Introduction
Lynch syndrome (LS) is an autosomal hereditary form
of colorectal cancer (CRC) with high penetrance and an incidence of
3-5% of all CRC cases (1-3).
This hereditary form of CRC is clinically distinguishable from
familial adenomatous polyposis (FAP), another hereditary form of
CRC that develops from numerous polyps, as FAP is associated with
APC mutations (4). Individuals
with LS are characterized by a high lifetime risk of development of
CRC, as well as endometrial, gastric and ovarian cancers, and other
extracolonic tumors (such as small intestine, brain, skin,
hepatobiliary, and urinary tract) (5,6).
This hereditary syndrome is due to germline mutations in DNA
mismatch repair (MMR) genes, mainly MLH1, MSH2, MSH6 and PMS2
(7-11).
The molecular characterization of these patients
involves germline testing to detect a deleterious mutation in MLH1,
MSH2, MSH6 or PMS25 (11), by
direct sequencing and multiplex ligation-dependent probe
amplification (MLPA), commonly used for the detection of large
rearrangements (12). Numerous
pathogenic mutations have been detected in MLH1 and MSH2 (13,14), while germline mutations in MSH6
and PMS2 are responsible for the disease only in a minority of
cases (15-18). DNA MMR deficiency determines the
presence of microsatellite instability (MSI) or loss of MMR protein
expression at the somatic level (19). To identify patients with LS
suitable for genetic testing, MSI testing and/or the lack of MMR
protein expression (dMMR) according to immunohistochemical staining
of colorectal or endometrial tumoral tissues is recommended for
universal tumor screening, and these should be conducted as the
first step (20,21). However, the Amsterdam criteria
(AC) still remain a valid tool to allow clinicians to raise a
clinical suspicion of LS in young patients with CRC and a
significant family history of cancer (22).
To date, however, a large proportion of patients
that exhibit the MSI/dMMR phenotype and undergo genetic testing do
not show germline pathogenic variants in any of the MMR genes
(MLH1, MSH2, MSH6, or PMS2). Unfortunately, the molecular
mechanisms underlying MSI/dMMR CRC remain poorly understood
(23). With the introduction of
a next-generation sequencing (NGS) approach (24,25) that allows the concurrent
investigation of multiple genes (25), the evaluation of inherited CRC is
changing. For example, previous high-throughput sequencing studies
suggested that tumor-suppressed genes such as TP53 and
cyclin-dependent kinase inhibitor 2A (CDKN2A) could be responsible
for genomic instability in numerous sporadic cancers (26,27). In the last few decades, NGS
applications have led to the discovery of mutations in several
predisposing CRC genes and to LS-related cancers. Germline
mutations in other DNA-repair genes, such as ATM, PALB2, MRE11, and
CHEK2, expressed throughout the cell cycle in response to
double-stranded DNA breaks, have been associated with
susceptibility to CRC (28,29). Furthermore, variants in CDH1, a
gene normally associated with gastric cancer, could be a risk
factor for CRC (30); a
reduction of its expression is associated with tumoral
dedifferentiation, lymphatic vessel invasion, and metastatic
processes in CRC (31). In
several previous studies, common MSH3 polymorphisms and rare
variants were found to be significantly associated with CRC and
prostate cancer as low-penetrance risk alleles (32-35). Moreover, biallelic MSH3 germline
mutations appear to cause an additional rare recessively-inherited
subtype of colorectal adenomatous polyposis (36). Evidence for an involvement of
MLH3 includes a recent publication by Olkinuora et al
(37) showing that a biallelic
MLH3-truncating variant causes classical or attenuated adenomatous
polyposis and possibly extracolonic tumors, while rare heterozygous
variants of the MLH3 gene are associated with the LS phenotype
(38). Finally, individuals with
constitutional MMR deficiency (CMMRD) often have a high risk of
developing a broad spectrum of malignancies and frequently display
features reminiscent of neurofibromatosis type 1 (NF1) (39).
However, despite the NGS applications involving the
multiple and simultaneous investigation of the various genes
hitherto associated with MSI/dMMR CRC, the number of
pathogenic variants identified following these analyses remains
almost unchanged. Instead the variants of uncertain significance
(VUS) that are detected in these patients who exhibit the MSI/dMMR
phenotype are increasingly numerous.
Therefore, the aim of the present study was to
elucidate the molecular basis of predisposition to the development
of hereditary LS-related cancers in a cohort of 73 patients with a
clinical suspicion of LS, using an NGS multigene panel designed and
standardized by our research group and evaluating the pathogenicity
of the numerous VUS identified, by applying criteria well known in
the literature in order to obtain conclusive interpretation.
Materials and methods
Patient selection
A total of 73 patients with suspected LS meeting AC
and/or Bethesda guidelines (BG) were recruited from Federico II
University Hospital, National Cancer Institute IRCCS G. Pascale
Foundation, and Luigi Vanvitelli University Hospital, all in
Napoli, in southern Italy between January 2006 and December 2019,
after study of their clinical characteristics and MSI and/or MMR
protein expression in tumors.
Personal and family histories were obtained from
each proband and written informed consent was provided by all
patients. The present study was approved (protocol no. 120/10) by
the local ethics committee 'Comitato Etico per le Attività
Biomediche 'Carlo Romano' ('Carlo Romano' Ethics Committee for
Biomedical Activities) at the University of Naples Federico II
(Napoli, Italy).
Genomic DNA extraction
Genomic DNA was isolated from 3 ml of peripheral
blood lymphocytes using the Nucleon BACC2 kit (Cytiva), according
to the manufacturer's protocol. DNA quantity was assessed using a
NanoDrop OneC spectrophotometer (reading at 260 nm and ratio
260/280 and 260/230 nm) and an Invitrogen Qubit 4 fluorometer (both
from Thermo Fisher Scientific, Inc.). DNA quality was evaluated by
1% agarose gel electrophoresis and visualized with ethidium
bromide.
Mutational analysis of coding regions of
MMR genes
Genomic rearrangements in the MMR genes were
analyzed by MLPA using SALSA-MSH6 P072 and SALSA-PMS2 P008 C1 kits
(MRC-Holland BV), according to the manufacturer's protocol.
Targeted NGS
Library construction
Patient DNA samples were examined using an AmpliSeq
Custom Panel (Illumina, Inc.), targeting 15 genes (Table I) involved in the MMR pathway or
associated with CRC and other well-characterized cancer syndromes.
This panel was developed based on the literature (26-39) to include genes associated with an
increased risk of developing colon cancer. The kit (cat. no.
20020495; Illumina, Inc.) includes 470 amplicon regions that cover
87,353 bp, all the exonic and flanking intronic regions of these
genes.
 | Table ILynch syndrome full-exome panel. |
Table I
Lynch syndrome full-exome panel.
| Gene name | RefSeq | Band Chr | Genomic size | RNA size | Exons |
|---|
| MLH1 | NM_000249.3 | 3p22.2 | 57497 | 2662 | 19 |
| MSH2 | NM_000251.2 | 2p21 | 80162 | 3226 | 16 |
| MSH6 | NM_000179.2 | 2p16.3 | 23872 | 4435 | 10 |
| PMS2 | NM_000535.5 | 7p22.1 | 35868 | 2851 | 15 |
| MLH3 | NM_001040108.1 | 14q24.3 | 37769 | 7911 | 13 |
| MSH3 | NM_002439.4 | 5q14.1 | 222168 | 4472 | 24 |
| EPCAM | NM_002354.2 | 2p21 | 17881 | 1731 | 9 |
| CDH1 | NM_004360.3 | 16q22.1 | 98250 | 4815 | 16 |
| TP53 | NM_000546.5 | 17p13.1 | 19149 | 2591 | 11 |
| ATM | NM_000051.3 | 1p34.1 | 11229 | 1945 | 16 |
| CHEK2 | NM_001005735.1 | 22q12.1 | 54092 | 1991 | 16 |
| PALB2 | NM_024675 | 16p12.2 | 38196 | 4069 | 13 |
| MRE11A | NM_005591 | 11q21 | 76572 | 5141 | 20 |
| NF1 | NM_001042492 | 17q11.2 | 282751 | 12444 | 58 |
| CDKN2A | NM_000077 | 9p21.3 | 7382 | 1267 | 3 |
DNA was diluted to a final concentration of 4
ng/µl using Low Tris-EDTA buffer (included in the kit) and
re-quantitated with the fluorometric quantification method
(Invitrogen Qubit 4). The standard input was 20 ng of DNA per
sample. Briefly, the workflow involved multiplex PCR to amplify
target regions of each DNA sample according to the procedure for
two primer pools from an AmpliSeq Illumina Custom DNA Panel. FuPa
reagent (included in the kit) was used to partially digest
amplicons and each library was mixed and ligated with a unique
index-specific combination. Subsequently, a second amplification
step ensured sufficient quantity for the final sequencing analysis.
The quality and quantity of the libraries obtained were assessed
using a TapeStation 4200 System (Agilent Technologies, Inc.) with
an Agilent DNA 1000 kit. The sequencing was performed on a
MiSeqSystem (Illumina, Inc.) with a Nano V.2 flow cell (300 cycles)
reagent kit according to the manufacturer's protocol. The raw data
generated by this analysis are available on site Mendeley
(Elsevier; https://data.mendeley.com/datasets/mxp536twnw/draft?a=a642b2f6-f2374e78-802c-c5cab21ef866.
Bioinformatics analysis
The sequencing data was analyzed using a BaseSpace
Sequence Hub (Illumina, Inc.). Primary data analysis involved the
detection and analysis of raw data (signal analysis), targeting
sequencing reads (base calling) and scoring base quality. FASTQ
files (generated by MiSeq Reporter Software 1.3.17; Integrative
Genomics Viewer) were the outputs from this primary analysis. A
demultiplexing process was subsequently required to produce
separated sequencing read files, according to the single index used
for each sample. In a secondary data analysis, FASTQ files for each
sample were aligned against an entire reference genome specified in
the manifest file with a DNA Amplicon Analysis App on BaseSpace
Sequence Hub (version 2.1.1).
In silico analysis of unclassified
variants
The following variant calling step had the main
objective of identifying variants using a post-processed BAM file
(https://basespace.illumina.com/analyses; BAM metrics
version 0.0.22). A default value of 10 was used to define the
Variant Caller Depth Filter level. Lower filter values may cause
further false positive variants to pass the filter.
Output VCard File (VCF) was finally used for
downstream analysis on a Variant Interpreter App (https://variantinterpreter.informatics.illumina.com;
version 2.16.0.235), integrated with a BaseSpace Sequence Hub, that
provided variant classification and reporting.
The BaseSpace Variant Interpreter is a cloud-based
platform that uses the following annotation sources: Single
Nucleotide Polymorphism Database (dbSNP) (https://www.ncbi.nlm.nih.gov/snp/?cmd=search),
Catalogue of Somatic Mutations in Cancer (COSMIC) (https://cancer.sanger.ac.uk/cosmic), ClinVar
(https://www.ncbi.nlm.nih.gov/clinvar/), 1000 Genomes
(https://www.internationalgenome.org),
Exome Variant Server (EVS) (https://evs.gs.washington.edu/EVS/), (ExAC)
(https://gnomad.broadinstitute.org),
Polymorphism Phenotyping v.2 (PolyPhen-2), and Sorting Intolerant
from Tolerant (SIFT). Such software classify germline variants as
pathogenic, likely pathogenic, VUS, likely benign and benign. These
categories follow The American College of Medical Genetics and
Genomics (ACMG) guidelines (40). A total of 7 different
complementary in silico programs were subsequently used for
functional impact predictions of the identified variants.
Human Splicing Finder (HSF) (www.umd.be/HSF/) for silent and intron variants is a
tool designed to predict the effects of mutations on splicing
signals or to identify splicing motifs in human sequences. It
contains all available matrices for auxiliary sequence prediction
and presents a novel position weight matrix to assess the strength
of 5′ and 3′ splice sites and branch points (41).
SIFT (http://blocks.fhcrc.org/sift/SIFT.html), PolyPhen
(http://genetics.bwh.harvard.edu/pph/)
(42), and PredictProtein server
(http://www.predictprotein.org) (43) are prediction tools based on a
combination of phylogenetic, structural and sequence annotation
information characterizing a substitution with its position in the
protein.
Mutation Taster (http://www.mutationtaster.org/) (44) and Align-Grantham Variation
Grantham deviation (A-GVGD) (http://agvgd.hci.utah.edu/) (45) were employed in the study of
missense variants. Briefly, Mutation Taster analyses comprise
evolutionary conservation, splice-site changes, loss of protein
features, and changes that may affect the amount of mRNA; moreover,
the A-GVGD method can be used to identify sets of missense
substitutions that are either enriched for deleterious variants or
enriched for neutral variants.
Finally, Protein Variation Effect Analyzer (PROVEAN)
is useful software for predicting whether nonsynonymous or indel
variants are functionally important (http://provean.jcvi.org/index.php); its performance is
comparable to that of SIFT or PolyPhen-2.
All variants identified were annotated according to
the nomenclature recommendations from the Human Genome Variation
Society (www.hgvs.org/mutnomen).
Variant analysis by Sanger
sequencing
The coding regions corresponding to 22 variants
(pathogenic variants and deleterious variants by in silico
analysis) were amplified using customized primer sets (Table II). The PCR products were
separated on a 1-2% agarose gel to check for unspecific amplicons.
Subsequently, the PCR products were sequenced in both the forward
and reverse directions using an ABI 3100 Genetic Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.).
| Table IIPrimers, annealing PCR and amplicon
sizes of PCR products corresponding to the pathogenic variants and
variants resulted deleterious by in silico analysis. |
Table II
Primers, annealing PCR and amplicon
sizes of PCR products corresponding to the pathogenic variants and
variants resulted deleterious by in silico analysis.
| Gene variants | Primer sequences
(5′→3′) | TM (°C) | Amplicon (bp) | Exon |
|---|
| MLH1
c.350C>T | F:
GTGACCCAGCAGTGAGTTTT | 58 | 245 | 4 |
| R:
AGCCTCACTTTTACCCTCTCT | | | |
| MSH6
c.3311_3312del | F:
AGCCTCACTTTTACCCTCTCT | 58 | 398 | 5 |
| R:
TGGCTGACTTTTATGTAACTGTG | | | |
| MSH6
c.892C>T | F:
TGGTGGCTCTGATGTGGAAT | 59 | 206 | 4 |
| R:
TTGCTTGTTTGGTGGCTGAG | | | |
| ATM c.3802delG | F:
TGCTACTGAACAAGGTCCCA | 59 | 448 | 26 |
| R:
CTCTCTTTGCTGTGCCATCC | | | |
| MLH1
c.376T>A | F:
GTGACCCAGCAGTGAGTTTT | 58 | 245 | 4 |
| R:
ACGTACTCAAGATCTCTGCCA | | | |
| EPCAM
c.332A>G | F:
TGATGAAGGCAGAAATGAATGG | 58 | 250 | 3 |
| R:
ACAAGTAGTATAGGCAGCCCC | | | |
| MSH3
c.1778G>A | F:
CCTGGGCATTAGAGTGGGAA | 58 | 264 | 13 |
| R:
TGTCCTCAAGCTGAAGAACAC | | | |
| MLH3
c.470T>C | F:
TATGGTTTCCGAGGAGAGGC | 59 | 232 | 2 |
| R:
CCAGTCTAGGGTCCATGCAT | | | |
| MLH3
c.3440A>T | F:
TCAGTTTGTGCAGAAAGAGGT | 58 | 250 | 3 |
| R:
GAGTTAGGTGGTACGATGTGT | | | |
| ATM
c.7475T>G | F:
AGGAAGGTGTGTGAATTGCA | 58 | 465 | 50 |
| R:
CCTGACATCAAGGGGCTTATG | | | |
| ATM
c.1178G>T | F:
GGCAACAACAGCGAAACTCT | 58 | 396 | 9 |
| R:
TGTCATGGCAATCACATATCCC | | | |
| ATM
c.8734A>G | F:
ATTAGCTGTCAAACCTCCTAACT | 58 | 221 | 60 |
| R:
TGCCCAGCCCATGTAATTTT | | | |
| CHEK2
c.911T>C | F:
TGTCTTCTGTCCAAGTGCGT | 59 | 245 | 9 |
| R:
GGTCCCTCGATTTCTGCCTA | | | |
| PMS2
c.1004A>G | F:
AAAGTGAATTTGGCTGGGCG | 59 | 498 | 10 |
| R:
TGGCTGCTGACTGACATTTAGCTTG | | | |
| PMS2
c.2249G>A | F:
TCTCAGGAAGTTTTGTGACACT | 59 | 295 | 13 |
| R:
CACCCAGCCGCTATAGTTCT | | | |
| PMS2
c.1253C>T | F:
GACCCTCTTCTCCGTCCAC | 58 | 491 | 11 |
| R:
GAGAGTCCACATGTTCCTGC | | | |
| MLH1
c.589-9_589-6delGTTT | F:
GTTTGCTGGTGGAGATAAGGT | 58 | 392 | 8 |
| R:
ACGCCACAGAATCTAGGAGA | | | |
| MRE11
c.1783+7A>G | F:
ACTTTCTCCTTCTTCTCCCTCT | 59 | 410 | 15 |
| R:
TGTCAGAACTGCCTTAAAGACTG | | | |
| CDH1
c.585A>C | F:
TTCTCTGGGAGGGATTTGGC | 59 | 293 | 5 |
| R:
CCCGGTGTCAACAAGCTTC | | | |
| CDH1
c.344C>T | F:
GAAGATTGCACCGGTCGAC | 58 | 250 | 3 |
| R:
CAACAGCGAACTTCTCAGAAAA | | | |
| NF1
c.4445T>C | F:
CTGGGTGTATCTGGTGTTGAAAA | 58 | 486 | 34 |
| R:
GGATCTATAACAATCTGCAAGCC | | | |
| CHEK2
c.688G>T | F:
CTTGAAGTGGACCCAGGAGT | 58 | 242 | 6 |
| R:
TGGGAAGTTATGAAGACGTGTT | | | |
Results
A total of 73 patients with a clinical suspicion of
LS were analyzed in the present study. The patients were selected
as follows: 42 patients based on the AC (5), and 31 patients according to MMR
deficiency detected in tumoral tissue with respect to the Bethesda
guidelines (BG) (20).
Individuals undergoing analysis were all affected by CRC, and two
had MMR-deficient endometrial cancer. These patients were also
affected by ovarian, bladder, breast, prostate, melanoma and renal
cancers. The mean age at diagnosis, MSI-high (MSI-H) status
analysis and sex of patients are outlined in Table III.
| Table IIIClinical and molecular
characteristics of the 73-patient cohort. |
Table III
Clinical and molecular
characteristics of the 73-patient cohort.
|
Characteristics | Amsterdam
criteria | Bethesda
guidelines | Total |
|---|
| Sex | | | |
| Female | 21 | 14 | 35 |
| Male | 21 | 17 | 38 |
| Age at diagnosis,
years (mean ± SD) | 33.47±11.96 | 47.59±16.93 | 44.89±14.26 |
| Tumor type | | | |
| CRC | 40 | 31 | 71 |
| Breast | 3 | 3 | 6 |
| Endometrium | 3 | 4 | 9 |
| Other tumors | | 2 | 3 |
| MSI status | | | |
| MSI | 9 | 31 | 40 |
| Unknown | 31 | | 31 |
All patients were previously shown to be negative
for pathogenic variants in the MLH1 and MSH2 genes detected by
single-gene analyses [denaturing high-pressure liquid
chromatography (dHPLC) followed by Sanger sequencing] and large
rearrangements (by MLPA).
Germline DNA samples from the patients were tested
with NGS methods using a multigene custom panel developed by our
research group that targets a set of 15 genes (Table I), MLH1, MSH2, MSH6, PMS2, MSH3,
MLH3, CHEK2, MRE11, EPCAM, ATM, TP53, CDKN2A, PALB2, CDH1 and NF1.
These genes are involved in the MMR pathway or associated with CRC
and/or other well-characterized cancer syndromes (26-39).
Paired-end NGS generated an average of 705,000
reads, 93.93% of which mapped against the human genome (version
GRCh38). Variants with 99.42% of exons covered were only selected,
labelled as 'PASS' by the filter applied, with an estimated average
amplicon depth of coverage of 1,350 reads. In this manner, an
overall number of 724 variants were identified, of which only four
(0.55%) were pathogenic variants already known, reported in the
international databases Insight-Group and ClinVar (http://insight-database.org; https://www.ncbi.nlm.nih.gov/clinvar/) as pathogenetic
variants. Most of the remaining variants (87.8%) were already known
in the literature as being benign or polymorphic variants; 86
variants (11.88%) had been classified as variants that are likely
benign or of uncertain pathogenic significance.
Clinically significant variants:
Pathogenic variants
At least one clinically significant variant was
identified in four patients (5.6%) of the cohort: One nonsense
variant in ATM, two nonsense variants in MSH6, and one missense
variant in MLH1 (Table IV).
These had not been previously found by traditional methods (dHPLC
and Sanger sequencing). These variants were validated by Sanger
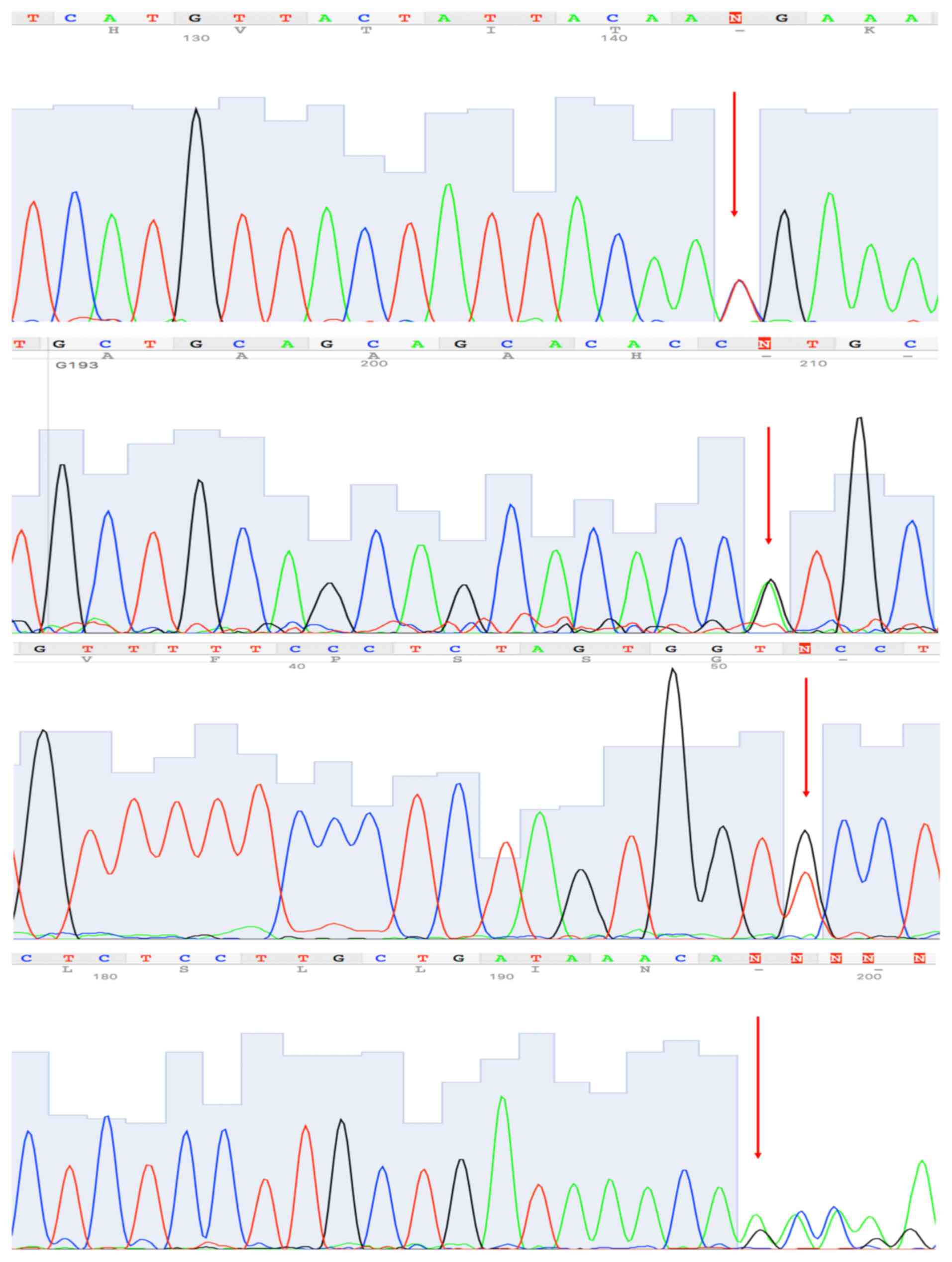
sequencing (Fig. 1).
| Table IVClinically significant variants
identified. |
Table IV
Clinically significant variants
identified.
| ID of patient | Gene | Variant | Variant
classification | InSiGHT | ClinVar | PHENOTYPE (age
onset) |
|---|
| 07.19 | MLH1 | c.350C>T p.
(Thr117Met) | Pathogenetic | Pathogenetic | Pathogenetic | Index case k-co
(34), MSI-H; father k-co
(37) and his father's brother
succumbed to k-co (65) |
| 07.13 | MSH6 | c.3311_3312del p.
(Phe1104TrpfsTer3) | Pathogenetic | Pathogenetic | Pathogenetic | Index case k-co
(49), MSI-H; father k-st
(50), died k-co (58), his
father's brother k-co (70), his cousin k-st (61) and colon
polyps |
| 14.07 | MSH6 | c.892C>T p.
(Arg298Ter) | Pathogenetic | Pathogenetic | Pathogenetic | Index case k-end
(32), MSI-H; father leukemia
(72), and her father's mother succumbed to leukemia (45) |
| 13.68 | ATM | c.3802delG p.
(Val1268Ter) | Pathogenetic | - | Pathogenetic | Index case k-co
(36) and several colon polyps;
Sister k-co (75), brother k-pro (60) and father died k-pan
(70) |
Unclassified and likely benign variants:
Pathogenicity assessment
For the present study, 86 variants were considered
as VUS. These were the variants that showed a minor allele
frequency (MAF) that was very low (<0.1%) or not reported, some
known in the international database (as ClinVar and Insight) and
others novel. At least one of these VUS was identified in 57
patients (80.3%) in the cohort. These variants were distributed
among the Lynch full-exome panel genes as follows: 4 in MLH1, 2 in
MSH2, 4 in MSH6, 12 in PMS2, 12 in MSH3, 4 in MLH3, 3 in CHEK2, 5
in MRE11, 3 in EPCAM, 18 in ATM, 3 in TP53, 5 in PALB2, 5 in CDH1
and 6 in NF1. For each VUS identified in the present study,
multiple bioinformatics analyses were performed using several
software programs described in Table
V and in the Materials and methods section. For silent and
intronic variants, an ad hoc in silico analysis was used (as
described in Table V and in the
Materials and methods). Most of these variants presented discordant
results from the computational algorithms applied. All lines of
computational evidence supported a deleterious effect for 13 of
these 86 variants. In addition, five others showed a deleterious
effect in all but one or two of the computational algorithms used
(Table V). These variants were
validated by Sanger sequencing (Fig.
1).
| Table VVariants identified in the panel
genes and predicted in silico to have a deleterious
effect. |
Table V
Variants identified in the panel
genes and predicted in silico to have a deleterious
effect.
| Variants | ID Cancer (age
onset) selected criteria | MAF% | Mutation
Taster | Poly Phen 2
[0-1] | Align GVGD | Predict
protein | Provean (c. off
2.5) | Sift (c.off
0.05) | HSF | Source |
|---|
| c.376T>A p.
(Tyrl26Asn) MLH1 | 19.39 endometrial
(26) BG | 0.004% |
Disease-causing | 0.911 (POSS
DAM) | CLASS C65 | 95 | DEL (-7.69) | DAM (0) | | Insight-group
ClmVar |
| c.332A>G p.
(AsnlllSer) EPCAM | 18.33 colon
(40) AC | |
Disease-causing | 1.00 (DAM) | CLASS C45 | 71 | DEL (-4.13) | DAM (0.004) | - | ClinVar |
| c,1778G>A p.
(Arg593Gln) MSH3 | 14.42 breast
(38) colon (38) AC | - |
Disease-causing | 1.00 (DAM) | CLASS C35 | 59 | DEL (-3.90) | DAM (0.001) | - | None |
| c.470T>C p.
(Vall57Ala) MLH3 | 18.17 breast
(37) colon (38), melanoma AC | |
Disease-causing | 1.00 (DAM) | CLASS C55 | 49 | DEL (-3.66) | DAM (0) | - | None |
| c.3440A>T p.
(Asnll47 Ile) MLH3 | 17.14 colon
(52) AC | 0.03% |
Disease-causing | 1.00 (DAM) | CLASS C65 | | DEL (-6.77) | DAM (0) | - | ClinVar |
| c.7475T>G p.
(Leu2492 Arg) ATM | 18.17 (as above)
06.10 colon (53) AC | 0.02% |
Disease-causing | 1.00 (DAM) ' | CLASS C6f | | DEL (-4.53) | DAM (0.003) | - | None |
| c,1178G>T p.
(Trp393Leu) ATM | 19.24 colon
(29) AC | 0.002% |
Disease-causing | 1.00 (DAM) | CLASS C55 | | DEL (-4.86) | DAM (0) | | None |
| c.8734A>G p.
(Arg2912 Gly)AW | 18.08 rectum 16
BG | 0.02% |
Disease-causing | 1.00 (DAM) | CLASS C65 | | DEL (-5.83) | DAM (0) | | ClinVar |
| c.911T>C p.
(Met304Thr) CHEK2 | 11.33 sigma
(51) AC | 0.0004% |
Disease-causing | 0.999 (DAM) | CLASS C65 | 86 | DEL (-3.76) | DAM (0.014) | - | ClinVar |
| c.1004a>g p.
(Asn335Ser) PMS2 | 18.21 breast
(49) colon (50) BG | 0.1% |
Disease-causing | 1.00 (DAM) | CLASS C45 | 48 | DEL (-4.47) | DAM (0.006) | | Insight-group
ClinVar |
| c.2149G>A p.
(Val717Met) PMS2 | 18.36 colon
(38) AC | 0.08% |
Disease-causing | 0.982 (DAM) | CLASS C15 | 14 | NEU (-1-79) | DAM (0.010) | - | ClinVar |
| c.1253C>T p.
(Ser418Phe) PMS2 | 10.17 colon
(49) AC | 0.0008% |
Disease-causing | 0.977 (DAM) | CLASS C65 | -6 | DEL (-3.04) | DAM (0.004) | | Insight-group
ClmVar |
|
c.589-9_589-6delGTTT MLH1 | 18.08 (as
above) | 0.01% | | | | | | | Activation of an
intronic cryptic acceptor site. Potential alteration of
spicing. | Insight-group
ClinVar |
| c. 1783+7A> G
MRE11 | 09.10 colon
(34) BG | 0.004% | - | - | - | - | - | - | Activation of an
intronic cryptic donor site. Potential alteratior of spicing. | None 1 |
| c.585A>C p.
(Gln195His) CDH1 | 11.25 colon
prostate (69) BG | - |
Disease-causing | 0.873 (POSS
DAM) | CLASS C 15 | -65 | DEL (-2.59) | TOL (0.059) | - | ClinVar |
| c.344C>T p.
(Thrll5Met) CDH1 | 19.59 colon, kidney
(36) AC | 0.0024% |
Disease-causing | 0.992 (DAM) | CLASS C 15 | -81 | NEUT (-1.93) | TOL (0.080) | - | ClinVar |
| c.4445t>c p.
(Ilel482Thr) NF1 | 06.08 colon
(28) AC | - |
Disease-causing | 0.393 (BEN) | CLASS C65 | | DEL (-4.15) | DAM (0.002) | - | ClinVar |
| c.688G>T p.
(Ala230Ser) CHEK2 | 18.30 sigma 24
BG | 0.0008% |
Disease-causing | 0.076 (BEN) | CLASS C 65 | 64 | DEL (-2.76) | DAM (0.006) | - | ClinVar |
Some of these rare variants are not present in
healthy controls (as they are not reported in The Genome
Aggregation Database, Exome Aggregation Consortium, or 1,000 Genome
Projects database) and they are present in genes for which an
association with a predisposition to developing colorectal tumors
(or LS-related cancers) is well known. Therefore, the criteria
reported in ACMG and the Association for Molecular Pathology
guidelines were applied for the interpretation of sequence
variants, (Table VI) (40). The MSI/dMMR status on tumor
tissue was evaluated as a strong evidence of pathogenicity
comparable with the results of well-established in vivo
functional studies supportive of a damaging effect of variant on
the gene or gene product only for the rare variants identified in
the MMR genes. In this manner, eight of these 18 variants could be
considered to be 'likely pathogenic' and for some of these variants
the analysis of segregation in family environment was also
performed (Table VI). Of these,
two variants, (in the MLH3 and ATM genes) are novel and not
previously reported in the literature. For the remaining 78
variants, the criteria for being benign and pathogenic were
contradictory; therefore, these variants remain classified as
having an uncertain significance.
| Table VIVariants defined as 'likely
pathogenic'. |
Table VI
Variants defined as 'likely
pathogenic'.
| Variants | Gene | ID Cancer (age
onset) Familial history (Segregation analysis) | Somatic dMMR | The Genome
Aggregation Database (GnomAD) | Exome Aggregation
Consortium (ExAC) | 1000 Genomes
Project | Additional
information | ACMG Criteria
(a) | New classification
variant |
INSIGHT/CLINVAR |
|---|
| c.376T>A
p.(Tyr126Asn) | MLH1 | 19.39 endometrial
(26) Not affected- (N.D.)
BG |
MS1-HNO
MLH1
NO PMS2 | 0.00004 | 0.00007 | 0.00020 | This variant is
located in the N-terminal ATPase domain | PS3.PM1, PP2, PP3,
PP4 | Likely
pathogenic | Reported as VUS, as
pathogenic and six times as benign/VUS |
| c.470T>C
p.(Val157Ala) | MLH3 | 18.17 breast and
colon (37, 38) other affected-breast and colon
(N.D.) AC | MS1-H | No frequency | No frequency | No frequency | The valine residue
is highly conserved. | PS3, PM2, PP2, PP3,
PP4 | Likely
pathogenic | Not
reported/VUS |
| c.3440A>T
p.(Asn1147Ile) | MLH3 | 17.14 colon
(52) other affected-
endometrial and colon (N.D.) AC | MS1-H | 0.00040 | 0.00044 | 0.00060 | No relevant
information | PS3, PP2, PP3,
PP4 | Likely
pathogenic | Not
reported/conflicting interpretation |
| c.1178G>T
p.(Trp393Leu) | ATM | 19.24 colon
(29) other affected-endometrial
and gastric (N.D.) AC | ND | No frequency | No frequency | No frequency | No information | PM2, PM5, PP2, PP3,
PP4 | Likely
pathogenic | Not reported |
| c.911T>C
p.(Met304Thr) | CHEK2 | 11.33 sigma
(51) other affected- colon
(also in affected brother) AC | MS1-H | No frequency | No frequency | No frequency | The methionine
residue is highly conserved. This variant has been reported to
affect CHEK2 protein function (PMID: 30851065). | PM2.PP1, PP2, PP3,
PP4 | Likely
pathogenic | Not
reported/VUS |
| c.1253C>T
p.(Ser418Phe) | PMS2 | 10.17 colon
(49) other affected-colon (also
in affected brother) AC | MSI-H
NO
MLH1 NO
PMS2 | No frequency | No frequency | No frequency | The seine residue
is moderately conserved. | PS3, PM2, PP2, PP3,
PP4 | Likely
pathogenic | Not
reported/VUS |
|
c.589-9_589-6delGTTT | MLH1 | 18.08 Colon
(18) other affected-colon and
pancreas (also in affected father and two uncle) BG | MSI-H | No frequency | No frequency | No frequency | This variant could
affect mRNA splicing (HSF software) | PS3, PM2, PP3,
PP4 | Likely
pathogenic | Reported seven
times as VUS/conflicting interpretation |
| c.2149G>A
p.Val717Met | PMS2 | 19.46 Colon
(38) not affected-(N.D.)
BG | MSI-H
NO
MLH1
NO PMS2 | 0.00076 | 0.00091 | 0.00020 | The variant is
located within the MutL C-terminal, and dimerisation functional
domain. | PS3, PM2, PP3,
PP4 | Likely
pathogenic | Not
reported/conflicting interpretation |
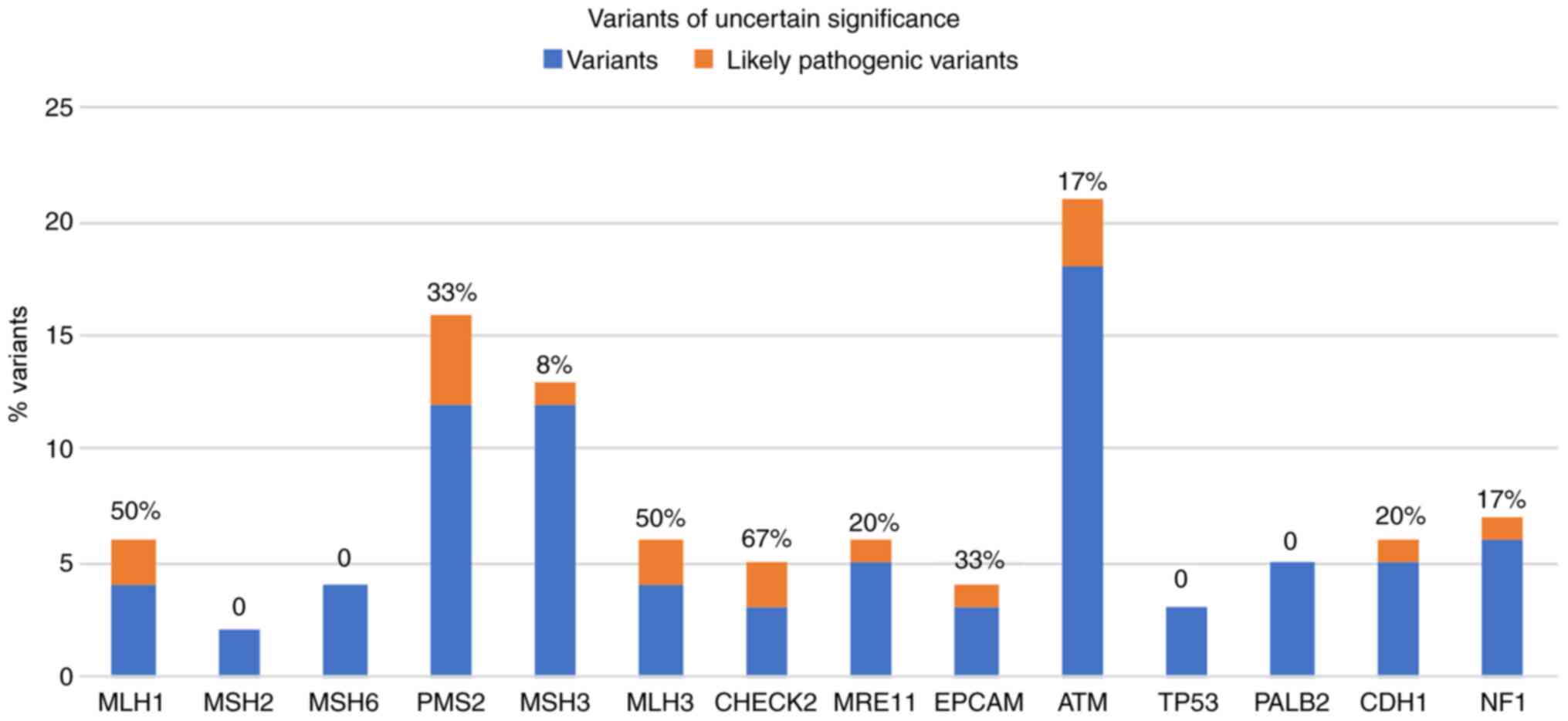
In Fig. 2, the
percentage of these 'likely pathogenic variants' of the total
uncertain variants identified for each gene analyzed, are
presented. Notably, 50% of the gene variants identified in MLH1 and
MLH3 are classified as likely pathogenic.
Discussion
The study of hereditary forms of CRC has increased
the importance of genetic testing. However, the limited capacity of
old genetic screening methods, due to their low sensitivity and
small number of genes studied, has left numerous gaps in
identifying variants conveying a predisposition to cancer. Indeed,
more than half of CRC cases with a clinical suspicion of LS that
are referred for genetic testing remain without a clear molecular
diagnosis. In the present study, an NGS multigene custom panel was
designed and standardized; it included, beyond the four MMR genes
classically associated with LS, other MMR genes such as MLH3 and
MSH3, and other genes that predispose to hereditary CRC or
LS-related cancer according to the literature, (26-39) such as CHEK2, MRE11, EPCAM, ATM,
TP53, PALB2, CDH1, NF1 and CDKN2A. This NGS multigene panel was
used to analyze 73 patients with a clinical suspicion of LS
selected according to the BG and AC. The patients had already been
shown to be negative for pathogenetic variants in the MLH1 and MSH2
genes. This is probably the reason why the number of pathogenic
variants clearly identified in the present study was so low. Only
four pathogenic variants reported in the literature were identified
in the cohort, of which one related to a gene not included among
the MMR genes. Indeed, one patient (ID 13.68) was found to carry a
variant, c.3802delG p. (Val1268Ter), in the ATM gene. This patient
developed a right colon adenocarcinoma at an early age of onset (36
years) and was selected by AC due to a strong family history of
early onset of colon tumors. Unfortunately for this patient, MSI
could not be performed on the tumor tissue; however, it was
certainly a case that could be confused with a clinical suspicion
of LS. Variants in the ATM gene are associated in the recessive
form with ataxia-telangiectasia and in the dominant form with
breast cancer susceptibility and more generally with hereditary
cancers (46). In the present
study, this pathogenetic variant, c.3802delG p. (Val1268Ter),
appeared to be associated with CRC.
The remaining pathogenic variants were identified in
MMR genes (MLH1 and MSH6) in patients who met the AC and showed a
typical LS phenotype with MSI-H status in cancer tissues (Table III). No other variant that was
already classified in the literature as pathogenic was identified
in the cohort, although the patients were selected according to
very specific criteria. However, numerous genetic variants beyond
these four clearly pathogenic variants were identified in the
present study. The majority of these were benign or polymorphic
variants, but numerous variants of unclear pathogenic significance
were also found. Unfortunately, these variants were difficult to
clinically interpret, which poses a significant barrier to the
broad utility of genetic testing and carrier screening. In LS,
nearly 90% of the identified genetic variants that are not included
as nonsense or indel variants are deemed 'variants of uncertain
significance' (47,48). To clarify the pathogenetic
significance of such variants, it would have been useful to perform
functional assays on proteins; recently, a massive parallel screen
in human cells has been proposed to identify loss of function
missense VUS in the MSH2 gene (49).
In the present study, 86 variants were identified
that were already classified in international databases
(Insight-group and/or ClinVar) as likely benign or of uncertain
pathogenic significance, and four novel variants that showed a MAF
that was very low or not reported. According to suggestions
reported in the ACMG guidelines for the classification of variants
(40), an interpretation of
these variants was performed using multiple bioinformatics
analyses, investigating seven different types of software for the
pathogenic prediction of each variant. When the results of the
in silico analyses were all in agreement as to the
pathogenicity of the variants, it was examined whether these
variants also respected the other ACMG criteria (population,
computational, functional and segregation data) for establishment
of the pathogenicity. Furthermore, the MSI/dMMR status (where this
had been determined), found on the tumor tissues of the patients
carrying these variants, was considered as a fundamental part for
the interpretation of the pathogenicity. To the best of our
knowledge, the MSI status in the majority of hereditary CRCs is
associated to pathogenic variants in MMR genes. Thus, for the rare
uncertain significance variants identified in the MMR genes that by
bioinformatics analyses were resulted as pathogenic variants, it
was hypothesized that the likely lack of function of corresponding
protein at the somatic level, could be confirmed by the MSI/dMMR
status showed on tumoral tissue of patients carrying these variants
(Table VI). Therefore, our
classification has arisen by the combination of the molecular and
clinical data of each patient, in particular data from segregation
and MSI analyses, applying the criteria provided from the
guidelines for the classification of variants established by the
ACMG (40). In this manner, it
was possible to classify eight of these 86 variants as 'likely
pathogenic' variants (Table
VI). Thus, applying the ACMG criteria (40) our variant interpretation differs
from classifications reported in public databases, such as ClinVar
which reports these variants in majority as uncertain significance.
Surely, further studies are needed to establish the real pathogenic
role of these variants; however, at present it can be hypothesized
that these variants could be the cause of disease in eight patients
of our cohort. Thus, in light of the results obtained in the
present study, the importance of establishing for the variants
identified in MMR genes, a correlation with a deficient MMR system
at the tumor level, is suggested, thus strengthening the evaluation
of pathogenicity (Table VI).
The interpretation of the VUS represents a crucial step in clinical
decision-making, improving risk assessment, and promoting
appropriate medical management, including variant-specific cascade
testing for relatives. Therefore, an accurate assessment of the
predictions of the clinical significance of the VUS is needed. The
rule-based classification of the ACMG as it was performed in the
present study, can represent a valid alternative to functional
studies of VUS, which remain the most reliable tool to support the
pathogenicity or benignity of the variant studied.
Furthermore, it is interesting to note that two of
the eight likely pathogenic variants were found in the MLH3 gene
(Table VI), the c.470T>C p.
(Val157Ala) and c.3440A> T p. (Asn1147Ile) variants. The first
is a novel variant that was identified in a woman (ID 18.17) with
adenocarcinoma in situ of the colon with an MSI-H phenotype
and with breast cancer. This patient was also negative for
pathogenetic variants in BRCA1 and 2, MutYH and APC. The second
variant was identified in a woman (ID 17.14) who developed
peritoneal adenocarcinoma with MSI-H at age 52. Both women were
selected for the present study since they met the AC. Both these
patients did not exhibit MLH1 hypermethylation, and were also
carriers of other two sequence variants in the MLH3 gene, the
c.2476A>G and c.2531C>T, already described as benign in the
ClinVar database. Finally, in these patients no other significant
variants were identified in the genes included in the panel. The
MLH3 gene is not routinely analyzed in patients with a clinical
suspicion of LS. However, a previous study on MLH3-knockout mice
highlighted the early onset of tumors in the abdominal sphere
(50). Moreover, previous
studies revealed a possible involvement of the MLH3 gene in LS
(17,51). Loukola et al (51) and Wu et al (17) reported data on missense mutations
and intronic substitutions in families meeting the AC, but without
germline mutations in the MLH1 and MSH2 genes. Nonetheless, this
gene is currently considered to be of low risk for a predisposition
to the development of tumors on the LS spectrum. Unfortunately,
functional assays were unable to be performed for either variant to
clarify the pathogenetic effects, and no family segregation studies
could be carried out due to a lack of interest from these patients.
However, it is important to point out that both patients showed an
MSI-H phenotype at the tumor level.
The results herein revealed that the use of the
custom panel allowed the identification of variants in genes not
routinely analyzed for cases with a clinical suspicion of LS,
mainly variants in the MLH3 gene, but also rare variants identified
in genes such as CHEK2, ATM, MSH3 and NF1. Although these results
do not offer any evidence for a disease-causing role, they
indicated the importance of deepening the study of all rare
variants, to define their pathogenicity and to clarify the
involvement of non-canonical genes in the pathogenesis of LS. The
assessment of rare uncertain variants in genetic counseling could
improve the risk estimate in those families that remain without a
clear molecular diagnosis to provide precision medicine for this
pathogenic condition (52).
Availability of data and materials
The data generated and/or analyzed during the
current study are available on site Mendeley (Elsevier; https://data.mendeley.com/datasets/z9gkb9vd9j/draft?a=5466cd6c-f760-4e6d-ba6d-3033b599b6bd.
Authors' contributions
FD conceptualized the study. FD, RL, ML and AN were
involved in the literature search, in the data interpretation and
critical reviewing of the manuscript. FD, AN and ML were involved
in the preparation of the draft of the manuscript. FD, MDR and PI
were involved in critically revising and editing the manuscript for
important intellectual content. FD, RL, ML and AN confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved (approval no. 120/10)
by the local ethics committee, 'Carlo Romano' Ethics Committee for
Biomedical Activities of the University of Naples, Federico II
(Napoli, Italy). Written informed consent was obtained by all
patients who participated in the present study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by 'Funding ATENEODuraturo
2018', Federico II Naples, Italy.
References
|
1
|
Jasperson KW, Tuohy TM, Neklason DW and
Burt RW: Hereditary and familial colon cancer. Gastroenterology.
138:2044–2058. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kohlmann W: Lynch syndrome and breast
cancer risk: Weighing the data. JCO Precis Oncol. Feb 26–2020.Epub
ahead of print. View Article : Google Scholar
|
|
3
|
Cerretelli G, Ager A, Arends MJ and
Frayling IM: Molecular pathology of lynch syndrome. J Pathol.
250:518–531. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dodaro C, Grifasi C, Florio J, Santangelo
ML, Duraturo F, De Rosa M, Izzo P and Renda A: The role of mutation
analysis of the APC gene in the management of FAP patients. A
controversial issue. Ann Ital Chir. 87:321–325. 2016.PubMed/NCBI
|
|
5
|
Duraturo F, Liccardo R, De Rosa M and Izzo
P: Genetics, diagnosis and treatment of lynch syndrome: Old lessons
and current challenges. Oncol Lett. 17:3048–3054. 2019.
|
|
6
|
Duraturo F, Liccardo R, Cavallo A, De Rosa
M, Rossi GB and Izzo P: Multivariate analysis as a method for
evaluating the pathogenicity of novel genetic MLH1 variants in
patients with colorectal cancer and microsatellite instability. Int
J Mol Med. 36:511–517. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liccardo R, De Rosa M, Rossi GB,
Carlomagno N, Izzo P and Duraturo F: Incomplete segregation of MSH6
frameshift variants with phenotype of lynch syndrome. Int J Mol
Sci. 18:9992017. View Article : Google Scholar :
|
|
8
|
Liccardo R, De Rosa M, Izzo P and Duraturo
F: Novel MSH2 splice-site mutation in a young patient with lynch
syndrome. Mol Med Rep. 17:6942–6946. 2018.
|
|
9
|
Liccardo R, Della Ragione C, Mitilini N,
De Rosa M, Izzo P and Duraturo F: Novel variants of unknown
significance in the PMS2 gene identified in patients with
hereditary colon cancer. Cancer. Manag Res. 18:6719–6725. 2019.
|
|
10
|
Liccardo R, De Rosa M, Izzo P and Duraturo
F: Novel implications in molecular diagnosis of lynch syndrome.
Gastroenterol Res Pract. 2017:25950982017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Giardiello FM, Allen JI, Axilbund JE,
Boland CR, Burke CA, Burt RW, Church JM, Dominitz JA, Johnson DA,
Kaltenbach T, et al: Guidelines on genetic evaluation and
management of lynch syndrome: A consensus statement by the US
multi-society task force on colorectal cancer. Gastroenterology.
147:502–526. 2014. View Article : Google Scholar
|
|
12
|
Duraturo F, Cavallo A, Liccardo R, Cudia
B, De Rosa M, Diana G and Izzo P: Contribution of large genomic
rearrangements in italian lynch syndrome patients: Characterization
of a novel Alu-mediated deletion. Biomed Res Int. 2013:2198972013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tognetto A, Pastorino R, Castorina S,
Condorelli DF, DeCensi A, De Vito C, Magnano A, Scaldaferri F,
Villari P, Genuardi M and Boccia S: The current practice of lynch
syndrome diagnosis and management in Italy: A qualitative
assessment. Public Health Genomics. 22:189–207. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lynch PM: The hMSH2 and hMLH1 genes in
hereditary nonpolyposis colorectal cancer. Surg Oncol Clin N Am.
18:611–624. 2009. View Article : Google Scholar
|
|
15
|
Hendriks YM, Wagner A, Morreau H, Menko F,
Stormorken A, Quehenberger F, Sandkuijl L, Møller P, Genuardi M,
Van Houwelingen H, et al: Cancer risk in hereditary nonpolyposis
colorectal cancer due to MSH6 mutations: Impact on counseling and
surveillance. Gastroenterology. 127:17–25. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Senter L, Clendenning M, Sotamaa K, Hampel
H, Green J, Potter JD, Lindblom A, Lagerstedt K, Thibodeau SN,
Lindor NM, et al: The clinical phenotype of lynch syndrome due to
germ-line PMS2 mutations. Gastroenterology. 135:419–428. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu Y, Berends MJ, Sijmons RH, Mensink RG,
Verlind E, Kooi KA, van der Sluis T, Kempinga C, van dDer Zee AG,
Hollema H, et al: A role for MLH3 in hereditary nonpolyposis
colorectal cancer. Nat Genet. 29:137–138. 2001. View Article : Google Scholar
|
|
18
|
Moreira L, Balaguer F, Lindor N, de la
Chapelle A, Hampel H, Aaltonen LA, Hopper JL, Le Marchand L,
Gallinger S, Newcomb PA, et al: Identification of lynch syndrome
among patients with colorectal cancer. JAMA. 17:1555–1565. 2012.
View Article : Google Scholar
|
|
19
|
Gupta S, Provenzale D, Llor X, Halverson
AL, Grady W, Chung DC, Haraldsdottir S, Markowitz AJ, Slavin TP Jr,
Hampel H, et al: NCCN guidelines insights: Genetic/familial
high-risk assessment: Colorectal, version 2. 2019.J Natl Compr Canc
Netw. 17:1032–1041. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vasen HF, Mecklin JP, Khan PM and Lynch
HT: The International collaborative group on hereditary
non-polyposis colorectal cancer (ICG-HNPCC). Dis Colon Rectum.
34:424–425. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vasen HF, Watson P, Mecklin JP and Lynch
HT: New clinical criteria for hereditary nonpolyposis colorectal
cancer (HNPCC, Lynch syndrome) Proposed By the International
Collaborative Group on HNPCC. Gastroenterology. 116:1453–1456.
1999. View Article : Google Scholar
|
|
22
|
Gonzaga-Jauregui C, Lupski JR and Gibbs
RA: Human genome sequencing in health and disease. Annu Rev Med.
63:35–61. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Turano M, Costabile V, Cerasuolo A,
Duraturo F, Liccardo R, Delrio P, Pace U, Rega D, Dodaro CA, Milone
M, et al: Characterisation of mesenchymal colon tumour-derived
cells in tumourspheres as a model for colorectal cancer
progression. Int J Oncol. 53:2379–2396. 2018.
|
|
24
|
Fecteau H, Vogel KJ, Hanson K and
Morrill-Cornelius S: The evolution of cancer risk assessment in the
era of next generation sequencing. J Genet Couns. 23:633–639. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mardis ER: A decade's perspective on DNA
sequencing technology. Nature. 470:198–203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin EI, Tseng LH, Gocke CD, Reil S, Le DT,
Azad NS and Eshleman JR: Mutational profiling of colorectal cancers
with microsatellite instability. Oncotarget. 6:42334–42344. 2015.
View Article : Google Scholar
|
|
27
|
Kim JC, Choi JS, Roh SA, Cho DH, Kim TW
and Kim YS: Promoter methylation of specific genes is associated
with the phenotype and progression of colorectal adenocarcinomas.
Ann Surg Oncol. 17:1767–1776. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu C, Wang QS and Wang YJ: The CHEK2
I157T variant and colorectal cancer susceptibility: A systematic
review and meta-analysis. Asian Pac J Cancer Prev. 13:2051–2055.
2012. View Article : Google Scholar
|
|
29
|
Han P, Liu G, Lu X, Cao M, Yan Y, Zou J,
Li X and Wang G: CDH1 rs9929218 Variant at 16q22.1 contributes to
colorectal cancer susceptibility. Oncotarget. 26:47278–47286. 2016.
View Article : Google Scholar
|
|
30
|
Shenoy S: CDH1 (E-Cadherin) mutation and
gastric cancer: Genetics, molecular mechanisms and guidelines for
management. Cancer Manag Res. 13:10477–10486. 2019. View Article : Google Scholar
|
|
31
|
Elzagheid A, Buhmeida A, Laato M,
El-Faitori O, Syrjänen K, Collan Y and Pyrhönen S: Loss of
E-cadherin expression predicts disease recurrence and shorter
survival in colorectal carcinoma. APMIS. 120:539–548. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Berndt SI, Platz EA, Fallin MD, Thuita LW,
Hoffman SC and Helzlsouer KJ: Mismatch repair polymorphisms and the
risk of colorectal cancer. Int J Cancer. 120:1548–1554. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Orimo H, Nakajima E, Yamamoto M, Ikejima
M, Emi M and Shimada T: Association between single nucleotide
polymorphisms in the hMSH3 gene and sporadic colon cancer with
microsatellite instability. J Hum Genet. 45:228–230. 2000.
View Article : Google Scholar
|
|
34
|
Jafary F, Salehi M, Sedghi M, Nouri N,
Jafary F, Sadeghi F, Motamedi S and Talebi M: Association between
mismatch repair gene MSH3 Codons 1036 and 222 polymorphisms and
sporadic prostate cancer in the Iranian population. Asian Pac J
Cancer Prev. 13:6055–6057. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Duraturo F, Liccardo R, Cavallo A, De Rosa
M, Grosso M and Izzo P: Association of low-risk MSH3 and MSH2
variant alleles with lynch syndrome: Probability of synergistic
effects. Int J Cancer. 129:1643–1650. 2011. View Article : Google Scholar
|
|
36
|
Adam R, Spier I, Zhao B, Kloth M, Marquez
J, Hinrichsen I, Kirfel J, Tafazzoli A, Horpaopan S, Uhlhaas S, et
al: Exome sequencing identifies biallelic MSH3 germline mutations
as a recessive subtype of colorectal adenomatous polyposis. Am J
Hum Genet. 99:337–351. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Olkinuora A, Nieminen TT, Mårtensson E,
Rohlin A, Ristimäki A, Koskenvuo L, Lepistö A; Swedish Extended
Genetic Analysis of Colorectal Neoplasia (SWEN) Study Group;
Gebre-Medhin S, Nordling M and Peltomäki P: Biallelic germline
nonsense variant of MLH3 underlies polyposis predisposition. Genet
Med. 21:1868–1873. 2019. View Article : Google Scholar
|
|
38
|
Duraturo F, Liccardo R and Izzo P:
Coexistence of MLH3 germline variants in colon cancer patients
belonging to families with lynch syndrome-associated brain tumors.
J Neurooncol. 129:577–578. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Titze S, Peters H, Währisch S, Harder T,
Guse K, Buske A, Tinschert S and Harder A: Differential MSH2
promoter methylation in blood cells of Neurofibromatosis type 1
(NF1) patients. Eur J Hum Genet. 18:81–87. 2010. View Article : Google Scholar :
|
|
40
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar :
|
|
41
|
Desmet FO, Hamroun D, Lalande M,
Collod-Béroud G, Claustres M and Béroud C: Human splicing finder:
An online bio-informatics tool to predict splicing signals. Nucleic
Acids Res. 37:e672009. View Article : Google Scholar
|
|
42
|
Ramensky V, Bork P and Sunyaev S: Human
non-synonymous SNPs: Server and survey. Nucleic Acids Res.
30:3894–3900. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rost B, Yachdav G and Liu J: The
predictprotein server. Nucleic Acids Res. 32:W321–W326. 2004.
View Article : Google Scholar :
|
|
44
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: Mutationtaster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar
|
|
45
|
Choi Y, Sims GE, Murphy S, Miller JR and
Chan AP: Predicting the functional effect of amino acid
substitutions and indels. PLoS One. 7:e466882012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhunussova G, Afonin G, Abdikerim S,
Jumanov A, Perfilyeva A, Kaidarova D and Djansugurova L: Mutation
spectrum of cancer-associated genes in patients with early onset of
colorectal cancer. Front Oncol. 9:6732019. View Article : Google Scholar :
|
|
47
|
Liccardo R, Nolano A, Lambiase M, Della
Ragione C, De Rosa M, Izzo P and Duraturo F: MSH2 overexpression
due to an unclassified variant in 3-Untranslated region in a
patient with colon cancer. Biomedicines. 8:1672020. View Article : Google Scholar
|
|
48
|
Arora S, Huwe PJ, Sikder R, Shah M, Browne
AJ, Lesh R, Nicolas E, Deshpande S, Hall MJ, Dunbrack RL Jr and
Golemis EA: Functional analysis of rare variants in mismatch repair
proteins augments results from computation-based predictive
methods. Cancer Biol Ther. 18:519–533. 2017. View Article : Google Scholar :
|
|
49
|
Jia X, Burugula BB, Chen V, Lemons RM,
Jayakody S, Maksutova M and Kitzman JO: Massively parallel
functional testing of MSH2 missense variants conferring lynch
syndrome risk. Am J Hum Genet. 108:163–175. 2021. View Article : Google Scholar
|
|
50
|
Hegan DC, Narayanan L, Jirik FR, Edelmann
W, Liskay RM and Glazer PM: Differing patterns of genetic
instability in mice deficient in the mismatch repair genes Pms2,
Mlh1, Msh2, Msh3 and Msh6. Carcinogenesis. 27:2402–5408. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Loukola A, Vilkki S, Singh J, Launonen V
and Aaltonen LA: Germline and somatic mutation analysis of MLH3 in
MSI-positive colorectal cancer. Am J Pathol. 157:347–352. 2000.
View Article : Google Scholar
|
|
52
|
Turano M, Delrio P, Rega D, Cammarota F,
Polverino A, Duraturo F, Izzo P and De Rosa M: Promising colorectal
cancer biomarkers for precision prevention and therapy. Cancers
(Basel). 11:19322019. View Article : Google Scholar
|