Every eukaryotic cell contends with various
intracellular and extracellular threats during DNA replication and
cellular metabolism, such as high-energy radiation, mutagenic
chemicals, free radicals and V(D)J recombination, as well as cell
type-specific challenges, such as immunoglobulin class-switching
recombination (CSR) in B-lymphocytes (1,2).
Failure to repair a DNA double-strand break (DSB) or restart
replication forks results in cell death, whereas DSB mis-repair and
catastrophic genome rearrangements are the major causes of genomic
instability and hence, carcinogenesis (3,4).
Thus, the fidelity and capacity of DSB repair needs to be clearly
elucidated. To date, four conserved and mechanistically distinct
pathways have been identified to be involved in the elimination of
DSBs from the genome: Homologous recombination (HR), non-homologous
end joining (NHEJ), alternative end joining (altEJ) and
single-strand annealing (SSA) (5). HR and NHEJ are the two major
DNA-repair pathways.
HR is the most accurate DSB repair mechanism and
also the default mechanism for replication fork repairs. HR occurs
following DSB end resection, which removes a few hundred or more
bases from the 5′-terminated strand to yield a 3′ single-stranded
DNA (ssDNA) tail, and this is achieved via the MRE11-RAD50-NBS1
(MRN) complex (6). The ssDNA
invades the template (the adjacent sister chromatid of 3′
overhangs) and this is mediated by the recombinase Rad51,
whereafter it displaces an intact strand to form a D-loop and
produces double Holliday junctions (7). However, since the HR machinery
requires an identical DNA template in the homologous sister
chromatid for DSB repair, it is most active in the mid-S phase and
mid-G2 phase of the cell cycle (8). altEJ was the second method to be
identified, and this is mediated by the microhomology of the 3′
ssDNA originating from end resection. In altEJ, DNA polymerase θ
(Pol θ)-associated helicase activity can displace the ssDNA-binding
protein, while its polymerase activity can stabilize the joint
between the two DNA ends (9).
Due to its apparent proclivity for connecting DSBs on different
chromosomes, the usage of altEJ for DSB repair has negative
ramifications for genomic integrity, resulting in chromosomal
translocations and mutagenic rearrangements (10). Third to be discovered was SSA,
which is considered to be an obligatorily error-prone pathway. At
the cost of deletion of the intervening sequences between the
repeats, SSA joins two homologous 3′ ssDNA ends (for example, at
tandem repeats) through annealing (11). Notably, both altEJ and SSA
require DNA end resection, and they are also primarily operational
in the S and G2 phases of the cell cycle (12). The error-prone DSB repair
pathways of alt-EJ and SSA operate in different biological contexts
and contribute to genome rearrangements and oncogenic
transformation, but do not serve as main DNA-repair pathways.
Alt-NHEJ and SSA are two additional DSB repair mechanisms that
primarily serve as backups when c-NHEJ and HR fail (13). In comparison, NHEJ is a
relatively simple repair process and remains active throughout the
entirety of the cell cycle, but is dominant in G0/G1 and G2 phases
of the cell cycle (14). NHEJ
takes place substantially at a more rapid rate than HR (several
hours), lasting ~30 min and accounting for >75% of repair
events, while HR repairs the remaining 25%, according to
fluorescent reporter structures integrated into the chromosomes of
human cell lines (15). NHEJ
repair involves the binding of the ring-shaped Ku70/80 heterodimer
to DSB ends and the recruitment of the DNA-dependent protein kinase
catalytic subunit (DNA-PKcs) to create the DNA-PK complex. DSBs are
then ligated by a complex involving DNA ligase IV and its
associated factors [e.g., X-ray repair cross complementing protein
4 (XRCC4) and XRCC4-like factor (XLF)] (16,17). Although NHEJ remains active
throughout the cell cycle, NHEJ can be inhibited by breast cancer
type 1 susceptibility protein (BRCA1) and other HR-related
molecules if DSBs contain 5′ or 3′ overhangs (18). As opposed to HR, altEJ and SSA,
which require a 3′ ssDNA tail, NHEJ acts first to attempt to repair
DSBs and is the only DSB repair pathway active in the G0 and G1
phases (14). Even within the G2
phase, NHEJ also repairs ≥80% of ionizing radiation-induced DSBs
(19,20). In general, when the DSB ends are
'clean' (have compatible or blunt ends), NHEJ is rapid, efficient,
yet mutagenic and is often accompanied by only short deletions and
fewer base changes. As the predominant DSB repair pathway in
mammalian cells, NHEJ deficiency can influence tumor sensitivity to
ionizing radiation and antineoplastic, and it can also cause
immunodeficiencies and other developmental abnormalities, including
dwarfism and defective neurogenesis associated with microcephaly
(21).
Two key players in the DSB repair process are tumor
protein 53-binding protein 1 (53BP1) that promotes NHEJ by
antagonizing DNA end overhang resection and BRCA1 that promotes HR
by promoting end-resection (22). In response to DSBs, 53BP1 rapidly
accumulates on the chromatin surrounding the DNA damage site to
form the irradiation-induced foci (IRIF), which is driven by a
signaling cascade that originates with the ataxia-telangiectasia
mutated (ATM) kinase-mediated phosphorylation of H2A histone family
member X (H2AX; known as γH2AX) (23,24). Similar to ATM deficiency
(ATM−/−), defective DNA damage responses (DDRs)
following treatment with ionizing radiation occur in
53BP1−/− cells, and 53BP1−/− mice exhibit
growth retardation, immune deficiency, increased radiation
sensitivity and an increased risk of developing cancers (25). For several decades, 53BP1 has
been described as a regulator and scaffold for DSB signaling, which
functions by recruiting other responsive proteins to DNA damage
sites to facilitate the NHEJ repair process. Therefore, the
identification of 53BP1 binding and the proteins it interacts with
has become an increasingly studied topic in an attempt to uncover
the biological functions of 53BP1-dependent NHEJ repair.
In the present review article, the structure,
functional characteristics and post-transcriptional modifications
(PTMs) of 53BP1 in the process of response to DSBs are discussed.
Progress on the identification of 53BP1 assembly and recruitment to
DSB sites, with a particular focus on the interactions of 53BP1 and
the reshaping of the chromatin architecture around DSB sites is
reviewed. The role of upstream factors in regulating 53BP1
recruitment, and the mechanisms through which 53BP1 interacts with
the downstream responsive effectors involved in the NHEJ signaling
pathways is also discussed. The present review also sheds light on
the challenges that remain to be overcome and the potential roles
of 53BP1 in cancer treatment and CRISPR/Cas9-induced HR repair,
providing a theoretical basis for the further study of 53BP1.
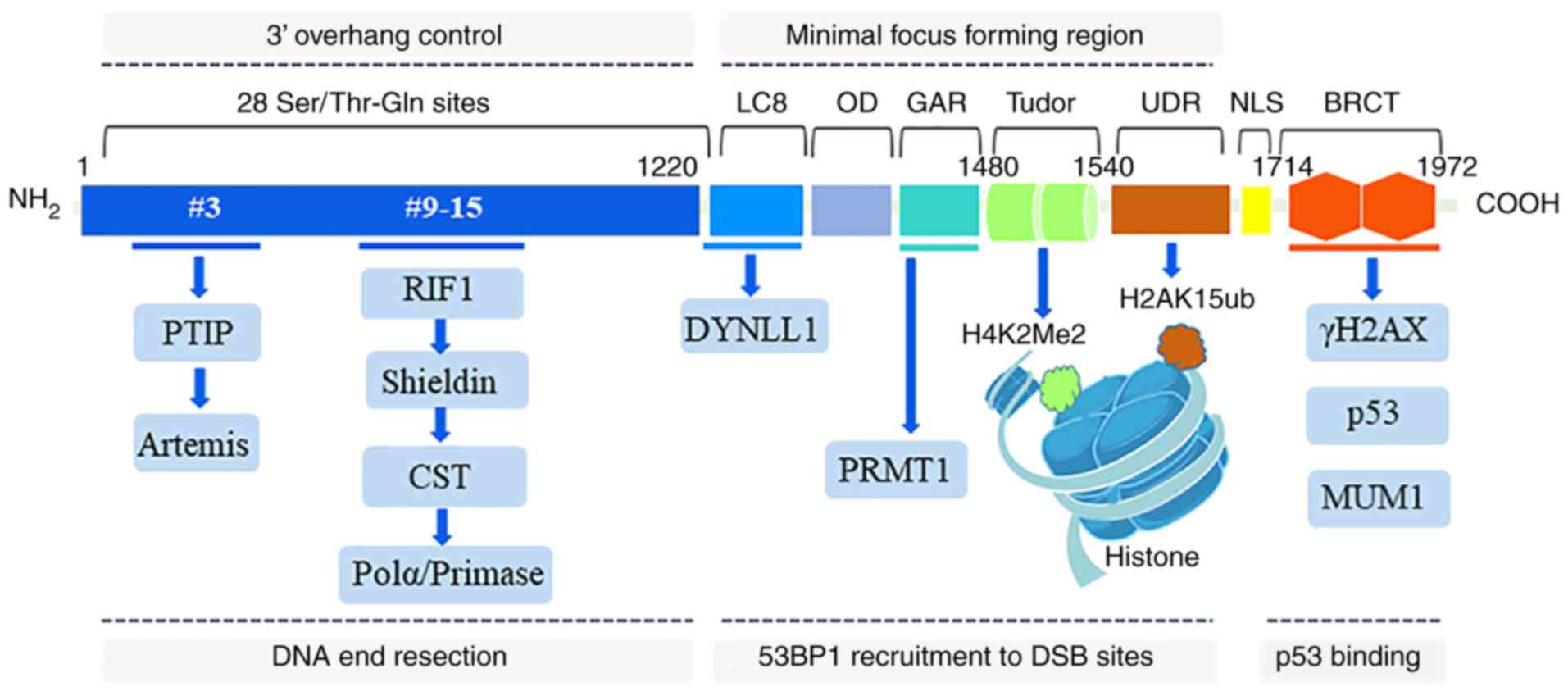
Human 53BP1 has 1,972 amino acids, a mass of ≥200
kDa, and is encoded by the TP53BP1 gene that is located on human
chromosome l5q15-12 (26,27).
As a large scaffolding protein that mediates the interactions with
modified histones and several effector proteins, 53BP1 consists of
multiple interaction surfaces for the DSB-response. Pivotal
structural regions of 53BP1 include the N-terminal region (1-1,220
aa), minimal focus forming region (1,220-1,711 aa) and the
C-terminal region (1,712-1,972 aa) (28). The 53BP1 N-terminal region
contains 28 amino-terminal Ser/Thr-Gln sites that are involved in
the interactions with the Pax transactivation domain-interacting
protein (PTIP) and RAP1-interacting factor 1 (RIF1) (29). The ability of 53BP1 to form IRIF
is attributable to its minimal focus forming region. This region
includes two dynein light chain (LC8) binding domains that bind to
dynein light chain 1 to promote 53BP1 oligomerization and
recruitment (30-32), an oligomerization domai that
mediates 53BP1 dimer and multimer formation and recruitment to a
DSB (33,34), a glycine-arginine-rich (GAR)
motif that is methylated by the protein arginine
N-methyltransferase 1 (PRMT1) to enhance DNA-binding function
(34,35), two tandem Tudor domains that bind
to H4K20me2 (36,37) and a ubiquitylation-dependent
recruitment (UDR) motif that interacts with H2AK15ub (38). The 53BP1 C-terminal region
contains two BRCA1 carboxyl-terminal (BRCT) domains that interact
with p53 and γH2AX, which is important for DSB repair in
heterochromatin (39,40). Overall, all interaction domains
of 53BP1 are indispensable for DSB repair in heterochromatin;
however, the contribution of these domains varies when the context
of DSB repair is altered (Fig.
1).
Canonical DNA (c-)NHEJ is the major DSB repair
pathway in mammalian cells due to its ability to function in all
phases of the cell cycle. c-NHEJ is a rapid kinetics-based repair
process involving the binding of the Ku heterodimer (Ku70/Ku80) to
dsDNA ends, the recruitment of the DNA-PKcs to create the DNA-PK
complex, and the DSB end ligation by XRCC4, XLF and DNA ligase IV
(LIG4) (41,42). Concomitant with DNA-PK binding to
DSB sites, the MRN (MRE11, RAD50 and NBS1) complex is also located
in the same region, and recruits ATM, which phosphorylates it
(43). ATM amplifies the damage
signal continuously via phosphorylation of the histone H2A variant
(H2AX; the Ser139 phosphorylated state is termed γH2AX) (44). γH2AX is located at DSB sites and
recruits the mediator of DNA damage checkpoint protein 1 (MDC1)
through a protein interaction network, and then E3 ubiquitin ligase
ring finger protein (RNF)8 and RNF168 are recruited by MDC1
(45,46). RNF8 and RNF168 cooperate with E2
ubiquitin-conjugating enzyme to ubiquitinate chromatin around DSB
sites. The histone H2A, serving as a key substrate of RNF168, is
ubiquitinated at Lys13 and Lys15 (H2AK13ub/15ub) (47). RNF8/RNF168-dependent
ubiquitination can produce a specific region on chromatin to allow
ubiquitin-dependent DSB-responding proteins (such as 53BP1) to
gather and generate IRIF (48,49). 53BP1 binds to residues of
H2AK15ub and H4K20me2 to form 53BP1 foci via its UDR motif and
Tudor domain, respectively (50). Although methylation transferase
may not be the primary driving force for the selective recruitment
of 53BP1, the space-time exposure of ubiquitin-regulated H4K20me2
modification is a vital factor mediating the accurate position of
53BP1 (51). The lethal 3
malignant brain tumor-like protein 1 (L3MBTL1) and Jumonji
domain-containing protein 2A (JMJD2A or KDM4A) competitively bind
to H4K20me2; thus this molecular marker is 'buried' under
physiological conditions. Following the occurrence of a DSB,
RNF8/NF168-mediated ubiquitination modification can rapidly degrade
these competitive proteins and promote the stable binding of the
53BP1 Tudor domain with H4K20Me2 (52,53). Additionally, point mutations of
the UDR motif (I1617A, L1619A, N1621A, L1622A and R1627A) hinder
53BP1 recruitment by inhibiting the binding of 53BP1 to H2AK15ub;
however, it does not affect the binding of 53BP1 to H4K20me2
(38). This suggests that
RNF168-mediated H4K20me2 competitive protein degradation and H2A
ubiquitin modification are mutually independent for 53BP1
recruitment. In a phosphorylation-independent pathway, 53BP1 serves
as a scaffold protein inducing mutated melanoma-associated antigen
1 (MUM1 or EXPAND1) to anchor at DSB sites through its BRCT domains
(54). Disrupting the nuclear
localization of MUM1 leads to a decrease in DNA damage repair
efficiency. As the primary downstream molecules, RIF1 and PTIP
interact with 53BP1 N-terminal Ser/Thr-Gln sites in an
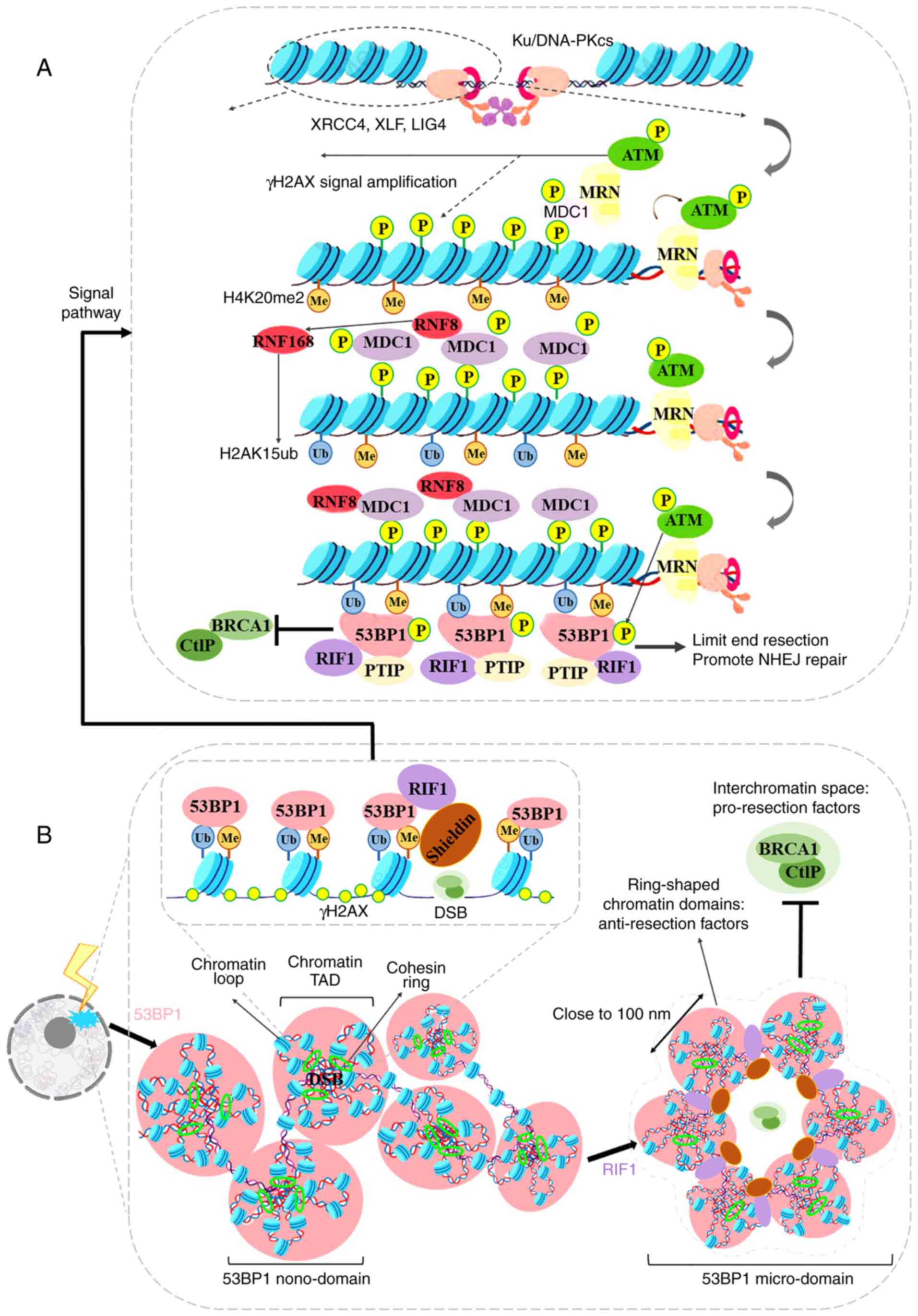
ATM-dependent phosphorylated manner (55,56) (Fig. 2A).
The tridimensional organization of chromatin in the
nuclear space controls 53BP1 foci accumulation, and the formation
of 53BP1 foci may in turn affect chromatin organization in the
vicinity of DSBs (Fig. 2B). Xie
et al (57) found that
following DNA damage induced by camptothecin, microrchidia family
CW-type zinc finger protein 2 (MORC2), an ATPase-dependent
chromatin remodeling enzyme, can form a homodimer through its
C-terminal coiled-coil (CC) domain. The homodimer is required for
nucleosome destabilization after DNA damage by promoting the
recruitment of the DNA repair proteins, BRCA1, 53BP1 and Rad51, to
sites of DNA damage. This suggests that the decondensation of the
highly compacted chromatin architecture is essential for efficient
DNA repair. Using single molecule localization microscopy (STORM),
Wu et al (58) observed
that the nuclear chromatin was relaxed from a 200-400 nm thick
irregular frame and remodeled to a disperse sub-100 nm structure
following X-ray irradiation. The relaxed nuclear chromatin is a
more feasible portion for the recruitment of DSB repair factors
(γH2AX, MDC1 and 53BP1) that were distributed as
microscale-colocalized and nanoscale interlaced substructures
(58). Notably, Ochs et
al (59), using 3D-SIM and
2D-stimulated emission depletion super-resolution microscopy
techniques, demonstrated that the 53BP1 and RIF1 proteins can form
an autonomous functional module, which can stabilize the chromatin
topological structure of DNA fragmentation sites. When DNA damage
occurs, 4-7 53BP1 sub-domains form ring structures (with a uniform
spherical body) in the DNA fragments. The diameter of the 53BP1
sub-domain is ~100 nm, and the center distance of the two 53BP1
sub-domains is close to 140 nm, which facilitates the reciprocal
association between the chromatin topology and the formation of
53BP1 foci in response to DSB. Further research (59) demonstrated that the chromatin
recruitment of 53BP1 foci occurs over megabases around the DSB and
corresponds to chromatin topologically associated domains (TADs).
Subsequently, RIF1 and the Cohesin complex [Shieldin/CTC1-STN1-TEN1
(CST)/polymerase-α (Polα)] are recruited to the boundary of the TAD
structure, and the alternating distribution of 53BP1 and RIF1
stabilizes several adjacent TAD structures into an ordered ring
arrangement. This ring-like structure can limit the recruitment of
BRCA1 and prevent excessive cleavage of DNA breaks. Recently,
Arnould et al (60)
verified the hypothesis that chromatin high-dimensional structure
regulates DSB repair, and proposed that 'loop extrusion' may be the
mechanism through which the DNA repair center is formed. Following
the occurrence of a DSB, ATM and the Cohesin complex mediate
roadblock for unilateral loop extrusion, in which ATM
phosphorylates H2AX constitutively. Divergent one-sided loop
extrusion and the bidirectional spreading of phosphorylated H2AX
induce the assembly of the full DDR reaction focus. Notably,
although RIF1 organizes 53BP1 foci and accumulates at the
boundaries between 53BP1 nano-domains, RIF1 does not colocalize
with these domains (60,61).
The generation of 53BP1 foci surrounding DNA lesions
is required to recruit downstream effectors. The time frame and
mechanisms through which the spatial and temporal confinement of
protein assemblies at DNA damaged sites is achieved requires
further investigation. 53BP1 dimers, a dimerization mediated by the
53BP1 oligomerization domain, relocate from the nucleoplasm to
sites of DSBs (33). At these
sites, the consecutive recognition of H2AK15ub and dH4K20me leads
to the assembly of 53BP1 oligomers and promotes the formation of
mature 53BP1 foci structures (62). Using state-of-the-art microscopy,
Kilic et al (63)
observed that the 53BP1 foci exhibit the hallmarks of
phase-separated compartments and exhibit droplet-like behavior.
Phase-separated proteins self-organize into liquid-like droplets,
allowing NHEJ-interrelated molecules to become concentrated, while
excluding NHEJ-irrelevant molecules (64). The droplet-like 53BP1 foci is
highly sensitive to changes in osmotic pressure, temperature, salt
concentration and to the disruption of hydrophobic interactions,
suggesting that the assembly of 53BP1 is reversibly abolished
(63). The liquid-like nature of
53BP1 assemblies verifies previous observed results that
demonstrated that 53BP1 undergoes phase separation and forms a
spatiotemporally spherical shape (65,66). Pessina et al (67) proposed that DNA damage-induced
transcriptional promoters drive molecular crowding off DDR proteins
and RNA synthesis, which stimulates the phase separation of 53BP1
in the shape of foci. Therefore, it is possible that the phase
separation of 53BP1 foci integrates the localized DNA damage
recognition and the assembly of repair. However, the forming speed
of droplet-like 53BP1 foci and the fidelity of DSB repair is
dependent on the complexity of the lesion. 53BP1 has been shown to
be recruited in a few seconds to complex DSB sites using live cell
imaging combined with heavy ion trackers (68). In almost half of the isolated DSB
sites, the recruitment of 53BP1 is delayed ~5 min (68). Following neocarzinostatin
treatment, 53BP1 foci is formed in ~60 min and observed to
co-localize with γH2AX at the sites of DSBs (~80% of 53BP1 foci
contain exactly one DSB) that are accompanied by the higher
chromatin compaction (69).
As aforementioned, 53BP1 is recruited to the DSB
sites to coordinate the chromatin architecture around DSB sites and
to promote NHEJ repair. Therefore, the upstream molecules that
regulate recruitment and functions of 53BP1 in DNA repair deserve
further investigation.
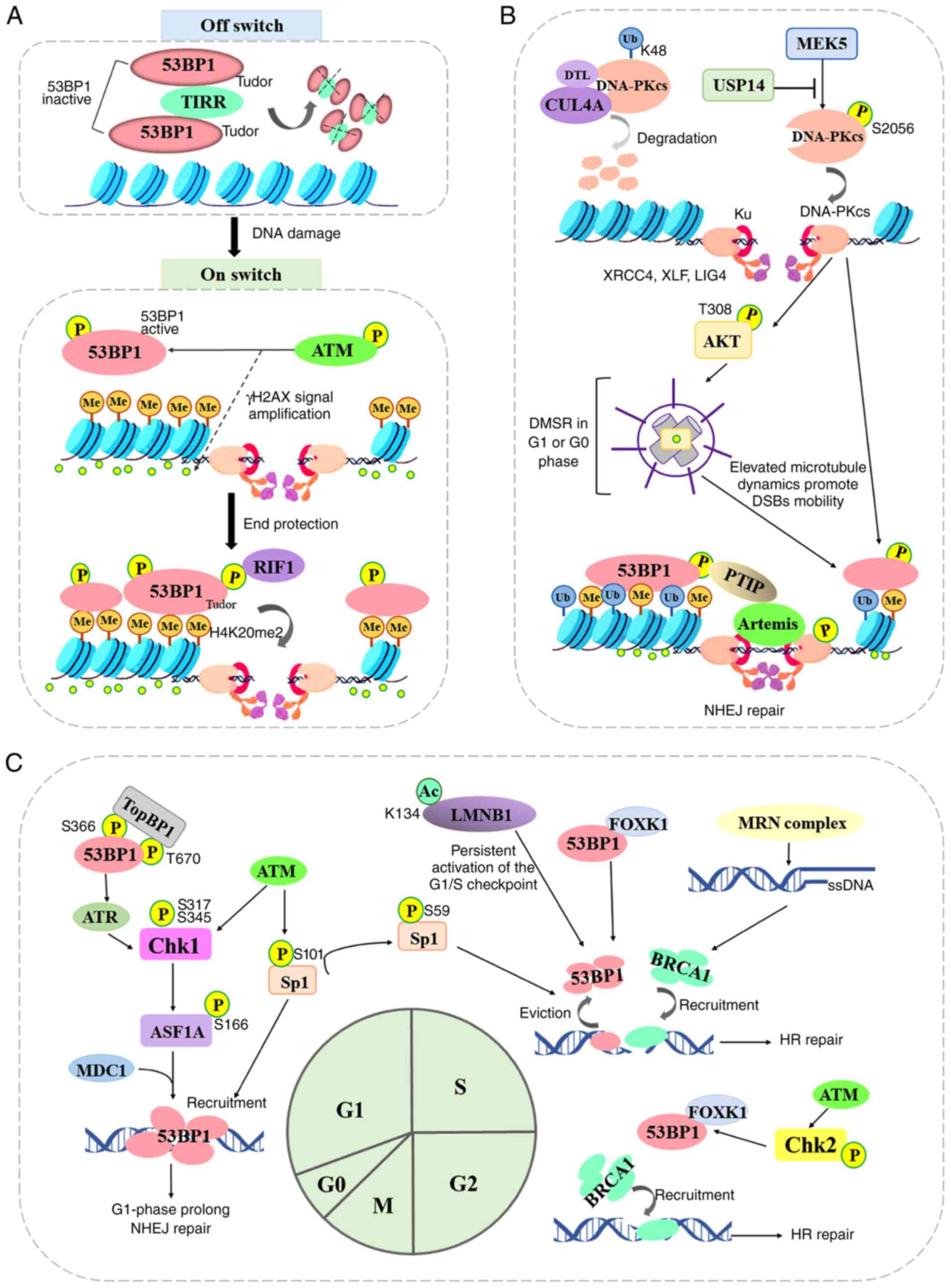
The DNA ends are marked with histones H4K20me2,
which is a specific binding target for the Tudor domain of 53BP1.
TIRR (or NUDT16L1), a member of the family of the nucleoside
diphosphate-linked moiety X (Nudix) hydrolases, was first
identified as an upstream molecule that inhibits this unique
binding in 2017 by Drané et al (70) and Zhang et al (71). Drané et al (70) demonstrated that TIRR directly
binds the tandem Tudor domain of 53BP1 and masks its H4K20me2
binding motif, and TIRR overexpression in the cells with low
expression of BRCA1 abrogated the development of resistance to
poly(ADP-Ribose) polymerase (PARP) inhibitors (PARPis), which may
be related to the loss of 53BP1 function. However, upon DNA damage,
ATM phosphorylates 53BP1 and recruits RIF1, thus inducing the
dissociation of the 53BP1-TIRR complex from chromatin. Thus, the
major function of TIRR is to serve as an off switch in the absence
of DNA damage, maintaining tandem Tudor domain in an inactive state
and keeping 53BP1 away from chromatin (Fig. 3A).
The recently reported crystal structures of TIRR in
complex with 53BP1 Tudor domain, together with supporting binding
assays using ubiquitinated modification and demethylated
modification nucleosomes, reveals that TIRR occludes the
methyl-lysine-binding site of Tudor domain (72-74). Guided by X-ray crystallography,
Botuyan et al (72)
revealed that a TIRR arginine (Arg107) residue could mask the
histone methyllysine-binding surface of 53BP1. They also found that
a mutation of a phenylalanine residue (F1553R) in 53BP1 abolished
the interaction with TIRR, but preserved interaction with H4K20me2,
which indicates that the two binding activities of the 53BP1 Tudor
domain could be functionally separated and independently explored
by mutagenesis. After analyzing the protein structure of 53BP1
Tudor and TIRR, Dai et al (74) revealed that the TIRR
amino-terminal region (residues 10-24) combined with the TIRR
L8-loop could prevent the methylation reader joining surface
(centered around Arg107) in the Tudor domain of 53BP1, which
inhibits 53BP1 recruitment to nucleosomes bearing H4K20me2.
Structural comparisons identified a TIRR histidine (H106 is absent
from the TIRR homolog NUDT16) that is essential for 53BP1 Tudor
binding. Wang et al (73)
demonstrated that three loops (α1-β1 loop, N-terminus loop and
β4-β5 loop) from TIRR interact with the 53BP1 Tudor domain and mask
the methylated lysine-binding pocket in tandem Tudor domain.
Additionally, TIRR inhibited the complex formation between the
Tudor domain of 53BP1 and a dimethylated form of p53 (K382me2),
which inhibited transcriptional activation of the p53 target genes
(75). Overall, these studies
elucidate the mechanisms by which TIRR recognizes the 53BP1 Tudor
domain and functions as a cellular inhibitor of the histone
methyl-lysine readers.
At DSB ends, the assembly of DNA-PK, a nuclear
serine/threonine protein kinase composed of a large catalytic
subunit (DNA-PKcs) and a heterodimeric DNA-targeting subunit Ku,
serves as a platform to recruit Artemis, DNA ligase IV and NHEJ
factors (such as 53BP1 and γH2AX), all of which are involved in
end-processing and ligation (76). Although the autophosphorylation
of DNA-PKcs occurs at numerous Ser/Thr residues throughout the
kinase, and this mediates NHEJ, certain molecules were confirmed to
function as a potential phosphorylase regulator (77,78). Mitogen-activated protein kinase
kinase 5 (MAPKK or MEK5) was found to promote phosphorylation of
the catalytic subunit of DNA-PK at serine 2,056 in response to
ionizing radiation or etoposide treatment by Broustas et al
(77). This revealed a
convergence between MEK5 upstream signaling and DNA repair by NHEJ
in conferring resistance to genotoxic stress in advanced prostate
cancer (77). Conversely, Sharma
and Almasan (78) identified
that ubiquitin-specific protease (USP)14, a proteasomal
deubiquitinase, decreased the IRIF formation of 53BP1 and
pS2056-DNA-PKcs, ultimately inhibiting NHEJ repair, promoting HR
repair, and suppressing the radiosensitization of non-small cell
lung cancer cells. Feng et al (79) demonstrated that the ubiquitin
ligase Cullin 4A binds to the DNA-PKcs protein in the NHEJ repair
pathway for nuclear degradation through its substrate receptor DTL.
CRL4ADTL is recruited to DSB sites and promotes the
ubiquitination of DNA-PKcs at K48 in the nucleus, inhibiting the
NHEJ repair pathway to increase cell genomic instability.
Similarly, as previously demonstrated, when cisplatin resistance
developed, DNA-PKcs activity and the formation of 53BP1 foci was
reduced, which antagonized cisplatin cytotoxicity for germ cell
tumor cells (80). Additionally,
Ma et al (81) found that
the activation of the DNA-PK-AKT cascade facilitated interphase
centrosome maturation and induced DSB-induced microtubule dynamics
stress response (DMSR), thus promoting DSB mobility and
53BP1-dependent NHEJ repair. DMSR occurs in G1 or G0 cells and
lasts around 6 h, providing an aggregated time for 53BP1 and its
partners. Although the mechanism by which DNA-PK promotes 53BP1
recruitment to DSB sites remains unclear, DNA-PK may serve as a
potential upstream regulatory molecule for 53BP1 (Fig. 3B).
The cell cycle phase is a critical determinant of
the choice of repair pathway at DSB sites (Fig. 3C). BRCA1-mediated HR repair is
restricted to the S and G2 phases of the cell cycle when a sister
chromatid is present, while 53BP1-mediated NHEJ repair is the
dominant process in the G1 phase. The checkpoint kinase 1 (Chk1),
activated by ATM kinase on DNA breaks in the G1 phase,
phosphorylates the histone chaperone, anti-silencing function 1A
histone chaperone (ASF1A) at Ser166 (82). The phosphorylation of ASF1A
interacts with the repair protein MDC1 and thus enhances its
downstream 53BP1 recruitment. Similarly, topoisomerase IIβ binding
protein 1 (TopBP1), a multi-domain 'scaffold' protein, has been
revealed to control the DNA damage checkpoint regulating S-phase
entry by binding to 53BP1 (83).
The BRCT domains of TopBP1 bind to conserved phosphorylation sites
(Ser366, Thr670) in the N-terminus of 53BP1, which promotes the
recruitment of TopBP1, ATR and Chk1 to 53BP1 damage foci, but does
not affect the formation of 53BP1 or ATM foci following DNA damage
(83,84). Chk1 is phosphorylated by ATR on
Ser317 and Ser345 in a DNA damage-dependent manner, thus prolonging
the G1 phase and inducing NHEJ repair by coordinating cell cycle
progression with DSB repair (84). Moreover, Ha et al
(85) found that DSB sites in
S/G2 cells can be processed by the Ku heterodimers and the MRN
complex. When a DSB site is bound by Ku heterodimers, the break is
then destined for 53BP1-mediated NHEJ. While DSB sites are bound by
an MRN complex, the ends are resected and ssDNA is generated,
leading to the activation of the ATR/Chk1/APCCdh1 axis,
and eventually the destruction of deubiquitinating enzyme USP1 and
the recruitment of BRCA1. Beishline et al (86) found that the transcription factor
Sp1, phosphorylated on serine 101 (pSp1) by ATM, was recruited to
DSBs 7.5 min following ionizing radiation-induced damage and
remained at the DSB site for at least 8 h. The same research group
researched further and revealed that Sp1 localized to DSBs in the
G1 phase and was necessary for the recruitment of 53BP1 to promote
NHEJ repair, while the phosphorylation of Sp1-S59 in the early S
phase evicted Sp1 and 53BP1 from the DSB site to allow BRCA1
binding (87). The forkhead box
K1 (FOXK1) associates with 53BP1 to negatively regulate 53BP1
function by inhibiting 53BP1 localization to DSB sites (88). The FOXK1-53BP1 interaction is
enhanced upon DNA damage during the S phase in an
ATM/CHK2-dependent manner, which reduces the association of 53BP1
with its downstream factors RIF1 and PTIP. The acetylation of lamin
B1 (LMNB1) at K134 negatively regulates canonical NHEJ repair by
impairing the recruitment of 53BP1 to DSB sites, and induces the
persistent activation of the G1/S checkpoint (89). Thus, given the apparent switching
effects of these regulators in integration of the cell cycle and
DSB repair pathway choice to favor NHEJ, a more complete
understanding of the function of these is required to validate the
aforementioned findings.
Notably, similar to how H4K20me2 promotes NHEJ
repair by presenting a binding site for the 53BP1 protein, H4K20me3
interactions with 53BP1 have been shown to be markedly pronounced
at DNA lesions in the G1 phase (90). Together, H4K20me3 and H3K9me3
represent epigenetic markers that are important for the function of
the 53BP1 recruitment in NHEJ repair, while the levels of these
histone markers are reduced in the very late S and G2 phases when
PCNA was recruited to locally micro-irradiated chromatin (90). Moreover, Nakamura et al
(91) reported that the ankyrin
repeat domain of BRCA1-associated RING domain protein 1 (BARD1)
promoted BRCA1 recruitment to DSB sites in the S and G2 phases by
recognizing and reading histone H4 unmethylated at lysine 20
(H4K20me0). The BARD1 recognition of H4K20me0 is required for HR
repair and resistance to PARPis, and opposes 53BP1 function and
NHEJ repair.
It has been shown that 53BP1 protein levels do not
significantly change in a DSB response, and that the expression of
53BP1 remains basically unaltered throughout the entirety of the
cell cycle (92,93). Therefore, 53BP1 is regulated by
multiple PTMs (Table I). The
first PTMs are phosphorylation and dephosphorylation. There are 28
ATM-regulated phosphorylation sites at the N-terminal phospho-SQ/TQ
domain of 53BP1 (29,94). The interaction between PTIP and
53BP1 is primarily dependent on the third phosphorylation site
(S25), which plays a role in pathological injury repair selection
and telomere fusion (56).
Interactions between RIF1 and 53BP1 are dependent on the
phosphorylation sites 9-15 (T302, S437, S452, S523, S543, S580 and
S625), which govern the processing of DNA ends by recruiting
Shieldin (55). Additionally,
the phosphorylation of 53BP1 is also involved in its recruitment
and cell cycle regulation: i) The vaccinia-related kinase 1 stably
phosphorylates 53BP1 at Ser25/29 without ATM, and is involved in
the formation of γH2AX, NBS1 and 53BP1 foci induced in NHEJ repair,
and the entry of the cell cycle into the G2/M phase (95,96). ii) The AMP-activated protein
kinase directly binds to 53BP1 and phosphorylates it at Ser1317,
and promotes 53BP1 recruitment, thus maintaining genomic stability
and diversity of the immune repertoire (97). iii) The glycogen synthesis kinase
3β was revealed to translocate from the cytoplasm to the nucleus
after exposure to ionizing radiation, where it induced DSB repair
in the nuclei of glioblastoma cells via the phosphorylation of
53BP1 at Ser166 (98). Moreover,
the dephosphorylation of 53BP1 plays a noteworthy role in DSB
repair pathway choice: i) The serine/threonine-protein phosphatase
4 catalytic subunit C (PP4C)/PP4CR3β complex dephosphorylates 53BP1
at T1609/T1618, and provides the structural basis for the normal
enrichment of 53BP1 in the G1 phase for NHEJ repair (99). ii) Both BRCA1 and PP4C can
promote the dephosphorylation of 53BP1 at T543 and the release of
the 53BP1-RIF1 complex from DSB sites to direct repair toward HR
(100). iii) The protein
phosphatase 2C δ (referred to as WIP1) decreases the 53BP1
positioning after IR by mediating 53BP1 dephosphorylation at Thr543
and inhibiting 53BP1 interaction with RIF1 (101).
Secondly, 53BP1 is also regulated by ubiquitination.
RNF168 modifies 53BP1 through the addition of a chain of
ubiquitin-polypeptides. Lysine 1268 of 53BP1 is important for this
ubiquitin modification, while the loss of this modification impairs
53BP1 recruitment to sites of DNA damage (47,102). Additionally, the UDR motifs of
53BP1 can recognize and bind to H2AK15ub (H2A monoubiquitination by
RNF168), which is crucial for recruiting 53BP1 to promote NHEJ
repair. However, the E3 ligase RNF168-mediated 53BP1 ubiquitination
and recruitment can be attenuated by lipolytic inhibitor G0/G1
switch gene 2 (103), ring
finger protein 126 (RNF126) (104), ubiquitin-editing enzyme
A20/TNFAIP3 (105) and the
phosphorylation of H2AK15ub at Thr12 (referred to as H2AK15pUbT12)
(106). Conversely, RNF169, an
uncharacterized E3 ubiquitin ligase paralogous to RNF168,
accumulates in DSB repair foci by recognizing RNF168-catalyzed
ubiquitylation products and acting as a molecular rheostat to limit
53BP1 deposition at DSBs (107,108). Hu et al (109) found that RNF169 induces 53BP1
disengagement from H2AK15ub-H4K20me2-53BP1 complex. RNF169 bridges
ubiquitin and histone surfaces, stabilizing a pre-existing
ubiquitin orientation in H2AK15ub-H4K20me2-53BP1 complex to form a
high-affinity complex (109).
This conformational selection mechanism contrasts with the
low-affinity binding mode of 53BP1, and it avails 53BP1
displacement.
Thirdly, 53BP1 is regulated by
methylation/acetylation. PRMT1, a protein that catalyzes substrates
to produce monomethylation or symmetric demethylated arginine,
methylates the GAR motif of 53BP1 to facilitate 53BP1
oligomerization and recruitment (34,35). Similarly, PRMT5, a homologous
protein of PRMT1, plays a parallel role to that of PRMT1 (110). Wild-type PRMT5 maintains 53BP1
stability and promotes NHEJ repair by methylating 53BP1 GAR motif,
while pY324 (phosphorylated by Src kinase) of PRMT5 inhibits its
activity during the DNA damage process and blocks NHEJ repair
(110). However, PRMT5
methylates RUVBL1 at R205, a cofactor of the TIP60 complex, which
promotes TIP60-dependent histone H4K16 acetylation and subsequently
facilitates 53BP1 displacement from DSB sites (111). Unlike methylation, recognition
or modification by acetylation appears to induce DSB repair towards
the HR pathway. As previously mentioned, the UDR motif mediates the
selective aggregation of 53BP1 by recognizing H2AK15Ub. Through a
histone reader domain for H4K20me1/2, the MBT domain-containing
protein 1 (MBTD1) allows TIP60 complex to associate with DSB sites
and acetylate H2AK15 (112,113). This acetylation blocks H2AK15
ubiquitylation that was regulated by RNF168, and inhibits 53BP1
recruitment through competitive bivalent binding. Additionally,
nuclear ATP-citrate lyase phosphorylation facilitates
TIP160-dependent histone acetylation at DSB sites, impairing 53BP1
localization and enabling BRCA1 recruitment (114,115). Notably, the acetylation of
53BP1 itself inhibits NHEJ and promotes HR by negatively regulating
its recruitment to DSB sites (116). Mechanistically,
acetyltransferase CBP acetylates the UDR motif of 53BP1 at
K1626/1628, thus disrupting the interaction between 53BP1 and
H2AK15ub, subsequently blocking the recruitment of 53BP1 and its
downstream factors PTIP and RIF1.
Finally, ADP-ribosylation can signal for
ubiquitination and promote the degradation of ADP-ribosylated
proteins (117,118). RNF146 contains a RING domain
that is an E3 ubiquitin ligase and a WWE domain that is a
PAR-binding domain, and it functions as an E3 ubiquitin ligase for
ADP-ribosylated 53BP1 (119,120). As the amount of DNA damage
increases, the C terminus (1043-1972aa) of 53BP1 is ADP-ribosylated
by PARP1, and ADP-ribosylated 53BP1 is targeted by RNF146, leading
to 53BP1 ubiquitination and degradation (121). NUDT16, member of Nudix proteins
that is characterized by a highly conserved 23-amino acid Nudix
motif, exhibits the hydrolase activity that removes the protein
ADP-ribosylation of 53BP1 (122), and inhibits 53BP1
ubiquitination and degradation, stabilizing 53BP1 protein and
allowing its recruitment to DSB sites (121). Together, the PTM status of
53BP1 plays key roles in its recruitment to DSB sites, and reveals
how specific 53BP1 modification and recognition modulate the
selection of DNA repair pathways.
There are other factors that contribute to the
regulation of 53BP1 recruitment and NHEJ repair. The nuclear basket
of nuclear pore complexes contains three nucleoporins Nup153, Nup50
and Tpr, and they play key roles in DSB repair by promoting the
nuclear import of 53BP1. Nup153 is required for the proper nuclear
import of 53BP1 and SENP1-dependent sumoylation of 53BP1, which
promotes the recruitment of 53BP1 to DNA repair foci (123,124). DROSHA, a miRNA biogenesis
enzyme, is required within minutes of a break occurring to control
the recruitment of NHEJ repair factors in a DROSHA-dependent manner
(125). DROSHA is recruited to
DSB sites without neither H2AX, nor ATM or DNA-PK kinase
activities, and interacts with RAD50 to promote its recruitment
(126). Indeed, DROSHA
knockdown and MRN complex inactivation (mirin treatment) increase
the association of downstream HR factors, such as RAD51 to DNA ends
and reduce NHEJ (125,126). Tripartite motif-containing
protein 29 (TRIM29) is required for the efficient recruitment of
53BP1 to facilitate the NHEJ pathway and thereby suppress the HR
pathway in response to DSB (127). The knockdown of histone lysine
demethylase PHF2 inhibits the resolution of 53BP1 foci, the
localization of C-terminal binding protein (CtBP)-interacting
protein (CtIP) and subsequent NHEJ repair (128). TNF receptor-associated death
domain (TRADD), an essential mediator of TNF receptor signaling,
facilitates NHEJ repair by recruiting 53BP1 and the Ku70/80 complex
(129). In contrast to the
depletion of the ubiquitin ligase HUWE1 increasing RAD51 levels to
partially restore HR, the depletion of histone acetyltransferase
KAT5 rewires DSB repair by promoting 53BP1 binding to DSBs
(130). KAT5 depletion can
promote PARPi sensitivity via the induction of imprecise NHEJ
repair in BRCA2-deficient cells. The chromodomain helicase
DNA-binding protein 1 (CHD1), a common genomic mutation found in
human prostate cancers associated with genomic instability,
disrupts 53BP1 stability and decreases error-prone NHEJ repair for
DSB repair (131). PARP2 limits
the accumulation of the resection barrier factor 53BP1 at DSB sites
independently of its PAR synthesis activity (132). PARP2 induces DSBs towards
resection-dependent repair pathways, which includes HR repair, SSA
and altEJ, rather than NHEJ repair.
The current research consensus is that BRCA1- and
53BP1-dependent pathways compete with each other during the early
stages of DSB repair, particularly for DNA end resection. In the G1
phase, 53BP1 is recruited to the DSB site where it forms a protein
complex that antagonizes BRCA1-mediated terminal modification (a
single stranded homologous arm of ~200 nt), thereby protecting the
terminal from excessive removal and determining the manner of cell
repair (133). Hence, it is
crucial to determine the effector molecules of 53BP1, and it is
beneficial for researchers to fully elucidate the effects of 53BP1
chromatin recruitment in DNA damage.
53BP1 phosphorylation, catalyzed by ATM on >25
sites that are concentrated in the N-terminal half of the protein,
leads to the activation of the DNA repair function of 53BP1 and
promotes its interaction with two proteins, PTIP and RIF1. These
two proteins are involved in limiting end resection at DSB sites
independently of each other (134). NHEJ repair is abolished in
53BP1−/− cells and in cells expressing
53BP128A (an allele harboring alanine substitutions in
all 28 N-terminal phosphorylation sites), while exerting a
considerably milder defect in RIF1−/− cells (135). Moreover, similar to the effect
of 53BP1 ablation, the conditional ablation of mouse RIF1 (not
PTIP) specifically in B-cells results in a profound defect in the
function of 53BP1 in several NHEJ-driven processes, such as
immunoglobulin CSR (55,136). Both processes of CSR and DSB
end ligation involve Ku70/80, DNA-PKcs, LIG4 and XRCC4/XLF of NHEJ
repair molecules (137). The
53BP1-RIF1 complex has indications for processing short overhangs,
and ssDNA longer than 20-30 nt is characteristic of resection
(16). Concerning the mechanism
by which the 53BP1-RIF1 complex limits the formation of ssDNA at
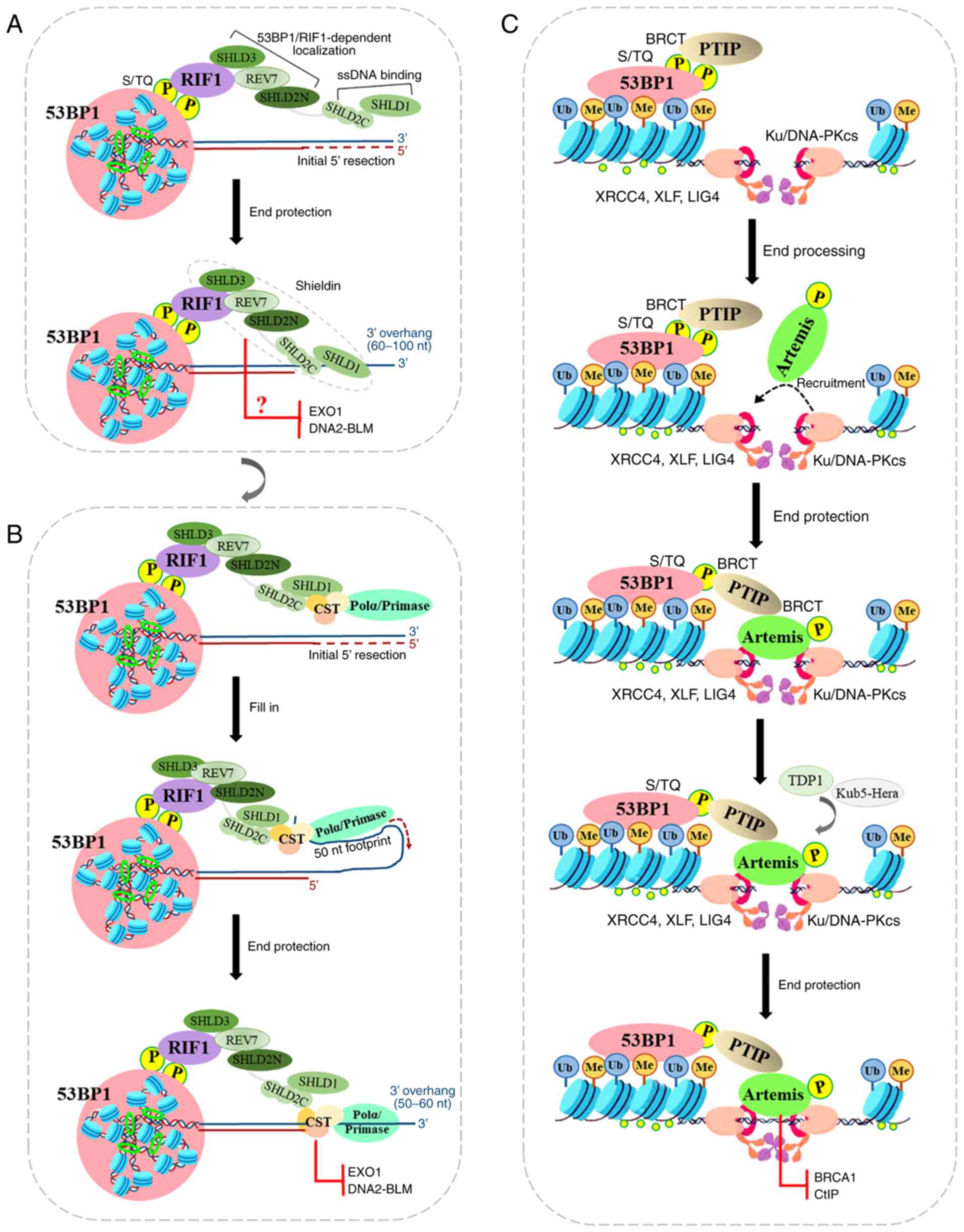
DNA breaks, there are two main models.
In the first model, 53BP1 uses the loading of
Shieldin onto the ssDNA to protect the 5′ end from resection. The
Shieldin complex is composed of REV7 plus SHLD3 (RINN1 or
CTC-534A2.2), SHLD2 (RINN2 or FAM35A) and SHLD1 (RINN3 or
C20ORF196), and is recruited to DSBs via the
ATM-RNF8-RNF168-53BP1-RIF1 axis, thus promoting NHEJ repair of
intrachromosomal breaks, CSR and the fusion of unprotected
telomeres (138,139). For the sake of clarity, the
SHLD1/2/3 nomenclature will be used herein. Shieldin localizes to
DSB sites in a 53BP1- and RIF1-dependent manner, and its SHLD3 and
REV7 subunits associate with the SHLD2 N-terminus to form the
53BP1-RIF1 complex localization module, while its SHLD1 subunit
associates with the SHLD2 C-terminus to form the ssDNA-binding
module (140). REV7 binds to
SHLD2/3 in the crystal structure of the SHLD3-REV7-SHLD2 ternary
complex by adopting two conformations with different topologies,
closed (C-REV7) and open (O-REV7) states (141). Therein, SHLD2 forms a β sheet
sandwich with O-REV7 and SHLD3 to promote NHEJ repair (141), while the conserved FXPWFP motif
of SHLD3 binds to C-REV7 and blocks REV7 binding to REV1, which
excludes Shieldin from the REV1/Pol ζ translesion synthesis complex
(141). Additionally, The
C-terminal half of SHLD2 is predicted at a high level of confidence
to form three tandem OB-folds to function as a ssDNA binding domain
(142). The OB-folds are
similar to those found in RPA1 (subunit of replication protein A)
and CTC1 (one of CTC1-STN1-TEN1 complex), and may provide a binding
site for these ssDNA-binding complexes (143,144). Hereby, the decision point of
the 53BP1-RIF1 complex in NHEJ repair revolves around Shieldin
(Fig. 3A). In order to ensure
that the 53BP1-RIF1-Shieldin complex induces 5′ ends to produce
sufficient resection to antagonize BRCA1-mediated HR repair, the
binding between Shieldin and ssDNA is worthy of further study. The
initiation of end resection occurs in a two-step process: Firstly,
the MRN resection complex induces endonuclease generated nicks on
the 5′-terminated strands on either side of the DSB site with the
aid of CtIP (145,146). These nicks are then expanded
through the 3′-5′ exonuclease activity of MRN and the 5′-3′
exonuclease activity of exonuclease 1 (EXO1) or DNA2-BLM (147,148). The resulting large tracts of
ssDNA are bound by RPA, which is then replaced by RAD51 to initiate
extensive degradation of the 5′ strands that are required for HR
repair. Although the SHLD2/SHLD1 complex binds to oligonucleotides
of 60-10 nt in vitro (149), the SHLD2/SHLD1 complex does not
completely inhibit BRCA1. Thus, these biochemical characterizations
of Shieldin presented above leave some unresolved questions: One
involves the mechanisms through which Shieldin prevents
end-resection prior to the initiation of resection by binding to
ssDNA. The other involves the mechanisms through which Shieldin
interrupts EXO1 or DNA2-BLM following the initiation of resection
by binding to ssDNA (Fig.
4A).
In the second model, Shieldin functions in
recruiting CST/Polα/Primase at resected ends, rather than blocking
end-resection nucleases per se, or by directly inducing resection.
The CST complex binds with high affinity to ssDNA and dsDNA
junctions, potentially allowing the complex to protect 5′ ends from
EXO1 and block access of the BLM and WRN helicases (150). The CST complex may function as
downstream molecules of 53BP1/RIF1 to protect DSBs from end
resection, which confers PARPi resistance in BRCA1-deficient cells
(151). 53BP1/RIF1/Shieldin/CST
complex binding at a DSB site requires a 3′ overhang (for CST, in
the range of 10-18 nt) (150).
As an accessory factor of Polα-primase, CST interacts with Shieldin
and localizes with Polα to DSB sites in a 53BP1- and
Shieldin-dependent manner (152). However, EXO1 and DNA2-BLM can
generate long ssDNA tracts, while Polα has limited ability and
usually synthesizes 20-25 nt overhangs (153). Therefore, the
Shieldin/CST/Polα/Primase fill-in reaction is predicted to leave a
considerable 3′ overhang that may be as long as 60 nt. During
telomere replication, CST-induced fill-in reactions allows for
retention of overhangs of at least 50 nt (154). The RIF1/Shieldin/CST axis has
the ability to protect 5′ ends from further resection, while 53BP1
action is predicted to carry a 3′ overhang. These results suggest
that CST/Polα/Primase-mediated fill-in reactions help to control
the repair of DSB by 53BP1, RIF1 and Shieldin (Fig. 4B).
PTIP is another critical factor acting as a
downstream effector of 53BP1, and it antagonizes BRCA1 function in
DNA repair by cooperating with RIF1. PTIP recruitment to DSB sites
depend on phosphorylated 53BP18A (the first eight
amino-terminal ATM sites), while PTFP depletion provides additional
or sustained end resection that is required for rescuing HR repair
in BRCA1-deficient cells (56).
PTIP, a large nuclear protein containing six BRCT (BRCA1
C-Terminal) domains, regulates gene transcription as part of the
MLL3-MLL4 methyltransferase complex that catalyzes H3K4me3
(155). PTIP interacts with
phosphorylated Ser 25 of 53BP1 through its tandem BRCT domains
(156,157) (Fig. 4C).
PTIP promotes NHEJ repair by recruiting proteins
required by NHEJ, Artemis, to sites of DNA damage (158). PTIP interacts with Artemis
through its second BRCT domain, while Artemis interacts with PTIP
through its damage-dependent phosphorylation of six S/T sites
(T656) at the very C-terminal end (158). Artemis, a nuclease with exo-
and endonuclease activity, cleaves a hairpin intermediate during
V(D)J recombination during DSB end processing (42,159). Artemis nuclease activity is
dependent on DNA-PKcs autophosphorylation, suggesting that DNA-PK
may remodel the end to allow Artemis cleavage (160). Ku70/80 protein binds to DSB end
and promotes Artemis recruitment, and DNA-PKcs also phosphorylates
Artemis. Artemis separates DNA-PKcs from end-joining complex
(Ku70/80, DNA ligase IV, XRCC4, XLF, and PAXX) by interacting with
XRCC4 (161). Therefore, the
endonuclease activity of Artemis permits it to trim DSB ends to
promote NHEJ, and consequently to prevent end resection and
RAD51-dependent HR repair (Fig.
4C).
Similar to Artemis nuclease, tyrosyl-DNA
phosphodiesterase (TDP1) is capable of resolving protruding
3′-phosphoglycolate termini of DSB sites to promote the C-NHEJ
pathway (162). Artemis
deficiency results in a fraction of unrepaired DSBs in 53BP1 foci,
while TDP1 deficiency tends to promote DSB end mis-joining. TDP1
and Artemis perform different but interrelated functions in the
repair of terminally blocked DSBs. Additionally, Kub5-Hera, the
human homolog of the yeast transcription termination factor Rtt103,
forms novel complexes with DSB repair factors (Ku70/Ku86, Artemis,
and others) and terminate transcription (RNA polymerase II) at DSB
sites (163). In
Kub5-Hera-deficient cells that are hypersensitive to cytotoxic
agents-induced DSBs, Artemis induces γ-H2AX and 53BP1
repair-related foci regression. 53BP1 promotes toxic end-joining
events (alt-NHEJ and c-NHEJ) via the retention of Artemis at DSB
sites, while BRCA2 antagonizes 53BP1, RIF1, and Artemis-dependent
NHEJ repair to prevent gross genomic instability in a
RAD51-independent manner (164). Thus, although these studies
highlight the importance of the 53BP1/PTIP/Artemis axis at DSB
repair, Artemis-related downregulation requires further research
(Fig. 4C).
As described above, RIF1 negatively regulates
resection through the effector Shieldin to prevent further
resection and HR repair. Isobe et al (165) found that RIF1 immediately
inhibited the accumulation of CtIP at DSB sites following damage,
suggesting that RIF1 has another effector in addition to Shieldin.
They found that protein phosphatase 1 localized to DSB sites in a
RIF1-dependent manner, and suppressed downstream CtIP accumulation
and limited MRN complex-mediated resection (165). Indeed, Cockayne syndrome (CS)
is a DNA repair impaired syndrome characterized by a broad mutation
of CS protein B (CSB), which is considered another RIF1 effector
(166). Batenburg et al
(167) found that CSB, a member
of the switch/sucrose non-fermentable (SWI2/SNF2) superfamily, was
phosphorylated by ATM (at S10) and cyclin A-CDK2 (at S158). In the
DNA DSB repair pathway choice in the S/G2 phases, CSB interacts
with RIF1 via its winged helix domain (WHD) and is recruited to
FokI-induced DSB sites in the S phase, limiting RIF1 and its
effector REV7, and evicting histones, but promoting BRCA1-mediated
HR repair (167). Further
research has found that the UV-induced disengagement of the
C-terminal region of CSB from the ATPase domain requires two
conserved amino acids (W1486 and L1488), and it contributes to the
hydrophobic core formation of WHD at its C-terminus (168). The dissociation of the CSB
domain interactions is a necessary step in repairing DNA damage.
Following RIF1 eviction, CSB interacts with the BRCT domain of
BRCA1 and this interaction is regulated by CDK-dependent
phosphorylation of CSB at S1276 in late S/G2 phase, mediating the
interaction of CSB with HR repair-related proteins consisting of
BRCA1, the MRN complex and CtIP (169).
Similar to CSB, the suppressor of cancer cell
invasion (SCAI) interacts with the tumor suppressing SWI/SNF
chromatin remodeling complex to promote changes in gene expression
(170). Hansen et al
(171) initially demonstrated
that SCAI is a mediator of 53BP1-dependent repair of
heterochromatin-associated DSBs and facilitates ATM kinase
signaling. SCAI undergoes prominent enrichment at DSB sites through
53BP1-dependent recruitment to DSB-surrounding chromatin, while
SCAI deficiency results in reduced NHEJ repair capacity. SCAI was
recently shown to stimulate HR repair through an interaction with
53BP1 phosphorylated at S/TQ sites in the S/G2 phases (172). SCAI inhibits and evicts RIF1 at
DSB sites via binding to 53BP1, thus facilitating BRCA1-mediated HR
repair (172). Inversely, LMO2
(also known as RBTN2, Rhombotin-2, or Ttg-2) inhibits BRCA1
recruitment to DSBs by interacting with 53BP1 during repair,
promoting error-prone NHEJ repair and increasing tumor cells'
sensitivity to PARPis in the G1 phase (173). Collectively, these molecules
are physiologically important components of both the NHEJ- and
HR-mediated pathways, in potentiating the DSB repair choice via
modulation of the downstream signaling of the 53BP1 axis.
53BP1 inhibits the formation of 3′ overhangs at DSB
sites and alters DSB chromatin dynamics; however, its selective
advantage remains an enigma. For this reason, 53BP1 may contribute
to the response to DSBs, but may also be potentially detrimental
for cells with multiple DSBs.
53BP1 not only affects resistance to cancer
treatments, such as chemotherapeutic agents, PARPis and radiation,
but is also a predictor of outcomes after undergoing treatment.
Studies have demonstrated that low levels of 53BP1 prolong the
overall survival of patients with non-small lung cancer cell
undergoing treatment with platinum to 19.3 months (high levels of
53BP1 to 8.2 months) (174).
However, in germ cell tumors, cisplatin-resistant cell lines have a
NHEJ-less phenotype characterized by a reduced basal expression of
53BP1 and DNA-PKcs (80).
Similarly, low levels of 53BP1 have an inferior response to
treatment with high-dose alkylating agents in breast cancer
(175), while 53BP1 is
upregulated in temozolomide-resistant glioblastoma cells (176). 53BP1−/− leads to
5-fluorouracil resistance in colorectal cancer cells by inhibiting
the ATM-CHK2-P53 pathway (177). It is hypothesized that the
reason for the ambiguous role of 53BP1 in cancer chemotherapy
resistance may be due to the fact that it is often studied in
isolation without taking the role of the ATM-CHK2-P53 pathway and
DNA repair into consideration. As previously demonstrated, a
53BP1−/− genotype increased resistance to PARPis in
BRCA1-deficient mice by promoting the re-emergence of HR repair.
BRCA1-deficient cancers prevent error-prone NHEJ-induced excessive
genomic alterations by downregulating RNF168 ubiquitin signaling
(178). The concept of
BRCA1−/−-affected HR repair is not an 'all-or-nothing'
concept. When the inhibition of RNF168-ub-H2AX signaling is not
sufficient to activate 53BP1 recruitment, PALB2, a partner and
localizer of BRCA2, potently stimulates the DNA strand-invasion
activity of RAD51 to prompt residual HR repair (178). In this process, 53BP1 binds to
the nucleosome acidic patch region via its UDR domain to block the
interaction between PALB2 chromatin-associated motif (ChAM) and the
nucleosome at the site of the DSB (179). It was previously demonstrated
that olaparib co-treatment with DNA synthesis-inhibiting agents
significantly increased 53BP1/γH2AX co-localization in anticancer
drug-treated cells to attenuate the toxicity of treatments
(180). In
BRCA1/53BP1-deficient cells, RAD51 foci are formed at resected DSBs
in a PALB2/BRCA2-dependent manner, and thereby induce HR repair
(179). As regards sensitivity
to PARPis, it is worth mentioning that targeting the upstream
signaling of 53BP1 is also an effective target.
The rapid and error-prone DSB repair of NHEJ in
cancer radiation therapy is considered to be the primary factor
involved in radiation resistance. Ward et al (25) demonstrated that 53BP1-deficient
mice were hypersensitive to radiation due to defects in NHEJ. Mu
et al (181) found that
the reduction in 53BP1 phosphorylation levels (not the levels of
53BP1 protein) induced the radiosensitization of glioblastoma cells
by inhibiting NHEJ repair. All ionizing radiation therapy, whether
it is multifraction radiotherapy (MFR) or single-dose radiotherapy
activates different DNA repair mechanisms (182). Compared with an equivalent
single dose of irradiation, both cancer cells and normal
fibroblasts exhibit an enhanced survival following MFR, and this
effect is entirely dependent on 53BP1/RIF1-mediated NHEJ repair
(183). These results are of
clinical significance as they can guide the selection of the most
effective ionizing radiation regimen by analyzing the expression
status of the 53BP1-regulated NHEJ repair in tumors. However,
although the mechanisms through which the 53BP1-mediated promotion
of cancer cell recovery and survival can reduce patient outcome are
understood, little is known regarding the DNA repair method that
occurs between different radiation fractions. Roobol et al
(184) monitored the
accumulation of the endogenous 53BP1 and replication protein A
using live-cell microscopy and found that low
linear-energy-transfer (LET) X-ray-induced 53BP1 foci were rapidly
and more dynamically resolved (184). Low-LET X-ray irradiation
triggers NHEJ repair, while high-LET α-particles induce multiple
replication protein A foci at closely interspaced DSB sites, thus
promoting HR-prone repair (184). Nevertheless, the γH2AX and
53BP1 foci size have been shown to increase with LET, suggesting
that the delay in repair kinetics was due to the occurrence of more
complex damage (185). These
findings appear to suggest that the biological effects of NHEJ or
HR repair choices may be significantly influenced by the dose, as
well as the type of radiation exposure. Therefore, current
knowledge regarding the importance of 53BP1-mediated NHEJ repair in
cancer therapy is at its early stages, and further studies focusing
on the selective advantage of NHEJ-prone repair are required.
In human mammary epithelial cells from older
individuals, the decreased activity of the primary DSB repair
pathways, which play crucial roles in maintaining genome integrity,
was found by Anglada et al (186). The deficient recruitment of
53BP1 to DSB sites in G1 cells from aged donors reveals a positive
association between age-associated DNA repair defects and the aging
process. As the expression levels of γH2AX and 53BP1 are promoted,
Li et al (187) found a
protective function of 53BP1-mediated NHEJ repair in premature
ovarian failure. In addition to DSB repair modulation, 53BP1
maintains heterochromatin integrity and genomic stability through
liquid-liquid phase separation (LLPS) with the heterochromatin
protein HP1α in a mutually dependent manner (188). The LLPS of 53BP1 rescues
heterochromatin de-repression and protects cells against
stress-induced DNA damage and senescence. If senescence is
bypassed, cells undergo crisis through the loss of checkpoints and
this results in mass cell death, concomitant with further telomere
shortening and spontaneous telomere fusions. Based on this, the
auxo-actions of 53BP1-dependent NHEJ repair in telomere fusions
cannot be ignored. Telomeres are protected by the six-subunit
shelterin complex [telomeric repeat binding factor (TRF)1, TRF2,
protection of telomeres 1 (POT1), TERF1 interacting nuclear factor
2 (TIN2), TINT1 and Rap1], which suppresses DNA damage signaling,
DNA repair, and 5′ end hyper-resection. In telomeres lacking TRF2,
telomere fusion boosts are due to several separable effects of
53BP1 promote: A promotion of mobility of unprotected telomeres
(189), the effects of
oligomerization and synapsis involving telomere clustering
(135), and the recruitment of
the RIF1/Shieldin/CST axis, which is involved in counteracting 5′
end resection. When telomeres are lost due to aging-associated
erosion, breakage, or failed replication, the telomere fusions
serve as a cell's final attempt to protect exposed chromosomal
ends. However, inappropriate end-to-end chromosomal rearrangements
and telomere fusions promote genomic instability and
carcinogenesis.
Although 53BP1 is most well-known for its
regulation of DNA damage repair mechanisms, it was initially
discovered via its binding to p53. During the differentiation of
human embryonic stem cells into neurons or into cortical organoids,
a transcriptional co-regulatory effect of 53BP1 and UTX, a
chromatin modifier, promotes human neurogenesis by upregulating key
neurodevelopmental genes (190). Additionally, the activation of
a 53BP1-USP28-p53 mitotic surveillance pathway facilitates
centrosome defect-induced neural progenitor cell (NPC) depletion
and microcephaly during development of the brain (191). In a p53-dependent pathway
underlying primary microcephaly, a delay of spindle assembly caused
by centrosome gene mutations triggers the activation of the
53BP1-USP28-p53 pathway, while 53BP1 deletion restores NPC
proliferation and brain size (192). In another p53-dependent
pathway, mutations in genes required for DNA repair or genomic
stability induce the accumulation of DNA lesions that trigger DNA
damage signaling in NPCs to activate p53 (192). Thus, the role of 53BP1 as a
regulator of DNA damage repair deserves further study. In the
developing epidermis, the activation of the 53BP1-USP28-p53 pathway
induced by genetically ablating centrosomes also cause a thinner
epidermis and hair follicle arrest (193). These studies provide insight
into 53BP1-related neurodevelopment and hyperproliferative diseases
that may recapitulate developmental programs.
Precise genomic editing based on programming
nucleases, such as the CRISPR/CRISPR-Cas system, are controlled by
HR repair and limited by the competing error-prone NHEJ repair
(194,195). As a critical regulator of the
method of repair between NHEJ and HR, 53BP1 deficiency induces an
increase in BRCA1-mediated HR repair, which suggests that the
inhibition of 53BP1 may be a promising tool to manipulate repair
method and promote genome-editing efficiency. Recently, Canny et
al (196) and Sun et
al (197) screened out
inhibitors of 53BP1, inhibitor 53 (i53) and DP308, and they
targeted the tandem Tudor domain of 53BP1. i53 blocked the
interaction between 53BP1 and H4K20Me2 at DSB sites and improved
gene targeting and chromosomal gene conversion by up to 5.6-fold.
Paulsen et al (198)
found that the ectopic expression of the dominant-negative murine
form of 53BP1 (mdn53BP1) competitively antagonized 53BP1
recruitment to DSB sites and improved Cas9-mediated HR repair
activity. Similarly, RAD18, a DNA damage response factor on
Cas9-induced HDR, competitively binds H2AK15ub with greater
affinity than 53BP1, thereby inhibiting 53BP1 recruitment to DSB
sites (199). Additionally,
researchers fused Cas9 nucleases and DN1S, a dominant-negative
mutant of 53BP1, and this fusion improved HR repair frequency,
reaching 86% in K562 cells, and almost 70% in leukocyte adhesion
deficiency (LAD) patient-derived immortalized B lymphocytes
(200). Therefore, the
inhibition of 53BP1 improves the efficiencies of
CRISPR-Cas9-mediated precise gene correction/insertion,
significantly reducing undesirable NHEJ repairs at the nuclease
cleavage site.
There are several open-ended questions remaining in
this field. First, ~80% of ionizing radiation- or drug-induced DSBs
are repaired by the NHEJ pathway, even in the G2 phase. However, in
the face of the different causes of DSBs, it would be beneficial to
determine the reasons why 53BP1-mediated NHEJ is beneficial or
harmful. The nucleolytic, polymerization and ligation steps of NHEJ
are flexible, as numerous different structural and chemical DNA end
configurations can be ligated at DSB sites. Based on the present
review, the mechanical or biochemical environment of chromatin,
cell cycle phases and PTMs of 53BP1 may explain the synergistic
effects of these ligated complexes. Second, the recruitment of
53BP1 on chromatin around the DSB form 53BP1 nano-domains that are
shaped by chromatin topology. However, it remains unknown as to
whether the regulation of 53BP1 recruitment by histone molecular
markers (such as H2AK15Ub and H4K20Me2) with binding specificity
and epigenetic modification enzymes (such as MMSET and KAT5) are
implemented in parallel, or whether they actually regulate
different stages of 53BP1 nanodomain formation. Third, 53BP1, in
conjunction with RIF1 and PTIP, promotes the restraints of end
resection to antagonize HR repair, and consequently promotes NHEJ
repair. As the cell cycle progresses, 53BP1 gradually loses its
dominant role in binding with its helper complex. However, the
mechanisms through which 53BP1 and HR-related proteins, such as
BRCA1 achieve a dynamic balance in damaged chromatin remain
unknown. Fourth, telomere protection in mammals is mediated by
TRF2, which binds chromosomal ends and ensures genomic integrity
through inhibiting NHEJ repair, which triggers chromosome fusion
end connection (201,202). 53BP1 disturbs telomere
stability, possibly through interaction with the TRF2 Shelterin
component, and induces telomere dysfunction and the aging process
(186). In future studies,
these questions regarding 53BP1 function need be addressed to
obtain a more complete and accurate understanding of DSB repair and
improve the clinical options available to patients of several
diseases.
Not applicable.
LC conceived and designed the review article. TL
wrote the manuscript, and prepared the table and figures. SD and ZP
reviewed and edited the manuscript. All authors have read and
approved the manuscript, and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was sponsored by the Natural Science
Foundation of Chongqing municipality (grant no.
cstc2021jcyj-msxmX0855).
|
1
|
Aparicio T, Baer R and Gautier J: DNA
double-strand break repair pathway choice and cancer. DNA Repair
(Amst). 19:169–175. 2014. View Article : Google Scholar
|
|
2
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alt FW and Schwer B: DNA double-strand
breaks as drivers of neural genomic change, function, and disease.
DNA Repair (Amst). 71:158–163. 2018. View Article : Google Scholar
|
|
4
|
Gorthi A and Bishop AJR: Ewing sarcoma
fusion oncogene: At the crossroads of transcription and DNA damage
response. Mol Cell Oncol. 5:e14650142018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Daley JM, Niu H, Miller AS and Sung P:
Biochemical mechanism of DSB end resection and its regulation. DNA
Repair (Amst). 32:66–74. 2015. View Article : Google Scholar
|
|
6
|
Heyer WD, Ehmsen KT and Liu J: Regulation
of homologous recombination in eukaryotes. Annu Rev Genet.
44:113–139. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matos J and West SC: Holliday junction
resolution: Regulation in space and time. DNA Repair (Amst).
19:176–181. 2014. View Article : Google Scholar
|
|
8
|
Ira G, Pellicioli A, Balijja A, Wang X,
Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth
NM, et al: DNA end resection, homologous recombination and DNA
damage checkpoint activation require CDK1. Nature. 431:1011–1017.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roerink SF, van Schendel R and Tijsterman
M: Polymerase theta-mediated end joining of replication-associated
DNA breaks in C. Elegans Genome Res. 24:954–962. 2014. View Article : Google Scholar
|
|
10
|
Zhang Y and Jasin M: An essential role for
CtIP in chromosomal translocation formation through an alternative
end-joining pathway. Nat Struct Mol Biol. 18:80–84. 2011.
View Article : Google Scholar
|
|
11
|
Paques F and Haber JE: Multiple pathways
of recombination induced by double-strand breaks in saccharomyces
cerevisiae. Microbiol Mol Biol Rev. 63:349–404. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Scully R, Panday A, Elango R and Willis
NA: DNA double-strand break repair-pathway choice in somatic
mammalian cells. Nat Rev Mol Cell Biol. 20:698–714. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mimori T and Hardin JA: Mechanism of
interaction between Ku protein and DNA. J Biol Chem.
261:10375–10379. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ceccaldi R, Rondinelli B and D'Andrea AD:
Repair pathway choices and consequences at the double-strand break.
Trends Cell Biol. 26:52–64. 2016. View Article : Google Scholar :
|
|
15
|
Mao Z, Bozzella M, Seluanov A and
Gorbunova V: Comparison of nonhomologous end joining and homologous
recombination in human cells. DNA Repair (Amst). 7:1765–1771. 2008.
View Article : Google Scholar
|
|
16
|
Pannunzio NR, Watanabe G and Lieber MR:
Nonhomologous DNA end-joining for repair of DNA double-strand
breaks. J Biol Chem. 293:10512–10523. 2018. View Article : Google Scholar :
|
|
17
|
Soutoglou E, Dorn JF, Sengupta K, Jasin M,
Nussenzweig A, Ried T, Danuser G and Misteli T: Positional
stability of single double-strand breaks in mammalian cells. Nat
Cell Biol. 9:675–682. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tarsounas M and Sung P: The
antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication.
Nat Rev Mol Cell Biol. 21:284–299. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Beucher A, Birraux J, Tchouandong L,
Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA
and Löbrich M: ATM and Artemis promote homologous recombination of
radiation-induced DNA double-strand breaks in G2. EMBO J.
28:3413–3427. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Karanam K, Kafri R, Loewer A and Lahav G:
Quantitative live cell imaging reveals a gradual shift between DNA
repair mechanisms and a maximal use of HR in mid S phase. Mol Cell.
47:320–329. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao B, Rothenberg E, Ramsden DA and
Lieber MR: The molecular basis and disease relevance of
non-homologous DNA end joining. Nat Rev Mol Cell Biol. 21:765–781.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hustedt N and Durocher D: The control of
DNA repair by the cell cycle. Nat Cell Biol. 19:1–9. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morales JC, Xia Z, Lu T, Aldrich MB, Wang
B, Rosales C, Kellems RE, Hittelman WN, Elledge SJ and Carpenter
PB: Role for the BRCA1 C-terminal repeats (BRCT) protein 53BP1 in
maintaining genomic stability. J Biol Chem. 278:14971–14977. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rappold I, Iwabuchi K, Date T and Chen J:
Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA
damage-signaling pathways. J Cell Biol. 153:613–620. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ward IM, Minn K, van Deursen J and Chen J:
p53 Binding protein 53BP1 is required for DNA damage responses and
tumor suppression in mice. Mol Cell Biol. 23:2556–2563. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Adams MM and Carpenter PB: Tying the loose
ends together in DNA double strand break repair with 53BP1. Cell
Div. 1:192006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Panier S and Boulton SJ: Double-strand
break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol.
15:7–18. 2014. View Article : Google Scholar
|
|
28
|
von Morgen P, Lidak T, Horejsi Z and
Macurek L: Nuclear localisation of 53BP1 is regulated by
phosphorylation of the nuclear localisation signal. Biol Cell.
110:137–146. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mirman Z and de Lange T: 53BP1: A DSB
escort. Genes Dev. 34:7–23. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
He YJ, Meghani K, Caron MC, Yang C, Ronato
DA, Bian J, Sharma A, Moore J, Niraj J, Detappe A, et al: DYNLL1
binds to MRE11 to limit DNA end resection in BRCA1-deficient cells.
Nature. 563:522–526. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Becker JR, Cuella-Martin R, Barazas M, Liu
R, Oliveira C, Oliver AW, Bilham K, Holt AB, Blackford AN,
Heierhorst J, et al: The ASCIZ-DYNLL1 axis promotes 53BP1-dependent
non-homologous end joining and PARP inhibitor sensitivity. Nat
Commun. 9:54062018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
West KL, Kelliher JL, Xu Z, An L, Reed MR,
Eoff RL, Wang J, Huen MSY and Leung JWC: LC8/DYNLL1 is a 53BP1
effector and regulates checkpoint activation. Nucleic Acids Res.
47:6236–6249. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zgheib O, Pataky K, Brugger J and
Halazonetis TD: An oligomerized 53BP1 Tudor domain suffices for
recognition of DNA double-strand breaks. Mol Cell Biol.
29:1050–1058. 2009. View Article : Google Scholar :
|
|
34
|
Adams MM, Wang B, Xia Z, Morales JC, Lu X,
Donehower LA, Bochar DA, Elledge SJ and Carpenter PB: 53BP1
oligomerization is independent of its methylation by PRMT1. Cell
Cycle. 4:1854–1861. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Boisvert FM, Rhie A, Richard S and Doherty
AJ: The GAR motif of 53BP1 is arginine methylated by PRMT1 and is
necessary for 53BP1 DNA binding activity. Cell Cycle. 4:1834–1841.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Botuyan MV, Lee J, Ward IM, Kim JE,
Thompson JR, Chen J and Mer G: Structural basis for the methylation
state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in
DNA repair. Cell. 127:1361–1373. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pellegrino S, Michelena J, Teloni F, Imhof
R and Altmeyer M: Replication-coupled dilution of H4K20me2 guides
53BP1 to pre-replicative chromatin. Cell Rep. 19:1819–1831. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fradet-Turcotte A, Canny MD,
Escribano-Diaz C, Orthwein A, Leung CC, Huang H, Landry MC,
Kitevski-LeBlanc J, Noordermeer SM, Sicheri F and Durocher D: 53BP1
is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark.
Nature. 499:50–54. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Derbyshire DJ, Basu BP, Serpell LC, Joo
WS, Date T, Iwabuchi K and Doherty AJ: Crystal structure of human
53BP1 BRCT domains bound to p53 tumour suppressor. EMBO J.
21:3863–3872. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cuella-Martin R, Oliveira C, Lockstone HE,
Snellenberg S, Grolmusova N and Chapman JR: 53BP1 Integrates DNA
repair and p53-dependent cell fate decisions via distinct
mechanisms. Mol Cell. 64:51–64. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Riballo E, Kühne M, Rief N, Doherty A,
Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, et al: A
pathway of double-strand break rejoining dependent upon ATM,
Artemis, and proteins locating to gamma-H2AX foci. Mol Cell.
16:715–724. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chang HHY, Pannunzio NR, Adachi N and
Lieber MR: Non-homologous DNA end joining and alternative pathways
to double-strand break repair. Nat Rev Mol Cell Biol. 18:495–506.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Uziel T, Lerenthal Y, Moyal L, Andegeko Y,
Mittelman L and Shiloh Y: Requirement of the MRN complex for ATM
activation by DNA damage. EMBO J. 22:5612–5621. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Panier S and Durocher D: Push back to
respond better: Regulatory inhibition of the DNA double-strand
break response. Nat Rev Mol Cell Biol. 14:661–672. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Stewart GS, Panier S, Townsend K, Al-Hakim
AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M,
et al: The RIDDLE syndrome protein mediates a ubiquitin-dependent
signaling cascade at sites of DNA damage. Cell. 136:420–434. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Stewart GS, Wang B, Bignell CR, Taylor AM
and Elledge SJ: MDC1 is a mediator of the mammalian DNA damage
checkpoint. Nature. 421:961–966. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mattiroli F, Vissers JH, van Dijk WJ, Ikpa
P, Citterio E, Vermeulen W, Marteijn JA and Sixma TK: RNF168
ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling.
Cell. 150:1182–1195. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gudjonsson T, Altmeyer M, Savic V, Toledo
L, Dinant C, Grøfte M, Bartkova J, Poulsen M, Oka Y, Bekker-Jensen
S, et al: TRIP12 and UBR5 suppress spreading of chromatin
ubiquitylation at damaged chromosomes. Cell. 150:697–709. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bekker-Jensen S, Rendtlew Danielsen J,
Fugger K, Gromova I, Nerstedt A, Lukas C, Bartek J, Lukas J and
Mailand N: HERC2 coordinates ubiquitin-dependent assembly of DNA
repair factors on damaged chromosomes. Nat Cell Biol. 12:80–86.
1–12. 2010. View Article : Google Scholar
|
|
50
|
Shibata A and Jeggo PA: Roles for 53BP1 in
the repair of radiation-induced DNA double strand breaks. DNA
Repair (Amst). 93:1029152020. View Article : Google Scholar
|
|
51
|
Pesavento JJ, Yang H, Kelleher NL and
Mizzen CA: Certain and progressive methylation of histone H4 at
lysine 20 during the cell cycle. Mol Cell Biol. 28:468–486. 2008.
View Article : Google Scholar :
|
|
52
|
Mallette FA, Mattiroli F, Cui G, Young LC,
Hendzel MJ, Mer G, Sixma TK and Richard S: RNF8- and
RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1
recruitment to DNA damage sites. EMBO J. 31:1865–1878. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Min J, Allali-Hassani A, Nady N, Qi C,
Ouyang H, Liu Y, MacKenzie F, Vedadi M and Arrowsmith CH: L3MBTL1
recognition of mono- and dimethylated histones. Nat Struct Mol
Biol. 14:1229–1230. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Huen MS, Huang J, Leung JW, Sy SM, Leung
KM, Ching YP, Tsao SW and Chen J: Regulation of chromatin
architecture by the PWWP domain-containing DNA damage-responsive
factor EXPAND1/MUM1. Mol Cell. 37:854–864. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chapman JR, Barral P, Vannier JB, Borel V,
Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD and
Boulton SJ: RIF1 is essential for 53BP1-dependent nonhomologous end
joining and suppression of DNA double-strand break resection. Mol
Cell. 49:858–871. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Callen E, Di Virgilio M, Kruhlak MJ,
Nieto-Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M,
Starnes LM, et al: 53BP1 mediates productive and mutagenic DNA
repair through distinct phosphoprotein interactions. Cell.
153:1266–1280. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xie HY, Zhang TM, Hu SY, Shao ZM and Li
DQ: Dimerization of MORC2 through its C-terminal coiled-coil domain
enhances chromatin dynamics and promotes DNA repair. Cell Commun
Signal. 17:1602019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wu R, Liu W, Sun Y, Shen C, Guo J, Zhao J,
Mao G, Li Y and Du G: Nanoscale insight into chromatin remodeling
and DNA repair complex in HeLa cells after ionizing radiation. DNA
Repair (Amst). 96:1029742020. View Article : Google Scholar
|
|
59
|
Ochs F, Karemore G, Miron E, Brown J,
Sedlackova H, Rask MB, Lampe M, Buckle V, Schermelleh L, Lukas J
and Lukas C: Stabilization of chromatin topology safeguards genome
integrity. Nature. 574:571–574. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Arnould C, Rocher V, Finoux AL, Clouaire
T, Li K, Zhou F, Caron P, Mangeot PE, Ricci EP, Mourad R, et al:
Loop extrusion as a mechanism for formation of DNA damage repair
foci. Nature. 590:660–665. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Caron P and Polo SE: Reshaping chromatin
architecture around DNA breaks. Trends Biochem Sci. 45:177–179.
2020. View Article : Google Scholar
|
|
62
|
Lou J, Priest DG, Solano A, Kerjouan A and
Hinde E: Spatiotemporal dynamics of 53BP1 dimer recruitment to a
DNA double strand break. Nat Commun. 11:57762020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kilic S, Lezaja A, Gatti M, Bianco E,
Michelena J, Imhof R and Altmeyer M: Phase separation of 53BP1
determines liquid-like behavior of DNA repair compartments. EMBO J.
38:e1013792019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Banani SF, Lee HO, Hyman AA and Rosen MK:
Biomolecular condensates: Organizers of cellular biochemistry. Nat
Rev Mol Cell Biol. 18:285–298. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Shin Y, Berry J, Pannucci N, Haataja MP,
Toettcher JE and Brangwynne CP: Spatiotemporal control of
intracellular phase transitions using light-activated optoDroplets.
Cell. 168:159–171.e14. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Piccinno R, Minneker V and Roukos V:
53BP1-DNA repair enters a new liquid phase. EMBO J. 38:e1028712019.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Pessina F, Giavazzi F, Yin Y, Gioia U,
Vitelli V, Galbiati A, Barozzi S, Garre M, Oldani A, Flaus A, et
al: Functional transcription promoters at DNA double-strand breaks
mediate RNA-driven phase separation of damage-response factors. Nat
Cell Biol. 21:1286–1299. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Jakob B, Dubiak-Szepietowska M, Janiel E,
Schmidt A, Durante M and Taucher-Scholz G: Differential repair
protein recruitment at sites of clustered and isolated DNA
double-strand breaks produced by high-energy heavy ions. Sci Rep.
10:14432020. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Eaton JA and Zidovska A: Structural and
dynamical signatures of local DNA damage in live cells. Biophys J.
118:2168–2180. 2020. View Article : Google Scholar :
|
|
70
|
Drané P, Brault ME, Cui G, Meghani K,
Chaubey S, Detappe A, Parnandi N, He Y, Zheng XF, Botuyan MV, et
al: TIRR regulates 53BP1 by masking its histone methyl-lysine
binding function. Nature. 543:211–216. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zhang A, Peng B, Huang P, Chen J and Gong
Z: The p53-binding protein 1-Tudor-interacting repair regulator
complex participates in the DNA damage response. J Biol Chem.
292:6461–6467. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Botuyan MV, Cui G, Drané P, Oliveira C,
Detappe A, Brault ME, Parnandi N, Chaubey S, Thompson JR,
Bragantini B, et al: Mechanism of 53BP1 activity regulation by
RNA-binding TIRR and a designer protein. Nat Struct Mol Biol.
25:591–600. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang J, Yuan Z, Cui Y, Xie R, Yang G,
Kassab MA, Wang M, Ma Y, Wu C, Yu X and Liu X: Molecular basis for
the inhibition of the methyl-lysine binding function of 53BP1 by
TIRR. Nat Commun. 9:26892018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Dai Y, Zhang A, Shan S, Gong Z and Zhou Z:
Structural basis for recognition of 53BP1 tandem Tudor domain by
TIRR. Nat Commun. 9:21232018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Parnandi N, Rendo V, Cui G, Botuyan MV,
Remisova M, Nguyen H, Drané P, Beroukhim R, Altmeyer M, Mer G and
Chowdhury D: TIRR inhibits the 53BP1-p53 complex to alter cell-fate
programs. Mol Cell. 81:2583–2595.e6. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Shibata A and Jeggo PA: Roles for the
DNA-PK complex and 53BP1 in protecting ends from resection during
DNA double-strand break repair. J Radiat Res. 61:718–726. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Broustas CG, Duval AJ, Chaudhary KR,
Friedman RA, Virk RK and Lieberman HB: Targeting MEK5 impairs
nonhomologous end-joining repair and sensitizes prostate cancer to
DNA damaging agents. Oncogene. 39:2467–2477. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Sharma A and Almasan A: USP14 regulates
DNA damage response and is a target for radiosensitization in
non-small cell lung cancer. Int J Mol Sci. 21:63832020. View Article : Google Scholar :
|
|
79
|
Feng M, Wang Y, Bi L, Zhang P, Wang H,
Zhao Z, Mao JH and Wei G: CRL4ADTL degrades DNA-PKcs to
modulate NHEJ repair and induce genomic instability and subsequent
malignant transformation. Oncogene. 40:2096–2111. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Caggiano C, Cavallo F, Giannattasio T,
Cappelletti G, Rossi P, Grimaldi P, Feldman DR, Jasin M and Barchi
M: Testicular germ cell tumors acquire cisplatin resistance by
rebalancing the usage of DNA repair pathways. Cancers (Basel).
13:7872021. View Article : Google Scholar
|
|
81
|
Ma S, Rong Z, Liu C, Qin X, Zhang X and
Chen Q: DNA damage promotes microtubule dynamics through a
DNA-PK-AKT axis for enhanced repair. J Cell Biol.
220:e2019110252021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lee KY and Dutta A: Chk1 promotes
non-homologous end joining in G1 through direct phosphorylation of
ASF1A. Cell Rep. 34:1086802021. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wardlaw CP, Carr AM and Oliver AW: TopBP1:
A BRCT-scaffold protein functioning in multiple cellular pathways.
DNA Repair (Amst). 22:165–174. 2014. View Article : Google Scholar
|
|
84
|
Bigot N, Day M, Baldock RA, Watts FZ,
Oliver AW and Pearl LH: Phosphorylation-mediated interactions with
TOPBP1 couple 53BP1 and 9-1-1 to control the G1 DNA damage
checkpoint. Elife. 8:e443532019. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ha K, Ma C, Lin H, Tang L, Lian Z, Zhao F,
Li JM, Zhen B, Pei H, Han S, et al: The anaphase promoting complex
impacts repair choice by protecting ubiquitin signalling at DNA
damage sites. Nat Commun. 8:157512017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Beishline K, Kelly CM, Olofsson BA, Koduri
S, Emrich J, Greenberg RA and Azizkhan-Clifford J: Sp1 facilitates
DNA double-strand break repair through a nontranscriptional
mechanism. Mol Cell Biol. 32:3790–3799. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Swift ML, Beishline K, Flashner S and
Azizkhan-Clifford J: DSB repair pathway choice is regulated by
recruitment of 53BP1 through cell cycle-dependent regulation of
Sp1. Cell Rep. 34:1088402021. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Tang M, Feng X, Pei G, Srivastava M, Wang
C, Chen Z, Li S, Zhang H, Zhao Z, Li X and Chen J: FOXK1
participates in DNA damage response by controlling 53BP1 function.
Cell Rep. 32:1080182020. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Murray-Nerger LA, Justice JL, Rekapalli P,
Hutton JE and Cristea IM: Lamin B1 acetylation slows the G1 to S
cell cycle transition through inhibition of DNA repair. Nucleic
Acids Res. 49:2044–2064. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Svobodová Kovaříková A, Legartová S,
Krejčí J and Bártová E: H3K9me3 and H4K20me3 represent the
epigenetic landscape for 53BP1 binding to DNA lesions. Aging
(Albany NY). 10:2585–2605. 2018. View Article : Google Scholar
|
|
91
|
Nakamura K, Saredi G, Becker JR, Foster
BM, Nguyen NV, Beyer TE, Cesa LC, Faull PA, Lukauskas S, Frimurer
T, et al: H4K20me0 recognition by BRCA1-BARD1 directs homologous
recombination to sister chromatids. Nat Cell Biol. 21:311–318.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Harding SM and Bristow RG: Discordance
between phosphorylation and recruitment of 53BP1 in response to DNA
double-strand breaks. Cell Cycle. 11:1432–1444. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Feng L, Li N, Li Y, Wang J, Gao M, Wang W
and Chen J: Cell cycle-dependent inhibition of 53BP1 signaling by
BRCA1. Cell Discov. 1:150192015. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Bothmer A, Robbiani DF, Di Virgilio M,
Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D,
Callen E, et al: Regulation of DNA end joining, resection, and
immunoglobulin class switch recombination by 53BP1. Mol Cell.
42:319–329. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Sanz-Garcia M, Monsalve DM, Sevilla A and
Lazo PA: Vaccinia-related kinase 1 (VRK1) is an upstream
nucleosomal kinase required for the assembly of 53BP1 foci in
response to ionizing radiation-induced DNA damage. J Biol Chem.
287:23757–23768. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Campillo-Marcos I, Garcia-Gonzalez R,
Navarro-Carrasco E and Lazo PA: The human VRK1 chromatin kinase in
cancer biology. Cancer Lett. 503:117–128. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Jiang Y, Dong Y, Luo Y, Jiang S, Meng FL,
Tan M, Li J and Zang Y: AMPK-mediated phosphorylation on 53BP1
promotes c-NHEJ. Cell Rep. 34:1087132021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Yang Y, Lei T, Du S, Tong R, Wang H, Yang
J, Huang J, Sun M, Wang Y and Dong Z: Nuclear GSK3β induces DNA
double-strand break repair by phosphorylating 53BP1 in
glioblastoma. Int J Oncol. 52:709–720. 2018.PubMed/NCBI
|
|
99
|
Lee DH, Acharya SS, Kwon M, Drane P, Guan
Y, Adelmant G, Kalev P, Shah J, Pellman D, Marto JA and Chowdhury
D: Dephosphorylation enables the recruitment of 53BP1 to
double-strand DNA breaks. Mol Cell. 54:512–525. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Isono M, Niimi A, Oike T, Hagiwara Y, Sato
H, Sekine R, Yoshida Y, Isobe SY, Obuse C, Nishi R, et al: BRCA1
directs the repair pathway to homologous recombination by promoting
53BP1 dephosphorylation. Cell Rep. 18:520–532. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Burdova K, Storchova R, Palek M and
Macurek L: WIP1 promotes homologous recombination and modulates
sensitivity to PARP inhibitors. Cells. 8:12582019. View Article : Google Scholar :
|
|
102
|
Bohgaki M, Bohgaki T, El Ghamrasni S,
Srikumar T, Maire G, Panier S, Fradet-Turcotte A, Stewart GS,
Raught B, Hakem A and Hakem R: RNF168 ubiquitylates 53BP1 and
controls its response to DNA double-strand breaks. Proc Natl Acad
Sci USA. 110:20982–20987. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Wang Y, Hou Y, Zhang W, Alvarez AA, Bai Y,
Hu B, Cheng SY, Yang K, Li Y and Feng H: Lipolytic inhibitor G0S2
modulates glioma stem-like cell radiation response. J Exp Clin
Cancer Res. 38:1472019. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Lee NS, Chang HR, Kim S, Ji JH, Lee J, Lee
HJ, Seo Y, Kang M, Han JS, Myung K, et al: Ring finger protein 126
(RNF126) suppresses ionizing radiation-induced p53-binding protein
1 (53BP1) focus formation. J Biol Chem. 293:588–598. 2018.
View Article : Google Scholar
|
|
105
|
Yang C, Zang W, Tang Z, Ji Y, Xu R, Yang
Y, Luo A, Hu B, Zhang Z, Liu Z and Zheng X: A20/TNFAIP3 regulates
the DNA damage response and mediates tumor cell resistance to
DNA-damaging therapy. Cancer Res. 78:1069–1082. 2018. View Article : Google Scholar
|
|
106
|
Walser F, Mulder MPC, Bragantini B, Burger
S, Gubser T, Gatti M, Botuyan MV, Villa A, Altmeyer M, Neri D, et
al: Ubiquitin phosphorylation at Thr12 modulates the DNA damage
response. Mol Cell. 80:423–436.e9. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Poulsen M, Lukas C, Lukas J, Bekker-Jensen
S and Mailand N: Human RNF169 is a negative regulator of the
ubiquitin-dependent response to DNA double-strand breaks. J Cell
Biol. 197:189–199. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
An L, Dong C, Li J, Chen J, Yuan J, Huang
J, Chan KM, Yu CH and Huen MSY: RNF169 limits 53BP1 deposition at
DSBs to stimulate single-strand annealing repair. Proc Natl Acad
Sci USA. 115:E8286–E8295. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Hu Q, Botuyan MV, Cui G, Zhao D and Mer G:
Mechanisms of ubiquitin-nucleosome recognition and regulation of
53BP1 chromatin recruitment by RNF168/169 and RAD18. Mol Cell.
66:473–487.e9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Hwang JW, Kim SN, Myung N, Song D, Han G,
Bae GU, Bedford MT and Kim YK: PRMT5 promotes DNA repair through
methylation of 53BP1 and is regulated by Src-mediated
phosphorylation. Commun Biol. 3:4282020. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Clarke TL, Sanchez-Bailon MP, Chiang K,
Reynolds JJ, Herrero-Ruiz J, Bandeiras TM, Matias PM, Maslen SL,
Skehel JM, Stewart GS and Davies CC: PRMT5-dependent methylation of
the TIP60 coactivator RUVBL1 is a key regulator of homologous
recombination. Mol Cell. 65:900–916.e7. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Zhang H, Devoucoux M, Song X, Li L, Ayaz
G, Cheng H, Tempel W, Dong C, Loppnau P, Côté J and Min J:
Structural basis for EPC1-mediated recruitment of MBTD1 into the
NuA4/TIP60 acetyltransferase complex. Cell Rep. 30:3996–4002.e4.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Jacquet K, Fradet-Turcotte A, Avvakumov N,
Lambert JP, Roques C, Pandita RK, Paquet E, Herst P, Gingras AC,
Pandita TK, et al: The TIP60 complex regulates bivalent chromatin
recognition by 53BP1 through direct H4K20me binding and H2AK15
acetylation. Mol Cell. 62:409–421. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Sivanand S, Rhoades S, Jiang Q, Lee JV,
Benci J, Zhang J, Yuan S, Viney I, Zhao S, Carrer A, et al: Nuclear
acetyl-CoA production by ACLY promotes homologous recombination.
Mol Cell. 67:252–265.e6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Chen Z, Zhong J, Ren X, Liu W, Wu D, Chen
C, Huang H, Huang X, Liu Y and Liu J: Involvement of a novel
regulatory cascade consisting of SET-H3K18ac/H3K27ac-53BP1 in
Cr(VI)-induced malignant transformation of 16HBE cells. Toxicol
Lett. 339:70–77. 2021. View Article : Google Scholar
|
|
116
|
Guo X, Bai Y, Zhao M, Zhou M, Shen Q, Yun
CH, Zhang H, Zhu WG and Wang J: Acetylation of 53BP1 dictates the
DNA double strand break repair pathway. Nucleic Acids Res.
46:689–703. 2018. View Article : Google Scholar :
|
|
117
|
Li N, Zhang Y, Han X, Liang K, Wang J,
Feng L, Wang W, Songyang Z, Lin C, Yang L, et al: Poly-ADP
ribosylation of PTEN by Tankyrases promotes PTEN degradation and
tumor growth. Genes Dev. 29:157–170. 2015. View Article : Google Scholar :
|
|
118
|
Guettler S, LaRose J, Petsalaki E, Gish G,
Scotter A, Pawson T, Rottapel R and Sicheri F: Structural basis and
sequence rules for substrate recognition by Tankyrase explain the
basis for cherubism disease. Cell. 147:1340–1354. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
DaRosa PA, Wang Z, Jiang X, Pruneda JN,
Cong F, Klevit RE and Xu W: Allosteric activation of the RNF146
ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature.
517:223–226. 2015. View Article : Google Scholar :
|
|
120
|
Zhang Y, Liu S, Mickanin C, Feng Y,
Charlat O, Michaud GA, Schirle M, Shi X, Hild M, Bauer A, et al:
RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin
degradation and Wnt signalling. Nat Cell Biol. 13:623–629. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zhang F, Lou L, Peng B, Song X, Reizes O,
Almasan A and Gong Z: Nudix hydrolase NUDT16 regulates 53BP1
protein by Reversing 53BP1 ADP-ribosylation. Cancer Res.
80:999–1010. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Palazzo L, Thomas B, Jemth AS, Colby T,
Leidecker O, Feijs KL, Zaja R, Loseva O, Puigvert JC, Matic I, et
al: Processing of protein ADP-ribosylation by Nudix hydrolases.
Biochem J. 468:293–301. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Mackay DR, Howa AC, Werner TL and Ullman
KS: Nup153 and Nup50 promote recruitment of 53BP1 to DNA repair
foci by antagonizing BRCA1-dependent events. J Cell Sci.
130:3347–3359. 2017.PubMed/NCBI
|
|
124
|
Duheron V, Nilles N, Pecenko S, Martinelli
V and Fahrenkrog B: Localisation of Nup153 and SENP1 to nuclear
pore complexes is required for 53BP1-mediated DNA double-strand
break repair. J Cell Sci. 130:2306–2316. 2017.PubMed/NCBI
|
|
125
|
Lu WT, Hawley BR, Skalka GL, Baldock RA,
Smith EM, Bader AS, Malewicz M, Watts FZ, Wilczynska A and Bushell
M: Drosha drives the formation of DNA:RNA hybrids around DNA break
sites to facilitate DNA repair. Nat Commun. 9:5322018. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Cabrini M, Roncador M, Galbiati A, Cipolla
L, Maffia A, Iannelli F, Sabbioneda S, d'Adda di Fagagna F and
Francia S: DROSHA is recruited to DNA damage sites by the MRN
complex to promote non-homologous end joining. J Cell Sci.
134:jcs2497062021. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Wikiniyadhanee R, Lerksuthirat T,
Stitchantrakul W, Chitphuk S, Sura T and Dejsuphong D: TRIM29 is
required for efficient recruitment of 53BP1 in response to DNA
double-strand breaks in vertebrate cells. FEBS Open Bio.
10:2055–2071. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Alonso-de Vega I, Paz-Cabrera MC, Rother
MB, Wiegant WW, Checa-Rodriguez C, Hernandez-Fernaud JR, Huertas P,
Freire R, van Attikum H and Smits VAJ: PHF2 regulates
homology-directed DNA repair by controlling the resection of DNA
double strand breaks. Nucleic Acids Res. 48:4915–4927. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Koo GB, Ji JH, Cho H, Morgan MJ and Kim
YS: Nuclear TRADD prevents DNA damage-mediated death by
facilitating non-homologous end-joining repair. Sci Rep.
7:33322017. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Clements KE, Schleicher EM, Thakar T, Hale
A, Dhoonmoon A, Tolman NJ, Sharma A, Liang X, Imamura Kawasawa Y,
Nicolae CM, et al: Identification of regulators of poly-ADP-ribose
polymerase inhibitor response through complementary CRISPR knockout
and activation screens. Nat Commun. 11:61182020. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Shenoy TR, Boysen G, Wang MY, Xu QZ, Guo
W, Koh FM, Wang C, Zhang LZ, Wang Y, Gil V, et al: CHD1 loss
sensitizes prostate cancer to DNA damaging therapy by promoting
error-prone double-strand break repair. Ann Oncol. 28:1495–1507.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Fouquin A, Guirouilh-Barbat J, Lopez B,
Hall J, Amor-Guéret M and Pennaneach V: PARP2 controls
double-strand break repair pathway choice by limiting 53BP1
accumulation at DNA damage sites and promoting end-resection.
Nucleic Acids Res. 45:12325–12339. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Zimmermann M and de Lange T: 53BP1: Pro
choice in DNA repair. Trends Cell Biol. 24:108–117. 2014.
View Article : Google Scholar :
|
|
134
|
Escribano-Diaz C and Durocher D: DNA
repair pathway choice-a PTIP of the hat to 53BP1. EMBO Rep.
14:665–666. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Lottersberger F, Bothmer A, Robbiani DF,
Nussenzweig MC and de Lange T: Role of 53BP1 oligomerization in
regulating double-strand break repair. Proc Natl Acad Sci USA.
110:2146–2151. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Manis JP, Morales JC, Xia Z, Kutok JL, Alt
FW and Carpenter PB: 53BP1 links DNA damage-response pathways to
immunoglobulin heavy chain class-switch recombination. Nat Immunol.
5:481–487. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
137
|
Methot SP and Di Noia JM: Molecular
mechanisms of somatic hypermutation and class switch recombination.
Adv Immunol. 133:37–87. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Gupta R, Somyajit K, Narita T, Maskey E,
Stanlie A, Kremer M, Typas D, Lammers M, Mailand N, Nussenzweig A,
et al: DNA repair network analysis reveals shieldin as a key
regulator of NHEJ and PARP inhibitor sensitivity. Cell.
173:972–988.e23. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Ghezraoui H, Oliveira C, Becker JR, Bilham
K, Moralli D, Anzilotti C, Fischer R, Deobagkar-Lele M,
Sanchiz-Calvo M, Fueyo-Marcos E, et al: 53BP1 cooperation with the
REV7-shieldin complex underpins DNA structure-specific NHEJ.
Nature. 560:122–127. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Noordermeer SM, Adam S, Setiaputra D,
Barazas M, Pettitt SJ, Ling AK, Olivieri M, Aacute;lvarez-Quilón A,
Moatti N, Zimmermann M, et al: The shieldin complex mediates
53BP1-dependent DNA repair. Nature. 560:117–121. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Liang L, Feng J, Zuo P, Yang J, Lu Y and
Yin Y: Molecular basis for assembly of the shieldin complex and its
implications for NHEJ. Nat Commun. 11:19722020. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Bochkarev A and Bochkareva E: From RPA to
BRCA2: Lessons from single-stranded DNA binding by the OB-fold.
Curr Opin Struct Biol. 14:36–42. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Findlay S, Heath J, Luo VM, Malina A,
Morin T, Coulombe Y, Djerir B, Li Z, Samiei A, Simo-Cheyou E, et
al: SHLD2/FAM35A co-operates with REV7 to coordinate DNA
double-strand break repair pathway choice. EMBO J. 37:e1001582018.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Gao S, Feng S, Ning S, Liu J, Zhao H, Xu
Y, Shang J, Li K, Li Q, Guo R and Xu D: An OB-fold complex controls
the repair pathways for DNA double-strand breaks. Nat Commun.
9:39252018. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Cannavo E and Cejka P: Sae2 promotes dsDNA
endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks.
Nature. 514:122–125. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Anand R, Ranjha L, Cannavo E and Cejka P:
Phosphorylated CtIP functions as a co-factor of the
MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol Cell.
64:940–950. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Nimonkar AV, Genschel J, Kinoshita E,
Polaczek P, Campbell JL, Wyman C, Modrich P and Kowalczykowski SC:
BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end
resection machineries for human DNA break repair. Genes Dev.
25:350–362. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Gravel S, Chapman JR, Magill C and Jackson
SP: DNA helicases Sgs1 and BLM promote DNA double-strand break
resection. Genes Dev. 22:2767–2772. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Dev H, Chiang TW, Lescale C, de Krijger I,
Martin AG, Pilger D, Coates J, Sczaniecka-Clift M, Wei W,
Ostermaier M, et al: Shieldin complex promotes DNA end-joining and
counters homologous recombination in BRCA1-null cells. Nat Cell
Biol. 20:954–965. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Bhattacharjee A, Wang Y, Diao J and Price
CM: Dynamic DNA binding, junction recognition and G4 melting
activity underlie the telomeric and genome-wide roles of human CST.
Nucleic Acids Res. 45:12311–12324. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Barazas M, Annunziato S, Pettitt SJ, de
Krijger I, Ghezraoui H, Roobol SJ, Lutz C, Frankum J, Song FF,
Brough R, et al: The CST complex mediates end protection at
double-strand breaks and promotes PARP inhibitor sensitivity in
BRCA1-deficient cells. Cell Rep. 23:2107–2118. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Mirman Z, Lottersberger F, Takai H, Kibe
T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D and de
Lange T: 53BP1-RIF1-shieldin counteracts DSB resection through CST-
and Polα-dependent fill-in. Nature. 560:112–116. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Pellegrini L: The Pol α-primase complex.
Subcell Biochem. 62:157–169. 2012. View Article : Google Scholar
|
|
154
|
Sfeir A and Symington LS:
Microhomology-mediated end joining: A back-up survival mechanism or
dedicated pathway? Trends Biochem Sci. 40:701–714. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Muñoz IM and Rouse J: Control of histone
methylation and genome stability by PTIP. EMBO Rep. 10:239–245.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Munoz IM, Jowsey PA, Toth R and Rouse J:
Phospho-epitope binding by the BRCT domains of hPTIP controls
multiple aspects of the cellular response to DNA damage. Nucleic
Acids Res. 35:5312–5322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Callen E, Faryabi RB, Luckey M, Hao B,
Daniel JA, Yang W, Sun HW, Dressler G, Peng W, Chi H, et al: The
DNA damage- and transcription-associated protein paxip1 controls
thymocyte development and emigration. Immunity. 37:971–985. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Wang J, Aroumougame A, Lobrich M, Li Y,
Chen D, Chen J and Gong Z: PTIP associates with Artemis to dictate
DNA repair pathway choice. Genes Dev. 28:2693–2698. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Ma Y, Pannicke U, Schwarz K and Lieber MR:
Hairpin opening and overhang processing by an Artemis/DNA-dependent
protein kinase complex in nonhomologous end joining and V(D) J
recombination. Cell. 108:781–794. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Goodarzi AA, Yu Y, Riballo E, Douglas P,
Walker SA, Ye R, Härer C, Marchetti C, Morrice N, Jeggo PA and
Lees-Miller SP: DNA-PK autophosphorylation facilitates Artemis
endonuclease activity. EMBO J. 25:3880–3889. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Park SJ, Gavrilova O, Brown AL, Soto JE,
Bremner S, Kim J, Xu X, Yang S, Um JH, Koch LG, et al: DNA-PK
promotes the mitochondrial, metabolic, and physical decline that
occurs during aging. Cell Metab. 26:4472017. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Kawale AS, Akopiants K, Valerie K, Ruis B,
Hendrickson EA, Huang SN, Pommier Y and Povirk LF: TDP1 suppresses
mis-joining of radiomimetic DNA double-strand breaks and cooperates
with Artemis to promote optimal nonhomologous end joining. Nucleic
Acids Res. 46:8926–8939. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Morales JC, Richard P, Rommel A, Fattah
FJ, Motea EA, Patidar PL, Xiao L, Leskov K, Wu SY, Hittelman WN, et
al: Kub5-Hera, the human Rtt103 homolog, plays dual functional
roles in transcription termination and DNA repair. Nucleic Acids
Res. 42:4996–5006. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Han J, Ruan C, Huen MSY, Wang J, Xie A, Fu
C, Liu T and Huang J: BRCA2 antagonizes classical and alternative
nonhomologous end-joining to prevent gross genomic instability. Nat
Commun. 8:14702017. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Isobe SY, Hiraga SI, Nagao K, Sasanuma H,
Donaldson AD and Obuse C: Protein phosphatase 1 acts as a RIF1
effector to suppress DSB resection prior to Shieldin action. Cell
Rep. 36:1093832021. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Spyropoulou Z, Papaspyropoulos A, Lagopati
N, Myrianthopoulos V, Georgakilas AG, Fousteri M, Kotsinas A and
Gorgoulis VG: Cockayne syndrome group B (CSB): The regulatory
framework governing the multifunctional protein and its plausible
role in cancer. Cells. 10:8662021. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Batenburg NL, Walker JR, Noordermeer SM,
Moatti N, Durocher D and Zhu XD: ATM and CDK2 control chromatin
remodeler CSB to inhibit RIF1 in DSB repair pathway choice. Nat
Commun. 8:19212017. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Batenburg NL, Qin J, Walker JR and Zhu XD:
Efficient UV repair requires disengagement of the CSB winged helix
domain from the CSB ATPase domain. DNA Repair (Amst). 68:58–67.
2018. View Article : Google Scholar
|
|
169
|
Batenburg NL, Walker JR, Coulombe Y,
Sherker A, Masson JY and Zhu XD: CSB interacts with BRCA1 in late
S/G2 to promote MRN- and CtIP-mediated DNA end resection. Nucleic
Acids Res. 47:10678–10692. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Kreßner C, Nollau P, Grosse R and Brandt
DT: Functional interaction of SCAI with the SWI/SNF complex for
transcription and tumor cell invasion. PLoS One. 8:e699472013.
View Article : Google Scholar
|
|
171
|
Hansen RK, Mund A, Poulsen SL, Sandoval M,
Klement K, Tsouroula K, Tollenaere MA, Räschle M, Soria R,
Offermanns S, et al: SCAI promotes DNA double-strand break repair
in distinct chromosomal contexts. Nat Cell Biol. 18:1357–1366.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Isobe SY, Nagao K, Nozaki N, Kimura H and
Obuse C: Inhibition of RIF1 by SCAI allows BRCA1-mediated repair.
Cell Rep. 20:297–307. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Parvin S, Ramirez-Labrada A, Aumann S, Lu
X, Weich N, Santiago G, Cortizas EM, Sharabi E, Zhang Y,
Sanchez-Garcia I, et al: LMO2 confers synthetic lethality to PARP
inhibition in DLBCL. Cancer Cell. 36:237–249.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Bonanno L, Costa C, Majem M, Sanchez JJ,
Gimenez-Capitan A, Rodriguez I, Vergnenegre A, Massuti B, Favaretto
A, Rugge M, et al: The predictive value of 53BP1 and BRCA1 mRNA
expression in advanced non-small-cell lung cancer patients treated
with first-line platinum-based chemotherapy. Oncotarget.
4:1572–1581. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Schouten PC, Vollebergh MA, Opdam M,
Jonkers M, Loden M, Wesseling J, Hauptmann M and Linn SC: High XIST
and low 53BP1 expression predict poor outcome after high-dose
alkylating chemotherapy in patients with a BRCA1-like breast
cancer. Mol Cancer Ther. 15:190–198. 2016. View Article : Google Scholar
|
|
176
|
Zhang T, Chai J and Chi L: Induction Of
XLF And 53BP1 expression is associated with temozolomide resistance
in glioblastoma cells. Onco Targets Ther. 12:10139–10151. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Yao J, Huang A, Zheng X, Liu T, Lin Z,
Zhang S, Yang Q, Zhang T and Ma H: 53BP1 loss induces
chemoresistance of colorectal cancer cells to 5-fluorouracil by
inhibiting the ATM-CHK2-P53 pathway. J Cancer Res Clin Oncol.
143:419–431. 2017. View Article : Google Scholar
|
|
178
|
Krais JJ, Wang Y, Bernhardy AJ, Clausen E,
Miller JA, Cai KQ, Scott CL and Johnson N: RNF168-mediated
ubiquitin signaling inhibits the viability of BRCA1-null cancers.
Cancer Res. 80:2848–2860. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Belotserkovskaya R, Raga Gil E, Lawrence
N, Butler R, Clifford G, Wilson MD and Jackson SP: PALB2 chromatin
recruitment restores homologous recombination in BRCA1-deficient
cells depleted of 53BP1. Nat Commun. 11:8192020. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Miyamoto K, Minegaki T, Hirano S, Hayashi
I, Tsujimoto M and Nishiguchi K: Olaparib potentiates anticancer
drug cytotoxicity via 53BP1 in oesophageal squamous cell carcinoma
cells. Anticancer Res. 40:813–823. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Mu F, Liu T, Zheng H, Xie X, Lei T, He X,
Du S, Tong R and Wang Y: Mangiferin induces radiosensitization in
glioblastoma cells by inhibiting nonhomologous end joining. Oncol
Rep. 40:3663–3673. 2018.PubMed/NCBI
|
|
182
|
Pustovalova M, Alhaddad L, Smetanina N,
Chigasova A, Blokhina T, Chuprov-Netochin R, Osipov AN and Leonov
S: The p53-53BP1-related survival of A549 and H1299 human lung
cancer cells after multifractionated radiotherapy demonstrated
different response to additional acute X-ray exposure. Int J Mol
Sci. 21:33422020. View Article : Google Scholar
|
|
183
|
Eke I, Zong D, Aryankalayil MJ, Sandfort
V, Bylicky MA, Rath BH, Graves EE, Nussenzweig A and Coleman CN:
53BP1/RIF1 signaling promotes cell survival after multifractionated
radiotherapy. Nucleic Acids Res. 48:1314–1326. 2020. View Article : Google Scholar :
|
|
184
|
Roobol SJ, van den Bent I, van Cappellen
WA, Abraham TE, Paul MW, Kanaar R, Houtsmuller AB, van Gent DC and
Essers J: Comparison of high- and low-LET radiation-induced DNA
double-strand break processing in living cells. Int J Mol Sci.
21:66022020. View Article : Google Scholar :
|
|
185
|
Oizumi T, Ohno R, Yamabe S, Funayama T and
Nakamura AJ: Repair kinetics of DNA double strand breaks induced by
simulated space radiation. Life (Basel). 10:3412020.
|
|
186
|
Anglada T, Genescà A and Martin M:
Age-associated deficient recruitment of 53BP1 in G1 cells directs
DNA double-strand break repair to BRCA1/CtIP-mediated DNA-end
resection. Aging (Albany NY). 12:24872–24893. 2020. View Article : Google Scholar
|
|
187
|
Li N, Wang J, Wang X, Sun J and Li Z:
Icariin exerts a protective effect against d-galactose induced
premature ovarian failure via promoting DNA damage repair. Biomed
Pharmacother. 118:1092182019. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Zhang L, Geng X, Wang F, Tang J, Ichida Y,
Sharma A, Jin S, Chen M, Tang M, Pozo FM, et al: 53BP1 regulates
heterochromatin through liquid phase separation. Nat Commun.
13:3602022. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Dimitrova N, Chen YC, Spector DL and de
Lange T: 53BP1 promotes non-homologous end joining of telomeres by
increasing chromatin mobility. Nature. 456:524–528. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Yang X, Xu B, Mulvey B, Evans M, Jordan S,
Wang YD, Pagala V, Peng J, Fan Y, Patel A and Peng JC:
Differentiation of human pluripotent stem cells into neurons or
cortical organoids requires transcriptional co-regulation by UTX
and 53BP1. Nat Neurosci. 22:362–373. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Lambrus BG, Daggubati V, Uetake Y, Scott
PM, Clutario KM, Sluder G and Holland AJ: A USP28-53BP1-p53-p21
signaling axis arrests growth after centrosome loss or prolonged
mitosis. J Cell Biol. 214:143–153. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Phan TP, Maryniak AL, Boatwright CA, Lee
J, Atkins A, Tijhuis A, Spierings DC, Bazzi H, Foijer F, Jordan PW,
et al: Centrosome defects cause microcephaly by activating the
53BP1-USP28-TP53 mitotic surveillance pathway. EMBO J.
40:e1061182021. View Article : Google Scholar
|
|
193
|
Damen M, Wirtz L, Soroka E, Khatif H,
Kukat C, Simons BD and Bazzi H: High proliferation and delamination
during skin epidermal stratification. Nat Commun. 12:32272021.
View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Turocy J, Adashi EY and Egli D: Heritable
human genome editing: Research progress ethical considerations, and
hurdles to clinical practice. Cell. 184:1561–1574. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Yang H, Ren S, Yu S, Pan H, Li T, Ge S,
Zhang J and Xia N: Methods favoring homology-directed repair choice
in response to CRISPR/Cas9 induced-double strand breaks. Int J Mol
Sci. 21:64612020. View Article : Google Scholar :
|
|
196
|
Canny MD, Moatti N, Wan LCK,
Fradet-Turcotte A, Krasner D, Mateos-Gomez PA, Zimmermann M,
Orthwein A, Juang YC, Zhang W, et al: Inhibition of 53BP1 favors
homology-dependent DNA repair and increases CRISPR-Cas9
genome-editing efficiency. Nat Biotechnol. 36:95–102. 2018.
View Article : Google Scholar :
|
|
197
|
Sun Y, Lu H, Fang X, Xiao S, Yang F, Chen
Y, Wang H, Li X, Lu J, Lin H, et al: Discovery of a novel 53BP1
inhibitor through AlphaScreen-based high-throughput screening.
Bioorg Med Chem. 34:1160542021. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Paulsen BS, Mandal PK, Frock RL, Boyraz B,
Yadav R, Upadhyayula S, Gutierrez-Martinez P, Ebina W, Fasth A,
Kirchhausen T, et al: Ectopic expression of RAD52 and dn53BP1
improves homology-directed repair during CRISPR-Cas9 genome
editing. Nat Biomed Eng. 1:878–888. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Nambiar TS, Billon P, Diedenhofen G,
Hayward SB, Taglialatela A, Cai K, Huang JW, Leuzzi G,
Cuella-Martin R, Palacios A, et al: Stimulation of CRISPR-mediated
homology-directed repair by an engineered RAD18 variant. Nat
Commun. 10:33952019. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Jayavaradhan R, Pillis DM, Goodman M,
Zhang F, Zhang Y, Andreassen PR and Malik P: CRISPR-Cas9 fusion to
dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically
at Cas9 target sites. Nat Commun. 10:28662019. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Markiewicz-Potoczny M, Lobanova A, Loeb
AM, Kirak O, Olbrich T, Ruiz S and Lazzerini Denchi E:
TRF2-mediated telomere protection is dispensable in pluripotent
stem cells. Nature. 589:110–115. 2021. View Article : Google Scholar
|
|
202
|
Vančevska A, Ahmed W, Pfeiffer V,
Feretzaki M, Boulton SJ and Lingner J: SMCHD1 promotes
ATM-dependent DNA damage signaling and repair of uncapped
telomeres. EMBO J. 39:e1026682020. View Article : Google Scholar
|