Introduction
Chronic obstructive pulmonary disease (COPD) is a
chronic airway inflammatory disorder; it is a common respiratory
illness and the third highest cause of mortality in China (1). COPD has attracted increased worldwide
attention due to its high incidence, morbidity and mortality rates
(2). It is characterized by an
irreversible expiratory airflow limitation and manifests as a
chronic inflammatory disorder and pulmonary emphysema (3). The primary risk factor for COPD is
exposure to cigarette smoke (CS). Research using animals has
revealed that frequent exposure to CS can enhance the number of
inflammatory factors, such as tumor necrosis factor (TNF), in the
alveolar space and lung parenchyma (4). A recent study reported that the
incidence rate of COPD among Chinese adults aged >40 years was
13.7%, and revealed that smoking and particulate matter 2.5 were
the main causes of COPD (5).
According to statistics, >70% of patients with COPD have a
history of smoking (6). The early
prevention and treatment of COPD remain a challenge worldwide.
Therefore, studying its pathogenesis is crucial for proposing novel
therapeutic strategies for the early prevention and treatment of
COPD.
Cell pyroptosis is a type of programmed cell death
mediated by Caspase-11 or −1. Under physiological conditions,
pyroptosis is crucial for organ development (7). When cell pyroptosis occurs, the cell
membrane rapidly ruptures and forms a pore-like membrane structure,
through which several inflammatory agents and the cell content are
released (8). When exogenous
microorganisms and stress result in the production of Caspase-1,
which is triggered and activated by the inflammasome, gasdermin D
(GSDMD) is cleaved to form the N-terminal fragment of GSDMD
(GSDMD-N), which further induces the formation of the membrane pore
and the release of inflammatory factors. Recent studies have
revealed that pyroptosis is a common occurrence in respiratory
illnesses; therefore, inhibiting the occurrence of pyroptosis in
the respiratory system may minimize the degree of lung cell injury,
as well as the production of the inflammatory response and its
associated factors (9–13). Tsai et al (14) reported that the NLR family pyrin
domain containing 3 (NLRP3) inflammasome promoted the apoptosis of
bronchial epithelial cells and induced airway epithelial injury and
airway remodeling, thereby promoting the occurrence of asthma and
COPD. It has been reported that CS extract (CSE) induces the
pyroptosis of human bronchial epithelial cells by activating
NLRP3/Caspase-1 signaling, thereby aggravating COPD development
(15). Similarly, adipose stem
cell-derived exosomes can mitigate CS-induced pyroptosis, and thus
inhibit COPD development (16).
Therefore, reducing pyroptosis may serve as an effective approach
for preventing the progression of COPD.
Hydrogen sulfide (H2S) is a toxic gas
with an odor of rotten egg (17).
H2S is now considered as the third gas signal molecule
after carbon monoxide and nitric oxide (18). Furthermore, it participates in a
variety of signal transduction pathways and plays a protective role
through its antioxidant, anti-apoptotic and anti-inflammatory
effects in various pathological cells, as well as organs. Studies
have demonstrated that H2S can protect cells from damage
in different disease models by activating different signaling
pathways. For example, H2S has been shown to attenuate
lipopolysaccharide (LPS)-induced acute lung injury in mice through
the activation of the PI3K/Akt/mTOR pathway (19). Similarly, H2S has been
found to inhibit thyroxin-induced myocardial fibrosis in rats by
activating the PI3K/Akt signaling pathway (20). It has also been demonstrated that
H2S alleviates age-related macular degeneration by
inhibiting inflammation and oxidative stress-mediated pyroptosis
(21). Furthermore, a previous
study by the authors revealed that H2S attenuated
CS-induced COPD by inhibiting the transforming growth
factor-β1/Smad pathway, suggesting that H2S may serve as
a potential therapeutic agent for COPD (22). However, whether the beneficial
effects of H2S are dependent on its impact on cell
pyroptosis remains unknown. Therefore, the aim of the present study
was to establish a rat model of CS-induced COPD to observe the
effects of H2S on cell pyroptosis. A 16HBE cell model
was also established to further examine the effects of
H2S on the Toll-like receptor (TLR)4/NF-κB signaling
pathway may be affected by and to elucidate the underlying
mechanisms.
Materials and methods
Animals
A total of 48 male Sprague-Dawley rats, weighing
250–270 g (9–10 weeks old), were purchased from the Hebei Chest
Hospital Animal Center. The animal procedures were approved by the
Animal Ethics Committee of the Hebei Chest Hospital Animal Center
and complied with the Guide of the Care and Use of Laboratory
Animals published by the National Institutes of Health (23). All rats were provided with water
and food ad libitum and were examined at the pathogen-free
barrier laboratory of the Hebei Chest Hospital Animal Center
(Shijiazhuang, China). The rats where maintained in a laboratory
with a 12-h light/dark cycle, a temperature of <23°C and a
humidity of 35%. Every attempt was made to reduce animal distress
and pain. The rats were housed in a holding house for adaptation
for 5 days following their arrival at the laboratory.
Model of CS-induced COPD
The rats were randomly assigned into different
groups (n=12 per group) as follows: The H2S, CS +
H2S, CS and control groups. The rat model of COPD was
established according to the study by Ke et al (24). CS inhalation by rats in the CS +
H2S and CS groups was performed using the Buxco
inhalation exposure system (Data Sciences International, Inc.) for
28 weeks. Derby cigarettes (Wuhu Cigarettes Factory) were used to
generate the smoke. Each cigarette contained 0.9 mg nicotine, 10 mg
tar and 12 mg carbon monoxide. The rats were treated with a
20-cigarette equivalent inhalation for 2 h and then permitted to
rest for 4 h on the same day. The rats breathed the CS for 6 days a
week. The rats in the sham (control group) inhaled air for 28 weeks
using the Buxco animal CS-exposure system, and then inhaled air
inside a plastic 20-liter container for 7 days.
H2S inhalation
During H2S inhalation, the rats were
placed in a plastic 20-liter container and permitted to inhale air
combined with H2S for 8 h every day for 7 days following
the establishment of the model of CS-induced COPD. H2S
flowed through a flowmeter and regulator, and blended into the air.
The flow of air, including H2S, was regulated to
maintain the H2S concentration at 40 ppm. Non-COPD model
rats also breathed 40 ppm H2S for 8 h every day for 7
days. H2S was purchased from the Shijiazhuang Zhongyuan
Specialty Gas Co., Ltd. The concentration of H2S was
measured using a H2S concentration monitor
(HG-BX-H2S, Haigu Co.; http://www.czhaigu.com/aboutus.html).
Histopathological analysis
The rats were anesthetized with pancuronium bromide
[0.6 mg/kg, intraperitoneal (i.p.)] and sodium pentobarbital (100
mg/kg, i.p.) following the inhalation of H2S. Cervical
dislocation was used as the method of sacrifice and a thoracotomy
was performed to expose the lungs. The lungs were perfused via the
right ventricle with 30 ml ice-cold sterile phosphate-buffered
saline (PBS). The left lung lobes were extracted and preserved at
25°C with 4% formalin for 48 h. The lung was then sliced into
4-µm-thick sections for further histological analysis.
For hematoxylin and eosin (H&E) staining, the
protocol used was as previously described (25). Briefly, the sections were blocked
with 3% hydrogen peroxide for 20 min, and non-specific binding
sites were blocked with QuickBlock™ immunostaining
blocking reagent (Beyotime Institute of Biotechnology) for 1 h. The
sections were then incubated with (H&E; Beyotime Institute of
Biotechnology) sequentially at room temperature for 20 min and
washed. All images were captured on an optical microscope (Carl
Zeiss AG).
Immunohistochemistry (IHC)
Paraffin-embedded sections, as well as block
fabrication, were conducted in the same manner as described above
for H&E staining (26). IHC
staining for GSDMD-N was performed on the lung tissue slides from
each group. The lung tissue slides were heated for 20 min in a
microwave (100°C) with 0.01 M citrate buffer (pH 6.0), and were
cooled down gently to room temperature. Following retrieval, the
slides were incubated for ~30 min at room temperature with 3%
H2O2 (Sigma-Aldrich, Merck KGaA). They were
then blocked with 5% BSA (Beyotime Institute of Biotechnology)
supplemented with 10% normal goat serum (Beyotime Institute of
Biotechnology) and PBS plus Tween-20 (Beyotime Institute of
Biotechnology) at room temperature for 1 h to inhibit non-specified
protein binding. This was followed by incubation with an
anti-GSDMD-N rat monoclonal primary antibody (dilution, 1:200;
PA5-104324, Invitrogen; Thermo Fisher Scientific, Inc.) at 4°C
overnight. An IgG secondary antibody (dilution, 1:500; BA-4000;
Vector Laboratories, Inc.) was used in the biopsy to examine the
expression of the primary antibody, and was incubated with the
sections for 30 min at 37°C. Avidin-biotin complex reagent (ABC
kit; Vector Laboratories, Inc.) was used to treat the slides for 30
min at room temperature. The sections were then incubated with
3,3′-diaminobenzidine (Sigma-Aldrich, Merck KGaA)
tetrahydrochloride hydrate solution for 2 min at room temperature.
Using the Slide view VS200 Scanner (Olympus Corporation), GSDMD-N
expression was detected and captured digitally.
Western blot analysis
First, RIPA buffer (Sigma-Aldrich, Merck KGaA) was
used to extract protein from the cultured cells or rat tissues. The
protein lysate was then separated using 15% sodium dodecyl
sulphate-polyacrylamide gel electrophoresis and transferred onto a
difluoride polyvinylidene membrane. The amount and quality of the
protein was examined using BCA assay (Beyotime Institute of
Biotechnology) in a Synergy H1 microplate reader (BioTek
Instruments, Inc.). Equal amounts of protein per lane (mass of 20
µl) were then loaded onto a 12% gel from a TGX Stain-Free FastCast
Acrylamide kit (Bio-Rad Laboratories, Inc.) and finally transferred
onto a 0.45 µm PVDF membrane (GE Healthcare, Inc.). The membrane
was blocked with 5% non-fat milk-PBS for 1 h at room temperature
and was then incubated overnight at 4°C with the following
antibodies: NLRP3 (dilution, 1:1,000; cat. no. sc-134306; Santa
Cruz Biotechnology, Inc.), cleaved Caspase-1 (dilution, 1:1,500;
cat. no. YC0002; ImmunoWay Biotechnology Company), Caspase-1
(dilution, 1:800; cat. no. PA5-99477; Invitrogen; Thermo Fisher
Scientific, Inc.), cleaved IL-1β (dilution, 1:1,000; cat. no.
83186; Cell Signaling Technology, Inc.), IL-1β (dilution, 1:1,000;
cat. no. P420B; Thermo Fisher Scientific, Inc.), cleaved GSDMD
(dilution, 1:1,000; cat. no. ab215203; Abcam, Inc.), pro-GSDMD
(dilution, 1:1,000; cat. no. sc-81868; Santa Cruz Biotechnology,
Inc.), TLR4 (dilution, 1:1,000; cat. no. sc-293072; Santa Cruz
Biotechnology, Inc.), inhibitor κB-α (IκBα, dilution, 1:1,000; cat.
no. sc-1643; Santa Cruz Biotechnology, Inc.), phosphorylated
(p)-IκBα (dilution, 1:1,000; cat. no. sc-8404; Santa Cruz
Biotechnology, Inc.), NF-κB p-p65 (dilution, 1:1,000; cat. no.
sc-166748; Santa Cruz Biotechnology, Inc.), NF-κB p50 (dilution,
1:1,000; cat. no. sc-8414; Santa Cruz Biotechnology, Inc.), GAPDH
(dilution, 1:1,000; cat. no. 5174; Cell Signaling Technology,
Inc.), Histone H3 (dilution, 1:1,000; cat. no. ab1791; Abcam, Inc.)
and NF-κB p65 (dilution, 1:1,000; cat. no. sc-8008; Santa Cruz
Biotechnology, Inc.). The membranes were washed and incubated with
with horseradish peroxidase-conjugated anti-rabbit secondary
antibodies (dilution, 1:10,000; cat. no. sc-2357; Santa Cruz
Biotechnology, Inc.) or anti-mouse secondary antibody (dilution,
1:10,000; cat. no. sc-2005; Santa Cruz Biotechnology, Inc.) for 2 h
at room temperature. Finally, the membranes were washed and
antibodies were identified using a SuperSignal West Pico
Chemilluminescent Substrate (Thermo Fisher Scientific, Inc.). The
band intensity was quantified using ImageJ software (version 1.6.0;
National Institutes of Health).
Cells, cell culture and treatment
Immortalized 16HBE cells were acquired from Procell
Life Science & Technology Co., Ltd. A total of 10 generations
of 16HBE cells were cultured in Dulbecco's modified Eagle's medium
(DMEM; Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10%
fetal bovine serum (Biological Industries) in room air with 5%
CO2 at 37°C. The preparation of CSE was performed as
previously described (24). The CS
was collected into a glass syringe containing 10 ml DMEM. The
serum-free DMEM (MilliporeSigma) was then filtered and titrated
with sodium hydroxide (NaOH) (Sangon Biotech, Inc.) at pH
7.35-7.45. The liquid was considered to yield the CSE at a
concentration of 100%. To achieve suitable concentrations, the
liquid was diluted again with DMEM. The CSE was prepared for ~30
min and then used. Subsequently, 5% CSE was used to challenge the
16HBE cells, and the GSDMD inhibitor, necrosulfonamide (NSA; 20 µM;
Cell Signaling Technology, Inc.), or the NLRP3 inhibitor, cytokine
release inhibitory drug 3 (CRID3; 10 µM; Cell Signaling Technology,
Inc.), were added to the culture medium 2 h prior to the CSE
challenge. Pyroptosis and cell viability were then measured using
lactate dehydrogenase (LDH) release assay and a water soluble
tetrazole salt-1 (WST-1) assay, as described below. To examine the
effects of H2S on the CSE-induced pyroptosis of 16HBE
cells, the cells were treated with 5% CSE and sodium hydrosulfide
(NaHS; an H2S donor; Thermo Fisher Scientific, Inc.) for
24 h. Pyroptosis, cell viability and the expression of
pyroptosis-related proteins were then measured.
Cell viability assay
The 16HBE cells were plated at a density of
2×103 cells/well in a 96-well culture plate and treated
with 0.5–10% CSE for 24 h. A WST-1 cell proliferation assay kit
(Beyotime Institute of Biotechnology) was used to evaluate 16HBE
cell viability. To fully dissolve the WST-1 powder and form its
solutions, 5 ml electronic coupling substance was added.
Subsequently, 100 µl solution containing 2,000 cells was added to
each well. Subsequently, 10 µl WST-1 solution was added to each
well followed by incubation for 2 h at room temperature. Finally, a
microplate reader (BioTek Instruments, Inc.) was used to measure
the optical density at a wavelength of 450 nm.
LDH release assay
The LDH Assay kit (Beyotime Institute of
Biotechnology) was used to detect the rate of pyroptosis. Briefly,
the culture supernatant of 16HBE cells (120 µl/well) was collected
and incubated for 30 min at 25°C with 60 µl LDH reaction buffer in
the dark. The absorbance was measured using a microplate reader
(BioTek Instruments, Inc.) at 490 nm.
Induction of TLR4 or NF-κB
overexpression via lentiviral vector
Similar to the study of Ding et al (27), TLR4, NF-κB and negative control
lentiviral vectors were synthesized by GeneChem, Inc. Briefly, 10
µl GV492-TLR4/NC-EGFP, 10 µl GV492-NF-κB/NC-EGFP or 10 µl packing
vector were transfected into the 293T cell line (The Cell Bank of
Type Culture Collection of the Chinese Academy of Sciences) using
Lipofectamine™ 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) for 5 min at room temperature. After 48 h, the
viral supernatant was harvested by ultracentrifugation for 1 h at
1,200 × g at 37°C.
Briefly, the 16HBE cells were cultured into 6-well
plates until the cells were ~50% confluent. The cells were then
transfected with the 10 µl GV492-TLR4/NC-EGFP, 10 µl
GV492-NF-κB/NC-EGFP or 10 µl packing vector (MOI: 50–80). The
medium was changed 6 h later, and the cells continued to be
cultured for 72 h. The lentivirus-treated 16HBE cells were then
treated with 5% CSE and NaHS for 24 h. The transfection efficiency
was measured using reverse transcription-quantitative PCR
(RT-qPCR).
RT-qPCR
The total RNA was extracted using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) following the
manufacturer's protocol. Reverse transcription was then performed
using the One-Step SYBR PrimeScript RT-PCR kit (Takara
Biotechnology Co., Ltd.). The reaction was performed using the ABI
PRISM 7500 Real-Time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) at 42°C for 5 min, 95°C for 10 sec, followed by
40 cycles of 95°C for 5 sec, 55°C for 30 sec and 72°C for 30 sec. A
total of three independent experiments were conducted each time.
The data were then analyzed by comparing the 2−ΔΔCq
value (28). The expression of
genes was normalized using GAPDH as a loading control. The primers
used were as follows: TLR4 upstream, 5′-TGGCATGAAACCCAGAGCTT-3′ and
downstream, 5′-ACCCGCAAGTCTGTGCAATA-3′; NF-κB upstream,
5′-GGGCAGGAAGAGGAGGTTTC-3′ and downstream,
5′-AATAGGCAAGGTCAGGGTGC-3′; GAPDH upstream,
5′-AATGGGCAGCCGTTAGGAAA-3′ and downstream,
5′-GCGCCCAATACGACCAAATC-3′.
TLR4 or NF-κB knockdown via lentiviral
vector
Similar to the study of Zhang et al (29), CRISPR/Cas9-mediated genome editing
methods were used in the present study. Briefly, TLR4 or NF-κB
single-guide RNAs (sgRNAs) and a scrambled control sequence were
cloned into a lentiviral plasmid GV393, separately. The 16HBE cells
were then seeded into 6-well plates at a density of
1×107 cells/well and subsequently infected with the
lentiviral constructs (MOI: 50–80) following the manufacturer's
instructions. The efficacy of TLR4 knockdown was confirmed using
western blot analysis. The medium was changed 6 h later, and the
cells continued to be cultured for 72 h. The lentivirus-treated
16HBE cells were then treated with 5% CSE and NaHS for 24 h.
Statistical analysis
All data were evaluated using SPSS 17.0. Software
(SPSS, Inc.). One-way ANOVA with the Bonferroni post hoc test was
performed to examine the quantitative data. Data are presented as
the mean ± standard deviation. P<0.05 was considered to indicate
a statistically significant difference.
Results
H2S attenuates CS-induced
lung injury and pyroptosis
The lung histopathological changes as detected using
H&E staining are presented in Fig.
1A. Exposure to CS induced inflammatory cell infiltration and
alveolar septum thickening, which were considerably alleviated in
the CS + H2S group. In addition, treatment with
H2S alone did not induce notable histopathological
changes. The IHC findings for GSDMD-N expression in the lungs are
illustrated in Fig. 1B. In the CS
group, the expression of GSDMD-N determined using IHC was markedly
increased, while it was markedly decreased by H2S.
Furthermore, GSDMD-N expression was not altered by H2S
alone. Representative results of the western blot analysis of
NLRP3, cleaved Caspase-1, Caspase-1, cleaved pro-IL-1β, IL-1β,
cleaved GSDMD and pro-GSDMD expression in lung tissue are shown in
Fig. 1C and D. Relative changes in
the levels of these proteins are presented in Fig. 1E-H. It was found that NLRP3
expression was markedly increased in the CS group, and the ratios
of cleaved IL-1β/pro-IL-1β, cleaved Caspase-1/Caspase-1 and cleaved
GSDMD/pro-GSDMD were all enhanced, while these were notably
decreased by H2S. The expression levels of these
proteins were not affected by H2S alone. Thus, these
results indicated that CS induced lung injury and pyroptosis, which
was partly reversed by H2S.
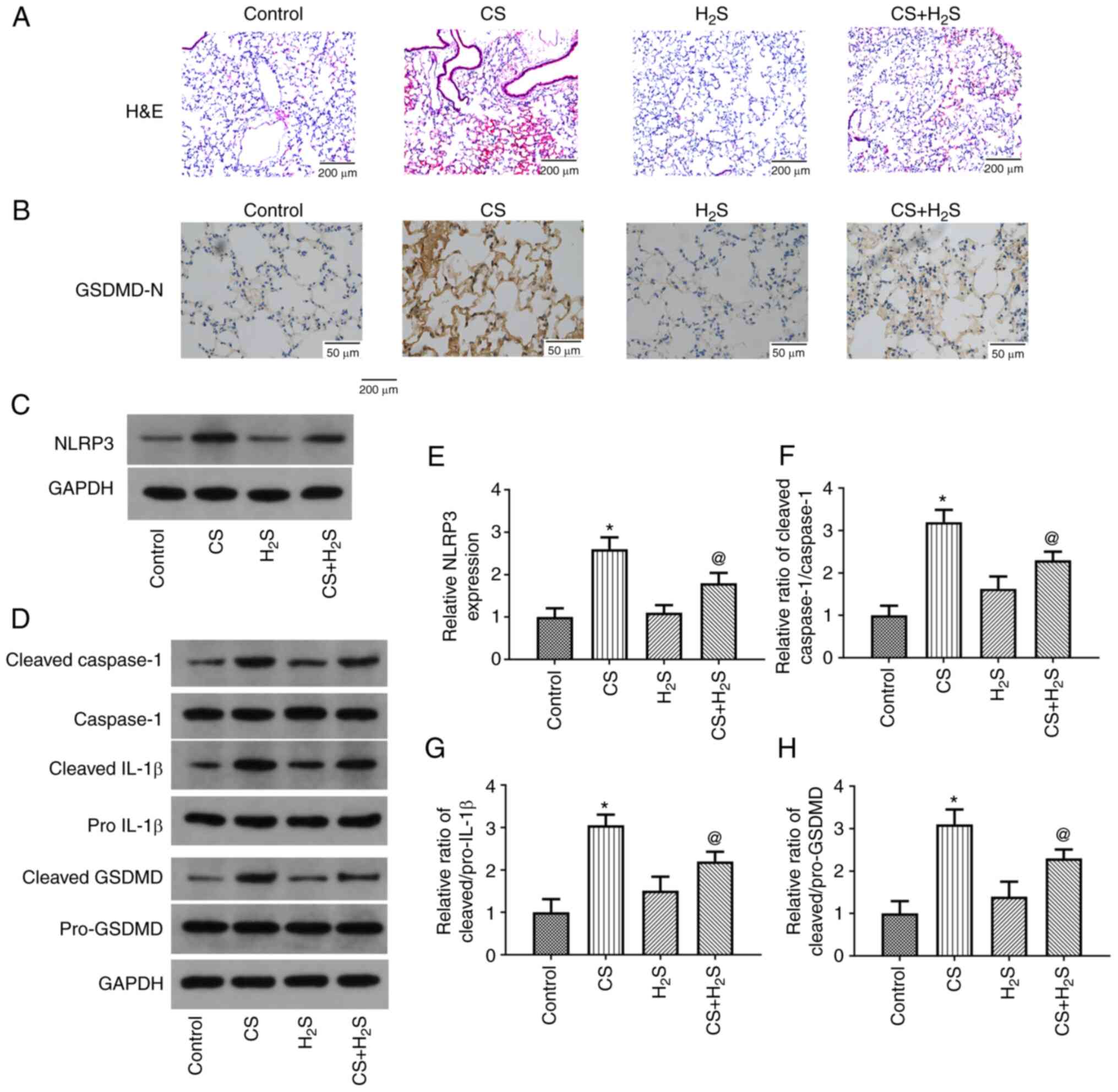 | Figure 1.Effect of H2S inhalation
on histopathological appearance of the lung tissue of rats and the
expression of pyroptosis-related proteins. (A) H&E staining
results. (B) GSDMD-N expression was detected in the lung using
immunohistochemistry. (C and D) Representative protein bands of
NLRP3, cleaved Caspase-1, Caspase-1, cleaved pro-IL-1β, IL-1β,
cleaved GSDMD and pro-GSDMD expression. (E-H) Quantitative results
of western blot analysis. Values are presented as the mean ± SD and
are representative of three independent experiments. *P<0.05
compared with the control group; @P<0.05 compared
with the CS group. H&E, hematoxylin and eosin; GSDMD, gasdermin
D; NLRP3, NLR family pyrin domain containing 3; CS, cigarette
smoke; H2S, hydrogen sulfide. |
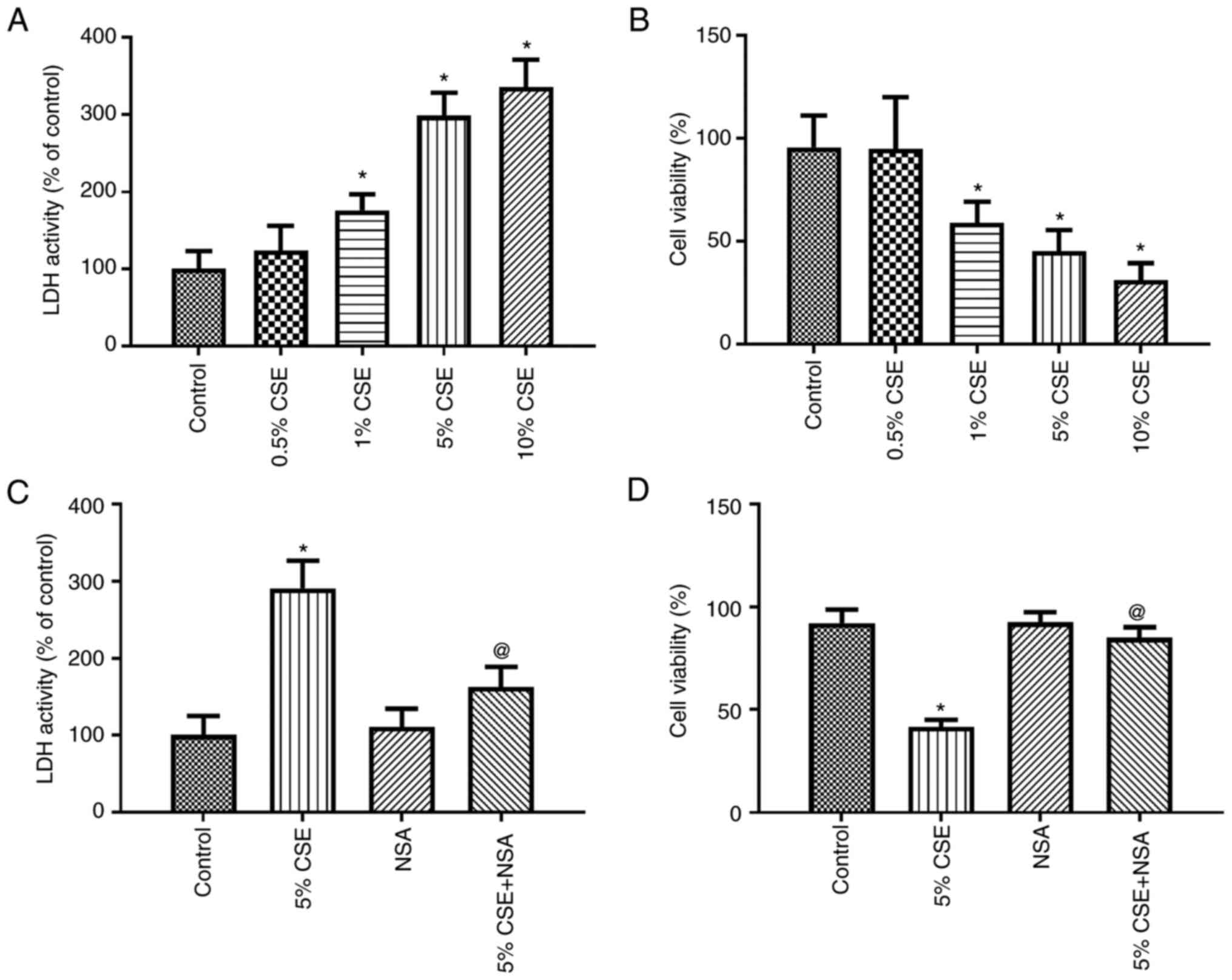
Effect of CSE on the pyroptosis of
16HBE cells
First, the 16HBE cells were exposed to various
concentrations of CSE (0.5–10% of the CSE) for 24 h, and pyroptosis
and cell viability were then measured using LDH and WST-1 assay,
respectively. The LDH activity was enhanced by CSE in a
concentration-dependent manner (Fig.
2A), while cell viability was decreased by CSE in a
concentration-dependent manner (Fig.
2B). Subsequently, 5% CSE was selected to treat the 16HBE
cells, and the GSDMD inhibitor, NSA, was added to the culture
medium. Pyroptosis and cell viability were then measured using LDH
and WST-1 assay, respectively. The results demonstrated that
pyroptosis was significantly inhibited by NSA (Fig. 2C). Moreover, 5% CSE induced a
significant decrease in cell viability, which was partially
attenuated by NSA (Fig. 2D). These
results revealed that pyroptosis played an essential role in the
cytotoxic effects of CSE on 16HBE cells.
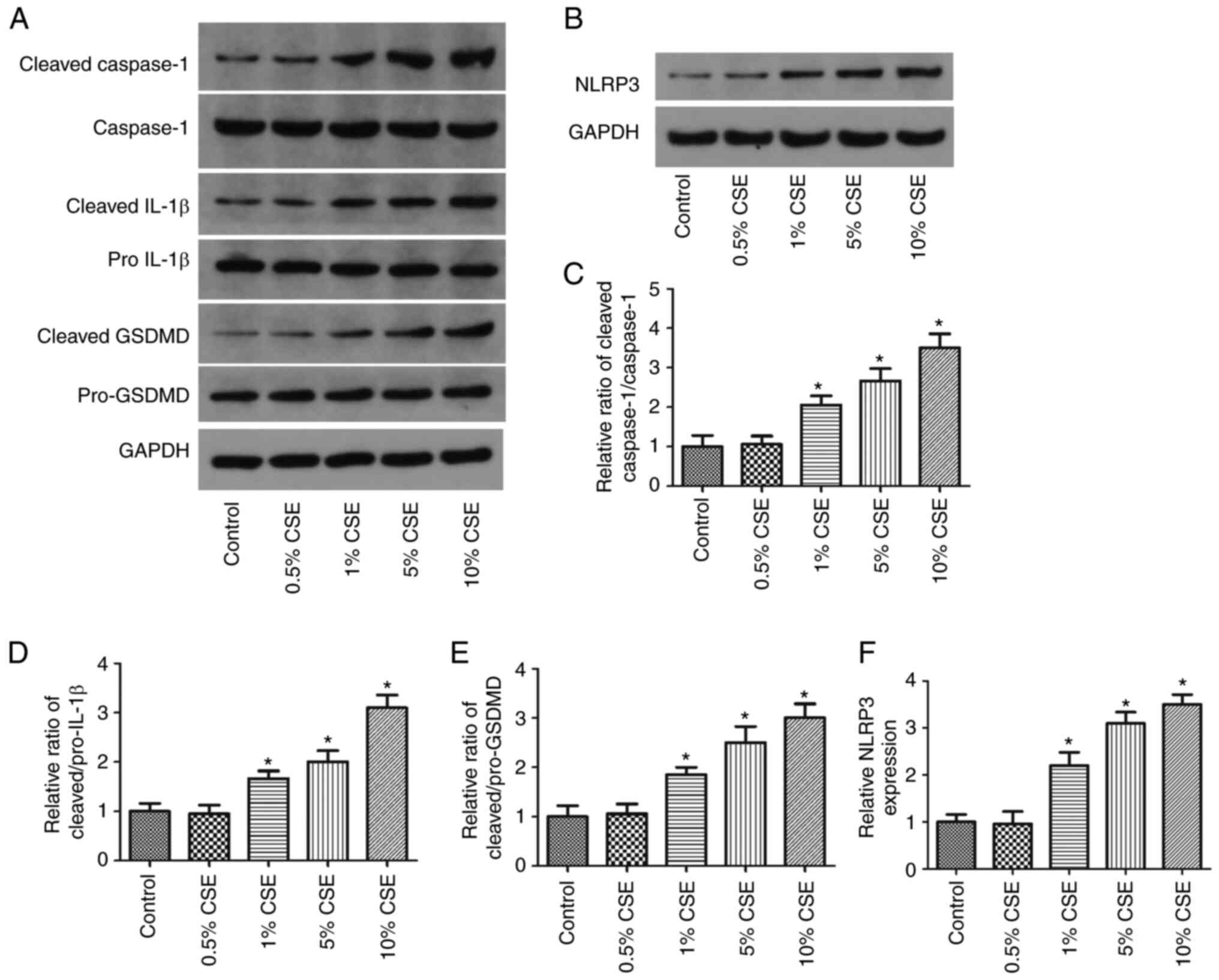
CSE increases the expression of
pyroptosis-related proteins in 16HBE cells in a
concentration-dependent manner
The 16HBE cells were then exposed to various
concentrations of CSE (0.5–10%) for 24 h to validate the effects on
pyroptosis. The expression of NLRP3, cleaved Caspase-1, cleaved
IL-1β, Caspase-1, pro-IL-1β, cleaved GSDMD and pro-GSDMD in the
cells was then detected using western blot analysis. Representative
results are shown in Fig. 3A and
B. The relative changes in the levels of these proteins are
shown in Fig. 3C-F. It was
demonstrated that NLRP3 expression and the ratios of cleaved
IL-1β/pro-IL-1β, cleaved Caspase-1/Caspase-1 and cleaved
GSDMD/pro-GSDMD were significantly increased by 1–10% CSE in a
concentration-dependent manner.
CSE-induced pyroptosis is inhibited in
16HBE cells by the NLRP3 inhibitor, CRID3
To examine the function of NLRP3 in 16HBE cells
undergoing CSE-induced pyroptosis, the cells were exposed to 5% CSE
and treated with the NLRP3 inhibitor, CRID3, for 24 h, and
pyroptosis and cell viability, as well as the expression levels of
pyroptosis-related proteins, were then examined. As shown in
Fig. 4A and B, CRID3 significantly
reduced the LDH activity and increased the viability of the 16HBE
cells. As shown in Fig. 4C-H,
CRID3 substantially decreased NLRP3 expression and the ratios of
cleaved IL-1β/pro-IL-1β, cleaved Caspase-1/Caspase-1 and cleaved
GSDMD/pro-GSDMD. These findings suggested that pyroptosis induced
by CSE was dependent on NLRP3 in the 16HBE cells.
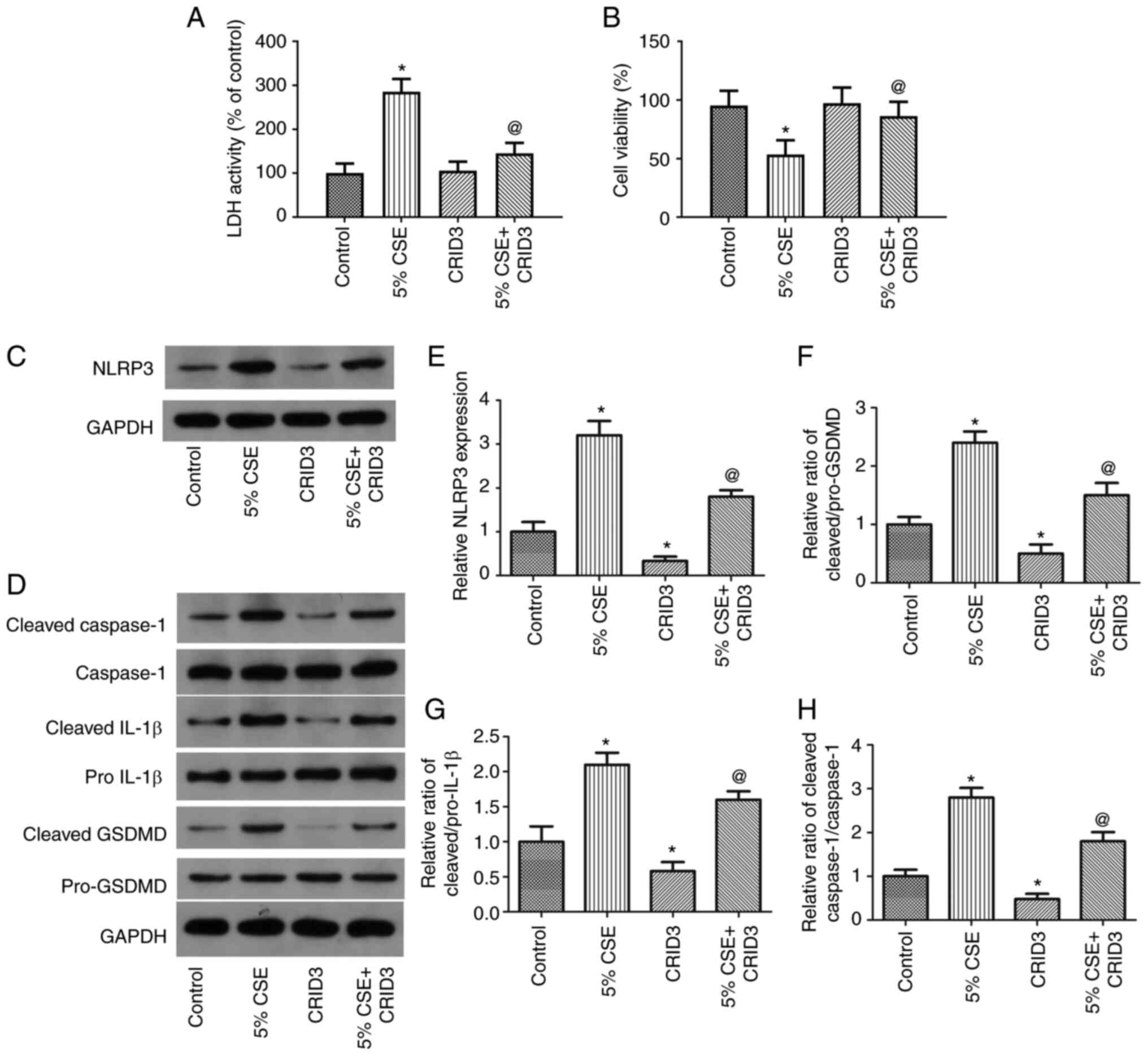 | Figure 4.The NLRP3 inhibitor, CRID3, reverses
the effects of CSE on the pyroptosis of 16HBE cells. To examine the
role of NLRP3 in the CSE-induced pyroptosis of 16HBE cells, the
cells were exposed to 5% CSE and treated with the NLRP3 inhibitor,
CRID3, for 24 h. (A and B) LDH activity and cell viability, (C-H)
as well as the expression levels of pyroptosis-related proteins
were examined. Data are presented as the mean ± SD and are
representative of three independent experiments. *P<0.05
compared with the control group; @P<0.05 compared
with the CSE group. NLRP3, NLR family pyrin domain containing 3;
GSDMD, gasdermin D; CRID3, cytokine release inhibitory drug 3; CSE,
cigarette smoke extract; LDH, lactate dehydrogenase. |
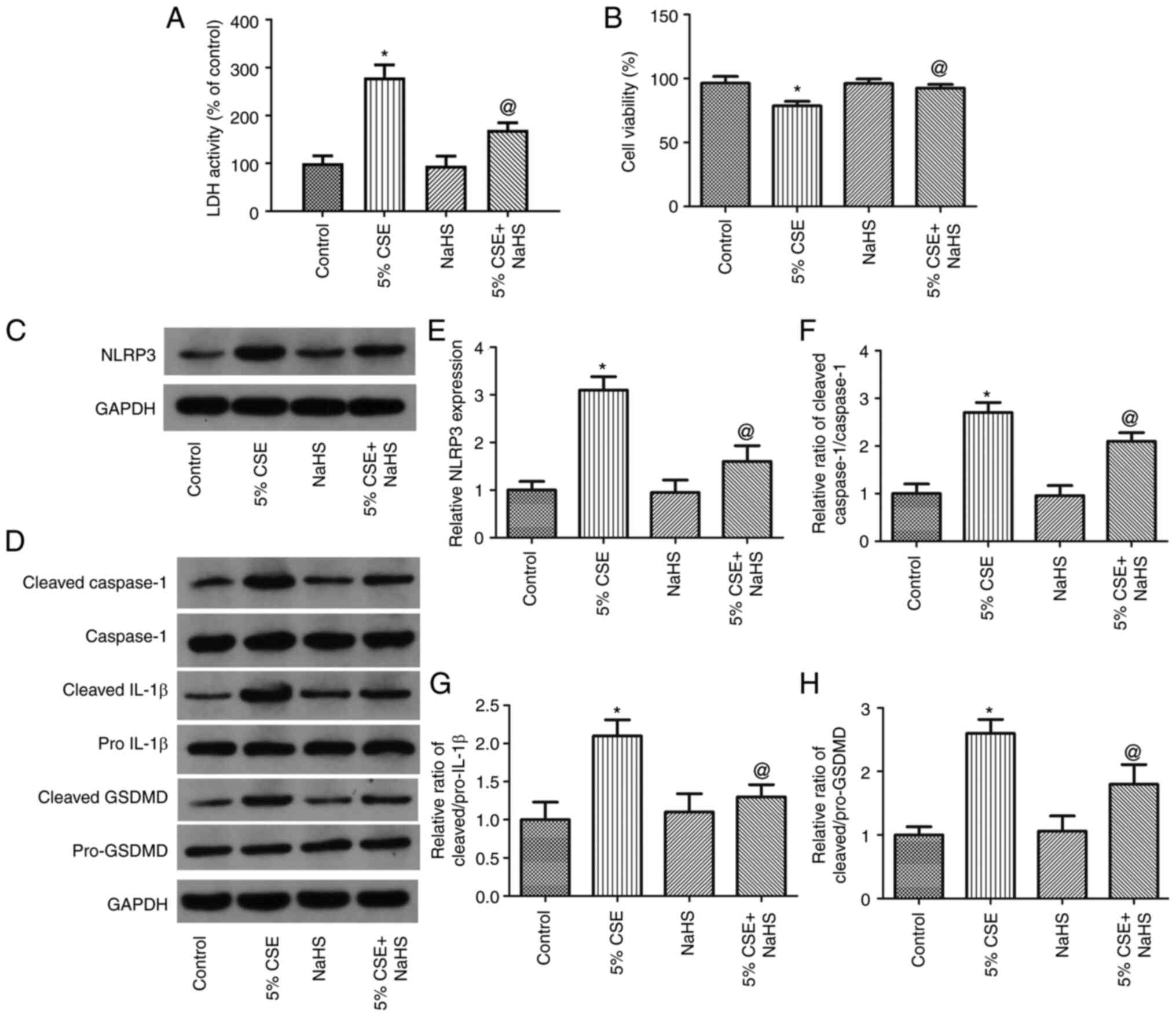
NaHS reverses the CSE-induced
pyroptosis of 16HBE cells
To examine the effects of H2S on the
CSE-induced pyroptosis of 16HBE cells, the cells were exposed to 5%
CSE and treated with NaHS (an H2S donor) for 24 h. The
LDH activity, cell viability and the expression levels of
pyroptosis-related proteins were then measured. As shown in
Fig. 5A and B, NaHS significantly
decreased LDH activity and increased 16HBE cell viability. As shown
in Fig. 5D-H, NaHS considerably
decreased NLRP3 expression and the ratios of cleaved
IL-1β/pro-IL-1β, cleaved Caspase-1/Caspase-1 and cleaved
GSDMD/pro-GSDMD. Thus, these findings demonstrated that
H2S decreased the pyroptosis of 16HBE cells induced by
CSE.
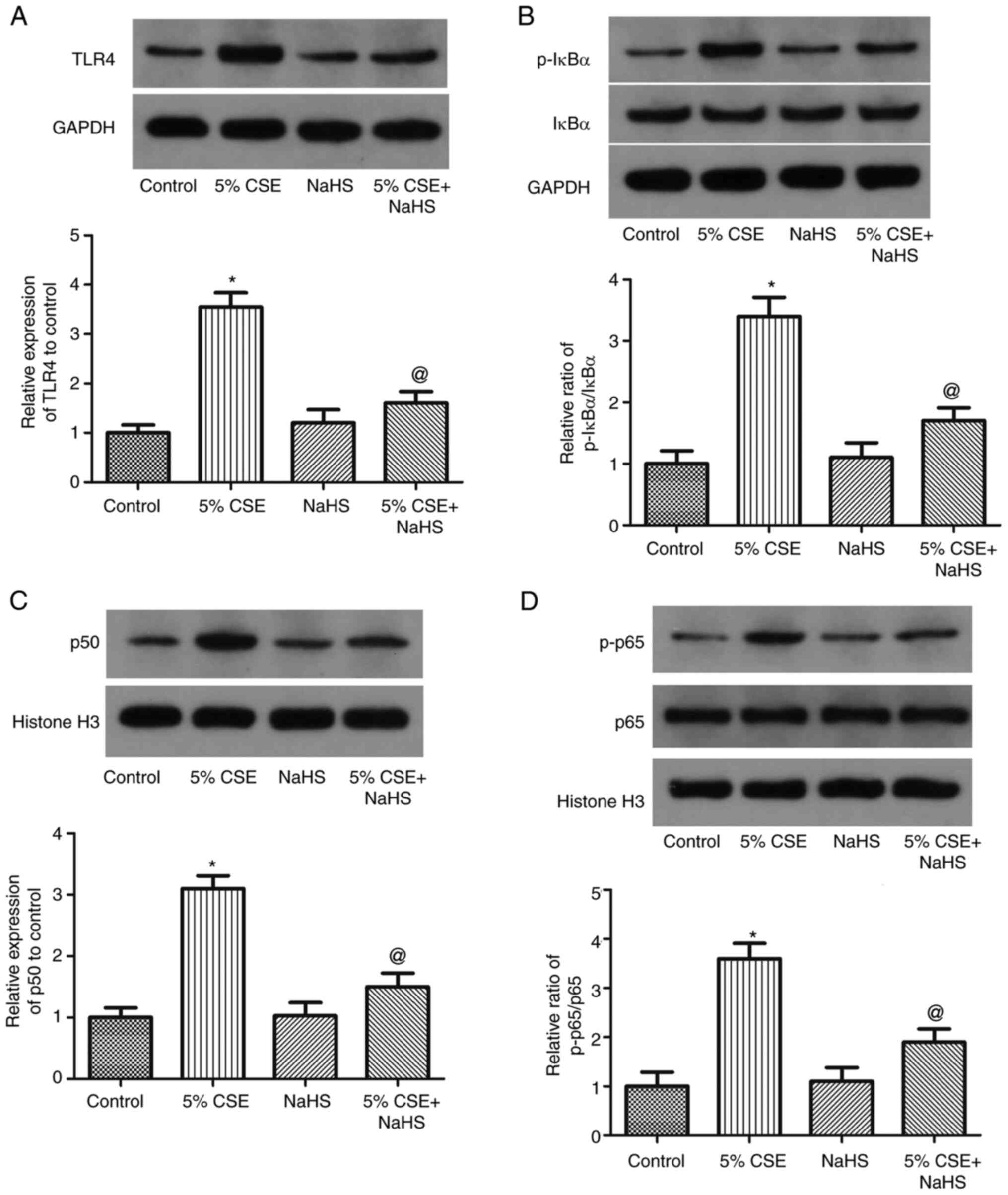
NaHS suppresses the activation of the
TLR4/NF-κB pathway in CSE-challenged 16HBE cells
To examine the role of the TLR4/NF-κB pathway in the
effects of H2S on the CSE-induced pyroptosis of 16HBE
cells, the cells were exposed to 5% CSE and NaHS (H2S
donor) for 24 h, and the expression levels of IκBα, p-IκBα, NF-κB
p50, NF-κB p65 and NF-κB p-p65 were measured (Fig. 6). The ratios of NF-κB p-p65/p65 and
p-IκBα/IκBα, as well as the expression levels of NF-κB p50 and TLR4
in 16HBE cells, were markedly increased by CSE; these effects were
partly reversed by NaHS. These findings thus revealed that CSE
promoted TLR4/NF-κB pathway activation in 16HBE cells, while this
was inhibited by H2S.
Effects of TLR4/NF-κB
overexpression/knockdown on the CSE-induced pyroptosis of 16HBE
cells
To examine the role of the TLR4/NF-κB pathway in the
effects of NaHS on CSE-induced pyroptosis, the 16HBE cells were
exposed to 5% CSE, and treated with NaHS and lentivirus
overexpressing TLR4 or NF-κB. The transfection efficiency was
determined using RT-qPCR (Fig. 7C and
D). LDH activity, cell viability and the expression levels of
pyroptosis-related proteins were then measured. As shown in
Fig. 7A and B, transfection with
TLR4 and NF-κB lentivirus significantly increased the LDH activity
and decreased 16HBE cell viability compared with the 5% CSE + NaHS
group. On the other hand, TLR4 and NF-κB lentivirus markedly
increased the ratios of cleaved Caspase-1/Caspase-1 and cleaved
GSDMD/pro-GSDMD (Fig. 7E-G). In
addition, when TLR4 or NF-κB expression was silenced in the 16HBE
cells by lentiviral transduction. The transfection efficiency was
determined using RT-qPCR (Fig. 7J and
K). It was found that TLR4 or NF-κB knockdown mimicked the
effects of NaHS in terms of LDH activity, cell viability and the
level of cleaved Caspase-1/Caspase-1 and cleaved GSDMD/pro-GSDMD
(Fig. 7H, I and L-N). These
findings revealed that the TLR4/NF-κB pathway was essential for the
protective effect of H2S against CSE-induced pyroptosis
in 16HBE cells.
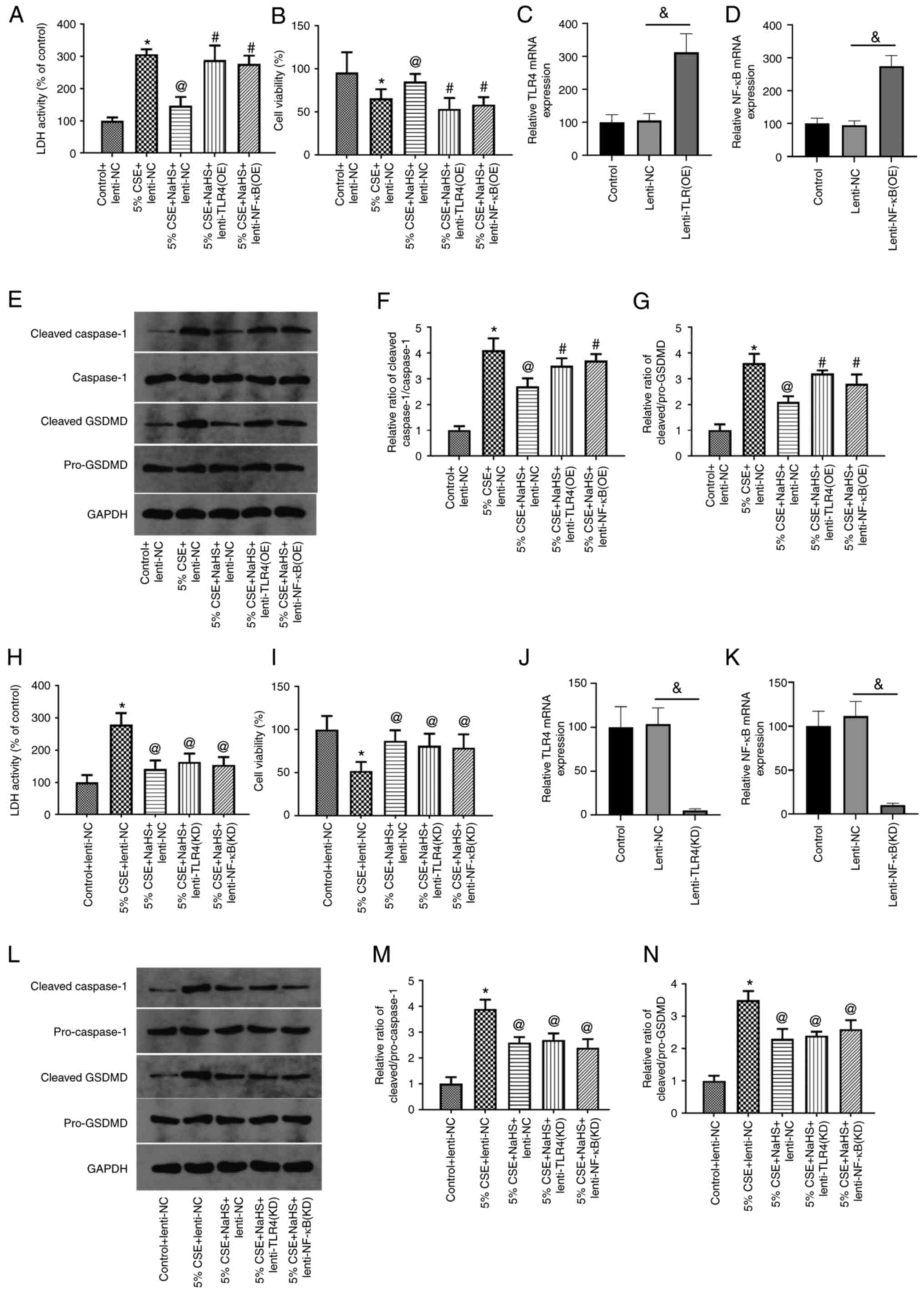 | Figure 7.Overexpression of TLR4/NF-κB
abolishes the suppressive effects of NaHS on the CSE-induced
pyroptosis of 16HBE cells. (A-G) 16HBE cells were exposed to 5%
CSE, and treated with NaHS and lentivirus overexpressing TLR4 or
NF-κB. (A) LDH activity, (B) cell viability and (E-G) the
expression levels of pyroptosis-related proteins were then
measured. (C and D) The transfection efficiency was measured using
RT-qPCR. (H-N) 16HBE cells were exposed to 5% CSE, and then treated
with NaHS and transfected with lentivirus to knockdown TLR4 or
NF-κB. (H) LDH activity, (I) cell viability and (L-N) the
expression levels of pyroptosis-related proteins were then
measured. (J and K) The transfection efficiency was measured using
RT-qPCR. Data are presented as the mean ± SD and are representative
of three independent experiments. #P<0.05 compared
with the 5% CSE + NaHS + Lenti-NC group; *P<0.05 compared with
the control + Lenti-NC group; @P<0.05 compared with
the 5% CSE + Lenti-NC group; &P<0.05 between
lenti-NC and transfection groups. TLR4, Toll-like receptor 4; CSE,
cigarette smoke extract; LDH, lactate dehydrogenase; GSDMD,
gasdermin D; NaHS, sodium hydrosulfide; RT-qPCR, reverse
transcription-quantitative PCR. |
Discussion
Pyroptosis is a type of Caspase-1-dependent
programmed cell death and is accompanied by an inflammatory
reaction (4). When exogenous
(bacteria and viruses) or endogenous (stress factors released when
the cell is attacked) danger signals stimulate the cells, NF-κB
activates NLRP3, which in turn activates Caspase-1 precursor,
cleaves GSDMD protein and releases N-terminal fragment. This
recognizes and binds the phospholipids on the cell membrane and
forms cell membrane pores, as well as loses the ability to control
the entry and exit of substances, leading to cell osmotic pressure
changes, cell lysis, the release of cell contents and cell death
(5). NLRP3 is the basic component
of the inflammasome in cell pyroptosis. Pyroptosis is closely
associated with inflammatory lung diseases. As previously
demonstrated in a mouse model of COPD, the activation of caspase-l,
the production of IL-18 and IL-1B, and the inflow of neutrophils in
bronchoalveolar lavage fluid of NLRP3-knockout mice were decreased,
indicating the involvement of NLRP3 in COPD pathogenesis (30). The findings of the present study
demonstrated that, in the CS-induced COPD model, cell pyroptosis
was significantly activated. Compared with the control group, the
expression level of GSDMD-N was increased in the CS group, as were
NLRP3 expression and the ratios of cleaved Caspase-1/Caspase-1,
cleaved IL-1β/pro-IL-1β and cleaved GSDMD/pro-GSDMD. The exposure
of 16HBE cells to CSE induced an increase in the pyroptosis (as
demonstrated by LDH leakage) and pyroptosis-related protein
expression (NLRP3, cleaved Caspase-1, cleaved GSDMD and cleaved
IL-1β) in a concentration-dependent manner. This finding is in
accordance with earlier research (15,16).
Previous animal studies have revealed that frequent exposure to CS
can lead to an increase in the number of inflammatory factors in
the alveolar space and lung parenchyma, leading to cell death
(31,32). Thioredoxin interacting protein
(TXNIP) combines with NLRP3 to form a complex in the process of
COPD, which can initiate pyroptosis and promote inflammatory
response through TXNIP overexpression, while the inhibition of
TXNIP expression can reduce the formation of inflammatory bodies,
improve cell viability and alleviate COPD progression (33,34).
GSDMD is the executor of cell pyroptosis. To
demonstrate that GSDMD was cleaved by Caspase-11 at the preserved
residual of d276, Kayagaki et al (35) used the CRISPR-Cas9 genome editing
technique to produce the GSDMD-C- and GSDMD-N-terminal domains,
which eventually led to cell pyroptosis. GSDMD is an essential
pyroptosis substance for all inflammatory caspases, and is a common
substrate. The results of the present study demonstrated that the
GSDMD inhibitor, NSA, reversed the increased pyroptosis (as
demonstrated by LDH leakage) and enhanced cell viability,
indicating that pyroptosis played an essential role in the
cytotoxic effects of CSE on 16HBE cells. This result also suggested
that the CSE-induced pyroptosis of 16HBE cells was
GSDMD-dependent.
NLRP3, as an important inflammatory body, can be
activated via two mechansims. On the one hand, it can stimulate
TLR4 and NF-κB and can modulate the expression levels of adhesion
and inflammatory factors, thus producing pro-IL-18 and pro-IL-1β
(36). On the other hand, NLRP3
binds with apoptosis-associated speck-like protein containing a
caspase activation and recruitment domain to form an activated
NLRP3 inflammasome, which then cleaves Caspase-1 to promote the
formation and maturation of pro-IL-18 and pro-IL-1β, thus mediating
pyroptosis (37). The cell
experimental results of the present study demonstrated that the
NLRP3 inhibitor, CRID3, significantly reduced pyroptosis (as
demonstrated by LDH leakage) and reduced the expression of
pyroptosis-related protein (NLRP3, cleaved IL-1β, Caspase-1 and
GSDMD) in 16HBE cells, indicating that the CSE-induced pyroptosis
of 16HBE cells was NLRP3-dependent. In combination, these results
support the conclusion that CS induces the pyroptosis of human
bronchial epithelial cells in a NLRP3/GSDMD-dependent manner.
Several studies have reported that H2S
plays a protective role against cell injury by deterring NLRP3
inflammasome activation. In human acute monocytic leukemia cells,
H2S has been shown to inhibit NLRP3 protein expression,
as well as the LPS-induced Caspase-1 expression (38). It has also been demonstrated that
H2S attenuates LPS-induced acute kidney damage in mice
by preventing the formation of the NLRP3 inflammasome (39). Furthermore, H2S reduces
ischemia-reperfusion injury by impeding the inflammatory response,
as well as TLR-mediated oxidative stress (40,41).
H2S has also been found to inhibit the inflammatory
response by inhibiting NF-κB activity, as well as by reducing the
expression level of TNF-α (42).
The effects of inhaled H2S on lung injury were
investigated in the present study, and its impact on CS-induced
pyroptosis in rats was then investigated. The results demonstrated
that inflammatory cell infiltration and alveolar septum thickening
were markedly alleviated by H2S, and it also decreased
GSDMD-N expression in the lungs. Furthermore, the expression levels
of pyroptosis-related proteins (NLRP3, cleaved IL-1β, cleaved
Caspase-1 and cleaved GSDMD) were markedly decreased by
H2S, indicating that inhaled H2S inhibited
pyroptosis in the model of CS-induced COPD. To further examine the
mechanisms of H2S on the CS-induced pyroptosis of lung
cells, the 16HBE cells were exposed to CSE and treated with NaHS
(H2S donor), and the pyroptosis (as demonstrated by LDH
leakage), viability and the expression levels of pyroptosis-related
proteins were then measured. Similar to the results of the in
vivo experiments, H2S significantly attenuated the
effects of CSE on pyroptosis, cell viability and pyroptosis-related
protein expression. It was thus indicated that H2S
inhibited pyroptosis by decreasing NLRP3 expression and GSDMD
activation.
The contribution of the TLR4/NF-κB signaling pathway
to the inflammatory response and other pathological changes has
been demonstrated (43). TLRs,
which can activate the innate immune system, are a family of
pattern recognition receptors. The primary function of TLR4 is to
recognize exogenous molecules from pathogens (such as LPS). TLR4
can be recognized by the metabolites of bacteria, viruses and other
pathogens (44). At the same time,
TLR4, as a specific recognition receptor of LPS, can be activated
in numerous immune cells, such as macrophages and B-lymphocytes
(36). Previous studies have
revealed that TLR4 is highly expressed in BV-2 cells, RAW264.7
macrophages and C57BL/6 mice exposed to LPS (45–47).
In the present study, to evaluate the role of TLR4 in the effects
of H2S on the CSE-induced pyroptosis of 16HBE cells, the
cells were treated with 5% CSE and NaHS (H2S donor) for
24 h. TLR4 expression in the 16HBE cells was then assessed. TLR4
expression in the 16HBE cells was increased by CSE, but was partly
reversed by NaHS. These findings suggested that in 16HBE cells, CSE
activated TLR4, while this was inhibited by H2S.
NF-κB is an ubiquitous transcription factor
mediating the cytoplasmic-nuclear signaling pathway. NF-κB exists
in the form of a dimer and its role in the development of different
inflammation-related illnesses has been proven, such as via its
involvement in cell apoptosis and proliferation (48). Liu et al (49) revealed that melatonin alleviated
inflammatory pyroptosis in mouse adipose tissue by modulating the
NF-κB/GSDMD signaling pathway, while Chen et al (50) reported that never in mitosis gene A
(NIMA)-related kinase 7 (NEK7) interacted with NLRP3 to regulate
pyroptosis through NF-κB signaling in inflammatory bowel disease.
The study by Shao et al (51) demonstrated that traumatic brain
injury-induced acute lung injury was alleviated by ghrelin through
the pyroptosis/NF-κB pathway, while Tian et al (52) found that exposure to ozone
stimulated pyroptosis in the lungs of rats through the
TLR2/4/NF-κB/NLRP3 signaling pathway. Therefore, it was
hypothesized that NF-κB may be the key effector molecule of
CSE-induced cell pyroptosis.
In the present study, to examine the function of the
NF-κB pathway, the cells were exposed to 5% CSE and treated with
NaHS, and the expression levels of IκBα, p-IκBα, NF-κB p50, NF-κB
p65 and NF-κB p-p65 were measured in the 16HBE cells. The ratios of
NF-κB p-p65/p65 and p-IκBα/IκBα, as well as the expression levels
of NF-κB p50 in 16HBE cells were increased by CSE, which these were
all partly reversed by NaHS. These findings suggested that CSE
induced the activation of the TLR4/NF-κB pathway in 16HBE cells,
while this was inhibited by H2S. A recent study reported
that NF-κB was the prominent GSDMD transcription factor (49). Under normal conditions, the
C-terminal of GSDMD automatically inhibits the pore-forming
activity of the N-terminal (53).
When NF-κB is activated, it activates the inflammasomes and
Caspase-1, and causes the separation of the N- and C-terminals of
GSDMD. Under external stimulation, activated TLR4 activates the IκB
kinase (IKK) complex through the myeloid differentiation primary
response 88-dependent pathway, leading to IKK phosphorylation.
Subsequently, IKK is degraded through the ubiquitin-proteasome
pathway, thus releasing NF-κB and promoting its entry into the
nucleus from the cytoplasm, thereby inducing the expression levels
of inflammation-related genes (54–56).
In the present study, to analyze the role of the TLR4/NF-κB
pathway, 16HBE cells were exposed to CSE, and treated with NaHS and
lentivirus overexpressing TLR4 or NF-κB; LDH activity, cell
viability and pyroptosis were then measured. NF-κB and TLR4
overexpression markedly increased pyroptosis (as demonstrated by
LDH leakage) and decreased the viability of 16HBE cells.
Furthermore, the ratios of cleaved GSDMD/pro-GSDMD and cleaved
Caspase-1/Caspase-1 were markedly increased, indicating that the
effect of H2S on cell pyroptosis was diminished by TLR4
and NF-κB overexpression. As TLR4 or NF-κB overexpression abrogated
the therapeutic effects of NaHS, it was suggested that NaHS reduced
pyroptosis by inhibiting TLR4/NF-κB signaling. Moreover, in order
to further strengthen the results, the expression of TLR4 and NF-κB
was silenced in 16HBE cells, followed by exposure to CSE and
treatment with NaHS. It was found that TLR4 or NF-κB knockdown
mimicked the effects of NaHS in terms of cell viability and the
level of cleaved Caspase-1/Caspase-1 and cleaved GSDMD/pro-GSDMD.
In combination, these results indicated that the TLR4/NF-κB pathway
may be essential for the protective effects of H2S
against the CSE-induced pyroptosis of 16HBE cells.
In conclusion, the present study demonstrated that
H2S alleviated lung injury and pyroptosis in a model of
CS-induced COPD by inhibiting the activation of the TLR4/NF-κB
signaling pathway. These findings suggest the importance of
pyroptosis in the development of COPD and provide an experimental
framework in which H2S and drugs targeting the
TLR4/NF-κB pathway may be utilized for protection against COPD.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CY and YL contributed to the conception of the
study. LW and JM performed all of the experiments. LW and JM
contributed significantly to the analysis of the results of the
experiments and to manuscript preparation. CW performed the data
analyses and wrote the manuscript. YW helped perform the analysis
of the proofs manuscript and provided constructive discussions. LW,
CW and JM confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
The animal procedures were approved by the Animal
Ethics Committee of the Hebei Chest Hospital Animal Center and
complied with the Guide of the Care and Use of Laboratory Animals
published by NIH (NIH Pub. no. 85-23, revised 1996).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhou M, Wang H, Zhu J, Chen W, Wang L, Liu
S, Li Y, Wang L, Liu Y, Yin P, et al: Cause-specific mortality for
240 causes in China during 1990–2013: A systematic subnational
analysis for the global burden of disease study 2013. Lancet.
387:251–272. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
López-Campos JL, Tan W and Soriano JB:
Global burden of COPD. Respirology. 21:14–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Saetta M, Turato G, Maestrelli P, Mapp CE
and Fabbri LM: Cellular and structural bases of chronic obstructive
pulmonary disease. Am J Respir Crit Care Med. 163:1304–1309. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Churg A, Wang RD, Tai H, Wang X, Xie C,
Dai J, Shapiro SD and Wright JL: Macrophage metalloelastase
mediates acute cigarette smoke-induced inflammation via tumor
necrosis factor-alpha release. Am J Respir Crit Care Med.
167:1083–1089. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang C, Xu J, Yang L, Xu Y, Zhang X, Bai
C, Kang J, Ran P, Shen H, Wen F, et al: Prevalence and risk factors
of chronic obstructive pulmonary disease in China (the China
Pulmonary Health [CPH] study): A national cross-sectional study.
Lancet. 391:1706–1717. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
US Preventive Services Task Force
(USPSTF), . Siu AL, Bibbins-Domingo K, Grossman DC, Davidson KW,
Epling JW Jr, García FA, Gillman M, Kemper AR, Krist AH, et al:
Screening for chronic obstructive pulmonary disease: US preventive
services task force recommendation statement. JAMA. 315:1372–1377.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shi J, Gao W and Shao F: Pyroptosis:
Gasdermin-mediated programmed necrotic cell death. Trends Biochem
Sci. 42:245–254. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vande Walle L and Lamkanfi M: Pyroptosis.
Curr Biol. 26:R568–R572. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dong ZW and Yuan YF: Juglanin suppresses
fibrosis and inflammation response caused by LPS in acute lung
injury. Int J Mol Med. 41:3353–3365. 2018.PubMed/NCBI
|
|
10
|
Li Y, Song D, Bo F, Deng M and Tang X:
Diazepam inhibited lipopolysaccharide (LPS)-induced pyroptotic cell
death and alleviated pulmonary fibrosis in mice by specifically
activating GABAA receptor α4-subunit. Biomed
Pharmacother. 118:1092392019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pinkerton JW, Kim RY, Robertson AAB,
Hirota JA, Wood LG, Knight DA, Cooper MA, O'Neill LAJ, Horvat JC
and Hansbro PM: Inflammasomes in the lung. Mol Immunol. 86:44–55.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang YC, Liu QX, Zheng Q, Liu T, Xu XE,
Liu XH, Gao W, Bai XJ and Li ZF: Dihydromyricetin alleviates
sepsis-induced acute lung injury through inhibiting NLRP3
inflammasome-dependent pyroptosis in mice model. Inflammation.
42:1301–1310. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu WJ, Wang XX, Jin JJ, Zou Q, Wu L, Lv
TF, Wan B, Zhan P, Zhu SH, Liu HB, et al: Inhibition of GGPPS1
attenuated LPS-induced acute lung injury and was associated with
NLRP3 inflammasome suppression. Am J Physiol Lung Cell Mol Physiol.
316:L567–L577. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsai YM, Chiang KH, Hung JY, Chang WA, Lin
HP, Shieh JM, Chong IW and Hsu YL: Der f1 induces pyroptosis in
human bronchial epithelia via the NLRP3 inflammasome. Int J Mol
Med. 41:757–764. 2018.PubMed/NCBI
|
|
15
|
Zhang MY, Jiang YX, Yang YC, Liu JY, Huo
C, Ji XL and Qu YQ: Cigarette smoke extract induces pyroptosis in
human bronchial epithelial cells through the ROS/NLRP3/caspase-1
pathway. Life Sci. 269:1190902021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu Z, Lian X, Su X, Wu W, Zeng Y and Chen
X: Exosomes derived from adipose-derived stem cells alleviate
cigarette smoke-induced lung inflammation and injury by inhibiting
alveolar macrophages pyroptosis. Respir Res. 23:52022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang R: Hydrogen sulfide: The third
gasotransmitter in biology and medicine. Antioxid Redox Signal.
12:1061–1064. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oláh G, Módis K, Törö G, Hellmich MR,
Szczesny B and Szabo C: Role of endogenous and exogenous nitric
oxide, carbon monoxide and hydrogen sulfide in HCT116 colon cancer
cell proliferation. Biochem Pharmacol. 149:186–204. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu X, Li H, Gong Y, Zheng H and Zhao D:
Hydrogen sulfide ameliorated lipopolysaccharide-induced acute lung
injury by inhibiting autophagy through PI3K/Akt/mTOR pathway in
mice. Biochem Biophys Res Commun. 507:514–518. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu M, Li Z, Liang B, Li L, Liu S, Tan W,
Long J, Tang F, Chu C and Yang J: Hydrogen sulfide ameliorates rat
myocardial fibrosis induced by thyroxine through PI3K/AKT signaling
pathway. Endocr J. 65:769–781. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
George AK, Singh M, Homme RP, Majumder A,
Sandhu HS and Tyagi SC: A hypothesis for treating inflammation and
oxidative stress with hydrogen sulfide during age-related macular
degeneration. Int J Ophthalmol. 11:881–887. 2018.PubMed/NCBI
|
|
22
|
Wang L, Meng J, Wang C, Yang C, Wang Y and
Li Y and Li Y: Hydrogen sulfide alleviates cigarette smoke-induced
COPD through inhibition of the TGF-β1/smad pathway. Exp Biol Med
(Maywood). 245:190–200. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
National Research Council (US) Institute
for Laboratory Animal Research, . Guide for the Care and Use of
Laboratory Animals. National Academies Press; Washington, DC:
1996
|
|
24
|
Ke Q, Yang L, Cui Q, Diao W, Zhang Y, Xu M
and He B: Ciprofibrate attenuates airway remodeling in cigarette
smoke-exposed rats. Respir Physiol Neurobiol. 271:1032902020.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hosseini A, Rasaie D, Soleymani Asl S,
Nili Ahmadabadi A and Ranjbar A: Evaluation of the protective
effects of curcumin and nanocurcumin against lung injury induced by
sub-acute exposure to paraquat in rats. Toxin Rev. 40:1233–1241.
2019. View Article : Google Scholar
|
|
26
|
Song B, Ye L, Wu S and Jing Z: Long
non-coding RNA MEG3 regulates CSE-induced apoptosis and
inflammation via regulating miR-218 in 16HBE cells. Biochem Biophys
Res Commun. 521:368–374. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ding Y, Liu P, Chen ZL, Zhang SJ, Wang YQ,
Cai X, Luo L, Zhou X and Zhao L: Emodin attenuates
lipopolysaccharide-induced acute liver injury via inhibiting the
TLR4 signaling pathway in vitro and in vivo. Front Pharmacol.
9:9622018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Y, Xia G, Zhang Y, Liu J, Liu X, Li
W, Lv Y, Wei S, Liu J and Quan J: Palmitate induces VSMC apoptosis
via toll like receptor (TLR)4/ROS/p53 pathway. Atherosclerosis.
263:74–81. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang W, Ni H, Wang H and Gu H: NLRP3
inflammasome is essential for the development of chronic
obstructive pulmonary disease. Int J Clin Exp Pathol.
8:13209–13216. 2015.PubMed/NCBI
|
|
31
|
Lee S, Suh GY, Ryter SW and Choi AM:
Regulation and function of the nucleotide binding domain
leucine-rich repeat-containing receptor, pyrin domain-containing-3
inflammasome in lung disease. Am J Respir Cell Mol Biol.
54:151–160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Eltom S, Belvisi MG, Stevenson CS, Maher
SA, Dubuis E, Fitzgerald KA and Birrell MA: Role of the
inflammasome-caspase1/11-IL-1/18 axis in cigarette smoke driven
airway inflammation: An insight into the pathogenesis of COPD. PLoS
One. 9:e1128292014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Barnes PJ: Inflammatory mechanisms in
patients with chronic obstructive pulmonary disease. J Allergy Clin
Immunol. 138:16–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Heo MJ, Kim TH, You JS, Blaya D,
Sancho-Bru P and Kim SG: Alcohol dysregulates miR-148a in
hepatocytes through FoxO1, facilitating pyroptosis via TXNIP
overexpression. Gut. 68:708–720. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kayagaki N, Stowe IB, Lee BL, O'Rourke K,
Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT,
et al: Caspase-11 cleaves gasdermin D for non-canonical
inflammasome signalling. Nature. 526:666–671. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ding J, Wang K, Liu W, She Y, Sun Q, Shi
J, Sun H, Wang DC and Shao F: Pore-forming activity and structural
autoinhibition of the gasdermin family. Nature. 535:111–116. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liang H and Liu Y: Gasdermins pore cell
membrane to pyroptosis. Sci China Life Sci. 59:1090–1092. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yue LM, Gao YM and Han BH: Evaluation on
the effect of hydrogen sulfide on the NLRP3 signaling pathway and
its involvement in the pathogenesis of atherosclerosis. J Cell
Biochem. 120:481–492. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen Y, Jin S, Teng X, Hu Z, Zhang Z, Qiu
X, Tian D and Wu Y: Hydrogen sulfide attenuates LPS-induced acute
kidney injury by inhibiting inflammation and oxidative stress. Oxid
Med Cell Longev. 2018:67172122018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tan Z, Shi Y, Yan Y, Liu W, Li G and Li R:
Impact of endogenous hydrogen sulfide on toll-like receptor pathway
in renal ischemia/reperfusion injury in rats. Ren Fail. 37:727–733.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Han SJ, Kim JI, Park JW and Park KM:
Hydrogen sulfide accelerates the recovery of kidney tubules after
renal ischemia/reperfusion injury. Nephrol Dial Transplant.
30:1497–1506. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen X, Xu W, Wang Y, Luo H, Quan S, Zhou
J, Yang N, Zhang T, Wu L, Liu J, et al: Hydrogen sulfide reduces
kidney injury due to urinary-derived sepsis by inhibiting NF-κB
expression, decreasing TNF-α levels and increasing IL-10 levels.
Exp Ther Med. 8:464–470. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fu Y, Liu B, Zhang N, Liu Z, Liang D, Li
F, Cao Y, Feng X, Zhang X and Yang Z: Magnolol inhibits
lipopolysaccharide-induced inflammatory response by interfering
with TLR4 mediated NF-κB and MAPKs signaling pathways. J
Ethnopharmacol. 145:193–199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Varshney D, Singh S, Sinha E, Mohanty KK,
Kumar S, Kumar Barik S, Patil SA and Katara P: Systematic review
and meta-analysis of human Toll-like receptors genetic
polymorphisms for susceptibility to tuberculosis infection.
Cytokine. 152:1557912022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhou P, Weng R, Chen Z, Wang R, Zou J, Liu
X, Liao J, Wang Y, Xia Y and Wang Q: TLR4 signaling in MPP+-induced
activation of BV-2 cells. Neural Plast. 2016:50767402016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Huang H, Park PH, McMullen MR and Nagy LE:
Mechanisms for the anti-inflammatory effects of adiponectin in
macrophages. J Gastroenterol Hepatol. 23 (Suppl 1):S50–S53. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Säfholm J, Lövdahl C, Swedin L, Boels PJ,
Dahlén SE, Arner A and Adner M: Inflammation-induced airway smooth
muscle responsiveness is strain dependent in mice. Pulm Pharmacol
Ther. 24:361–366. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Soleimani A, Rahmani F, Ferns GA, Ryzhikov
M, Avan A and Hassanian SM: Role of the NF-κB signaling pathway in
the pathogenesis of colorectal cancer. Gene. 726:1441322020.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu Z, Gan L, Xu Y, Luo D, Ren Q, Wu S and
Sun C: Melatonin alleviates inflammasome-induced pyroptosis through
inhibiting NF-κB/GSDMD signal in mice adipose tissue. J Pineal Res.
63:2017. View Article : Google Scholar
|
|
50
|
Chen X, Liu G, Yuan Y, Wu G, Wang S and
Yuan L: NEK7 interacts with NLRP3 to modulate the pyroptosis in
inflammatory bowel disease via NF-κB signaling. Cell Death Dis.
10:9062019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shao XF, Li B, Shen J, Wang QF, Chen SS,
Jiang XC and Qiang D: Ghrelin alleviates traumatic brain
injury-induced acute lung injury through pyroptosis/NF-κB pathway.
Int Immunopharmacol. 79:1061752020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tian L, Yan J, Li K, Zhang W, Lin B, Lai
W, Bian L, Liu H, Xi Z and Liu X: Ozone exposure promotes
pyroptosis in rat lungs via the TLR2/4-NF-κB-NLRP3 signaling
pathway. Toxicology. 450:1526682021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liu Z, Wang C, Rathkey JK, Yang J, Dubyak
GR, Abbott DW and Xiao TS: Structures of the gasdermin D C-terminal
domains reveal mechanisms of autoinhibition. Structure.
26:778–784.e3. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ayala-Cuellar AP, Cho J and Choi KC:
Toll-like receptors: A pathway alluding to cancer control. J Cell
Physiol. 234:21707–21715. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lei J, Fu Y, Zhuang Y, Zhang K and Lu D:
miR-382-3p suppressed IL-1β induced inflammatory response of
chondrocytes via the TLR4/MyD88/NF-κB signaling pathway by directly
targeting CX43. J Cell Physiol. 234:23160–23168. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen Z, Liu Q, Zhu Z, Xiang F, Wu R and
Kang X: Toll-like receptor 4 contributes to uterine activation by
upregulating pro-inflammatory cytokine and CAP expression via the
NF-κB/P38MAPK signaling pathway during pregnancy. J Cell Physiol.
235:513–525. 2020. View Article : Google Scholar : PubMed/NCBI
|