Introduction
Acute kidney injury (AKI) is a serious syndrome
characterized by a rapid decline in kidney function, with high
mortality and no effective therapy currently. Renal
ischemia-reperfusion (I/R) injury is the most common cause of AKI,
where the proximal tubule is the mainstay of injury (1). Increasing evidence indicates that
severity of proximal tubule injury determines renal prognosis
(2). More importantly, promoting
tubular repair protects from the ischemic AKI (3). However, the exact mechanism of
tubule injury remains poorly understood.
Histologically, tubule injury and infiltration of
immune cells are the characteristic pathological changes of I/R
injury. Among immune cell populations, the macrophage is the major
contributor to renal injury. For example, pharmacologic strategies
with deletion of macrophages effectively protects the kidney from
injury induced by ischemic, obstructive or toxic insults (4). Interestingly, in addition to the
change in quantity, phenotype switching of macrophages also plays a
crucial role in the prognosis of kidney diseases (5). However, the molecular signals
through which macrophages induce tubule cell injury during AKI
remains to be elucidated.
Exosomes are nanometer-scale, membrane-enclosed
extracellular vesicles, which are released by almost all cell types
under physiological and pathological conditions. Over the past
decades, increasing studies have determined that exosomes can
mediate intracellular communication by transferring cell-specific
cargos, including proteins, lipids and genetic information (such as
DNA, mRNA, and microRNA (miR), to target cells, even at a distance
from the parent cells (6,7). Recently, convincing studies
demonstrated that under some specific conditions, increased
production of exosomes is induced and content of exosomes is also
modulated to regulate the key biological functions of recipient
cells (8,9). Interestingly, exosomes are also
found to mediate the cross-talk between tubular epithelial cells
(TECs) and fibroblasts in kidney fibrosis (10). In addition, exosomes derived from
injured TECs can transfer specific mRNAs into macrophages to alter
the biological functions of macrophages in both AKI and chronic
kidney disease (11). However,
whether exosomes derived from macrophages influence the function of
TECs has not been reported.
miRNAs are a class of epigenetic regulators with the
capability of modulating gene expression at the
post-transcriptional level, which plays important roles in kidney
diseases. A previous study revealed that miRNAs are commonly
enriched in exosomes (12).
Interestingly, the miRNA class was observed to be the largest and
most consistent proportional change in exosomes using total
RNA-sequencing in a model of I/R (12), suggesting that miRNAs are emerging
as crucial regulators of cellular function through exosome-mediated
cellular communication. Notably, Li et al found that miR-23a
which transfers from tubules to immune cells through exosomes could
promote kidney injury (13). In
addition, tubular cells could also take up exosomal miRNAs to
regulate acute tubular injury as previously reported (14). However, the role of
macrophage-derived exosomal miRNAs in tubular injury remains
largely unclear.
Recently, increasing evidence demonstrated that
miR-155 expression in the M1 polarized macrophage was significantly
upregulated (GSE33453). Notably, the level of miR-155 was
significantly enhanced in the exosomes derived from M1 macrophages
as well. Furthermore, transfer of miR-155 via exosomes could
modulate various pathophysiological functions (15,16). In the present study, it is
suggested that exosomal miR-155 released by macrophages could
promote tubular injury by conveying the injury signals. Elucidating
the exact mechanism underlying tubular injury in AKI not only
provides novel insights into the pathophysiology of tubular injury
but also offers a new therapeutic strategy for kidney diseases.
Materials and methods
Reagents
Lipopolysaccharides (LPS) from Escherichia
coli 0111:B4 (product no. L3012) were purchased from
Sigma-Aldrich; Merck KGaA. Recombinant mouse interferon-γ (IFN-γ;
cat. no. 485-MI-100; R&D Systems, Inc.) was used for macrophage
intervention. The levels of plasma blood urea nitrogen (BUN; cat.
no. C013-2-1) and serum creatinine (SCr; cat. no. C011-2-1) were
measured using commercial kits (Nanjing Jiancheng Bioengineering
Institute) according to the manufacturer's instructions.
Animals
The experimental animal procedures were approved
(approval no. N2017-078) by the Ethics Committee of Soochow
University (Suzhou, China). All the mice were housed under
pathogen-free conditions in a standard laboratory with a controlled
room temperature (22±1°C) and humidity (65-70%), and a 12:12-h
light-dark cycle, with free access to food and water. For the
ischemic AKI model (n=6), bilateral I/R injury was induced in mice
by clamping the renal pedicle for 30 min as previously described
(17). Mice were sacrificed by
cervical dislocation at 24 h after I/R injury, and the renal cortex
was harvested. Mice in the GW4869 group received intraperitoneal
injection of GW4869 (2.5 mg/kg; Sigma-Aldrich; Merck KGaA; n=6) 2 h
before I/R surgery. The intraparenchymal injection (n=6) was
performed as previously described (18). Briefly, the inferior pole of the
left kidney was exposed. Exosomes derived from M1 or M0 macrophages
(20 µg in 60 µl of PBS) were injected into 2 sites
(the left and right sides of the inferior pole of the kidney) via a
50-G needle. The in vivo miRNA-155 inhibitor (Suzhou
GenePharma Co., Ltd.) was transfected into the mouse kidneys
through tail vein injections using the in vivo-jetPEI
(Polyplus-transfection SA). Briefly, the miR-155 inhibitor (50
µg) or negative control (NC; 50 µg) dissolved in 5%
glucose solution was injected into each mouse at 24 h before
surgery as instructed by the manufacturer's protocols. Mice were
sacrificed at 24 h after I/R, and their serum and renal cortex were
harvested. To ameliorate the suffering of mice throughout the
experimental period, the mice were euthanized with isoflurane
inhalation followed by the dislocation of the cervical vertebra,
where isoflurane was used at 4% for induction and 2% for
maintenance of anesthesia. Animal sacrifice was confirmed by
respiratory and cardiac arrest, and no righting reflex.
Kidney histology and tubular injury
scoring
Periodic Acid-Schiff (PAS) staining was performed.
Kidneys were fixed in 10% buffered formalin at room temperature for
24 h, embedded in paraffin, cut into 3-µm sections, stained
with Periodic Acid for 20 min and Schiff for 20 min at room
temperature, and visualized at a magnification of ×400 under an
optical microscope (Olympus Optical Co., Ltd.). To evaluate the
tubular injury score, 10 random tissue section images per animal
were assessed on PAS-stained sections in a blinded manner by
nephropathologists, and semi-quantitatively scored as previously
described (19).
Immunohistochemical staining
For immunohistochemical staining, 10% buffered
formalin-fixed at room temperature for 24 h, and paraffin-embedded
tissue sections (4 µm) were blocked with 10% goat serum
(Wuhan Servicebio Technology Co., Ltd.) for 2 h at room temperature
and were incubated with primary antibodies against F4/80 (1:200;
product code ab6640; Abcam) and kidney injury molecule-1 (KIM-1)
(1:200; cat. no. MA5-28211; Invitrogen; Thermo Fisher Scientific,
Inc.) for 12 h at 4°C and then analyzed using a streptavidin
peroxidase detection system (50 µl; cat. no. KIT-9720;
Fuzhou Maixin Biotech Co., Ltd.) at room temperature according to
the manufacturer's protocol. DAB (Fuzhou Maixin Biotech Co., Ltd.)
was used as an HRP-specific substrate. The images were visualized
under an optical microscope (Olympus Corporation).
Cell culture
TECs were isolated for primary culture using an
established method (20) and then
were cultured in DMEM-Ham's-F12 medium (Hyclone; Cytiva)
supplemented with 10% fetal bovine serum (FBS; ScienceCell Research
Laboratories, Inc.), and 1% penicillin-streptomycin (Gibco; Thermo
Fisher Scientific, Inc.). Ischemia/reperfusion injury (I/RI)
conditions of TECs were modeled in vitro (1% oxygen, 94%
N2 and 5% CO2 with glucose-free and FBS-free
for 12 h and then regular culture medium with 21% oxygen for 2 h of
reoxygenation). The mouse RAW264.7 macrophage cell line (American
Type Culture Collection; ATCC no. TIB-71) was used for this study.
RAW264.7 cells were cultured in RPMI-1640 (Hyclone; Cytiva)
supplemented with 10% FBS and 1% penicillin-streptomycin. Both cell
lines (TECs and RAW264.7 macrophages) were cultured in an
atmosphere of 5% CO2 and 95% air at 37°C.
Isolation and characterization of
exosomes
For kidney exosome extraction, 100 mg of the kidney
cortex derived from the ischemic kidney was collected. Renal
exosomes were isolated using ultracentrifugation as previously
reported (21). For in
vitro experiments, RAW264.7 macrophages were cultured with the
presence or absence of LPS (100 ng/ml) plus IFN-γ (20 ng/ml) in
serum-free RPMI-1640 medium. The medium was then used for exosome
purification using differential ultracentrifugation (Type 70 Ti
Rotor; Beckman Coulter Optima L-80 XP) as previously described
(21). Briefly, the medium was
centrifuged at 2,000 × g for 20 min at 4°C to eliminate the cells
and debris and at 13,500 × g for 20 min at 4°C to eliminate the
microvesicles, followed by ultracentrifugation at 200,000 × g for
120 min at 4°C. The exosome pellet was washed in 20 ml of PBS and
collected by subsequent ultracentrifugation at 200,000 × g for 120
min at 4°C.
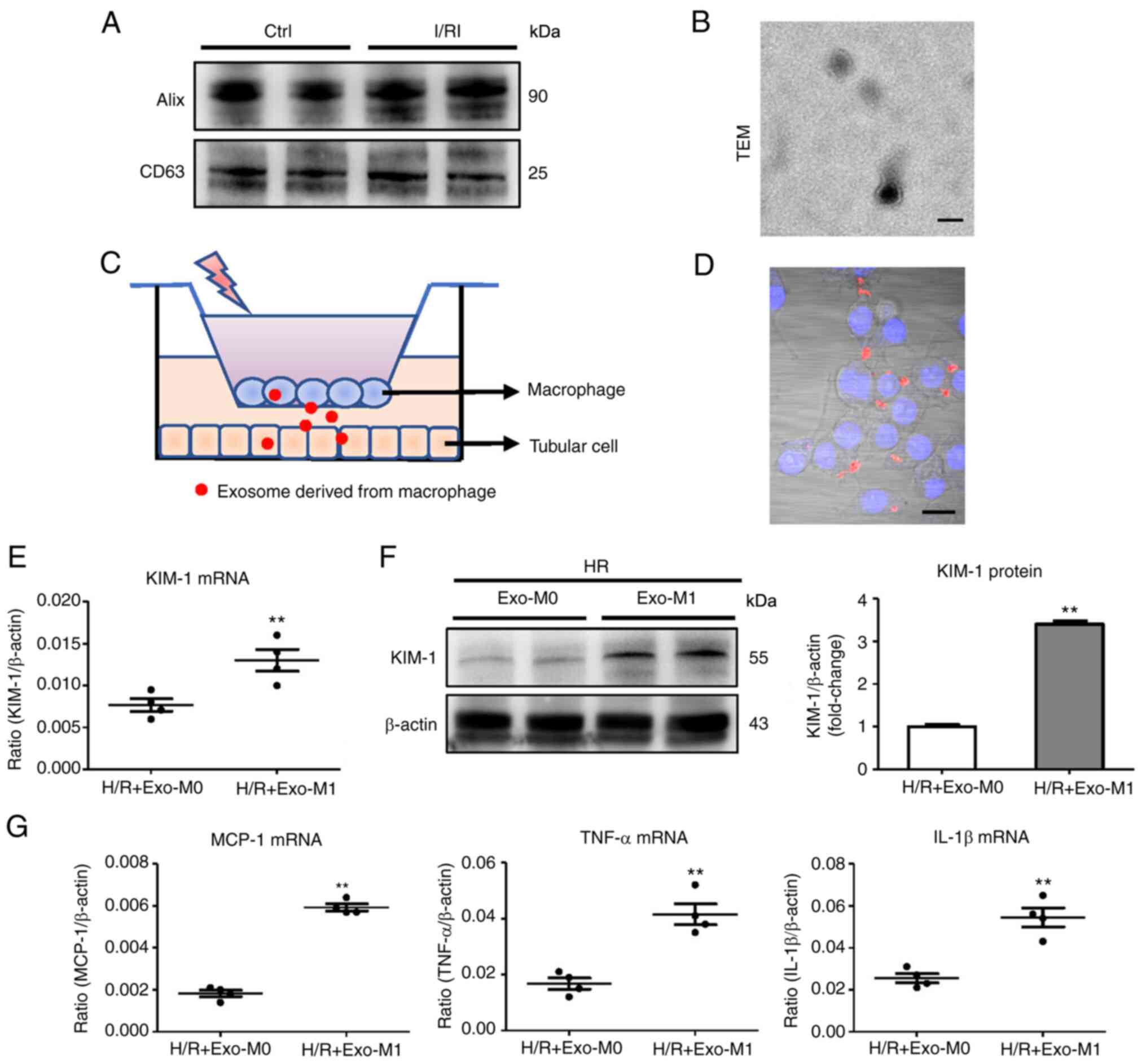
The size and morphology were detected by electron
microscopy and the specific surface markers (Alix, CD63, CD9) of
the isolated exosomes were also detected by western blotting for
characterization of the exosomes.
Transmission electron microscope
(TEM)
The exosome sample was diluted 10 times with PBS and
then stained with 2% phosphotungstic acid for 10 min at room
temperature. The samples were detected using a TEM (Hitachi HT
7700; Hitachi, Ltd.) at 80 kV.
Transwell study
To study the process of macrophage-derived exosomes
communicating with TECs, a Transwell Permeable Support system
(Corning, Inc.) with a 0.4-µm pore-size filter was used
according to the manufacturer's protocol. Macrophages
(2×106) were seeded into the upper chamber containing
RPMI-1640 medium supplemented with 10% FBS. Subsequently, recipient
TECs were then seeded in 12-well plates (lower chamber) in
DMEM-Ham's-F12 medium supplemented with 10% FBS. Both cell lines
were cultured for 12 h at an atmosphere of 5% CO2 and
95% air at 37°C. Macrophages with LPS plus IFN-γ treatment were
then assessed in the upper chamber followed by DiO-labeling for 6 h
at 37°C. All co-cultured experiments were then conducted in hypoxia
or normoxia for 12 h. Uptake of DiO-labeled exosomes by TECs was
visualized by the confocal microscope (FV1000; Olympus
Corporation).
Bioinformatics analysis
m iR NA ta rgets were predicted using 5 online
databases: TargetScan (Human 8.0/Mouse 8.0; https://www.targetscan.org/mmu_80/), miRDB (http://www.mirdb.org/), miRanda (http://www.microrna.org/microrna/home.do),
DIANA-TarBase (v7.0; http://www.microrna.gr/tarbase), and PicTar
(http://www.pictar.org/).
miRNA inhibitor interference studies
RAW264.7 cells were transfected with NC inhibitor
(forward, 5′-CCC CCC CCC CCC CCC CCC CC-3′ and reverse, 5′-CCC CCC
CCC CCC CCC CCC CC-3′) or miR-155 inhibitor (forward, 5′-ACC CCU
AUC ACG AUU AGC AUU AA-3′ and reverse, 5′-ACG UGA CAC GUU CGG AGA
ATT-3′) at a concentration of 100 nM using Lipofectamine 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer′s protocol. Following transfection for 6 h at 37°C,
the medium was removed and fresh medium was added. The subsequent
experiments were performed after 18 h of transfection.
RNA isolation and reverse
transcription-quantitative (RT-q) PCR
Total RNA was extracted using TRIzol following the
manufacturer's instructions (Takara Bio, Inc.). mRNA was
reverse-transcribed using PrimeScript RT reagent kit (Takara Bio,
Inc.) according to the manufacturer's instructions and PCR was
performed using SYBR Premix Ex Taq and 7300 Real-Time PCR System
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The
amplification level was programmed with a denaturation step at 95°C
for 30 sec, followed by 40 cycles at 95°C for 10 sec and 60°C for
30 sec. miRNA was reverse-transcribed and detected with All-in-One
miRNA First-Strand cDNA Synthesis kit and All-in-One miRNA qPCR kit
(GeneCopoeia, Inc.) according to the manufacturer's instructions.
The housekeeping genes β-actin and U6 were used as controls. The
2−ΔΔCq method was used as previously reported (22). All the primers for RT-qPCR are
listed in Table I.
 | Table IPrimers used for
quantitative-PCR. |
Table I
Primers used for
quantitative-PCR.
| Genes | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| Mouse IL-1β |
TGCCACCTTTTGACAGTGATG |
AAGGTCCACGGGAAAGACAC |
| Mouse TNF-α |
TCTTCTCATTCCTGCTTGTGG |
GGTCTGGGCCATAGAACTGA |
| Mouse MCP-1 |
CATCCACGTGTTGGCTCA |
GATCATCTTGCTGGTGAATGAGT |
| Mouse SOCS-1 |
CACTCACTTCCGCACCTTCC |
CAGCCGGTCAGATCTGGAAG |
| Mouse KIM-1 |
CGGTACAACTTAAAGGGGCA |
GACGTGTGGGAATCTCTGGT |
| Mouse β-actin |
GAGACCTTCAACACCCCAGC |
ATGTCACGCACGATTTCCC |
| Mouse miR-155 |
GGGGGTTAATGCTAATTGTGAT | AGTGCGTGTCGTGG |
| Mouse U6 |
CTCGCTTCGGCAGCACA |
AACGCTTCACGAATTTGCGT |
Western blot analysis
Samples of cells and cortical tissues were lysed in
cold RIPA lysis buffer (Thermo Fisher Scientific, Inc.)
supplemented with protease inhibitor cocktail and the protein
concentration was determined using a BCA protein assay kit (Nanjing
KeyGen Biotech Co., Ltd.). Proteins (20-40 µg) were
subjected to 10% SDS-PAGE (Thermo Fisher Scientific, Inc.) and
transferred to PVDF membranes (EMD Millipore). The membranes were
blocked with 5% non-fat milk for 2 h at room temperature. The
primary antibodies used were anti-Alix (1:500; cat. no. sc-53540;
Santa Cruz Biotechnology, Inc.), anti-CD63 (1:1,000; product code
ab213090; Abcam), anti-suppressor of cytokine signaling-1 (SOCS-1;
1:1,000; product code ab62584; Abcam), and anti-β-actin (1:3,000;
cat. no. sc-47778; Santa Cruz Biotechnology, Inc.) and the
membranes were incubated with the primary antibodies for 12 h at
4°C. Goat anti-mouse or anti-rabbit horseradish
peroxidase-conjugated secondary antibodies (1:3,000; product nos.
7076 and 7074, respectively; Cell Signaling Technology, Inc.) were
used for detection for 2 h at room temperature. The signals were
then detected using an enhanced chemiluminescent kit (GE
Healthcare; Cytiva). Finally, ImageJ software (v1.8.0; National
Institutes of Health) was used for densitometry.
Luciferase reporter assay
The plasmids were all obtained from Shanghai
Genechem Co., Ltd. TECs were transfected with 3′UTR luciferase
reporter constructs (3′UTR-NC and 3′UTR-SOCS-1), miRNA (miR-155
mimic, 5′-TTA AUG CTA ATC GTG ATA GGG GT-3′; and miR-NC, 5′-CCC CCC
CCC CCC CCC CCC CC-3′) and Renilla luciferase using
Lipofectamine 3000, according to the manufacturer′s instructions
(Invitrogen; Thermo Fisher Scientific, Inc.). Following
transfection for 6 h at 37°C, the medium was removed and fresh
medium was added. To assess the binding specificity, the sequences
in the 3′UTR-SOCS-1 (3′UTR-SOCS-1-mut) that interact with the
miR-155 seed sequence were mutated. After 48 h of transfection, the
luciferase activity of cells was detected using a Dual Luciferase
Assay kit (cat. no. E1910; Promega Corporation). Renilla
luciferase activity was normalized to Firefly luciferase
activity.
Statistical analyses
Data were obtained from at least three independent
experiments and expressed as the means ± SEM. Statistical analysis
was performed using unpaired Student's t-tests or one-way analysis
of variance (ANOVA) followed by Bonferroni correction. Statistical
analyses were performed using SPSS version 20.0 (IBM Corp.). A
2-sided P-value of <0.05 was considered to indicate a
statistically significant difference.
Results
Tubular injury and macrophage
infiltration are observed in I/R-induced AKI
In the present study, markedly elevated levels of
SCr and BUN were observed in the I/R-treated mice (Fig. 1A). Histologically, TEC injury
(necrosis, detachment, cellular debris, and cast formation) and
increased inflammatory cell infiltration were observed in the
I/R-treated kidneys (Fig. 1B). In
addition, KIM-1, a marker of tubular injury, was detected to reveal
tubular injury (Fig. 1C and D).
Concomitantly, there were significant increases in the mRNA
expression of renal inflammatory cytokines [monocyte
chemoattractant protein-1 (MCP-1), tumor necrosis factor-α (TNF-α),
and interleukin-1β (IL-1β)] (Fig.
1E). Notably, the number of F4/80+ macrophages was
also revealed to be significantly increased (Fig. 1F). Thus, these results suggested
that macrophage infiltration was associated with tubular injury in
I/R-induced AKI.
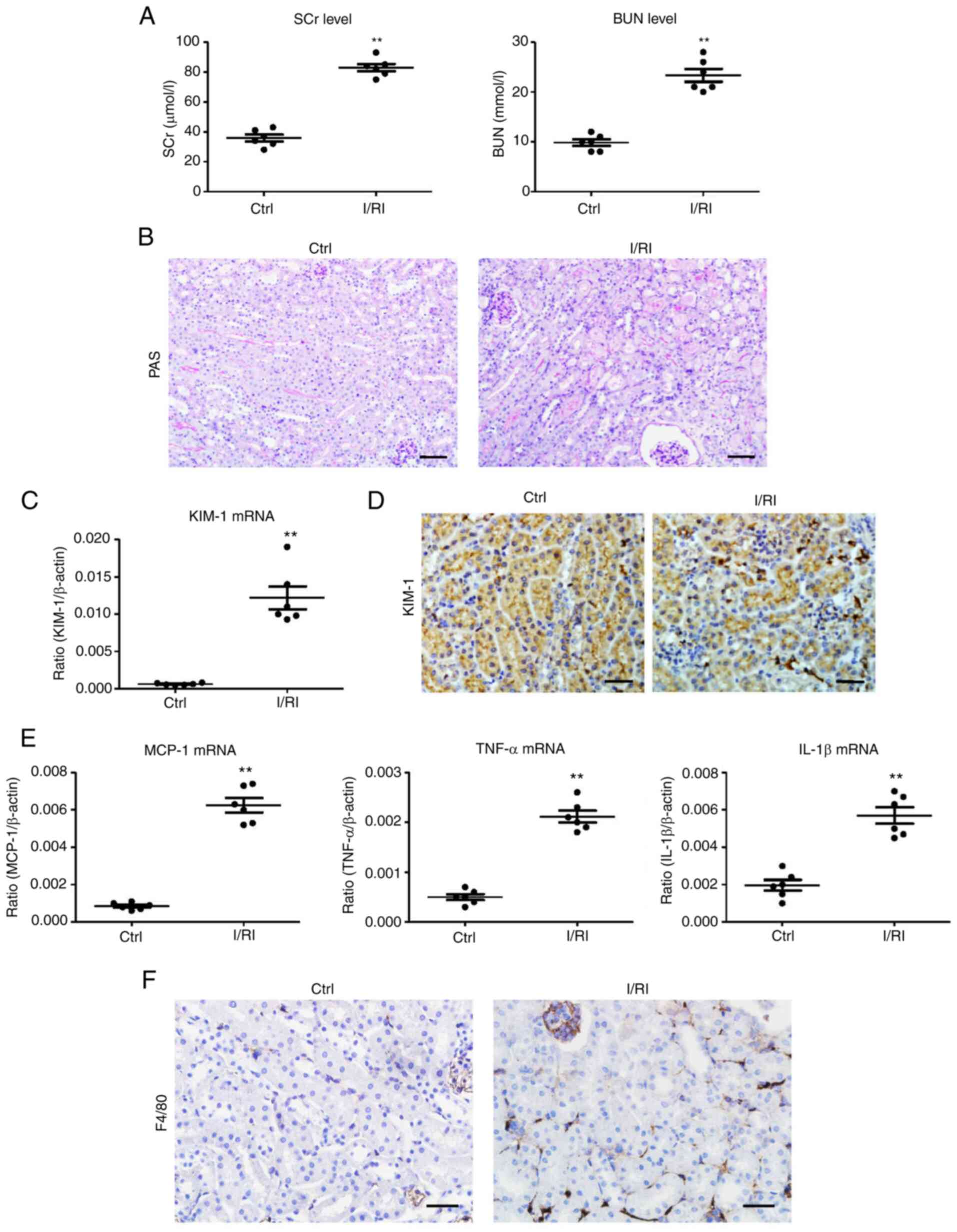 | Figure 1Tubule injury and macrophage
infiltration are observed during I/RI of the kidney. (A) The levels
of SCr and BUN in I/R-induced renal injury. (B) Histological
changes (PAS staining; scale bar, 100 µm). (C) RT-qPCR
analysis of the mRNA level of KIM-1 in kidney tissues. (D)
Representative images of KIM-1 expression in kidney tissues from
I/R-injured mice assessed by immunohistochemistry (scale bar, 50
µm). (E) Representative images of F4/80 expression in kidney
tissues from I/R-injured mice assessed by immunohistochemistry
(scale bar, 50 µm). (F) RT-qPCR analysis of inflammatory
cytokines, MCP-1, TNF-α, and IL-1β mRNA levels in kidney tissues
(n=6). Data are presented as the means ± SEM.
**P<0.01 vs. the Ctrl group. I/RI,
ischemia/reperfusion injury; SCr, serum creatinine; BUN, blood urea
nitrogen; I/R, ischemia/reperfusion; PAS, periodic acid-Schiff;
RT-qPCR, reverse transcription quantitative-PCR; KIM-1, kidney
injury molecule-1; MCP-1, monocyte chemoattractant protein-1;
TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; Ctrl,
control. |
Blocking exosome production ameliorates
tubular injury in I/R-induced AKI
In this experiment, GW4869 was used to block exosome
production (23). Notably, it was
determined that the levels of SCr and BUN were markedly decreased
in GW4869-treated mice (Fig. 2A).
Histologically, ameliorated TEC injury was observed in
GW4869-treated kidneys (Fig. 2B).
In addition, the results of PCR and immunohistochemical analysis of
KIM-1 also confirmed similar results (Fig. 2C and D). Concomitantly, the renal
mRNA expression of inflammatory factors (MCP-1, TNF-α, and IL-1β)
was significantly decreased in the GW4869-treated group compared
with the vehicle (Fig. 2E). Thus,
the findings suggested that exosome secretion is an important
mechanism of tubular injury in I/R-induced AKI.
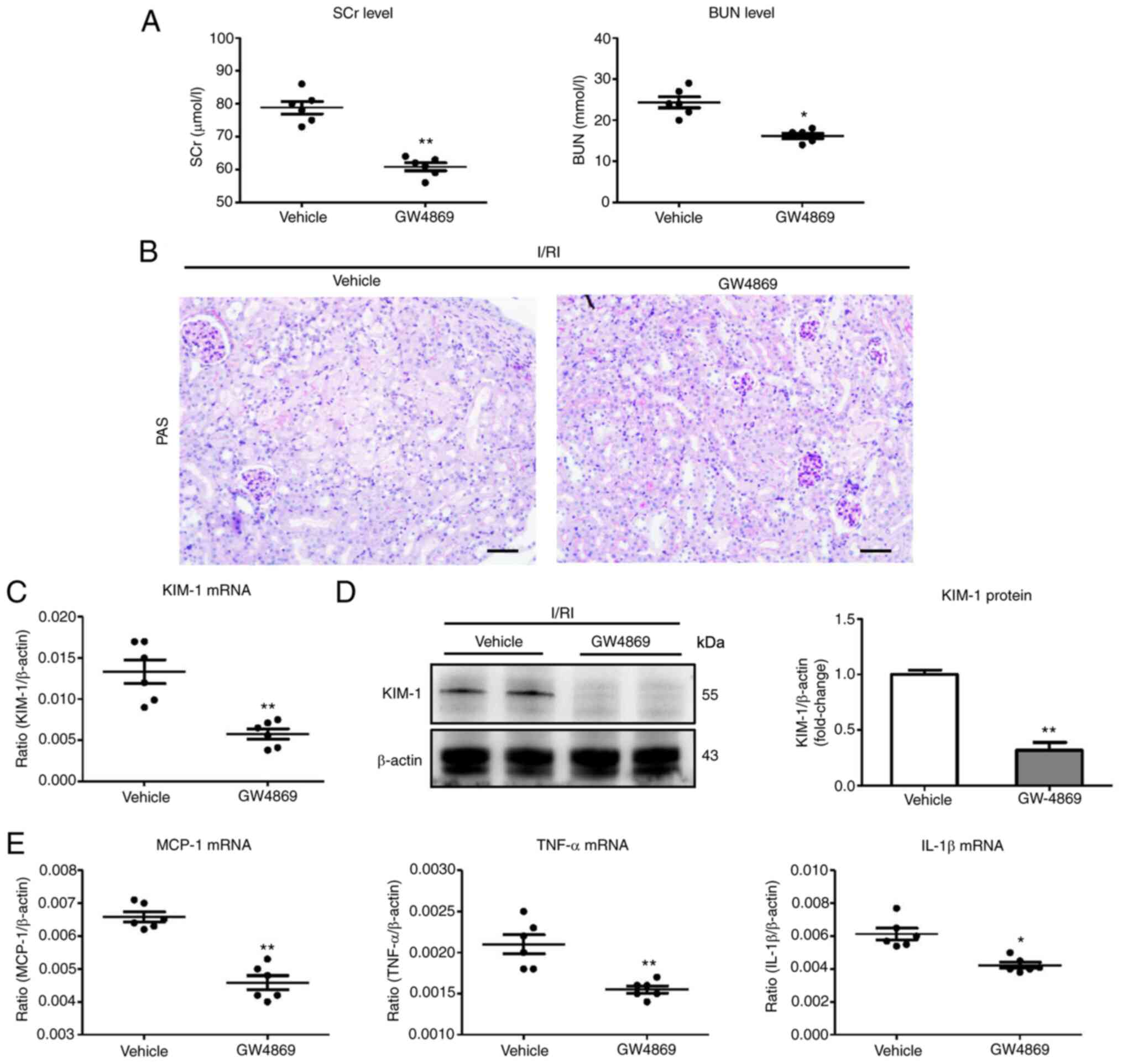 | Figure 2Blocking exosome production using
GW-4869 suppresses tubule injury in I/R-induced kidney injury. (A)
The SCr and BUN levels in I/R-treated mice with GW-4869
administration. (B) Histological changes (PAS staining; scale bar
100 µm). (C and D) RT-qPCR and western blot analysis of
KIM-1 expression in I/R-induced kidney injury with GW-4869
administration. (E) RT-qPCR analysis of inflammatory cytokine mRNA
levels in kidney tissues (n=6). Data are presented as the means ±
SEM. *P<0.05 and **P<0.01 vs. the
vehicle group. I/R, ischemia/reperfusion; SCr, serum creatinine;
BUN, blood urea nitrogen; PAS, periodic acid-Schiff; RT-qPCR,
reverse transcription quantitative-PCR; KIM-1, kidney injury
molecule-1; MCP-1, monocyte chemoattractant protein-1; TNF-α, tumor
necrosis factor-α; IL-1β, interleukin-1β; I/RI,
ischemia/reperfusion injury. |
Exosomes derived from activated
macrophages promote tubular cell injury
To explore the potential mechanism for the effect of
exosomes on tubular injury, exosomes from I/R-injured kidneys were
first isolated and characterized via western blotting (using Alix
and CD63, as exosome markers) and TEM (Fig. 3A and B). To mimic the
microenvironment in which exosomes are released from the activated
macrophages to promote tubular injury, a schematic diagram
depicting the process of the experiment is provided in Fig. 3C. As anticipated, it was
determined that exosomes released from macrophages with LPS and
IFN-γ treatment (activated macrophages, namely M1 macrophages)
transferred to TECs (Fig. 3D). To
study the effect of exosomes derived from M1 macrophages on tubular
cells, TECs with exosomes derived from M1 macrophages were cultured
under hypoxia (exo-M1). It was determined that exo-M1 promoted
tubular injury compared with the exosomes derived from
non-activated macrophages (exo-M0) (Fig. 3E and F). Concomitantly, the mRNA
expression of the inflammatory factors was significantly increased
in the exo-M1-treated group (Fig.
3G). These findings indicated that exosomes secreted from
activated macrophages promote tubular injury.
Exosomal miR-155 level is associated with
tubular injury
For this experiment, the role of exosomal miR-155 in
ischemia-induced kidney injury was explored. Interestingly, the
kidneys with I/R injury secreted exosomes that were highly enriched
in miR-155 (Fig. 4A). M1
macrophage exosomes were then isolated for miR-155 detection. As
expected, exosomes derived from M1 macrophages were highly enriched
in miR-155 (Fig. 4B). To examine
the effects of exosomal miRNA-155, miR-155 was silenced in M1
macrophages, and exosomes derived from M1 macrophages were
isolated. The miR-155-silenced exosomes (Exo-miR-155 inhibitor)
were then used to treat TECs. Notably, compared with the Exo-miR-NC
group, Exo-miR-155 inhibitor administration significantly
ameliorated tubular injury (Fig. 4C
and D). In addition, the mRNA expression of inflammatory
factors (MCP-1, TNF-α, and IL-1β) was decreased in the Exo-miR-155
inhibitor-treated group (Fig.
4E). Therefore, the level of miR-155 in exosomes parallels
tubular injury, suggesting that the secretion of miR-155-laden
exosomes by M1 macrophages may be associated with the pathological
mechanism of tubular injury.
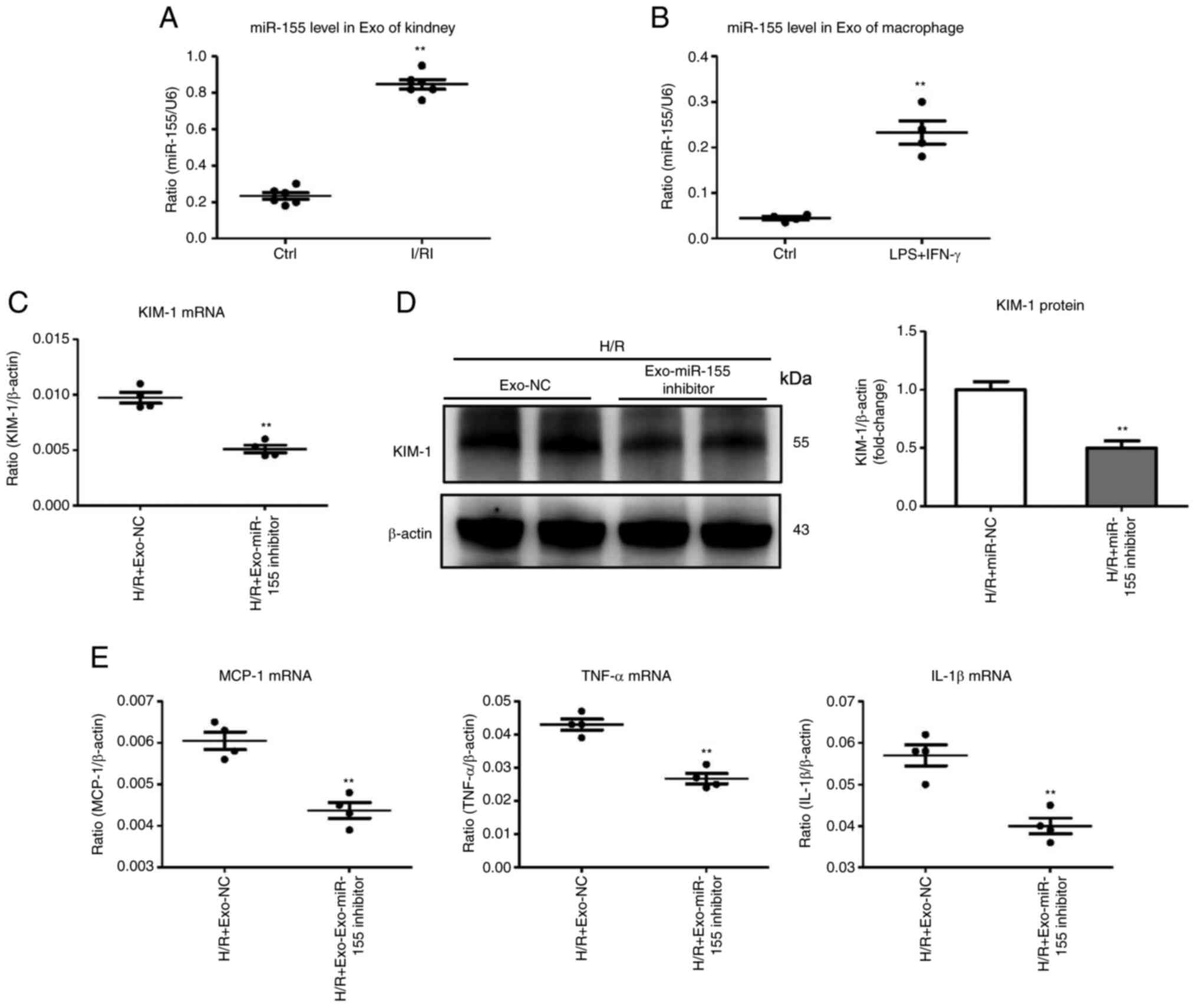 | Figure 4Exosomal miR-155 is associated with
TEC injury. (A) miR-155 expression in exosomes derived from the
I/R-injured kidneys was assessed by RT-qPCR. Data are expressed as
the mean ± SEM for groups of 6 mice. (B) miR-155 in exosomes
derived from the activated macrophages was detected by RT-qPCR. (C
and D) mRNA and protein expression of KIM-1 in TECs after treatment
with Exo-miR-155-inhibitor/Exo-NC (the exosomes derived from
activated macrophages transfected with miR-155 inhibitor/miR-NC).
(E) RT-qPCR analysis of inflammatory cytokine mRNA levels in TECs
treated with Exo-miR-155-inhibitor/Exo-NC. **P<0.01
vs. the H/R + Exo-NC. miR-155, microRNA-155; TEC, tubular
epithelial cell; I/R, ischemia/reperfusion; RT-qPCR, reverse
transcription quantitative-PCR; KIM-1, kidney injury molecule-1;
Exo, exosome; NC, negative control; H/R, hypoxia/reoxygenation;
LPS, lipopolysaccharides; IFN-γ, interferon-γ; MCP-1, monocyte
chemoattractant protein-1; TNF-α, tumor necrosis factor-α; IL-1β,
interleukin-1β. |
miR-155 inhibition alleviates tubular
injury in I/R-induced kidney injury
To determine the effect of miR-155 on I/R-induced
kidney injury, miR-155 inhibitor or scrambled NC was administered
before I/R injury to the kidney. Interestingly, compared with the
scrambled NC-treated mice, the miR-155 inhibitor efficiently
reversed the upregulation of SCr and BUN levels (Fig. 5A). Notably, miR-155 inhibitor
could ameliorate TEC injury and significantly reduce protein casts
(Fig. 5B). The results of PCR and
immunohistochemical analysis of KIM-1 also confirmed similar
results (Fig. 5C and D). In
addition, the mRNA expression of inflammatory factors was similarly
attenuated in the I/RI + miR-155 inhibitor group (Fig. 5E). Thus, these data indicated that
miR-155 inhibition could attenuate I/R-induced tubular injury.
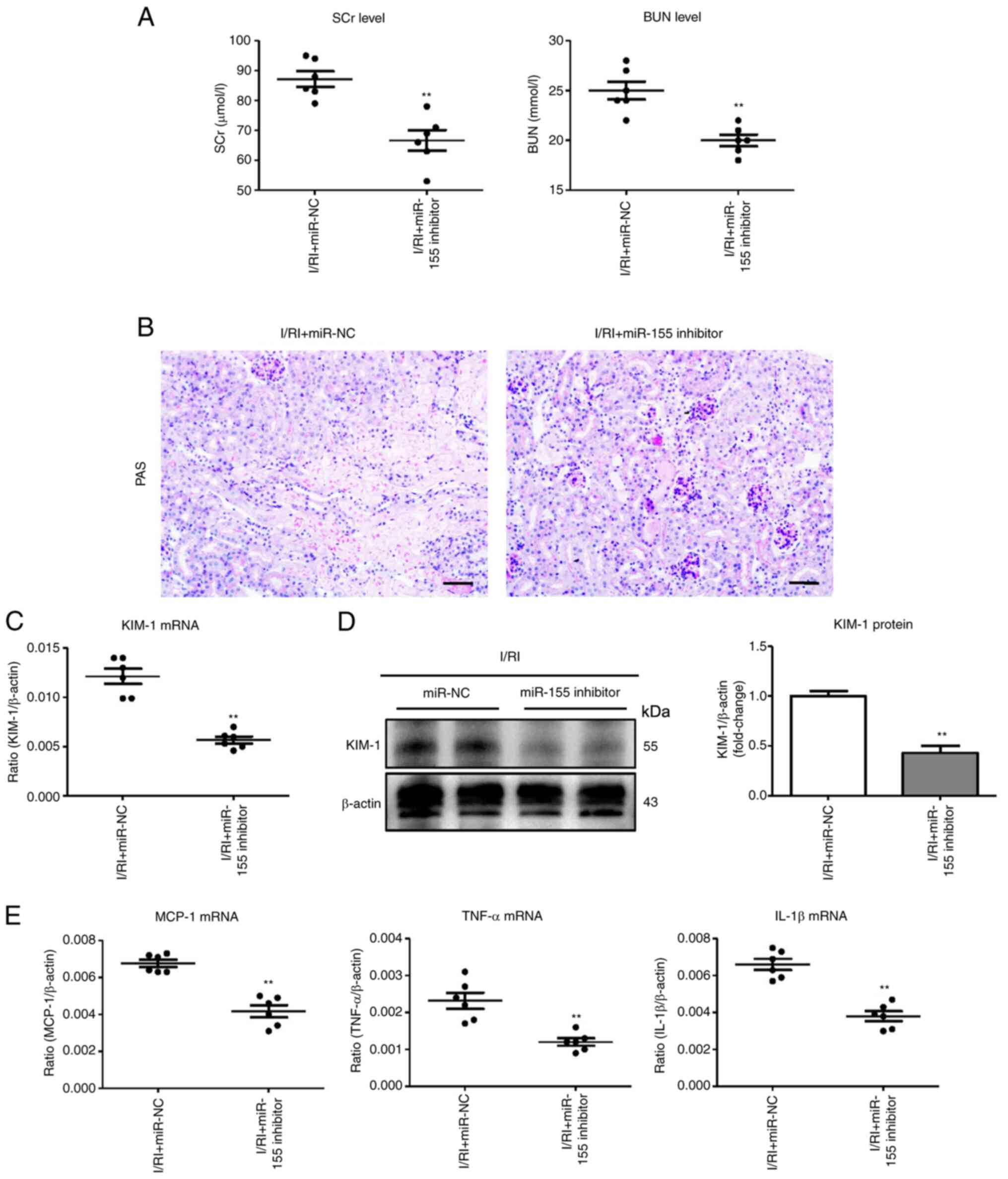 | Figure 5miR-155 inhibition ameliorates tubule
injury in I/R-induced kidney injury. (A) The SCr and BUN levels in
I/R-injured mice following miR-155 inhibitor administration. (B)
Histological changes (PAS staining; scale bar, 100 µm). (C
and D) RT-qPCR and western blot analysis of KIM-1 expression in
I/R-treated kidneys with miR-155 inhibitor administration. (E) mRNA
expression of inflammatory cytokines in I/R-treated kidneys with
miR-155 inhibitor administration. n=6. Data are presented as the
means ± SEM. **P<0.01 vs. the I/RI + miR-NC group.
miR-155, microRNA-155; I/R, ischemia/reperfusion; SCr, serum
creatinine; BUN, blood urea nitrogen; PAS, periodic acid-Schiff;
RT-qPCR, reverse transcription quantitative-PCR; KIM-1, kidney
injury molecule-1; I/RI, ischemia/reperfusion injury; NC, negative
control; MCP-1, monocyte chemoattractant protein-1; TNF-α, tumor
necrosis factor-α; IL-1β, interleukin-1β. |
Exosomal miR-155 promotes tubular injury
by targeting SOCS-1
To further investigate the exact molecular mechanism
of exosomal miR-155 on tubular cells, possible miR-155 targets that
contribute to tubular injury were predicted using 5 online
databases: TargetScan (Human 8.0/Mouse 8.0), miRDB, miRanda,
DIANA-TarBase (v7.0), and PicTar. Overlap analysis revealed that
the possible target of miR-155 was SOCS-1 (Fig. 6A), a negative regulatory factor of
NF-κB. It was determined that exosomes released from M1 macrophages
decreased the levels of SOCS-1 protein in tubule cells (Fig. 6B and C). In addition, the in
vitro study revealed that the miR-155-silenced exosomes
(Exo-miR-155 inhibitor) were used to treat TECs. As expected,
SOCS-1 expression was increased in the Exo-miR-155 inhibitor group
(Fig. 6D and E), indicating that
SOCS-1 was modulated by exosome-containing miR-155. To examine
whether miR-155 could regulate the expression of SOCS-1 in
I/R-induced kidney injury, as revealed in Fig. 6F, miR-155 inhibitor significantly
increased SOCS-1 protein expression. Notably, luciferase reporter
assay (Fig. 6G) showed that the
activity of luciferase reporters was markedly reduced by miR-155
overexpression as compared with the control. Furthermore, the
activity of SOCS-1-3′-UTR-mut luciferase reporter was not affected
by the miR-155 overexpressed vector compared with the control,
demonstrating that miR-155 could directly interact with the 3′-UTR
of SOCS-1. These results indicated that increased miR-155 in
macrophage-derived exosomes contributed to tubular injury via
targeting SOCS-1.
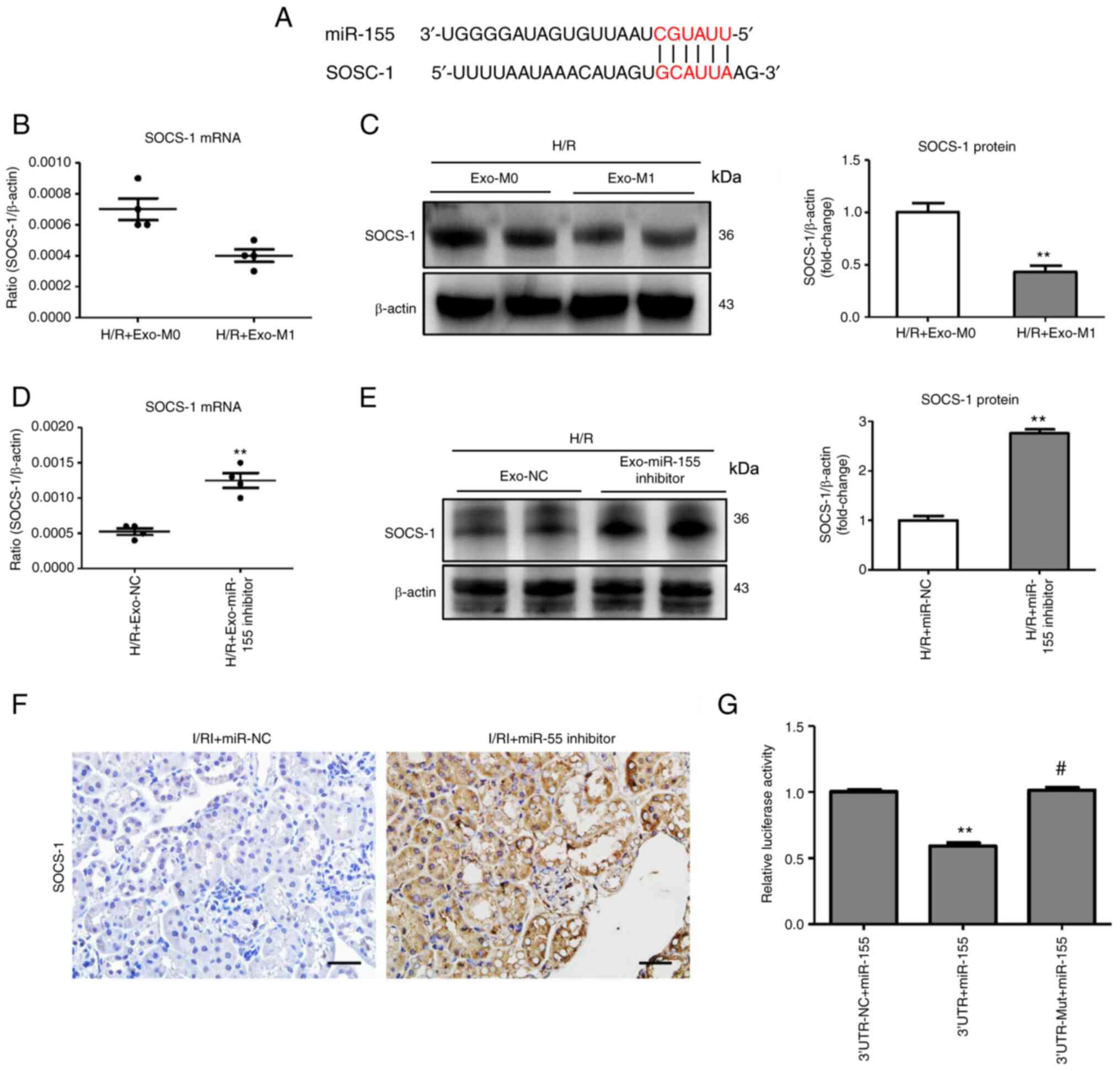 | Figure 6Exosomal miR-155 promotes tubule
injury by targeting SOCS-1. (A) Schematic diagram depicting the
predicted mmu-miR-155 targets using TargetScan, release 7.1
(http://www.targetscan.org/mmu_71/).
(B and C) RT-qPCR and western blot analysis of SOCS-1 expression in
H/R-treated TECs treated with exo-M1. Data are presented as the
means ± SEM. **P<0.01 vs. H/R + Exo-M0. (D and E)
mRNA and protein expression of SOCS-1 in TECs after treatment with
Exo-miR-155-inhibitor/Exo-NC. Data are presented as the means ±
SEM. **P<0.01 vs. the H/R + Exo-NC group. (F)
Representative images of SOCS-1 expression in I/R-treated kidney
with miR-155 inhibitor administration (scale bar, 50 µm).
(G) A luciferase reporter assay was performed using constructs with
SOCS-1 3′-UTR or SOCS-1 3′-UTR-mut. TECs were co-transfected with
these constructs along with the miR-155 overexpressed plasmid.
**P<0.01 vs. the 3′-UTR-NC + miR-155 group;
#P<0.01 vs. the 3′-UTR + miR-155 group. miR-155,
microRNA-155; SOCS-1, suppressor of cytokine signaling-1; RT-qPCR,
reverse transcription quantitative-PCR; H/R, hypoxia/reoxygenation;
Exo, exosomes; TECs, tubular epithelial cells; NC, negative
control; UTR, untranslated region; mut, mutated; I/RI,
ischemia/reperfusion injury. |
Exosomes derived from M1 macrophages
promote tubular injury in mice
To determine the role of exosomes derived from M1
macrophages in vivo, exosomes derived from activated
macrophages (exo-M1) were transferred into the kidneys by direct
renal parenchyma injection. As anticipated, it was determined that
exo-M1 treatment promoted the damage of kidney function (Fig. 7A). Histologically, increased TEC
injury was observed in the exo-M1-treated kidneys (Fig. 7B). In addition, the results of PCR
and immunohistochemical analysis for KIM-1 also confirmed the
effect of tubular injury promotion (Fig. 7C and D). Furthermore, the renal
mRNA expression of inflammatory factors (MCP-1, TNF-α, and IL-1β)
was similarly elevated in the exo-M1-treated group (Fig. 7E). Additionally,
immunohistochemical analysis revealed a reduction of SOCS-1
expression in the exo-M1 recipients (Fig. 7D). Thus, the findings demonstrated
that exosomes derived from M1 macrophages contribute to tubular
injury.
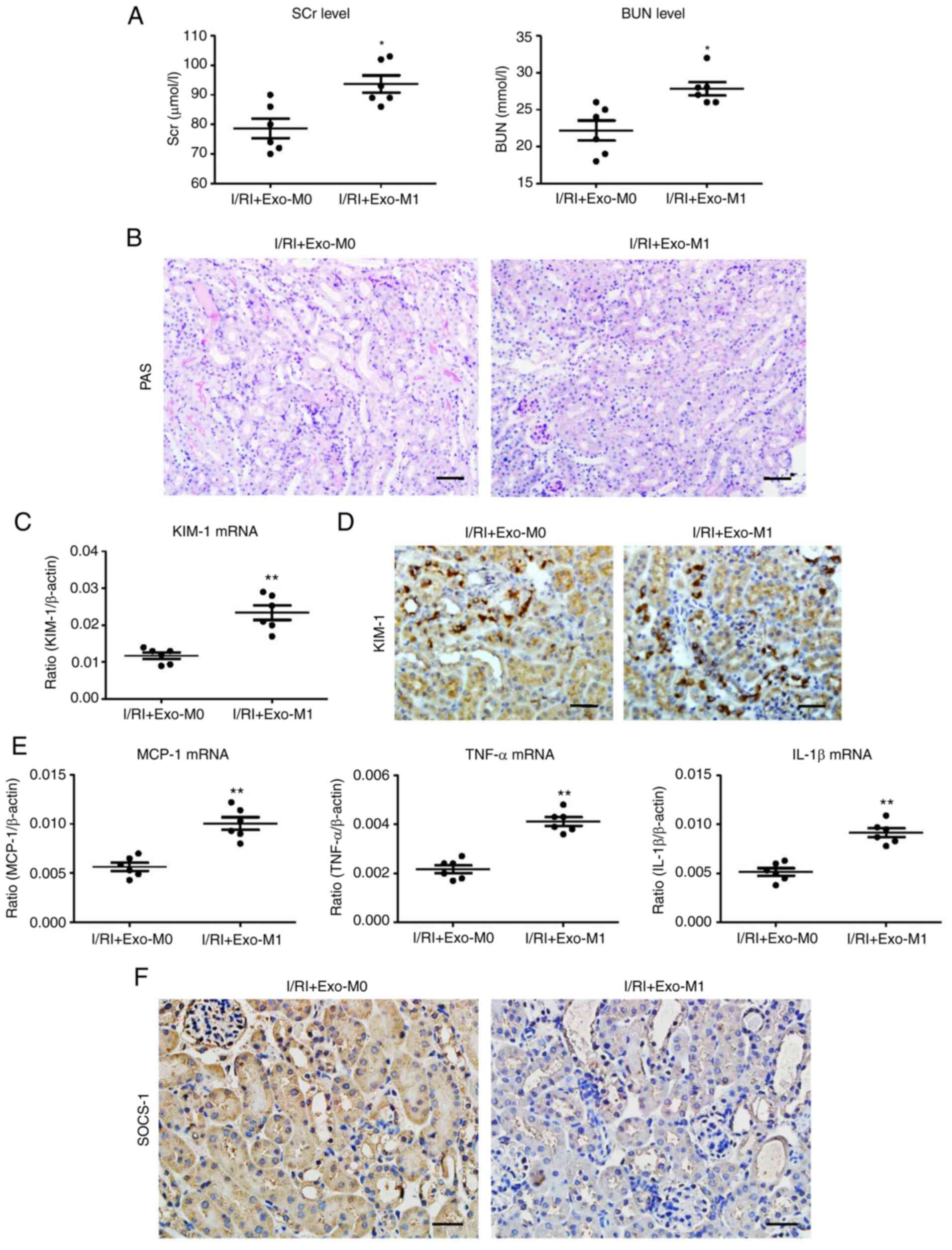 | Figure 7Exosomes secreted from M1 macrophages
promote tubule injury in I/R-induced kidney injury. (A) The SCr and
BUN levels in I/R-injured mice after exo-M1 administration. (B)
Representative PAS-stained kidney sections after exo-M1 injection
(scale bar, 100 µm). (C) RT-qPCR analysis of KIM-1
expression in I/R-treated kidney with Exo-M1 injection. (D)
Representative images of KIM-1 expression assessed by
immunohistochemistry (scale bar, 50 µm). (E) mRNA expression
of inflammatory cytokines in the exosome-injected kidney. (F)
Representative images of SOCS-1 expression in the exosome-injected
kidney, assessed by immunohistochemistry (scale bar, 50 µm).
Data are expressed as the mean ± SEM. n=6. *P<0.05
and **P<0.01 vs. I/RI + Exo-M0 (Ctrl). I/R,
ischemia/reperfusion; SCr, serum creatinine; BUN, blood urea
nitrogen; PAS, periodic acid-Schiff; RT-qPCR, reverse transcription
quantitative-PCR; KIM-1, kidney injury molecule-1; Exo, exosome;
SOCS-1, suppressor of cytokine signaling-1; I/RI,
ischemia/reperfusion injury; MCP-1, monocyte chemoattractant
protein-1; TNF-α, tumor necrosis factor-α; IL-1β,
interleukin-1β. |
Discussion
Renal tubule injury is a characteristic pathological
feature of AKI and determines the outcome of kidney disease.
However, the mechanism of TEC injury remains unclear. In the
present study, a novel mechanism through which the
macrophage-derived exosomal miR-155/SOCS-1 axis mediated renal
tubule injury was demonstrated. The findings not only provide novel
insights into the pathophysiology of AKI but also offer a new
therapeutic strategy for kidney diseases.
Renal tubules, which are packed with mitochondria
and dependent on oxidative phosphorylation, are particularly
vulnerable to a variety of injuries including obstructive,
ischemic, hypoxic, toxins, and metabolic, and determine the
prognosis of kidney diseases (24). There are several studies that
revealed that TECs could undergo changes and function as
inflammatory cells, with the consequent production of various
inflammatory molecules (MCP-1, TNF-α, and IL-1β) that drive
progression of kidney diseases (25,26). Thus, elucidating the exact
mechanism underlying tubular injury is urgent.
As a type of principal innate immune cell, the
macrophage plays a central role in the maintenance of tissue
homeostasis (27). Increasing
studies have demonstrated that macrophages exhibit high diversity
and plasticity in response to different microenvironments, thereby
exerting diverse functions. For example, classically activated
macrophages (M1) and alternatively activated macrophages (M2) could
perform a variety of biological functions via the production of
large amounts of cytokines (28).
Recently, convincing evidence revealed that macrophages also play a
critical role in innate immunity and adaptive immune response
through the secretion of exosomes (29). More interestingly, the function of
exosomes derived from macrophages is gaining increasing attention
in the course of various diseases (30,31). With regard to kidney diseases,
Huang et al (32) recently
reported that macrophage-derived exosomes improved high
glucose-induced podocyte injury by transferring specific miRNA.
However, the roles of exosomes derived from macrophages in AKI
remains poorly understood. In the present study, it was determined
that exosomes derived from M1 macrophages mediated tubule injury
during AKI. Notably, blocking exosome production ameliorated
tubular injury in I/R-induced AKI, suggesting that exosomes play a
crucial role in the pathogenesis of tubule injury. To the best of
our knowledge, this is the first time the function of exosomes
derived from macrophages during the AKI was explored. Therefore,
the present study plays a vital role in understanding
ischemia-induced kidney injury.
To further investigate the exact mechanism of
exosomes derived from M1 macrophages on renal tubule injury, the
specific cargos of exosomes was analyzed. Given that miRNAs are
commonly enriched in exosomes and the miRNA class was observed to
be the largest and most consistent proportional change in exosomes
using total RNA-sequencing in a model of I/R (12), it was hypothesized that exosomes
could regulate tubular cells by transferring cell-specific miRNA.
Notably, it was determined that it was miR-155 that was
specifically assembled into the exosomes and miR-155 inhibition
alleviated tubular injury in vitro and in vivo. In a
previous study by Ying et al, it was reported that miR-155
was loaded into macrophage exosomes (33). In addition, convincing evidence
has shown that macrophage-derived miR-155-containing exosomes
promoted cardiac hypertrophy and fibrosis by targeting FoxO3a in
uremic mice (34). Therefore,
these results indicated that exosomes derived from M1 macrophages
are implicated in tubule injury via transferring of miR-155.
Finally, the exact mechanism through which
macrophage-derived exosomal miR-155 regulated tubule injury was
further investigated. Notably, it was determined that exosomal
miR-155 was implicated in the pathogenesis of tubule injury by
suppression of SOCS-1, a dual guanine nucleotide exchange factor
that plays an important role in regulating cellular hemostasis
(35). Silencing of miR-155
markedly increased the levels of SOCS-1 expression in vitro
and in vivo and resulted in reduced tubule injury. SOCS-1
has previously been demonstrated as an inflammation suppressor that
normally functions as a negative regulator of NF-κB signaling
pathways (36), and which
aggravates tubular injury during ischemic AKI (37). More interestingly, a previous
study indicated that SOSC-1 is a target of miR-155 involved in the
regulation of immune response (38). Thus, the findings of the present
study, demonstrated that SOCS-1, a target of miR-155, played a new
molecular role in regulating the function of tubular cells.
In summary, it was demonstrated that exosomal
miR-155 mediated the cross-talk between tubules and macrophages and
contributed to tubule injury in ischemia-induced AKI. The
exosome/miR-155/SOCS-1 axis played a critical role in renal tubule
injury. The findings suggested that macrophage-derived exosomal
miR-155 provides a novel understanding of the molecular mechanisms
of renal tubule injury and represents a new therapeutic target for
the treatment of AKI.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
ZZ, HC, LZ, GL, and LW designed the study, carried
out experiments, analyzed the data, and wrote the paper. CL carried
out the experiments and analyzed the data. ZZ, GL, and LW prepared
the figures and edited the manuscript. ZZ and HC confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
The experimental animal procedures were approved
(approval no. N2017-078) by the Ethics Committee of Soochow
University (Suzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was funded by the Project of Precision
Medicine of Wuxi Municipal Health Commission (grant no. J202004),
the Wuxi Medical Leadership Talent and Innovation Team (grant no.
CXTDJS001), the Wuxi Medical Innovation Team Project (grant no.
CXTD2021010) and the Scientific Research Project of Wuxi Health
Committee (grant no. Z201914).
References
|
1
|
Ronco C, Bellomo R and Kellum JA: Acute
kidney injury. Lancet. 394:1949–1964. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Takaori K, Nakamura J, Yamamoto S, Nakata
H, Sato Y, Takase M, Nameta M, Yamamoto T, Economides AN, Kohno K,
et al: Severity and frequency of proximal tubule injury determines
renal prognosis. J Am Soc Nephrol. 27:2393–2406. 2016. View Article : Google Scholar :
|
|
3
|
Cao JY, Wang B, Tang TT, Wen Y, Li ZL,
Feng ST, Wu M, Liu D, Yin D, Ma KL, et al: Exosomal miR-125b-5p
deriving from mesenchymal stem cells promotes tubular repair by
suppression of p53 in ischemic acute kidney injury. Theranostics.
11:5248–5266. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tang PM, Nikolic-Paterson DJ and Lan HY:
Macrophages: Versatile players in renal inflammation and fibrosis.
Nat Rev Nephrol. 15:144–158. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huen SC and Cantley LG: Macrophages in
renal injury and repair. Annu Rev Physiol. 79:449–469. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mathieu M, Martin-Jaular L, Lavieu G and
Théry C: Specificities of secretion and uptake of exosomes and
other extracellular vesicles for cell-to-cell communication. Nat
Cell Biol. 21:9–17. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ding H, Li LX, Harris PC, Yang J and Li X:
Extracellular vesicles and exosomes generated from cystic renal
epithelial cells promote cyst growth in autosomal dominant
polycystic kidney disease. Nat Commun. 12:45482021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mori MA, Ludwig RG, Garcia-Martin R,
Brandão BB and Kahn CR: Extracellular miRNAs: From biomarkers to
mediators of physiology and disease. Cell Metab. 30:656–673. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van Niel G, D'Angelo G and Raposo G:
Shedding light on the cell biology of extracellular vesicles. Nat
Rev Mol Cell Biol. 19:213–228. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu X, Miao J, Wang C, Zhou S, Chen S, Ren
Q, Hong X, Wang Y, Hou FF, Zhou L and Liu Y: Tubule-derived
exosomes play a central role in fibroblast activation and kidney
fibrosis. Kidney Int. 97:1181–1195. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lv LL, Feng Y, Wen Y, Wu WJ, Ni HF, Li ZL,
Zhou LT, Wang B, Zhang JD, Crowley SD and Liu BC: Exosomal CCL2
from tubular epithelial cells is critical for albumin-induced
tubulointerstitial inflammation. J Am Soc Nephrol. 29:919–935.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
de Couto G, Gallet R, Cambier L,
Jaghatspanyan E, Makkar N, Dawkins JF, Berman BP and Marbán E:
Exosomal MicroRNA transfer into macrophages mediates cellular
postconditioning. Circulation. 136:200–214. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li ZL, Lv LL, Tang TT, Wang B, Feng Y,
Zhou LT, Cao JY, Tang RN, Wu M, Liu H, et al: HIF-1α inducing
exosomal microRNA-23a expression mediates the cross-talk between
tubular epithelial cells and macrophages in tubulointerstitial
inflammation. Kidney Int. 95:388–404. 2019. View Article : Google Scholar
|
|
14
|
Yu W, Zeng H, Chen J, Fu S, Huang Q, Xu Y,
Xu A, Lan HY and Tang Y: miR-20a-5p is enriched in hypoxia-derived
tubular exosomes and protects against acute tubular injury. Clin
Sci (Lond). 134:2223–2234. 2020. View Article : Google Scholar
|
|
15
|
Wang C, Zhang C, Liu L, A X, Chen B, Li Y
and Du J: Macrophage-derived mir-155-containing exosomes suppress
fibroblast proliferation and promote fibroblast inflammation during
cardiac injury. Mol Ther. 25:192–204. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ge X, Tang P, Rong Y, Jiang D, Lu X, Ji C,
Wang J, Huang C, Duan A, Liu Y, et al: Exosomal miR-155 from
M1-polarized macrophages promotes EndoMT and impairs mitochondrial
function via activating NF-κB signaling pathway in vascular
endothelial cells after traumatic spinal cord injury. Redox Biol.
41:1019322021. View Article : Google Scholar
|
|
17
|
Wei Q, Liu Y, Liu P, Hao J, Liang M, Mi
QS, Chen JK and Dong Z: MicroRNA-489 induction by hypoxia-inducible
factor-1 protects against ischemic kidney injury. J Am Soc Nephrol.
27:2784–2796. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gusella GL, Fedorova E, Hanss B, Marras D,
Klotman ME and Klotman PE: Lentiviral gene transduction of kidney.
Hum Gene Ther. 13:407–414. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weidemann A, Bernhardt WM, Klanke B,
Daniel C, Buchholz B, Câmpean V, Amann K, Warnecke C, Wiesener MS,
Eckardt KU and Willam C: HIF activation protects from acute kidney
injury. J Am Soc Nephrol. 19:486–494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li ZL, Wang B, Lv LL, Tang TT, Wen Y, Cao
JY, Zhu XX, Feng ST, Crowley SD and Liu BC: FIH-1-modulated HIF-1α
C-TAD promotes acute kidney injury to chronic kidney disease
progression via regulating KLF5 signaling. Acta Pharmacol Sin.
42:2106–2119. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lv LL, Feng Y, Wu M, Wang B, Li ZL, Zhong
X, Wu WJ, Chen J, Ni HF, Tang TT, et al: Exosomal miRNA-19b-3p of
tubular epithelial cells promotes M1 macrophage activation in
kidney injury. Cell Death Differ. 27:210–226. 2020. View Article : Google Scholar :
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Catalano M and O'Driscoll L: Inhibiting
extracellular vesicles formation and release: A review of EV
inhibitors. J Extracell Vesicles. 9:17032442019. View Article : Google Scholar
|
|
24
|
Chevalier RL: The proximal tubule is the
primary target of injury and progression of kidney disease: Role of
the glomerulotubular junction. Am J Physiol Renal Physiol.
311:F145–F161. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu BC, Tang TT, Lv LL and Lan HY: Renal
tubule injury: A driving force toward chronic kidney disease.
Kidney Int. 93:568–579. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baek JH, Zeng R, Weinmann-Menke J,
Valerius MT, Wada Y, Ajay AK, Colonna M and Kelley VR: IL-34
mediates acute kidney injury and worsens subsequent chronic kidney
disease. J Clin Invest. 125:3198–3214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wen Y and Crowley SD: The varying roles of
macrophages in kidney injury and repair. Curr Opin Nephrol
Hypertens. 29:286–292. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Meng XM, Tang PM, Li J and Lan HY:
Macrophage phenotype in kidney injury and repair. Kidney Dis
(Basel). 1:138–146. 2015. View Article : Google Scholar
|
|
29
|
Wang Y, Zhao M, Liu S, Guo J, Lu Y, Cheng
J and Liu J: Macrophage-derived extracellular vesicles: Diverse
mediators of pathology and therapeutics in multiple diseases. Cell
Death Dis. 11:9242020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu R, Gao W, Yao K and Ge J: Roles of
exosomes derived from immune cells in cardiovascular diseases.
Front Immunol. 10:6482019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu J, Wu F and Zhou H: Macrophage-derived
exosomes in cancers: Biogenesis, functions and therapeutic
applications. Immunol Lett. 227:102–108. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang H, Liu H, Tang J, Xu W, Gan H, Fan Q
and Zhang W: M2 macrophage-derived exosomal miR-25-3p improves high
glucose-induced podocytes injury through activation autophagy via
inhibiting DUSP1 expression. IUBMB Life. 72:2651–2662. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ying W, Riopel M, Bandyopadhyay G, Dong Y,
Birmingham A, Seo JB, Ofrecio JM, Wollam J, Hernandez-Carretero A,
Fu W, et al: Adipose tissue macrophage-derived exosomal miRNAs can
modulate in vivo and in vitro insulin sensitivity. Cell.
171:372–384.e12. 2017. View Article : Google Scholar
|
|
34
|
Wang B, Wang ZM, Ji JL, Gan W, Zhang A,
Shi HJ, Wang H, Lv L, Li Z, Tang T, et al: Macrophage-derived
exosomal Mir-155 regulating cardiomyocyte pyroptosis and
hypertrophy in uremic cardiomyopathy. JACC Basic Transl Sci.
5:148–166. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yoshimura A, Naka T and Kubo M: SOCS
proteins, cytokine signalling and immune regulation. Nat Rev
Immunol. 7:454–465. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Park SH, Kim KE, Hwang HY and Kim TY:
Regulatory effect of SOCS on NF-kappaB activity in murine
monocytes/macrophages. DNA Cell Biol. 22:131–139. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Markó L, Vigolo E, Hinze C, Park JK, Roël
G, Balogh A, Choi M, Wübken A, Cording J, Blasig IE, et al: Tubular
epithelial NF-κB activity regulates ischemic AKI. J Am Soc Nephrol.
27:2658–2669. 2016. View Article : Google Scholar
|
|
38
|
Heymans S, Corsten MF, Verhesen W, Carai
P, van Leeuwen RE, Custers K, Peters T, Hazebroek M, Stöger L,
Wijnands E, et al: Macrophage microRNA-155 promotes cardiac
hypertrophy and failure. Circulation. 128:1420–1432. 2013.
View Article : Google Scholar : PubMed/NCBI
|