Introduction
The deubiquitinase CYLD lysine 63
deubiquitinase (CYLD) was first described in cylindromatosis
(1) and its tumor suppressive
properties have been investigated in various types of cancer, such
as pancreas, breast and liver cancer (2-4).
In malignant melanoma, the expression of CYLD is
downregulated by elevated expression of the transcription factor
SNAIL1, which results in increased proliferative and migratory
potential of melanoma cells (5,6).
Recently, our group generated a novel transgenic (Tg) melanoma
mouse model, Tg(Grm1) Cyld, showing enhanced tumor
development and growth in Cyld-knockdown
(Cyld−/−) mice compared with Cyld-wild
type (Cyld+/+) mice (7). Our previous study described a novel
regulatory role of CYLD in vasculogenic mimicry and lymph-
and angiogenesis (7). To the
best of our knowledge, however, the underlying mechanisms resulting
in accelerated tumor growth in Cyld−/− mice have
not yet been determined.
Besides transcriptomic changes, epigenetic
dysregulation, including DNA methylation and post-translational
modification of histones, has been shown to be associated with
cancer in a number of studies (8,9).
These changes lead to organization of DNA into chromatin and ensure
genomic integrity (10). Since
epigenetic processes are reversible, they are currently the focus
of research for novel therapeutic approaches to circumvent drug
resistance in cancer (11,12). The present study aimed to
determine the underlying mechanism of accelerated melanoma
development and the role of CYLD in epigenetic processes of
chromatin formation and histone methylation.
Materials and methods
Murine melanoma cell lines
No animals were sacrificed as murine melanoma cell
lines generated in 2019 were used (7). In brief, tissue samples from
primary tumors (ear and tail) from two
Tg(Grm1)Cyld−/− and two
Tg(Grm1)Cyld+/+ male mice (age, 153 and
217 days) were collected and washed with Braunol® (7.5%;
B Braun Meisungen AG), followed by 1X PBS, 70% ethanol and 1X PBS.
Tumor tissue was added to a mixture of DMEM (Sigma-Aldrich; Merck
KGaA) and collagenase. Following incubation for 3 h at 37°C, the
cell suspension was centrifuged (4 min at 600 g, room temperature)
and the cells were seeded in T25 flasks, as previously described
(7,13).
Cell culture
Murine melanoma cell lines
(mCyld+/+: EPv24 and EPv40 ear;
mCyld−/−: EC36 and EC111 ear) derived from tumor
tissue of Tg(Grm1) model animals (13) were cultivated in DMEM (4,500 mg
glucose/l, 110 mg sodium pyruvate/l and l-glutamine) with 10% fetal
bovine serum, amphotericin B (2.5 μg/ml) and 5%
penicillin/streptomycin (all Sigma-Aldrich; Merck KGaA) in a
humidified atmosphere containing 8% CO2 at 37°C. Cells
were reseeded at a ratio 1:3 to 1:5 twice weekly.
Human primary melanoma cell line Mel Juso [provided
by Dr Judith Johnson (Ludwig Maximilian University of Munich,
Munich, Germany), stably transfected with control green fluorescent
protein [human (h)CYLD−], CYLD
(hCYLD+) or CYLDC/S
(catalytically inactive mutant of CYLD) vector (5,6)
were cultivated (8% CO2 at 37°C) in RPMI-1640
(Sigma-Aldrich; Merck KGaA) with NaHCO3 and the
aforementioned supplements. For cell counting the light microscope
AE2000 from Motic Incorporation Ltd. was used (×4 or ×20
magnification).
For inhibitor studies, murine and human melanoma
cells were treated with Chaetocin (inhibitor of histone
methyltransferase SUV39H1; 10 nM; Absource Diagnostics GmbH), the
Jumonji histone demethylase inhibitor JIB04 (500 nM; Sigma-Aldrich;
Merck KGaA) and CM272 [dual EHMT2/DNA methyltransferase (DNMTs)
inhibitor; 200 nM; kindly provided by Matías A Ávila (14)] for 24 h at 37°C.
For simplification, the following nomenclature has
been used in the rest of the text: murine Cyld-expressing
(mCyld+/+) and Cyld-deficient
(mCyld−/−) cells; human CYLD-expressing
(hCYLD+) and CYLD-deficient
(hCYLD−) and CYLD mutant
(hCYLDC/S) cells.
RNA isolation, reverse
transcription-quantitative (RT-q) PCR
Total RNA and cDNA of the murine and human melanoma
cells were generated as previously described (7). RT-qPCR was performed with
LightCycler 480 system (Roche Diagnostics GmbH) to analyze mRNA
expression, as previously described (15). For each reaction, 1.0 cDNA
template, 0.5 forward and reversed primers each (20 μM each)
and 10.0 μl SYBR Green I (Roche Diagnostics GmbH) were used
in a total volume of 20 μl. The primers were as follows:
β-actin forward, 5′-TGG AAT CCT GTG GCA TCC ATG AAA C-3′ and
reverse, 5′-TAA AAC GCA GCT CAG TAA CAG TCC G-3′; cadherin (CDH)1
forward, 5′-ACG TAT CAG GGT CAA GTG CC-3′ and reverse, 5′-CCT GAC
CCA CAC CAA AGT CT-3′; suppressor of cytokine signaling (SOCS)1
forward, 5′-TAA CCC GGT ACT CCG TGA CT-3′ and reverse, 5′-CTC ACC
CTC CAC AAC CAC TC-3′; methylthioadenosine phosphorylase (MTAP)
forward, 5′-ATC GTG ACC ACA GCT TGC GGG-3′ and reverse, 5′-TCT GCC
CGG GAG CTG AA-3′ and euchromatic histone lysine methyltransferase
2 (EHMT2) forward, 5′-TAC CCA TCC CCT GTG TCA AT-3′ and reverse,
5′-TCC TTG TCA TAC CAG CAT CG-3′. All samples were analyzed in
duplicate and normalized to β-actin.
Protein isolation and western
blotting
Murine and human melanoma cells were lysed in RIPA
buffer (Roche Diagnostics GmbH) for 15 min at 4°C and cell debris
was separated via centrifugation at 600 x g and 4°C for 10 min.
Protein concentration was determined using the Pierce BCA Protein
Assay kit (Thermo Fisher Scientific, Inc.). A total of 35 μg
total lysate per lane was separated on 10.00-12.75% SDS-PAGE gels
and subsequently transferred onto a PVDF membrane. Following
blocking for 1 h at room temperature with 5% BSA/0.1% TBS-T
(Sigma-Aldrich; Merck KGaA) or 5% non-fat dry milk/TBS-T (0.1%
Tween-20), the membrane was incubated overnight at 4°C with the
following antibodies: β-actin (1:3,000; cat. no. A5441;
Sigma-Aldrich; Merck KGaA), histone 3 (H3) K9me2 (1:1,000; cat. no.
ab3251; Abcam), H3K9me3 (1:1,000; cat. no. 07-442; Merck KGaA), H3
(1:1,000; cat. no. 4499; Cell Signaling, Technology, Inc.), EHMT2
(1:1,000; cat. no. 3306; Cell Signaling Technology, Inc.) and CYLD
[1:1,000; cat. nos. 12797 (mCYLD) and 4495 (hCYLD), Cell Signaling
Technology, Inc.].
After washing three times with TBS-T (0.1%
Tween-20), membranes were incubated for 1 h with horseradish
peroxidase-conjugated secondary anti-rabbit (cat. no. 7074) or
anti-mouse (7076) antibody (both 1:3,000; both Cell Signaling
Technology, Inc.). Finally, the membrane was washed three times in
TBS-T and the immunoreaction was visualized using Clarity™ Western
ECL Substrate (Bio-Rad Laboratories, Inc.) and Chemostar
chemiluminescence imager (Intas Pharmaceuticals, Ltd.). Signal
intensity was quantified using LabImage software Version 4.2.1
(Kapelan Bio-Imaging GmbH).
Histone extraction
For isolation of histone extracts, Histone
Extraction kit was used (cat. no. ab113476; Abcam). A total of
1x107 melanoma cells (murine or human) were seeded in
T75 flasks and harvested after 24 h incubation (8% CO2
at 37°C) with a cell scraper. Following isolation according to the
manufacturer's instructions, protein concentration was quantified
using the Pierce BCA Protein Assay kit (Thermo Fisher Scientific,
Inc.). For western blot analysis, 10 μg purified histone was
used. Aliquots were stored at -80°C.
5-Aza-deoxycytidine (AZA) treatment
Murine melanoma cells were seeded in a T75 flask
(750,000 cells/flask) 24 h (8% CO2 at 37°C) before
treatment. Cells were treated with 5 μM AZA (Sigma-Aldrich;
Merck KGaA) for 72 h (8% CO2 at 37°C) and harvested for
qPCR and western blot analysis.
Clonogenic assay
To investigate proliferation, colony formation
ability was assessed. A total of 300 cells (murine and human
melanoma cell lines) was seeded in a 6-well plate and cultivated
for 10 days (8% CO2 at 37°C). After washing twice with
PBS, cells were fixed and stained with 6.0 glutaraldehyde and 0.5%
crystal violet (Sigma-Aldrich; Merck KGaA), respectively, for 1 h
at room temperature). The colony (>50 cells) size was analyzed
using cellSens dimension-software (V1.12) and an IX83 fluorescence
microscope (both Olympus Corporation).
Chromatin accessibility assay
The EpiQuik™ Chromatin Accessibility Assay kit was
used according to the manufacturer's instructions (BioCat GmbH).
Briefly, 1×106 cells (murine or human melanoma cells)
were collected 24 h after seeding. Following cell lysis and
chromatin isolation, one half of the lysed cell was digested with a
nuclease mix (Nse) and the other was left untreated (No-Nse).
Subsequently, DNA was purified and analyzed via qPCR as
aforementioned. Fold enrichment (FE) was calculated as follows:
FE=2(Nse Ct-No-Nse Ct)/-2(Nse Ct-No-Nse Ct)
×100%. FE>400% indicated closed chromatin and FE<1,600%
indicated open chromatin (values between 400 and 1,600% represent
partially open chromatin). FE was normalized to Cyld-deficient
cells and the control.
H3 modification multiplex assay
Histone H3 Modification Multiplex Assay kit (cat.
no. ab185910, Abcam) was used to quantify 21 H3 modifications
according to the manufacturer's instructions. A total of 50 ng/well
histone extract from Cyld-expressing
(Cyld+/+) and -deficient
(Cyld−/−) murine cell lines, 50 ng/well histone
extract was used. H3 lysine mono-di- and trimethylation was
measured at sites K4, K9, K27, K36 and K79.
DNA sequencing (seq) and copy number
variation (CNV) analysis
DNA-seq was performed using input samples of
corresponding chromatin immunoprecipitation (ChIP) experiments.
Chromatin from 1×107 cells (mCyld−/−
and mCyld+/+) of each sample was crosslinked in
1:10 volume of fixation buffer (50 mM HEPES/KOH, pH 7.9, 11%
formaldehyde) for 10 min at room temperature and quenched by 0.125
M glycine. Following two washes with PBS and PMSF, cells were
scraped and centrifuged at 4°C and 3,500 x g for 10 min. The
supernatant was discarded and the pellet was resuspended in 15 ml
lysis buffer 1 (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP-40, 1X Roche
Complete, EDTA-free protease inhibitor) and incubated for 10 min on
ice. Following centrifugation of 5 min at 4°C and 3,500 x g, the
supernatant was discarded and the pellet resuspended in 15 ml lysis
buffer 2 (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 1% SDS, 1X Roche
Complete, EDTA-free protease inhibitor). The suspension was
incubated on ice for an additional 10 min and examined under the
light microscope (magnification: x200) for quality assessment of
isolated nuclei from cytoplasmic fractions. The nuclei were
pelleted at 4°C and 3,500 x g for 5 min. The supernatant was
discarded and the pellet was resuspended in sonication buffer
(1x107 cells/450 μl sonication buffer: 16.7 mM
Tris-HCl, pH 8.0, 1.2 mM EDTA, 16,7 mM NaCl, 0.01% SDS, 1.10%
Triton X-100, 1X Roche Complete, EDTA-free protease inhibitor).
Cross-linked chromatin was sheared to an average DNA fragment size
of 200-700 bp using the Covaris ME220 Focused-ultrasonicator
(Covaris, Inc.). Then, 20 μl supernatant was used for DNA
purification using the QIAquick PCR purification kit (Qiagen GmbH).
Library preparation was performed with purified DNA using the
TruSeq® ChIP Sample Preparation kit according to the
manufacturer's instructions (Illumina, Inc.). Quality and quantity
of the final libraries were checked for size (200-500 bp) by
TapeStation 4200 using the High-Sensitivity DNA kit (Agilent
Technologies, Inc.) and concentration was determined by Qubit 4
Fluorometer (Thermo Fisher Scientific, Inc.). Each library was
diluted to 4 nM and pooled to a final concentration of 5 nM. DNA
libraries were sequenced for 50 cycles on an Illumina HiSeq4000
with single-end module (HiSeq 3000/4000 SBS kit, 50 cycles; cat.
no. FC-410-1001; llumina, Inc.). Sequence tags of all experiments
were mapped to the current mouse reference sequence (GRCm38/mm10)
using Bowtie 2 (v 2.2.7) (16,17). Only uniquely mapped tags were
used for CNV determination via Control-FREEC (v11.6) (18).
RNA-seq and mutational analysis
Total RNA samples were isolated from
mCyld−/− and mCyld+/+ cell
lines using Total RNA kit I (Omega Bio-Tek, Inc.) according to
manufacturer's instructions. All RNA samples were examined for
integrity and purity by TapeStation 4200 (Agilent Technologies,
Inc.).
Library preparation was performed with two
biological replicates using the TruSeq® Stranded Total
RNA Library Prep Human/Mouse/Rat kit (cat. no. 20020596, Illumina,
Inc.) according to the manufacturer's instructions. The resulting
libraries were checked for size (200-500 bp) by TapeStation 4200
using the High-Sensitivity DNA kit (Agilent Technologies, Inc.) and
concentration was determined by Qubit 4 Fluorometer (Thermo Fisher
Scientific, Inc.). Each library was diluted to 4 nM and pooled to a
final concentration of 5 nM. Paired-end seq was performed using a
HiSeq4000 with paired-end module (Illumina, Inc.) according to the
manufacturer's instructions. The samples were sequenced from each
side of a fragment ~75 bp long with an average number 20 million
reads per sample. Following quality check using FastQC (Babraham
Bioinformatics; bioinformatics.babraham. ac.uk/projects/fastqc/)
paired-end reads were aligned to the mouse reference genome
(GRCm38, mm10) using the STAR alignment software (v 2.5.2a)
(17,19). Following mapping, only reads that
mapped to a single unique location were selected for further
analysis. The mapped reads were used for variant calling by Genome
Analysis Toolkit (GATK; v4.1; gatk. broadinstitute.org) (20). Single nucleotide variants (SNVs)
were identified with a cut-off of 1% minor allele frequency and a
minimum of 10 variant reads. Variants were annotated using SnpEff
5.0e (21). Intronic variants
were discarded and all exon variants were analyzed to determine
their effect on the resulting coding sequence (non-synonymous or
synonymous). Plots of contingency tables were produced using
variant calling output and R script Genotype-Variants (v1.1;
github. com/cfarkas/Genotype-variants) of the genotype-variant
pipeline as described by Farkas et al (22).
Schematic illustrations
Schematic illustrations were abstracted and modified
from 'Les Laboratoires Servier-smart' Medical Art (smart.servier.com/). Servier Medical Art by Servier is
licensed under a Creative Commons Attribution 3.0 Unported
License.
Statistical analysis
Data are presented as the mean ± SEM of ≥3
independent experiments, unless otherwise stated. Statistical
significance was determined using Student's unpaired t-test or
one-way ANOVA followed by Bonferroni's post hoc test. Data were
analyzed using GraphPad Prism Version 9 (GraphPad Software, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
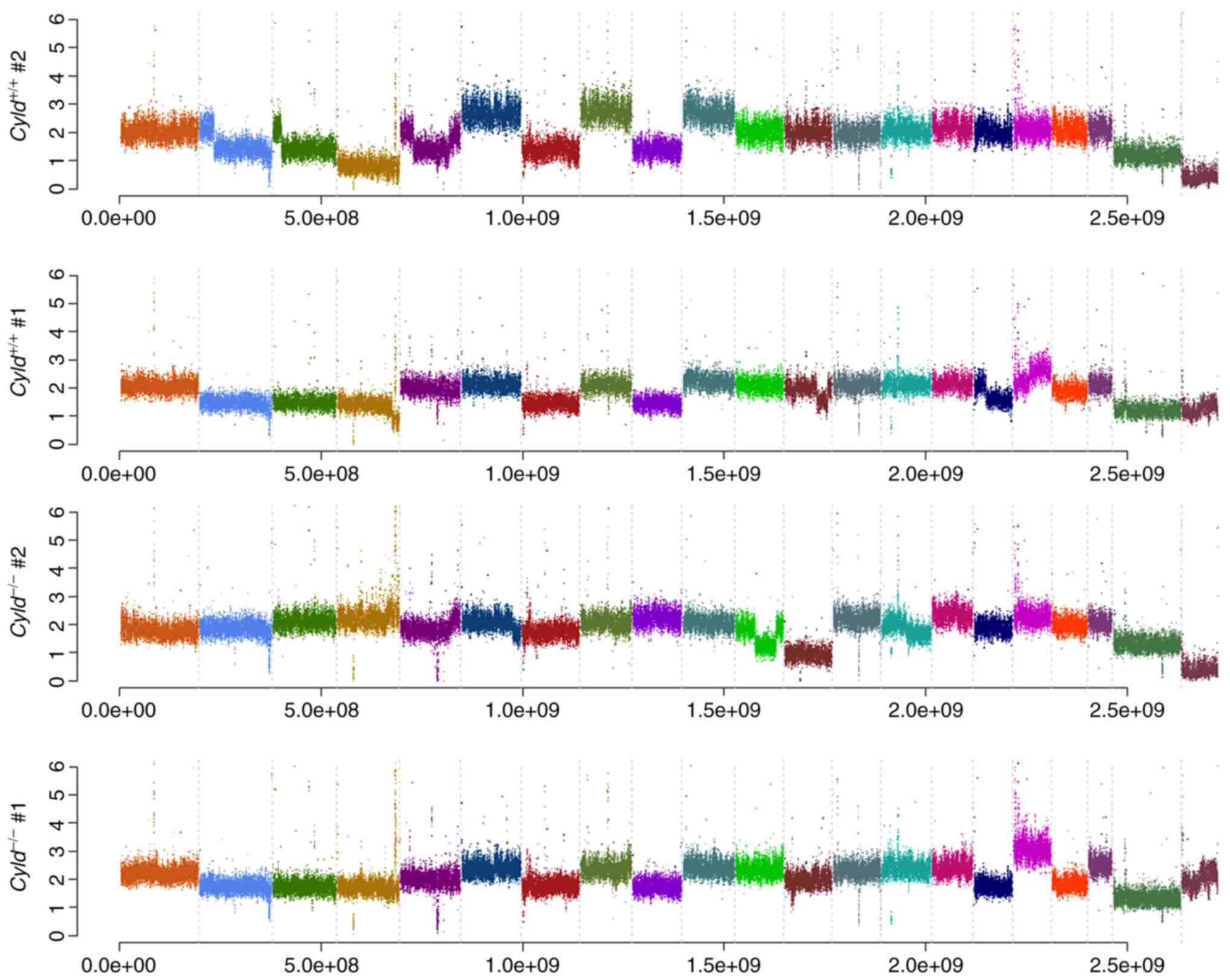
Absence of CNVs and common mutations in
Cyld−/− and Cyld+/+ cells
Melanoma is one of the tumors with the highest
mutation burden among solid tumors (23). To investigate whether CYLD
affects melanoma-associated mutations, sequencing analyses were
performed. No common CNVs were detected in
mCyld−/− cell and mCyld+/+
cells (Fig. 1). Therefore, SNVs
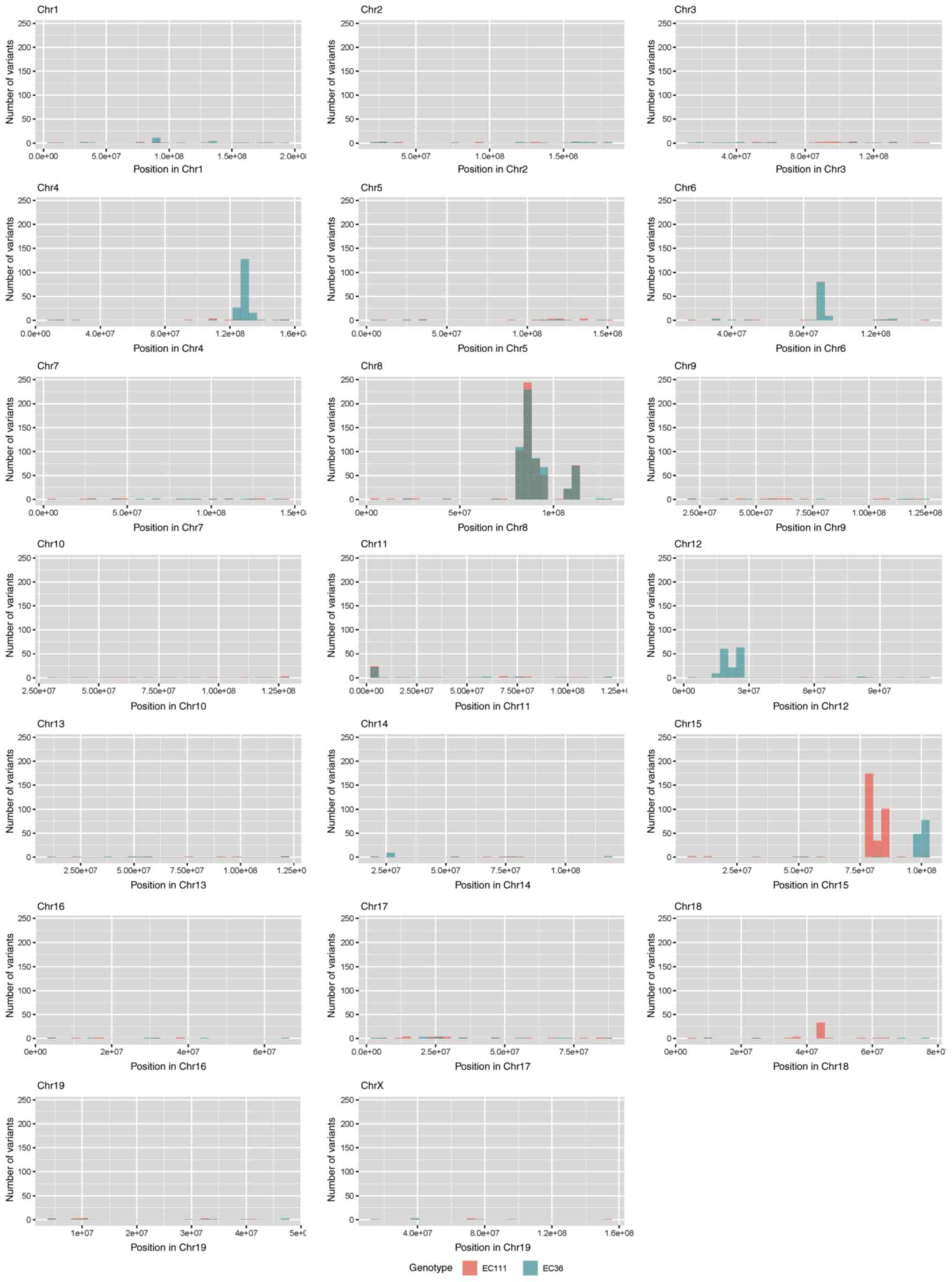
were investigated to identify potential mutations underlying
CYLD-dependent changes in tumor development of
Tg(Grm1)Cyld mice. Variant calling was performed
using Mutect2 of GATK (v4.2) to discover somatic variants in the
RNA-seq dataset mCyld−/− vs.
mCyld+/+ cells. Few common somatic mutations were
determined and all but one variants (pre-mRNA processing factor 39
on chromosome 12) in mCyld−/− were located on
chromosome 8 near the CYLD genomic location (Fig. 2). In summary, no
Cyld-dependent significant copy number gains or losses and
just a few somatic mutations were detected.
CYLD does not affect DNA methylation
Besides genomic mutations, epigenetic dysregulation
has also been linked to tumor development and progression in a
large number of studies (9-12). To investigate the role of
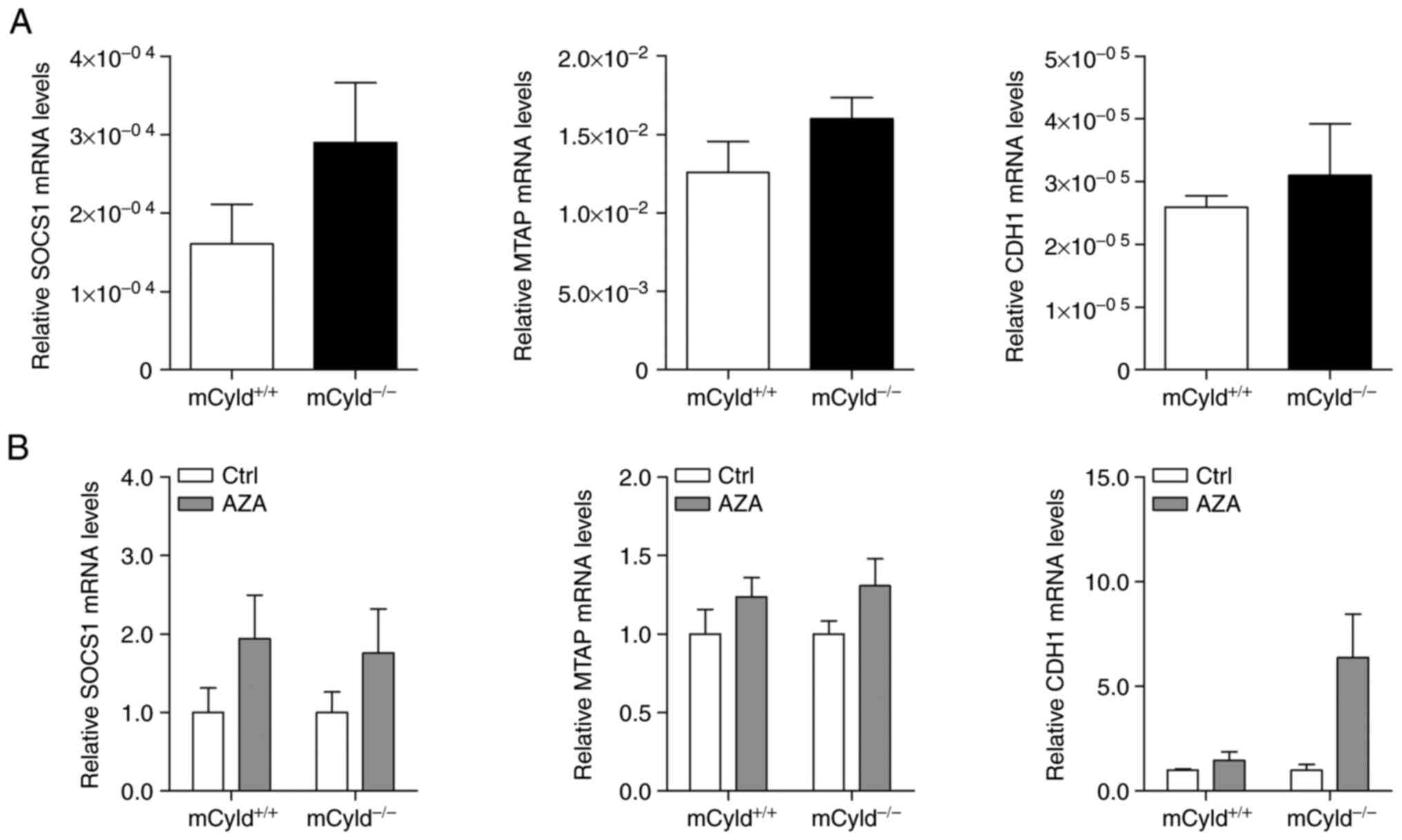
CYLD in controlling methylation of CpG islands, expression
of known hypermethylated genes (MTAP, CDH1 and SOCS1) in human
melanoma was investigated (24,25). No significant differences in
MTAP, CDH1 and SOCS1 mRNA expression were observed between
mCyld+/+ and mCyld−/− cell
lines (Fig. 3A), indicating that
CYLD was not involved in DNA methylation. DNA demethylation
was induced by treatment with AZA to investigate
CYLD-dependent alterations in gene expression. AZA treatment
was associated with a notable (but not significant) increase in
CYLD protein expression (Fig. S1). mRNA expression of
hypermethylated genes was not significantly altered in
mCyld+/+ and mCyld−/− cells
following AZA treatment (Fig.
3B), confirming the aforementioned results.
In summary, these results indicated that CYLD
had no effect on DNA methylation.
CYLD deficiency results in more compact
chromatin structure and affects H3 modification associated with
heterochromatin
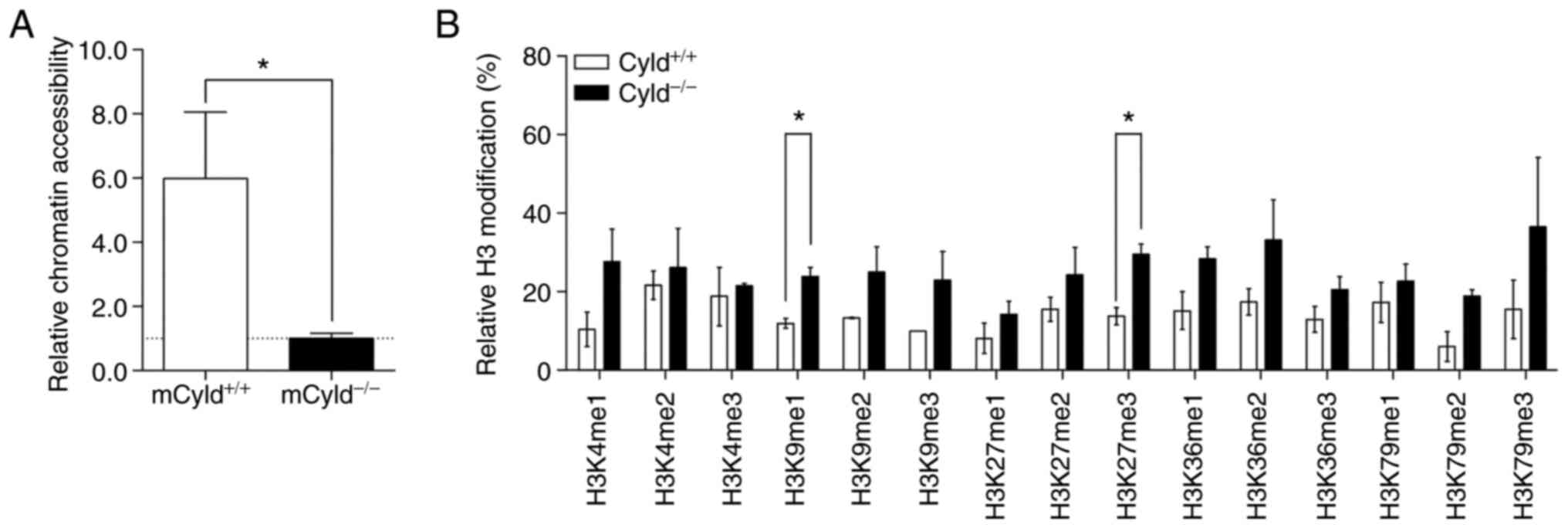
Epigenetic events affect the accessibility of
chromatin via formation of heterochromatin and euchromatin
(26). To determine whether
CYLD affected chromatin structure, chromatin accessibility
was assessed using the EpiQuik chromatin accessibility kit. This
assay showed that mCyld+/+ cells exhibited a more
open chromatin structure compared with mCyld−/−
cells (Fig. 4A).
Heterochromatin is associated with certain H3
modifications (27); to the best
of our knowledge, however, there are no studies regarding H3
modification and CYLD. Therefore, H3 modification assay was
performed. Although modifications were notably higher in
mCyld−/− compared with mCyld+/+
cells, this was only significant for H3K9me1- and H3K27me3-
modifications (Fig. 4B). Taken
together, these data suggested that CYLD was involved in
chromatin structure and histone methylation.
CYLD-dependent effects of inhibitors
Chaetocin, JIB04 and CM272 on H3 methylation and cell
proliferation
The aforementioned results suggested an association
between CYLD, chromatin structure and histone methylation.
Because aberrant histone modification disrupts epigenetic balance
and contributes to melanoma progression (28), the effect of histone
demethylase/methyltransferase inhibitors on CYLD-dependent
proliferation were analyzed.
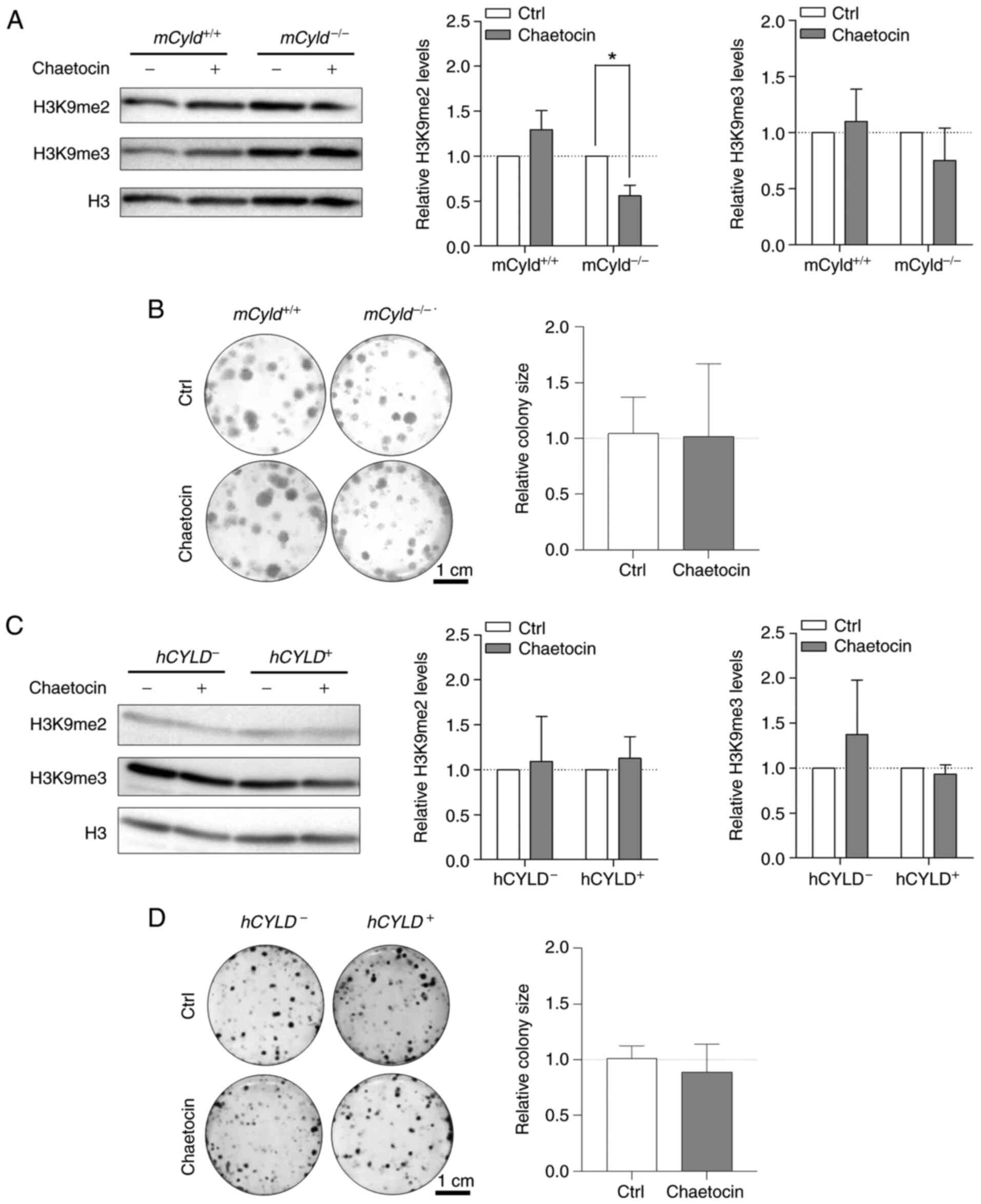
Treatment with Chaetocin, a lysine-specific histone
methyltransferase, leads to inhibition of the histone
methyltransferase suppressor of variegation 3-9 homolog 1 (SUV39H1,
also known as KMT1A), which is involved in formation of
hetero-chromatin by di-and trimethylating H3K9 (29,30). Here, treatment resulted in
significantly decreased H3K9 dimethylation only in
mCyld−/− cells; trimethylation of H3K9 was not
significantly affected (Fig.
5A). A previous study has demonstrated that Chaetocin exerts
anti-proliferative effects in vivo in melanoma cells
(31). However, clonogenic assay
revealed no significant change in colony size between
mCyld−/− and mCyld+/+ cells
(Fig. 5B).
Using the established human CYLD melanoma system, no
significant changes in H3K9 di- and trimethylation levels were
observed via western blot analysis after treatment with Chaetocin
(Fig. 5C). Consistent with the
results in the murine cell lines, proliferation was not
significantly altered by Chaetocin in human cell lines (Fig. 5D). Treatment with Chaetocin had
no significant effect on CYLD protein expression in murine and
human melanoma cells (Fig. S2A and
B).
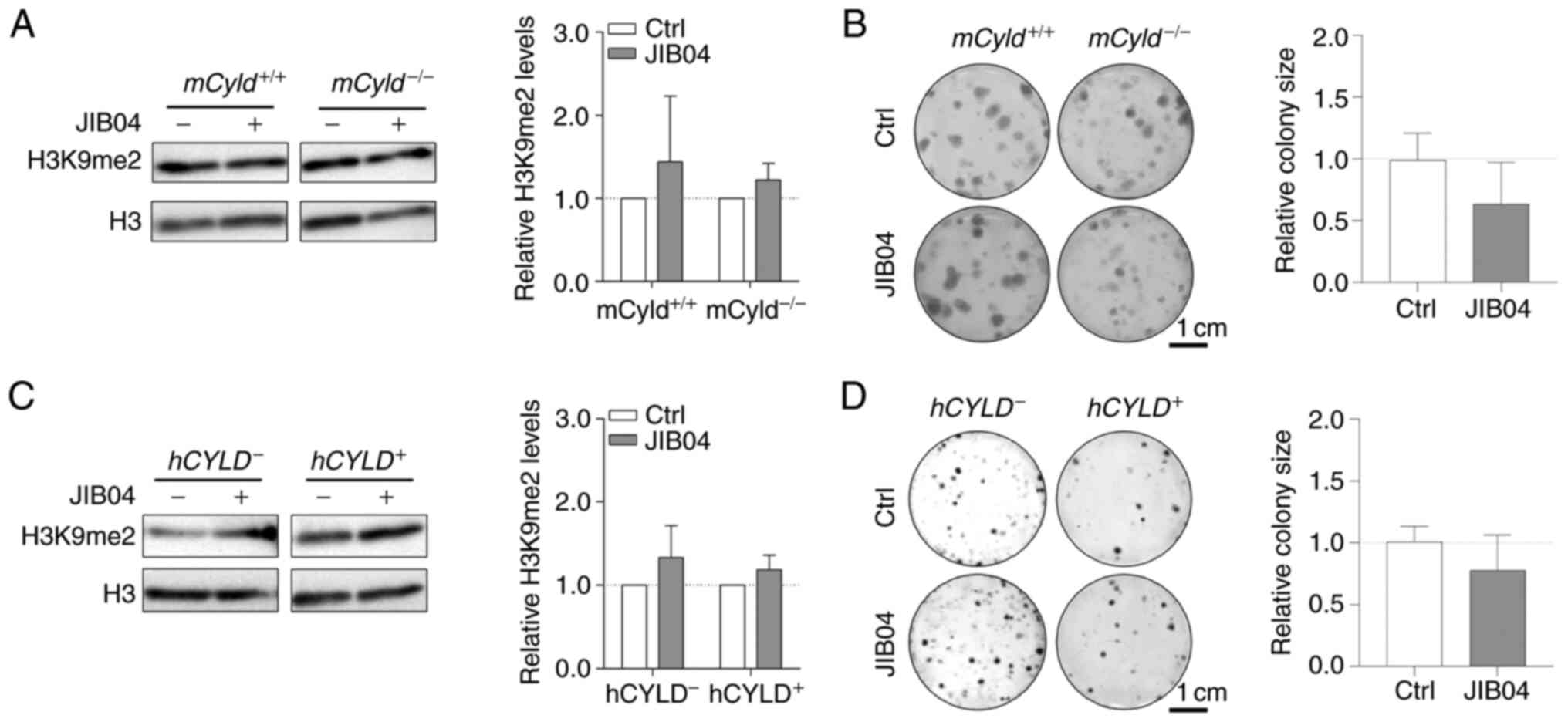
The counterpart of histone lysine methyltransferases
are histone lysine demethylases (KDMs), including family members
KDM3 [Jumonji Domain-Containing Protein 1A (JMJD1)], KDM4 (JMJD2)
and KDM2 (JmjC domain-containing histone demethylation protein 1)
(32). To the best of our
knowledge, there are no inhibitors targeting specific KDMs.
Nevertheless, there are broad-spectrum KDM inhibitors, such as the
small molecule JIB04, which increases H3K9me2 levels (33). Since the aforementioned results
excluded an effect of CYLD on H3K9 trimethylation on
proliferation, cells were treated with JIB04; this treatment had no
significant effect on levels of H3K9me2 in
mCyld−/− or mCyld+/+ cells
(Fig. 6A). Clonogenic assay
revealed no significant changes in colony size following JIB04
treatment in mCyld−/− compared with
mCyld+/+ cells (Fig. 6B). There was no significant
change in H3K9me2 levels (Fig.
6C) or colony size (Fig. 6D)
in hCYLD− compared with hCYLD+
cells. CYLD protein expression was not significantly affected by
treatment with JIB04 (Fig. S2C and
D).
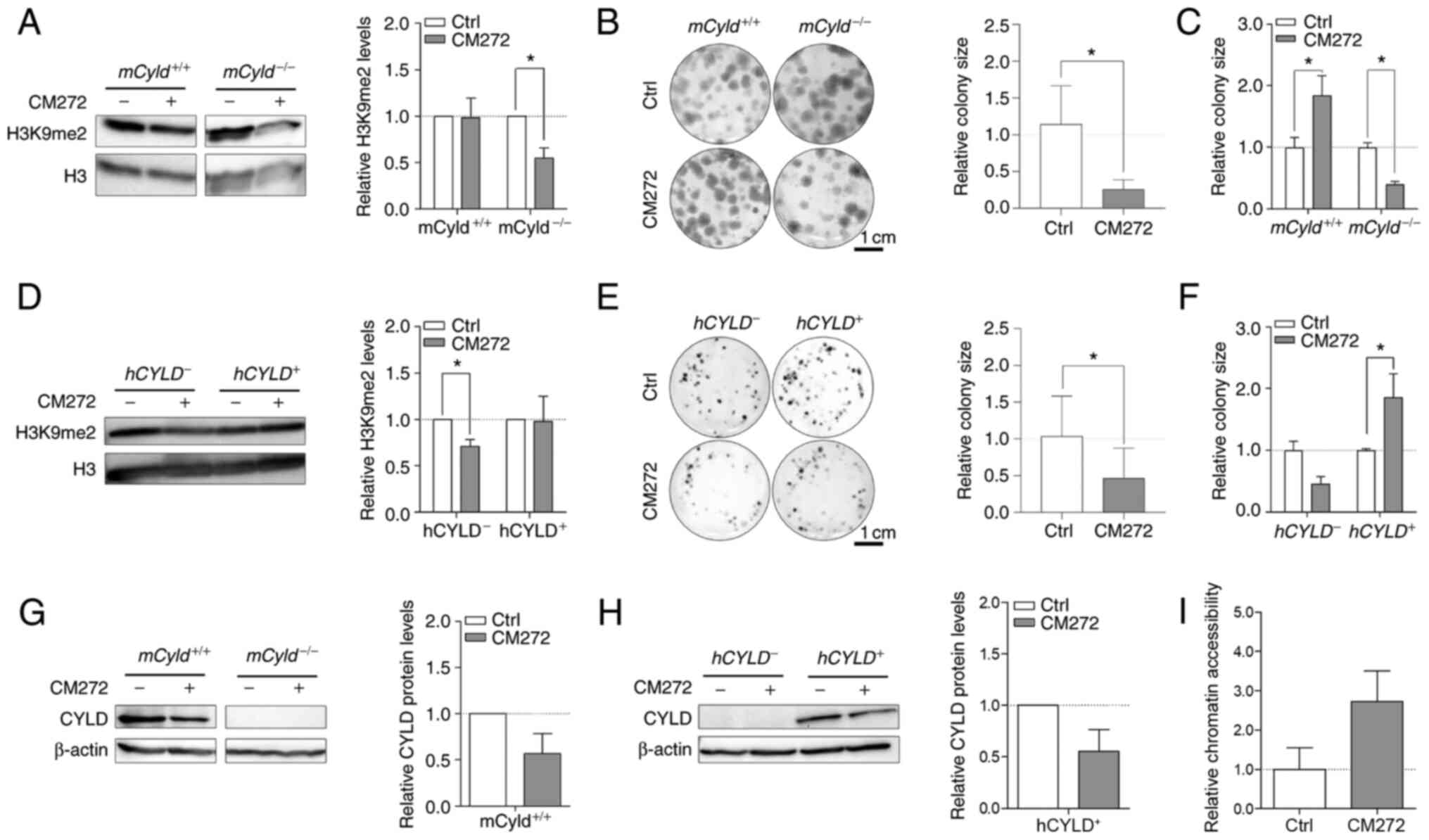
Selective inhibitor, CM272 suppresses activity of
EHMT2 (also known as G9a) (14).
This inhibitor contributes to decreased dimethylation of H3K9 and
may hinder melanoma growth (14). To the best of our knowledge,
however, no data concerning the role of CYLD in this context
are available. Western blot analysis confirmed a significant
decrease H3K9me2 levels following CM272 treatment only in
mCyld−/− cell lines (Fig. 7A). Clonogenic assay revealed a
significant decrease in colony size in mCyld−/−
cells, which was consistent with H3K9me2 western blot results
(Fig. 7B and C).
mCyld+/+ cells exhibited a significantly
increased colony size (Fig. 7C),
as well as significantly decreased CYLD expression following
CM272 treatment (Fig. 7D). In
conclusion, CM272 treatment of CYLD-expressing cells led to
decreased expression and tumor suppressive function of CYLD.
These results were confirmed in human cells:
hCYLD− cells exhibited significantly decreased
H3K9me2 levels (Fig. 7E) and
proliferation following CM272 treatment (Fig. 7F and G). hCYLD+
cells showed decreased CYLD expression following CM272
treatment (Fig. 7H) but this was
not significant.
It was hypothesized that treatment of
Cyld-deficient cells with CM272 would lead to looser
chromatin structure. Chromatin accessibility analysis of
mCyld−/− cells showed more open chromatin
structure following CM272 treatment compared with control cells
(Fig. 7I) but this was not
significant.
Catalytically inactive CYLD mutant cells
(hCYLDC/S) exhibited similar trends to
hCYLD− cells. There was a significant decrease in
H3K9me2 levels (Fig. S3A) and
colony size (Fig. S3B), whereas
CYLD expression was not significantly altered following CM272
treatment (Fig. S3C). Moreover,
hCYLDC/S cells exhibited more compact chromatin
structure, similar to hCYLD− cells, whereas
hCYLD+ cells exhibited a more open chromatin
structure (Fig. S3D).
Together, these data showed that the inhibitors
Chaetocin and JIB04 exerted no CYLD-dependent effect on cell
proliferation. The inhibitor CM272 had anti-proliferative effects
on Cyld-deficient cells but pro-proliferative effects on
Cyld-expressing cells. The pro-proliferative effects were
dependent on enzymatic function of CYLD.
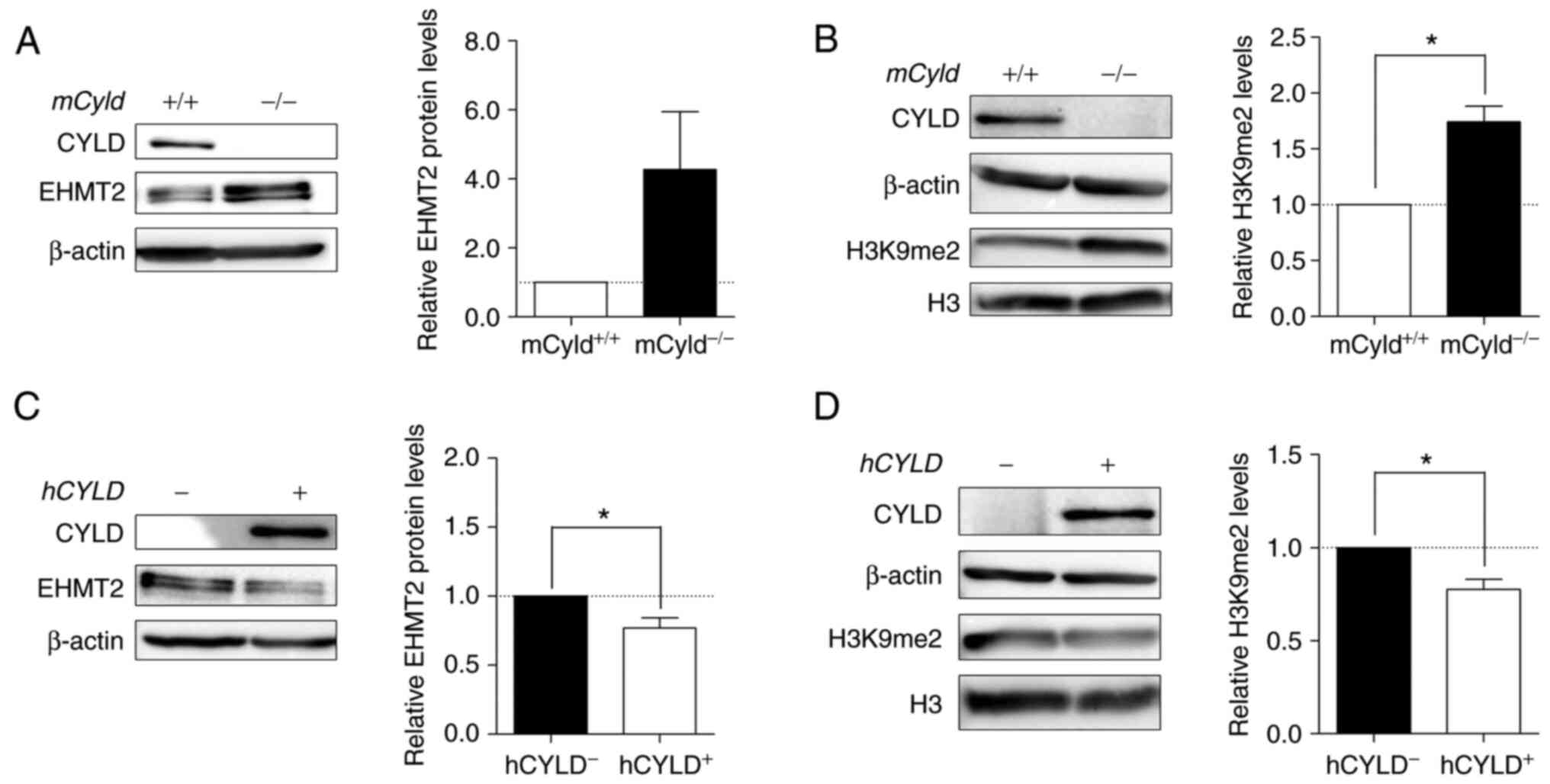
High levels of EHMT2 in Cyld-deficient
cells increase H3K9 dimethylation
Histone lysine methyltransferase EHMT2 specifically
mono- and dimethylates H3K9 and catalyzes trimethylation of H3K27
(34). Thus, the present study
investigated expression levels of this methyltransferase and its
primary methylation site H3K9me2. Western blot analysis
demonstrated notably enhanced EHMT2 expression in
mCyld−/− cells (Fig. 8A). mCyld−/−
cells showed a significant increase in H3K9me2 compared with
Cyld+/+ cells (Fig. 8B). Same investigations in our
human cell system verified the negative association between
CYLD and EHMT2 expression as well as a positive association
between H3K9me2 and EHMT2 (Fig. 8C
and D). hCYLDC/S cells showed similar H3K9me2
and EHMT2 expression to hCYLD− cells (Fig. S3E and F).
Discussion
The deubiquitinase CYLD is downregulated in
different types of cancer, including hepatocellular carcinoma,
breast cancer and malignant melanoma (6,35,36). Using the melanoma mouse model
Tg(Grm1) and murine cell lines generated from tumors, our
previous study demonstrated the tumor suppressive function of
CYLD in proliferation, migration and lymph- and angiogenesis
(7). Nevertheless, the
underlying molecular mechanism of early tumor onset in
Cyld−/− mice has not yet been uncovered.
Chromosomal aberrations or nucleotide polymorphisms
affect pathways involved in tumor formation and progression in
breast cancer or cutaneous melanoma (37,38). However, the present study was
unable to identify chromosomal alterations and detected only a few
nucleotide variations in mCyld−/− compared with
mCyld+/+ cells. Most mutations occurred in
proximity to CYLD. To the best of our knowledge, clustering
of mutations near knockout sites has not previously been
investigated but it is hypothesized that the sequence in which the
CYLD gene locus is located is genetically unstable or
particularly susceptible to single nucleotide polymorphisms
(39). CYLD interacts
with and deubiquitinates p53 in response to DNA damage (40) and therefore affects genomic
instability. To investigate this, mutation analysis of normal mouse
tissue is required. The present study identified few mutations but
did not determine a link with melanoma-associated mutations or
early tumor onset in CYLD knockout mice.
In addition to genomic instability and abnormal
gene expression, dysregulation of epigenetic mechanisms, such as
DNA methylation and histone modification are hallmarks of cancer
(8). Certain studies have shown
that CYLD expression is suppressed by epigenetic regulatory
mechanisms, such as DNA methylation or histone deacetylation
(41,42). The present western blot analysis
demonstrated enhanced CYLD expression following AZA
treatment. This was consistent with CYLD regulation in
gastric adenocarcinoma, where hypermethylation of the CYLD
promoter results in decreased expression of CYLD (41). Colon and hepatocellular carcinoma
show no expression changes of CYLD following AZA treatment
(35). Although, we describe a
transcriptional control of the CYLD promoter via
overexpression of the transcriptional factor SNAIL1 (6), epigenetic regulatory mechanisms may
also contribute to downregulation of CYLD. However, the present
analysis suggested that CYLD serves no key role in DNA
methylation in a mouse model.
In addition to DNA methylation, histone tails
undergo post-translational modifications that alter chromatin
structure and dynamics (26).
CYLD interacts with certain histone deacetylases (HDACs); in
hepatic stellate cells, CYLD regulates hepatocyte growth
factor expression via interaction with HDAC7, leading to lower
hepatocellular damage and liver fibrosis (43). Moreover, Wickström et al
(44) showed negative regulation
of cell cycle by via inhibitory interaction of CYLD and
HDAC6 in keratinocytes and melanoma cells. To the best of our
knowledge, there is no evidence of a direct effect of the tumor
suppressor CYLD on other types of histone modification, such
as methylation. The present data revealed that the overall
accessibility of chromatin is decreased in
Cyld−/− melanoma cells, suggesting that
CYLD deficiency favors the formation of more condensed
heterochromatin and associated histone modifications, such as H3
methylation. H3 modification assay revealed higher levels of
heterochromatin-specific histone modifications H3K9me and H3K27me
in Cyld−/− compared with
Cyld+/+ cells.
Histone-modifying enzymes affect the structure of
chromatin and are deregulated in cancer, including prostate,
breast, colon, skin, and lung cancers (45,46). Cancer drug discovery has focused
on development of competitive analogs of co-factors, such as JIB04
and CM272, which modulate activity of epigenetic enzymes
upregulated in tumors (47,48). Although inhibitors Chaetocin and
JIB04 had no influence on proliferation of melanoma cells,
treatment with dual-inhibitor CM272 showed CYLD-dependent
effects. Previously, anti-proliferative effects of CM272 were
observed in hematological tumor (14). Moreover, CM272 inhibits
fibrogenesis and proliferation in cholangiocarcinoma and
hepatocellular carcinoma (49-51). Furthermore, decreased cell
viability and induction of type I interferon response have been
observed in murine melanoma cells (52). The present study revealed
decreased proliferation of Cyld−/− melanoma cells
following CM272 treatment, suggesting that this inhibitor may be a
potential therapy for melanoma. Notably, Cyld+/+
cells showed enhanced proliferation following treatment,
potentially due to decreased CYLD protein expression.
Therefore, CM272 should be used in clinical trials to investigate
its effect on CYLD expression. Downregulation of CYLD in
patients with melanoma has been described in previous studies
(6,53). Nevertheless, a recent study
reported heterogeneous expression between melanoma cell lines and
melanoma in vivo (54).
This shows the importance of defining CYLD status when using CM272
inhibitor in melanoma therapy, as treatment of
Cyld-expressing cells results in an increase in
proliferation. Moreover, Rodriguez-Madoz et al (55) showed that CM272 treatment leads
to heterochromatin relaxation. This was also observed in
Cyld−/− melanoma cells in the present study,
suggesting that this inhibitor reverses silencing of tumor
suppressors, which decreases proliferation when CYLD is
lost. In summary, in vitro analysis of the effect of
inhibitors revealed that CYLD is involved in histone
methylation modification associated with chromatin structure.
There was an inverse association between
CYLD and expression of the potential target of CM272, EHMT2.
The methyltransferase EHMT2 is deregulated in many tumor entities
and is associated with increased migration, invasion and poor
prognosis for patients, demonstrating its important role in
tumorigenesis (33,56,57). Additionally, this lysine
methyltransferase contributes to H3K9 and H3K27 methylation, which
are associated with gene silencing and chromatin condensation
(58). In a zebrafish melanoma
model, histone methyltransferase (HKMT) SET domain bifurcated
histone lysine methyltransferase 1 (SETDB1) promotes melanoma
formation (59). This enzyme
also methylates H3K9 and serves as an oncogene in melanoma
(59). Moreover, a multimeric
complex of four H3K9 HKMTs, including EHMT2/G9a/KMT1C, GLP/KMT1D,
SETDB1/KMT1E and Suv39h1/KMT1A, is involved in regulating gene
expression and heterochromatin assembly (60). Thus, the role of these enzymes
and their association with CYLD in development of melanoma
should be further investigated. However, the Suv39h1 inhibitor
Chaetocin did not affect proliferation of Cyld+/+
or Cyld−/− cells, suggesting no effect of this
HKMT on melanoma cells. Nevertheless, it is unclear whether there
is a direct or indirect connection between CYLD and EHMT2.
The present results indicated direct interaction due to the
negative association between expression of CYLD and EHMT2 and
effect of the inhibitor CM272 on proliferation and chromatin
structure. To demonstrate this potential association between CYLD
and EHMT2, rescue experiments should be performed in follow-up
studies.
In the present study, analysis of human melanoma
cells showed that hCYLDC/S behaved like
hCYLD-deficient cells (Fig.
S3). This suggested involvement of the deubiquitinase function
of CYLD in epigenetic processes. Certain studies have demonstrated
the role of deubiquitinases in crosstalk between different histone
post-translational modifications (61). For example, ubiquitin specific
peptidase (USP28) enhances tumorigenicity of breast cancer cells
via stabilization of lysine-specific demethylase 1 (62). Moreover, knockdown of USP24
increases Suv39h1 expression, which results in enhanced H3K9me
levels and prevents the progression of malignant lung cancer
(63). Furthermore, H3
polyubiquitination suppresses transcription of fetal and cell cycle
genes in postnatal mouse liver by crosstalk with H3K9 methylation
and loss of ubiquitin ligase CRL4 inhibit H3K9 methylation
(64). The present data and
existing literature suggest that CYLD may inhibit H3K9
dimethylation via deubiquitination of upstream histone modifiers,
such as EHMT2 or H3K9, and therefore suppress cell proliferation
due to activation of silenced tumor suppressors.
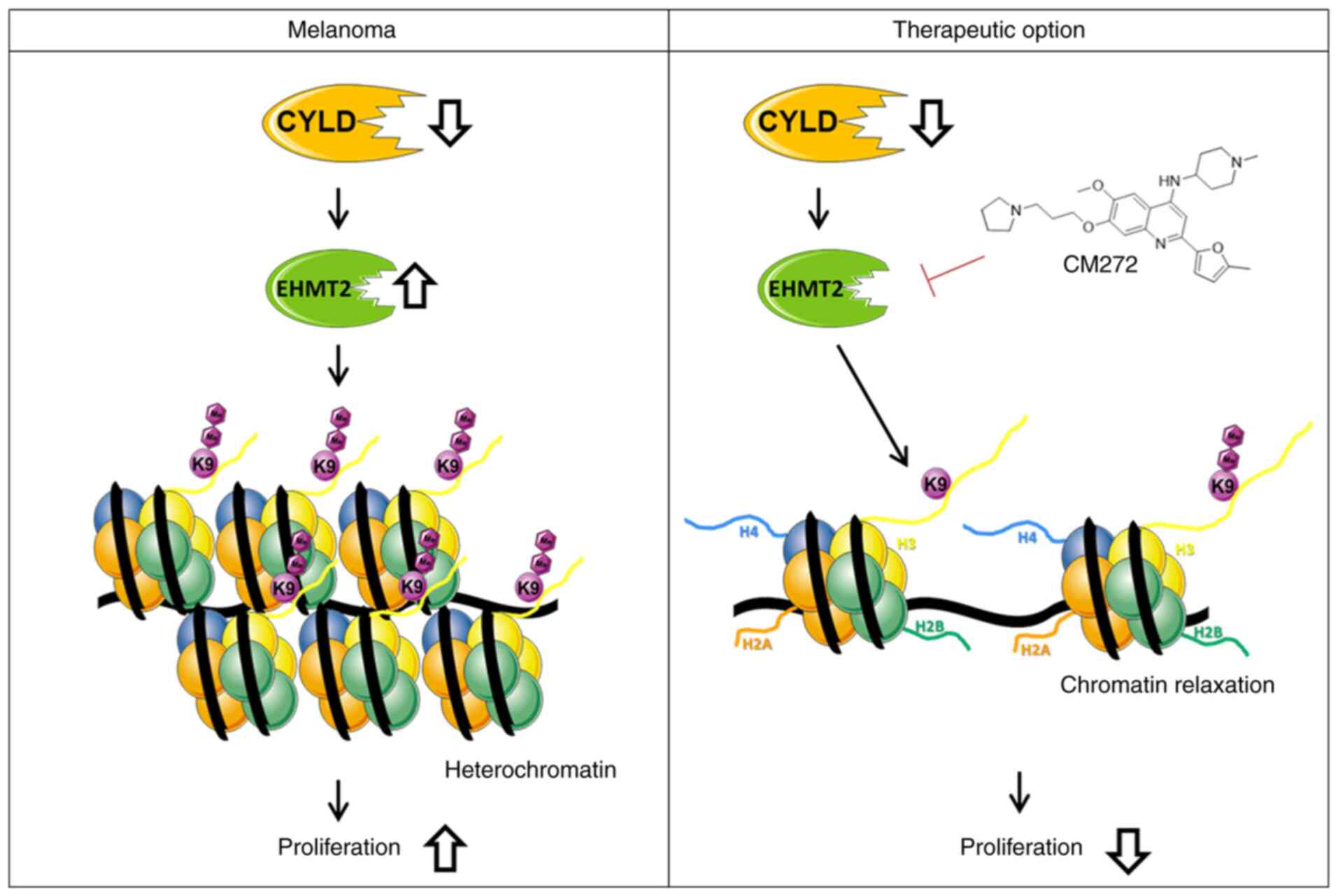
The present study revealed modulation of chromatin
structure in a CYLD-dependent manner in melanoma. The
present results indicated that increased expression of EHMT2 led to
increased H3K9me2, subsequently promoting a more compact
heterochromatin structure when CYLD is lost (Fig. 9). These results may explain
enhanced tumor onset of CYLD knockout mice. Further studies
should investigate the role of CYLD in epigenetic processes
as the present results indicated other H3 modifications (such as
H3K27me) might be CYLD-dependent. To the best of our
knowledge, the present study is the first to demonstrate an
association between tumor suppressor CYLD, chromatin
structure and histone methylation. Moreover, suppressing
proliferation of the higher H3K9me2 expressing
Cyld−/− cells via CM272 may be a promising
strategy for melanoma treatment (Fig. 9). The high similarity of mouse
and human melanoma cells illustrates the relevance of our generated
cells for the study of molecular mechanisms in melanoma.
Supplementary Data
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the NCBI BioProject database
(accession no. PRJNA796721; (dataview.ncbi.nlm.nih.gov/object/PRJNA796721?reviewer=q05a8apg3kla63kggpoh5gi6dt).
Authors' contributions
AKB, SK and MS conceived the project, analyzed the
data and wrote the manuscript. MS designed and performed the
experiments. AKB, SK, MGFB, APL, MAA, SF and MKF provided
materials, analyzed the data and wrote the manuscript. All authors
have read and approved the final manuscript. MS and AKB confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like thank Professor Suzie Chen
(Rutgers University, Chemical Biology, Piscataway, USA) for
providing Tg(Grm1) animals and Dr Ramin Massoumi (Department
of Laboratory Medicine, Lund University, Sweden) for providing
C57Bl/6 Cyld−/− mice. The authors would like to
thank Mr. Alexander-Oliver Matthies
(Friedrich-Alexander-University; FAU) for preparation of RNA
samples and RNA-Seq libraries and Professor Christoph Bock
(Biomedical Sequencing Facility, Research Center for Molecular
Medicine of the Austrian Academy of Sciences), Vienna, Austria) for
sequencing samples. The authors would also like to thank Mr. Ingmar
Henz for technical assistance and Mrs. Valerie Fritz for critical
discussion (both FAU).
Funding
The present study was supported by the German Research
Foundation (grant nos. DFG FOR2127 and TRR305, B12),
FEDER/Ministerio de Ciencia, Innovación y Universidades-Agencia
Estatal de Investigación, Spain (grant nos.
PID2019-104878RB-100/AEI/10.13039/501100011033 and
PID2020-PID2020-120387RB-I00), Fundación Fuentes Dutor and the
Ramón y Cajal Program (grant no. RYC-2018-024475-1).
References
|
1
|
Bignell GR, Warren W, Seal S, Takahashi M,
Rapley E, Barfoot R, Green H, Brown C, Biggs PJ, Lakhani SR, et al:
Identification of the familial cylindromatosis tumour-suppressor
gene. Nat Genet. 25:160–165. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hellerbrand C and Massoumi R:
Cylindromatosis-a protective molecule against liver diseases. Med
Res Rev. 36:342–359. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Orfanidou T, Xanthopoulos K, Dafou D,
Pseftogas A, Hadweh P, Psyllaki C, Hatzivassiliou E and Mosialos G:
Down-regulation of the tumor suppressor CYLD enhances the
transformed phenotype of human breast cancer cells. Anticancer Res.
37:3493–3503. 2017.PubMed/NCBI
|
|
4
|
Xie S, Wu Y, Hao H, Li J, Guo S, Xie W, Li
D, Zhou J, Gao J and Liu M: CYLD deficiency promotes pancreatic
cancer development by causing mitotic defects. J Cell Physiol.
234:9723–9732. 2019. View Article : Google Scholar
|
|
5
|
Massoumi R, Chmielarska K, Hennecke K,
Pfeifer A and Fassler R: Cyld inhibits tumor cell proliferation by
blocking Bcl-3-dependent NF-kappaB signaling. Cell. 125:665–677.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Massoumi R, Kuphal S, Hellerbrand C, Haas
B, Wild P, Spruss T, Pfeifer A, Fässler R and Bosserhoff AK:
Down-regulation of CYLD expression by Snail promotes tumor
progression in malignant melanoma. J Exp Med. 206:221–232. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
de Jel MM, Schott M, Lamm S, Neuhuber W,
Kuphal S and Bosserhoff AK: Loss of CYLD accelerates melanoma
development and progression in the Tg(Grm1) melanoma mouse model.
Oncogenesis. 8:562019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Strub T, Ballotti R and Bertolotto C: The
'ART' of epigenetics in melanoma: From histone 'alterations, to
resistance and therapies'. Theranostics. 10:1777–1797. 2020.
View Article : Google Scholar :
|
|
9
|
Nebbioso A, Tambaro FP, Dell'Aversana C
and Altucci L: Cancer epigenetics: Moving forward. PLoS Genet.
14:e10073622018. View Article : Google Scholar :
|
|
10
|
Putiri EL and Robertson KD: Epigenetic
mechanisms and genome stability. Clin Epigenetics. 2:299–314. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roesch A, Vultur A, Bogeski I, Wang H,
Zimmermann KM, Speicher D, Körbel C, Laschke MW, Gimotty PA,
Philipp SE, et al: Overcoming intrinsic multidrug resistance in
melanoma by blocking the mitochondrial respiratory chain of
slow-cycling JARID1B(high) cells. Cancer Cell. 23:811–825. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Strub T, Ghiraldini FG, Carcamo S, Li M,
Wroblewska A, Singh R, Goldberg MS, Hasson D, Wang Z, Gallagher SJ,
et al: SIRT6 haploinsufficiency induces BRAFV600E
melanoma cell resistance to MAPK inhibitors via IGF signalling. Nat
Commun. 9:34402018. View Article : Google Scholar
|
|
13
|
Schiffner S, Braunger BM, de Jel MM,
Coupland SE, Tamm ER and Bosserhoff AK: Tg(Grm1) transgenic mice: A
murine model that mimics spontaneous uveal melanoma in humans? Exp
Eye Res. 127:59–68. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
San José-Enériz E, Agirre X, Rabal O,
Vilas-Zornoza A, Sanchez-Arias JA, Miranda E, Ugarte A, Roa S,
Paiva B, Estella-Hermoso de Mendoza A, et al: Discovery of
first-in-class reversible dual small molecule inhibitors against
G9a and DNMTs in hematological malignancies. Nat Commun.
8:154242017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dietrich P, Koch A, Fritz V, Hartmann A,
Bosserhoff AK and Hellerbrand C: Wild type Kirsten rat sarcoma is a
novel microRNA-622-regulated therapeutic target for hepatocellular
carcinoma and contributes to sorafenib resistance. Gut.
67:1328–1341. 2018. View Article : Google Scholar
|
|
16
|
Langmead B: Aligning short sequencing
reads with Bowtie. Curr Protoc Bioinformatics Chapter 11. Unit
11.7. 2010.
|
|
17
|
Frankish A, Diekhans M, Ferreira AM,
Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J,
Armstrong J, et al: GENCODE reference annotation for the human and
mouse genomes. Nucleic Acids Res. 47(D1): D766–D773. 2019.
View Article : Google Scholar :
|
|
18
|
Boeva V, Popova T, Bleakley K, Chiche P,
Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O and
Barillot E: Control-FREEC: A tool for assessing copy number and
allelic content using next-generation sequencing data.
Bioinformatics. 28:423–425. 2012. View Article : Google Scholar :
|
|
19
|
Dobin A, Davis CA, Schlesinger F, Drenkow
J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR: STAR:
Ultrafast universal RNA-seq aligner. Bioinformatics. 29:15–21.
2013. View Article : Google Scholar
|
|
20
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high confidence variant
calls: The genome analysis toolkit best practices pipeline. Curr
Protoc Bioinformatics. 43:11.10.1–11.10.33. 2013.
|
|
21
|
Cingolani P, Platts A, Wang le L, Coon M,
Nguyen T, Wang L, Land SJ, Lu X and Ruden DM: A program for
annotating and predicting the effects of single nucleotide
polymorphisms, SnpEff: SNPs in the genome of Drosophila
melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6:80–92.
2012. View Article : Google Scholar
|
|
22
|
Farkas C, Fuentes-Villalobos F,
Rebolledo-Jaramillo B, Benavides F, Castro AF and Pincheira R:
Streamlined computational pipeline for genetic background
characterization of genetically engineered mice based on next
generation sequencing data. BMC Genomics. 20:1312019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cancer Genome Atlas Network: Genomic
classification of cutaneous melanoma. Cell. 161:1681–1696. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schinke C, Mo Y, Yu Y, Amiri K, Sosman J,
Greally J and Verma A: Aberrant DNA methylation in malignant
melanoma. Melanoma Res. 20:253–265. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Micevic G, Theodosakis N and Bosenberg M:
Aberrant DNA methylation in melanoma: Biomarker and therapeutic
opportunities. Clin Epigenetics. 9:342017. View Article : Google Scholar :
|
|
26
|
Morgan MA and Shilatifard A: Chromatin
signatures of cancer. Genes Dev. 29:238–249. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sharakhov IV and Sharakhova MV:
Heterochromatin, histone modifications, and nuclear architecture in
disease vectors. Curr Opin Insect Sci. 10:110–117. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vardabasso C, Hake SB and Bernstein E:
Histone variant H2A.Z.2: A novel driver of melanoma progression.
Mol Cell Oncol. 3:e10734172016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Greiner D, Bonaldi T, Eskeland R, Roemer E
and Imhof A: Identification of a specific inhibitor of the histone
methyltransferase SU(VAR) 3-9. Nat Chem Biol. 1:143–145. 2005.
View Article : Google Scholar
|
|
30
|
Rea S, Eisenhaber F, O'Carroll D, Strahl
BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD
and Jenuwein T: Regulation of chromatin structure by site-specific
histone H3 methyltransferases. Nature. 406:593–599. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Han X, Han Y, Zheng Y, Sun Q, Ma T, Zhang
J and Xu L: Chaetocin induces apoptosis in human melanoma cells
through the generation of reactive oxygen species and the intrinsic
mitochondrial pathway, and exerts its anti-tumor activity in vivo.
PLoS One. 12:e01759502017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cloos PA, Christensen J, Agger K and Helin
K: Erasing the methyl mark: Histone demethylases at the center of
cellular differentiation and disease. Genes Dev. 22:1115–1140.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim MS, Cho HI, Yoon HJ, Ahn YH, Park EJ,
Jin YH and Jang YK: JIB-04, a small molecule histone demethylase
inhibitor, selectively targets colorectal cancer stem cells by
inhibiting the Wnt/β-catenin signaling pathway. Sci Rep.
8:66112018. View Article : Google Scholar
|
|
34
|
Zylicz JJ, Dietmann S, Gunesdogan U,
Hackett JA, Cougot D, Lee C and Surani MA: Chromatin dynamics and
the role of G9a in gene regulation and enhancer silencing during
early mouse development. Elife. 4:e095712015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hellerbrand C, Bumes E, Bataille F,
Dietmaier W, Massoumi R and Bosserhoff AK: Reduced expression of
CYLD in human colon and hepatocellular carcinomas. Carcinogenesis.
28:21–27. 2007. View Article : Google Scholar
|
|
36
|
Hayashi M, Jono H, Shinriki S, Nakamura T,
Guo J, Sueta A, Tomiguchi M, Fujiwara S, Yamamoto-Ibusuki M,
Murakami K, et al: Clinical significance of CYLD downregulation in
breast cancer. Breast Cancer Res Treat. 143:447–457. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bauer M, Kantelhardt EJ, Stiewe T, Nist A,
Mernberger M, Politt K, Hanf V, Lantzsch T, Uleer C, Peschel S, et
al: Specific allelic variants of SNPs in the MDM2 and MDMX genes
are associated with earlier tumor onset and progression in
Caucasian breast cancer patients. Oncotarget. 10:1975–1992. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Elefanti L, Sacco G, Stagni C, Rastrelli
M, Menin C, Russo I and Alaibac M: TLR7 Gln11Leu single nucleotide
polymorphism and susceptibility to cutaneous melanoma. Oncol Lett.
12:275–280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jenner MW, Leone PE, Walker BA, Ross FM,
Johnson DC, Gonzalez D, Chiecchio L, Dachs Cabanas E, Dagrada GP,
Nightingale M, et al: Gene mapping and expression analysis of 16q
loss of heterozygosity identifies WWOX and CYLD as being important
in determining clinical outcome in multiple myeloma. Blood.
110:3291–3300. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fernández-Majada V, Welz PS, Ermolaeva MA,
Schell M, Adam A, Dietlein F, Komander D, Büttner R, Thomas RK,
Schumacher B and Pasparakis M: The tumour suppressor CYLD regulates
the p53 DNA damage response. Nat Commun. 7:125082016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ghadami E, Nikbakhsh N, Fattahi S,
Kosari-Monfared M, Ranaee M, Taheri H, Amjadi-Moheb F, Godazandeh
G, Shafaei S, Nosrati A, et al: Epigenetic alterations of CYLD
promoter modulate its expression in gastric adenocarcinoma: A
footprint of infections. J Cell Physiol. 234:4115–4124. 2019.
View Article : Google Scholar
|
|
42
|
Zhong S, Fields CR, Su N, Pan YX and
Robertson KD: Pharmacologic inhibition of epigenetic modifications,
coupled with gene expression profiling, reveals novel targets of
aberrant DNA methylation and histone deacetylation in lung cancer.
Oncogene. 26:2621–2634. 2007. View Article : Google Scholar
|
|
43
|
Pannem RR, Dorn C, Hellerbrand C and
Massoumi R: Cylindromatosis gene CYLD regulates hepatocyte growth
factor expression in hepatic stellate cells through interaction
with histone deacetylase 7. Hepatology. 60:1066–1081. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wickström SA, Masoumi KC, Khochbin S,
Fässler R and Massoumi R: CYLD negatively regulates cell-cycle
progression by inactivating HDAC6 and increasing the levels of
acetylated tubulin. EMBO J. 29:131–144. 2010. View Article : Google Scholar
|
|
45
|
Chi P, Allis CD and Wang GG: Covalent
histone modifications-miswritten, misinterpreted and mis-erased in
human cancers. Nat Rev Cancer. 10:457–469. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kampranis SC and Tsichlis PN: Histone
demethylases and cancer. Adv Cancer Res. 102:103–169. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang L, Chang J, Varghese D, Dellinger M,
Kumar S, Best AM, Ruiz J, Bruick R, Peña-Llopis S, Xu J, et al: A
small molecule modulates Jumonji histone demethylase activity and
selectively inhibits cancer growth. Nat Commun. 4:20352013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rabal O, San José-Enériz E, Agirre X,
Sánchez-Arias JA, Vilas-Zornoza A, Ugarte A, de Miguel I, Miranda
E, Garate L, Fraga M, et al: Discovery of reversible DNA
methyltransferase and lysine methyltransferase G9a inhibitors with
antitumoral in vivo efficacy. J Med Chem. 61:6518–6545. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fernández-Barrena MG, Arechederra M, Colyn
L, Berasain C and Avila MA: Epigenetics in hepatocellular carcinoma
development and therapy: The tip of the iceberg. JHEP Rep.
2:1001672020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Barcena-Varela M, Caruso S, Llerena S,
Aacute;lvarez-Sola G, Uriarte I, Latasa MU, Urtasun R, Rebouissou
S, Alvarez L, Jimenez M, et al: Dual targeting of histone
methyltransferase G9a and DNA-methyltransferase 1 for the treatment
of experimental hepatocellular carcinoma. Hepatology. 69:587–603.
2019. View Article : Google Scholar
|
|
51
|
Colyn L, Bárcena-Varela M,
Aacute;lvarez-Sola G, Latasa MU, Uriarte I, Santamaría E, Herranz
JM, Santos-Laso A, Arechederra M, Ruiz de Gauna M, et al: Dual
targeting of G9a and DNA methyltransferase-1 for the treatment of
experimental cholangiocarcinoma. Hepatology. 73:2380–2396. 2021.
View Article : Google Scholar
|
|
52
|
De Beck L, Prosper F, Maes K, Vanderkerken
K and Breckpot K: Can CM272, a dual G9a/DNMT1 inhibitor, be used as
an immunomodulating agent to enhance the efficacy of existing
immunotherapies in melanoma? In: Presented at BACR: Novel
combination strategies for cancer treatment (poster session);
Antwerp. 2019
|
|
53
|
Ke H, Augustine CK, Gandham VD, Jin JY,
Tyler DS, Akiyama SK, Hall RP and Zhang JY: CYLD inhibits melanoma
growth and progression through suppression of the JNK/AP-1 and
β1-integrin signaling pathways. J Invest Dermatol. 133:221–229.
2013. View Article : Google Scholar
|
|
54
|
La T, Jin L, Liu XY, Song ZH, Farrelly M,
Feng YC, Yan XG, Zhang YY, Thorne RF, Zhang XD and Teng L:
Cylindromatosis is required for survival of a subset of melanoma
cells. Oncol Res. 28:385–398. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Rodriguez-Madoz JR, San Jose-Eneriz E,
Rabal O, Zapata-Linares N, Miranda E, Rodriguez S, Porciuncula A,
Vilas-Zornoza A, Garate L, Segura V, et al: Reversible dual
inhibitor against G9a and DNMT1 improves human iPSC derivation
enhancing MET and facilitating transcription factor engagement to
the genome. PLoS One. 12:e01902752017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Dang NN, Jiao J, Meng X, An Y, Han C and
Huang S: Abnormal overexpression of G9a in melanoma cells promotes
cancer progression via upregulation of the Notch1 signaling
pathway. Aging (Albany NY). 12:2393–2407. 2020. View Article : Google Scholar
|
|
57
|
Kato S, Weng QY, Insco ML, Chen KY,
Muralidhar S, Pozniak J, Diaz JMS, Drier Y, Nguyen N, Lo JA, et al:
Gain-of-function genetic alterations of G9a drive oncogenesis.
Cancer Discov. 10:980–997. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Pan MR, Hsu MC, Chen LT and Hung WC: G9a
orchestrates PCL3 and KDM7A to promote histone H3K27 methylation.
Sci Rep. 5:187092015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ceol CJ, Houvras Y, Jane-Valbuena J,
Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ,
Ferré F, et al: The histone methyltransferase SETDB1 is recurrently
amplified in melanoma and accelerates its onset. Nature.
471:513–517. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Fritsch L, Robin P, Mathieu JR, Souidi M,
Hinaux H, Rougeulle C, Harel-Bellan A, Ameyar-Zazoua M and
Ait-Si-Ali S: A subset of the histone H3 lysine 9
methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a
multimeric complex. Mol Cell. 37:46–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Pinto-Fernandez A and Kessler BM: DUBbing
cancer: Deubiquitylating enzymes involved in epigenetics, DNA
damage and the cell cycle as therapeutic targets. Front Genet.
7:1332016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wu Y, Wang Y, Yang XH, Kang T, Zhao Y,
Wang C, Evers BM and Zhou BP: The deubiquitinase USP28 stabilizes
LSD1 and confers stem-cell-like traits to breast cancer cells. Cell
Rep. 5:224–236. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang YC, Wang SA, Chen PH, Hsu TI, Yang
WB, Chuang YP, Su WC, Liaw HJ, Chang WC and Hung JJ: Variants of
ubiquitin-specific peptidase 24 play a crucial role in lung cancer
malignancy. Oncogene. 35:3669–3680. 2016. View Article : Google Scholar
|
|
64
|
Li G, Ji T, Chen J, Fu Y, Hou L, Feng Y,
Zhang T, Song T, Zhao J, Endo Y, et al: CRL4DCAF8
ubiquitin ligase targets histone H3K79 and promotes H3K9
methylation in the liver. Cell Rep. 18:1499–1511. 2017. View Article : Google Scholar : PubMed/NCBI
|