Introduction
Traumatic brain injury (TBI) has a high mortality and disability rate, and is a major cause of mortality and disability worldwide. The main pathogenic causes of TBI are traffic accidents or falling from a height (1). Excessive activation of the neuroinflammatory response mediated by microglial activation serves an important role in secondary TBI, which is associated with inflammation and edema, and results in neurovascular unit damage and neurological dysfunction (2). Recent studies have reported that microglial activation is dependent on the polarization of microglia towards either the proinflammatory phenotype or the anti-inflammatory phenotype (3,4). The former promotes neuroinflammatory responses and aggravates brain damage, whereas the latter inhibits neuroinflammation and promotes nerve tissue repair in the surrounding region (5). Therefore, modulation of microglial phenotypic transformation following TBI may serve to be a potential therapeutic target.
C-type lectin-like receptors (CLRs) represent a family of transmembrane pattern recognition receptors (6,7), which are expressed primarily in myeloid cells (8). CLRs have been demonstrated to function not only as mock pathogen moieties for host defense, but to also interact with self-antigens, such as damage-associated molecular patterns (DAMPs) released from dead cells (9). Dysfunction in these receptors may lead to the deterioration of inflammatory diseases, such as multiple sclerosis (10). In particular, microglial macrophage-inducible C-type lectin (Mincle) is a type of transmembrane, germline-encoded pattern recognition receptor (11), which is an important member of CLRs and is widely expressed in antigen-presenting cells, including macrophages, neutrophils, dendritic cells and B cells (11), where it mediates damaged cell-induced innate immune responses in various pathological processes (12,13). In addition, Mincle has previously been reported to be expressed on the membranes of neurons (14). As well as recognizing glycolipids on pathogens, Mincle can also bind to ligands released by dead cells, including spliceosome-associated protein 130 (SAP130), β-glucosylceramide, cholesterol sulfate or cholesterol crystals (15,16). SAP130 released from damaged neurons in the brain has been found to act as a DAMP and activate Mincle signaling in cortical neurons in culture (16). Additionally, SAP130 may bind to Mincle on the microglia to mediate the crosstalk between neuronal necroptosis and the microglial immune response following subarachnoid hemorrhage (SAH)-related injury (17,18).
Following stimulation, Mincle and its downstream effector spleen tyrosine kinase (Syk) can activate a number of signaling pathways to increase inflammatory cytokine production, such as IL-1β, TNF-α, IL-6 and cytokines (such as ASC and NLR family pyrin domain-containing protein 3) (18). These cytokines then promote microglial activation and aggravate neurological dysfunction through hyperactivation of the inflammatory response during SAH (18). Activation of neuroinflammation following TBI serves a key role in the clearance of necrotic tissues from the surrounding regions (19); however, excessive neuroinflammation can cause increased damage to the neuronal system (19). This process has been reported to involve the phosphorylation and activation of the Mincle/SYK pathway, NF-κB and MAPK (20), activator protein 1 (21) or nuclear factor of activated T cells (22), which then mediates the transcription of downstream inflammatory genes, such as IL-18, IL-1β and IL-6 (23,24). Mincle/Syk signaling has previously been shown to be associated with NF-κB, which can activate proinflammatory factors and inflammasomes, such as ASC and caspase-1, resulting in the potentiation of the neuroinflammatory response (25). Syk is a transcription factor that serves as a bridge between Mincle/Syk and NF-κB signaling (23,24,26). Therefore, the Mincle/Syk pathway may be considered a potential novel target for the treatment of neuroinflammatory damage after TBI.
In the present study, primary cultured microglial cells treated with cerebral cortex extract (CCE) were used to mimic the TBI model in vitro. In addition, an in vivo controlled cortical impact (CCI) mouse model was used to evaluate the effects of the Syk inhibitor BAY61-3606 (BAY) on microglial activation and phenotypic transformation after TBI (27). BAY (C20H20Cl20N6O6) is a potent selective Syk inhibitor that competes with ATP, which has a Ki and IC50 of 7.5 and 10 nM, respectively (28). Furthermore, the possible functions of BAY on neuroinflammation and neurovascular unit damage were explored to identify the prospects for post-TBI treatment.
Materials and methods
Patient specimens and ethics
The present study was performed in accordance with the Declaration of Helsinki, and the study protocol and written informed consent for the use of human samples were approved by the Medical Ethics Committee of Tongji Hospital (approval no. TJH-IRB 20200103; Wuhan, China). Written informed consent was signed by the next of kin of the patients and control individuals. TBI was diagnosed using the World Health Organization criteria (29). The brain biopsies used in the present study were obtained from 10 patients with severe TBI who underwent surgery at the Department of Neurosurgery, Tongji Hospital. Data were collected between January and December 2020; the mean age of patients was 56.40±12.91 years (range 40-80 years), seven (70.0%) were male and three (30.0%) were female; seven patients improved and three patients succumbed to subfalcine hernia induced by brain edema. These biopsy tissues were either necrotic brain tissues or tissues with severe edema around the lesion site that must be removed. The control group comprised specimens from patients with epilepsy who required necessary surgical excision of the epilepsy lesion; the mean age of the control patients was 31.71±20.73 years (range, 11-62 years); five (71.4%) were male and two (28.6%) were female.
Animals
Animal ethics approval for the use of adult and newborn mice was received from The Experimental Animal Ethics Committee of Huazhong University of Science and Technology (approval no. TJH-202012001; Wuhan, China). Male C57BL/10ScNJ mice (age, 8-10 weeks; weight, 23.02±1.64 g) were obtained from GemPharmatech Co., Ltd. The mice were fed in The Experimental Animal Center of Tongji Scientific Building, Tongji Hospital. All mice were maintained in a controlled environment at 22±3°C and 60% relative humidity under a 12-h light/dark cycle, and were given free access to standard rodent nutrition and water.
CCI model of TBI
A total of 120 mice were randomly divided into the following four groups: i) Control group (n=30); ii) TBI group (n=30); iii) TBI + BAY group (n=30); and iv) BAY group (n=30). The control group mice received only vehicle solution [10% DMSO (cat. no. D2650; Hybri-Max™; Sigma-Aldrich; Merck KGaA)]. Mice in the TBI group and TBI + BAY group received severe CCI. Briefly, after anesthesia with 5% isoflurane in oxygen with a delivery rate of 0.5 l/min for induction and 2% isoflurane for maintenance, the mice were placed onto a stereotaxic apparatus platform. Sterile procedures were adhered to and an incision was first made along the midline. Subsequently, a 3-mm2 craniotomy was performed at the following specific location: Left hemisphere; 1 mm anterior-λ; 1 mm left-midline; and 1 mm post-bregma. In total, 3 mm2 of the skull was removed avoiding damage to the dura mater. The CCI (YHCI99; Wuhan Yihong Technology Co., Ltd.) parameters were as follows: i) Deformation depth, 1 mm; ii) impact speed, 3.5 m/sec and ⅲ) duration, 400 msec (3). The incision was then sutured and the mice were placed on a 36-37°C heat pad. Mice in the TBI + BAY and BAY groups were administered with BAY (3 mg/kg) intraperitoneally (30) for 7 days after TBI insult. Mice in the BAY group did not receive CCI insult.
Primary microglia, PC12 cell culture and CCE generation
Primary microglia and astrocytes were extracted from the brains of neonatal male C57BL/10ScNJ mice (n=48; age, 1-2 days) as previously described (31-35). Briefly, all mice were euthanized via injection with an overdose of ketamine (180 mg/kg) + xylazine (30 mg/kg), as previously described (31-35). To obtain CCE, the mouse cortex samples were placed into a glass grinder for grinding tissue, after which the samples were centrifugated at 350 × g for 5 min at 4°C to remove cells and filtered through a 0.22-µm filter unit (cat. no. SLGP033RB; MilliporeSigma). The weight of the neonatal mice at the time of sacrifice was 1.31±0.13 g and death was verified by the lack of breathing, pulse, corneal reflex and response to firm toe pinch. Subsequently, the brain tissues of the neonatal mice were collected for primary microglia culture. The cerebrum was cut into 1-mm3 pieces and digested with 0.125% trypsin at room temperature for 4 min and then at 37°C in a 5% CO2 incubator for 4 min, before the digestion process was terminated by the addition of 2X the volume of culture medium [DMEM (cat. no. G4510; Wuhan Servicebio Technology Co., Ltd.) supplemented with 10% FBS (cat. no. 10091148; Gibco; Thermo Fisher Scientific, Inc.) and 1% penicillin-streptomycin (cat. no. C100C5; Newcell & Molecular Biotech Co., Ltd.)]. The cerebrum extract was filtered using a 70-µm nylon mesh. This extract was then centrifugated at 59 × g for 5 min at 4°C (cat. no. 3K15; MilliporeSigma), the cell pellet was collected and cultured in a T-75 culture flask with 15 ml culture medium; each T-75 flask contained extracts from four neonatal mouse brain tissues. Microglia and astrocytes were divided using a constant temperature culture oscillator (cat. no. ZWY-103B; ZHCHENG Co., Ltd.) at 100 rpm for 40 min at 37.0°C after 10 days, and were then cultured in 6- or 24-well plates at 37°C in a 5% CO2 incubator for subsequent experiments. Microglia cells were divided into the following four groups: i) Control group; ii) CCE group; iii) CCE + BAY group; and iv) BAY group. In the control group, microglia were incubated with vehicle solution (10% DMSO) for 24 h. In the CCE group, microglial cells were solely stimulated with CCE (100 µl/ml) for 24 h, whereas in the CCE + BAY group, BAY (2 µM) was added together with CCE (100 µl/ml) to the microglial cells for 24 h. In the BAY group, microglial cells were treated with BAY (2 µM) for 24 h.
The PC12 cell line, which was purchased from The Cell Bank of Type Culture Collection of The Chinese Academy of Sciences, was recovered rapidly in a 37°C water bath, then centrifugated at 59 × g for 5 min at 4°C. The cell pellet was collected and cultured in 6- or 24-well plates with culture medium (DMEM supplemented with 10% FBS and 1% penicillin-streptomycin) at 37°C in a 5% CO2 incubator for subsequent experiments. PC12 cells were divided into the following five groups: i) Control group; ii) microglia-coculture group; iii) microglia + CCE co-culture group; iv) microglia + CCE + BAY co-culture group; and v) microglia + BAY co-culture group. In the control group, PC12 cells were treated with 10% DMSO for 24 h. For microglia stimulation, CCE was obtained by filtering the supernatant from murine brain cortex samples. Microglia were first stimulated with CCE (100 µl/ml) and/or BAY (2 µM) for 24 h and the medium was then changed. Subsequently, microglial cells were co-cultured with PC12 cells for another 48 h.
Cell Counting Kit-8 (CCK-8) assay
The viability of PC12 cells was evaluated using CCK-8 (cat. no. ST1008; Sunbao-Biotech Co., Ltd.) in accordance with the manufacturer's instructions. PC12 cells were divided into the aforementioned five groups. After co-culture, PC12 cells were seeded into 96-well plates (cat. no. 3599; Costar; Corning, Inc.) at a density of 1×105 cells/ml (5,000/well). Subsequently, CCK-8 regent was added to each well at 10 µl/ml for 4 h at 37°C. The viability of PC12 cells was evaluated using a microplate reader (Infinite F50; Tecan Group Ltd.) at a wavelength of 450 nm (36).
Morris water maze (MWM)
Mice were divided into four groups: i) Control group (n=12); ii) TBI group (n=12); iii) TBI + BAY group (n=12); and iv) BAY group (n=12). The spatial learning and memory of the mice were tested using the MWM starting on day 7 after surgery. The apparatus comprised of a swimming pool (diameter, 1.5 m; height, 60 cm) filled with water (temperature, 21-25°C). Mice were trained to find a submerged platform (depth, 1.5 cm) using the marker around the side wall. During the 6-day training period, the platform was placed in the third quadrant of the testing arena. At each time trial every day, all mice were allowed 60 sec to find the platform and were given a 30-sec stay on the platform. At the start of each trial the mice were randomly placed into one of the three quadrants (first, second and fourth quadrant). The average latency, moving distance and swimming speed before locating the platform at each time trial on the test day was recorded using the WMT-100 Morris software (Chengdu Taimeng Technology Co., Ltd.). On day 7, a space exploration experiment was performed, where the platform was removed and the mice were given 60 sec to locate the area where the platform used to be; the duration of time spent in the target quadrant and the number of times that the mice crossed the previous platform location were recorded (37).
Modified neurological severity scores (mNSS) test
The mice were separated into the same groups as prior to the Morris water maze assay. Neurological damage was evaluated using the mNSS system on days 1-28 after the induction of TBI by CCI; this system assesses abnormal movements, and motor, sensory and reflex deficits. The total score is 18 points; the higher the score, the more severe the neurological damage. This system is widely used to assess the degree of nerve injury in central nerve system disease. The test was operated on days 1, 3, 7, 14 and 28 by three individuals through a triple-blind manner, and the scores were averaged (38,39).
Rotarod test
The mice were separated into the same groups as prior to the Morris water maze assay. Mouse coordination, strength and balance was assessed from day 7 post-surgery. Mice were placed on a swivel bar fatigue tester, the rotation speed was set at 30 rpm and the mice were recorded for 5 min. Mice were trained twice daily for 6 days with 300 sec as the cut-off value, and then tested, as previously described (40,41). The latency to fall was recorded.
Tissue preparation for immunofluorescence
Mice were anesthetized with 5% isoflurane in oxygen with a delivery rate of 0.5 l/min for induction and 2% isoflurane for maintenance. All mice were then immediately injected with an overdose of ketamine (180 mg/kg) + xylazine (30 mg/kg) for euthanasia (31-35) 7 days post-TBI. The mice were transcardially perfused with PBS and 4% paraformaldehyde (PFA). Brain tissues were then isolated and placed in 4% PFA for 1 day at room temperature. The tissues were fixed with 30% sucrose for 48 h at room temperature. Frozen brain sections (40 µm) were sliced using a sliding microtome; the freezing platform and freezing chamber were set at -35 and -20°C, respectively.
Immunofluorescence
Mice were divided into four groups: i) Control group (n=6); ii) TBI group (n=6); iii) TBI + BAY group (n=6); and iv) BAY group (n=6). All mice were injected with an overdose of ketamine (180 mg/kg) + xylazine (30 mg/kg) immediately for euthanasia 7 days post-TBI. The frozen brain sections were blocked in 5% BSA (cat. no. G5001-100G; Wuhan Servicebio Technology Co., Ltd.) with 0.1% Triton X-100 in TBS for 2 h at room temperature. Subsequently, tissues were incubated with the following primary antibodies diluted in 5% BSA in TBS overnight at 4°C: Goat anti-ionized calcium binding adaptor molecule 1 (1:200; cat. no. ab5076; Abcam), rabbit anti-CD31 (1:200; cat. no. ab28364; Abcam), mouse anti-neuronal nuclear protein (1:200; cat. no. 94403S; Cell Signaling Technology, Inc.), rabbit anti-myelin basic protein (1:5,000; cat. no. ab218011; Abcam) and rabbit anti-glial fibrillary acidic protein (GFAP; cat. no. A0237; ABclonal Biotech Co., Ltd.). The sections were subsequently washed three times for 10 min each in TBS and then incubated for 2 h at room temperature with the following secondary antibodies: Cy3-conjugated donkey anti-goat (1:500; cat. no. GB21404; Wuhan Servicebio Technology Co., Ltd.), Cy3-conjugated goat anti-rabbit (1:500; cat. no. GB21303; Wuhan Servicebio Technology Co., Ltd.), FITC-conjugated donkey anti-rabbit (1:200; cat. no. GB22403; Wuhan Servicebio Technology Co., Ltd.), FITC-conjugated donkey anti-goat (1:200; cat. no. GB22404; Wuhan Servicebio Technology Co., Ltd.) and FITC-conjugated donkey anti-mouse (1:200; cat. no. GB22301; Wuhan Servicebio Technology Co., Ltd.) diluted in 5% BSA and DAPI (50%; cat. no. G1012; Wuhan Servicebio Technology Co., Ltd.) in TBS. The sections were washed four times (10 min/wash) with TBS before being sealed with fluorescent antifade solution (cat. no. G1401; Wuhan Servicebio Technology Co., Ltd.) and a cover glass. Images were captured using a fluorescence microscope (cat. no. CKX53; Olympus Corporation). ImageJ 1.8.0 (National Institutes of Health) software was used for data analysis.
ELISA
Microglial cells were randomly divided into the following four groups: i) Control group; ii) CCE group; iii) CCE + BAY group; and iv) BAY group. After 24 h stimulation, the supernatant of each group was centrifuged at 350 × g for 5 min at 4°C to remove cells and filtered through a 0.22-µm filter (cat. no. SLGP033RB; MilliporeSigma). The inflammation-associated proteins IL-1β, IL-6 and TNF-α were detected in the cellular supernatants of different groups using IL-1β (cat. no. 70-EK201B/3-96), IL-6 (cat. no. 70-EK206/3-96) and TNF-α (cat. no. 70-EK282/4-96) ELISA kits [Multisciences (Lianke) Biotech, Co., Ltd.] kits, according to the manufacturer's protocols. The absorbance values of the samples were detected using a microplate reader at 450 nm and the final concentrations of these inflammatory cytokines were calculated according to the absorbance (42).
Reverse transcription-quantitative PCR (RT-qPCR)
Mice were divided into four groups: i) Control group (n=6); ii) TBI group (n=6); iii) TBI + BAY group (n=6); and iv) BAY group (n=6). Microglial cells were grouped as aforementioned. All mice were injected with an overdose of ketamine (180 mg/kg) + xylazine (30 mg/kg) immediately for euthanasia 7 days post-TBI (31-35). The weight of adult mice at the time of sacrifice was at 21.05-25.71 g. Total RNA was extracted from the cortex or cells using TRIzol® reagent (cat. no. 15596018; Invitrogen; Thermo Fisher Scientific, Co., Ltd.) according to the manufacturer's protocol, and RNA levels and quality were detected using a NanoDrop™ (Thermo Fisher Scientific, Co., Ltd.). mRNA was reverse transcribed into cDNA using Hifair® III 1st Strand cDNA Synthesis Supermix (cat. no. 11141ES10; Shanghai Yeasen Biotechnology Co., Ltd.); RT was performed in a 20-µl system, under the following thermocycling conditions: 25°C for 5 min, 55°C for 15 min and 85°C for 5 min. qPCR was performed using Hieff® qPCR SYBR® Green Master mix (cat. no. 11202ES03; Shanghai Yeasen Biotechnology Co., Ltd.) and Quant Studio-1™ Design & Analysis Software (cat. no. A40426; Thermo Fisher Scientific, Inc.), under the following thermocycling conditions: One cycle at 95°C for 5 min, followed by 40 cycles at 95°C for 10 sec and 60°C for 30 sec, and a final melt curve stage at 60°C for 1 min and 95°C for 1 sec (12). The specific primers used for qPCR were as follows: CD86, forward, 5′-TGTTTCCGTGGAGACGCAAG-3′ and reverse, 5′-TTGAGCCTTTGTAAATGGGCA-3′; inducible nitric oxide synthase (iNOS), forward, 5′-CATTCAGATCCCGA AACGCT-3′ and reverse, 5′-TGTAGGACAATCCACAACT CGC-3′; IL-1β, forward, 5′-TGCCACCTTTTGACAGTGATG-3′ and reverse, 5′-AAGGTCCACGGGAAAGACAC-3′; IL-6, forward, 5′-AAAGAGTTGTGCAATGGCAATTCT-3′ and reverse, 5′-AAGTGCATCATCGTTGTTCATACA-3′; TNF-α, forward, 5′-CATCTTCTCAAAATTCGAGTGACAA-3′ and reverse, 5′-TGGGAGTAGACAAGGTACAACCC-3′; CD206, forward, 5′-CGTTTCGGT GGACTGTGGA-3′ and reverse, 5′-GTTGTGGGCTCTGGTGGG-3′; CD163, forward, 5′-CTG GCCTCTGAGTTTAGGGTC-3′ and reverse, 5′-CCCTTGGTG TCGAACCAGC-3′; IL-10, forward, 5′-ACCTGGTAGAAGTG ATGCCC-3′ and reverse, 5′-ACACCTTGGTCTTGGAGCTT-3′; arginase-1 (Arg1), forward, 5′-CTCCAAGCCAAAGTCCTTA GAG-3′ and reverse, 5′- AGGAGCTGTCATTAGGGACATC-3′; Bax, forward, 5′- TGAAGACAGGGGCCTTTTTG -3′ and reverse, 5′- AATTCGCCGGAGACACTCG -3′; Bim, forward, 5′-CCAAATGGCAAAGCAACCTTCTG-3′ and reverse, 5′- CTGTCAATGCATTCTCCACACC-3′; and GAPDH (housekeeping gene) forward, 5′-CGGAGTCAACGGATTTGG TCGTAT-3′ and reverse, 5′-AGCCTTCTCCATGGTGGTGAA GAG-3′ (3). Relative target gene expression to the geometric mean of the reference gene GAPDH was determined by the formula: 2−ΔΔCq, where ΔCq = Cq (target gene)−Cq (GAPDH) and ΔΔCq = ΔCq (target gene) −ΔCq (control) (43). Changes in gene expression are presented as fold changes compared to gene expression in the control group. Heatmaps were generated using the 'Pheatmaps' package in R version 4.1.1 (https://www.r-project.org/).
Western blotting
Mice were divided into four groups: i) Control group (n=6); ii) TBI group (n=6); iii) TBI + BAY group (n=6); and iv) BAY group (n=6). All mice were injected with an overdose of ketamine (180 mg/kg) + xylazine (30 mg/kg) immediately for euthanasia 7 days post-TBI. Microglial cells were grouped as aforementioned. RIPA (cat. no. G2002; Wuhan Servicebio Technology Co., Ltd.) lysis buffer (RIPA:PMSF:phosphoproteins inhibitor A:B=100:1:1:1) was used to extract total proteins from primary microglial cells, PC12 cells, mouse brain tissues and patient biopsy tissues. Protein concentration was determined using the BCA (cat. no. G2026-200T; Wuhan Servicebio Technology Co., Ltd.) reaction system via a microplate reader (Infinite F50; Tecan Group Ltd.) at a wavelength of 570 nm. The final protein concentration was 5 µg/µl. Equal amounts (30 µg/lane) of protein lysates were separated by SDS-PAGE on 10% gels and transferred onto Immobilon®-P PVDF membranes (cat. no. IPVH00010; MilliporeSigma). After blocking with 5% BSA for 2 h at RT, membranes were incubated with the following primary antibodies overnight at 4°C: Anti-Mincle (1:1,000; cat. no. A20156; ABclonal Biotech Co., Ltd.), anti-Claudin-5 (1:1,000; cat. no. 34-1600; Thermo Fisher Scientific Co., Ltd.), anti-Syk (1:1,000; cat. no. A2123; ABclonal Biotech Co., Ltd.), anti-phosphorylated (p)-Syk (1:1,000; cat. no. 2717S; Cell Signaling Technology, Inc.), anti-p-NF-κB (1:1,000; cat. no. AP0123; ABclonal Biotech Co., Ltd.), anti-NF-κB (1:1,000; cat. no. A19653; ABclonal Biotech Co., Ltd.), anti-ASC (1:1,000; cat. no. A16672; ABclonal Biotech Co., Ltd.), anti-caspase-1 (1:1,000; cat. no. A0964; ABclonal Biotech Co., Ltd.), anti-GAPDH (1:1,000; cat. no. A19056; ABclonal Biotech Co., Ltd.), anti-β-actin (1:1,000; cat. no. AC026; ABclonal Biotech Co., Ltd.) and anti-Bax (1:1,000; cat. no. 50599-2-1g; ProteinTech Group, Inc.). Subsequently, the membranes were incubated with the following corresponding secondary anti-bodies at room temperature for 2 h: HRP-conjugated goat anti-rabbit IgG (1:5,000; cat. no. GB23303; Wuhan Servicebio Technology Co., Ltd.). The protein bands were visualized using BeyoECL reagent (cat. no. P0018S; Beyotime Institute of Biotechnology) and a GeneGnome XRQ Chemiluminescence Imaging System (GeneGnome XRQ; Syngene) exposure instrument. Gray intensity of the bands was calculated using ImageJ 1.8.0 software. Finally, the expression levels of the proteins were standardized by densitometric analysis to β-actin or GAPDH levels.
Agarose spot assay
Cellular chemotaxis activity was examined using an agarose spot test by modifying the protocol of the chemotactic invasion assay described previously (44). Microglial cells were divided into the following four groups: i) Control group; ii) lipopolysaccharide (LPS) group; iii) LPS + BAY group; and iv) BAY group. In the control group, microglia were incubated with vehicle solution (10% DMSO). In the LPS group, microglial cells were solely stimulated with LPS (100 ng/ml), whereas in the LPS + BAY group, BAY (2 µM) was added together with LPS (100 ng/ml) to the microglial cells. In the BAY group, microglial cells were treated with BAY (2 µM). Briefly, low-melting point agarose (Shanghai Yeasen Biotechnology Co., Ltd.) was diluted in 0.5% PBS. Subsequently, LPS (cat. no. SMB00610; MilliporeSigma) or BAY was mixed with 0.5% agarose solution to final concentrations of 100 ng/ml and 10 nM, respectively. The mixed agarose solution for each spot (10 µl) was then pipetted into 20-mm glass-bottomed dishes (Thermo Fisher Scientific, Inc.) to generate four spots, two containing PBS as the negative control and two containing BAY, which were allowed to cool for 15 min at 4°C. Subsequently, primary cultured microglial cells (3×105/ml) were plated into the dishes and incubated at 37°C for 3 h. Images were captured using the Ocular 2.0 software (Thermo Fisher Scientific, Inc.) with a light microscope (cat. no. CKX53; Olympus Corporation). The number of microglial cells that appeared underneath the agarose spots were counted using a microscope.
Transwell assay
The Transwell assay was used to analyze cell migration as previously described (44). Microglial cells were randomly divided into the following four groups: i) Control group; ii) LPS group; iii) LPS + BAY group; and iv) BAY group. Briefly, the upper and lower chambers were separated with a polycarbonate filter (pore size, 8 µm; Corning, Inc.). Culture medium (DMEM) without FBS supplemented with LPS (100 ng/ml) or LPS (100 ng/ml) + BAY (2 µM) (30) was added into the upper chamber, and culture medium with 10% FBS was added into the lower chamber. Subsequently, primary cultured microglia (5×104/ml) were seeded into the upper chamber and both chambers were incubated at 37°C for 24 h. Subsequently, the upper side of the membrane was fixed with 4% paraformaldehyde for 30 min at room temperature, then wiped with a cotton bud and cells that migrated to the lower chamber were stained with a crystal violet solution (cat. no. G1014; Wuhan Servicebio Technology Co., Ltd.) for 30 min at room temperature. Finally, the rate of microglial migration was calculated by counting cells in four random fields in each well under a bright-field microscope at ×20 magnification.
Statistical analysis
All statistical analyses were performed using Graph Pad Prism 6 software (GraphPad Software, Inc.). Data are presented as the mean ± SD and experiments were repeated at least three times. Statistical differences were determined using either an unpaired two-tailed Student's t-test or one-way ANOVA followed by Tukey's post-hoc test, and the Kruskal-Wallis test was performed when analyzing non-parametric data. P<0.05 was considered to indicate a statistically significant difference.
Results
Mincle/Syk proteins are expressed in TBI tissues from patients and mice, and in a CCE-induced microglia model
The protein expression levels of Mincle and Syk in TBI tissues and microglial cells were detected in samples isolated from patients with TBI, mice that underwent TBI in vivo by CCI and microglial cells that were exposed to CCE in vitro. The expression levels of Mincle and p-Syk were significantly increased in the brain tissues of patients following TBI compared with in the control group (brain tissues from patients with epilepsy) (Fig. 1A) and in the mouse TBI model (Fig. 1B). Similar observations were observed in activated microglia in the CCE model compared with in the control group (Fig. 1C).
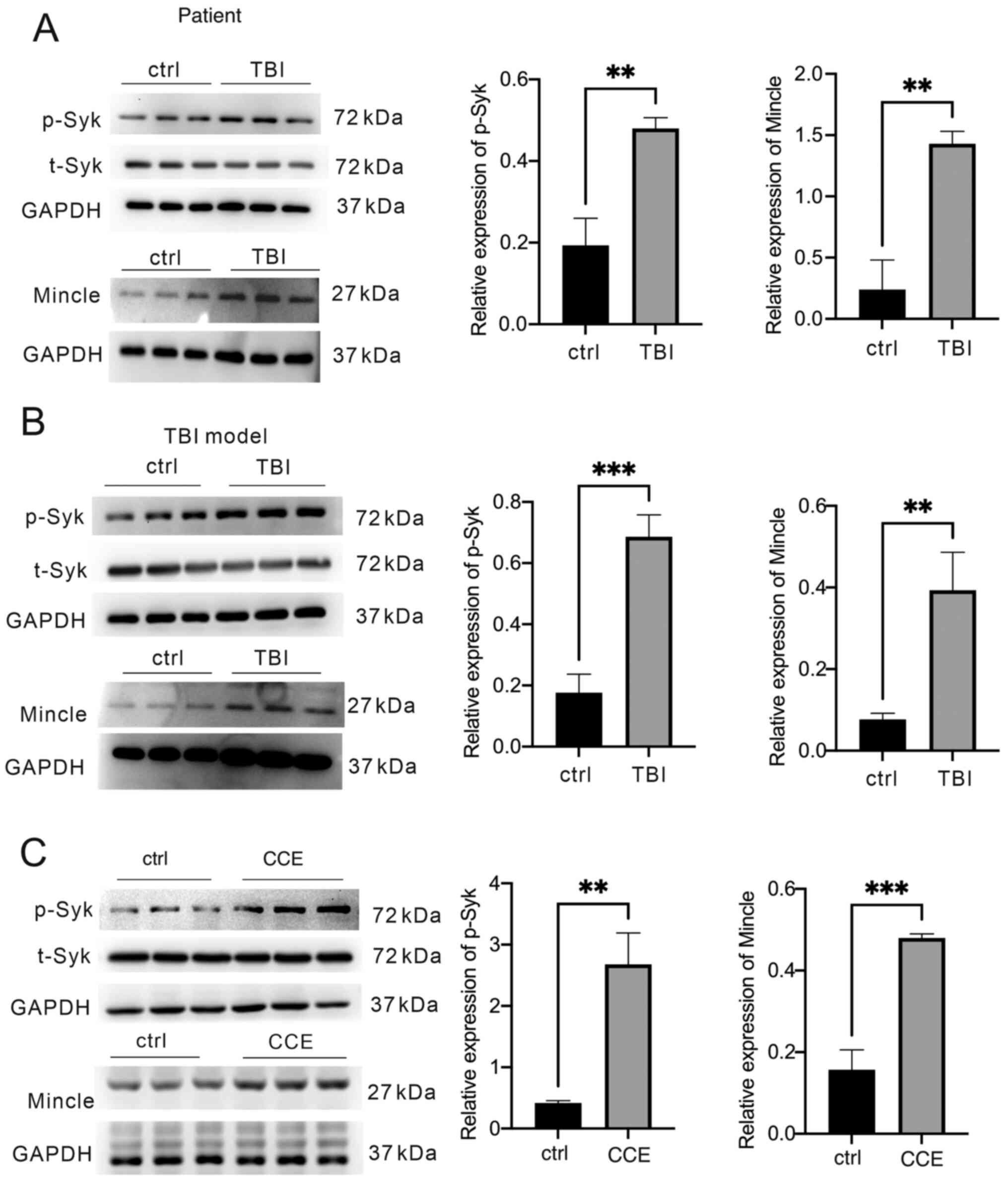 |
Figure 1
Analysis of Mincle/Syk pathway activation in patients with TBI, and mouse TBI models in vivo or in vitro. Brain tissues were collected from patients with TBI, mice following TBI induction and microglial cells. The levels of p-Syk, total Syk and Mincle expression were determined via western blot analysis of (A) patient samples, (B) mouse samples after CCI and (C) primary microglia following treatment with CCE in vitro. **P<0.01, ***P<0.001. TBI, traumatic brain injury; Mincle, macrophage-inducible C-type lectin; p, phosphorylated; t, total; Syk, spleen tyrosine kinase; CCI, controlled cortical impact; CCE, cerebral cortex extract.
|
BAY alleviates microglia-induced neuroinflammation after TBI
To evaluate the potential effects of BAY on microglia-induced neuroinflammation, the mRNA expression levels of proinflammatory and anti-inflammatory factors were determined via RT-qPCR in TBI brain tissues and microglial cells. BAY significantly suppressed the expression levels of proinflammatory factors, such as CD86, TNF-α, IL-6, iNOS and IL-1β, but promoted those of anti-inflammatory factors, including CD206, Arg1 and IL-10, in brain tissues from mice in the TBI + BAY group compared with in the TBI group (Fig. 2A). A similar effect was also observed in BAY-treated microglia following CCE in the primary microglial cells (Fig. 2C). Expression levels of the inflammation-related proteins ASC and caspase-1 were higher in the TBI mouse model in vivo compared with in the TBI + BAY group (Fig. 2B). To assess the activation of the signaling pathway downstream, the effects of BAY on the expression and activation of proteins in the Mincle/Syk/NF-κB signaling pathway were assessed in microglia in the CCE in vitro model. BAY attenuated CCE-induced Mincle/Syk/NF-κB upregulation (Fig. 2D), including Mincle expression and the phosphorylation of Syk and NF-κB. To verify the effect of BAY on the secretory function of microglia of inflammatory factors, the culture supernatant of stimulated microglia was collected to measure the production levels of TNF-α, IL-1β and IL-6. BAY significantly attenuated CCE-induced proinflammatory factor release (Fig. 2E).
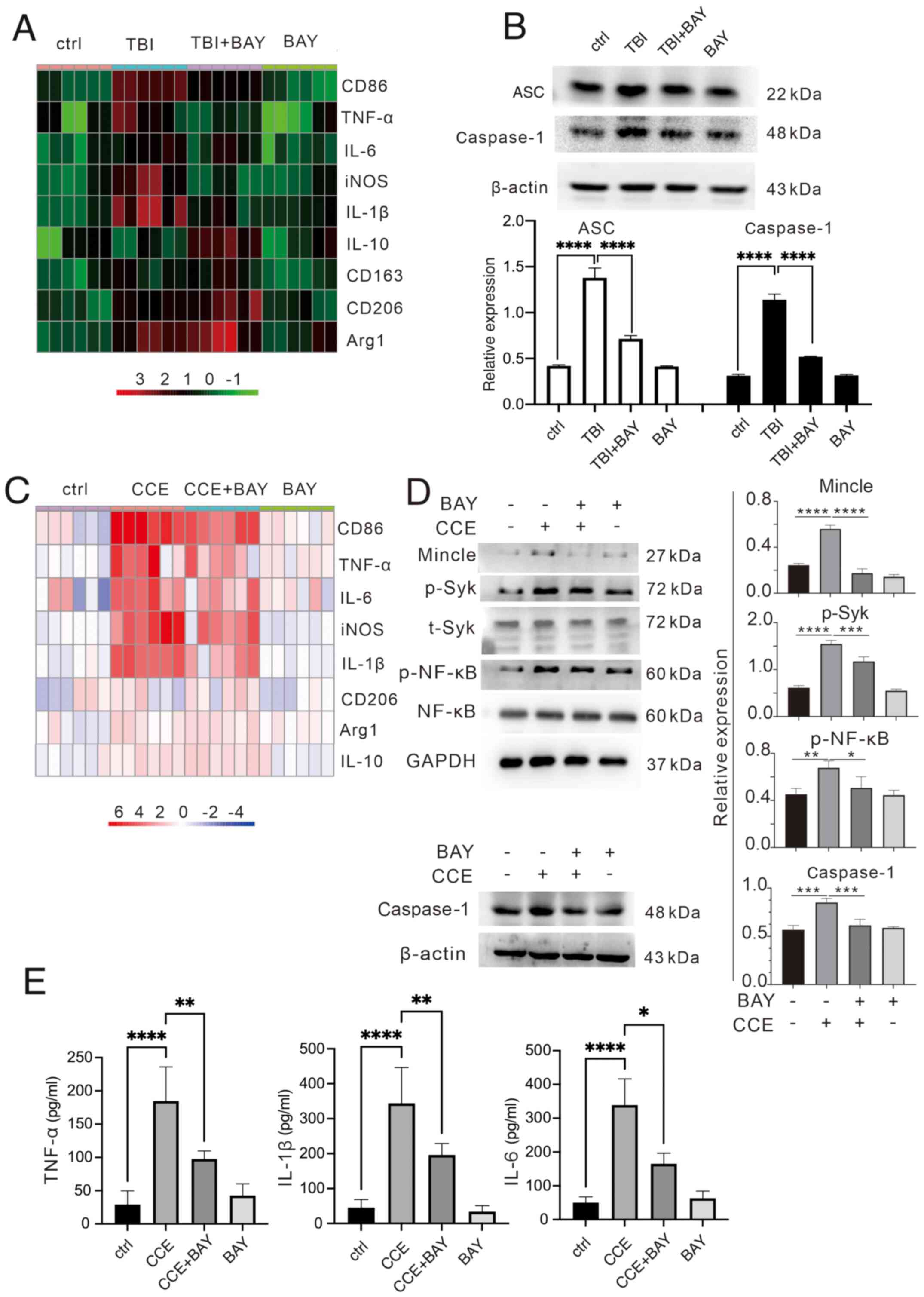 |
Figure 2
BAY can modulate the immunological function of microglia after TBI insult. (A) Inflammatory factors (CD86, TNF-α, IL-6, iNOS, IL-1β, IL-10, CD163, CD206 and Arg1) in mouse brain tissues after TBI were measured using RT-qPCR. (B) Expression levels of ASC and caspase-1 proteins in vivo were determined via western blotting. (C) Inflammatory factors (CD86, TNF-α, IL-6, iNOS, IL-1β, IL-10, CD163, CD206 and Arg1) in microglial cells treated with CCE were measured using RT-qPCR. (D) Expression levels of caspase-1 and proteins in the Mincle/Syk/NF-κB signaling pathway associate proteins (Mincle, p-Syk, t-Syk, p-NF-κB and NF-κB) in vitro were determined via western blotting. (E) Production of TNF-α, IL-1β and IL-6 in the supernatant of microglia treated with CCE was determined using ELISA. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. BAY, BAY61-3606; TBI, traumatic brain injury; CCE: cerebral cortex extract; Mincle, macrophage-inducible C-type lectin; Syk, spleen tyrosine kinase; p, phosphorylated; t, total; iNOS, inducible nitric oxide synthase; Arg1, arginase-1; RT-qPCR, reverse transcription-quantitative PCR.
|
BAY modulates the immunological function of microglia following TBI
Microglia accumulation and infiltration into the injury area is considered an important characteristic that promotes neuroinflammation (45). In addition, the Mincle/Syk signaling cascade has previously been reported to affect macrophage accumulation in the tumor microenvironment (46). The effects of the inhibition of Mincle/Syk signaling by BAY (2 µM) on microglial migration following TBI stimulation were therefore examined. As shown in Fig. 3A, in vivo, BAY treatment markedly reduced primary microglial density in the area surrounding the lesion compared with in the TBI group, suggesting that BAY may improve the morphology and phagocytic function of microglia, reducing the production of inflammatory factors following TBI to preserve neurological function. Subsequently, primary microglial cell migration/chemotaxis was evaluated using Transwell and agarose spot assays. BAY could markedly attenuate LPS-induced microglial migration (Fig. 3B). In addition, chemotaxis of microglial cells induced by LPS was assessed using an agarose spot test. The chemotactic capabilities of the microglial cells were significantly reduced by BAY compared with those in the LPS group (Fig. 3C).
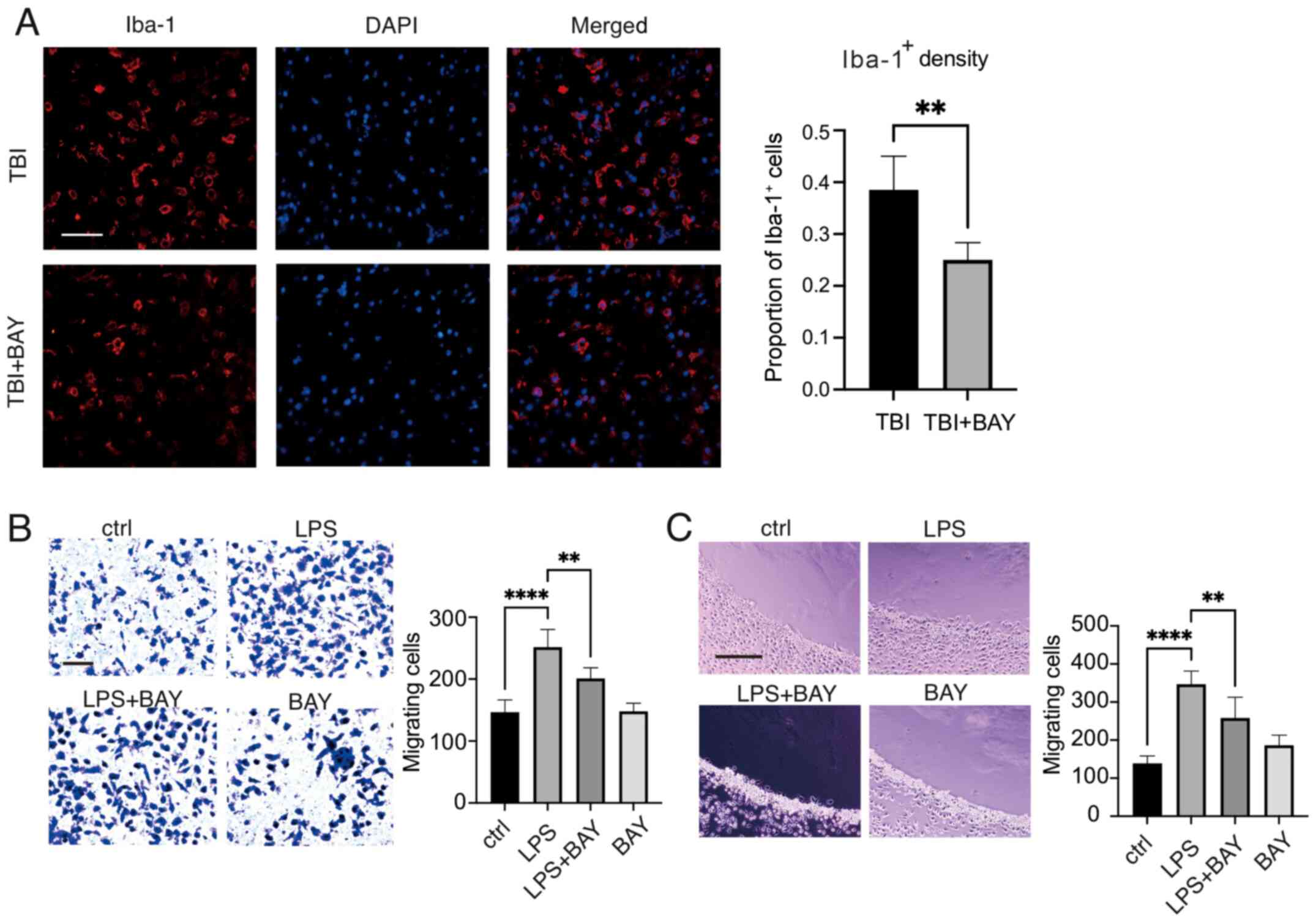 |
Figure 3
BAY regulates the function of microglia after TBI. (A) Microglial density was measured in mouse brain tissues following TBI using Iba-1 immunofluorescence staining. Scale bar, 50 µm (Iba-1, red; DAPI, blue). Microglial migration and chemotaxis were examined using. (B) Transwell assay and (C) agarose spot test. Scale bar, 50 µm. **P<0.01, ****P<0.0001. BAY, BAY61-3606; TBI, traumatic brain injury; Iba-1, ionized calcium binding adaptor molecule 1; LPS, lipopolysaccharide.
|
BAY protects the neurovascular unit in a mouse CCI model
Brain tissues were harvested from mice after TBI induction and BAY treatment for 7 days for neurological structure assessment. The area of peripheral edema surrounding the lesions following TBI were assessed. As shown in Fig. 4A and B, BAY treatment markedly increased neuronal cell and oligodendrocyte density in the surrounding injured area. Since endothelial cells form a core part of the cellular structure of the neurovascular unit (47), the density of endothelial cells was also determined. There was a significantly higher density of CD31-positive cells in the surrounding injured area in mouse brain tissues from the TBI + BAY group compared with that in the TBI group (Fig. 4C), suggesting increased neurovascular unit recovery or preservation. Furthermore, the degree of astrogliosis was assessed by semi-quantifying GFAP-positive cells; however, no notable differences were observed between the groups (Fig. 4D).
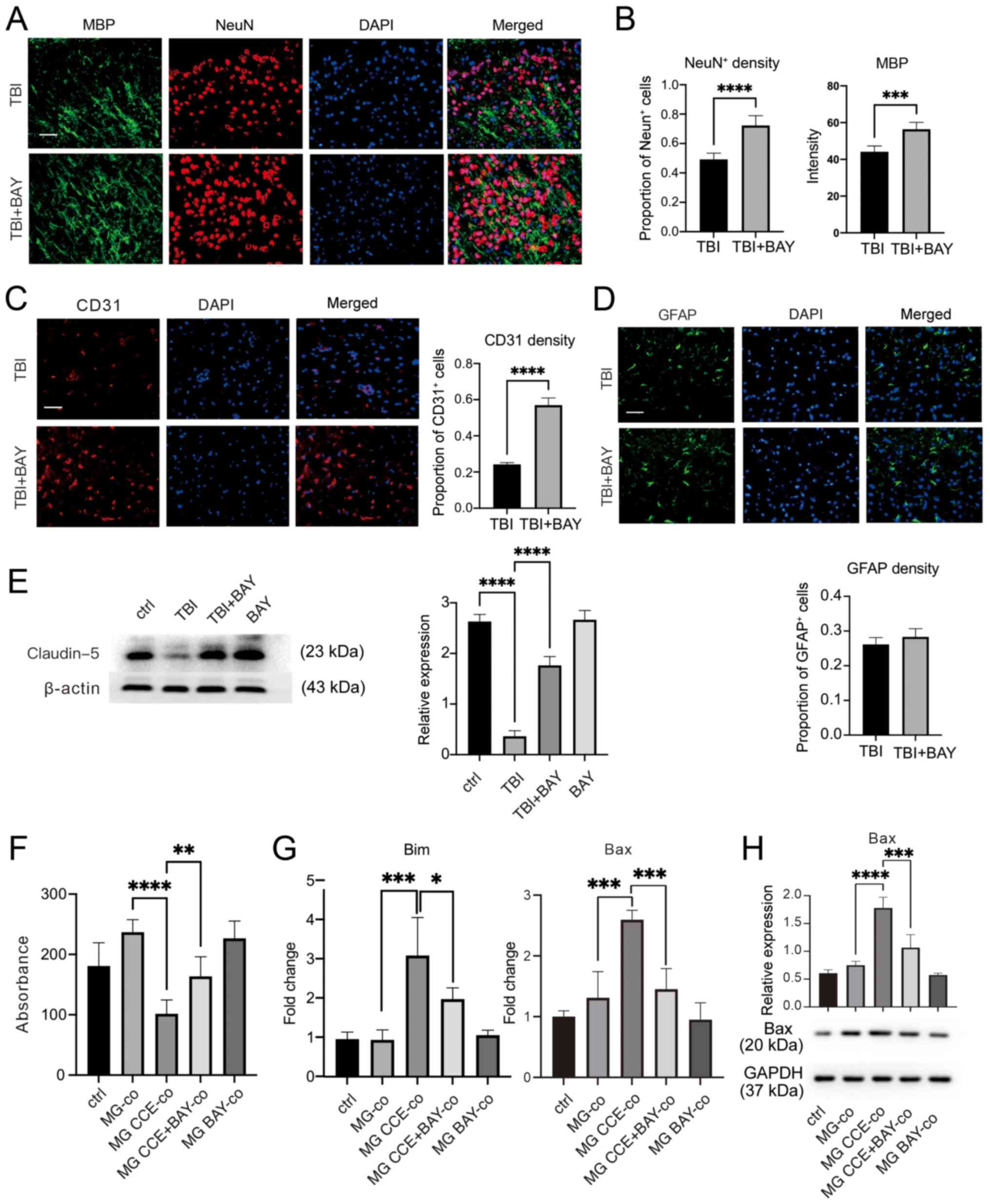 |
Figure 4
BAY protects the neurovascular unit in the mouse controlled cortical impact-indued TBI model. (A) TBI mice were administrated with BAY (3 mg/kg) for 7 days before the density of neuronal cells and oligodendrocytes in the surrounding lesion area were assessed by IF staining. Scale bar, 50 µm (MBP, green; NeuN, red; DAPI, blue). (B) Quantification of NeuN and MBP expression. (C) Cerebrovascular damage in the surrounding lesions in mice after TBI induction was assessed by IF staining. Scale bar, 50 µm (CD31, red; DAPI, blue). (D) Changes in astrocytes in the surrounding lesions in TBI mice were tested using IF. Scale bar, 50 µm (GFAP, green; DAPI, blue). (E) Expression of the tight junction marker protein Claudin-5 in mouse brain tissues was detected via western blotting. (F) Viability of PC12 cells co-cultured with MG treated with CCE was determined using Cell Counting Kit-8. (G) mRNA expression levels of apoptosis markers Bax and Bim were detected via reverse transcription-quantitative PCR. (H) Bax protein expression was detected via western blotting. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. BAY, BAY61-3606; TBI, traumatic brain injury; IF, immunofluorescence; NeuN, neuronal nuclear protein; GFAP, glial fibrillary acidic protein; MBP: myelin basic protein; MG, microglia; CCE, cerebral cortex extract.
|
Tight junctions serve a key role in maintaining the function of the neurovascular structure (48); therefore, the expression levels of the tight junction-associated protein Claudin-5 were determined. Increased protein expression levels of Claudin-5 were detected in BAY-treated TBI brain tissues compared with those in TBI brain tissues (Fig. 4E). Microglia have previously been shown to exert essential effects on neurological damage after TBI (49). To further investigate if the neuroprotective properties of BAY after TBI were mediated by affecting microglial function, primary cultured microglial cells were first treated with CCE to mimic TBI. Subsequently, these microglia were co-cultured with neuronal PC12 cells, which is a pheochromocytoma differentiated cell line from the adrenal medulla with general characteristics of neuroendocrine cells, with or without BAY treatment. The viability of neuronal cells was determined using the Cell Counting Kit-8 assay; BAY treatment markedly attenuated activated microglia-induced neuronal cell damage compared with in the microglia + CCE co-culture (Fig. 4F). In addition, the mRNA of PC12 cells was harvested and the expression levels of pro-apoptosis markers Bax and Bim (Fig. 4G) were detected via RT-qPCR, and Bax protein expression levels were detected via western blot (Fig. 4H). BAY markedly decreased activated microglia-induced Bax and Bim transcription in PC12 cells.
BAY attenuates neuroglial dysfunction in the mouse CCI model
The effects of BAY on neurological function after TBI were detected in the mouse CCI model. C57BL/10ScNJ mice were administered BAY (3 mg/kg, intraperitoneal) (30) for 7 days after TBI insult. The mice were then tested using the MWM. The mice underwent 6 days of training prior to the final test on day 7 (Fig. 5A). The distance moved, swimming speed and time spent crossing the platform were recorded during the 6 days. BAY markedly promoted the improvements in these parameters over time compared with in the TBI group (Fig. 5B-D). On day 7, the mice were examined further in the MWM (Fig. 5E). BAY-treated TBI mice also required less time to cross the platform and the respective quadrant (Fig. 5F). In addition, compared with those in the TBI group, BAY-treated TBI mice successfully crossed the platform and the effective zone more times (Fig. 5G and H). To assess the motor and balance function of BAY-treated TBI mice further, the rotarod test was applied to the mice after TBI and BAY treatment. BAY treatment significantly prolonged the latency on the examiner device compared with in the TBI group (Fig. 5I). Furthermore, the neurological function of the TBI model mice was tested using the mNSS system on days 1, 3, 7, 14 and 28. BAY-treated TBI mice achieved notable reductions in neurological deficit compared with the TBI mice on day 7 (Fig. 5J). Overall, these results suggested that BAY was able to improve the neurological function of TBI model mice.
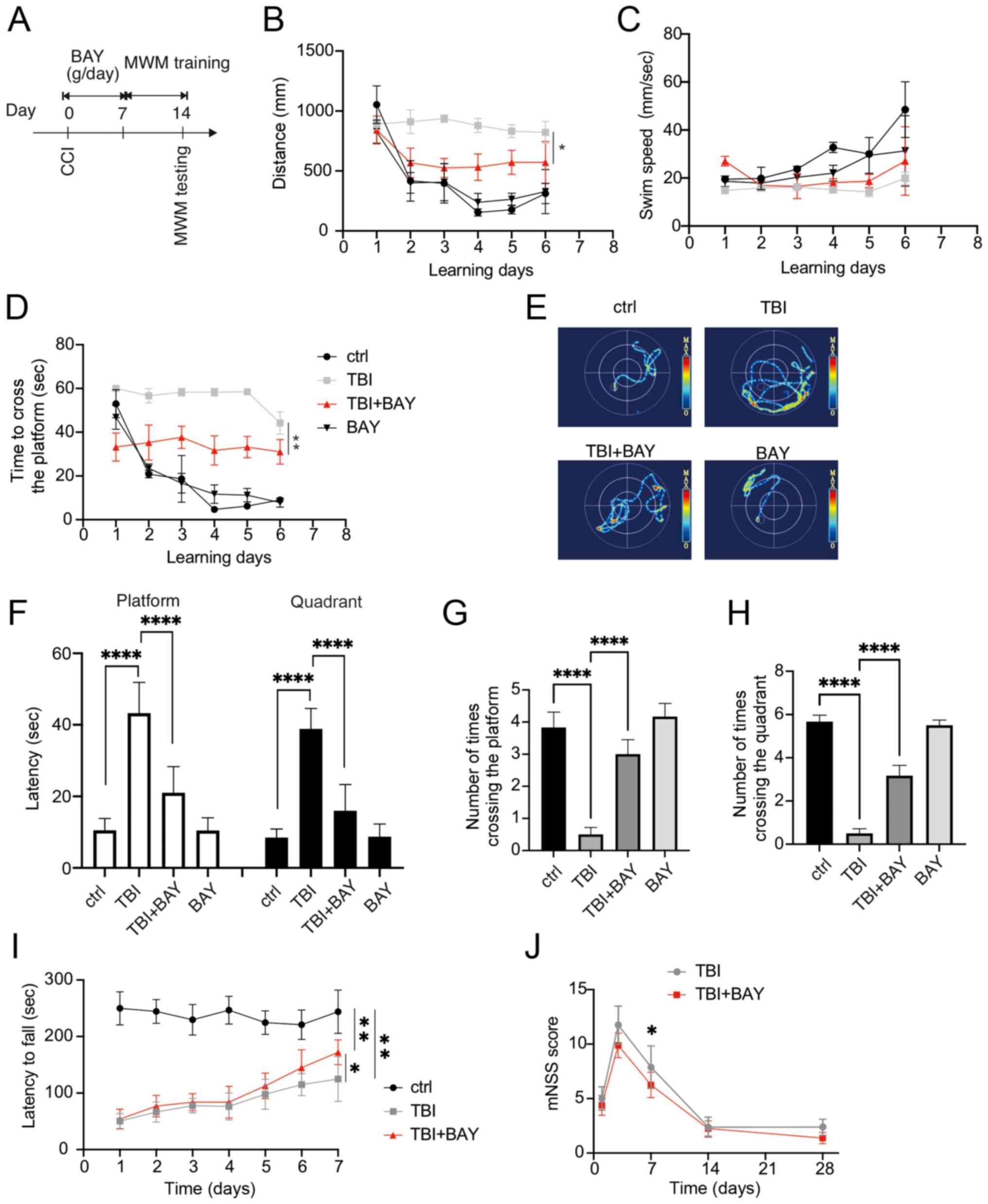 |
Figure 5
BAY attenuates neuroglial dysfunction in the mouse CCI model of TBI. (A) Overview of the experimental design. BAY (3 mg/kg) treatment was initiated immediately post-TBI for 7 days. Spatial learning training began from days 8 to 13 before the spatial memory test was performed on day 14 post-surgery in the MWM. (B) Distance of swam before finding the platform in spatial learning test, (C) swimming speed and (D) time spent to find the platform were recorded during the spatial learning test using the MWM on training days. (E) Representative heatmaps of spatial learning. (F) Time spent crossing the platform and zone. Number of times crossing (G) the platform and (H) zone during the spatial memory test as assessed using MWM on the testing day. (I) Rotarod motor test in TBI mice. (J) mNSS analysis of TBI mice. *P<0.05, **P<0.01, ****P<0.0001 as indicated or vs. TBI + BAY. CCI, controlled cortical impact; BAY, BAY61-3606; TBI, traumatic brain injury; MWM, Morris water maze; mNSS, modified neurological severity score.
|
Discussion
The present study explored a potential approach for the regulation of microglial phenotypic transformation, which serves an important role in neuroinflammatory injury and functional impairment following TBI. The Mincle/Syk/NF-κB signaling pathway was revealed to be involved in the process of microglial activation. As a blocker of Syk, BAY significantly promoted the transformation of microglia from a proinflammatory phenotype to an anti-inflammatory phenotype. In addition, BAY attenuated damage to components of the neurovascular unit, such as inflammatory factors ASC and caspase-1, and consequently protected brain function following TBI induction.
Although Mincle activation leads to inflammation, which serves to contain injury and initiate tissue repair (50), excessive activation can amplify collateral tissue damage, and has previously been implicated in numerous inflammatory diseases, including SAH and multiple sclerosis (51). As it was previously reported that Mincle can sense dead cells and potentiate inflammatory damage by promoting M1-type macrophage polarization in an acute kidney injury model (30), it was hypothesized that Mincle may also regulate microglial phenotypic transformation and the subsequent neuroinflammatory response in TBI. Expression and activation of the protein components in the Mincle/Syk/NF-κB signaling pathway were observed to be increased in the brain tissues isolated from human patients with TBI and from mice following CCI in the present study. M1-type microglial markers (CD86, TNF-α, IL-6, iNOS and IL-1β) were also detected in mouse brain tissue post-TBI and in CCE-stimulated microglia. This excessive activation of M1 microglia-induced inflammation upstream of neuroinflammatory injury may block neuroregeneration and revascularization.
Since a Mincle-specific inhibitor remains elusive, BAY was chosen to inhibit the downstream Syk/NF-κB pathway (8). BAY is a highly specific selective inhibitor of Syk that can block the activation of Syk; therefore, the Mincle signaling pathway cannot be further activated, leading to inactivation/attenuation of the downstream NF-κB pathway (45). NF-κB signaling has been largely revealed to serve crucial roles in pro-inflammatory processes (52), and downregulating the Mincle/Syk pathway could block NF-κB activation, which has been shown to inhibit inflammation in macrophages (53). Therefore, it was hypothesized that BAY may effectively induce the transformation of microglia from a pro-inflammatory to an anti-inflammatory phenotype by inhibiting the Mincle/Syk pathway. In the present study, BAY exhibited potent inhibitory effects on the phosphorylation of Syk and NF-κB, which may suppress the inflammatory response after TBI. BAY also promoted the microglial phenotypic transformation from M1 to M2, which reduced the expression of inflammation-related proteins, caspase-1 and ASC, and the production of proinflammatory cytokines, IL-1β, IL-6, TNF-α and caspase-1. In addition, BAY could protect the neurovascular unit, including preservation of the viability of neurons and oligodendrocytes, whilst promoting blood-brain barrier (BBB) integrity, as determined by detecting the BBB-related protein Claudin-5 in the surrounding lesion area. This may explain the underlying mechanism of BAY-mediated protection against neuroinflammation and neurological damage, by inhibiting microglial Mincle/Syk/NF-κB signaling after TBI in vivo and in vitro.
In conclusion, results from the present study indicated a role for Mincle/Syk/NF-κB in microglial activation and phenotypic transformation post-TBI. BAY, an inhibitor of Syk, may serve to be a potential therapeutic drug for attenuating neurovascular unit injury and improving neurological function after TBI.
Availability of data and materials
The datasets used and/or analyzed in the current study are available from the corresponding author upon reasonable request.
Authors' contributions
HZ and CG designed the study. XH performed the in vivo and in vitro experiments. YH was responsible for data analysis. YL, XZ, PY and ZM were responsible for tissue processing. XM, XL, YY, XW and JL performed the rest of the experiments. KS and TL made substantial contributions to conception and design of the study. HZ, XH and YH confirm the authenticity of all the raw data. All authors approved the final version of the manuscript.
Ethics approval and consent to participate
Ethics approval for animal experiments was received from the Experimental Animal Ethics Committee of Huazhong University of Science and Technology (approval no. TJH-202012001). With regard to patient-derived specimens, the present study was performed in accordance with the Declaration of Helsinki, and the study protocol and written informed consent were approved by the Medical Ethics Committee of Tongji Hospital (approval no. TJH-IRB 20200103). Written informed consent was signed by the next of kin of the patients and control individuals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Natural Science Foundation of Hubei province (grant no. WJ2019Z008), the Natural Science Foundation of Tongji Hospital (grant no. 2020JZKT651) and the Natural Science Foundation of China (grant no. 81801235). In addition, The First Affiliated Hospital, School of Medicine, Shihezi University provided a stipend for Xuejun He.
References
|
1
|
Rauen K, Reichelt L, Probst P, Schäpers B, Müller F, Jahn K and Plesnila N: Decompressive craniectomy is associated with good quality of life up to 10 years after rehabilitation from traumatic brain injury. Crit Care Med. 48:1157–1164. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fouad K, Popovich PG, Kopp MA and Schwab JM: The neuroanatomical-functional paradox in spinal cord injury. Nat Rev Neurol. 17:53–62. 2021. View Article : Google Scholar
|
|
3
|
Long X, Yao X, Jiang Q, Yang Y, He X, Tian W, Zhao K and Zhang H: Astrocyte-derived exosomes enriched with miR-873a-5p inhibit neuroinflammation via microglia phenotype modulation after traumatic brain injury. J Neuroinflammation. 17:892020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu H, Zheng J, Xu S, Fang Y, Wu Y, Zeng J, Shao A, Shi L, Lu J, Mei S, et al: Mer regulates microglial/macrophage M1/M2 polarization and alleviates neuroinflammation following traumatic brain injury. J Neuroinflammation. 18:22021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim H, Wang SY, Kwak G, Yang Y, Kwon IC and Kim SH: Exosome-guided phenotypic switch of M1 to M2 macrophages for cutaneous wound healing. Adv Sci (Weinh). 6:19005132019. View Article : Google Scholar
|
|
6
|
Cueto FJ, Del Fresno C and Sancho D: DNGR-1, a dendritic cell-specific sensor of tissue damage that dually modulates immunity and inflammation. Front Immunol. 10:31462020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Drouin M, Saenz J and Chiffoleau E: C-type lectin-like receptors: Head or tail in cell death immunity. Front Immunol. 11:2512020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Loane DJ and Kumar A: Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp Neurol. 275:316–327. 2016. View Article : Google Scholar
|
|
9
|
Hong X, Jiang F, Li Y, Fang L, Qian Z, Chen H and Kong R: Treatment with 5-methoxytryptophan attenuates microglia-induced neuroinflammation in spinal cord trauma. Int Immunopharmacol. 88:1069882020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
N'diaye M, Brauner S, Flytzani S, Kular L, Warnecke A, Adzemovic MZ, Piket E, Min JH, Edwards W, Mela F, et al: C-type lectin receptors Mcl and Mincle control development of multiple sclerosis-like neuroinflammation. J Clin Invest. 130:838–852. 2020. View Article : Google Scholar
|
|
11
|
Tanaka M, Saka-Tanaka M, Ochi K, Fujieda K, Sugiura Y, Miyamoto T, Kohda H, Ito A, Miyazawa T, Matsumoto A, et al: C-type lectin Mincle mediates cell death-triggered inflammation in acute kidney injury. J Exp Med. 217:e201922302020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
He Y, Xu L, Li B, Guo ZN, Hu Q, Guo Z, Tang J, Chen Y, Zhang Y, Tang J, et al: Macrophage-inducible C-type lectin/spleen tyrosine kinase signaling pathway contributes to neuroinflammation after subarachnoid hemorrhage in rats. Stroke. 46:2277–2286. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Iizasa E, Chuma Y, Uematsu T, Kubota M, Kawaguchi H, Umemura M, Toyonaga K, Kiyohara H, Yano I, Colonna M, et al: TREM2 is a receptor for non-glycosylated mycolic acids of mycobacteria that limits anti-mycobacterial macrophage activation. Nat Commun. 12:22992021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee EJ, Brown BR, Vance EE, Snow PE, Silver PB, Heinrichs D, Lin X, Iwakura Y, Wells CA, Caspi RR, et al: Mincle activation and the Syk/Card9 signaling axis are central to the development of autoimmune disease of the eye. J Immunol. 196:3148–3158. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arumugam TV, Manzanero S, Furtado M, Biggins PJ, Hsieh YH, Gelderblom M, MacDonald KP, Salimova E, Li YI, Korn O, et al: An atypical role for the myeloid receptor Mincle in central nervous system injury. J Cereb Blood Flow Metab. 37:2098–2111. 2017. View Article : Google Scholar :
|
|
16
|
Richardson MB and Williams SJ: MCL and Mincle: C-type lectin receptors that sense damaged self and pathogen-associated molecular patterns. Front Immunol. 5:2882014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xie Y, Guo H, Wang L, Xu L, Zhang X, Yu L, Liu Q, Li Y, Zhao N, Zhao N, et al: Human albumin attenuates excessive innate immunity via inhibition of microglial Mincle/Syk signaling in subarachnoid hemorrhage. Brain Behav Immun. 60:346–360. 2017. View Article : Google Scholar
|
|
18
|
Kim JW, Roh YS, Jeong H, Yi HK, Lee MH, Lim CW and Kim B: Spliceosome-associated protein 130 exacerbates alcohol-induced liver injury by inducing NLRP3 inflammation related proteins-mediated IL-1β in mice. Am J Pathol. 188:967–980. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kezić A, Stajic N and Thaiss F: Innate immune response in kidney ischemia/reperfusion injury: Potential target for therapy. J Immunol Res. 2017:63054392017. View Article : Google Scholar
|
|
20
|
Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, Avanzi A, Barilla R, Daley D, Greco SH, et al: The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 532:245–249. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dzharullaeva AS, Tukhvatulin AI, Erokhova AS, Bandelyuk AS, Polyakov NB, Solovyev AI, Nikitenko NA, Shcheblyakov DV, Naroditsky BS, Logunov DY, et al: Stimulation of dectin-1 and dectin-2 during parenteral immunization, but not Mincle, induces secretory IgA in intestinal mucosa. J Immunol Res. 2018:38357202018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Khan A, Braganza CD, Kodar K, Timmer MS and Stocker BL: Stereochemistry, lipid length and branching influences Mincle agonist activity of monoacylglycerides. Org Biomol Chem. 18:425–430. 2020. View Article : Google Scholar
|
|
23
|
Lv LL, Tang PMK, Li CJ, You YK, Li J, Huang XR, Ni J, Feng M, Liu BC and Lan HY: The pattern recognition receptor, Mincle, is essential for maintaining the M1 macrophage phenotype in acute renal inflammation. Kidney Int. 91:587–602. 2017. View Article : Google Scholar
|
|
24
|
Pandori WJ, Lima TS, Mallya S, Kao TH, Gov L and Lodoen MB: Toxoplasma gondii activates a Syk-CARD9-NF-κB signaling axis and gasdermin D-independent release of IL-1β during infection of primary human monocytes. PLoS Pathog. 15:e10079232019. View Article : Google Scholar
|
|
25
|
Liu HD, Li W, Chen ZR, Hu YC, Zhang DD, Shen W, Zhou ML, Zhu L and Hang CH: Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res. 38:2072–2083. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
de Rivero Vaccari JC, Brand FJ III, Berti AF, Alonso OF, Bullock MR and de Rivero Vaccari JP: Mincle signaling in the innate immune response after traumatic brain injury. J Neurotrauma. 32:228–236. 2015. View Article : Google Scholar
|
|
27
|
Tümmler C, Dumitriu G, Wickström M, Coopman P, Valkov A, Kogner P, Johnsen JI, Moens U and Sveinbjörnsson B: SYK inhibition potentiates the effect of chemotherapeutic drugs on neuroblastoma cells in vitro. Cancers (Basel). 11:2022019. View Article : Google Scholar
|
|
28
|
Kim SY, Park SE, Shim SM, Park S, Kim KK, Jeong SY, Choi EK, Hwang JJ, Jin DH, Chung CD, et al: Bay 61-3606 sensitizes TRAIL-induced apoptosis by downregulating Mcl-1 in breast cancer cells. PLoS One. 10:e01460732015. View Article : Google Scholar
|
|
29
|
Stenberg M, Godbolt AK, Nygren De Boussard C, Levi R and Stålnacke BM: Cognitive impairment after severe traumatic brain injury, clinical course and impact on outcome: A swedish-icelandic study. Behav Neurol. 2015:6803082015. View Article : Google Scholar
|
|
30
|
Tan RZ, Li JC, Liu J, Lei XY, Zhong X, Wang C, Yan Y, Linda Ye L, Darrel Duan D, Lan HY, et al: BAY61-3606 protects kidney from acute ischemia/reperfusion injury through inhibiting spleen tyrosine kinase and suppressing inflammatory macrophage response. FASEB J. 34:92020. View Article : Google Scholar
|
|
31
|
Xiong J, Ni J, Chen C and Wang K: miR-148a-3p regulates alcoholic liver fibrosis through targeting ERBB3. Int J Mol Med. 46:1003–1012. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tighe RM, Birukova A, Yaeger MJ, Reece SW and Gowdy KM: Euthanasia- and lavage-mediated effects on bronchoalveolar measures of lung injury and inflammation. Am J Respir Cell Mol Biol. 59:257–266. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Prinz M and Hanisch UK: Murine microglial cells produce and respond to interleukin-18. J Neurochem. 72:2215–2218. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Markovic DS, Glass R, Synowitz M, Rooijen N and Kettenmann H: Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J Neuropathol Exp Neurol. 64:754–762. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ifuku M, Hinkelmann L, Kuhrt LD, Efe IE, Kumbol V, Buonfiglioli A, Krüger C, Jordan P, Fulde M, Noda M, et al: Activation of Toll-like receptor 5 in microglia modulates their function and triggers neuronal injury. Acta Neuropathol Commun. 8:1592020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li Q, Cheng K, Wang AY, Xu QG, Fu ZF, He SY and Xu PX: microRNA-126 inhibits tube formation of HUVECs by inter-acting with EGFL7 and down-regulating PI3K/AKT signaling pathway. Biomed Pharmacother. 116:1090072019. View Article : Google Scholar
|
|
37
|
Barnhart CD, Yang D and Lein PJ: Using the Morris water maze to assess spatial learning and memory in weanling mice. PLoS One. 10:e01245212015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu ZH, Chen NY, Tu PH, Wu CT, Chiu SC, Huang YC, Lim SN and Yip PK: DHA attenuates cerebral edema following traumatic brain injury via the reduction in blood-brain barrier permeability. Int J Mol Sci. 21:62912020. View Article : Google Scholar
|
|
39
|
Shen M, Wang S, Wen X, Han XR, Wang YJ, Zhou XM, Zhang MH, Wu DM, Lu J and Zheng YL: Dexmedetomidine exerts neuroprotective effect via the activation of the PI3K/Akt/mTOR signaling pathway in rats with traumatic brain injury. Biomed Pharmacother. 95:885–893. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nam HS, Kwon I, Lee BH, Kim H, Kim J, An S, Lee OH, Lee PH, Kim HO, Namgoong H, et al: Effects of mesenchymal stem cell treatment on the expression of matrix metalloproteinases and angiogenesis during ischemic stroke recovery. PLoS One. 10:e01442182015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shen C, Sun FL, Zhang RY, Zhang L, Li YL, Zhang L and Li L: Tetrahydroxystilbene glucoside ameliorates memory and movement functions, protects synapses and inhibits α-synuclein aggregation in hippocampus and striatum in aged mice. Restor Neurol Neurosci. 33:531–541. 2015.
|
|
42
|
Winston CN, Romero HK, Ellisman M, Nauss S, Julovich DA, Conger T, Hall JR, Campana W, O'Bryant SE, Nievergelt CM, et al: Assessing neuronal and astrocyte derived exosomes from individuals with mild traumatic brain injury for markers of neurodegeneration and cytotoxic activity. Front Neurosci. 13:10052019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔC(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
44
|
Huang Y, Li Q, Tian H, Yao X, Bakina O, Zhang H, Lei T and Hu F: MEK inhibitor trametinib attenuates neuroinflammation and cognitive deficits following traumatic brain injury in mice. Am J Transl Res. 12:6351–6365. 2020.PubMed/NCBI
|
|
45
|
Willis EF, MacDonald KPA, Nguyen QH, Garrido AL, Gillespie ER, Harley SB, Bartlett PF, Schroder WA, Yates AG, Anthony DC, et al: Repopulating microglia promote brain repair in an IL-6-dependent manner. Cell. 180:833–846.e16. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li C, Xue VW, Wang QM, Lian GY, Huang XR, Lee TL, To KF, Tang PM and Lan HY: The Mincle/Syk/NF-κB signaling circuit is essential for maintaining the protumoral activities of tumor-associated macrophages. Cancer Immunol Res. 8:1004–1017. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Begum G, Song S, Wang S, Zhao H, Bhuiyan MI, Li E, Nepomuceno R, Ye Q, Sun M, Calderon MJ, et al: Selective knockout of astrocytic Na+/H+ exchanger isoform 1 reduces astrogliosis, BBB damage, infarction, and improves neurological function after ischemic stroke. Glia. 66:126–144. 2018. View Article : Google Scholar
|
|
48
|
Vázquez-Rosa E, Shin MK, Dhar M, Chaubey K, Cintrón-Pérez CJ, Tang X, Liao X, Miller E, Koh Y, Barker S, et al: P7C3-A20 treatment one year after TBI in mice repairs the blood-brain barrier, arrests chronic neurodegeneration, and restores cognition. Proc Natl Acad Sci USA. 117:27667–27675. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bellver-Landete V, Bretheau F, Mailhot B, Vallières N, Lessard M, Janelle ME, Vernoux N, Tremblay ME, Fuehrmann T, Shoichet MS, et al: Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat Commun. 10:5182019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kawata K, Illarionov P, Yang GX, Kenny TP, Zhang W, Tsuda M, Ando Y, Leung PSC, Ansari AA and Gershwin ME: Mincle and human B cell function. J Autoimmun. 39:315–322. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Inoue T: M1 macrophage triggered by Mincle leads to a deterioration of acute kidney injury. Kidney Int. 91:526–529. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Roth S, Bergmann H, Jaeger M, Yeroslaviz A, Neumann K, Koenig PA, Prazeres da Costa C, Vanes L, Kumar V, Johnson M, et al: Vav proteins are key regulators of Card9 signaling for innate antifungal immunity. Cell Rep. 17:2572–2583. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Schierwagen R, Uschner FE, Ortiz C, Torres S, Brol MJ, Tyc O, Gu W, Grimm C, Zeuzem S, Plamper A, et al: The role of macrophage-inducible C-type lectin in different stages of chronic liver disease. Front Immunol. 11:13522020. View Article : Google Scholar : PubMed/NCBI
|














