Introduction
It is well known that cerebral neurons have
different suscep-tibilities to deleterious conditions, such as
ischemic injury and neurodegeneration, depending on brain area
(1). The hippocampus is one of
the brain areas vulnerable to ischemic insults, and hippocampal
neurons respond differently to ischemic damage according to its
subfields (CA1-3) and impact of ischemia. A short period (5 min) of
transient forebrain ischemia (TFI) in gerbils induces death of
pyramidal neurons located in hippocampal CA1 at 4 or 5 days after
TFI, but pyramidal neurons of CA2/3 are resistant (2,3).
Hippocampal neuronal vulnerability to cerebral ischemia can be
altered by body temperature or ischemic duration (4,5).
Higher temperatures cause more severe ischemic damage. It was
reported that in gerbils, hyperthermia before and during a brief
episode (5 min) of TFI resulted in the accelerated death of
hippocampal CA1 pyramidal neurons (6). Clinically, increased body
temperature during the first day after stroke is related to poor
outcomes and leads to devastating effects on mortality (7).
Hypoxia-inducible factor 1α (HIF-1α), a well-known
isoform of hypoxia-inducible factors, exerts hypoxia-inducible
transcriptional activity triggered by low oxygen conditions,
whereas HIF-1β is an oxygen-insensitive subunit (8). HIF-1α expression is localized in
the cytosol of pyramidal neurons located in all hippocampal
subfields (CA1-3) of rats and gerbils (9-11). HIF-1α plays a key role in
regulating cellular adaptation to low oxygen conditions by
modulating gene expression, targeting cell survival, angiogenesis,
and glucose metabolism, which may contribute to alleviating
ischemic neuronal damage (12,13). Increased HIF-1α expression
significantly attenuates post-ischemic damage of hippocampal CA1
pyramidal neurons by increasing the expression of nuclear factor-κB
(NF-κB) and vascular endothelial growth factor (VEGF) following
ischemic preconditioning (9). In
addition, systemic administration of a HIF-1α activator (ML228) was
found to alleviate neuronal apoptosis in hippocampal CA1 by
attenuating the amplified expression of pro-inflammatory cytokines
and their receptors following cardiac arrest-induced cerebral
ischemia (14).
Although previous studies have reported the effects
of HIF-1α-mediated neuroprotection and its related signaling
pathways, few studies have been conducted on changes in HIF-1α
expression in hippocampal subfields induced by TFI under
hyperthermic conditions that cause more severe neuronal damage.
Therefore, the present study aimed to investigate the pattern of
neuronal death and expression of HIF-1α in hippocampal subfields
(CA1-3) after 5 min TFI under hyperthermic conditions in
gerbils.
Materials and methods
Experimental animals and groups
In the present study, a total of 240 male gerbils
(age, 6 months; body weight, 70±5 g) were used at the start of the
experiment. The gerbils were bred and kept in the Experimental
Animal Center at Kangwon National University located in Chuncheon
(Korea). The animals were housed in a conventional room
(temperature, 22±2°C; humidity, 57±5%; light/dark cycle, 12:12)
with freely accessible water and food. All experimental procedures
were approved (approval no. KW-200113-1) by the Institutional
Animal Care and Use Committee on January 13, 2020. The Animal care
and handling conformed to the NIH Guide for the Care and Use of
Laboratory Animals (The National Academies Press, 8th edition,
2011) (15). All efforts were
taken at each stage of the experiments to minimize the numbers of
animals used and to limit any discomfort to which the animals might
be exposed.
Experimental gerbils (total n=240) were divided into
four groups: i) sham-operated gerbils under normothermia (NT/sham
group; n=36), ii) TFI-operated gerbils under normothermia
(NT/ischemia group; n=84), iii) sham-operated gerbils under
hyperthermia (HT/sham group; n=36), iv) TFI-operated gerbils under
hyperthermia (HT/ischemia group; n=84).
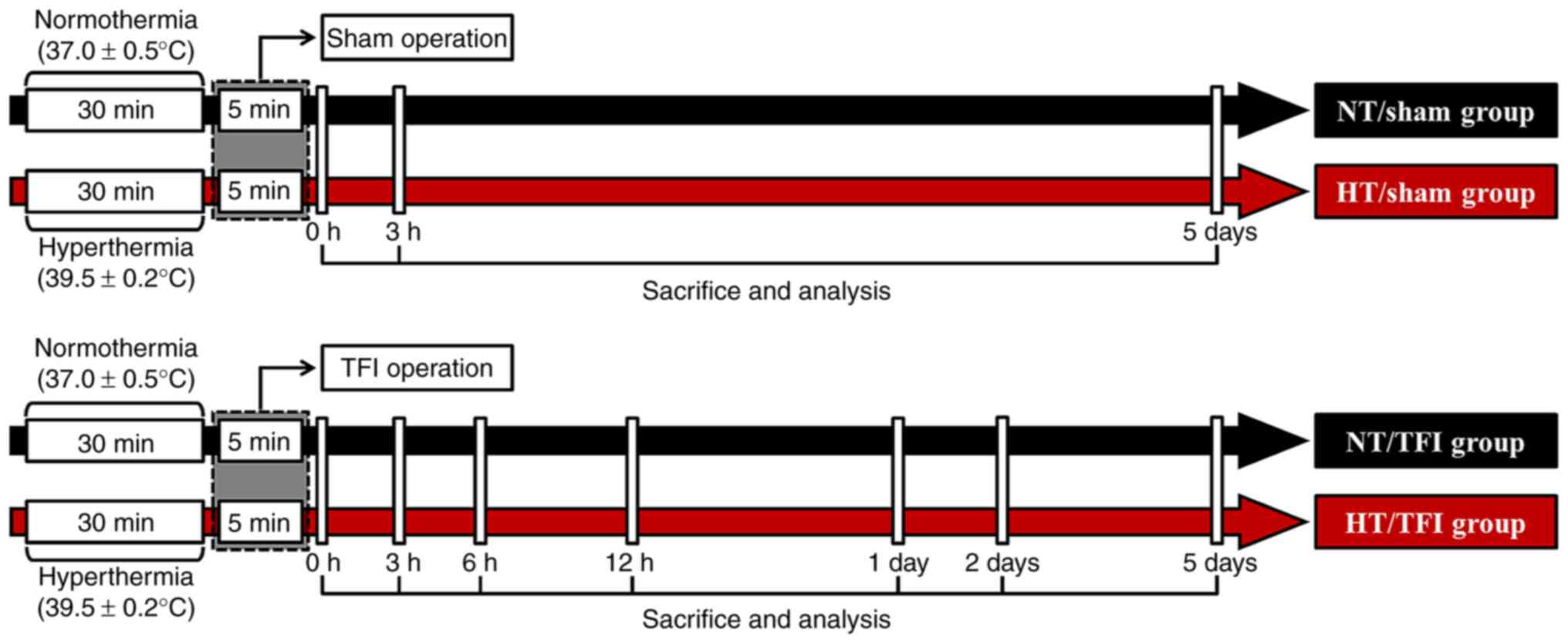
In each group, 12 gerbils (n=5 for western blotting;
n=7 for histology) were sacrificed at 0 h (immediately after 5 min
of TFI operation), 3, 6 and 12 h, 1, 2 and 5 days after TFI
(Fig. 1).
Induction of TFI under normothermia or
hyperthermia
TFI induction was performed according to previously
described methods (16,17) with minor modification. The
gerbils were anesthetized using inhalation anesthesia machine
(Harvard Apparatus) with isoflurane (2.5%; Baxtor) in oxygen (33%)
and nitrous oxide (67%). Under anesthesia, the bilateral common
carotid arteries, which are located in the ventral neck, were
ligated with aneurysm clips for 5 min. Before and during the TFI
surgery, body temperature was controlled using a heating pad, which
was connected to a rectal thermistor (Harvard Apparatus) (Fig. 1). Rectal temperature was
controlled at 37.0±0.5°C for normothermia and elevated up to
39.5±0.2°C for hyperthermia for 30 min. After the TFI surgery, the
gerbils were kept in thermal incubators (temperature, 22±2°C;
humidity, 57±5%) to adjust body temperature on a normothermic
level. The gerbils in the sham groups with normothermia or
hyperthermia received the same TFI surgery without ligation of the
arteries.
Western blot analysis
To analyze HIF-1α protein level in CA1 and CA2/3,
the gerbils of each group were sacrificed under profound anesthesia
by intraperitoneal (i.p.) injection of pentobarbital sodium (200
mg/kg) (18) (JW Pharmaceutical
Co., Ltd.) at the designated time (0, 3, 6 and 12 h, 1, 2 and 5
days after TFI). The death of animals was confirmed according to
vital signs including heart beats, pupillary response, and
respiratory pattern (lack of cardiac activity for 5 min by cardiac
palpation, unresponsiveness to light with dilated pupils using
light into the eyes of the animal, and lack of spontaneously
breathing pattern with shallow and irregular breathing pattern).
CA1 and CA2/3 tissues were respectively collected from the
hippocampi using brain matrix. Proteins of CA1 and CA2/3 were
extracted according to a previously published method (9) using rabbit anti-HIF-1α (cat. no.
ab228649, diluted 1:5,000, Abcam) and anti-β-actin (cat. no.
ab8227, diluted 1:20,000, Abcam) as primary antibodies.
For quantification of the bands, densitometry
analysis was performed using Scion Image software, Ver 4.0 (Scion
Corp.). The ratio, as relative density, of HIF-1α protein was
calibrated with the corresponding expression rate of β-actin and
normalized to that in NT/sham group at 0 h.
Tissue processing for histology
For histology, the gerbils of each group were
profoundly anesthetized with 200 mg/kg (i.p.) of pentobarbital
sodium (18) (JW Pharmaceutical
Co., Ltd.) at the designated time (0, 3, 6 and 12 h, 1, 2 and 5
days after TFI) and sacrificed after confirmation of death
according to the method described above. As previously described
(16,17), in short, the gerbils were
perfused transcardially with 4% paraformaldehyde and their brains
were extracted from the skulls. The brains were post-fixed in the
same fixative for 5 h. The fixed brain tissues were infiltrated
with 30% sucrose as cryoprotective agent when the brain tissues
were cut. To prepare brain sections containing the hippocampus,
using a cryostat, the frozen tissues were frontally cut into a
30-µm thickness. Thereafter, the sections were stored in
6-well plates containing PBS for histological staining.
Histochemistry using cresyl violet
(CV)
To examine the morphology and distribution of
hippocampal cells in all groups, CV histochemical staining was
conducted as previously described (9). In brief, the sections were stained
with 1.0% CV acetate (Sigma-Aldrich; Merck KGaA) containing 0.28%
glacial acetic acid. The stained sections were dehydrated using
ethanol series and mounted with Canada balsam.
For analysis of hippocampal change following TFI,
CV-stained structures were observed using AxioM1 light microscope
at ×4 and ×20 magnifications (Carl Zeiss). In this examination, six
sections per gerbil were selected at 120-µm intervals.
Histofluorescence using Fluoro-Jade B
(FJB)
To examine TFI-induced neuronal death (loss) in CA1
and CA2/3, histofluorescence using FJB, a representative marker of
neuronal degeneration/death, was conducted according to previously
published studies (16,17). In brief, the brain tissues were
soaked in 0.06% potassium permanganate and incubated in 0.0004% FJB
(Histochem). Thereafter, the stained sections were prepared for
observation.
For analysis of FJB-positive cells, images of
FJB-positive cells were captured using a fluorescence microscope
(Carl Zeiss) with blue excitation fluorescence filter (450-490 nm).
The captured images (FJB-positive cells) were analyzed using image
analyzing software (NIH Image J 1.59). Cell count was performed in
400 µm2 at middle of the stratum pyramidale of
CA1 and CA2/3.
Immunohistochemistry
To examine changes in neurons and HIF-1α expression,
immunohistochemistry was performed according to previously
published methods (9,19,20). In short, the brain sections were
immersed in 0.3% hydrogen peroxide (H2O2) and
soaked in 5% normal goat serum. Thereafter, the brain sections were
incubated with each primary antibody, rabbit anti-neuronal nuclei
(cat. no. MAB377, NeuN; diluted 1:1,100, Chemicon International)
and rabbit anti-HIF-1α (cat. no. ab8366, diluted 1:500, Abcam)
overnight at 4°C. Subsequently, they were incubated in secondary
antibodies, biotinylated goat anti-rabbit IgG (cat. no.
BA-1000-1.5, diluted 1:200, Vector Laboratories, Inc.) and
streptavidin peroxidase complex (diluted 1:200, Vector
Laboratories, Inc.). Finally, the sections were visualized using
0.05% 3,3′-diaminobenzidine tetrahydrochloride.
Analyses of the numbers of NeuN immunoreactive
neurons and HIF-1α immunoreactivity in CA1 and CA2/3 were conducted
according to previously published methods (9,19-21). In short, images of NeuN
immunoreactive structures and HIF-1α immunoreactive structures were
captured using an AxioM1 light microscope at ×20 magnification. To
quantitatively analyze the numbers of NeuN immunoreactive neurons,
the neurons were counted in 400 µm2 at the middle
of CA1 and CA2/3. To quantitatively analyze HIF-1α
immunoreactivity, the immunoreactive structures were captured at
the same area of interest and calibrated into an array of 512×512
pixels. Finally, the density of the HIF-1α immunoreactive
structures was relatively evaluated using Adobe Photoshop version
8.0 and NIH Image J 1.59 software.
Statistical analysis
Data obtained in this study are expressed as the
mean ± SEM. We statistically analyzed the differences of the means
between all groups by analysis of variance (ANOVA) with a post hoc
Bonferroni's multiple comparison tests using SPSS program (version
18.0, IBM SPSS Statistics). For all statistical analyses, P-values
<0.05 were considered statistically significant.
Results
Differences in neuronal damage/death
Findings by CV histochemistry
CV-stained cells were easily identified in all
subfields of the gerbil hippocampus of the NT/sham group. In
particular, large pyramidal cells as principal neurons consisted of
the stratum pyramidale (SP) (Fig.
2A-C). In the NT/ischemia group, the distribution of CV-stained
cells was not altered until 2 days after TFI in all subfields
(Fig. 2D-F). However, on day 5
after TFI, CV-stained pyramidal cells of the SP were apparently
damaged in CA1, but those located in CA2/3 were similar to those in
the NT/sham group (Fig.
2G-I).
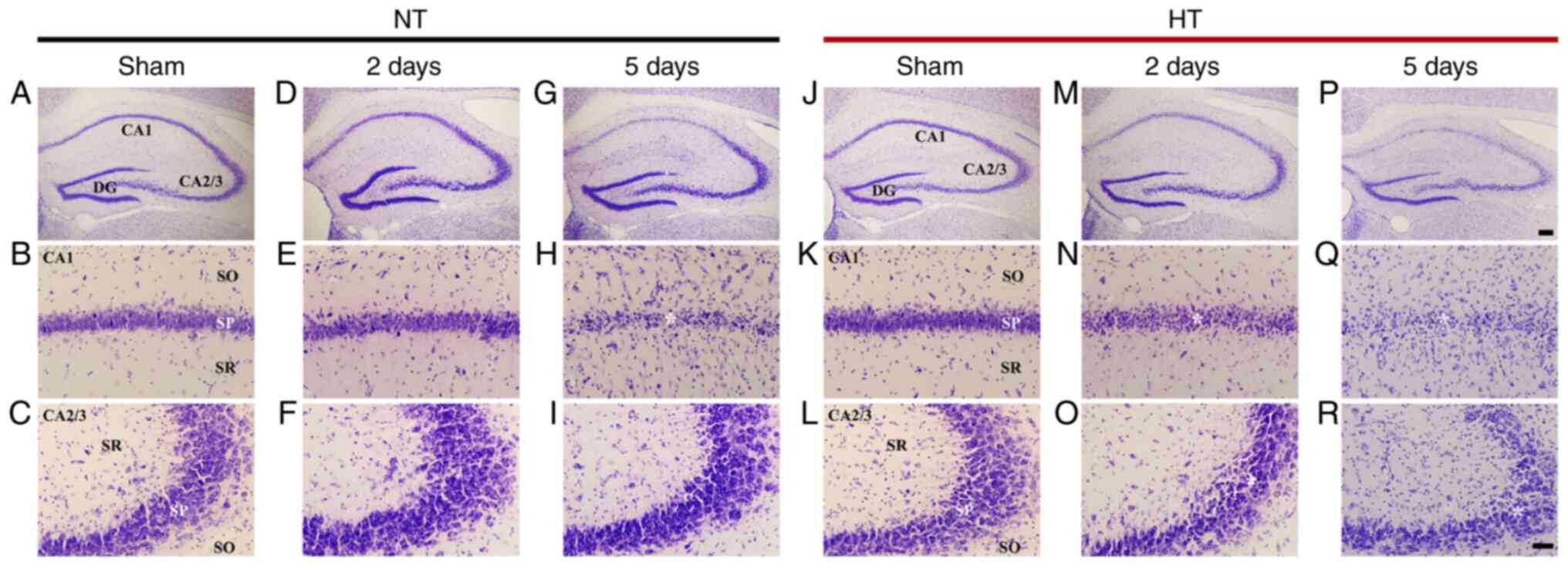 | Figure 2(A-R) Cresyl violet (CV) staining in
CA1 and CA2/3 of the NT/sham (A-C), NT/ischemia (D-I), HT/sham
(J-L), and HT/ischemia (M-R) groups on day 2 and 5 after TFI.
CV-stained cells (asterisk in H) in the stratum pyramidale (SP) of
CA1 of the NT/ischemia group were severely damaged on day 5 after
TFI. In the HT/ischemia group, decreased stainability in CV-stained
cells (asterisks in N, O) is shown in the SP of both CA1 and CA2/3
on day 2 after TFI. On day 5 after TFI, CV-stained cells (asterisks
in Q, R) were apparently damaged in the SP of both CA1 and CA2/3.
Scale bar, 200 µm (upper row) and 50 µm (middle and
lower rows). DG, dentate gyrus; SO, stratum oriens; SR, stratum
radiatum; NT, normothermia; HT, hyperthermia. |
In the HT/sham group, the distribution of CV-stained
cells in all subfields was not different from that in the NT/sham
group (Fig. 2J-L). In the
HT/ischemia group, on day 2 after TFI, most of the pyramidal cells
in CA1 were weakly stained by CV (Fig. 2N), and CV-stained cells in CA2/3
were not significantly different from those of the HT/sham group
(Fig. 2O). On day 5 after TFI,
most of the CV-stained pyramidal cells in CA1 were severely
damaged, and CA2/3 pyramidal cells showed weak CV staining or were
damaged (Fig. 2P-R).
Findings by FJB histofluorescence
CA1
FJB fluorescence staining revealed no FJB-stained
degenerating (or dead) cells in the SP of CA1 in the NT/sham group
(Fig. 3A-a). In the NT/ischemia
group, FJB-stained cells were not found in the SP on days 1 and 2
after TFI (Fig. 3A-c and e).
However, a significant increase in the number of FJB-stained cells
(36.2±2.7) was observed in the SP on day 5 after TFI (Fig. 3A-g and q).
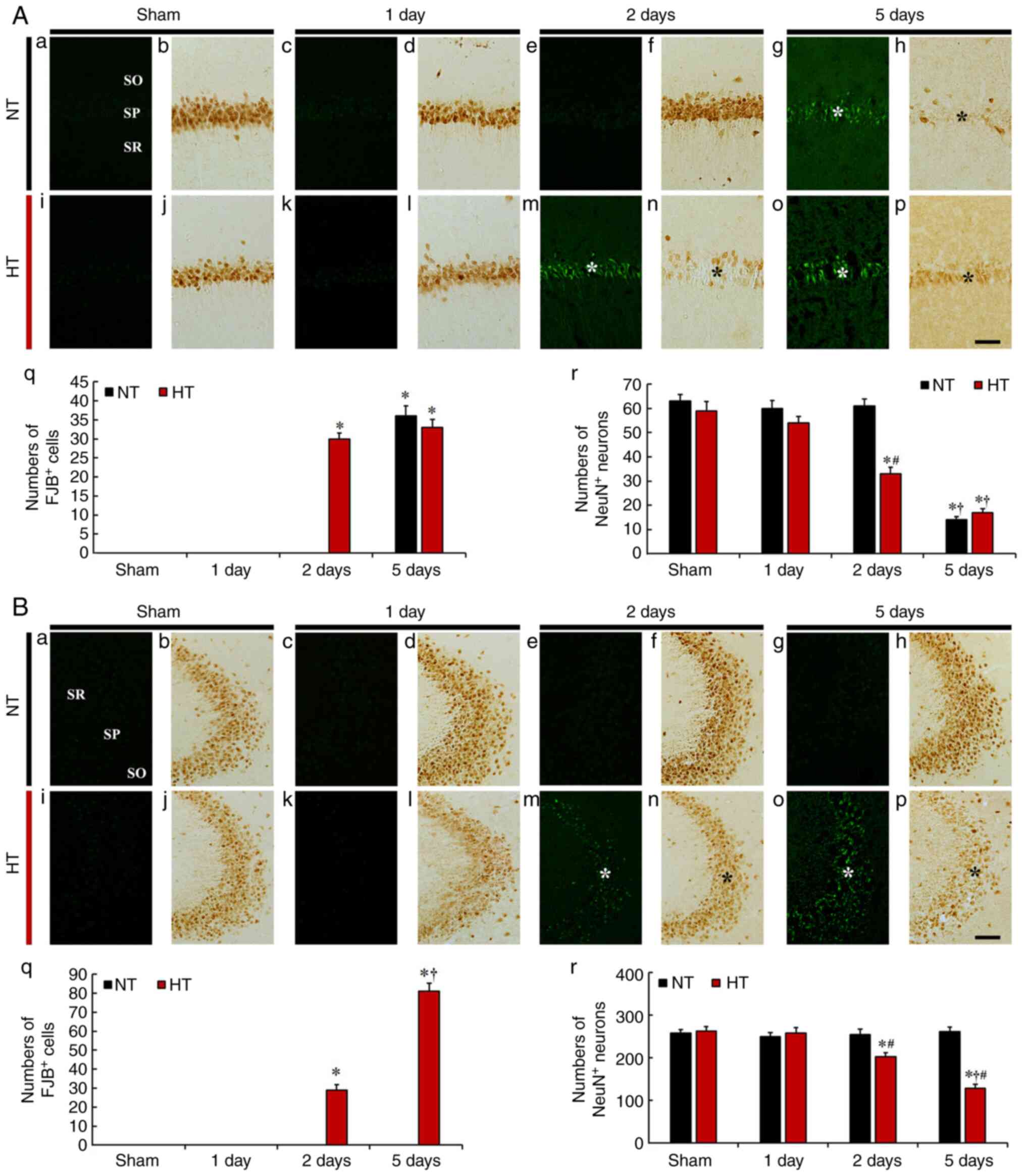 | Figure 3(A and B) Fluoro-Jade B (FJB)
histofluorescence staining and anti-neuronal nuclei (NeuN)
immunohistochemistry in CA1 (A) and CA2/3 (B) of the NT/sham (a and
b), NT/ischemia (c-h), HT/sham (i and j) and HT/ischemia (k-p)
groups on day 1, 2 and 5 after TFI. In the NT/ischemia group, many
FJB-stained cells (white asterisk in A-g) were observed in the SP
on day 5 after TFI in CA1, but not in CA2/3. However, in the
HT/ischemia group, many FJB-stained cells (white asterisks in m and
o) were detected in both CA1 and CA2/3 from 2 days after TFI. The
numbers of NeuN-immunostained pyramidal cells of the NT/ischemia
group were significantly reduced (black asterisk in A-h) only in
CA1 on day 5 after TFI. In the HT/ischemia group, the numbers of
NeuN-immunostained pyramidal cells were decreased (black asterisks
in n and p) in both CA1 and CA2/3 from 2 days after TFI. Scale bar,
50 µm. (q) Numbers of FJB-stained cells in CA1 (A) and CA2/3
(B). (r) Numbers of NeuN-immunostained cells in CA1 (A) and CA2/3
(B). *P<0.05 vs. NT/sham group; †P<0.05
vs. pre-time point group; #P<0.05 vs. NT/ischemia
group. The bars indicate the means ± SEM (n=7, respectively). Note:
the use of only small letters indicates these panels in both A and
B. |
In the HT/sham group, similar to the NT/sham group,
FJB-stained cells were not found in the SP (Fig. 3A-i). In the HT/ischemia group, a
few FJB-stained cells were detected in the SP on day 1 after TFI
(Fig. 3A-k and q). In addition,
abundant FJB-stained cells were found in the SP on days 2 and 5
(30±1.5 and 33.9±2.1, respectively) after TFI (Fig. 3A-m, o and q).
CA2/3
In the NT/sham group, FJB-stained cells were not
observed in the SP of CA2/3 (Fig.
3B-a). In the NT/ischemia group, no FJB-stained cells were
observed at any point in time after TFI in CA2/3 (Fig. 3B-c, e, g and q).
In the HT/sham group, FJB-stained cells were not
detected in the SP of CA2/3 (Fig.
3B-i). In the HT/ischemia group, FJB-stained cells were not
found in the SP on day 1 after TFI (Fig. 3B-k and q). However, many
FJB-stained cells were found in the SP on day 2 after TFI
(29.4±2.8) (Fig. 3B-m and q).
Furthermore, on day 5 after TFI, the number of FJB-stained cells
was significantly increased (81.7±4.2) (Fig. 3B-o and q).
Findings by NeuN
immunohistochemistry
CA1
In the NT/sham group, NeuN immunoreactivity was
observed in the pyramidal cells of the SP in CA1 (Fig. 3A-b). In the NT/ischemia groups,
the distribution of NeuN-immunostained pyramidal cells in CA1 was
not different from that of the NT/sham group on days 1 and 2 after
TFI (Fig. 3A-d and f). However,
on day 5 after TFI, NeuN-immunostained pyramidal cells were rarely
observed in the SP. The percentage of remaining NeuN-immunostained
pyramidal cells was 22.2% in the NT/sham group (Fig. 3A-h and r).
In the HT/sham group, the distribution of
NeuN-immunostained pyramidal cells was similar to that observed in
the NT/sham group (Fig. 3A-j).
In the HT/ischemia group, the NeuN immunoreactivity of
NeuN-immunostained pyramidal cells was weak on day 1 after TFI
(Fig. 3A-l). On days 2 and 6
after TFI, NeuN-immunostained pyramidal cells were significantly
decreased (55.9 and 28.8% of sham, respectively) (Fig. 3A-n, p and r), showing that
NeuN-immunostained pyramidal cells had significant morphological
alterations (pyknotic and tangle-like appearance) on day 5 after
TFI (Fig. 3A-p).
CA2/3
In the NT/sham group, NeuN-immunostained pyramidal
cells were typically distributed in the SP of CA2/3 (Fig. 3B-b). In the NT/ischemia group,
the distribution and number of NeuN-immunostained pyramidal cells
were similar to those of the NT/sham group on days 1, 2 and 5 after
TFI (Fig. 3B-d, f, h and r).
In the HT/sham group, the distribution of
NeuN-immunostained pyramidal cells was not different from that of
the NT/sham group (Fig. 3B-j).
In the HT/ischemia group, the NeuN immunoreactivity of the SP was
slightly weak on day one after TFI (Fig. 3B-l). On days 2 and 5 after TFI,
the number of NeuN-immunostained pyramidal cells was significantly
reduced (78.7 and 49.9%, respectively) compared to those in the
HT/sham group (Fig. 3B-n, p and
r).
Differences in HIF-1α protein level and
immunoreactivity HIF-1α protein level
The temporal pattern of HIF-1α protein levels in CA1
and CA2/3 after TFI normothermia or hyperthermia were observed to
be different, as shown in Fig. 4A
and C. Small amounts of HIF-1α protein were detected in CA1 and
CA2/3 of the NT/sham group; the levels at 0 and 3 h, and 5 days
after TFI were not altered. In the NT/ischemia group, the level of
HIF-1α protein was significantly increased from 12 h after TFI
(Fig. 4B and D). In the HT/sham
group, at 0 h after TFI, HIF-1α protein levels in CA1 and CA2/3
were significantly higher (236 and 218% of the NT/sham group,
respectively) than that in the NT/sham group, and the increased
HIF-1α protein level was maintained until 6 h post-TFI, peaked at
12 h post-TFI, and after that decreased (Fig. 4B and D).
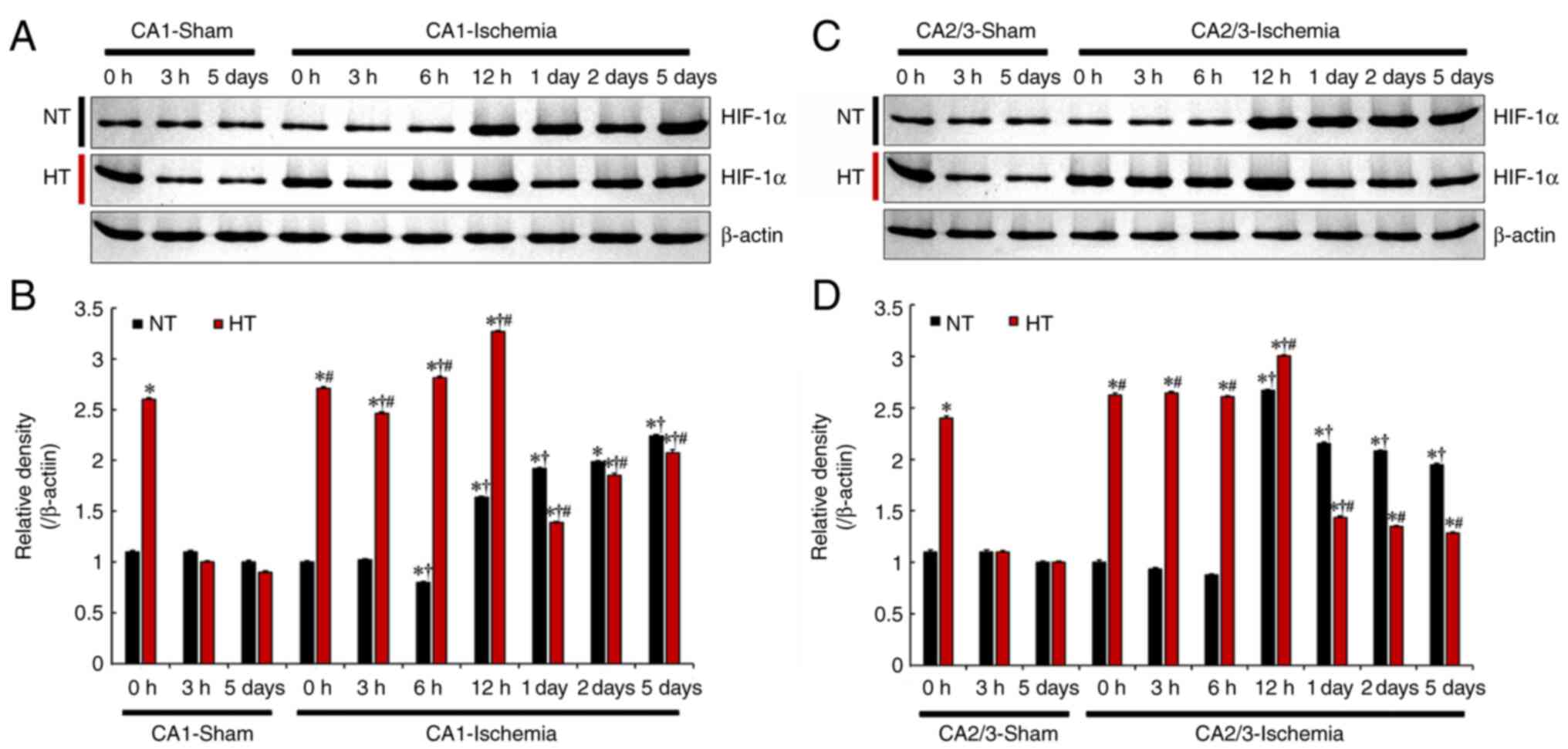 | Figure 4(A and C) Western blot analysis of
HIF-1α in CA1 (A) and CA2/3 (C) of the NT/sham, NT/ischemia,
HT/sham, and HT/ischemia groups at 0, 3, 6 and 12 h, 1, 2 and 5
days after TFI. (B and D) Relative density of the immunoblot bands
is represented. Protein levels of HIF-1α normalized to β-actin in
CA1 (B) and CA2/3 (D) *P<0.05 vs. NT/sham group at 0
h; †P<0.05 vs. pre-time point group;
#P<0.05 vs. NT/ischemia group. The bars indicate the
means ± SEM (n=5, respectively). HIF-1α, hypoxia-inducible factor
1α; NT, normothermia; HT, hyperthermia; TFI, transient forebrain
ischemia. |
HIF-1α immunoreactivity
CA1
In the NT/sham group, HIF-1α immunoreactivity was
hardly detected in CA1 pyramidal cells at 0 and 3 h, and 5 days
after TFI (Fig. 5A-a-c and C).
In the NT/ischemia group, HIF-1α immunoreactivity in the CA1
pyramidal cells was not significantly altered at 3 and 6 h after
TFI (Fig. 5Ba-c and C),
significantly increased at 12 h and 1 day after TFI (534.6 and
593.3% of the NT/sham group, respectively) (Fig. 5B-d, e and C), returned to
baseline level (113.5% of the NT/sham group) at 2 days after TFI
(Fig. 5B-f and C), and again
increased (208.7% of the NT/sham group) at 5 days after TFI
(Fig. 5B-g and C).
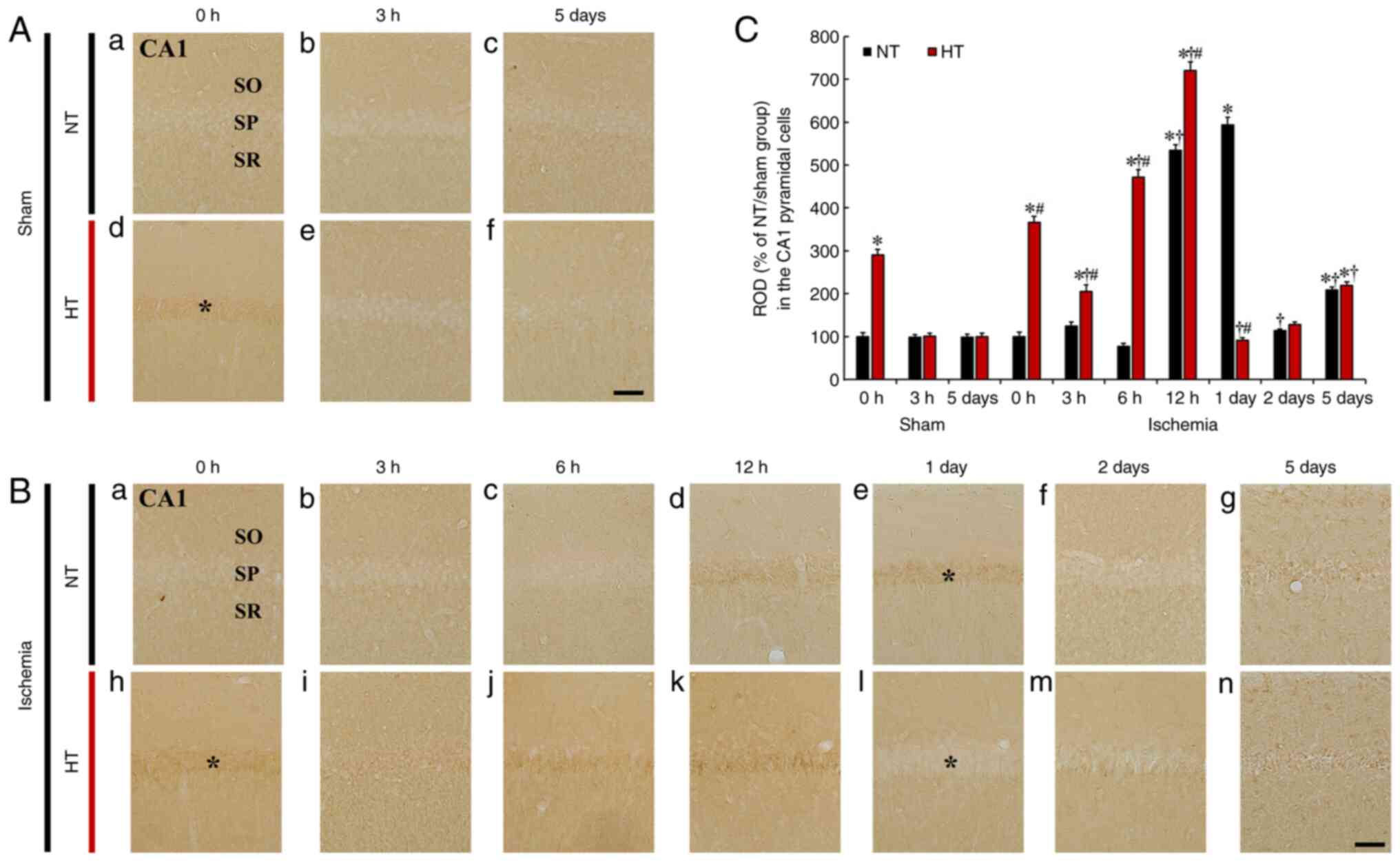 | Figure 5(A and B) HIF-1α immunohistochemistry
in CA1 of the (A) NT/sham (a-c), (B) NT/ischemia (a-g), (A) HT/sham
(d-f) and (B) HT/ischemia (h-n) groups at 0, 3, 6 and 12 h, 1, 2
and 5 days after TFI. In the NT/sham group, HIF-1α immunoreactivity
in the SP was very weak. In the NT/ischemia group, HIF-1α
immunoreactivity in the SP was significantly increased at 12 h and
1 day (asterisk), markedly reduced at 2 days and again increased at
5 days after TFI. In the HT/sham group, HIF-1α immunoreactivity was
very high (asterisk) at 0 h and similar to that of the NT/sham
group from 3 h after sham TFI. In the HT/ischemia group, increased
HIF-1α immunoreactivity was gradually increased until 12 h after
TFI and thereafter was lower (asterisk) than that in the HT/sham
group. Scale bar, 50 µm. (C) Relative optical density (ROD)
(% of NT/sham group) of HIF-1α immunoreactivity in CA1.
*P<0.05 vs. NT/sham group at 0 h;
†P<0.05 vs. pre-time point group;
#P<0.05 vs. NT/ischemia group. The bars indicate the
means ± SEM (n=7, respectively). HIF-1α, hypoxia-inducible factor
1α; NT, normothermia; HT, hyperthermia; TFI, transient forebrain
ischemia. |
In the HT/sham group, HIF-1α immunoreactivity was
significantly increased (290.6% of the NT/sham group) in the
cytoplasm of the CA1 pyramidal cells at 0 h after sham TFI
(Fig. 5A-d and C) and returned
to the baseline from 3 h after TFI (Fig. 5A-e, f and C). In the HT/ischemia
group, increased HIF-1α immunoreactivity in the CA1 pyramidal cells
was shown at 0, 3, 6 and 12 h after TFI (365.6, 205.2, 471.3 and
719.9% of the NT/sham group, respectively) (Fig. 5B-h-k and C). However, HIF-1α
immunoreactivity was dramatically decreased (15.4% of the
NT/ischemia group) at 1 day after TFI (Fig. 5B-l and C), and, at 2 and 5 days
after TFI, HIF-1α immunoreactivity was similar to that observed in
the NT/ischemia group (Fig. 5B-m, n
and C).
CA2/3
In the NT/sham group, very weak HIF-1α
immunoreactivity was observed in CA2/3 pyramidal cells, showing
that the immunoreactivity was not altered at 0 and 3 h, and 5 days
after TFI (Fig. 6Aa-c). In the
NT/ischemia groups, HIF-1α immunoreactivity in the CA2/3 pyramidal
cells was slightly increased at 3 and 6 h (Fig. 6B-b, c and C), peaked (580.6% of
NT/sham group) at 12 h (Fig. 6B-d
and C), and after that gradually decreased (526.1% at 1 day,
411.8% at 2 days, and 412.6% at 5 days vs. the NT/sham group) until
5 days after TFI (Fig. 6Be-g and
C).
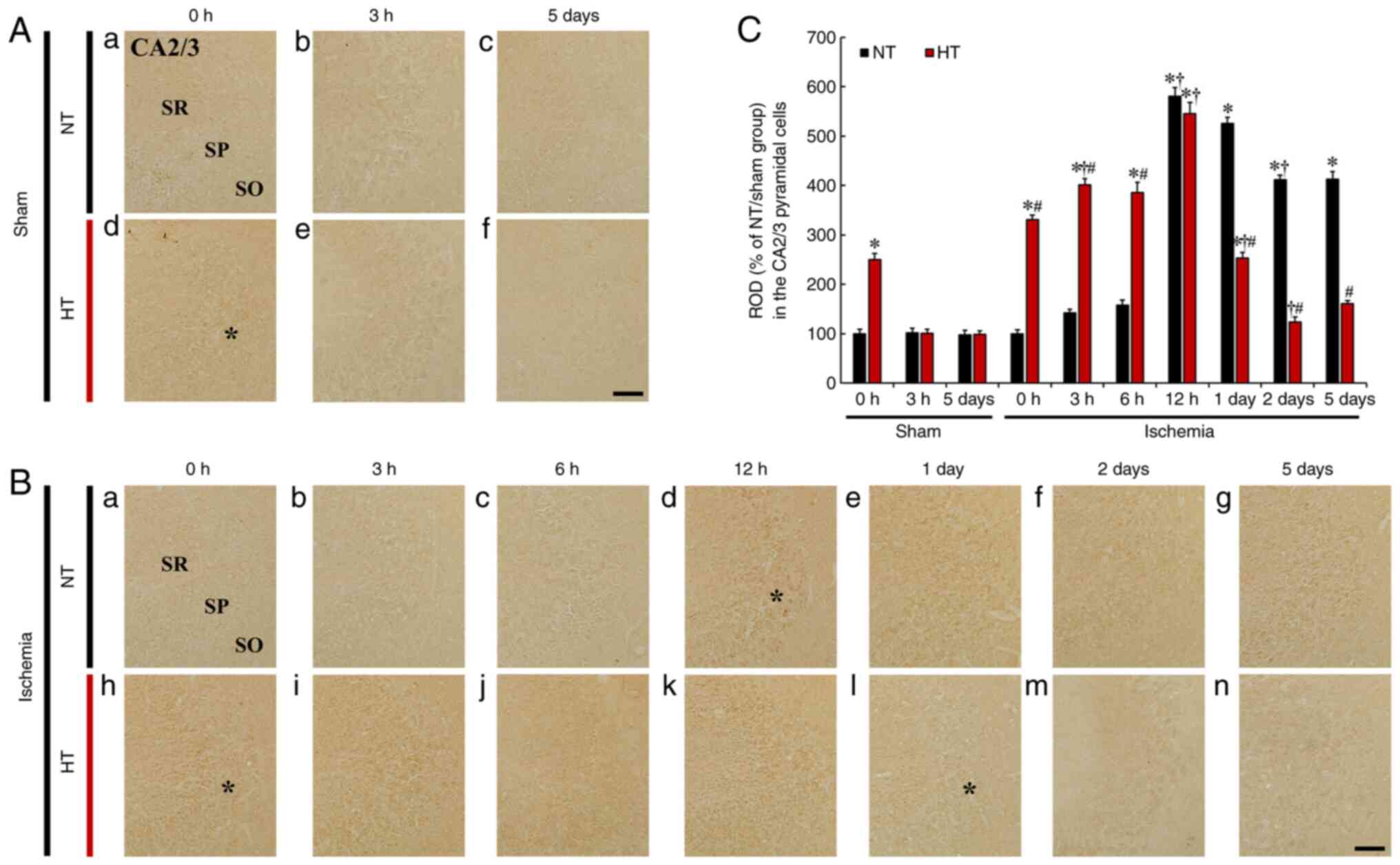 | Figure 6(A and B) HIF-1α immunohistochemistry
in CA2/3 of the (A) NT/sham (a-c), (B) NT/ischemia (a-g), (A)
HT/sham (d-f) and (B) HT/ischemia (h-n) groups at 0, 3, 6 and 12 h,
1, 2 and 5 days after TFI. In the NT/sham group, HIF-1α
immunoreactivity in the SP was very weak. In the NT/ischemia group,
HIF-1α immunoreactivity was significantly increased at 12 h
(asterisk) and thereafter slightly reduced until 5 days after TFI.
In the HT/sham group, HIF-1α immunoreactivity was very high at 0 h
and thereafter reduced to the level of the NT/sham group. In the
HT/ischemia group, increased HIF-1α immunoreactivity was
continuously increased until 1 day, and thereafter HIF-1α
immunoreactivity was significantly decreased until 5 days after
TFI. Scale bar, 50 µm. (C) Relative optical density (ROD) (%
of NT/sham group) of HIF-1α immunoreactivity in CA2/3.
*P<0.05 vs. NT/sham group at 0 h;
†P<0.05 vs. pre-time point group;
#P<0.05 vs. NT/ischemia group. The bars indicate the
means ± SEM (n=7, respectively). HIF-1α, hypoxia-inducible factor
1α; NT, normothermia; HT, hyperthermia; TFI, transient forebrain
ischemia. |
In the HT/sham group, HIF-1α immunoreactivity in the
CA2/3 pyramidal cells was significantly increased (250.3% of the
NT/sham group) only at 0 h after sham TFI when compared with the
NT/sham group (Fig. 6A-d and C).
In the HT/ischemia group, HIF-1α immunoreactivity in the CA2/3
pyramidal cells was gradually increased (330.3% at 0 h, 401.1% at 3
h and 385.6% at 6 h vs. the NT/sham group) (Fig. 6Bh-j and C), peaked (544.9% of the
NT/sham group) at 12 h (Fig. 6B-k
and C), and after that significantly decreased (253.3% at 1
day, 123.1% at 2 days, and 161.0% at 5 days vs. the NT/sham group)
after TFI, showing that each immunoreactivity was significantly
lower than that observed in the NT/ischemia group (Fig. 6Bl-n and C).
Discussion
Using a gerbil model of 5 min transient forebrain
ischemia (TFI), we investigated the effect of TFI on neuronal death
(loss) in the hippocampal CA1 and CA2/3 under hyperthermic
conditions and the relationship between neuronal death and changes
in hypoxia-inducible factor 1α (HIF-1α) expression following TFI
under hyperthermia.
In the present study, TFI under normothermia induced
selective death of pyramidal cells, which are principal neurons, in
the CA1 on day 5 after TFI, but no loss of pyramidal cells occurred
in CA2/3 on day 5 after TFI. This finding is consistent with the
results of previous studies (2,3,22). These results indicate that the
pyramidal cells of CA1 are susceptible to brief transient ischemia,
while the pyramidal cells of CA2/3 are resistant to brief transient
ischemia, showing that there are differences in neuronal
sensitivity to ischemic stress according to subfields in the
hippocampus.
It has been reported that an elevation in body
temperature (observed in more than 25% of patients with stroke)
during or after ischemia induction is associated with more severe
symptoms, poorer prognosis and mortality in patients with acute
stroke (23,24). It is known that an increase in
core temperature during or after ischemic insults induces an
increase in metabolic rate (25)
and production of oxygen radicals (26), which may aggravate ischemic
neuronal damage. In our present study, hyperthermia for 30 min
before TFI (HT/ischemia group) resulted in the earlier death of
pyramidal cells in CA1 on day 2 after TFI and induced death in
CA2/3 pyramidal cells on day 2 after TFI, which was not observed in
the NT/ischemia group. These results are similar to the findings of
previous studies showing that, in rats and gerbils,
ischemia-reperfusion under hyperthermic conditions induces more
accelerated neuronal cell death in the hippocampal CA1 from
approximately 1 day after transient ischemia (4-6),
and disruption of ischemic resistance of pyramidal neurons in the
hippocampal CA2/3 (4,6) and granular cells in the hippocampal
dentate gyrus (19).
Furthermore, in a rat model of focal ischemic stroke, hyperthermia
was found to increase infarct size and worsen behavioral outcomes
when ischemic duration was maintained for more than 2 h (7). Based on these results, it can be
suggested that hyperthermic conditions before and during transient
ischemia augment and accelerate ischemic damage of pyramidal cells
in the hippocampus compared to that under normothermia.
It has been demonstrated that HIF-1α is related to
neuronal damage/death or survival in the brain following ischemic
insults. Zhu et al reported that neuronal death in the
hippocampal CA1 was observed, and HIF-1α immunoreactivity was
reduced following transient global ischemia in rats. They showed
that the neuroprotective effect induced by hypoxia
post-conditioning against ischemia was abolished by
2-methoxyestradiol (a specific HIF-1α inhibitor) (11). In addition, Kopach et al
reported the role of HIF-1α in regulating neuronal protection. The
activation of HIF-1α decreased Ca2+-dependent
excitotoxicity in CA1 neurons in situ through significant
upregulation of endoplasmic reticulum Ca2+-ATPase mRNA
levels after oxygen-glucose deprivation (27).
In the present study, we found that, in the
NT/ischemia group, HIF-1α expression was significantly elevated in
the pyramidal cells of CA1 and CA2/3 at 12 h, and 1 day after TFI
the expression was differently decreased between CA1 and CA2/3. In
CA1, the expression was significantly reduced on day 2 and
increased again on day 5 after TFI, but, in CA2/3, the expression
on days 2 and 5 after TFI was slightly decreased (significantly
higher than that in CA1). Based on this finding, in the NT/ischemic
group, survival of the CA2/3 pyramidal cells from TFI may be due to
the difference in HIF-1α expression on day 2 after TFI. This result
is supported by a previous report showing that increased HIF-1α
immunoreactivity in ischemic CA1 was decreased before neuronal
death in a gerbil model of TFI (9). In addition, Lushnikova et al
reported that HIF-1α mRNA levels were downregulated only in CA1
neurons, but not in CA3 neurons, along with the significant death
of CA1 neurons in situ after oxygen-glucose deprivation
(28), showing that the degree
of HIF-1α expression was different between the CA1 and CA2/3
pyramidal cells. Taken together, it can be speculated that a
decrease in elevated HIF-1α expression in the hippocampal CA1 is
closely related to ischemic neuronal cell death and persistence of
high HIF-1α expression in CA2/3 may contribute to ischemic
resistance of CA2/3 pyramidal neurons.
On the other hand, under hyperthermic conditions,
the basal level of HIF-1α expression at 0 h in the HT/sham and
HT/ischemia groups was significantly increased in both CA1 and
CA2/3 pyramidal cells when compared to the NT/sham and NT/ischemia
groups. Similar to this early increase in HIF-1α expression under
hyperthermia in the present study, an earlier increase in HIF-1α
expression was reported in previous studies using an animal model
that caused relatively severe damage compared to 5 min of TFI under
normothermia. Upregulated HIF-1α was observed immediately after 10
min of TFI in the rat hippocampal CA1 region (11) and at 30 min after
hypoxia-ischemia injury in the mouse cortex (29). In the present study, HIF-1α
expression peaked at 12 h in both CA1 and CA2/3 pyramidal cells of
the HT/ischemia group and then significantly decreased on day 1
after TFI (just before neuronal cell death), showing earlier
neuronal death despite the initial high expression of HIF-1α as
compared with the NT/ischemia group. Although the exact reason for
the early increase in HIF-1α expression remains unclear, it may be
due to a protective mechanism against greater damage, or another
mechanism may be involved in hyperthermia-induced severe neuronal
damage. Therefore, based on our and previous studies, it is likely
that the reduction of increased HIF-1α expression in the
hippocampus precedes ischemic neuronal cell death under
normothermic and hyperthermic conditions.
In summary, the present study showed that pyramidal
cells in CA1 and CA2/3 had differences in ischemia sensitivity
under normothermia, and the relatively high resistance of CA2/3
pyramidal cells may be closely related to the maintenance of high
expression of HIF-1α after TFI. In addition, hyperthermia
accelerated ischemic neuronal death in both CA1 and CA2/3, showing
that hyperthermia-induced neuronal death is closely related to a
reduction in increased HIF-1α expression in both ischemic CA1 and
CA2/3. Therefore, we should consider the environment or conditions
of the brain in which ischemic insults occur because ischemic
damage in the brain is not the same in all patients with ischemic
stroke.
Availability of data and materials
The data presented in this study are available on
request from the corresponding author.
Authors' contributions
Conceptualization of the study concept was achieved
by MHW and JHA. Investigations in regards to western blot analysis
and immunohistochemistry were carried out by TKL, DWK and HS. The
study methodology, use of the software, validation of the data
collected and supervision of the study were the responsibility of
JCL, HIK, MCS, JHC, JHP, CHL, JHP and JHC. Data curation was
carried out by TKL, DWK and JHA. Writing of the original draft was
carried out by TKL, DWK, MHW and JHA. Writing of the review and
editing were MHW and JHA. Project administration was the
responsibility of TKL DWK, MHW and JHA. Funding acquisition was
carried out by JHA, MHW and TKL. All authors have read and agreed
to the published version of the manuscript.
Ethics approval and consent to
participate
Experimental procedures for the present study were
approved by the Institutional Animal Care and Use Committee at
Kangwon National University (approval no. KW-200113-1). The
procedures of handling and caring animals conformed to the
guidelines of the current international laws and policies (NIH
Guide for the Care and Use of Laboratory Animals, The National
Academies Press, 8th ediiton, 2011).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
Professor Moo-Ho Won: ORCID: 0000-0002-7178-6501;
Professor Ji Hyeon Ahn, ORCID: 0000-0002-5304-0714.
Acknowledgments
The authors would like to acknowledge Mr. Seung Uk
Lee and Ms. Hyun Sook Kim for their technical assistance in this
study.
Funding
This research was supported by Basic Science Research Program
through the National Research Foundation of Korea (NRF) funded by
the Ministry of Education (NRF-2021R1A2C1094224,
NRF-2020R1F1A1052380, and NRF-2020R1I1A1A01070897).
References
|
1
|
Wang X and Michaelis EK: Selective
neuronal vulnerability to oxidative stress in the brain. Front
Aging Neurosci. 2:122010.PubMed/NCBI
|
|
2
|
Kirino T: Delayed neuronal death in the
gerbil hippocampus following ischemia. Brain Res. 239:57–69. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu DK, Yoo KY, Shin BN, Kim IH, Park JH,
Lee CH, Choi JH, Cho YJ, Kang IJ, Kim YM and Won MH: Neuronal
damage in hippocampal subregions induced by various durations of
transient cerebral ischemia in gerbils using Fluoro-Jade B
histofluorescence. Brain Res. 1437:50–57. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Minamisawa H, Smith ML and Siesjo BK: The
effect of mild hyperthermia and hypothermia on brain damage
following 5, 10 and 15 min of forebrain ischemia. Ann Neurol.
28:26–33. 1990. View Article : Google Scholar
|
|
5
|
Dietrich WD, Busto R, Valdes I and Loor Y:
Effects of normothermic versus mild hyperthermic forebrain ischemia
in rats. Stroke. 21:1318–1325. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim MJ, Cho JH, Park JH, Park JH, Ahn JH,
Tae HJ, Cho GS, Yan BC, Hwang IK, Lee CH, et al: Impact of
hyperthermia before and during ischemia-reperfusion on neuronal
damage and gliosis in the gerbil hippocampus induced by transient
cerebral ischemia. J Neurol Sci. 348:101–110. 2015. View Article : Google Scholar
|
|
7
|
de Jonge JC, Wallet J and van der Worp HB:
Fever worsens outcomes in animal models of ischaemic stroke: A
systematic review and meta-analysis. Eur Stroke J. 4:29–38. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Albadari N, Deng S and Li W: The
transcriptional factors HIF-1 and HIF-2 and their novel inhibitors
in cancer therapy. Expert Opin Drug Discov. 14:667–682. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee JC, Tae HJ, Kim IH, Cho JH, Lee TK,
Park JH, Ahn JH, Choi SY, Bai HC, Shin BN, et al: Roles of HIF-1α,
VEGF, and NF-κB in ischemic preconditioning-mediated
neuroprotection of hippocampal CA1 pyramidal neurons against a
subsequent transient cerebral ischemia. Mol Neurobiol.
54:6984–6998. 2017. View Article : Google Scholar
|
|
10
|
Long Q, Fan C, Kai W, Luo Q, Xin W, Wang
P, Wang A, Wang Z, Han R, Fei Z, et al: Hypoxia inducible factor-1α
expression is associated with hippocampal apoptosis during
epileptogenesis. Brain Res. 1590:20–30. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu T, Zhan L, Liang D, Hu J, Lu Z, Zhu X,
Sun W, Liu L and Xu E: Hypoxia-inducible factor 1α mediates
neuroprotection of hypoxic postconditioning against global cerebral
ischemia. J Neuropathol Exp Neurol. 73:975–986. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shi H: Hypoxia inducible factor 1 as a
therapeutic target in ischemic stroke. Curr Med Chem. 16:4593–4600.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chatzi C, Schnell E and Westbrook GL:
Localized hypoxia within the subgranular zone determines the early
survival of newborn hippocampal granule cells. Elife. 4:e087222015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xing J and Lu J: HIF-1α activation
attenuates IL-6 and TNF-α pathways in hippocampus of rats following
transient global ischemia. Cell Physiol Biochem. 39:511–520. 2016.
View Article : Google Scholar
|
|
15
|
National Research Council (US) Committee
for the Update of the Guide for the Care and use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press; Washington, DC: 2011
|
|
16
|
Kim B, Ahn JH, Kim DW, Lee TK, Kim YS,
Shin MC, Cho JH, Kim YM, Park JH, Kang IJ, et al: Transient
forebrain ischemia under hyperthermic condition accelerates memory
impairment and neuronal death in the gerbil hippocampus by
increasing NMDAR1 expression. Mol Med Rep. 23:2562021. View Article : Google Scholar :
|
|
17
|
Ohk TG, Ahn JH, Park YE, Lee TK, Kim B,
Lee JC, Cho JH, Park JH, Won MH and Lee CH: Comparison of neuronal
death and expression of TNF-α and MCT4 in the gerbil hippocampal
CA1 region induced by ischemia/reperfusion under hyperthermia to
those under normothermia. Mol Med Rep. 22:1044–1052. 2020.
View Article : Google Scholar :
|
|
18
|
Kanda I: Exotic animal formulary 4
edition. Can Vet J. 56:7362015.
|
|
19
|
Kim DW, Cho JH, Cho GS, Kim IH, Park JH,
Ahn JH, Chen BH, Shin BN, Tae HJ, Hong S, et al: Hyperthermic
preconditioning severely accelerates neuronal damage in the gerbil
ischemic hippocampal dentate gyrus via decreasing SODs expressions.
J Neurol Sci. 358:266–275. 2015. View Article : Google Scholar
|
|
20
|
Yang GE, Tae HJ, Lee TK, Park YE, Cho JH,
Kim DW, Park JH, Ahn JH, Ryoo S, Kim YM, et al: Risperidone
treatment after transient ischemia induces hypothermia and provides
neuroprotection in the gerbil hippocampus by decreasing oxidative
stress. Int J Mol Sci. 20:46212019. View Article : Google Scholar :
|
|
21
|
Lee JC, Cho JH, Lee TK, Kim IH, Won MH,
Cho GS, Shin BN, Hwang IK, Park JH, Ahn JH, et al: Effect of
hyperthermia on calbindin-D 28k immunoreactivity in the hippocampal
formation following transient global cerebral ischemia in gerbils.
Neural Regen Res. 12:14582017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bartsch T and Wulff P: The hippocampus in
aging and disease: From plasticity to vulnerability. Neuroscience.
309:1–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Azzimondi G, Bassein L, Nonino F, Fiorani
L, Vignatelli L, Re G and D'Alessandro R: Fever in acute stroke
worsens prognosis: A prospective study. Stroke. 26:2040–2043. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Reith J, Jørgensen HS, Pedersen PM,
Nakayama H, Raaschou HO, Jeppesen LL and Olsen TS: Body temperature
in acute stroke: Relation to stroke severity, infarct size,
mortality, and outcome. Lancet. 347:422–425. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Busija DW, Leffler CW and Pourcyrous M:
Hyperthermia increases cerebral metabolic rate and blood flow in
neonatal pigs. Am J Physiol. 255(2 Pt 2): H343–H346.
1988.PubMed/NCBI
|
|
26
|
Corbett D and Thornhill J: Temperature
modulation (hypothermic and hyperthermic conditions) and its
influence on histological and behavioral outcomes following
cerebral ischemia. Brain Pathol. 10:145–152. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kopach O, Maistrenko A, Lushnikova I,
Belan P, Skibo G and Voitenko N: HIF-1α-mediated upregulation of
SERCA2b: The endogenous mechanism for alleviating the
ischemia-induced intracellular Ca(2+) store dysfunction in CA1 and
CA3 hippocampal neurons. Cell Calcium. 59:251–261. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lushnikova I, Orlovsky M, Dosenko V,
Maistrenko A and Skibo G: Brief anoxia preconditioning and HIF
prolyl-hydroxylase inhibition enhances neuronal resistance in
organotypic hippocampal slices on model of ischemic damage. Brain
Res. 1386:175–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jeon GW, Sheldon RA and Ferriero DM:
Hypoxia-inducible factor: Role in cell survival in superoxide
dismutase overexpressing mice after neonatal hypoxia-ischemia.
Korean J Pediatr. 62:444–449. 2019. View Article : Google Scholar : PubMed/NCBI
|