Introduction
Our previous studies have demonstrated the
detrimental effect of iron on osteoarthritis (OA) chondrocytes
(1), and that arresting iron
influx or decreasing iron concentration using iron chelators may
inhibit iron deposition-induced OA progression (2). Iron chelators are drugs that may be
beneficial for patients with iron overload (3). However, considering the severe side
effects of iron chelators, such as renal damage and neurological
dysfunction, new strategies for inhibiting the iron influx into
cells must be investigated (4).
As the most abundant trace metal in the human body, iron is crucial
for the majority of cellular metabolic processes (5). Iron homeostasis is delicately
regulated in humans, and iron overload may damage cells by
promoting the production of reactive oxygen species (ROS) (6). Iron overload can generate highly
toxic hydroxyl radicals and result in ferroptosis (7). As the human body lacks effective
ways for excreting excess iron, iron overload is common among
elderly individuals and has been reported to be implicated in
several diseases or pathological conditions (8). OA is one of the most common
complications in a variety of diseases characterized by abnormal
intracartilaginous hemorrhage or iron deposition, such as
hereditary hemochromatosis, hemophilic arthropathy and rheumatoid
arthritis, in which cartilage degeneration and OA progression are
common. Moreover, iron deposition has also been observed in
traumatic and age-related OA (9). Iron is considered to be the main
cause of continual degeneration of the knee joint in traumatic
arthritis (10). Iron has also
been reported to induce crystal deposition in the supersaturated
synovial fluid through the process of nucleation and cartilage
damage in OA (11). Moreover,
recent findings suggested that cellular iron overload-induced
oxidative stress and mitochondrial dysfunction play pivotal roles
in OA progression (12,13). Oxidative stress and mitochondrial
dysfunction were also reported to play important roles during OA
development (14). Iron
metabolism has been demonstrated to be an important contributor to
normal mitochondrial function. By converting from Fe2+
to Fe3+ and vice versa, iron can participate in the
mitochondrial oxidative respiratory chain and exchange a single
electron with other substrates. Excess iron can lead to increased
ROS production and destroy mitochondrial structure; mitochondrial
dysfunction, in turn, can produce excess ROS and result in MMP
activation and decreased collagen synthesis (15).
Iron in the diet is taken up by enterocytes through
the divalent metal transporter 1 (DMT1) and exported via
ferroportin to the general circulation. Then, plasma iron is
delivered into tissues or organs via transferrin (TF) receptor 1
(TfR1)-mediated endocytosis after binding to TF in most types of
cells (16). As Ca2+
and Fe2+ have equally positive charges, it is possible
that Ca2+ may affect Fe2+ influx. It was
recently demonstrated that the L-type and T-type calcium channels
are involved in iron uptake into the heart under conditions of iron
overload (17). Sripetchwandee
et al (18) reported that
blockade of the mitochondrial calcium uniporter prevents cardiac
mitochondrial dysfunction caused by iron overload. Kumfu et
al (19) found that
combining an iron chelator and a T-type calcium channel blocker
exhibited greater cardioprotective efficacy compared with iron
chelator alone in an iron overload-induced thalassemia mouse model.
Moreover, Zhang et al (20) reported that calcium channel
blockers (CCBs) ameliorated iron overload-associated hepatic
fibrosis by altering iron transport, and indicated that CCBs are
potential therapeutic agents for iron overload-induced diseases.
Calcium was reported to modulate receptor-mediated endocytosis and,
as TfR1 modulates iron influx through this pathway, it was further
demonstrated that calcium could antagonize iron-TF-TfR1 complex
internalization and, thus, modulate iron uptake (21).
The present study was undertaken to investigate the
effect of the calcium chelator BAPTA acetoxymethyl ester (BAPTA-AM)
on iron overload-induced chondrocyte apoptosis and MMP expression.
Primary chondrocytes were treated with various concentrations of
ferric ammonium citrate (FAC) to mimic iron overload in
vitro, and the effects of BAPTA-AM on iron influx into
chondrocytes, intracellular iron concentration and ROS production
were investigated. Moreover, mitochondrial depolarization and
mitochondrial morphology were examined to determine the
mitochondrial function using immunofluorescence and flow cytometry
assays. The aim was to determine whether calcium chelators may be
potential therapeutic agents in treating iron metabolism diseases
and iron overload-induced OA progression.
Materials and methods
Cell isolation and culture
A total of 30 male C57/BL6 mice (aged 5 days and
weighing <5 g) were used in the present study. The mice were
anesthetized by intraperitoneal injection of 2% pentobarbital
sodium (35 mg/kg body weight) and sacrificed via cervical
dislocation. All animal protocols were approved by the
Institutional Animal Care of the Shandong Provincial Hospital
Affiliated to Shandong First Medical University (no. 2020-526).
Briefly, cartilage was obtained from bilateral knee joints and
minced into pieces. The cartilage pieces were then washed with cold
PBS, followed by the addition of 0.25% trypsin-EDTA for 30 min at
37°C. After washing with PBS, samples were digested with 0.25%
collagenase solution at 37°C for 7 h. After collection and
centrifugation at 100 × g for 10 min at 37°C, chondrocytes were
resuspended in DMEM/F12 (HyClone; Cytiva) supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.) and cultured in a
humidified atmosphere of 5% CO2 at 37°C. Chondrocytes at
passages 1 or 2 were used in the following experiments.
Cell Counting Kit-8 (CCK-8) assay
CCK-8 was obtained from Wuhan Boster Biological
Technology, Ltd. and used to detect cell viability according to the
manufacturer's instructions. Briefly, cells were collected and
resuspended in DMEM/F12 supplemented with 10% FBS at a density of
5×104 cells/ml, and 200 µl medium containing
1×104 cells were added into each well of a 96-well
plate. Increasing concentrations of FAC (1-200 µm) were then
added and cultured for different time periods (0-5 days). The
culture medium was changed every other day. CCK-8 solution (10
µl) was added into each well on days 0, 1, 3 and 5 and
incubated at 37°C in the dark for 1 h. The absorbance at 450 nm was
recorded using spectral IMAX190 absorbance microplate reader
(Molecular Devices, LLC). Optical density values at 450 nm were
recorded to evaluate cell viability.
Western blot analysis
Chondrocytes were seeded in 6-well plates at a
density of 1×105 cells per well. After being subjected
to various treatments, chondrocytes were collected and lysed with
100 µl RIPA lysis buffer (Wuhan Boster Biological
Technology, Ltd.) on ice for 1 h. The lysates were then collected
and centrifuged at 300 × g for 20 min at 4°C, the supernatant was
collected and the protein concentration was detected using the BCA
assay method. Equal amounts of protein (25 µg/lane) were
electrophoresed using 12% SDS-PAGE and transferred to PVDF
membranes (MilliporeSigma). The PVDF membranes were then blocked
with 5% skimmed milk for 1 h at room temperature and incubated with
targeted primary antibodies against MMP3 (1:1,000; cat. no.
17873-1-AP; ProteinTech Group, Inc.), MMP13 (1:1,000; cat. no.
ab39012; Abcam), mitochondrial fission 1 protein (FIS1; 1:1,000;
cat. no. 10956-1-AP; ProteinTech Group, Inc.), dynamin-related
protein 1 (DRP1; 1:1,000; cat. no. 8570; Cell Signaling Technology,
Inc.), mitochondrial fission factor (MFF; 1:1,000; cat. no. 84580;
Cell Signaling Technology, Inc.), BAX (1:1,000; cat. no.
50599-2-Ig; ProteinTech Group, Inc.), cytochrome c (1:1,000;
cat. no. ab13575; Abcam) and β-actin (1:500; cat. no. BM0627; Wuhan
Boster Biological Technology, Ltd.) at 4°C overnight. After washing
with Tris-buffered saline with Tween-20 [50 mM Tris (pH 7.6), 150
mm Nacl and 0.1% Tween-20] three times, the membranes were
incubated with a horseradish peroxidase-conjugated secondary
antibody (1:2,000; cat. no. BA1055; Wuhan Boster Biological
Technology, Ltd.) at room temperature for 1 h. The protein bands
were visualized with enhanced chemiluminescence reagent (Wuhan
Boster Biological Technology, Ltd.) and images were captured. The
density of each band was quantified by ImageJ software (version
1.8.0; National Institutes of Health).
Reverse transcription-quantitative
(RT-q)PCR analysis
Chondrocytes were seeded in 12-well plates at a
density of 5×104 cells per well and treated with 100
µm FAC (MilliporeSigma) for 24 h. Chondrocytes were then
treated with TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) and total RNA was extracted using the Toyobo
Total RNA Extraction kit (Toyobo Life Science) following the
manufacturer's instructions. Complementary DNA (cDNA) was
synthesized from RNA using the First Strand cDNA Synthesis kit
(Toyobo Life Science) and amplified using SYBR Green Real-time PCR
Master Mix (Toyobo Life Science) under the following cycling
conditions: 30 sec of polymerase activation at 95°C, followed by 40
cycles at 95°C for 5 sec and 60°C for 30 sec. Relative expression
levels were calculated using the 2−ΔΔcq method (22). GAPDH was used as an internal
control. Each cDNA sample was assayed in triplicate. The sequences
of the primers for the genes of interest are listed as follows: A
disintegrin and metalloproteinase with thrombospondin motifs 5
(ADAMTS5): Forward, 5′-TAT GAC AAG TGC GGA GTA TG-3′ and reverse,
5′-TTC AGG GCT AAA TAG GCA GT-3′; MMP3: Forward, 5′-ATG CCC ACT TTG
ATG ATG ATG AAC-3′ and reverse, 5′-CCA CGC CTG AAG GAA GAG ATG-3′;
MMP13: Forward, 5′-GCT GGA CTC CCT GTT G-3′ and reverse, 5′-TCG GAG
CCT GTC AAC T-3′; and GAPDH: Forward, 5′-CTC CCA CTC TTC CAC CTT
CG-3′ and reverse, 5′-TTG CTG TAG CCG TAT TCA TT-3′.
Immunofluorescence staining
Chondrocytes at passage 1 or 2 were seeded in
24-well plates at a density of 1×104 cells per well.
Following treatment with 100 µm FAC (MilliporeSigma) for 24
h, cells were washed with serum-free DMEM/F12 (HyClone; Cytiva) and
incubated with diluted Mito-Tracker Red solution (1:1,000;
Invitrogen; Thermo Fisher Scientific, Inc.) for 30 min at 37°C in
the dark. Cells were then washed with PBS three times and fixed
with 4% paraformaldehyde for 20 min at room temperature. Cells were
then washed with PBS three times and treated with 0.1% Triton X-100
for 15 min. After blocking with 10% FBS (Wuhan Boster Biological
Technology, Ltd.) for 30 min at room temperature, the cells were
then incubated with mixed rabbit polyclonal BAX primary antibody
(1:1,000; cat. no. 50599-2-Ig; ProteinTech Group, Inc.) and mouse
monoclonal cytochrome c antibody (1:1,000; cat. no. ab13575;
Abcam) at 4°C overnight. Following rinsing with PBS three times,
mixed goat anti-rabbit FITC-conjugated secondary antibody (1:200;
cat. no. BA1105; Wuhan Boster Biological Technology, Ltd.) and goat
anti-mouse Dylight 405 secondary antibody (1:400; Beyotime
Institute of Biotechnology) was added and incubated for 1 h at room
temperature in the dark. Finally, cells were washed with PBS and
analyzed at a magnification of ×200 under a fluorescence microscope
(EVOS™ FL Auto; Thermo Fisher Scientific, Inc.).
Measurement of intracellular iron
levels
Briefly, primary chondrocytes were treated with 100
µm FAC, with or without 10 µm BAPTA-AM (Selleck
Chemicals), for 6 h. Then, cells were loaded with 0.5 µmol/l
calcein-AM for 30 min at 37°C. Chondrocytes were then washed with
PBS three times. Free calcein (non-metal-bound) was measured for
fluorescence (excitation: 488 nm; emission: 517 nm) using a
fluorescence microscope (EVOS™ FL Auto; Thermo Fisher Scientific,
Inc.) at a magnification of ×200. To quantify the fluorescence
values, four separate fields monitored were randomly selected and
the mean fluorescence signal was processed with Image Pro Plus
software (version 6.0; National Institutes of Health).
Evaluation of intracellular ROS
Chondrocytes were seeded in 12-well plates at a
density of 1×104 cells per well and treated with 100
µm FAC, with or without 10 µm BAPTA-AM, for 24 h.
After treatment, the intracellular ROS level was determined using
the Reactive Oxygen Species Assay kit (cat. no. S0033; Beyotime
Institute of Biotechnology) according to manufacturer's
instructions. Briefly, chondrocytes were washed with serum-free
DMEM/F12 (HyClone; Cytiva) three times, then 10 µm
dichloro-dihydro-fluorescein diacetate (DCFH-DA) was added to the
culture medium and incubated in the dark for 20 min. Chondrocytes
were then washed with DMEM/F12 (HyClone; Cytiva) and examined at a
magnification of ×100 under a fluorescence microscope (EVOS™ FL
Auto; Thermo Fisher Scientific, Inc.) at an excitation wavelength
of 488 nm and an emission wavelength of 525 nm.
Measurement of mitochondrial membrane
potential
Cells were seeded in 12-well plates at a density of
1×104 per well and treated with 100 µm FAC, with
or without 10 µm BAPTA-AM, for 24 h. After washing with
DMEM/F12 three times, chondrocytes were incubated with JC-1
staining solution (Beyotime Institute of Biotechnology) for 30 min
at room temperature. After incubation, the culture medium was
changed and the chondrocytes were washed with washing buffer. The
fluorescence was examined using a fluorescence microscope.
Aggregated JC-1 in mitochondria with high mitochondrial membrane
potential levels produced red fluorescence, whereas when
mitochondrial membrane potential decreased, JC-1 was found in its
monomer form and produced green fluorescence.
To quantify mitochondrial membrane potential in
chondrocytes, cells were incubated with JC-1 staining solution as
described above. After incubation, cells were re-suspended in 500
ml DMEM/F12 and the fluorescence intensity was recorded with a
FACSCalibur flow cytometer (BD Biosciences).
Mitochondrial-specific fluorescence
staining
Mito-Tracker Green (Beyotime Institute of
Biotechnology) was used to evaluate the morphological changes of
mitochondria. Mito-Tracker Green is an mitochondrial membrane
potential-independent mitochondrial staining reagent. Briefly,
chondrocytes were treated with FAC (100 µm), with or without
10 µm BAPTA-AM, for 24 h; subsequently, cells were washed
twice with FBS-free DMEM/F12 and incubated with diluted
Mito-Tracker Green solution (1:1,000) for 30 min at 37°C in the
dark. The shapes of the mitochondria were imaged at a magnification
of ×400 under a fluorescence microscope (EVOS™ FL Auto; Thermo
Fisher Scientific, Inc.).
Evaluation of apoptosis
An Annexin V-FITC/PI Apoptosis Detection kit (cat.
no. C1063; Beyotime Institute of Biotechnology) was used to detect
the cell apoptosis rate according to the manufacturer's
instructions. Chondrocytes were seeded in 6-well plates at a
density of 1×105 per well and treated with 100 µm
FAC, with or without 10 µm BAPTA-AM, for 24 h. After the
aforementioned treatment, cells were washed with PBS and then
stained with Annexin V-FITC/PI for 20 min in the dark. A
FACSCalibur flow cytometer (BD Biosciences) was used to analyze the
results. Annexin V+/PI− and Annexin
V+/PI+ cells were considered as early and
late apoptotic cells, respectively.
Statistical analysis
The results are presented as mean ± SD. All
experiments were repeated three times independently. Unpaired
Student's t-test was used to evaluate differences between two
groups. Differences among multiple groups were determined using
one-way analysis of variance followed by Tukey's post hoc test.
P<0.05 was considered to indicate statistically significant
differences.
Results
Iron overload promotes chondrocyte
apoptosis and MMP expression
First, primary chondrocytes were treated with
various concentrations of FAC to mimic iron overload in
vitro, and then CCK-8 assay was performed to verify the
cytotoxic effects of iron on chondrocytes. As shown in Fig. 1A, FAC decreased chondrocyte
viability in a dose-dependent manner. FAC at a concentration of 1
µm slightly enhanced chondrocyte proliferation, while FAC at
concentrations >50 µm significantly decreased chondrocyte
viability. Western blotting demonstrated that FAC promoted the
expression of the cell apoptosis marker cleaved caspase-3 in a
dose-dependent manner, and FAC at 100 µm was associated with
significantly elevated cleaved caspase-3 expression. Next, the
expression of the OA-related markers, MMP3 and MMP13, was examined
using western blot and RT-qPCR analyses. As shown in Fig. 1C and D, FAC promoted MMP3 and
MMP13 protein expression in a dose-dependent manner, with the
protein levels peaking at 100 µm FAC. Similar results were
obtained using RT-qPCR analysis, with 100 µm FAC
significantly promoting ADAMTS5, MMP3 and MMP13 mRNA expression
(Fig. 1B).
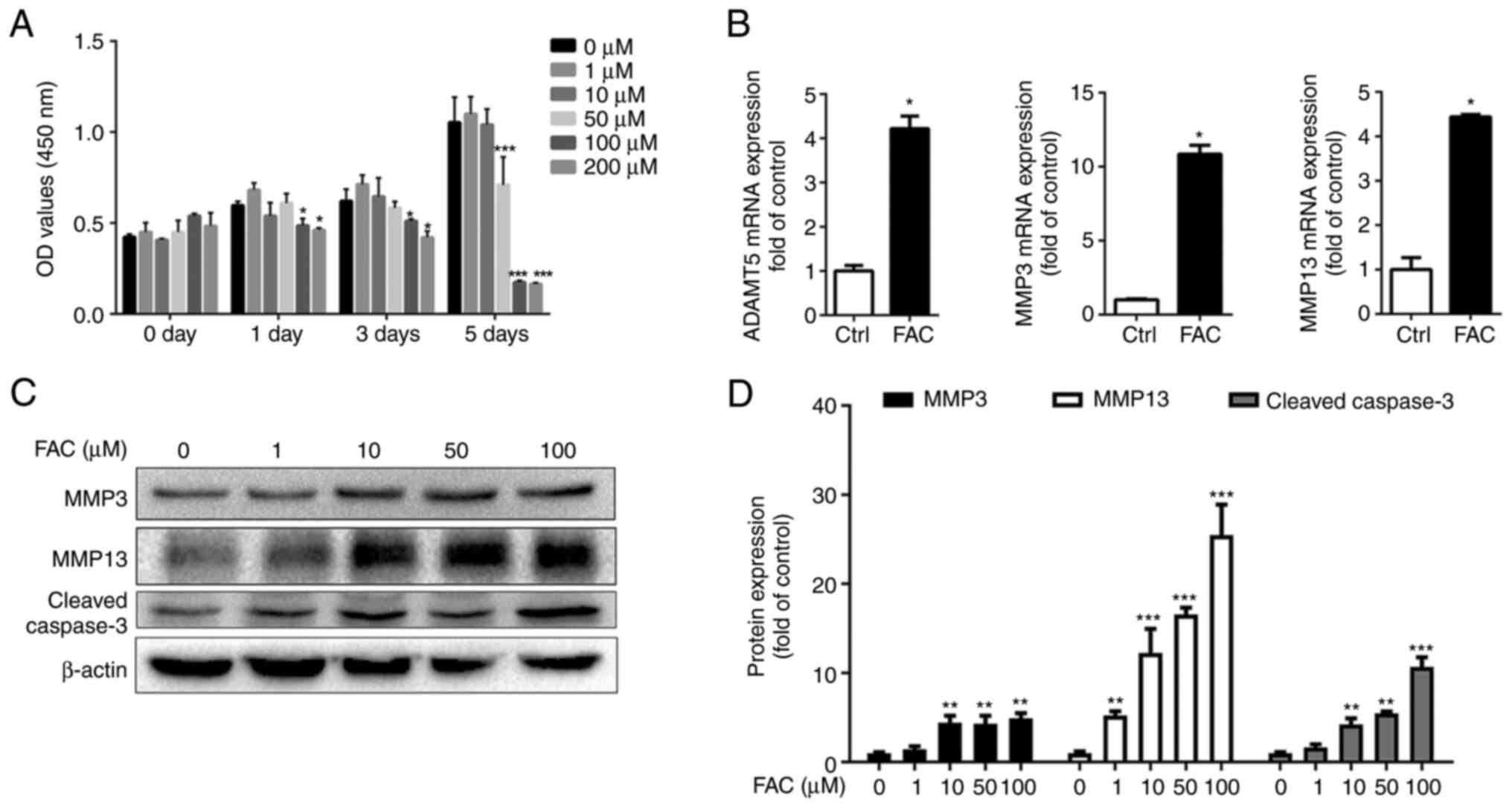 | Figure 1Iron overload promotes chondrocyte
apoptosis and osteoarthritis-related marker expression. (A)
Chondrocytes were treated with various concentrations of FAC (0, 1,
10, 50, 100 and 200 µm) for different time periods (0, 1, 3
and 5 days). Cell viability was determined using the Cell Counting
Kit-8 assay. Data are presented as mean ± SD.
*P<0.05; ***P<0.001 vs. 0 µM
FAC. (B) Chondrocytes were treated with 100 µm FAC for 24 h
and reverse transcription-quantitative PCR analysis was conducted
to examine ADAMTS5, MMP3 and MMP13 gene expression. Data are
presented as mean ± SD. *P<0.05 vs. Ctrl. (C and D)
Chondrocytes were treated with increasing concentrations of FAC for
24 h and western blotting was conducted to examine MMP3, MMP13 and
cleaved caspase-3 protein expression. The band density of MMP3,
MMP13 and cleaved caspase-3 was quantified and normalized to
control. Data are presented as mean ± SD. **P<0.01;
***P<0.001 vs. 0 µM FAC. ADAMTS5, A
disintegrin and metalloproteinase with thrombospondin motifs 5;
FAC, ferric ammonium citrate; Ctrl, control group; OD, optical
density. |
Iron overload impairs mitochondrial
dynamics in chondrocytes
Mitochondrial function plays an important role in OA
progression (15). To further
investigate the mechanisms underlying iron overload-induced
accelerated progression of OA, chondrocytes at passage 1 or 2 were
stimulated with FAC to mimic iron overload in vitro.
Immunofluorescence staining was then performed to examine the
localization of cytochrome c and BAX proteins. It was
observed that FAC promoted mitochondrial BAX expression and
translocation of cytochrome c from the mitochondria to the
cytoplasm, indicating that mitochondrial structure was destroyed
after FAC treatment (Fig. 2A).
Mitochondrial fission and fusion play important roles in
maintaining the function and morphology of mitochondria (23). The results of the present study
demonstrated that the expression of mitochondrial fission proteins
(DRP1, MFF and FIS1) was upregulated following treatment with 100
µm FAC in a time-dependent manner (Fig. 2B and C). In addition, the protein
expression of BAX and cytochrome c was upregulated following
treatment with 100 µm FAC. These results indicated that FAC
promoted mitochondrial fission and caused destruction of
mitochondrial morphology and leakage of cytochrome c, a
classic caspase-dependent apoptosis inducer in mitochondria,
further promoting mitochondria-dependent apoptosis.
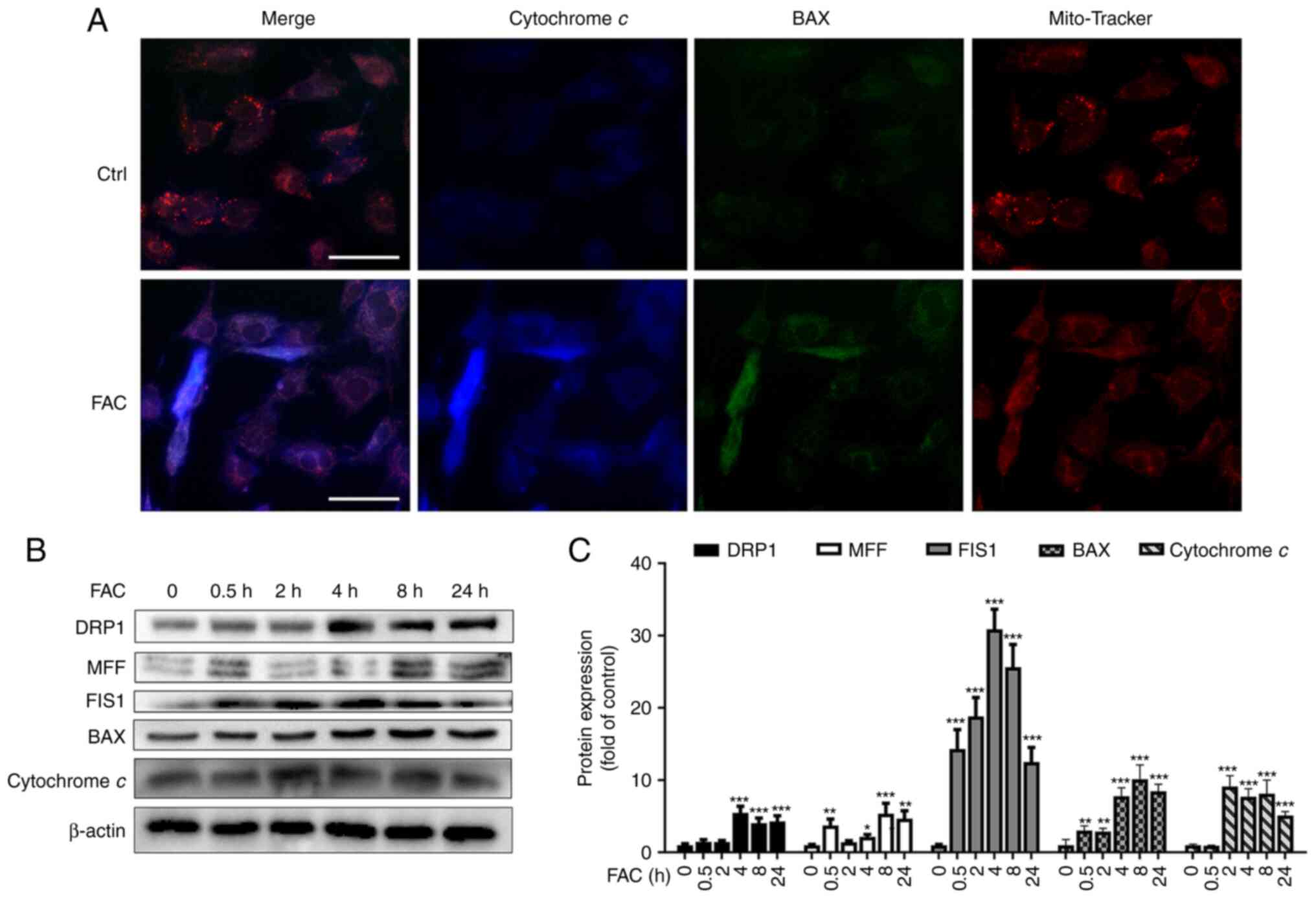 | Figure 2Iron overload impairs mitochondrial
dynamics in chondrocytes. (A) Chondrocytes were treated with 100
µm FAC for 24 h and immunofluorescence staining was
conducted to examine the expression and localization of BAX (green)
and cytochrome c (blue). Mito-Tracker Red was used to stain
mitochondria. Scale bar, 50 µm (B) Chondrocytes were treated
with 100 µm FAC for 24 h and western blotting was conducted
to examine the expression of the mitochondrial fission proteins
MFF, DRP1, FIS1, BAX and cytochrome c. Representative bands
of western blots were from the same sample; for FIS1, BAX and
cytochrome c proteins, the molecular weights of which were
very close, images were derived from three or four gels of the same
sample. (C) The band density of DRP1, MFF, FIS1, BAX and cytochrome
c was quantified and normalized to the control. Data are
presented as mean ± SD. *P<0.05;
**P<0.01; ***P<0.001 vs. 0 h. MFF,
mitochondrial fission factor; DRP1, dynamin-related protein 1;
FIS1, mitochondrial fission 1 protein; FAC, ferric ammonium
citrate; Ctrl, control group. |
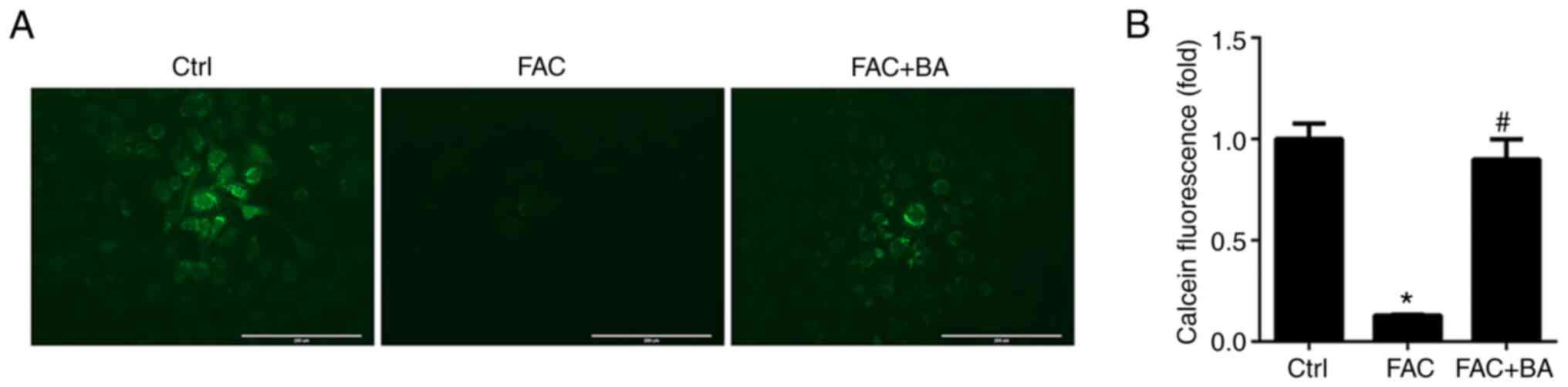
Calcium chelator decreases intracellular
iron concentration
Calcium chelator was reported to inhibit iron influx
(21). To test this hypothesis,
chondrocytes were treated with 100 µm FAC, with or without
BAPTA-AM. The fluorescence dye calcein-AM was used to assess the
ferrous iron uptake and outflow in chondrocytes. As shown in
Fig. 3, the fluorescence
intensity significantly decreased in the FAC treatment group,
indicating elevated iron content in chondrocytes. By contrast, the
fluorescence intensity significantly increased in the BAPTA-AM
co-treatment group, indicating that the BAPTA-AM co-treatment
decreased iron concentration in chondrocytes.
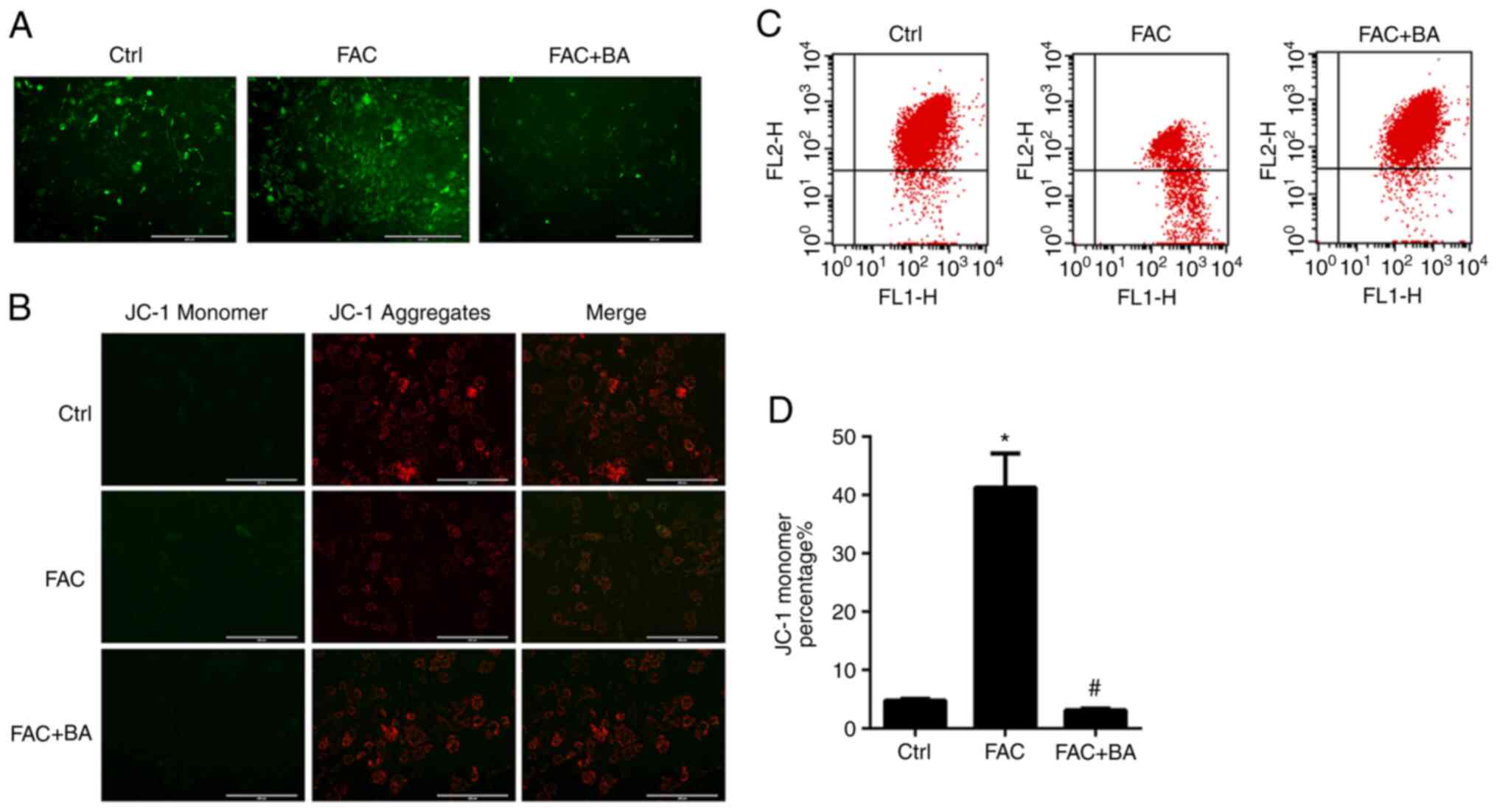
Calcium chelator inhibits ROS production
and protects against collapse of mitochondrial membrane potential
induced by iron overload
It was reported that excess iron in mitochondria
could generate ROS via the Fenton reaction (5). As shown in Fig. 4A, DCFH-DA staining demonstrated
that FAC promoted ROS production and this effect was inhibited by
the calcium chelator BAPTA-AM. Iron overload impairs normal
mitochondrial morphology and function, whereas mitochondrial
dysfunction, in turn, affects cellular iron influx and increases
ROS production (24). Next, JC-1
staining was utilized to investigate whether calcium chelator could
inhibit the collapse of the mitochondrial membrane potential. As
shown in Fig. 4B-D, the ratio of
green JC-1 monomers to red JC-1 aggregates was increased in the FAC
treatment group, while BAPTA-AM treatment suppressed the changes in
mitochondrial membrane potential induced by iron. These results
indicated that calcium chelators may protect against iron
overload-induced ROS production and mitochondrial dysfunction.
Calcium chelator protects chondrocytes
against iron overload-induced mitochondrial damage
Mitochondria in healthy chondrocytes display a
wire-like shape. As shown in Fig.
5A, FAC treatment (100 µm) significantly increased the
number of granulated mitochondria in chondrocytes, while BAPTA-AM
restored the normal mitochondrial shape in chondrocytes.
Mitochondrial fission and fusion are known to regulate the function
and morphology of mitochondria. A number of studies have shown that
mitochondrial fission is an early event during the process of cell
death (25). As shown in
Fig. 5A, the expression of the
mitochondrial fission proteins, DRP1, MFF and FIS1, was found to be
upregulated by treatment with 100 µm FAC, and it was
decreased by treatment with 10 µm BAPTA-AM. Moreover, it was
observed that the pro-inflammatory cytokine IL-1β increased
FAC-induced mitochondrial fission protein expression, whereas this
effect was also reversed by BAPTA-AM. In conclusion, the
aforementioned results indicated that iron overload significantly
promoted mitochondrial destruction and dysfunction, and calcium
chelator treatment could effectively preserve the normal
mitochondrial morphology and function.
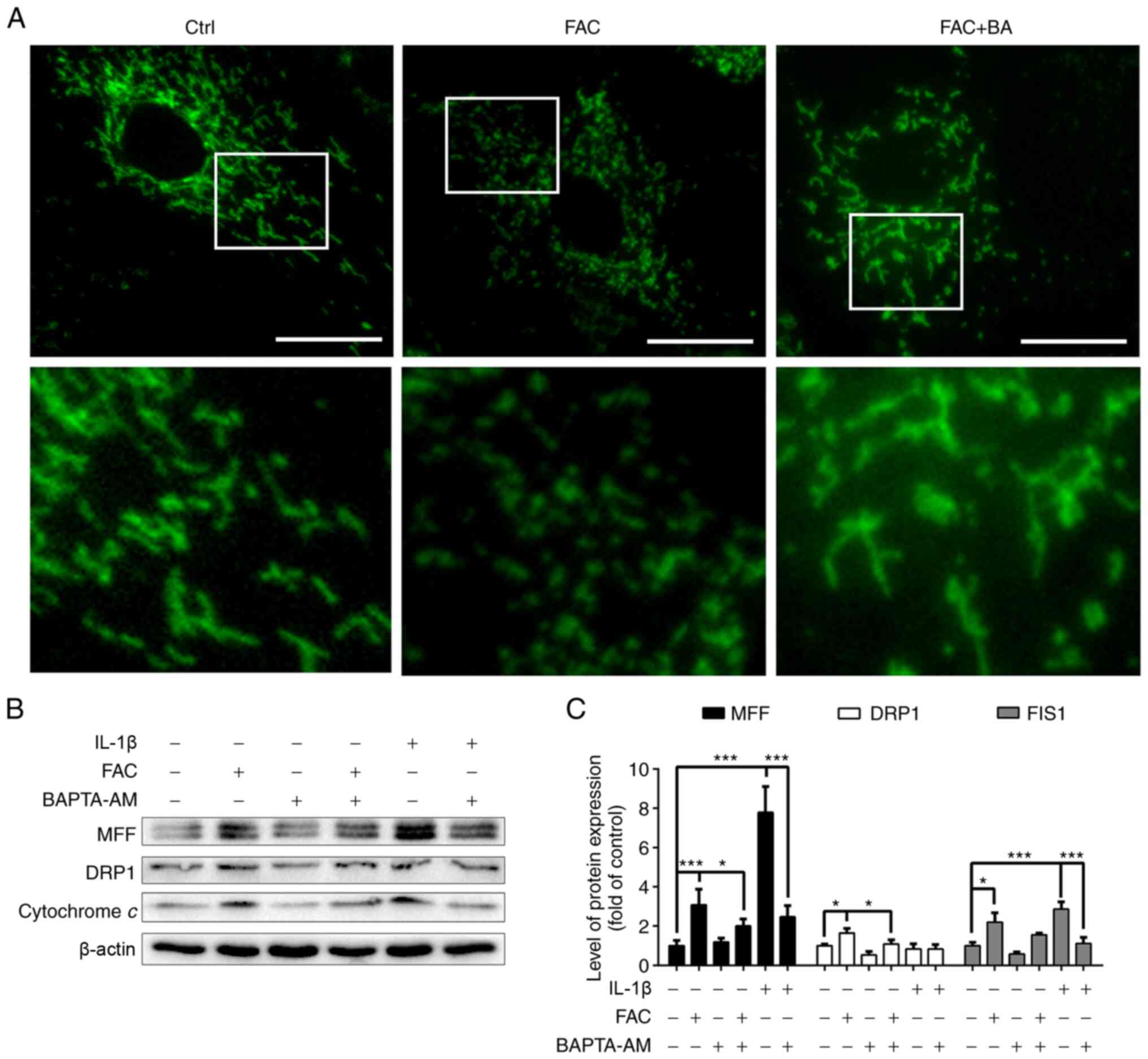 | Figure 5Calcium chelator BAPTA-AM protects
chondrocytes against iron overload-induced mitochondrial damage.
(A) Representative fluorescence images of mitochondria and local
amplification images of the selected area. Chondrocytes were
treated with 100 µm FAC in the presence or absence of 10
µm of the calcium chelator BAPTA-AM and the morphology of
the mitochondria was visualized using Mito-Tracker Green staining.
Scale bars, 25 µm. (B) Chondrocytes were treated with 100
µm FAC or 10 ng/ml IL-1 β in the presence or absence of 10
µm of the calcium chelator BAPTA-AM. Western blotting was
conducted to examine the protein expression levels of MFF, DRP1 and
FIS1. (C) Densitometric analysis of MFF, DRP1 and FIS1 protein
expression normalized to β-actin. Data are presented as mean ± SD.
*P<0.05; ***P<0.001. MFF, mitochondrial
fission factor; DRP1, dynamin-related protein 1; FIS1,
mitochondrial fission 1 protein; FAC, ferric ammonium citrate;
BAPTA-AM, BAPTA acetoxymethyl ester; Ctrl, control group; FAC + BA,
FAC + BAPTA-AM group. |
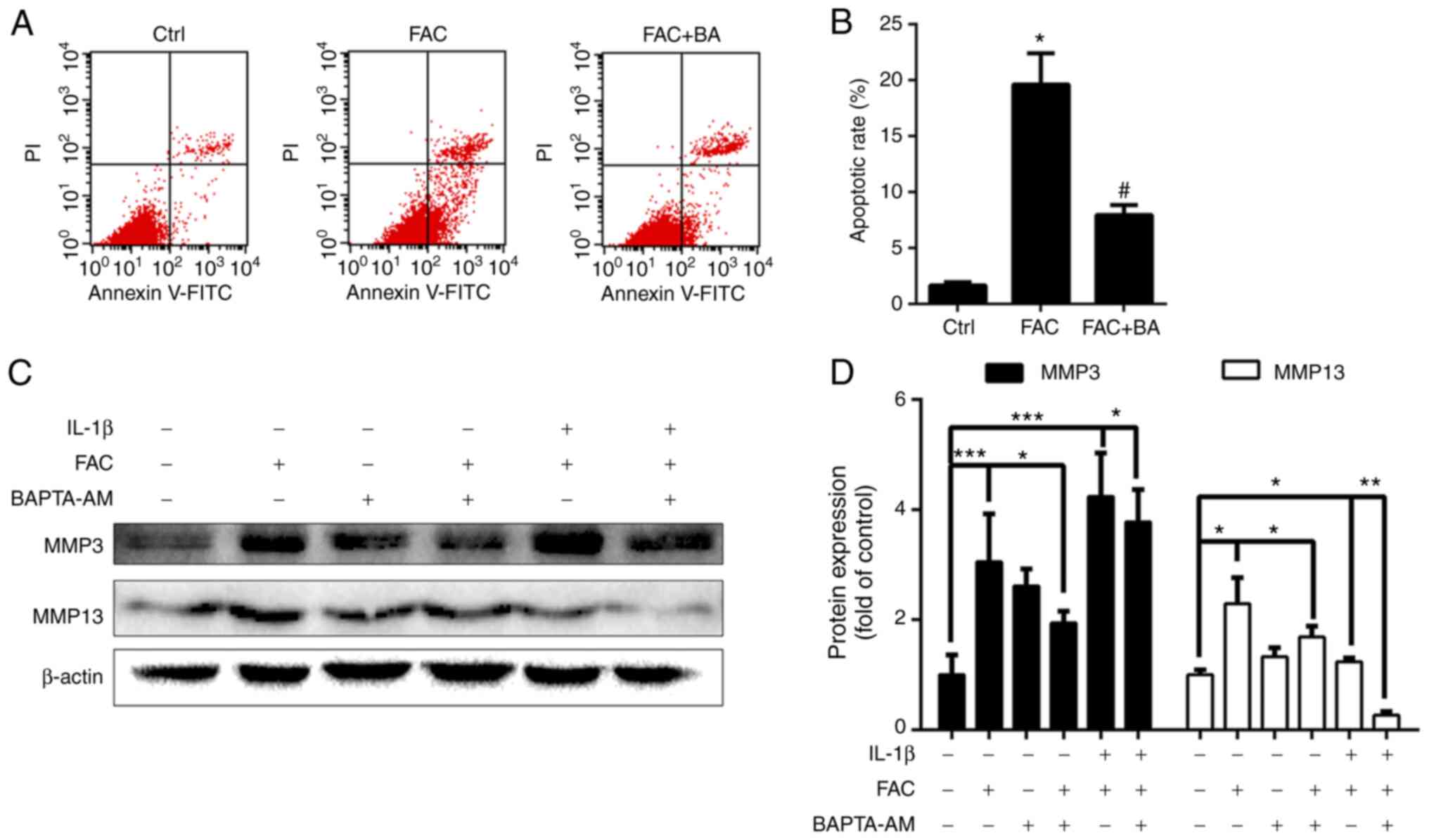
Calcium chelator protects chondrocytes
against iron overload-induced apoptosis and lowers the expression
of MMPs
Our previous findings demonstrated the association
between iron overload and OA progression (26). In the present study, it was
investigated whether calcium chelator treatment could protect
chondrocytes against iron overload-induced apoptosis and suppress
the expression of MMPs. As shown in Fig. 6A and B, FAC treatment
significantly promoted chondrocyte apoptosis, whereas BAPTA-AM
decreased the chondrocyte apoptosis rate induced by 100 µm
FAC. A hallmark of OA chondrocytes is the increased production of
MMPs. As shown in Fig. 6C and D,
BAPTA-AM treatment decreased chondrocyte MMP3 and MMP13 expression,
which was induced by 100 µm FAC. Moreover, it was observed
that FAC and IL-1β co-treatment significantly promoted MMP3
expression compared with the FAC group, and this effect was also
inhibited by BAPTA-AM. In summary, these results indicated that
calcium chelator treatment may protect chondrocytes against iron
overload-induced apoptosis and lower OA-related marker
expression.
Discussion
As the most abundant trace metal in the body, iron
participates in a variety of vital functions, including DNA
synthesis, oxygen transport, energy metabolism and cellular
respiration. However, excess iron accumulation is common in certain
tissues or organs, such as the bone marrow, heart and brain, and
may lead to the development of pathological conditions or diseases
(12,27). By participating in the
mitochondrial respiratory chain, iron leads to the generation of
ROS through exchanging single electrons with substrates. This
triggers oxidative stress, lipid peroxidation and DNA damage, which
may result in genomic instability and DNA repair defects,
ultimately compromising cell viability and promoting programmed
cell death (28). Our previous
studies have demonstrated a link between iron overload and OA. Iron
overload is common in OA cartilage, as indicated by the presence of
hemosiderin deposition. Abnormal cartilage iron accumulation is not
only found in hemophilic arthropathy and hereditary
hemochromatosis, but also in traumatic arthritis and rheumatoid
arthritis (29,30). A potential cause of increased
iron deposition in the afflicted joints of patients with OA may be
the microbleeding due to the compromised vasculature, which may
result in intra-articular iron loading. Iron chelation is the main
treatment of patients with systemic iron overload. Therefore, it is
important to investigate new treatment strategies to inhibit iron
overload-induced detrimental effects.
Cellular iron homeostasis is delicately regulated by
several iron metabolism-related proteins, among which TfR1 plays
the most important role in controlling cellular iron levels
(31). Chondrocytes take up iron
mainly through TfR1 mediated iron-TF-TfR1 complex endocytosis.
Ca2+ was reported to regulate iron-TF-tfR1 complex
internalization and promote iron influx (21,32). In the present study, the calcium
chelator BAPTA-AM was used to test the efficacy of calcium chelator
treatment in iron overload-induced chondrocyte damage. BAPTA-AM
significantly reduced iron levels in chondrocytes and inhibited
iron overload-induced cell apoptosis and the expression of MMPs,
thus providing new insights into the treatment of iron
overload-induced diseases.
Recently, mitochondria have been shown to play a
pivotal role in the progression of OA (33,34). However, the role of mitochondrial
dysfunction in iron overload-induced chondrocyte apoptosis and MMP
expression remains to be fully elucidated. In the present study,
the immunofluorescence results demonstrated that mitochondrial
destruction increased along with leakage of cytochrome c
from the mitochondria to the cytoplasm and an increase in the
expression of BAX. These results indicated that excess iron may
lead to disrupted mitochondrial function and morphology.
Mitochondrial fission and fusion play important roles in the
adaptation of mitochondria to harmful stimuli (35). Mitochondrial fission occurs at
the early stages of mitochondrial dysfunction and morphology
disruption (36). The results of
the present study demonstrated that FAC promoted the expression of
mitochondrial fission-related proteins in a time-dependent manner.
Mitochondrial morphological assays produced similar results.
Following treatment with 100 µm FAC for 24 h, the
mitochondrial shape changed from a long wire-like to a short,
granular shape, while the detrimental effects of iron overload were
reversed and the number of wire-like shaped mitochondria was
increased following calcium chelator treatment. These results
indicated that calcium chelators may protect chondrocytes against
iron overload-induced mitochondrial dysfunction.
Iron is required for mitochondria to synthesize heme
and iron-sulfur clusters, which are essential for mitochondrial
function (37). However, excess
iron accumulation in mitochondria may catalyze the generation of
highly reactive OH-radicals by reacting with
H2O2 and eventually causing ROS
overproduction. Mitochondria are major organelles that generate
ROS, but excessive ROS production induces mitochondrial
dysfunction. Elevated ROS levels could further destroy
mitochondrial structure and function, subsequently activating the
mitochondrial apoptotic pathway (38). The results of the present study
demonstrated that ROS production was markedly reduced by calcium
chelator treatment. The results also revealed that FAC treatment
decreased mitochondrial membrane potential and lead to
mitochondrial membrane depolarization, whereas these effects were
notably attenuated by BAPTA-AM treatment.
Oxidative stress has been identified as an important
contributor to OA progression. Increased ROS generation does not
only activate the mitochondrial apoptotic pathway, but also
promotes the expression of MMPs (39). The present study demonstrated
that iron reduced cell viability and increased chondrocyte
apoptotic rate, which was also reversed by calcium chelator
treatment. Our previous study demonstrated that pro-inflammatory
cytokines could disrupt iron homeostasis and promote iron influx
via promoting the expression of the iron influx regulators TfR1 and
DMT1 (26). In the present
study, it was observed that the pro-inflammatory cytokine IL-1β
promoted mitochondrial fission protein and MMP expression, which
was induced by iron overload. This effect was also shown to be
inhibited by calcium chelator treatment.
In conclusion, using an in vitro iron
overload model, the present study demonstrated that excess iron is
toxic to chondrocytes. Iron overload did not only promote
chondrocyte apoptosis, but also promoted MMP expression via
catalyzing ROS production and mitochondrial dysfunction. Calcium
chelator treatment decreased intracellular iron concentration and
iron overload-induced ROS overproduction and mitochondrial
dysfunction, thus protecting against iron overload-induced
chondrocyte apoptosis and MMP expression. These findings indicate
that calcium chelators may hold promise in the prevention or
treatment of iron overload-related OA progression. This was a
preliminary study designed to demonstrate whether a calcium
chelator could protect against iron overload-induced chondrocyte
degeneration, as compared to iron chelators. There are numerous
calcium chelator agents that have been approved by FDA; in
addition, the present study is potentially limited by the lack of
in vivo experiments, and further studies are required to
fully elucidate the exact mechanism underlying the role of the
calcium chelator BAPTA-AM in modulating chondrocyte iron
influx.
Availability of data and materials
The data that support the findings of this study are
available from the corresponding author upon reasonable
request.
Authors' contributions
XJ designed the experiments. XJ and QW wrote the
manuscript and analyzed the data. TD, WZ, XL, TL, GW and FC carried
out the experiments. QL, TD and QW made substantial contributions
to analysis and interpretation of data, supervised the present
study, searched the literature and revised the manuscript. XC and
TD confirm the authenticity of all the raw data. All the authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
All animal protocols were approved by the
Institutional Animal Care of the Shandong Provincial Hospital
Affiliated to Shandong First Medical University (no. 2020-526).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82002325) and the Natural Science
Foundation of Shandong Province (grant nos. ZR2020QH075 and
ZR2020QH264).
References
|
1
|
Jing X, Du T, Li T, Yang X, Wang G, Liu X,
Jiang Z and Cui X: The detrimental effect of iron on OA
chondrocytes: Importance of pro-inflammatory cytokines induced iron
influx and oxidative stress. J Cell Mol Med. 25:5671–5680. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jing X, Lin J, Du T, Jiang Z, Li T, Wang
G, Liu X, Cui X and Sun K: Iron overload is associated with
accelerated progression of osteoarthritis: The role of DMT1
mediated iron homeostasis. Front Cell Dev Biol. 8:5945092020.
View Article : Google Scholar
|
|
3
|
Jeney V: Clinical impact and cellular
mechanisms of iron overload-associated bone loss. Front Pharmacol.
8:772017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sheth S: Iron chelation: An update. Curr
Opin Hematol. 21:179–185. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gozzelino R and Arosio P: Iron homeostasis
in health and disease. Int J Mol Sci. 17:1302016. View Article : Google Scholar :
|
|
6
|
Leipuviene R and Theil E: The family of
iron responsive RNA structures regulated by changes in cellular
iron and oxygen. Cell Mol Life Sci. 64:2945–2955. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differe. 23:369–379. 2016. View Article : Google Scholar
|
|
8
|
Siddique A and Kowdley KV: Review article:
The iron overload syndromes. Aliment Pharmacol Ther. 35:876–893.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ogilvie-Harris DJ and Fornaiser VL:
Synovial iron deposition in osteoarthritis and rheumatoid
arthritis. J Rheumatol. 7:30–36. 1980.PubMed/NCBI
|
|
10
|
Roosendaal G, Tekoppele JM, Vianen ME, van
den Berg HM, Lafeber FP and Bijlsma JW: Articular cartilage is more
susceptible to blood induced damage at young than at old age. J
Rheumatol. 27:1740–1744. 2000.PubMed/NCBI
|
|
11
|
Naughton DP: Iron(III)-mediated
intra-articular crystal deposition in arthritis: A therapeutic role
for iron chelators. Med Hypotheses. 57:120–122. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nakamura T, Naguro I and Ichijo H: Iron
homeostasis and iron-regulated ROS in cell death, senescence and
human diseases. Biochim Biophys Acta. 1863:1398–1409. 2019.
View Article : Google Scholar
|
|
13
|
Kennish L, Attur M, Oh C, Krasnokutsky S,
Samuels J, Greenberg JD, Huang X and Abramson SB: Age-dependent
ferritin elevations and HFE C282Y mutation as risk factors for
symptomatic knee osteoarthritis in males: A longitudinal cohort
study. BMC Musculoskelet Disord. 15:82014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Suantawee T, Tantavisut S, Adisakwattana
S, Tanavalee A, Yuktanandana P, Anomasiri W, Deepaisarnsakul B and
Honsawek S: Oxidative stress, vitamin e, and antioxidant capacity
in knee osteoarthritis. J Clin Diagn Res. 7:1855–1859.
2013.PubMed/NCBI
|
|
15
|
Blanco FJ, Rego I and Ruiz-Romero C: The
role of mitochondria in osteoarthritis. Nat Rev Rheumatol.
7:161–169. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kühn LC: Iron regulatory proteins and
their role in controlling iron metabolism. Metallomics. 7:232–243.
2015. View Article : Google Scholar
|
|
17
|
Kumfu S, Chattipakorn SC, Fucharoen S and
Chattipakorn N: Dual T-type and L-type calcium channel blocker
exerts beneficial effects in attenuating cardiovascular dysfunction
in iron-overloaded thalassaemic mice. Exp Physiol. 101:521–539.
2016. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sripetchwandee J, KenKnight SB, Sanit J,
Chattipakorn S and Chattipakorn N: Blockade of mitochondrial
calcium uniporter prevents cardiac mitochondrial dysfunction caused
by iron overload. Acta Physiol (Oxf). 210:330–341. 2014. View Article : Google Scholar
|
|
19
|
Kumfu S, Khamseekaew J, Palee S,
Srichairatanakool S, Fucharoen S, Chattipakorn SC and Chattipakorn
N: Combined iron chelator and T-type calcium channel blocker exerts
greater efficacy on cardioprotection than monotherapy in
iron-overload thalassemic mice. Eur J Pharmacol. 822:43–50. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Y, Zhao X, Chang Y, Zhang Y, Chu X,
Zhang X, Liu Z, Guo H, Wang N, Gao Y, et al: Calcium channel
blockers ameliorate iron overload-associated hepatic fibrosis by
altering iron transport and stellate cell apoptosis. Toxicol Appl
Pharmacol. 301:50–60. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cui R, Choi SE, Kim TH, Lee HJ, Lee SJ,
Kang Y, Jeon JY, Kim HJ and Lee KW: Iron overload by transferrin
receptor protein 1 regulation plays an important role in
palmitate-induced insulin resistance in human skeletal muscle
cells. FASEB J. 12:fj201800448R2018.
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Meyer JN, Leuthner TC and Luz AL:
Mitochondrial fusion, fission, and mitochondrial toxicity.
Toxicology. 391:42–53. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang H, Liu C, Zhao Y and Gao G:
Mitochondria regulation in ferroptosis. Eur J Cell Biol.
99:1510582019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tilokani L, Nagashima S, Paupe V and
Prudent J: Mitochondrial dynamics: Overview of molecular
mechanisms. Essays Biochem. 62:341–360. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jing X, Lin J, Du T, Jiang Z, Li T, Wang
G, Liu X, Cui X and Sun K: Iron overload is associated with
accelerated progression of osteoarthritis: The role of DMT1
mediated iron homeostasis. Front Cell Dev Biol. 8:5945092021.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chung JY, Kim HS and Song J: Iron
metabolism in diabetes-induced Alzheimer's disease: A focus on
insulin resistance in the brain. Biometals. 31:705–714. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao M, Monian P, Pan Q, Zhang W, Xiang J
and Jiang X: Ferroptosis is an autophagic cell death process. Cell
Res. 26:1021–1032. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nieuwenhuizen L, Schutgens RE, van Asbeck
BS, Wenting MJ, van Veghel K, Roosendaal G, Biesma DH and Lafeber
FP: Identification and expression of iron regulators in human
synovium: Evidence for upregulation in haemophilic arthropathy
compared to rheumatoid arthritis, osteoarthritis, and healthy
controls. Haemophilia. 19:e218–e227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Camacho A, Simao M, Ea HK, Cohen-Solal M,
Richette P, Branco J and Cancela ML: Iron overload in a murine
model of hereditary hemochromatosis is associated with accelerated
progression of osteoarthritis under mechanical stress.
Osteoarthritis Cartilage. 24:494–502. 2016. View Article : Google Scholar
|
|
31
|
Gammella E, Buratti P, Cairo G and
Recalcati S: The transferrin receptor: The cellular iron gate.
Metallomics. 9:1367–1375. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen MP, Cabantchik ZI, Chan S, Chan GC
and Cheung YF: Iron overload and apoptosis of HL-1 cardiomyocytes:
Effects of calcium channel blockade. PLoS One. 9:e1129152014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lopez de Figueroa P, Lotz MK, Blanco FJ
and Carames B: Autophagy activation and protection from
mitochondrial dysfunction in human chondrocytes. Arthritis
Rheumatol. 67:966–976. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang Z, Xu T, Chen J, Shao Z, Wang K, Yan
Y, Wu C, Lin J, Wang H, Gao W, et al: Parkin-mediated mitophagy as
a potential therapeutic target for intervertebral disc
degeneration. Cell Death Dis. 9:9802018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tsuchiya M, Ichiseki T, Ueda S, Ueda Y,
Shimazaki M, Kaneuji A and Kawahara N: Mitochondrial stress and
redox failure in steroid-associated osteonecrosis. Int J Med Sci.
15:205–209. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Suwanjang W, Wu KL, Prachayasittikul S,
Chetsawang B and Charngkaew K: Mitochondrial dynamics impairment in
dexamethasone-treated neuronal cells. Neurochem Res. 44:1567–1581.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Anderson CP, Shen M, Eisenstein RS and
Leibold EA: Mammalian iron metabolism and its control by iron
regulatory proteins. Biochim Biophys Acta. 1823:1468–1483. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Esposito G, Vos M, Vilain S, Swerts J,
Valadas JD, Van Meensel S, Schaap O and Verstreken P: Aconitase
causes iron toxicity in Drosophila pink1 mutants. PLoS Genet.
9:e10034782013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bresgen N and Eckl PM: Oxidative stress
and the homeodynamics of iron metabolism. Biomolecules. 5:808–847.
2015. View Article : Google Scholar : PubMed/NCBI
|