
wild-type cells (Fig. 6C-E).
After treating GAK-KO cells with HCQ for 8 h, the TEM results also
indicated the accumulation of incompletely fused autolysosomes,
which presented as snowman-like structures in which spheres were
connected (Fig. 6F). Taken
together, these results indicated that autophagosome-lysosome
fusion was delayed in GAK-KO cells and that GAK kinase activity was
required to regulate these steps.
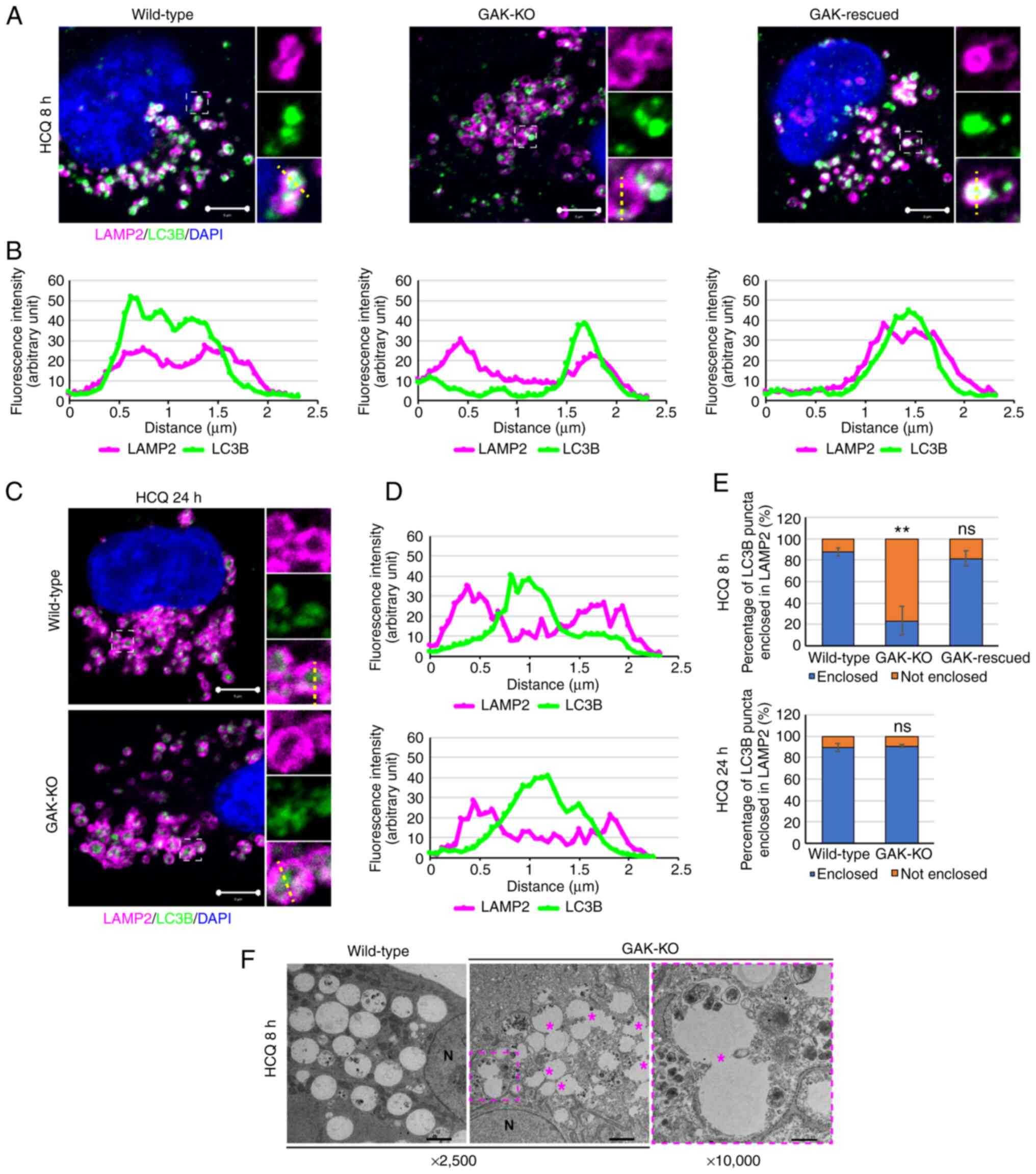 | Figure 6GAK disruption delays
autophagosome-lysosome fusion. (A) Immunofluorescence analysis of
LAMP2 (magenta) and LC3B (green) in wild-type, GAK-KO and
GAK-rescued cells after treatment with 50 µM HCQ for 8 h.
The dashed boxed regions are shown at a high magnification (×3) on
the right. Scale bar, 5 µm. (B) Intensity profiles of LAMP2
(magenta) and LC3B (green) along the dashed yellow line shown in
the magnified image in (A). (C) Immunofluorescence analysis of
LAMP2 (magenta) and LC3B (green) in wild-type and GAK-KO cells
after treatment with 50 µM HCQ for 24 h. The dashed boxed
regions are shown at a high magnification (×3) on the right. Scale
bar, 5 µm. (D) Intensity profiles of LAMP2 (magenta) and
LC3B (green) along the dashed yellow line shown in the magnified
image in (C). (E) Quantification of LC3B-positive puncta enclosed
by LAMP2-positive vesicles and those not enclosed. In the upper
panel, data were analyzed using one-way ANOVA followed by Dunnett's
post hoc test. **P<0.01 vs. wild-type. In the lower
panel, data were analyzed using an unpaired two-tailed Student's
t-test. Data are presented as the mean ± SD (HCQ 8 h; wild-type,
n=330; GAK-KO, n=330; GAK-rescued, n=316; and HCQ 24 h; wild-type,
n=476; GAK-KO, n=321). (F) Representative transmission electron
microscopy images of wild-type and GAK-KO cells after treatment
with 50 µM HCQ for 8 h. The dashed boxed regions are shown
at a high magnification (scale bar, 500 nm) on the right.
Incompletely fused autolysosomes are indicated by *.
Scale bar, 2 µm. GAK, cyclin G-associated kinase; LAMP,
lysosomal associated membrane protein; KO, knockout; HCQ,
hydroxychloroquine; ns, not significant. |
GAK disruption impairs autophagic
lysosome reformation
Subsequently, ALR in GAK-KO cells was analyzed.
mTOR, which is a key negative regulator of autophagy, is
reactivated upon prolonged starvation, triggering ALR (19). Phosphorylation of S6K (Thr389),
an indicator of mTOR activity, was reduced by starvation, but
induced after 8 h of starvation in wild-type cells. By contrast,
S6K phosphorylation remained decreased even after 8 h of starvation
in GAK-KO cells (Fig. 7A and B).
Therefore, mTOR was reactivated in wild-type cells but not in
GAK-KO cells.
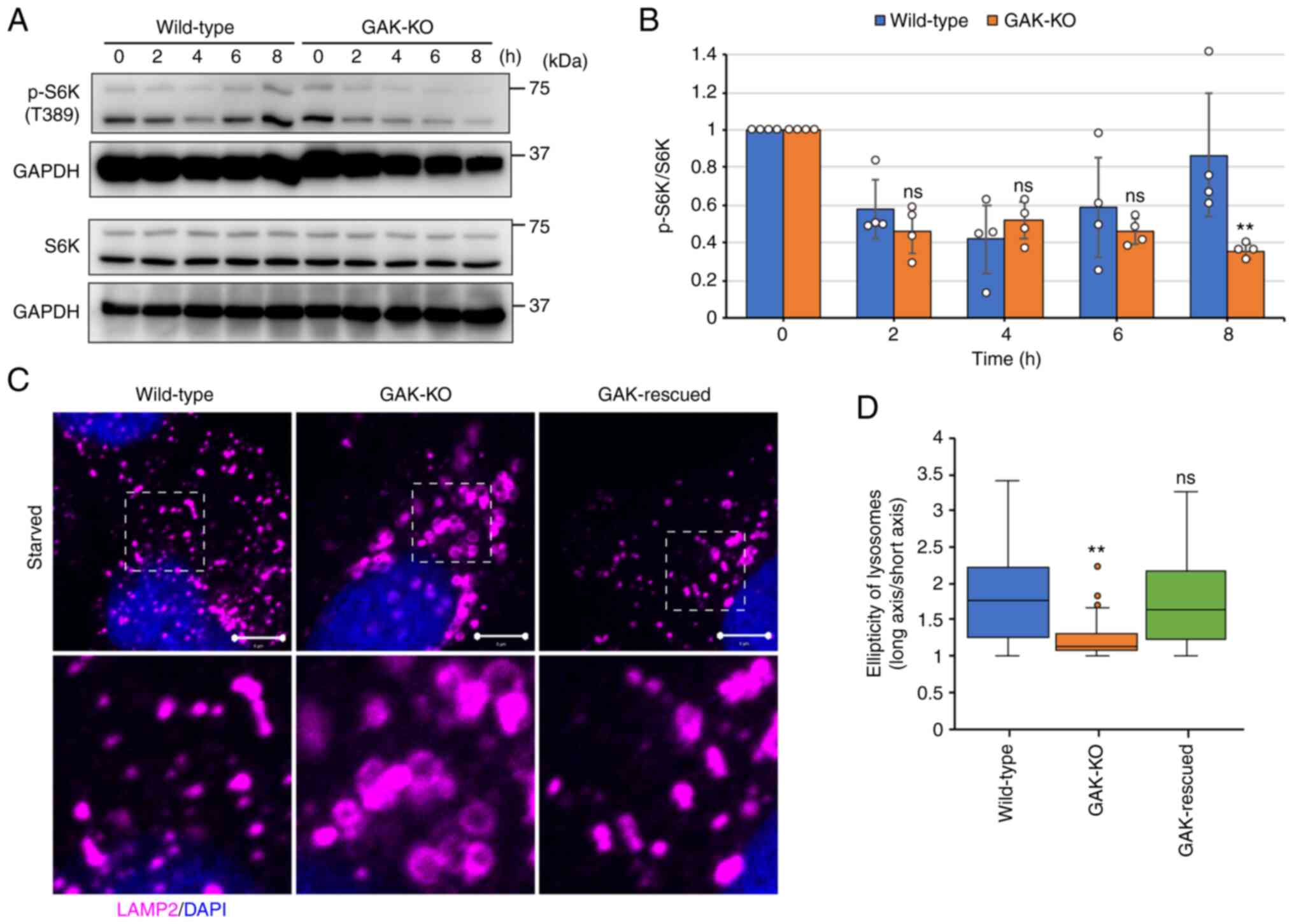 | Figure 7GAK disruption impairs
autophagic lysosome reformation. (A) Cell lysates were prepared
from wild-type and GAK-KO cells after starvation using RPMI-Gln for
the indicated time. Immunoblot analysis of the phosphorylation
levels of S6K (Thr389). (B) Densitometry was performed to determine
the ratio of p-S6K (Thr389)/total S6K. Data were analyzed using
two-way ANOVA followed by Bonferroni's post hoc test. Data are
presented as the mean ± SD (n=4). **P<0.01 vs.
wild-type. (C) Immunofluorescence analysis of LAMP2 in wild-type,
GAK-KO and GAK-rescued cells under starvation conditions (RPMI-Gln
for 8 h). The dashed boxed regions are shown at a high
magnification (×3) at the bottom. Scale bar, 5 µm. (D) Long
and short axes of lysosomes were measured using ZEN 2012 software
and the ratio (long axis/short axis) was calculated. Data were
analyzed using one-way ANOVA followed by Dunnett's post hoc test.
The box extends from the lower to the upper quartile, the middle
line indicates the median, the X indicates the mean and the
whiskers represent the minimum to maximum values, except for
outliers, which are indicated by dots (n=50).
**P<0.01 vs. wild-type. GAK, cyclin G-associated
kinase; KO, knockout; Gln, glutamine; S6, ribosomal protein S6;
S6K, S6 kinase; p, phosphorylated; LAMP, lysosomal associated
membrane protein; ns, not significant. |
Prolonged starvation has been reported to induce the
formation of reformation tubules from the autolysosomes, which is a
characteristic feature of ALR, and the impairment of ALR results in
the enlargement of autolysosomes (19,54-56). The accumulation of enlarged
LAMP2-positive vesicles in GAK-KO cells under starvation conditions
was observed (Figs. 2D, 4A and 7C). Although significant tubule
formation, as reported in previous studies (19,54,56), was not confirmed in A549 cells,
the deformation of LAMP2-positive vesicles was observed in
wild-type and GAK-rescued cells, but not in GAK-KO cells (Fig. 7C and D). These results suggested
that ALR was delayed or impaired in GAK-KO cells, which was
consistent with the observation that there were fewer
LAMP2-positive vesicles in GAK-KO cells compared with in wild-type
cells (Fig. 2G).
ROCK inhibition mitigates GAK
disruption-induced stagnation of autophagic flux
Actin cytoskeleton and myosin motor proteins work
together to serve essential roles in each stage of autophagy, such
as the formation and transport of vesicular cargoes, the fusion
between autophagosomes and lysosomes, and the formation of
reformation tubules (20,21).
GAK disruption stagnated autophagosome-lysosome fusion and impaired
ALR, suggesting the dysregulation of actomyosin in GAK-KO cells.
Therefore, the ROCK inhibitor Y-27632 was used to suppress the
function of ROCK (ROCK1 and ROCK2) (57), which mediates actin-myosin
contractility and regulates actin cytoskeleton dynamics (58). Treatment with Y-27632 attenuated
the accumulation of enlarged LC3B-positive puncta detected in
GAK-KO cells under starvation conditions (Figs. 8A, 8B, S10A and S10B). ROCK1 knockdown also
attenuated the accumulation of enlarged LC3B-positive puncta
detected in GAK-KO cells under starvation conditions (Fig. 9A-C). Furthermore, the addition of
Baf caused enlargement of the autophagosomes in ROCK1-knockdown
GAK-KO cells, which indicated that ROCK1 knockdown in GAK-KO cells
restored the fusion between autophagosomes and lysosomes to normal
rather than impairing autophagosome formation (Fig. S11). However, no interaction was
detected between GAK and ROCK1 (Fig. S12). In addition, treatment with
Y-27632 (Fig. 8A and C) or ROCK1
knockdown (Fig. 9B and D)
reversed the enlargement of LAMP2-positive vesicles in GAK-KO cells
under starvation conditions. Collectively, these results indicated
that the disturbance of lysosomal dynamics and the stagnation of
autophagic flux due to GAK loss involved a pathway for
ROCK-mediated cytoskeletal control (Fig. S13).
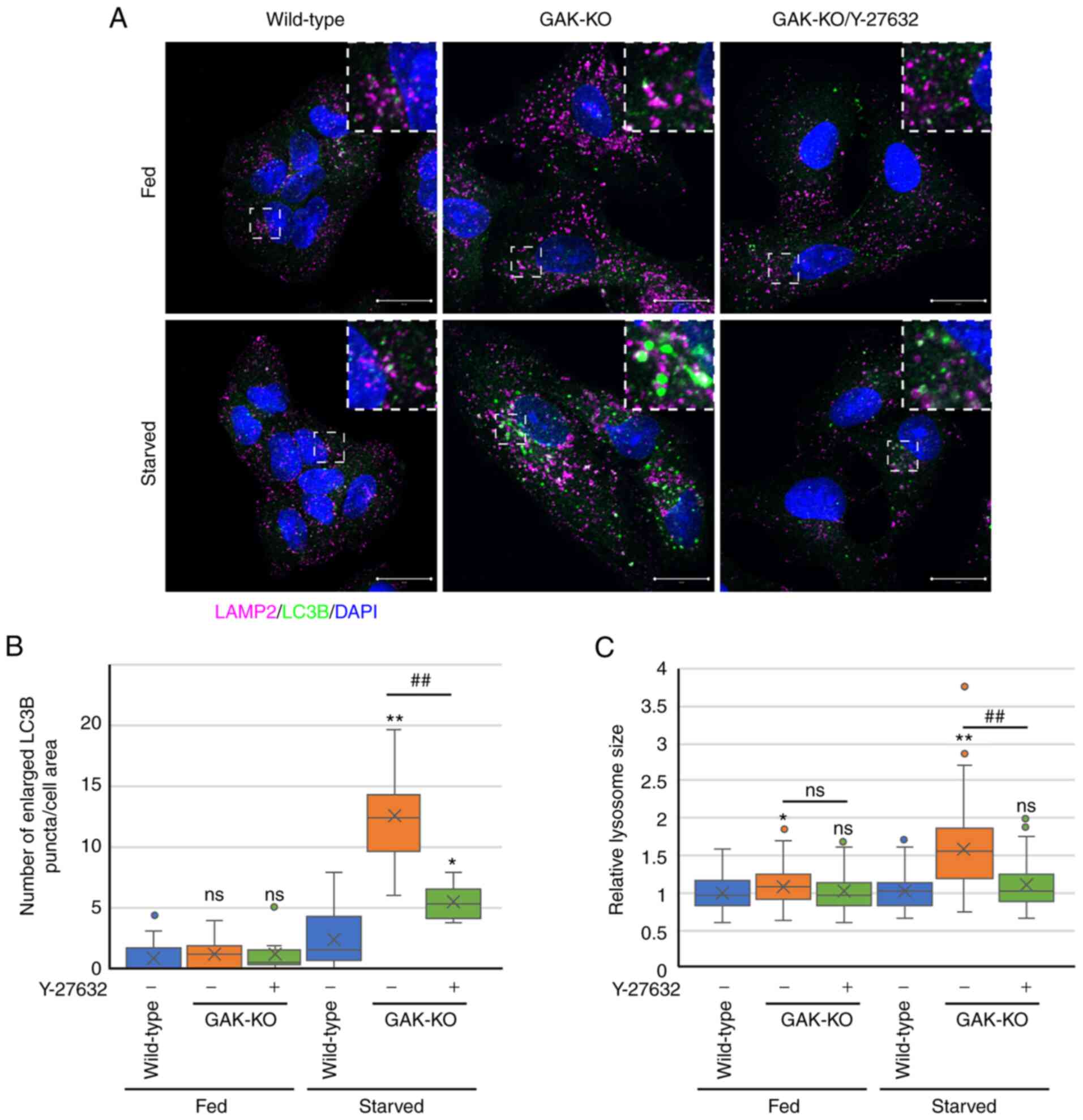 | Figure 8Rho-associated protein kinase
inhibition eliminates the accumulation of autophagosomes and
autolysosomes induced by GAK disruption. (A) Immunofluorescence
analysis of LAMP2 (magenta) and LC3B (green) in wild-type cells,
GAK-KO cells and GAK-KO cells treated with Y-27632 under basal-fed
or starvation (RPMI-Gln for 8 h) conditions. Dashed boxed regions
are shown at a high magnification (×3) in the inset. Scale bar, 20
µm. (B) Number of enlarged LC3B-positive puncta in wild-type
cells, GAK-KO cells and GAK-KO cells treated with Y-27632 was
quantified using Imaris. Data were analyzed using one-way ANOVA
followed by Tukey's post hoc test. The box extends from the lower
to the upper quartile, the middle line indicates the median, the X
indicates the mean and the whiskers represent the minimum to
maximum values, except for outliers, which are indicated by dots
(wild-type/fed, n=16; GAK-KO/fed, n=10; GAK-KO/Y-27632/fed, n=7;
wild-type/starved, n=27; GAK-KO/starved, n=26;
GAK-KO/Y-27632/starved, n=25). *P<0.05 and
**P<0.01 vs. wild-type. ##P<0.01 vs.
GAK-KO. (C) Lysosomal size in wild-type cells, GAK-KO cells and
GAK-KO cells treated with Y-27632 quantified using ZEN 2012
software. Data were analyzed using one-way ANOVA followed by
Tukey's post hoc test. The box extends from the lower to the upper
quartile, the middle line indicates the median, the X indicates the
mean and the whiskers represent the minimum to maximum values,
except for outliers, which are indicated by dots (n=100).
*P<0.05 and **P<0.01 vs. wild-type.
##P<0.01 vs. GAK-KO. GAK, cyclin G-associated kinase;
LAMP, lysosomal associated membrane protein; KO, knockout; Gln,
glutamine; ns, not significant. |
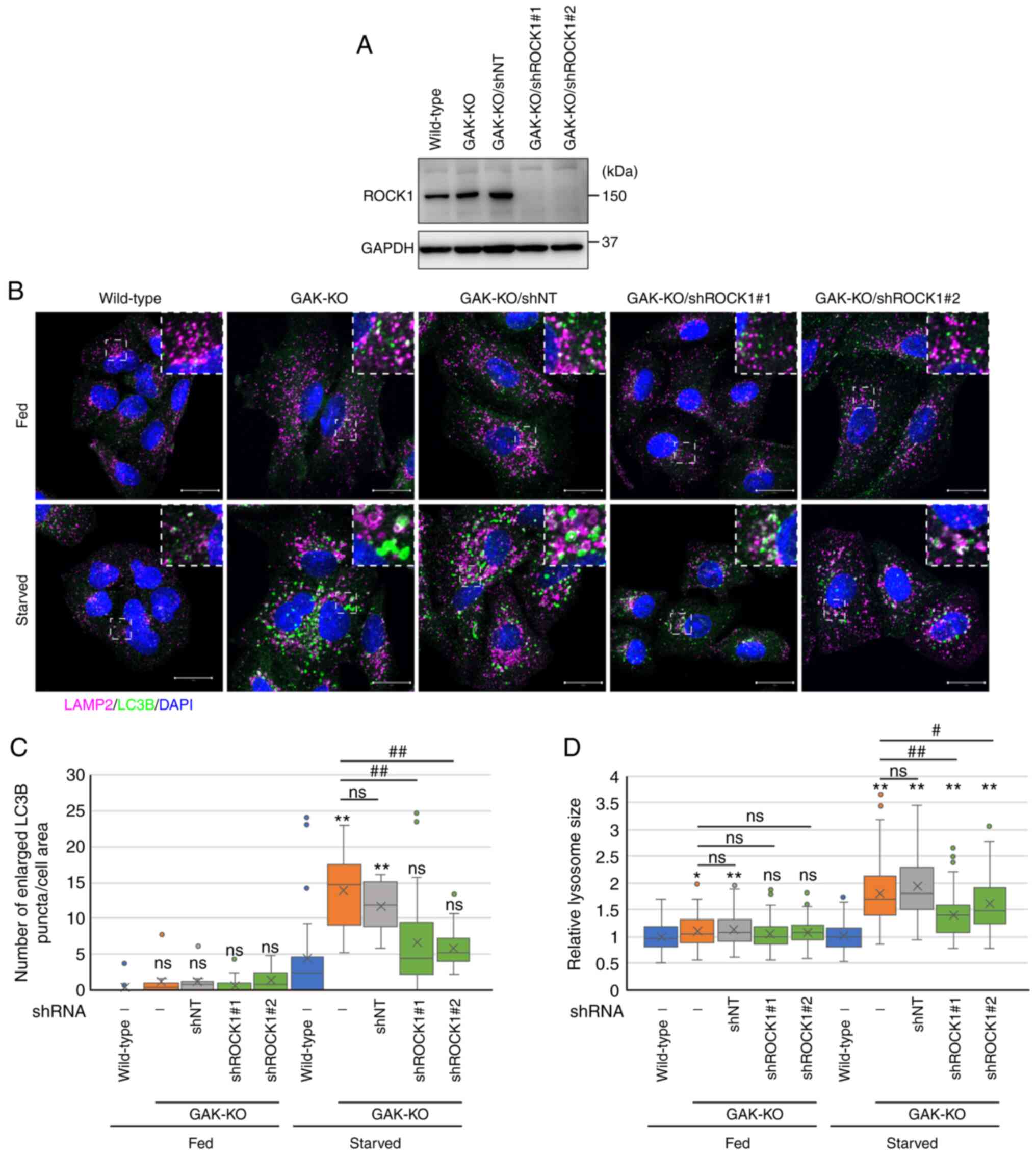 | Figure 9ROCK1 knockdown eliminates the
accumulation of autophagosomes and autolysosomes induced by GAK
disruption. (A) ROCK1 expression in wild-type, GAK-KO, GAK-KO/shNT
and GAK-KO/shROCK1 cells was confirmed by immunoblotting. (B)
Immunofluorescence analysis of LAMP2 (magenta) and LC3B (green) in
wild-type, GAK-KO, GAK-KO/shNT and GAK-KO/shROCK1 cells under
basal-fed or starvation (RPMI-Gln for 8 h) conditions. The dashed
boxed regions are shown at a high magnification (×3) in the inset.
Scale bar, 20 µm. (C) Number of enlarged LC3B-positive
puncta in wild-type, GAK-KO, GAK-KO/shNT and GAK-KO/shROCK1 cells
was quantified using Imaris. Data were analyzed using one-way ANOVA
followed by Tukey's post hoc test. The box extends from the lower
to the upper quartile, the middle line indicates the median, the X
indicates the mean and the whiskers represent the minimum to
maximum values, except for outliers, which are indicated by dots
(wild-type/fed, n=16; GAK-KO/fed, n=11; GAK-KO/shNT/fed, n=9;
GAK-KO/shROCK1#1/fed, n=18; GAK-KO/shROCK1#2/fed, n=22;
wild-type/starved, n=27; GAK-KO/starved, n=13; GAK-KO/shNT/starved,
n=13; GAK-KO/shROCK1#1/starved, n=28; GAK-KO/shROCK1#2/starved,
n=22). **P<0.01 vs. wild-type. ##P<0.01
vs. GAK-KO. (D) Lysosomal size in wild-type, GAK-KO, GAK-KO/shNT
and GAK-KO/shROCK1 cells was quantified using ZEN 2012 software.
Data were analyzed using one-way ANOVA followed by Tukey's post hoc
test. The box extends from the lower to the upper quartile, the
middle line indicates the median, the X indicates the mean and the
whiskers represent the minimum to maximum values, except for
outliers, which are indicated by dots (n=100).
*P<0.05 and **P<0.01 vs. wild-type;
#P<0.05 and ##P<0.01 vs. GAK-KO. ROCK,
Rho-associated protein kinase; GAK, cyclin G-associated kinase; KO,
knockout; sh, short hairpin RNA; NT, non-targeting; LAMP, lysosomal
associated membrane protein; Gln, glutamine; ns, not
significant. |
Discussion
Lysosomes, which are critical for the terminal
degradative stage of autophagy, are highly dynamic organelles that
fuse with autophagosomes to form autolysosomes and are reformed
from autolysosomes via fission (9,12). The present study demonstrated
that GAK may serve an essential role in the autophagy-lysosome
system. GAK disruption led to the stagnation of autophagic flux,
accompanied by a marked accumulation of enlarged autolysosomes and
autophagosomes under starvation conditions. Moreover, the results
indicated that ROCK-mediated actomyosin regulation was involved in
the regulation of lysosomal dynamics by GAK.
GAK has been reported to be involved in transporting
acid hydrolases, such as CTSD, from the trans-Golgi network
to lysosomes via interaction with adaptor protein 1 (31,33), which is a critical step in the
maturation of acid hydrolases and is required to degrade the cargo
carried to lysosomes. The present study did not detect immature
CTSD in wild-type and GAK-KO A549 cells (data not shown), and there
was no marked change in the amount of mature CTSD between wild-type
and GAK-KO cells. Regarding CTSB, both mature and immature forms
were detected in A549 wild-type cells and GAK-KO cells, and it was
shown that GAK disruption suppressed the maturation of CTSB, as
previously reported (31,33).
Therefore, in addition to the increase in intra-lysosomal pH,
suppression of CTSB maturation was observed, and GAK deficiency
altered the intra-lysosomal conditions. However, the degradation
rates of p62/SQSTM1 protein, which is degraded by the
autophagy-lysosomal pathway, did not change between wild-type cells
and GAK-KO cells. Furthermore, no accumulation of undigested
content was observed in the enlarged autolysosomes in GAK-KO cells
in the TEM images. Therefore, the results suggested that the
proteolytic ability of lysosome/autolysosomes was maintained as
much as necessary, and alterations to intra-lysosomal conditions
are unlikely to be considered as the major cause of the stagnation
of autophagic flux.
GAK binds to clathrin and is involved in the control
of clathrin-dependent endocytosis (22,23,29-31). Moreover, it has been reported
that clathrin is involved in ALR control (54); therefore, GAK may control ALR by
binding to clathrin. WASP homolog-associated protein with actin,
membranes and microtubules (WHAMM), which serves as a
nucleation-promoting factor that stimulates Arp2/3-mediated actin
polymerization, is involved in autophagosome biogenesis and ALR
(59-61). In addition, the Src substrate
cortactin (CTTN), which contributes to the organization of the
actin cytoskeleton, is involved in autophagosome-lysosome fusion
(62,63). Since the present study
demonstrated a relationship between GAK and actomyosin regulation,
GAK may have a functional interaction with WHAMM and CTTN; however,
to the best of our knowledge, there have been no reports so far. It
is worth mentioning that yeast actin regulating kinase 1 and p53
regulating kinase 1, which have homology in the amino acid sequence
in the kinase domain with GAK, have been reported to be involved in
actin organization and endocytosis (64-66).
GAK has been reported to form a complex indirectly
with LRRK2 (34), which has been
linked to its role in the autophagy pathway and lysosomal activity
in neurons (37). However, the
role of GAK in the autophagy pathway has not been elucidated.
Mutations in LRRK2 cause inherited PD, and common variants around
LRRK2 are risk factors for sporadic PD (67-70). Genome-wide association studies
have identified an SNP in GAK as a risk factor for sporadic PD
(39-41). The pathophysiology of PD involves
mitochondrial dysfunction and oxidative stress (71,72), but increasing evidence suggests
that lysosomal dysfunction is also involved (73-75). The present study showed that GAK
was involved in the regulation of lysosomal dynamics. Therefore,
considering the role of LRRK2 shown in previous reports (34,37,67-70), it is possible to speculate that
GAK may serve a role in regulating the autophagy-lysosome system in
cooperation with LRRK2 and is involved in the development of
PD.
EGFR tyrosine kinase inhibitors (TKIs), such as
gefitinib and erlotinib, are used for cancer treatment (76). Previous studies have reported
that autophagic flux changes in a cell type- or context-dependent
manner when EGFR-TKIs are administered to cancer cells (44,45,77,78). Furthermore, our previous study
demonstrated that LC3B-II accumulation after gefitinib
administration even in EGFR-KO cells (78). The main target of gefitinib and
erlotinib is EGFR, but several molecules, including GAK, have been
reported as secondary targets of these drugs (79,80). The present study revealed that
GAK inhibition stagnated autophagic flux, which suggested that at
least one of the possible causes of the changes in autophagic flux
induced by gefitinib administration might be its inhibitory effect
on GAK. GAK is overexpressed in various cancer cells, such as
osteosarcoma and hormone-refractory prostate cancer cells (38,81). Since autophagy in cancer cells is
considered to act in a cytoprotective manner (82-84), gefitinib or erlotinib may be
highly effective against cancer cells with high GAK expression.
It is also necessary to consider several additional
factors. First, it has been suggested that GAK controls autophagic
flux through actomyosin regulation, but the underlying molecular
mechanism remains elusive. Autophagy stagnation and accompanying
phenomena were observed not only when GAK is knocked out, but also
when a GAK kinase inhibitor was applied, suggesting that the kinase
activity of GAK might be necessary for this regulation. However,
further analysis is needed to identify the GAK substrates involved
in regulating the autophagy-lysosome system. Furthermore, it is
necessary to clarify the functional relationship between GAK and
ROCK in actomyosin regulation. Second, in the present study, only
in vitro analyses were performed, and analysis using animal
models, such as mice or zebrafish, will be required in the future
to confirm these phenomena in vivo. Third, numerous splicing
variants have been reported in the GAK (85) and there may be splicing variants
that have not yet been identified. In the present study, gRNAs were
designed for two different exons of GAK for the production of
GAK-KO cells. Similar phenomena were observed in both types of
GAK-KO cells. However, these two exons are not commonly used in all
splicing variants; therefore, it cannot be completely ruled out
that several splicing variants of GAK may remain in each type of
GAK-KO cell.
In the present study, GAK, a regulator of
clathrin-mediated endocytosis, was found to be involved in the
regulation of lysosomal dynamics and autophagic flux. Both
endocytosis and autophagy are intracellular membrane vesicle
transport systems that are interconnected and influence each other
(86,87). Therefore, we propose that GAK may
serve as a critical regulator of intracellular membrane traffic and
could be a vital coordinator. Further studies on the molecular
functions of GAK regarding the regulation of actomyosin should
provide important insights into the mechanisms of interconnection
and integration of the intracellular membrane vesicle transport
system.
Supplementary Data
Availability of data and materials
All data generated or analyzed during this study
are available from the corresponding author upon reasonable
request.
Authors' contributions
MH, MM and KM designed the study. MM, HKo, JT, MT,
HH, HKa, NT and MH performed the experiments and analyzed the data.
MM and MH confirm the authenticity of all the raw data. MH, MM, NT
and KM majorly contributed to drafting the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patent consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr A. Abe, Mr S.
Moriya, Ms A. Hirota and Ms Y. Yamada (Department of Biochemistry,
Tokyo Medical University, Japan) for their technical
assistance.
Funding
The present study was supported by the Japan Society for the
Promotion of Science KAKENHI (grant no. 18K06901), the
MEXT-Supported Program of the Strategic Research Foundation at
Private Universities (grant nos. S1411011 and 2014e2018) from the
Ministry of Education, Culture, Sports, Science and Technology of
Japan and the Tokyo Medical University Research Grant (2020).
Abbreviations:
|
ALR
|
autophagic lysosome reformation
|
|
CTSB
|
cathepsin B
|
|
CTSD
|
cathepsin D
|
|
EGFR
|
epidermal growth factor receptor
|
|
GAK
|
cyclin G-associated kinase
|
|
Gln
|
glutamine
|
|
HBSS
|
Hanks' balanced salt solution
|
|
HCQ
|
hydroxychloroquine
|
|
KO
|
knockout
|
|
Baf
|
Bafilomycin A1
|
|
LAMP
|
lysosomal associated membrane
protein
|
|
LRRK2
|
leucine-rich repeat kinase 2
|
|
NSCLC
|
non-small cell lung cancer
|
|
OE
|
overexpression
|
|
PD
|
Parkinson's disease
|
|
ROCK
|
Rho-associated protein kinase
|
|
S6
|
ribosomal protein S6
|
|
S6K
|
ribosomal protein S6 kinase
|
References
|
1
|
Morishita H and Mizushima N: Diverse
cellular roles of autophagy. Annu Rev Cell Dev Biol. 35:453–475.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
King KE, Losier TT and Russell RC:
Regulation of autophagy enzymes by nutrient signaling. Trends
Biochem Sci. 46:687–700. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hara T, Nakamura K, Matsui M, Yamamoto A,
Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I,
Okano H and Mizushima N: Suppression of basal autophagy in neural
cells causes neurodegenerative disease in mice. Nature.
441:885–889. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Alers S, Löffler AS, Wesselborg S and
Stork B: Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy:
Cross talk, shortcuts, and feedbacks. Mol Cell Biol. 32:2–11. 2012.
View Article : Google Scholar :
|
|
5
|
Galluzzi L, Baehrecke EH, Ballabio A, Boya
P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P,
Colombo MI, et al: Molecular definitions of autophagy and related
processes. EMBO J. 36:1811–1836. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mercer TJ, Gubas A and Tooze SA: A
molecular perspective of mammalian autophagosome biogenesis. J Biol
Chem. 293:5386–5395. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nishimura T and Tooze SA: Emerging roles
of ATG proteins and membrane lipids in autophagosome formation.
Cell Discov. 6:322020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Parzych KR and Klionsky DJ: Vacuolar
hydrolysis and efflux: Current knowledge and unanswered questions.
Autophagy. 15:212–227. 2019. View Article : Google Scholar :
|
|
9
|
Yim WW and Mizushima N: Lysosome biology
in autophagy. Cell Discov. 6:62020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kawabata T and Yoshimori T: Autophagosome
biogenesis and human health. Cell Discov. 6:332020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mizushima N and Levine B: Autophagy in
human diseases. N Engl J Med. 383:1564–1576. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ballabio A and Bonifacino JS: Lysosomes as
dynamic regulators of cell and organismal homeostasis. Nat Rev Mol
Cell Biol. 21:101–118. 2020. View Article : Google Scholar
|
|
13
|
Pu J, Guardia CM, Keren-Kaplan T and
Bonifacino JS: Mechanisms and functions of lysosome positioning. J
Cell Sci. 129:4329–4339. 2016.PubMed/NCBI
|
|
14
|
de Araujo ME, Liebscher G, Hess MW and
Huber LA: Lysosomal size matters. Traffic. 21:60–75. 2020.
View Article : Google Scholar :
|
|
15
|
Chen Y and Yu L: Recent progress in
autophagic lysosome reformation. Traffic. 18:358–361. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Korolchuk VI, Saiki S, Lichtenberg M,
Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M,
Menzies FM, et al: Lysosomal positioning coordinates cellular
nutrient responses. Nat Cell Biol. 13:453–460. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dalle Pezze P, Ruf S, Sonntag AG,
Langelaar-Makkinje M, Hall P, Heberle AM, Razquin Navas P, van
Eunen K, Tölle RC, Schwarz JJ, et al: A systems study reveals
concurrent activation of AMPK and mTOR by amino acids. Nat Commun.
7:132542016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu GY and Sabatini DM: mTOR at the nexus
of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol.
21:183–203. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu L, McPhee CK, Zheng L, Mardones GA,
Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al: Termination of
autophagy and reformation of lysosomes regulated by mTOR. Nature.
465:942–946. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kruppa AJ, Kendrick-Jones J and Buss F:
Myosins, actin and autophagy. Traffic. 17:878–890. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kast DJ and Dominguez R: The
cytoskeleton-autophagy connection. Curr Biol. 27:R318–R326. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Greener T, Zhao X, Nojima H, Eisenberg E
and Greene LE: Role of cyclin G-associated kinase in uncoating
clathrin-coated vesicles from non-neuronal cells. J Biol Chem.
275:1365–1370. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee DW, Wu X, Eisenberg E and Greene LE:
Recruitment dynamics of GAK and auxilin to clathrin-coated pits
during endocytosis. J Cell Sci. 119:3502–3512. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kanaoka Y, Kimura SH, Okazaki I, Ikeda M
and Nojima H: GAK: A cyclin G associated kinase contains a
tensin/auxilin-like domain. FEBS Lett. 402:73–80. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shimizu H, Nagamori I, Yabuta N and Nojima
H: GAK, a regulator of clathrin-mediated membrane traffic, also
controls centrosome integrity and chromosome congression. J Cell
Sci. 122:3145–3152. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Naito Y, Shimizu H, Kasama T, Sato J,
Tabara H, Okamoto A, Yabuta N and Nojima H: Cyclin G-associated
kinase regulates protein phosphatase 2A by phosphorylation of its
B'γ subunit. Cell Cycle. 11:604–616. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fukushima K, Wang M, Naito Y, Uchihashi T,
Kato Y, Mukai S, Yabuta N and Nojima H: GAK is phosphorylated by
c-Src and translocated from the centrosome to chromatin at the end
of telophase. Cell Cycle. 16:415–427. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yabuno Y, Uchihashi T, Sasakura T, Shimizu
H, Naito Y, Fukushima K, Ota K, Kogo M, Nojima H and Yabuta N:
Clathrin heavy chain phosphorylated at T606 plays a role in proper
cell division. Cell Cycle. 18:1976–1994. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao X, Greener T, Al-Hasani H, Cushman
SW, Eisenberg E and Greene LE: Expression of auxilin or AP180
inhibits endocytosis by mislocalizing clathrin: Evidence for
formation of nascent pits containing AP1 or AP2 but not clathrin. J
Cell Sci. 114:353–365. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee DW, Zhao X, Zhang F, Eisenberg E and
Greene LE: Depletion of GAK/auxilin 2 inhibits receptor-mediated
endocytosis and recruitment of both clathrin and clathrin adaptors.
J Cell Sci. 118:4311–4321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang CX, Engqvist-Goldstein AE, Carreno
S, Owen DJ, Smythe E and Drubin DG: Multiple roles for cyclin
G-associated kinase in clathrin-mediated sorting events. Traffic.
6:1103–1113. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang L, Gjoerup O and Roberts TM: The
serine/threonine kinase cyclin G-associated kinase regulates
epidermal growth factor receptor signaling. Proc Natl Acad Sci USA.
101:10296–10301. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kametaka S, Moriyama K, Burgos PV,
Eisenberg E, Greene LE, Mattera R and Bonifacino JS: Canonical
interaction of cyclin G associated kinase with adaptor protein 1
regulates lysosomal enzyme sorting. Mol Biol Cell. 18:2991–3001.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Beilina A, Rudenko IN, Kaganovich A,
Civiero L, Chau H, Kalia SK, Kalia LV, Lobbestael E, Chia R, Ndukwe
K, et al: Unbiased screen for interactors of leucine-rich repeat
kinase 2 supports a common pathway for sporadic and familial
Parkinson disease. Proc Natl Acad Sci USA. 111:2626–2631. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Plowey ED, Cherra SJ III, Liu YJ and Chu
CT: Role of autophagy in G2019S-LRRK2 -associated neurite
shortening in differentiated SH-SY5Y cells. J Neurochem.
105:1048–1056. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gómez-Suaga P, Luzón-Toro B, Churamani D,
Zhang L, Bloor-Young D, Patel S, Woodman PG, Churchill GC and
Hilfiker S: Leucine-rich repeat kinase 2 regulates autophagy
through a calcium-dependent pathway involving NAADP. Hum Mol Genet.
21:511–525. 2012. View Article : Google Scholar :
|
|
37
|
Madureira M, Connor-Robson N and
Wade-Martins R: LRRK2: Autophagy and lysosomal activity. Front
Neurosci. 14:4982020. View Article : Google Scholar
|
|
38
|
Susa M, Choy E, Liu X, Schwab J, Hornicek
FJ, Mankin H and Duan Z: Cyclin G-associated kinase is necessary
for osteosarcoma cell proliferation and receptor trafficking. Mol
Cancer Ther. 9:3342–3350. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rhodes SL, Sinsheimer JS, Bordelon Y,
Bronstein JM and Ritz B: Replication of GWAS associations for GAK
and MAPT in Parkinson's disease. Ann Hum Genet. 75:195–200.
2011.
|
|
40
|
Dumitriu A, Pacheco CD, Wilk JB,
Strathearn KE, Latourelle JC, Goldwurm S, Pezzoli G, Rochet JC,
Lindquist S and Myers RH: Cyclin-G-associated kinase modifies
α-synuclein expression levels and toxicity in Parkinson's disease:
Results from the GenePD Study. Hum Mol Genet. 20:1478–1487. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ma ZG, He F and Xu J: Quantitative
assessment of the association between GAK rs1564282 C/T
polymorphism and the risk of Parkinson's disease. J Clin Neurosci.
22:1077–1080. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cong L, Ran FA, Cox D, Lin S, Barretto R,
Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA and Zhang F:
Multiplex genome engineering using CRISPR/Cas systems. Science.
339:819–823. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ran FA, Hsu PD, Wright J, Agarwala V,
Scott DA and Zhang F: Genome engineering using the CRISPR-Cas9
system. Nat Protoc. 8:2281–2308. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tanaka H, Hino H, Moriya S, Kazama H,
Miyazaki M, Takano N, Hiramoto M, Tsukahara K and Miyazawa K:
Comparison of autophagy inducibility in various tyrosine kinase
inhibitors and their enhanced cytotoxicity via inhibition of
autophagy in cancer cells in combined treatment with azithromycin.
Biochem Biophys Rep. 22:1007502020.PubMed/NCBI
|
|
45
|
Kaizuka T, Morishita H, Hama Y, Tsukamoto
S, Matsui T, Toyota Y, Kodama A, Ishihara T, Mizushima T and
Mizushima N: An autophagic flux probe that releases an internal
control. Mol Cell. 64:835–849. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Spangle JM, Ghosh-Choudhury N and Munger
K: Activation of cap-dependent translation by mucosal human
papillomavirus E6 proteins is dependent on the integrity of the
LXXLL binding motif. J Virol. 86:7466–7472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Stewart SA, Dykxhoorn DM, Palliser D,
Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et
al: Lentivirus-delivered stable gene silencing by RNAi in primary
cells. RNA. 9:493–501. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yokota A, Hiramoto M, Hino H, Tokuhisa M,
Miyazaki M, Kazama H, Takano N and Miyazawa K: Sequestosome 1 (p62)
accumulation in breast cancer cells suppresses progesterone
receptor expression via argonaute 2. Biochem Biophys Res Commun.
531:256–263. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lamb CA, Joachim J and Tooze SA:
Quantifying autophagic structures in mammalian cells using confocal
microscopy. Methods Enzymol. 587:21–42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
51
|
Poole B and Ohkuma S: Effect of weak bases
on the intralysosomal pH in mouse peritoneal macrophages. J Cell
Biol. 90:665–669. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Amaravadi RK, Yu D, Lum JJ, Bui T,
Christophorou MA, Evan GI, Thomas-Tikhonenko A and Thompson CB:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Maclean KH, Dorsey FC, Cleveland JL and
Kastan MB: Targeting lysosomal degradation induces p53-dependent
cell death and prevents cancer in mouse models of lymphomagenesis.
J Clin Invest. 118:79–88. 2008. View Article : Google Scholar
|
|
54
|
Rong Y, Liu M, Ma L, Du W, Zhang H, Tian
Y, Cao Z, Li Y, Ren H, Zhang C, et al: Clathrin and
phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome
reformation. Nat Cell Biol. 14:924–934. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chang J, Lee S and Blackstone C: Spastic
paraplegia proteins spastizin and spatacsin mediate autophagic
lysosome reformation. J Clin Invest. 124:5249–5262. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
McGrath MJ, Eramo MJ, Gurung R, Sriratana
A, Gehrig SM, Lynch GS, Lourdes SR, Koentgen F, Feeney SJ, Lazarou
M, et al: Defective lysosome reformation during autophagy causes
skeletal muscle disease. J Clin Invest. 131:e1351242021. View Article : Google Scholar :
|
|
57
|
Uehata M, Ishizaki T, Satoh H, Ono T,
Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M
and Narumiya S: Calcium sensitization of smooth muscle mediated by
a Rho-associated protein kinase in hypertension. Nature.
389:990–994. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
58
|
Julian L and Olson MF: Rho-associated
coiled-coil containing kinases (ROCK): Structure, regulation, and
functions. Small GTPases. 5:e298462014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kast DJ, Zajac AL, Holzbaur EL, Ostap EM
and Dominguez R: WHAMM Directs the Arp2/3 Complex to the ER for
autophagosome biogenesis through an Actin comet tail mechanism.
Curr Biol. 25:1791–1797. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Mathiowetz AJ, Baple E, Russo AJ, Coulter
AM, Carrano E, Brown JD, Jinks RN, Crosby AH and Campellone KG: An
Amish founder mutation disrupts a PI(3)P-WHAMM-Arp2/3
complex-driven autophagosomal remodeling pathway. Mol Biol Cell.
28:2492–2507. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Dai A, Yu L and Wang HW: WHAMM initiates
autolysosome tubulation by promoting actin polymerization on
autolysosomes. Nat Commun. 10:36992019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Nakamura S, Hasegawa J and Yoshimori T:
Regulation of lysosomal phosphoinositide balance by INPP5E is
essential for autophagosome-lysosome fusion. Autophagy.
12:2500–2501. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nakamura S and Yoshimori T: New insights
into autophagosome-lysosome fusion. J Cell Sci. 130:1209–1216.
2017.PubMed/NCBI
|
|
64
|
Henry KR, D'Hondt K, Chang JS, Nix DA,
Cope MJ, Chan CS, Drubin DG and Lemmon SK: The actin-regulating
kinase Prk1p negatively regulates Scd5p, a suppressor of clathrin
deficiency, in actin organization and endocytosis. Curr Biol.
13:1564–1569. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sekiya-Kawasaki M, Groen AC, Cope MJ,
Kaksonen M, Watson HA, Zhang C, Shokat KM, Wendland B, McDonald KL,
McCaffery JM and Drubin DG: Dynamic phosphoregulation of the
cortical actin cytoskeleton and endocytic machinery revealed by
real-time chemical genetic analysis. J Cell Biol. 162:765–772.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Toshima J, Toshima JY, Martin AC and
Drubin DG: Phosphoregulation of Arp2/3-dependent actin assembly
during receptor-mediated endocytosis. Nat Cell Biol. 7:246–254.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Paisán-Ruíz C, Jain S, Evans EW, Gilks WP,
Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM,
Khan N, et al: Cloning of the gene containing mutations that cause
PARK8-linked Parkinson's disease. Neuron. 44:595–600. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zimprich A, Biskup S, Leitner P, Lichtner
P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB,
et al: Mutations in LRRK2 cause autosomal-dominant parkinsonism
with pleomorphic pathology. Neuron. 44:601–607. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zimprich A, Müller-Myhsok B, Farrer M,
Leitner P, Sharma M, Hulihan M, Lockhart P, Strongosky A, Kachergus
J, Calne DB, et al: The PARK8 locus in autosomal dominant
parkinsonism: Confirmation of linkage and further delineation of
the disease-containing interval. Am J Hum Genet. 74:11–19. 2004.
View Article : Google Scholar
|
|
70
|
Usmani A, Shavarebi F and Hiniker A: The
Cell Biology of LRRK2 in Parkinson's disease. Mol Cell Biol.
41:e00660–20. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Clark EH, Vázquez de la Torre A, Hoshikawa
T and Briston T: Targeting mitophagy in Parkinson's disease. J Biol
Chem. 296:1002092020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Malpartida AB, Williamson M, Narendra DP,
Wade-Martins R and Ryan BJ: Mitochondrial dysfunction and mitophagy
in Parkinson's disease: From mechanism to therapy. Trends Biochem
Sci. 46:329–343. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Plotegher N and Duchen MR: Crosstalk
between Lysosomes and Mitochondria in Parkinson's disease. Front
Cell Dev Biol. 5:1102017. View Article : Google Scholar
|
|
74
|
Nguyen M, Wong YC, Ysselstein D, Severino
A and Krainc D: Synaptic, Mitochondrial, and Lysosomal dysfunction
in Parkinson's disease. Trends Neurosci. 42:140–149. 2019.
View Article : Google Scholar
|
|
75
|
Vidyadhara DJ, Lee JE and Chandra SS: Role
of the endolysosomal system in Parkinson's disease. J Neurochem.
150:487–506. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Oxnard GR, Janjigian YY, Arcila ME, Sima
CS, Kass SL, Riely GJ, Pao W, Kris MG, Ladanyi M, Azzoli CG and
Miller VA: Maintained sensitivity to EGFR tyrosine kinase
inhibitors in EGFR-mutant lung cancer recurring after adjuvant
erlotinib or gefitinib. Clin Cancer Res. 17:6322–6328. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Han W, Pan H, Chen Y, Sun J, Wang Y, Li J,
Ge W, Feng L, Lin X, Wang X, et al: EGFR tyrosine kinase inhibitors
activate autophagy as a cytoprotective response in human lung
cancer cells. PLoS One. 6:e186912011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Sugita S, Ito K, Yamashiro Y, Moriya S,
Che XF, Yokoyama T, Hiramoto M and Miyazawa K: EGFR-independent
autophagy induction with gefitinib and enhancement of its cytotoxic
effect by targeting autophagy with clarithromycin in non-small cell
lung cancer cells. Biochem Biophys Res Commun. 461:28–34. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Brehmer D, Greff Z, Godl K, Blencke S,
Kurtenbach A, Weber M, Müller S, Klebl B, Cotton M, Kéri G, et al:
Cellular targets of gefitinib. Cancer Res. 65:379–382.
2005.PubMed/NCBI
|
|
80
|
Yamamoto N, Honma M and Suzuki H:
Off-target serine/threonine kinase 10 inhibition by erlotinib
enhances lymphocytic activity leading to severe skin disorders. Mol
Pharmacol. 80:466–475. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ray MR, Wafa LA, Cheng H, Snoek R, Fazli
L, Gleave M and Rennie PS: Cyclin G-associated kinase: A novel
androgen receptor-interacting transcriptional coactivator that is
overexpressed in hormone refractory prostate cancer. Int J Cancer.
118:1108–1119. 2006. View Article : Google Scholar
|
|
82
|
Sooro MA, Zhang N and Zhang P: Targeting
EGFR-mediated autophagy as a potential strategy for cancer therapy.
Int J Cancer. 143:2116–2125. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wu M and Zhang P: EGFR-mediated autophagy
in tumourigenesis and therapeutic resistance. Cancer Lett.
469:207–216. 2020. View Article : Google Scholar
|
|
84
|
Rehman SK, Haynes J, Collignon E, Brown
KR, Wang Y, Nixon AM, Bruce JP, Wintersinger JA, Singh Mer A, Lo
EB, et al: Colorectal cancer cells enter a Diapause-like DTP state
to survive chemotherapy. Cell. 184:226–242.e221. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Nagle MW, Latourelle JC, Labadorf A,
Dumitriu A, Hadzi TC, Beach TG and Myers RH: The 4p163 Parkinson
disease risk locus is associated with GAK expression and genes
involved with the synaptic vesicle membrane. PLoS One.
11:e01609252016. View Article : Google Scholar
|
|
86
|
Tumbarello DA, Kendrick-Jones J and Buss
F: Myosin VI and its cargo adaptors-linking endocytosis and
autophagy. J Cell Sci. 126:2561–2570. 2013.PubMed/NCBI
|
|
87
|
Bilanges B, Posor Y and Vanhaesebroeck B:
PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev
Mol Cell Biol. 20:515–534. 2019. View Article : Google Scholar : PubMed/NCBI
|














